

Abstract
Lung cancer is the leading cause of cancer mortality worldwide and KRAS is the most commonly mutated gene in lung adenocarcinoma (LUAD). The 78-kDa glucose-regulated protein GRP78/BiP is a key endoplasmic reticulum (ER) chaperone protein and a major pro-survival effector of the unfolded protein response (UPR). Analysis of the Cancer Genome Altas (TCGA) database and immunostain of patient tissues revealed that compared to normal lung, GRP78 expression is generally elevated in human lung cancers, including tumors bearing the KRASG12D mutation. To test the requirement of GRP78 in human lung oncogenesis, we generated mouse models containing floxed Grp78 and Kras Lox-Stop-Lox G12D (KrasLSL-G12D) alleles. Simultaneous activation of the KrasG12D allele and knockout of the Grp78 alleles were achieved in the whole lung or selectively in lung alveolar epithelial type 2 cells known to be precursors for adenomas which progress to LUAD. Here we report that GRP78 haploinsufficiency is sufficient to suppress KrasG12D-mediated lung tumor progression and prolong survival. Furthermore, GRP78 knockdown in human lung cancer cell line A427 (KrasG12D/+) leads to activation of UPR and apoptotic markers and loss of cell viability. Our studies provide evidence that targeting GRP78 represents a novel therapeutic approach to suppress mutant KRAS-mediated lung tumorigenesis.
Introduction
Lung cancer is the leading cause of cancer mortality worldwide with limited therapeutic options (1). Non-small cell lung cancer (NSCLC) accounts for the majority (~85%) of all lung cancers and lung adenocarcinoma (LUAD) is the most common type of lung cancer in the United States. Activating mutations of the KRAS oncogene are found in one-quarter to one-half of human LUAD cases, resulting in constitutive activation of KRAS signaling (2, 3). KRAS4B is the predominant splice variant of KRAS, and hereafter will be referred to as KRAS (4). KRAS is a membrane-associated GTPase signaling protein that promotes proliferation and cell survival. Newly synthesized KRAS is cytosolic and inactive, and it undergoes a series of post-translational modifications at the cytosolic surface of the ER mediated by enzymes that are transmembrane ER proteins (5, 6). Thus, the ER is a major site for the maturation of KRAS and perturbation of ER homeostasis and dysregulated protein quality control could be detrimental to KRAS-driven LUAD.
The 78-kDa glucose regulated protein (GRP78), also referred to as BiP and encoded by the HSPA5 gene, is an essential ER chaperone and a master regulator of ER functions (7, 8, 9). GRP78 exerts critical quality control of proteins processed in the ER impacting a wide range of human diseases including cancer (7, 10, 11). While GRP78 is primarily a luminal ER protein, upon stress, subfractions of GRP78 can be localized on the cell surface and other cellular compartments (12, 13). Through direct or indirect complex formation at the ER/cytosol interface, GRP78 regulates the activation of key proteins localized to the outer surface of the ER, such as caspase-7 and BCL interacting killer (BIK) (14, 15). Cancer cells, in response to intrinsic and extrinsic stress, often mount the adaptive unfolded protein response (UPR) (7, 10, 16). GRP78 is a key regulator of the UPR through complex formation with the transmembrane ER stress sensors and is a key pro-survival component of the UPR. GRP78 exhibits potent anti-apoptotic and pro-tumorigenic properties and is commonly over-expressed in human cancers (10, 17), In contrast to an earlier report (18), multiple recent studies have shown that lung cancer patients with LUAD expressing higher GRP78 levels had considerably shorter survival rates and worse prognosis compared to those with low levels (19–21). These findings suggest a potential role of GRP78 in promoting lung tumorigenesis, although the requirement of GRP78 in KRAS-driven lung cancer is not known.
Conditional expression of oncogenic KRAS in genetically modified mouse models utilizing the floxed mouse line Kras lox-stop-lox G12D (KrasG12D) mimics human LUAD and has been used successfully to analyze lung tumor initiation and progression (2). To examine the role of GRP78 in mutant Kras-driven LUAD, we crossed the KrasG12D mice with floxed Grp78 (Grp78f/f) mice (22). Simultaneous activation of the KrasG12D allele and knockout of the Grp78 alleles were achieved in the whole lung through intratracheal injection of adenovirus-Cre (adeno-Cre), or selectively in lung alveolar epithelial type 2 (AT2) cells known to be precursors for lung adenomas which progress to LUAD (23), by breeding with an inducible human surfactant protein C (SPC)-Cre mouse model (24). Here we establish that GRP78 haploinsufficiency in these mouse models is sufficient to suppress KrasG12D-driven LUAD and that knockdown of GRP78 in a human lung cancer cell line A427 (KrasG12D/+) leads to activation of UPR and apoptotic markers and loss of cell viability.
Results
Suppression of Mutant Kras-driven Pulmonary Tumorigenesis by GRP78 Insufficiency.
Analysis of the Cancer Genome Altas (TCGA) database showed that GRP78 mRNA expression in human LUAD is significantly higher than in normal lungs (Fig. S1A). Further analysis of the LUAD tissues by mutation status of both KRAS and another commonly altered in LUAD, epidermal growth factor receptor (EGFR), revealed increased GRP78 mRNA expression in LUAD tissues regardless of mutant or wild type status of KRAS or EGFR, in line with general upregulation of GRP78 in human tumors (Fig. S1B). Activating mutations in KRAS codon 12 are common in human LUAD (3) and all KRAS mutations in the TCGA analysis including the G12 type had increased GRP78 mRNA expression over normal lung tissues (Fig. S1B). Furthermore, immunhistochemical (IHC) staining of patient tissues showed that compared to normal pulmonary alveoli and admixed non-neoplastic stromal cells, brown cytosolic staining of GRP78 is much more abundant in lung carcinomas bearing the KRASG12D mutation, as well as KRAS WT tumors (Fig. S1C).
