

Graphical abstract

Keywords: PTEN, PHTS, Genotype-phenotype correlations, Mutation analysis, Machine learning
Highlights
-
•
This work links mechanistic effects of mutations and different phenotypes in PTEN.
-
•
Distinct PTEN diseases lie on a spectrum of changes in protein stability and function.
-
•
Computational analysis could accurately distinguish between PTEN phenotypes..
Abstract
Phosphate and tensin homolog on chromosome ten (PTEN) germline mutations are associated with an overarching condition known as PTEN hamartoma tumor syndrome. Clinical phenotypes associated with this syndrome range from macrocephaly and autism spectrum disorder to Cowden syndrome, which manifests as multiple noncancerous tumor-like growths (hamartomas), and an increased predisposition to certain cancers. It is unclear, however, the basis by which mutations might lead to these very diverse phenotypic outcomes. Here we show that, by considering the molecular consequences of mutations in PTEN on protein structure and function, we can accurately distinguish PTEN mutations exhibiting different phenotypes. Changes in phosphatase activity, protein stability, and intramolecular interactions appeared to be major drivers of clinical phenotype, with cancer-associated variants leading to the most drastic changes, while ASD and non-pathogenic variants associated with more mild and neutral changes, respectively. Importantly, we show via saturation mutagenesis that more than half of variants of unknown significance could be associated with disease phenotypes, while over half of Cowden syndrome mutations likely lead to cancer. These insights can assist in exploring potentially important clinical outcomes delineated by PTEN variation.
1. Introduction
Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) is a dual-specificity phosphatase, and powerful tumor suppressor, with additional lipid dephosphorylation properties within the PI3K/AKT/mTOR signalling pathway. It is responsible for the dephosphorylation of PIP3 to PIP2, ultimately blocking cell division mediated by AKT. Independent of its PIP3 dephosphorylation activity, it is associated with the regulation of transcription, cell proliferation and genome maintenance [1]. PTEN activity is closely regulated by its subcellular localization, which is mediated by post-translational modifications (PTMs) including phosphorylation, SUMOylation and ubiquitination, as well as protein–protein interactions [1], [2].
Structurally, PTEN is 403aa long and composed of two main domains (Fig. 1): (i) the phosphatase domain (N-terminus; residues 1–185), which contains the protein tyrosine phosphatase (PTP) conserved signature motif (HCXXGXXR) responsible for its dual-specificity phosphatase activity and lipid binding site [3], [4], and (ii) the C2 domain (C-terminus; residues 186–403), which contains a disordered loop spanning residues 286–309 [4], [5] (Fig. 1). The active site is essentially formed by the P-loop, which contains the HCXXGXXR motif, and the WPD- and TI-loop backbone atoms [4] (Fig. 1B). The TI loop is uniquely inserted in PTEN and is responsible for a large active site volume which permits PIP3 binding [4]. Parts of the WPD and TI loops are also present in the Phosphatase-C2 domain interface, which have been suggested to be important for overall folding, are highly conserved, and mutated in different cancers [4]. The C-terminal domain harbors the C2 domain, which contains the CBR3 loop responsible for PTEN attachment to the phospholipid membrane, with adequate phosphatase domain orientation to enable membrane-associated PIP3 binding [4]. This property is a result of the net + 5 charge, and two hydrophobic residues at the CBR3 tip, as well as a basic patch within the neighboring cɑ2 helix (Fig. 1B) [4]. Mutations in this domain were also associated with a reduction in PTEN’s tumor suppressor activity [4].
Fig. 1.

Main domains and subdomains present in PTEN. PTEN is primarily made up of two domains (A), the phosphatase domain (light orange) which comprises the P-, TI- and WPD- loops and the C2 domain (green) which comprises the membrane binding CBR3 tip and cɑ2 helix basic patch. The phosphatase is the site for PIP3 binding, shown in (B) bound to tartrate ion (black). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Missense mutations across the entire structure of PTEN have been associated with PTEN hamartoma tumor syndrome (PHTS) [6], an overarching condition with a broad range of phenotypes including different cancers and other tumorigenic states like Cowden syndrome [7], Bannayan-Riley-Ruvalcaba syndrome [8], Proteus [9] and Proteus-like [10] syndromes, to brain-related disorders such as macrocephaly, developmental delay and autism spectrum disorder (ASD) [11]. While tumor-related phenotypes have been attributed to changes in PIP3 dephosphorylation, the molecular consequences leading to ASD-related phenotypes remain unclear.
Initial efforts to understand PTEN-related disease mechanisms have focused on evaluating the effects of missense mutations on cellular fitness through an in vivo measurement of lipid phosphatase activity [12]. This revealed that the fitness effects of ClinVar pathogenic mutations and gnomAD population variants clustered in two distributions [12]. While this provided insight into the distinction between pathogenic and non-pathogenic mutations, differentiating between cancer-causing and ASD-causing phenotypes within the pathogenic class remains a challenge, primarily because of the limited numbers of reported ASD-causing mutations.
In an effort to address this, Smith and colleagues [13] looked at the effects of a limited subset of cancer-and ASD-causing mutations on PTEN’s conformational dynamics. They suggested that cancer-causing mutations (n = 6) exhibited higher connectivity to core PTEN nodes, and greater effects on interdomain interactions; while the ASD-causing mutations (n = 6) were focused at nodes near the CBR3 loop [11]. While this showed the potential for structural insights to delineate the different phenotypic outcomes of mutations in PTEN, it was based on a very limited subset of known PTEN disease mutations, and it was unclear how this might translate beyond those twelve mutations. Therefore, a more thorough analysis of the underlying molecular mechanisms across all clinically characterized variants is needed to provide a better understanding of overall disease etiology and how it can be treated.
We have previously shown that by considering the diversity of potential molecular consequences of a mutation on protein structure and function, it is possible to accurately predict mutations leading to cancer [14], [15], [16], [17], [18], different genetic diseases [19], [20], [21], [22], [23] and drug resistance [24], [25], [26], [27], [28], [29], [30], [31], [32], [33], [34], [35], [36]. Here, we therefore investigated the effects of mutations on protein stability, dynamics, activity, and molecular interactions across all clinically observed PTEN mutations till date, in order to identify molecular mechanisms driving the different clinical phenotypes in PTEN. Our analysis suggested that protein stability plays an important role in PTEN function and disease, where different pathologies displayed different residue-level interaction profiles and localized at different protein backbone environments. This suggests that PTEN stability and backbone conformation determines the subsequent interactions within biological pathways. A similar pattern was observed in lipid phosphatase activity, which is considered a proxy measure for cellular fitness, suggesting that protein stability and local residue interactions also mediate this functional effect.
2. Materials and methods
2.1. Dataset curation
Due to the wide spectrum of phenotypes manifested clinically resulting from missense PTEN mutations, data curation was carried out in sequential steps. Mutations conferring pathogenicity were initially extracted from ClinVar [37] (accessed July 2020), which classified them as ‘Pathogenic’ and ‘Likely Pathogenic’. To ascertain clinical involvement and increase confidence of assigned phenotypes, each mutation was cross checked with the literature, where only mutations identified directly from patients were kept for analysis. During this literature check, any mutations outside of the ClinVar dataset, which were similarly identified in clinical patients were also collected. Finally, to ensure that curation of pathogenic mutations was as comprehensive as possible, specific studies detailing large clinical PHTS, Cowden Syndrome (CS), or Bannayan-Riley-Ruvalcaba syndrome (BRRS) cohorts obtained from the Cleveland Clinic [38], [39], and a list of ASD and cancer mutations curated by Spinelli et al. [40], were used as a final check.
When present in the literature, clinical manifestations brought about by mutations were noted and used to assign a specific class (Suppl. Table 1). For the purposes of this study, the main pathogenic classes analyzed were ‘Cancer’ and ‘ASD’. Therefore, these phenotypes were prioritized even when co-occurring with other PHTS, CS and BRRS symptoms such as macrocephaly, gastro-intestinal polyps, café-au-lait marks, and thyroid dysfunction. While CS and its debated pediatric manifestation BRRS are linked to increased cancer risk, only mutations which were found in clinical cancer cases were assigned the ‘Cancer’ phenotype. Similarly, mutations in patients clinically presenting with ASD, developmental or speech delay or mental retardation were assigned the ‘ASD’ class. To further exhaust the search for PTEN mutations in ASD, mutations present in ASD-dedicated databases VariCarta [41] and SFARI [42] were similarly compared with the literature [11], [43], [44], [45], [46], [47], [48], [49], [50], [51], [52], [53], [54], [55], [56], [57], [58] and assigned the ‘ASD’ phenotype. Notably, during machine learning, a subset of ASD mutations which were only identified in ASD cases, without PHTS symptoms, were kept as a clinical validation test set.
Data curation revealed specific phenotypic manifestations within the PHTS condition. Specifically, patients having PHTS, CS or BRRS symptoms were sometimes observed to manifest neither cancer nor ASD (considered as ‘mild PHTS’), or both diseases (considered as ‘severe PHTS’). Ultimately, following consolidation of data from different sources, the interim ‘PHTS’ class was composed of ‘mild PHTS’ mutations not otherwise reported in a specific disease, and those which caused ‘severe PHTS’. A subset of ‘mild PHTS’ mutations identified in studies on CS and BRRS patient families were kept aside as a separate ‘CS’ class. Despite the lack of data accompanying these mutations with respect to cancer development, these mutations are considered ‘likely cancer-causing’ as it is known that CS is associated with increased cancer risk. The curation of data from different sources also identified overlaps across diseases. Mutations leading to both cancer and ASD in separate patients (identified from separate sources), were labelled ‘Both’.