In mice, the KrasG12D mutation induces spontaneous LUAD (2). To investigate the requirement of GRP78 in LUAD development, we conditionally depleted GRP78 in the lungs of KrasG12D/+ mice containing either two floxed alleles of Grp78 (Grp78f/f) for homozygous deletion, or one floxed and one wild type allele (Grp78 f/+) for heterozygous deletion, with KrasG12D/+ mice containing two wild type alleles (Grp78+/+) serving as control. These mice, referred to as K78f/f (KrasG12D;Grp78f/f), K78f/+ (KrasG12D;Grp78f/+), K78+/+ (KrasG12D;Grp78+/+) respectively, were generated as described in the breeding scheme (Fig. S2A). For the first mouse model, referred to as the adeno-Cre mice, the mice were subjected to intratracheal adeno-Cre administration to simultaneously activate the mutant Kras allele and delete the Grp78 floxed allele (Fig. 1A). Polymerase chain reaction (PCR) of mouse tail and lung DNA confirmed the genotypes of the mice cohorts (Fig. 1B). Based on previous reports, three time points (12, 16, and 22 wk post-Cre-activation) were chosen for analysis and the lung tissues were subjected to immunohistochemistry (IHC).
Fig. 1. Characterization of the adenovirus-Cre mouse model.
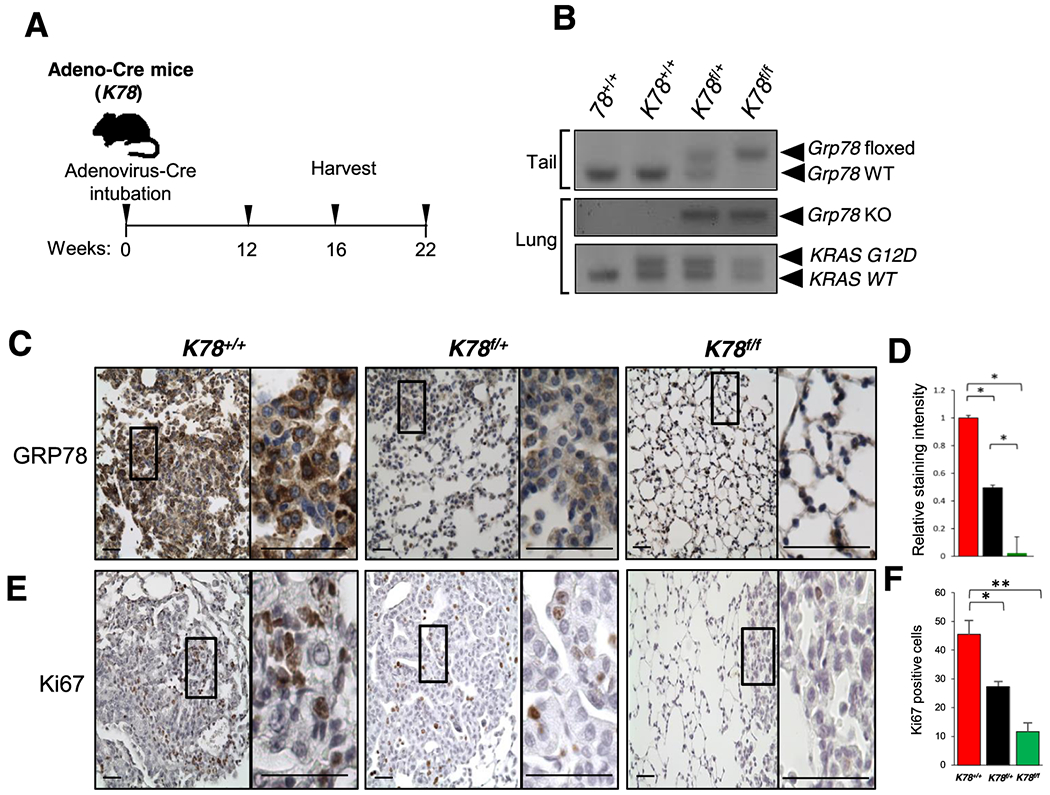
a Timeline of mice analysis. 10 wk old adult mice were intubated with adenovirus-Cre (adeno-Cre) and the lungs were harvested and processed for analysis at 12, 16 and 22 wk post adeno-Cre treatment. b Representative genotyping results of the adeno-Cre mice using DNA extracted from tail (top panel) and from lung (bottom two panels) after Cre treatment. c Immunohistochemistry (IHC) of GRP78 in lungs of the indicated genotypes 12 wk post adeno-Cre treatment by intratracheal intubation (Scale bar: 100 μm). Enlarged views of the boxed area are shown on the right (Scale bar: 100 μm). d Quantitation of GRP78 staining from (c). The values represent the average staining of the sampled area ± s.e., *p<0.05. Number of images that were randomly chosen to be quantified are the following K78+/+ (n=20), K78f/+ (n=20) and K78f/f (n=20). At least three to four mice were analyzed per genotype. e Representative immunostains for Ki67 in lungs of mice of the indicated genotypes 22 wk post adeno-Cre treatment. Magnifications same as (c). f Quantitation of Ki67 positive cells using 5 random high-power fields of lung tissues 22 wk after adeno-Cre treatment from (e). Data are presented as mean ± s.e., *p<0.05 and **p<0.01.