Finally, following a thorough identification of pathogenic mutations within PTEN, any missense mutations present in the general population, as obtained from gnomAD [59] (accessed July 2020), which were not identified as pathogenic during data curation, were considered as ‘Non-Pathogenic’. This class also included one mutation in ClinVar which was classified as ‘likely Benign’. Further to that, ClinVar mutations which were classified as variants of unknown significance (‘VUS’) and were not identified in the clinical literature were kept aside as the ‘VUS’ class. A total of 229 missense mutations were grouped in specific pathogenic or non-pathogenic classes, while 294 mutations were retained in the VUS class.
For the purpose of this study, the main classes being compared were those describing ASD, Cancer, and Non-Pathogenic mutations. However, interim classes (PHTS, Both and CS) were also used in specific analyses, for possible insight into biological effects describing different mutation profiles, which can help delineate understandings of disease. A summary of the classes, description and use across different analyses is detailed in Table 1 and Fig. 2, while an account of all mutations curated in this work is detailed in Suppl. Table 1.
Table 1.
Data curation and in silico analyses. The curation of data from different sources identified subsets of pathogenic mutations apart from ASD and Cancer, which were the main pathogenic classes of interest in this study. To best characterize the biological effects mediated by these mutations, all classes and subclasses served a purpose in our analyses, which consisted of qualitative structural analysis, statistical t-test, data visualization techniques and supervised machine learning (ML). The use of the subsets within each analysis is summarized.
| Class | n | Description | Analyses |
|---|---|---|---|
| Cancer | 59 | Mutations present in cancer cases, irrespective of ‘mild PHTS’ and which have not been identified in ASD | Qualitative structural, statistical, data visualization, supervised ML |
| ASD | 65 | Mutations present in ASD patients, irrespective of ‘mild PHTS’ and which have not been identified in Cancer | Qualitative structural, statistical, data visualization, supervised ML: n = 43 for model development; n = 22 for clinical validation |
| PHTS | 26 | Mutations which either presented with overall PHTS symptoms, including CS and BRRS and no cancer/ASD (“mild PHTS”), or mutations manifesting in both diseases within the same patient (“severe PHTS”) | Qualitative structural, statistical, data visualization |
| Both | 31 | Mutations causing both Cancer and ASD, identified from separate patients | Qualitative structural, statistical, data visualization |
| CS | 26 | ‘Mild PHTS’ mutations identified in CS/BRRS patients with no other phenotype identified | Qualitative structural, statistical, data visualization, supervised ML: identification of mutations increasing cancer risk |
| VUS | 294 | ClinVar classified ‘variants of unknown significance’ or ‘conflicting interpretations of pathogenicity’ which have not been identified in the pathogenic classes | Supervised ML: suggesting reclassification of VUS |
| Non-Pathogenic | 22 | Mutations present in the general population which have not been identified in the pathogenic datasets | Qualitative structural, statistical, data visualization, supervised ML |
Fig. 2.

Methodology Pipeline followed in this study. Initial mutation curation was carried out to obtain Cancer (n = 59), ASD (n = 65) and Non-Pathogenic (n = 22; labelled as Benign in figure) mutations from five different sources. Data curation also involved processing of the experimental crystal structure to fill in missing residues and model the missing loop (286–309). In silico biophysical tools were then used to measure the effects of mutations on protein structure and function (Feature Generation), which was followed by different structural and statistical analyses and the development of a three-class prediction model.
2.2. PTEN structural curation
The experimental crystal structures of PTEN bound to tartrate ion (TLA; PDB ID: 5BZZ [5]), and the vanadate ion (VO4; PDB ID: 5BZX [5]) were obtained from the RCSB Protein Data Bank. Both structures were of the full-length protein and had a good resolution (2.20–2.50 Å), unresolved N- and C- termini (1–13 and 352–403) and an unresolved flexible loop (residues 286–309). Prior to mutational analysis, the structures were preprocessed using Maestro (Schrodinger suites), and MODELLER [60] to fill in missing atoms and model the missing loop. The TLA-bound structure was used for all structural analyses, while the VO4-bound structure was only used to calculate changes in affinity to VO4 upon mutation, and associated distance between mutated residues and ion binding site.
2.3. Feature engineering
In order to quantify the different potential mechanistic effects of mutations to protein structure and function, a range of sequence- and structure-based properties were calculated using in silico biophysical tools (Fig. 2) in a manner previously described [26], [61]. A total of 101 features were calculated on the curated structures, which can be categorized into four classes describing (i) the local residue environment, (ii) non-covalent interactions, (iii) changes in active, binding and conserved sites, and (iv) predicted changes on protein stability and dynamics. A list of calculated features per category is summarized in Suppl. Table 2. For feature calculation requiring both wildtype and mutant structures (e.g., differences in non-covalent interactions), homology modelling (using MODELLER [60]) was performed for every single-point mutation individually.
Local residue environment. We calculated features describing the local residue environment, including backbone psi and phi angles, secondary structure (SST [62]), residue depth and relative solvent accessibility (using BioPython [63]), levels of disorder (using IUPRED [64]), protein fluctuation and deformation energies (using Bio3D [65]). As a measure of residue environment geometry and physicochemical properties, graph-based signatures were also calculated [66]. We have previously shown that graph-based signatures are a powerful approach to represent a protein 3D structure in order to predict the effects of mutations on protein stability [66], [67], [68], [69], [70] and interactions [66], [71], [72], [73], [74], [75], [76], [77].
Interactions. Features describing PTEN interactions included changes in ligand affinity to TLA and VO4, and associated distances to ligand, which were calculated using mCSM-lig [76]. We also measured changes in local interactions upon mutation using Arpeggio [78] and described relevant molecular interactions as frequencies such as hydrogen bonds, pi-interactions and hydrophobic interactions. Changes in residue pharmacophore such as hydrogen bond donors and acceptors, were also calculated to reflect residue-level changes which can affect interactions.
Functional changes. Since PTEN function is related to its conserved sites, we measured the rate of residue evolution through ConSurf [79], and analyzed the functional effect of each mutation using conservation-based features SIFT [80], SNAP2 [81] and PROVEAN [82] protein. Further to these, we measured the Missense Tolerance Ratio [83], [84], which accounts for rate of mutation under neutrality and evolutionary substitution matrices PAMs and BLOSUMs, which measure the statistical likelihood of a mutation to occur. Finally, an additional biological feature was obtained from Mighell et al. [12] which described the lipid phosphatase activity of each mutation, which is a function of cellular fitness.
Changes in stability and dynamics. Changes in protein stability and dynamics upon mutation can play an important role in the emergence of different phenotypes [21], [23], [85], [86]. In this work we quantified these changes, also referred here as in silico biophysical measurements, using a range of well-established computational methods including mCSM-Stability [66], DUET [69], SDM [87], Dynamut [70] and ENCoM [88].
2.4. Qualitative structural and statistical analyses
The mutations within each phenotypic subset (ASD, Cancer and Non-pathogenic) were assigned to major molecular mechanisms of disease in a method similar to ones previously described [26], [89]. The in silico biophysical measurements of changes in ligand affinity, protein stability and protein dynamics were quantitatively compared for every mutation, and classified based on direction of change (e.g., increased or decreased stability) and intensity (measured as the change in Gibbs Free Energy of folding or binding, ΔΔG, given in kcal/mol, and labelled as mild: 0.5 <= |ΔΔG| < 1; moderate: 1 <= |ΔΔG| < 2 or high: |ΔΔG| >=2) [89]. The overall mechanism assigned depended on the extent of mutational change across all mutational measurements. Proportions of overall mechanisms across the datasets were obtained to possibly shed light on the patterns underlying different phenotypic classes. Finally, a two-tailed Welch sample t-test [90] was carried out on all calculated features to identify stratifying features between all pathogenic (n = 207) and Non-Pathogenic (n = 22) mutations, and cancer-causing (n = 59) and autism-causing (n = 65) mutations using the t.test function in R (v.3.6.1) [91]. Similarly, to identify possible differences between interim classes, a two-tailed Welch sample t-test [90] was also carried out between the classes PHTS (n = 26) and ‘Both’ (n = 31), and PHTS (n = 26) and CS (n = 26). Features were considered significant if their associated p-value was < 0.05.
2.5. Data visualization techniques
A visual discernment between classes can highlight phenotype-distinguishing features. This was particularly important considering that a large number of features (n = 101), or dimensions, were generated to describe a small number of data points spread across the three main phenotype classes: ASD (n = 65), Cancer (n = 59) and Non-Pathogenic (n = 22). As the purpose of this analysis was to highlight potentially distinguishing features between these three main classes in lower dimensions, data visualization techniques were used on all features describing ASD, Cancer and Non-Pathogenic mutations. To visually compare the interim classes PHTS (n = 26), CS (n = 26) and ‘Both’ (n = 31) to the main phenotypes, these data points were plotted on the same 2D axes, and their clustering patterns observed.
Two different techniques were tested: Principal Component Analysis (PCA) [92] and uniform manifold approximation and projection (UMAP) [93], using R (v.3.6.1) [91] packages “cluster” and “umap”, respectively. These methods were chosen as they are based on different approaches: PCA is a linear approach, which focuses on maintaining data variance [92], while UMAP is non-linear, where the distances between individual data points are maintained in the visualization [93]. Testing out two fundamentally different approaches ensured that the data could be visually represented as comprehensibly as possible, while accounting for underlying correlations between data points. These techniques were carried out at different feature levels. When using all features, features contributing to the two visualized dimensions could help suggest protein properties underlying mechanisms of disease, particularly if the classes could be distinguished visually. This process was also carried out on the subset of features which presented high class stratification from the statistical analysis (n = 54, i.e., features that presented a significant distribution difference between classes), where a visual distinction between classes can again prioritize which features are most important. Based on this similar rationale, these techniques were also carried out on the final features identified following greedy feature selection. Visually inspecting how classes cluster at different feature levels could be considered as validation for the results from other methods, where an improvement in class distinction is expected upon lowering the number of features statistically, and through greedy feature selection.
2.6. Supervised learning
A predictive model was developed using supervised learning aiming to accurately distinguish between three classes of missense mutations arising in PTEN: ASD, Cancer and Non-pathogenic. This composes a multiclass classification problem, which can be tackled by different approaches. For simplicity, we opted to use the “transformation into binary” technique, assessing both OneVsOne (which performs a pairwise comparison of all classes) and OneVsRest (which accounts for the performance of one class compared to the remaining two) strategies during model development, both available within the scikit-learn (v.0.23.2) [94] “multiclass” package.
The predictive model was trained on the curated mutations describing clinical presence of ASD (n = 43) and Cancer (n = 59), and the Non-pathogenic (n = 22) mutations were derived from residual population variation. A subset (n = 22) of ASD mutations, which described mutations identified in ASD patients without other PHTS symptoms, was kept aside as a second clinical validation test, to verify the clinical utility of the final model. The remaining ASD data used (n = 43) described mutations causing ASD symptoms in conjunction with other PHTS symptoms, excluding cancer manifestation.
ASD (n = 43), Cancer (n = 59) and Non-pathogenic (n = 22) mutations were divided into a training set (70%; ASD: 32; Cancer: 39; Non-Pathogenic: 17) and a non-redundant blind test (30%; ASD: 11; Cancer: 20; Non-Pathogenic: 5), using the GroupShuffleSplit function within scikit-learn (v.0.23.2) [94], which retained the relative proportions of classes. Due to the smaller dataset curated for Non-Pathogenic mutations, training was also carried out at one level of oversampling for this class (n = 34), to establish a more balanced training set [95].
A range of classification algorithms available within the Python scikit-learn toolkit (v.0.23.2) [94] were assessed using default parameters: Gaussian Naïve Bayes, Support Vector Machines (kernel = ‘rbf’), K-nearest neighbor (k = 3), XGBoost (n_estimators = 300), Multilayer Perceptron, and the ensemble classifiers: Gradient Boosting (n_estimators = 300), ExtraTrees (n_estimators = 100), Random Forest (n_estimators = 300) and AdaBoost (n_estimators = 300). To minimize risk of overfitting, internal model validation was carried out during training through k-fold cross validation, at k = 3, 5 and 10. Briefly, this cross-validation splits the training dataset into k number of folds, and iteratively leaves one fold out as a test set. Due to the relatively small number of data points used for training, cross validation was carried out using the StratifiedKfold function within scikit-learn (v.0.23.2) [94], which ensured that each fold retained class proportions representative of the whole dataset, and that the final metrics were representative of the whole data. A bottom-up greedy feature selection approach was employed to minimize model complexity, as prevoiusly described [67], [68], [71], [72], [77]. Best performing models were selected based on the Matthew's Correlation Coefficient (MCC), which is a well-established and balanced metric not affected by class sizes. The best classifier was chosen out of 108 resultant models (Suppl. Table 6), based on consistent performance between cross-validation and blind test, and number of features. The final model was subjected to an additional clinical ASD dataset, which permitted the assessment of model applicability in the clinic.
3. Results
3.1. Curation of PTEN disease mutations reveals that cancer mutations cluster at the phosphatase domain
The final curated dataset was obtained from ClinVar, gnomAD, SFARI and through the literature (in total 80 papers manually curated) and consisted of 65 ASD-causing, 59 cancer-causing and 22 Non-pathogenic mutations. In addition, two interim classes were defined, one labelled ‘PHTS’ (n = 26), which had the same phenotype in ClinVar with no additional details, and another labelled ‘Both’ (n = 31), which contained mutations associated with both ASD and cancer across the different sources. It is worth noting, however, that the major class of mutations obtained through ClinVar were variants of unknown significance (‘VUS’, n = 294), suggesting that disease etiology within PTEN is very complex and still poorly understood. Observing the spatial distribution of the main mutation classes within the gene and subsequent structure (Fig. 3) shows that, while mutations associated with either cancer or ASD were widely distributed across the whole gene, those causing cancer were more enriched within the phosphatase domain, which mediates its tumor suppressive function.
Fig. 3.

Mutation distribution. Differences in distribution of mutations across the three main phenotypes: ASD (yellow), Cancer (red) and non-pathogenic (blue) across protein structure (A), and gene (B). Cancer mutations are observed in higher concentrations within the phosphatase region, suggesting a direct effect on PIP3 dephosphorylation and subsequent tumor suppression. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
While it was previously proposed that ASD-causing variant effects were concentrated around the CBR3 tip, we observed that the spatial distribution of ASD-causing variants was not localized to a specific domain. This included ASD-causing variants in the PIP3-binding site residues: D92, H93 and Q171. Of these, D92 is required for protonation, while the other three residues are important for PIP3 binding [4]. When considering the phosphatase-C2 interdomain region, two ASD variants were observed in the C2 domain residue D252, which is involved in interdomain hydrogen-bond networks, and lies in a highly conserved region.
Cancer-causing mutations were predominantly found in the phosphatase domain, which is associated with the tumor-suppressing activity of PTEN, particularly in residues D92, H123, G127, K128, R130 and T167 which lie within the PIP3 binding site. Of these, K128 was reported to bind directly to PIP3, R130 is required for catalysis, H123 and G127 determine the conformation, and D92 is required for protonation. Some cancer mutations were also observed in the phosphatase interdomain region, in position Y174. This region is highly conserved within the protein, with neighboring residues S170 and R173 involved in interdomain hydrogen-bonding [4].
Finally, non-pathogenic mutations were observed at a lower frequency across the gene, with no specific domain localization. Only one variant was observed within the PIP3 binding site (at T167), and only two in the membrane binding region (at L265 and D268). Interestingly, no Non-pathogenic variants were observed in interdomain residues, suggesting that these residues play an important role in disrupting PTEN function and leading to both ASD and cancer.
3.2. Exploring the molecular consequences of PTEN mutations leading to disease
Analyzing the molecular and structural properties of mutations associated with disease (n = 207) to those identified as non-pathogenic (n = 22; Suppl. Table 3) revealed that most measures of conservation (as described in Section 2.3) showed a significant distinction between the two classes (ConSurf: p = 3.3e−4, PROVEAN: p = 2.0e−5, SNAP2: p = 1.6e−6, SIFT: p = 1.7e−3), suggesting that pathogenic mutations are more likely to be found at highly conserved regions, and would be associated with more deleterious fitness consequences. Consistent with this, pathogenic mutations were also more likely to be buried within the protein core (ResDepth: p = 1.1e−4, RSA: p = 2.2e−3), and to significantly destabilize the PTEN protein structure (SDM: p = 2.6e−4, DUET: p = 8.4e−3, mCSM-Stability: p = 0.03).
This disruption in protein stability may be explained through local environmental changes, where pathogenic mutations tended to localize at residues having a smaller backbone Psi angle (p = 0.04) and were enriched in mutations occurring from a wildtype Glycine (p = 4.8e-5), and to a mutant Proline (p = 2.7e−5). This suggests that a disruption in normal PTEN function in disease is mediated through changes in backbone conformation, consistent with a previous study by Smith et al. [13], suggesting different patterns of connectivity between cancer- and ASD-causing mutations. Other measures of local environment which highlight molecular differences underlying pathogenicity include graph-based signature features. Specifically, features describing the presence of polar residue atoms at varying distances from other polar (e.g. PP:11.00: p = 1.1e−7; PP:2.00: p = 9.8 e−6) or hydrophobic atoms (e.g. HP:11.00: p = 1.7e−4; HP:2.00: p = 1.6 e−6), were enriched for within the pathogenic mutation class. As polar residues mediate fundamental and specific interactions during molecular recognition, this distinct pattern observed for the pathogenic class suggests that these mutations are clustered at sites involved in specific molecular interactions.
Additionally, pathogenic mutations also exhibited differential interaction patterns at the residue level when compared to the non-pathogenic mutations. These residue-mediated interactions included wildtype residue polar interaction counts (p = 1.4e−3), which were higher at pathogenic mutation sites. These differences also explain the functional profiles observed through conservation-based features. In addition, while considering specific PTEN functions, we also observed that pathogenic mutations tended to be located close to the PIP3 binding site (distance to tartrate: p = 0.01) and leading to a significant reduction in ligand binding affinity (mCSM-lig, p = 0.02) and lipid phosphatase activity (p = 3.0e−13). This is consistent with a previous study [12] that suggested that lipid phosphatase activity was reduced by cancer and ASD mutations, and not by non-pathogenic ones.