Consistent with upregulation of GRP78 in human LUAD, strong immunostaining for GRP78 was detected in the tumor tissues of these mouse cohorts, as compared to the surrounding lung tissues (Fig. S3). As expected with Cre-mediated excision of the floxed Grp78 allele, at all three time points, the staining intensity for GRP78 decreased by about 50% in the lungs of K78f/+ as compared to the K78+/+ mice, and was further decreased in K78f/f mice (Fig. 1C and D and Fig. S4A and B). Correspondingly, the proliferation marker Ki67 showed a 45% and 80% decrease respectively in lung tissues of K78f/+ and in K78f/f mice compared to K78+/+ mice (Fig. 1E and F).
Histological examination of tissue sections stained with hematoxylin and eosin (H&E) showed decreased lung tumor burden in the K78f/+ and even more so in K78f/f mice compared to K78+/+ mice for all three time points analyzed (Fig. 2A and B). We next compared the frequency of pulmonary lesions [atypical adenomatous hyperplasia (AAH), adenoma, and LUAD] in each genotype by histological examination. Examples of each of these lesions as well as of adjacent normal tissues are shown in Fig. 2C. Consistent with the tumor burden measurements, pulmonary lesions appeared earlier in the K78+/+ mice compared to the K78f/+ and K78f/f mice. Lung lesions in K78+/+ mice showed LUAD as early as 16 wk post-Cre-activation compared to 22 wk in K78f/+ mice while LUAD was not observed in any of the K78f/f mice analyzed. Lungs of K78f/f mice were either histologically normal or showed AAH 12 wk post-intubation. In contrast, 50% of the K78+/+ mice and 20% of the K78f/+ mice showed adenomas at 12 wk post-intubation. Only adenomas were seen in the lungs of K78f/f mice at the 22 wk time point (Fig. 2D). Weight loss was detected in K78+/+ mice starting at 18 wk, which was not observed in the K78f/+ or K78f/f mice (Fig. S2B).
Fig. 2. Comparative analysis of the lungs of K78+/+, K78f/+ and K78f/f mice following adenovirus-Cre treatment.
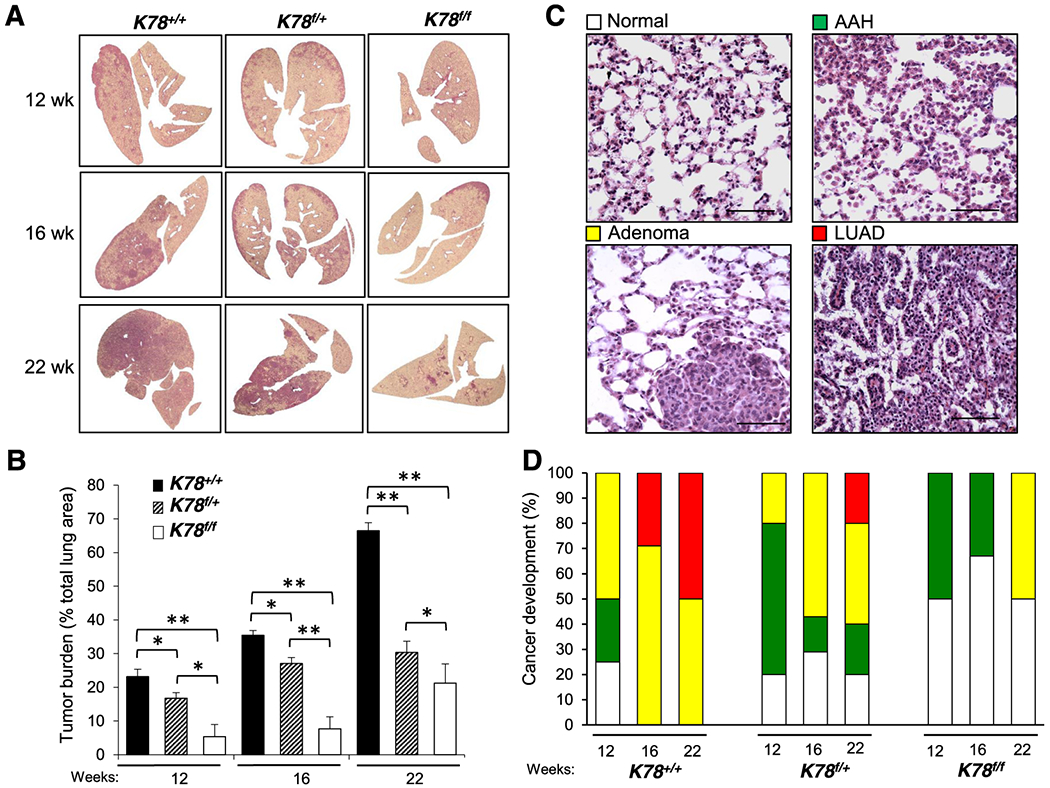
a Panels showing cross sections of whole lungs from mice of the indicated genotypes at 12, 16, or 22 wk post adenovirus-Cre injection. b Quantitation of tumor burden for the K78+/+, K78f/+, K78f/f mice at 12 wk (n=9, 8 and 9, respectively), 16 wk (n=8, 9 and 11, respectively), and 22 wk (n=9, 8 and 8, respectively). The p-values compare the two genotypes under each bracket. Data are presented as mean ± s.e., *p<0.05 and **p<0.01. c Panels show representative examples of hematoxylin and eosin (H&E) staining of lung tissues exhibiting normal morphology, atypical adenomatous hyperplasia (AAH), adenoma, or adenocarcinoma (LUAD) (Scale bar: 200 μm). d Quantitation of the histological grades of lungs of the mice of the indicated genotypes at the indicated time points. The number of mice analyzed was the same as in (b).
These histological observations were further confirmed through imaging of the mouse lungs at 12, 16, and 22 wk. Contrast-enhanced, computerized tomography (CT) segmentation was aided by 18F-fluorodeoxyglucose (FDG) positron emission tomography (PET)/CT signal to determine the progression of lung tumors in the three genoytypes. K78+/+ and K78f/+, but not K78f/f mouse, showed FDG uptake in the lung regions (Fig. 3A). Consistent with detection of LUAD in the K78+/+ mouse, PET scans revealed earlier and higher FDG uptake in the K78+/+ mouse compared to the K78f/+ mouse. Three-dimensional visualization of FDG corroborated contrast-enhanced CT and confirmed that the tumor burden was highest in the K78+/+ mouse, which was reduced in the K78f/+ mouse and hardly detectable in the K78f/f mouse at all three time points (Fig. 3B).
Fig. 3. Imaging of K78+/+, K78f/+, K78f/f mouse lungs.
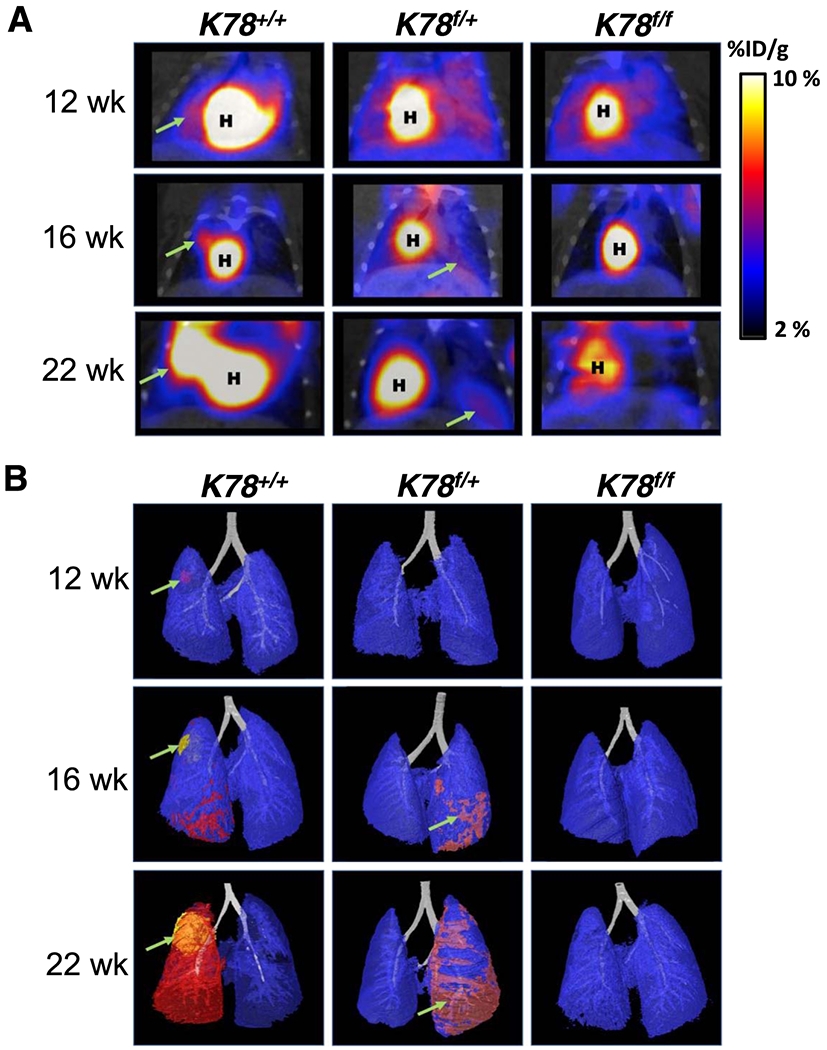
a FDG PET/CT scan of K78+/+, K78f/+, K78f/f mice for the respective genotype at 12, 16, and 22 wk post adenovirus-Cre intubation. Scans denote the heart (H) and tumorigenic areas (green arrow). On right is the color gradient representing injected dose per gram (% ID/g). b Three dimensional contrast-enhanced CT images of lungs of K78+/+, K78f/+ and K78f/f mice from (a). Tumorigenic progression (green arrow) from corroborated 18F-FDG PET/CT signal was segmented as pink, red, and yellow to indicate respectively increasing 18F-FDG avidity in the same mouse.
GRP78 Haploinsufficiency in Lung Alveolar Epithelial Type 2 Cells is Sufficient to Halt Lung Tumor Progression and Prolong Survival.
The presence of oncogenic KrasG12D in lung AT2 cells leads to multifocal clonal adenomas progressing to LUAD (23). Thus, in our second approach, referred to below as the SPC-Cre model, we tested the effect of an inducible conditional knockout of Grp78 targeted to AT2 cells carrying a KrasG12D mutation to induce lung tumorigenesis. This approach utilized a previously described SPC-CreERT2 construct that includes the SPC promoter enabling Cre expression in AT2 cells and the estrogen receptor portion allowing inducibility by tamoxifen (tam) (24). The breeding scheme to generate mouse cohorts carrying a single copy of SPC-CreERT2, a single copy of the KrasG12D knock-in allele, and either wild type (CK78+/+), 1 or 2 copies of the floxed Grp78 allele (CK78f/+ and CK78f/f, respectively) is summarized in Fig. S5A. At 10 wk of age, mice were injected with tam and lungs were genotyped and examined at various time points (Fig. 4A and B). We noted that the mortality of the CK78+/+ mice started at around 3 wk post-tam injection and that none of these mice survived more than 8 wk post-injection, whereas CK78f/+ and CK78f/f mice survived over 30 wk post injection (Fig. 4C). Necropsy examination of the CK78+/+ mice revealed that the abnormalities were confined to the lungs which showed multiple tumor nodules; in comparison, lungs and other organs of tam-injected C78+/+ mice carrying only the SPC-Cre-allele appeared to be normal, consistent with the notion that tumorigenesis observed in the CK78+/+ mice was dependent on mutant Kras (Fig. S5B).