3.3. Exploring the molecular differences of PTEN mutations leading to different pathogenicities
To better understand how pathogenic mutations in PTEN specifically lead to cancer (n = 59) or ASD (n = 65; Suppl. Table 3), we looked closer at the molecular and structural features describing these mutations. Interestingly, further to our observation on the role of backbone conformation in mediating pathogenicity, mutations causing cancer were observed to be significantly enriched (p = 0.05) in changes to Proline, while ASD mutations were significantly enriched in mutating from a Proline residue (p = 0.04). Further to this, at the residue interaction level, cancer mutations were observed to occur in residues mediating ionic interactions (p = 0.02) while ASD mutations clustered at ones mediating aliphatic amide-ring interactions (p = 6.0 e−3). These opposing properties highlight the involvement of mutant residue interactions in distinct molecular pathways and PTEN functions. These interactions and protein backbone profiles may further possibly explain the significantly different level of lipid phosphatase activity (p = 0.02) between classes, observed to be more disrupted via cancer mutations.
The trends underlying general PTEN pathogenicity, studied through the comparison of interim classes PHTS (n = 26) and ‘Both’ (n = 31; Suppl. Table 3) were less specific to those observed for the main pathogenic mutation classes, ASD and cancer. Interestingly, this comparison highlighted that mutations observed to lead to both diseases in separate patients, given by the ‘Both’ class, were present in more conserved regions (ConSurf: p = 0.05) and were enriched in aromatic substitutions (p = 0.04). On the other hand, mutations within the PHTS class, where the main phenotypes, if present, were occurring in the same patient, were observed to significantly occur from a Proline (p = 0.04), and lead to neutrally charged residues (p = 6.9 e−3). Similarly to what was observed when comparing pathogenic and non-pathogenic mutations, mutations in the ‘Both’ class were observed to occur closer to the PIP3 binding site (p = 4.1e−3), and were more enriched in neighboring polar residues given by graph-based signature features (p = 0.03), where mutations significantly increased hydrophobic interactions counts (p = 0.02) at the residue level. Collectively these results suggest that mutations found in both diseases separately (‘Both’) are more detrimental to fitness than those present in PHTS, given by a molecular profile closely related to pathogenicity effects. In comparing mutations causing CS with those present in the general condition PHTS, similar trends towards fitness costs were observed through the CS class, where these mutations reduced protein stability (DUET: p = 0.02; mCSM-Stability: p = 0.03; SDM: p = 0.02) and occurred at a greater backbone Phi angle (p = 3.5e-3) than PHTS mutations, again suggesting the role of backbone conformation in mediating disease.
When further considering the structural effects of mutations on protein stability (DUET), protein dynamics (Dynamut), ligand affinity and lipid phosphatase activity (Suppl. Table 4), we observed a general trend where non-pathogenic mutations were associated with neutral or mild effects, followed by mutations linked to ASD, which had a mix of mild and large molecular consequences, and finally Cancer mutations, which overall had the largest molecular effects. This suggests that PTEN mutations leading to cancer have higher fitness costs compared to those leading to ASD. The pattern was evident, for example, in changes in protein stability (Suppl. Tables 4 and 5), with 54.2% of Cancer, 52.3% of ASD and only 27.3% of non-pathogenic mutations predicted to decrease stability. By contrast, only 5.1% of cancer mutations increased protein stability as an overall effect, while 9.2% of ASD and 18.2% of non-pathogenic mutations were estimated to increase protein stability. When the same analysis was carried out on the interim classes ‘Both’, PHTS and CS, it was again observed that mutations present in the PHTS had milder fitness effects, particularly on protein stability, where 61.5% of mutations, compared to 71.0% in the ‘Both’ class, and 80.8% within the CS class were observed to reduce protein stability.
Collectively, these observations suggest that different pathogenic phenotypes within PTEN, even interim disorders, lie on a spectrum, where the main protein property involved seems to affect function through mutations in conserved regions, changes in core residues and lipid phosphatase activity. It is known that stability is also regulated by protein–protein interactions [1], suggesting that different stages of PTEN stability play a role in different biological pathways, hence leading to different diseases.
3.4. Using the structural consequences of PTEN mutations to distinguish distinct disease outcomes
To better understand the interplay of protein properties between the distinct disease states associated with PTEN mutations, we used data visualization techniques to analyze property distributions across both main (ASD, Cancer, Non-pathogenic) and interim classes (Both, PHTS and CS) and supervised machine learning approaches to assess our ability to predict the three main phenotypes observed.
Two different data visualization techniques: 2-component PCA and U-MAP, were evaluated across mutations labelled as ASD, Cancer and Non-Pathogenic and applied to the interim classes ‘Both’, PHTS and CS for observation. While U-MAP offered no visual insight into possible mutation distribution patterns across different protein properties (Suppl. Fig. 1), 2-component PCA consistently showed a distinction of the non-pathogenic class (Fig. 4, Suppl. Fig. 2; blue). The two principal components accounted for 34.2% of the variance observed within all the features. Despite this small variance, the main component PC1 (24.0%) was consistent with our previous analyses, and had significant contributions from mutation properties such as relative solvent accessibility (RSA), changes in protein stability (calculated by DUET, mCSM-Stability and SDM), lipid phosphatase activity, measures of conservation (ConSurf, PROVEAN, SIFT), changes in ligand affinity and distance to active site. These measures were all significant in distinguishing pathogenic from non-pathogenic mutations, which accounts for the visual distinction of the Non-pathogenic class from the rest. The second principal component (PC2; 10.21%) was composed of a measure of vibrational entropy change (ENCoM), which accounts for dynamic effects, but was primarily composed of residue level interactions such as changes in Polar, hydrogen bond and hydrophobic interaction counts. While interesting to note, these specific interaction types did not account for the distinct residue level interactions mediated by ASD and Cancer mutations observed from the statistical analysis, which can be visually observed in the plot through a direct overlap of these two classes. Interestingly, when plotting the interim classes on the same axes, common patterns between these classes, previously observed through other methods, have emerged. Specifically, mutations in the PHTS class (Fig. 4B; Suppl. Fig. 2; green), which were suggested by the structural analyses to have the mildest effects, mapped close to the Non-pathogenic mutations (Fig. 4A; blue). On the other hand, mutations in the ‘Both’ class (Fig. 4B; purple) shared a similar molecular property distribution to the Cancer mutations (Fig. 4A; Suppl. Fig. 2; red). This was in line with structural and statistical findings which suggested that mutations in the ‘Both’ class were the most disruptive. These results further highlight that different pathogenic classes within PTEN have spectral effects, where the resultant phenotype is an interplay between different pathway level effects.
Fig. 4.
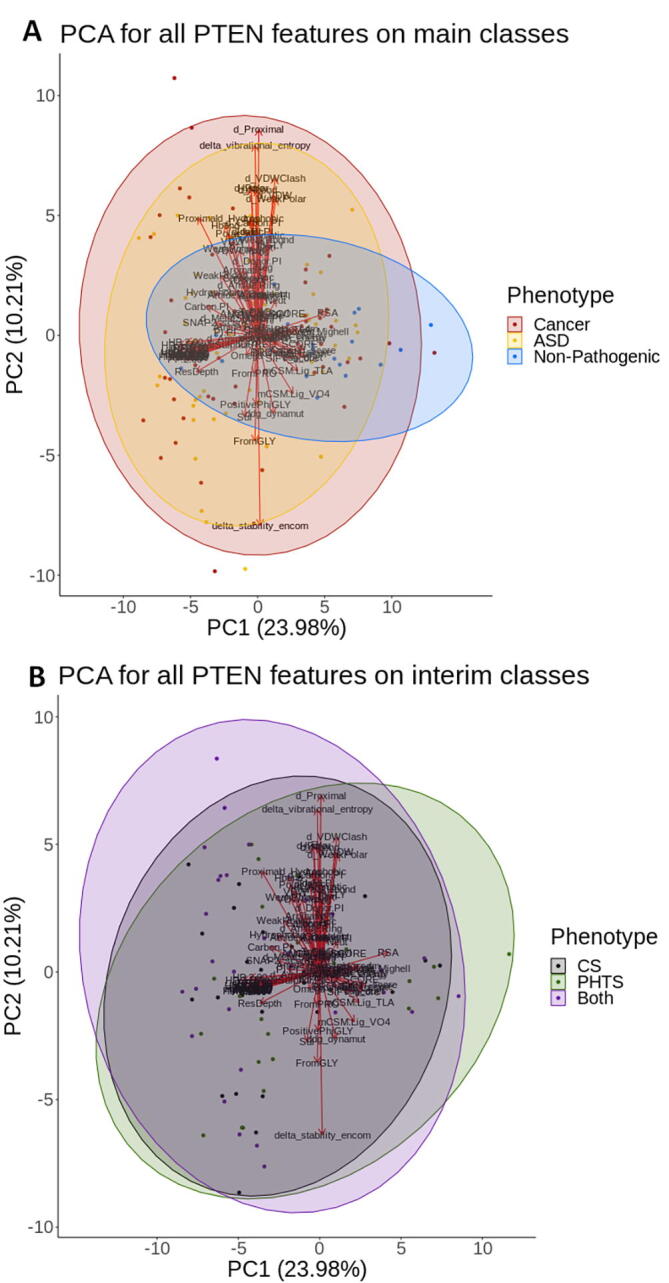
Principal component analysis plot on all features. When considering the main phenotypes (A), pathogenic classes ASD (yellow) and Cancer (red), were observed to overlap, while Non-pathogenic mutations (blue) mapped at distinct regions on the plot. A comparison of the interim classes (B) shows slight distinctions between Both (purple) and PHTS (green), while CS (grey) mutations lied in between. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Based on the consistent observations across different techniques, we employed supervised machine learning to assess the extent to which combinations of specific protein features can be used to distinguish between ASD, Cancer and Non-pathogenic mutations. Machine learning was carried out on all features generated, which included a graph-based signature representation of the 3-dimensional protein environment around the mutated residue [96]. The atoms, labelled as either hydrophobic or polar, were represented as nodes, with the edges capturing the molecular interactions between atoms. We have previously shown that graph-based signature representation of the mutation environment is a powerful and accurate approach to predicting the effects of mutations [61], [66].