Fig. 4. Analysis and characterization of CK78+/+, CK78f/+ and CK78f/f mice following SPC-Cre activation and effect of GRP78 knockdown in a human lung cancer cell line.
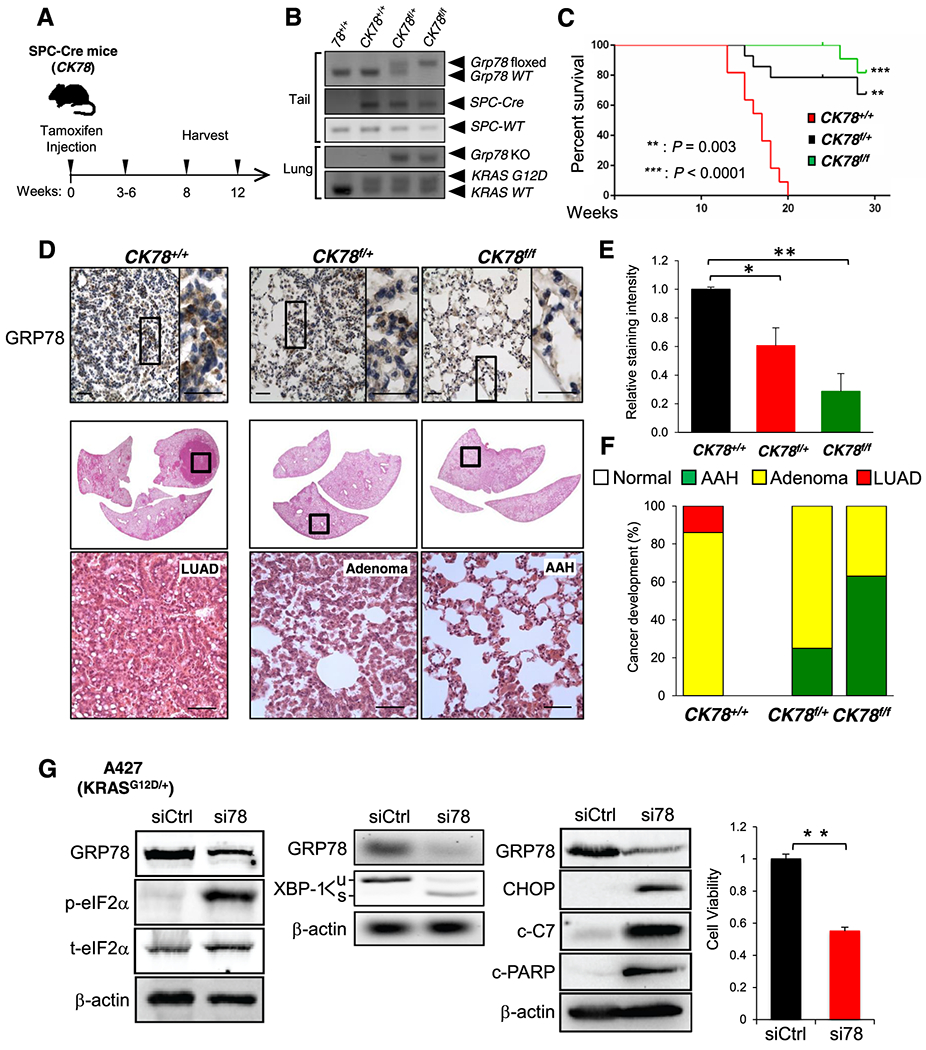
a Harvesting schedule for the SPC-Cre, floxed Grp78 mouse cohorts. To induce the SPC-Cre-recombinase, 10 wk old mice were subjected to intraperitoneal (i.p.) tamoxifen injection and the mouse lungs were harvested at the indicated time points. b Representative genotyping results of the SPC-Cre mice using DNA extracted from tail (top three panels) and from lung (bottom two panels) after Cre activation. c Kaplan-Meier survival curves of CK78+/+ (n=11), CK78f/+ (n=11) and CK78f/f (n=13) mice. Male and female mice were combined as there were no differences between the two groups. CK78f/+ and CK78f/f mice exhibited prolonged survival compared to CK78+/+ mice (**: p=0.003) and (***: p<0.0001), one-sided log-rank test, respectively. d Immunohistochemistry (IHC) staining of GRP78 in lungs of CK78+/+ mice at 8 wk post-tamoxifen i.p. injection. (Scale bar: 100 μm). Enlarged view of the boxed area is shown to the right. (Scale bar: 100 μm). Below is a cross section of the whole lungs with magnified boxed area showing H&E staining of lung adenocarcinoma (LUAD) lesions. (Scale bar: 200 μm). Lungs of CK78f/+ and CK78f/f mice at 12 wk post-tamoxifen injection are also represented. Representative H&E staining images are shown below with examples of adenoma and atypical adenomatous hyperplasia (AAH) lesions in the CK78f/+ and CK78f/f lungs respectively. e Quantitation of GRP78 staining from (d). The values represent the average staining of the sampled area ± s.e., *p<0.05 and **p<0.01. Number of images that were randomly chosen to be quantified are the following CK78+/+ (n=10), CK78f/+ (n=10) and CK78f/f (n=10). At least three to four mice were analyzed per genotype. f Quantitation of the histological grades exhibiting normal morphology (N), AAH, adenoma, or LEI AD of lungs of CK78+/+ mice at 8 wk and CK78f/+ and CK78f/f mice at 12 wk post-tamoxifen i.p. injection. The number of mice analyzed is as follows: CK78+/+ (n=7), CK78f/+ (n=4) and CK78f/f (n=8). g A427 cells were transfected with control siRNA (siCtrl) or siRNA specifically targeting GRP78 (si78) for 48 hr and analyzed for levels of phospho (p) and total (t) eIF2α, unspliced (u) and spliced (s) form of XBP-1 mRNA, in parallel with GRP78 and β-actin mRNA, CHOP and cleaved form of Caspase-7 (c-C7) and PARP (c-PARP) as well as cell viability measured by WST-1 assay.
Representative GRP78 immunostains and histological images of the lung sections of the three genotypes are shown in Fig. 4D. For example, CK78+/+ mice exhibited robust GRP78 staining of adenomas and LUAD at 8 wk following tam injection (Fig. 4D–F). CK78f/+ mice showed reduction of GRP78 expression, which was further decreased in CK78f/f mice (Fig. 4E). Tumor prevalence and grade were much reduced in both genotypes, with CK78f/+ mice showing predominantly adenoma while CK78f/f lesions were primarily AAH even at 12 wk after tam injection (Fig. 4D and F). In comparison, the lung cells of the C78f/f mice carrying only the SPC-Cre and Grp78 floxed alleles appeared normal at 12 wk after tam injection (Fig. S5C). Similar to the adeno-Cre model, weight loss was observed in CK78+/+ mice but not in the CK78f/+ and CK78f/f mice (Fig. S5D), and the staining intensity of GRP78 was higher in the tumor compared to surrounding lung tissues (Fig. S6). Collectively, these results indicate that GRP78 haploinsufficiency in AT2 cells is sufficient to suppress mutant Kras-driven lung tumorigenesis and prolong survival.
Towards understanding potential mechanisms whereby GRP78 deficiency impedes lung tumorigenesis, we utilized the human lung cancer cell line A427, which harbors the same KRASG12D mutation as our mouse models and offers an experimental system for biochemical analysis. Consistent with GRP78 being a key regulator of the UPR, knockdown of GRP78 by siRNA led to UPR activation (Fig. 4G), as exemplified by upregulation of phosphorylated eIF2α (p-eIF2α), which is a downstream effector of the ER stress sensor protein kinase R-like ER kinase (PERK), as well as the splicing of X-box binding protein 1 (XBP-1) mRNA, a downstream effector of the ER stress sensor inositol-requiring enzyme 1 α (IRE1α). The activation of apoptotic markers, including C/EBP homologous protein (CHOP), which is downstream of PERK activation, cleaved caspase-7 (c-C7), and cleaved poly (ADP-ribose) polymerase (PARP) (c-PARP), were also detected, corresponding with decrease in cell viability (Fig. 4G).
Discussion
Curbing mutant KRAS-driven tumorigenesis remains elusive as clinical responses to most inhibitors can be relatively short-lived due to compensatory mechanisms leading to acquired resistance. Here, we discovered a new approach to suppress KRAS oncogenesis by targeting a critical ER chaperone, GRP78. Upregulation of GRP78 is generally observed in human tumors due to intrinsic and extrinsic stress (10). This study further reveals that GRP78 mRNA and protein levels are upregulated in human LUAD bearing KRAS mutations, including the G12D mutation used in our mouse models. This, coupled with our finding that knockdown of GRP78 in the human lung cancer A427 cells bearing the KRASG12D mutation reduced their viability, in agreement with similar observations in A549 cells bearing KRASG12S mutation (17), suggests GRP78 could be a novel therapeutic target for LUAD, including those harboring KRAS mutations. Tissue-specific ablation of GRP78 using genetically engineered mouse models established the requirement of GRP78 in Pten-null driven cancers and PI3K/AKT signaling (10, 22, 25). GRP78 haploinsufficiency suppresses acinar-to-ductal metaplasia, proliferative signaling and mutant Kras-driven pancreatic tumorigenesis in mice (26). Based on the established utility of the KrasG12D mouse model for monitoring lung cancer initiation and progression (2), here, we created new mouse models where heterozygous activation of mutant KrasG12D is coupled with mono- or bi-allelic deletion of Grp78 either in whole lung or in AT2 cells. Our studies uncover several exciting findings. In the first mouse model where adeno-Cre was injected into the whole lung of K78+/+, K78f/+ and K78f/f mice, lung tumor burden and progression, as well as cancer cell proliferation, correlates with GRP78 levels. Importantly, in K78f/+ mice with loss of only one Grp78 allele, the partial reduction of GRP78 expression was sufficient to impede tumorigenesis as confirmed by histological grade evaluations and further confirmed using 18F-FDG PET/CT and contrast-enhanced CT. The suppression of tumorigenesis via heterozygous knockout of Grp78 is even more pronounced in our second mouse model, where the tam-inducible SPC promoter driven Cre-recombinase was used to activate Kras and delete Grp78 in AT2 cells. Both the heterozygous CK78f/+ and the homozygous knockout CK78f/f showed prolonged survival compared to CK78+/+. Our Grp78 mouse model results directly support a recent study showing that in vivo deletion of the deubiquitylase OTUD3 slowed down KrasG12D-driven LUAD initiation and progression and markedly increased survival in mice via destabilization of GRP78 (27). Our observation that rare tumors formed in the K78f/f and CK78f/f mice showed positive GRP78 staining suggests that there could be minute percentage of cells in which Kras is activated but Grp78 is not deleted and get selected such that tumors are ultimately GRP78 positive. Alternatively, these tumors in the f/f mice could arise non-autonomously due to inflammatory or other unknown signals stimulating transformation and tumor growth in cells in which the Cre-recombinase was never expressed.