During model development, different model parameters were tested in parallel across 108 runs (Suppl. Table 6) which included the presence or absence of oversampling for the non-pathogenic class, multiclass approaches OneVsOne and OneVsRest, different classification algorithms and different cross validation schemes (k = 3, 5, 10). When choosing the best performing model, greedy feature selection cut-offs prioritized consistent values between MCC cross-validation result and blind test. During this process, the number of features was kept to a minimum, in order to limit model complexity, with the AdaBoost algorithm being the best performing model under 10-fold cross validation, using oversampling of the least frequent class.
When comparing the two models obtained by the different multiclass approaches (OneVsOne and OneVsRest; Suppl. Table 7), it was observed that both models prioritized experimental lipid phosphatase activity, which describes PTEN function, a change in cation-Pi interaction counts and a graph-based signature feature describing the presence of two polar atoms within interacting distance (2 Å; Suppl. Table 7). Despite having more features (n = 11 compared to n = 7 in OneVsRest), the model based on the OneVsOne multiclass approach was chosen, as the MCC values obtained following cross-validation and subjection to a blind test were consistent and robust (MCC: 0.68). This model was additionally based on another graph-based signature detailing two polar residues within 6.5 Å of each other, relative solvent accessibility, MTR score, which is a measure of missense intolerance at a specific residue, and different residue level interaction counts involving Pi interactions (hydrogen bond donor-Pi, change in Pi-Pi interactions), aliphatic (amide-ring interactions) and aromatic residues. Further to that, this model accounted for mutations leading to Proline, which is known to change backbone conformation. Interestingly, most of these features were highlighted to be significant when distinguishing between pathogenic and non-pathogenic mutations (graph-based signatures, RSA), and between ASD and cancer-causing mutations (mutation to Proline, amide-ring interactions), while the functional lipid phosphatase activity was observed to contribute significantly to both stratifications from our t-test [90] analysis. These consistencies between analyses, further suggest model robustness, based on biologically discerning features among classes.
Further model assessment using other metrics, including those accounting for data imbalances: balanced accuracy and F1 score (with micro, macro and weighted averaging) confirmed model robustness through similar metrics obtained across the different validation methods. This is particularly important considering the small dataset used, and despite requiring oversampling for the non-pathogenic class. Finally, to quantify how well the model can detect each class, we calculated Recall per class via the confusion matrices (Fig. 5A, Table 2), which showed inconsistencies between cross validation and blind test, particularly for the ASD cohort. This suggests that the features, despite identified as significant through other methods within this study, may not fully encompass the complexities underlying ASD-mediating mutations. To further test the applicability of the model to predict ASD mutations, we checked the model performance on a held-out dataset (n = 22), which was not used in model development. This test also gave poor results, suggesting low confidence in clinical applicability. One possible reason beneath these ASD metrics could be the combination of PHTS phenotypes co-occurring with ASD in the dataset used for model development, particularly since the clinical validation test represented ASD-only mutations.
Fig. 5.

Metrics for the chosen model. Model was obtained after greedy feature selection, where the confusion matrices (A) calculated the three-class classifier using the OneVsOne method on the Adaboost algorithm, validated through 10-fold cross validation, with prediction performance of up to 0.68 MCC. Confusion matrices show correctly predicted data points per class, across the diagonal. (B) Observing the contribution of each feature within the estimators of our final model shows that MTR score and RSA are important in identification of disease variants, while changes in lipid phosphatase activity plays an important role in distinguishing between disease outcomes.
Table 2.
Balanced metrics observed in final model. The final model performed similarly between cross validation and blind test, suggesting there is no inherent bias underlying predictions.
| Validation method | MCC | B. acc | F1 (micro) | F1 (macro) | F1 (weighted) | Recall ASD | Recall cancer | Recall NP |
|---|---|---|---|---|---|---|---|---|
| 10-fold CV | 0.68 | 0.77 | 0.78 | 0.76 | 0.77 | 0.37 | 0.54 | 0.77 |
| Blind test | 0.68 | 0.77 | 0.81 | 0.77 | 0.81 | 0.91 | 0.41 | 0.60 |
| ASD test | – | 0.32 | 0.32 | 0.16 | 0.48 | 0.32 | – | – |
As our final model is based on OneVsOne binarization, three pairwise estimators contribute to the final phenotype prediction: ASD vs Cancer; ASD vs Non-Pathogenic and Cancer vs Non-Pathogenic. As a final analysis, we then observed the extent to which our final features (Suppl. Table 7) contribute to each pairwise problem. Fig. 5B shows that lipid phosphatase activity contributes highly to all pairwise estimators for classification. On the other hand, MTR score was a particularly important contributor to estimators involving the Non-Pathogenic class, reconfirming that pathogenic mutations are localized at functionally important regions of the gene. Interestingly, RSA and polar atom pairs within interacting distances were also involved in distinguishing either pathogenic class from the Non-pathogenic.
3.5. Exploring the potential disease landscape of PTEN using in silico saturation mutagenesis
In order to help guide analysis of novel variants, we performed in silico saturation mutagenesis using our best machine learning-trained model (Suppl. Tables 6 and 8; Suppl. Fig. 3). Looking at the distributions of the different predicted phenotypes across the gene, distinct patterns have emerged: predicted Cancer mutations predominantly occupied the phosphatase domain, non-pathogenic mutations were concentrated at the C2 domain, while ASD mutations distributed across the whole gene, concentrating at the interdomain region. This distribution is thought to reflect specific domain functions, particularly considering the predicted Cancer cluster at the phosphatase domain, which includes the PIP3 binding site. This also suggested that mutations within the dynamic loop of the C2 domain are most likely to be Non-pathogenic mutations. This suggests that our model might be able to detect specific mutations affecting membrane binding and consequent tumor suppressor activity.
Using the results from the saturation mutagenesis, we explored the predicted phenotypes of the Variants of Unknown Significance (VUS, n = 294) that had been curated. We observed that our model predicted 26.5% of these as ASD, 39.1% as Cancer and 34.4% as Non-Pathogenic. This suggests that nearly half of the previously uncharacterized variants could be associated with a disease phenotype, suggesting that further follow up work is needed to explore the potential clinical implications of these. Similarly, we also wanted to assess the cancer risk for a subset of mutations which were associated with CS/BRRS (n = 26), and found that our predictor considers 57.7% of these mutations to be cancer-causative. Clinically, this risk could be considered as part of patient management strategies.
4. Discussion
Missense mutations in PTEN lead to very diverse disease states, collectively referred to as PHTS. The main challenge in PTEN disease management is to differentiate and predict the effect of specific mutations within this gene, as treatment options and patient monitoring across cancer and ASD is very diverse. Further to this, we have observed that some mutations have been associated with both phenotypes across different clinical sources, highlighting that underlying mechanisms are affected by different traits and the interactions between them. Despite being germline mutations, manifestations may also differ between members of the same family, making the distinction between ASD and cancer phenotypes more complex, and suggesting a reason behind our lower predictive performance for ASD mutations.
Previous efforts to distinguish between disease phenotypes have looked at in vitro lipid phosphatase activity [12], a measure traditionally correlated with the cancer phenotype, and conformational dynamics approaches [13] on only a very small sample of cancer (n = 6) and ASD (n = 6) mutations. Despite the hypotheses presented, neither work offers an in-depth comparison of different mutational effects using multivariate protein properties, which can lead to a better, more holistic understanding of the biological problem. In this work, we have manually curated our mutations from different sources into three clinically confirmed phenotypes: ASD (n = 65), Cancer (n = 59) and Non-pathogenic (n = 22), making our dataset comprehensible enough to draw rational conclusions from our different analyses.
Through in silico biophysical measurements describing different protein properties, we have sought to identify the molecular basis behind why specific mutations lead to one phenotype and not the other. Using different data analysis techniques, the effect of mutations on protein stability, as well as their localization in buried and conserved regions have consistently been observed to lead to pathogenicity. In comparing ASD-causing and cancer-causing mutations, we observed that different interaction profiles at the residue level correlated with protein backbone conformation effects, highlighting that protein conformation may be responsible for different diseases. A similar pattern was observed when considering in vitro lipid phosphatase activity, which could differentiate across all classes: ranging from the most detrimental (cancer) to hypomorphic (ASD), to wild type (non-pathogenic) function. We also analyzed mutations leading to both ASD and Cancer in different patients (‘Both’ class), it was observed that these mutations were a more disruptive ‘interim’ class, suggesting a molecular basis for age-related mutational penetrance between those who develop ASD and those who develop cancer. On the other hand, mutations in patients with co-occurring PHTS symptoms, including both ASD and cancer within the same patient, were observed to have less disruptive effects on protein properties in this study. These results collectively show that pathogenicity within PTEN may occur as a spectrum, consistent with the hypothesis proposed by Mighell et al. [12], with the most intense pathogenic mutations leading to cancer, and ASD causing mutations lying between the two extremes (cancer and non-pathogenic).