With regard to the effect of GRP78 deficiency on normal lung cells, it should be noted that in mice bearing the Grp78 floxed allele, the extent of Grp78 allele excision will depend on the efficiency of Cre-vector delivery in the adeno-Cre model, and activation of Cre-recombinase in the SPC-Cre model. We observed that the gross appearance of the lungs and the morphology of the lung cells of C78f/f mice 12 wk following tam injection appeared normal. It is possible that GRP78 deficiency triggered compensatory mechanisms such as upregulation of other ER chaperones (28), or that a low residual amount of GRP78 in the lung cells is sufficient to allow for maintenance of basal functions while cancer progression is thwarted. Furthermore, since prolonged GRP78 haploinsufficiency in mouse models of various genetic backgrounds showed no major deleterious effects (29), therapeutic agents that are able to partially suppress GRP78 expression or activity can potentially block LUAD development without harming normal organs. Here we showed that knockdown of GRP78 induces the UPR and apoptotic markers, associating with loss of cell viability in a human lung cancer cell line bearing the same KRAS mutation. Various GRP78 inhibitors have shown efficacy in suppressing tumor growth in mouse cancer models and human cancer cell lines (10, 11, 30). Of note, HA15, a small molecule that overcomes BRAF inhibitor resistance, targets GRP78 and kills many types of cancer cells with no observable toxicity in normal cells (31). IT-139, a ruthenium-based small molecule with anticancer activities lowers GRP78 levels in multiple cancer models but not in adjacent normal cells, and shown efficacy and manageable toxicity in a completed Phase I clinical trial (32–35). Since KRAS-mutants have been reported to harbor proteotoxic or ER stress (36, 37), these and other anti-GRP78 agents warrant vigorous investigations to develop drug combinations to combat mutant KRAS-driven lung cancer.
Materials and Methods
Mouse models, Cre activation and tissue processing, 18F-FDG PET/CT imaging, tissue staining, quantitation of tumor burden, grading of pulmonary tumors, cell culture, Western blot analysis, cell viability assay, gene expression data, human specimens and immunostains as well as statistical analysis can be found in Supplemental Information.
Supplementary Material
Acknowledgements
We thank Hal Chapman for the SPC-Cre mice, Peter Conti and Jennifer Choi for assistance with PET/CT, and Jorge Nieva and Robert Hsu for tumor samples. The work was supported by NIH grants R01 CA027607 and the Judy and Larry Freeman Chair (A.S. Lee), NIH Diversity Supplements (D. F. Rangel), the Hastings Foundation (B. Zhou, Z. Borok) and NIH grant R35 HL135747 and Ralph Edgington Chair (Z. Borok). We thank the USC Norris Comprehensive Cancer Translational Pathology Core and the USC Molecular Imaging Center (supported by P30 CA014089, 1S10OD012371 and 1S10OD18500) for technical assistance.
Footnotes
Conflict of interest. The authors declare that they have no conflict of interest.
Ethical approval. All protocols for animal use and euthanasia were reviewed and approved by the University of Southern California Institutional Animal Care and Use Committee. Patient lung tissues were obtained in accordance with a protocol approved by the Institutional Review Board of the University of Southern California. Confirmed consent was obtained from all participants.
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015; 65: 87–108. [DOI] [PubMed] [Google Scholar]
- 2.Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001; 15: 3243–3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cancer Genome Atlas Research Network, Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014; 511: 543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hancock JF, Magee AI, Childs JE, Marshall CJ. All ras proteins are polyisoprenylated but only some are palmitoylated. Cell. 1989; 57: 1167–1177. [DOI] [PubMed] [Google Scholar]
- 5.Hancock JF, Paterson H, Marshall CJ. A polybasic domain or palmitoylation is required in addition to the CAAX motif to localize p21ras to the plasma membrane. Cell. 1990; 63:133–139. [DOI] [PubMed] [Google Scholar]
- 6.Dai Q, Choy E, Chiu V, Romano J, Slivka SR, Steitz SA, et al. Mammalian prenylcysteine carboxyl methyltransferase is in the endoplasmic reticulum. J Biol Chem. 1998; 273:15030–15034. [DOI] [PubMed] [Google Scholar]
- 7.Luo B, Lee AS. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene. 2013; 32: 805–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ni M, Lee AS. ER chaperones in mammalian development and human diseases. FEBS Lett. 2007; 581: 3641–3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pobre KFR, Poet GJ, Hendershot LM. The endoplasmic reticulum (ER) chaperone BiP is a master regulator of ER functions: Getting by with a little help from ERdj friends. J Biol Chem. 2019; 294: 2098–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee AS. Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat Rev Cancer. 2014; 14: 263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang M, Kaufman RJ. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nature Rev Cancer. 2014; 14: 581–597. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y, Liu R, Ni M, Gill P, Lee AS, Cell surface relocalization of the endoplasmic reticulum chaperone and unfolded protein response regulator GRP78/BiP. J Biol Chem. 2010; 285: 15065–15075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ni M, Zhang Y, Lee AS. Beyond the endoplasmic reticulum: atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem J. 2011; 434: 181–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reddy RK, Mao C, Baumeister P, Austin RC, Kaufman RJ, Lee AS. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: role of ATP binding site in suppression of caspase-7 activation. J Biol Chem. 2003: 278: 20915–20924. [DOI] [PubMed] [Google Scholar]