Using these structural insights, we have developed a three-class prediction model, trained through supervised machine learning. During development, our model was better able to detect cancer-causing and non-pathogenic mutations, where a reduced applicability to ASD may be due to co-occurrent (mild) PHTS conditions. Subjecting the model to two sets of validation: 10-fold cross validation and validation through a blind test showed comparable results across different balanced metrics, implying that the model has not been overfit on the data it has been trained on. Our final model is primarily composed of local, functional and interaction-describing features, suggesting that differences in phenotype manifestation lie predominantly at the molecular level. One possible avenue for further improvement in our final model, however, is the inclusion of epistatic effects through protein–protein interaction features, as these are known to be regulatory mechanisms driving PTEN action, and further optimizing the model towards detection of ASD-only mutations in younger patients.
Applying this model to an in silico saturation mutagenesis approach described a phenotypic landscape which linked back to the specific domain functions, primarily in the localization of the predicted cancer-causing mutations. In observing the predictions on the VUS data points we saw that there is potential for reclassification of up to 65.6% of these mutations into cancer or ASD, warranting more specific patient monitoring for those patients. Further to that, we also tested for cancer-risk in a subset of mutations from CS/BRRS patients, where 58% of mutations were predicted to lead to cancer. Despite the small dataset used for training, the metrics describing our final model strongly suggest a potential for clinical utility, particularly at guiding protocols for patients with VUS and CS. Finally, while PHTS is also commonly mediated through truncating mutations, a distinction of the different phenotypes brought about by missense mutations could inform therapeutic development for the underlying pathologies.
5. Conclusions
Mutations in PTEN are associated with a range of complex disease phenotypes, which have proven hard to untangle. By considering the structural and functional consequences of mutations in PTEN, we have shown that disease phenotypes can be accurately predicted. This also revealed key underlying molecular drivers of disease outcomes, with decreases in protein stability and phosphatase activity associated with disease. Interestingly, the severity of these effects appeared to correlate with phenotype, with the most drastic effects linked to cancer, mild reductions linked to ASD and non-pathogenic mutations showing minimal changes. Using these insights, we have identified that more than half of currently assigned VUS could be disease associated, and used our model to predict the phenotypic outcomes of all possible mutations. This will be a valuable resource to further explore the role of mutations in PTEN and their links to patient outcomes and treatments.
CRediT authorship contribution statement
S.P. was responsible for data curation, structural and statistical analysis, machine learning and manuscript preparation. L.B. assisted with data curation. A.G.C.d.S. assisted with machine learning. D.E.V.P. assisted with supervision of machine learning. D.B.A. conceived, designed and supervised all aspects of the project. All authors assisted with manuscript writing.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
S.P. was funded by a Melbourne Research Scholarship. A.G.C.d.S. acknowledges the Joe White Bequest Fellowship for its support. D.B.A. and D.E.V.P. were funded by a Newton Fund RCUK-CONFAP Grant awarded by The Medical Research Council (MR/M026302/1). D.B.A. was supported by the Wellcome Trust (grant 093167/Z/10/Z) and an Investigator Grant from the National Health and Medical Research Council (NHMRC) of Australia (GNT1174405). Supported in part by the Victorian Government’s Operational Infrastructure Support Program.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2021.05.028.
Contributor Information
Douglas E.V. Pires, Email: douglas.pires@unimelb.edu.au.
David B. Ascher, Email: david.ascher@unimelb.edu.au.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Lee Y.-R., Chen M., Pandolfi P.P. The functions and regulation of the PTEN tumour suppressor: new modes and prospects. Nat Rev Mol Cell Biol. 2018;19(9):547–562. doi: 10.1038/s41580-018-0015-0. [DOI] [PubMed] [Google Scholar]
- 2.Hopkins B.D., Hodakoski C., Barrows D., Mense S.M., Parsons R.E. PTEN function: the long and the short of it. Trends Biochem Sci. 2014;39(4):183–190. doi: 10.1016/j.tibs.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Myers, M. P.; Stolarov Jp Fau - Eng, C.; Eng C Fau - Li, J.; Li J Fau - Wang, S. I.; Wang Si Fau - Wigler, M. H.; Wigler Mh Fau - Parsons, R.; Parsons R Fau - Tonks, N. K.; Tonks, N. K., P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. (0027-8424 (Print)). [DOI] [PMC free article] [PubMed]
- 4.Lee J.O., Yang H., Georgescu M.M., Di Cristofano A., Maehama T., Shi Y. Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell. 1999;99(3):323–334. doi: 10.1016/s0092-8674(00)81663-3. [DOI] [PubMed] [Google Scholar]
- 5.Lee C.U., Hahne G., Hanske J., Bange T., Bier D., Rademacher C. Redox Modulation of PTEN Phosphatase Activity by Hydrogen Peroxide and Bisperoxidovanadium Complexes. Angew Chem Int Ed Engl. 2015;54(46):13796–13800. doi: 10.1002/anie.201506338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yehia, L.; Keel, E.; Eng, C., The Clinical Spectrum of PTEN Mutations. Annu Rev Med 2020, 71 (1545-326X (Electronic)), 103-116. [DOI] [PubMed]
- 7.Liaw D., Marsh D.J., Li J., Dahia P.L., Wang S.I., Zheng Z. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet. 1997;16(1):64–67. doi: 10.1038/ng0597-64. [DOI] [PubMed] [Google Scholar]
- 8.Hendriks Y.M., Verhallen J.T., van der Smagt J.J., Kant S.G., Hilhorst Y., Hoefsloot L. Bannayan-Riley-Ruvalcaba syndrome: further delineation of the phenotype and management of PTEN mutation-positive cases. Fam Cancer. 2003;2(2):79–85. doi: 10.1023/a:1025713815924. [DOI] [PubMed] [Google Scholar]
- 9.Biesecker L.G., Rosenberg M.J., Vacha S., Turner J.T., Cohen M.M. PTEN mutations and Proteus syndrome. Lancet. 2001;358(9298) doi: 10.1016/S0140-6736(01)07109-4. [DOI] [PubMed] [Google Scholar]
- 10.Zhou X.P., Marsh D.J., Hampel H., Mulliken J.B., Gimm O., Eng C. Germline and germline mosaic PTEN mutations associated with a Proteus-like syndrome of hemihypertrophy, lower limb asymmetry, arteriovenous malformations and lipomatosis. Hum Mol Genet. 2000;9(5):765–768. doi: 10.1093/hmg/9.5.765. [DOI] [PubMed] [Google Scholar]
- 11.Buxbaum J.D., Cai G., Chaste P., Nygren G., Goldsmith J., Reichert J. Mutation screening of the PTEN gene in patients with autism spectrum disorders and macrocephaly. Am J Med Genet B Neuropsychiatr Genet. 2007;144B(4):484–491. doi: 10.1002/ajmg.b.30493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mighell T.L., Evans-Dutson S., O'Roak B.J. A Saturation Mutagenesis Approach to Understanding PTEN Lipid Phosphatase Activity and Genotype-Phenotype Relationships. Am J Hum Genet. 2018;102(5):943–955. doi: 10.1016/j.ajhg.2018.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith I.N., Thacker S., Seyfi M., Cheng F., Eng C. Conformational dynamics and allosteric regulation landscapes of germline PTEN mutations associated with autism compared to those associated with cancer. Am J Hum Genet. 2019;104(5):861–878. doi: 10.1016/j.ajhg.2019.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bayley J.P., Bausch B., Rijken J.A., van Hulsteijn L.T., Jansen J.C., Ascher D. Variant type is associated with disease characteristics in SDHB, SDHC and SDHD-linked phaeochromocytoma-paraganglioma. J Med Genet. 2020;57(2):96–103. doi: 10.1136/jmedgenet-2019-106214. [DOI] [PubMed] [Google Scholar]
- 15.Casey R.T., Ascher D.B., Rattenberry E., Izatt L., Andrews K.A., Simpson H.L. SDHA related tumorigenesis: a new case series and literature review for variant interpretation and pathogenicity. Mol Genet Genomic Med. 2017;5(3):237–250. doi: 10.1002/mgg3.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jafri M., Wake N.C., Ascher D.B., Pires D.E., Gentle D., Morris M.R. Germline mutations in the CDKN2B tumor suppressor gene predispose to renal cell carcinoma. Cancer Discov. 2015;5(7):723–729. doi: 10.1158/2159-8290.CD-14-1096. [DOI] [PubMed] [Google Scholar]
- 17.Andrews K.A., Ascher D.B., Pires D.E.V., Barnes D.R., Vialard L., Casey R.T. Tumour risks and genotype-phenotype correlations associated with germline variants in succinate dehydrogenase subunit genes SDHB SDHC and SDHD. J Med Genet. 2018;55(6):384–394. doi: 10.1136/jmedgenet-2017-105127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hnizda A., Fabry M., Moriyama T., Pachl P., Kugler M., Brinsa V. Relapsed acute lymphoblastic leukemia-specific mutations in NT5C2 cluster into hotspots driving intersubunit stimulation. Leukemia. 2018;32(6):1393–1403. doi: 10.1038/s41375-018-0073-5. [DOI] [PubMed] [Google Scholar]
- 19.Nemethova M., Radvanszky J., Kadasi L., Ascher D.B., Pires D.E., Blundell T.L. Twelve novel HGD gene variants identified in 99 alkaptonuria patients: focus on 'black bone disease' in Italy. Eur J Hum Genet. 2016;24(1):66–72. doi: 10.1038/ejhg.2015.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Soardi, F. C.; Machado-Silva, A.; Linhares, N. D.; Zheng, G.; Qu, Q.; Pena, H. B.; Martins, T. M. M.; Vieira, H. G. S.; Pereira, N. B.; Melo-Minardi, R. C.; Gomes, C. C.; Gomez, R. S.; Gomes, D. A.; Pires, D. E. V.; Ascher, D. B.; Yu, H.; Pena, S. D. J., Familial STAG2 germline mutation defines a new human cohesinopathy. NPJ Genom Med 2017, 2 (2056-7944 (Electronic)), 7. [DOI] [PMC free article] [PubMed]
- 21.Trezza A., Bernini A., Langella A., Ascher D.B., Pires D.E.V., Sodi A. A computational approach from gene to structure analysis of the human ABCA4 transporter involved in genetic retinal diseases. Invest Ophthalmol Vis Sci. 2017;58(12):5320–5328. doi: 10.1167/iovs.17-22158. [DOI] [PubMed] [Google Scholar]
- 22.Usher J.L., Ascher D.B., Pires D.E., Milan A.M., Blundell T.L., Ranganath L.R. Analysis of HGD gene mutations in patients with alkaptonuria from the united kingdom: identification of novel mutations. JIMD Rep. 2015;24 (2192–8304 doi: 10.1007/8904_2014_380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ascher D.B., Spiga O., Sekelska M., Pires D.E.V., Bernini A., Tiezzi M. Homogentisate 1,2-dioxygenase (HGD) gene variants, their analysis and genotype-phenotype correlations in the largest cohort of patients with AKU. Eur J Hum Genet. 2019;27(6):888–902. doi: 10.1038/s41431-019-0354-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Albanaz A.T.S., Rodrigues C.H.M., Pires D.E.V., Ascher D.B. Combating mutations in genetic disease and drug resistance: understanding molecular mechanisms to guide drug design. Expert Opin Drug Discov. 2017;12(6):553–563. doi: 10.1080/17460441.2017.1322579. [DOI] [PubMed] [Google Scholar]
- 25.Phelan J., Coll F., McNerney R., Ascher D.B., Pires D.E., Furnham N. Mycobacterium tuberculosis whole genome sequencing and protein structure modelling provides insights into anti-tuberculosis drug resistance. BMC Med. 2016;14(1):31. doi: 10.1186/s12916-016-0575-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Portelli S., Myung Y., Furnham N., Vedithi S.C., Pires D.E.V., Ascher D.B. Prediction of rifampicin resistance beyond the RRDR using structure-based machine learning approaches. Sci Rep. 2020;10(1):18120. doi: 10.1038/s41598-020-74648-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vedithi S.C., Malhotra S., Skwark M.J., Munir A., Acebron-Garcia-De-Eulate M., Waman V.P. HARP: a database of structural impacts of systematic missense mutations in drug targets of Mycobacterium leprae. Comput Struct Biotechnol J. 2020;18:3692–3704. doi: 10.1016/j.csbj.2020.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vedithi S.C., Rodrigues C.H.M., Portelli S., Skwark M.J., Das M., Ascher D.B. Computational saturation mutagenesis to predict structural consequences of systematic mutations in the beta subunit of RNA polymerase in Mycobacterium leprae. Comput Struct Biotechnol J. 2020;18:271–286. doi: 10.1016/j.csbj.2020.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vedithi S.C., Malhotra S., Das M., Daniel S., Kishore N., George A. Structural implications of mutations conferring rifampin resistance in mycobacterium leprae. Sci Rep. 2018;8(1):5016. doi: 10.1038/s41598-018-23423-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tunstall T., Portelli S., Phelan J., Clark T.G., Ascher D.B., Furnham N. Combining structure and genomics to understand antimicrobial resistance. Comput Struct Biotechnol J. 2020;18:3377–3394. doi: 10.1016/j.csbj.2020.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Portelli S., Olshansky M., Rodrigues C.H.M., D'Souza E.N., Myung Y., Silk M. Exploring the structural distribution of genetic variation in SARS-CoV-2 with the COVID-3D online resource. Nat Genet. 2020;52(10):999–1001. doi: 10.1038/s41588-020-0693-3. [DOI] [PubMed] [Google Scholar]
- 32.Ascher D.B., Wielens J., Nero T.L., Doughty L., Morton C.J., Parker M.W. Potent hepatitis C inhibitors bind directly to NS5A and reduce its affinity for RNA. Sci Rep. 2014;4:4765. doi: 10.1038/srep04765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karmakar M., Globan M., Fyfe J.A.M., Stinear T.P., Johnson P.D.R., Holmes N.E. Analysis of a Novel pncA mutation for susceptibility to pyrazinamide therapy. Am J Respir Crit Care Med. 2018;198(4):541–544. doi: 10.1164/rccm.201712-2572LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karmakar M., Rodrigues C.H.M., Holt K.E., Dunstan S.J., Denholm J., Ascher D.B. Empirical ways to identify novel Bedaquiline resistance mutations in AtpE. PLoS One. 2019;14(5) doi: 10.1371/journal.pone.0217169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karmakar M., Rodrigues C.H.M., Horan K., Denholm J.T., Ascher D.B. Structure guided prediction of Pyrazinamide resistance mutations in pncA. Sci Rep. 2020;10(1):1875. doi: 10.1038/s41598-020-58635-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hawkey J., Ascher D.B., Judd L.M., Wick R.R., Kostoulias X., Cleland H. Evolution of carbapenem resistance in Acinetobacter baumannii during a prolonged infection. Microb Genom. 2018;4(3):-. doi: 10.1099/mgen.0.000165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Landrum, M. J.; Lee, J. M.; Riley, G. R.; Jang, W.; Rubinstein, W. S.; Church, D. M.; Maglott, D. R., ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 2014, 42 (Database issue), D980-5. [DOI] [PMC free article] [PubMed]
- 38.Tan M.H., Mester J., Peterson C., Yang Y., Chen J.L., Rybicki L.A. A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands. Am J Hum Genet. 2011;88(1):42–56. doi: 10.1016/j.ajhg.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mighell T.L., Thacker S., Fombonne E., Eng C., O'Roak B.J. An Integrated Deep-Mutational-Scanning Approach Provides Clinical Insights on PTEN Genotype-Phenotype Relationships. Am J Hum Genet. 2020;106(6):818–829. doi: 10.1016/j.ajhg.2020.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spinelli L., Black F.M., Berg J.N., Eickholt B.J., Leslie N.R. Functionally distinct groups of inherited PTEN mutations in autism and tumour syndromes. J Med Genet. 2015;52(2):128–134. doi: 10.1136/jmedgenet-2014-102803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Belmadani M., Jacobson M., Holmes N., Phan M., Nguyen T., Pavlidis P. VariCarta: A comprehensive database of harmonized genomic variants found in autism spectrum disorder sequencing studies. Autism Res. 2019;12(12):1728–1736. doi: 10.1002/aur.2236. [DOI] [PubMed] [Google Scholar]
- 42.Abrahams B.S., Arking D.E., Campbell D.B., Mefford H.C., Morrow E.M., Weiss L.A. SFARI Gene 2.0: a community-driven knowledgebase for the autism spectrum disorders (ASDs) Mol Autism. 2013;4(1):36. doi: 10.1186/2040-2392-4-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bubien V., Bonnet F., Brouste V., Hoppe S., Barouk-Simonet E., David A. French Cowden Disease, N., High cumulative risks of cancer in patients with PTEN hamartoma tumour syndrome. J Med Genet. 2013;50(4):255–263. doi: 10.1136/jmedgenet-2012-101339. [DOI] [PubMed] [Google Scholar]
- 44.Butler M.G., Dasouki M.J., Zhou X.P., Talebizadeh Z., Brown M., Takahashi T.N. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet. 2005;42(4):318–321. doi: 10.1136/jmg.2004.024646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Frazier T.W., Embacher R., Tilot A.K., Koenig K., Mester J., Eng C. Molecular and phenotypic abnormalities in individuals with germline heterozygous PTEN mutations and autism. Mol Psychiatry. 2015;20(9):1132–1138. doi: 10.1038/mp.2014.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hobert J.A., Embacher R., Mester J.L., Frazier T.W. 2nd; Eng, C., Biochemical screening and PTEN mutation analysis in individuals with autism spectrum disorders and macrocephaly. Eur J Hum Genet. 2014;22(2):273–276. doi: 10.1038/ejhg.2013.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klein S., Sharifi-Hannauer P., Martinez-Agosto J.A. Macrocephaly as a clinical indicator of genetic subtypes in autism. Autism Res. 2013;6(1):51–56. doi: 10.1002/aur.1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McBride K.L., Varga E.A., Pastore M.T., Prior T.W., Manickam K., Atkin J.F. Confirmation study of PTEN mutations among individuals with autism or developmental delays/mental retardation and macrocephaly. Autism Res. 2010;3(3):137–141. doi: 10.1002/aur.132. [DOI] [PubMed] [Google Scholar]
- 49.Mester J., Eng C. Estimate of de novo mutation frequency in probands with PTEN hamartoma tumor syndrome. Genet Med. 2012;14(9):819–822. doi: 10.1038/gim.2012.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.O'Roak B.J., Vives L., Fu W., Egertson J.D., Stanaway I.B., Phelps I.G. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012;338(6114):1619–1622. doi: 10.1126/science.1227764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Orrico A., Galli L., Buoni S., Orsi A., Vonella G., Sorrentino V. Novel PTEN mutations in neurodevelopmental disorders and macrocephaly. Clin Genet. 2009;75(2):195–198. doi: 10.1111/j.1399-0004.2008.01074.x. [DOI] [PubMed] [Google Scholar]