- 15.Fu Y, Li J, Lee AS. GRP78/BiP inhibits endoplasmic reticulum BIK and protects human breast cancer cells against estrogen starvation-induced apoptosis. Cancer Res. 2007; 67:3734–3740. [DOI] [PubMed] [Google Scholar]
- 16.Rutkowski DT, Arnold SM, Miller CN, Wu J, Li J, Gunnison KM, et al. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 2006; 4: doi: 10.137/journal.pbio.0040374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chae YC, Caino MC, Lisanti S, Ghosh JC, Dohi T, Danial NN, et al. Control of tumor bioenergetics and survival stress signaling by mitochondrial HSP90s. Cancer Cell. 2012; 22: 331–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uramoto H, Uchiumi T, Izumi H, Kohno K, Oyama T, Sugio K, et al. A new mechanism for primary resistance to gefitinib in lung adenocarcinoma: the role of a novel G796A mutation in exon 20 of EGFR. Anticancer Res. 2007; 27: 2297–2303. [PubMed] [Google Scholar]
- 19.Ma X, Guo W, Yang S, Zhu X, Xiang J, Li H. Serum GRP78 as a tumor marker and its prognostic significance in non-small cell lung cancers: a retrospective study. Dis Markers. 2015; doi:. 10.1155/2015/814670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kwon D, Koh J, Kim S, Go H, Min HS, Kim YA, et al. Overexpression of endoplasmic reticulum stress-related proteins, XBP1s and GRP78, predicts poor prognosis in pulmonary adenocarcinoma. Lung Cancer. 2018; 122: 131–137. [DOI] [PubMed] [Google Scholar]
- 21.Imai H, Kaira K, Minato K. Clinical significance of post-progression survival in lung cancer. Thorac Cancer. 2017; 8: 379–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fu Y, Wey S, Wang M, Ye R, Liao CP, Roy-Burman P, et al. Pten null prostate tumorigenesis and AKT activation are blocked by targeted knockout of ER chaperone GRP78/BiP in prostate epithelium. Proc Natl Acad Sci U S A. 2008; 105: 19444–19449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Desai TJ, Brownfield DG, Krasnow MA. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature. 2014; 507: 190–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chapman HA, Li X, Alexander JP, Brumwell A, Lorizio W, Tan K, et al. Integrin alpha6beta4 identifies an adult distal lung epithelial population with regenerative potential in mice. J Clin Invest. 2011; 121: 2855–2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wey S, Luo S, Tseng CC, Ni M, Zhou H, Fu Y, et al. Inducible knockout of GRP78/BiP in the hematopoietic system suppresses Pten-null leukemogenesis and AKT oncogenic signaling. Blood. 2012; 119; 817–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen J, Ha DP, Zhu G, Rangel DF, Kobielak A, Gill PS, et al. GRP78 haploinsufficiency suppresses acinar-to-ductal metaplasia, signaling, and mutant Kras-driven pancreatic tumorigenesis in mice. Proc Natl Acad Sci U S A. 2017; 114: E4020–E4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Du T, Li H, Fan Y, Yuan L, Guo X, Zhu Q, et al. The deubiquitylase OTUD3 stabilizes GRP78 and promotes lung tumorigenesis. Nat Commun. 2019; 10: 2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ye R, Jung DY, Jun JY, Li J, Luo S, Ko HJ, et al. Grp78 heterozygosity promotes adaptive unfolded protein response and attenuates diet-induced obesity and insulin resistance. Diabetes. 2010; 59: 6–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee AS, Brandhorst S, Rangel DF, Navarrete G, Cohen P, Longo VD, et al. Effects of prolonged GRP78 haploinsufficiency on organ homeostasis, behavior, cancer and chemotoxic resistance in aged mice. Sci Rep. 2017; 7: 40919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang X, He Z, Xiang L, Li L, Zhang H, Lin F, et al. Codelivery of GRP78 siRNA and docetaxel via RGD-PEG-DSPE/DOPA/CaP nanoparticles for the treatment of castration-resistant prostate cancer. Drug Des Devel Ther. 2019; 13: 1357–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cerezo M, Lehraiki A, Millet A, Rouaud F, Plaisant M, Jaune E, et al. Compounds triggering ER stress exert anti-melanoma effects and overcome BRAF inhibitor resistance. Cancer Cell. 2016; 29: 805–819. [DOI] [PubMed] [Google Scholar]
- 32.Bakewell SJ, Rangel DF, Ha DP, Sethuraman J, Crouse R, Hadley E, et al. Suppression of stress induction of the 78-kilodalton glucose regulated protein (GRP78) in cancer by IT-139, an anti-tumor ruthenium small molecule inhibitor. Oncotarget. 2018; 9: 29698–29714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burris HA, Bakewell S, Bendell JC, Infante J, Jones SF, Spigel DR, et al. Safety and activity of IT-139, a ruthenium-based compound, in patients with advanced solid tumours: a first-in-human, open-label, dose-escalation phase I study with expansion cohort. ESMO Open. 2016; 1: dx.doi. 10.1136/esmoopen-2016-000154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gifford JB, Huang W, Zeleniak AE, Hindoyan A, Wu H, Donahue TR, et al. Expression of GRP78, Master Regulator of the Unfolded Protein Response, Increases Chemoresistance in Pancreatic Ductal Adenocarcinoma. Mol Cancer Ther. 2016; 15: 1043–1052. [DOI] [PubMed] [Google Scholar]
- 35.Lizardo MM, Morrow JJ, Miller TE, Hong ES, Ren L, Mendoza A, et al. Upregulation of Glucose-Regulated Protein 78 in metastatic cancer cells is necessary for lung metastasis progression. Neoplasia. 2016; 18: 699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Denoyelle C, Abou-Rjaily G, Bezrookove V, Verhaegen M, Johnson TM, Fullen DR, et al. Anti-oncogenic role of the endoplasmic reticulum differentially activated by mutations in the MAPK pathway. Nat Cell Biol. 2006; 8: 1053–1063. [DOI] [PubMed] [Google Scholar]
- 37.De Raedt T, Walton Z, Yecies JL, Li D, Chen Y, Malone CF, et al. Exploiting cancer cell vulnerabilities to develop a combination therapy for ras-driven tumors. Cancer Cell. 2011; 3: 400–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.