- 52.Rodriguez-Escudero I., Oliver M.D., Andres-Pons A., Molina M., Cid V.J., Pulido R. A comprehensive functional analysis of PTEN mutations: implications in tumor- and autism-related syndromes. Hum Mol Genet. 2011;20(21):4132–4142. doi: 10.1093/hmg/ddr337. [DOI] [PubMed] [Google Scholar]
- 53.Saskin A., Fulginiti V., Birch A.H., Trakadis Y. Prevalence of four Mendelian disorders associated with autism in 2392 affected families. J Hum Genet. 2017;62(6):657–659. doi: 10.1038/jhg.2017.16. [DOI] [PubMed] [Google Scholar]
- 54.Schwerd T., Khaled A.V., Schurmann M., Chen H., Handel N., Reis A. A recessive form of extreme macrocephaly and mild intellectual disability complements the spectrum of PTEN hamartoma tumour syndrome. Eur J Hum Genet. 2016;24(6):889–894. doi: 10.1038/ejhg.2015.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vanderver A., Tonduti D., Kahn I., Schmidt J., Medne L., Vento J. Characteristic brain magnetic resonance imaging pattern in patients with macrocephaly and PTEN mutations. Am J Med Genet A. 2014;164A(3):627–633. doi: 10.1002/ajmg.a.36309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Varga E.A., Pastore M., Prior T., Herman G.E., McBride K.L. The prevalence of PTEN mutations in a clinical pediatric cohort with autism spectrum disorders, developmental delay, and macrocephaly. Genet Med. 2009;11(2):111–117. doi: 10.1097/GIM.0b013e31818fd762. [DOI] [PubMed] [Google Scholar]
- 57.Wong C.W., Or P.M.Y., Wang Y., Li L., Li J., Yan M. Identification of a PTEN mutation with reduced protein stability, phosphatase activity, and nuclear localization in Hong Kong patients with autistic features, neurodevelopmental delays, and macrocephaly. Autism Res. 2018;11(8):1098–1109. doi: 10.1002/aur.1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yeung K.S., Tso W.W.Y., Ip J.J.K., Mak C.C.Y., Leung G.K.C., Tsang M.H.Y. Identification of mutations in the PI3K-AKT-mTOR signalling pathway in patients with macrocephaly and developmental delay and/or autism. Mol Autism. 2017;8(1):66. doi: 10.1186/s13229-017-0182-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q. Genome Aggregation Database, C., The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sali A., Blundell T.L. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234(3):779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 61.Pires D.E., Chen J., Blundell T.L., Ascher D.B. In silico functional dissection of saturation mutagenesis: Interpreting the relationship between phenotypes and changes in protein stability, interactions and activity. Sci Rep. 2016;6:19848. doi: 10.1038/srep19848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Konagurthu A.S., Lesk A.M., Allison L. Minimum message length inference of secondary structure from protein coordinate data. Bioinformatics. 2012;28(12):i97–i105. doi: 10.1093/bioinformatics/bts223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cock P.J., Antao T., Chang J.T., Chapman B.A., Cox C.J., Dalke A. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009;25(11):1422–1423. doi: 10.1093/bioinformatics/btp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Meszaros B., Erdos G., Dosztanyi Z. IUPred2A: context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018;46(W1):W329–W337. doi: 10.1093/nar/gky384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Grant B.J., Rodrigues A.P., ElSawy K.M., McCammon J.A., Caves L.S. Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics. 2006;22(21):2695–2696. doi: 10.1093/bioinformatics/btl461. [DOI] [PubMed] [Google Scholar]
- 66.Pires D.E., Ascher D.B., Blundell T.L. mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics. 2014;30(3):335–342. doi: 10.1093/bioinformatics/btt691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pires D.E.V., Rodrigues C.H.M., Ascher D.B. mCSM-membrane: predicting the effects of mutations on transmembrane proteins. Nucleic Acids Res. 2020;48(W1):W147–W153. doi: 10.1093/nar/gkaa416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rodrigues C.H.M., Pires D.E.V., Ascher D.B. DynaMut2: Assessing changes in stability and flexibility upon single and multiple point missense mutations. Protein Sci. 2021;30(1):60–69. doi: 10.1002/pro.3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pires, D. E.; Ascher, D. B.; Blundell, T. L., DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res 2014, 42 (Web Server issue), W314-9. [DOI] [PMC free article] [PubMed]
- 70.Rodrigues C.H., Pires D.E., Ascher D.B. DynaMut: predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. 2018;46(W1):W350–W355. doi: 10.1093/nar/gky300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Myung Y., Pires D.E.V., Ascher D.B. mmCSM-AB: guiding rational antibody engineering through multiple point mutations. Nucleic Acids Res. 2020;48(W1):W125–W131. doi: 10.1093/nar/gkaa389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Myung Y., Rodrigues C.H.M., Ascher D.B., Pires D.E.V. mCSM-AB2: guiding rational antibody design using graph-based signatures. Bioinformatics. 2020;36(5):1453–1459. doi: 10.1093/bioinformatics/btz779. [DOI] [PubMed] [Google Scholar]
- 73.Pires D.E., Ascher D.B. mCSM-AB: a web server for predicting antibody-antigen affinity changes upon mutation with graph-based signatures. Nucleic Acids Res. 2016;44(W1):W469–W473. doi: 10.1093/nar/gkw458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nemethova M., Radvanszky J., Kadasi L., Ascher D.B., Pires D.E.V., Blundell T.L. Twelve novel HGD gene variants identified in 99 alkaptonuria patients: focus on ‘black bone disease’ in Italy. Eur J Hum Genet. 2016;24(1):66–72. doi: 10.1038/ejhg.2015.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pires D.E.V., Ascher D.B. mCSM-NA: predicting the effects of mutations on protein-nucleic acids interactions. Nucleic Acids Res. 2017;45(W1):W241–W246. doi: 10.1093/nar/gkx236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pires D.E., Blundell T.L., Ascher D.B. mCSM-lig: quantifying the effects of mutations on protein-small molecule affinity in genetic disease and emergence of drug resistance. Sci Rep. 2016;6:29575. doi: 10.1038/srep29575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rodrigues C.H.M., Myung Y., Pires D.E.V., Ascher D.B. mCSM-PPI2: predicting the effects of mutations on protein-protein interactions. Nucleic Acids Res. 2019;47(W1):W338–W344. doi: 10.1093/nar/gkz383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jubb H.C., Higueruelo A.P., Ochoa-Montano B., Pitt W.R., Ascher D.B., Blundell T.L. Arpeggio: A Web Server for Calculating and Visualising Interatomic Interactions in Protein Structures. J Mol Biol. 2017;429(3):365–371. doi: 10.1016/j.jmb.2016.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ashkenazy H., Abadi S., Martz E., Chay O., Mayrose I., Pupko T. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016;44(W1):W344–W350. doi: 10.1093/nar/gkw408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vaser R., Adusumalli S., Leng S.N., Sikic M., Ng P.C. SIFT missense predictions for genomes. Nat Protoc. 2016;11(1):1–9. doi: 10.1038/nprot.2015.123. [DOI] [PubMed] [Google Scholar]
- 81.Hecht M., Bromberg Y., Rost B. Better prediction of functional effects for sequence variants. BMC Genom. 2015;16 Suppl 8 (8):S1. doi: 10.1186/1471-2164-16-S8-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Choi Y., Chan A.P. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31(16):2745–2747. doi: 10.1093/bioinformatics/btv195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Silk M., Petrovski S., Ascher D.B. MTR-Viewer: identifying regions within genes under purifying selection. Nucl Acids Res. 2019;47(W1):W121–W126. doi: 10.1093/nar/gkz457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Traynelis J., Silk M., Wang Q., Berkovic S.F., Liu L., Ascher D.B. Optimizing genomic medicine in epilepsy through a gene-customized approach to missense variant interpretation. Genome Res. 2017;27(10):1715–1729. doi: 10.1101/gr.226589.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hildebrand J.M., Kauppi M., Majewski I.J., Liu Z., Cox A.J., Miyake S. A missense mutation in the MLKL brace region promotes lethal neonatal inflammation and hematopoietic dysfunction. Nat Commun. 2020;11(1):3150. doi: 10.1038/s41467-020-16819-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jatana N., Ascher D.B., Pires D.E.V., Gokhale R.S., Thukral L. Human LC3 and GABARAP subfamily members achieve functional specificity via specific structural modulations. Autophagy. 2020;16(2):239–255. doi: 10.1080/15548627.2019.1606636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pandurangan A.P., Ochoa-Montano B., Ascher D.B., Blundell T.L. SDM: a server for predicting effects of mutations on protein stability. Nucleic Acids Res. 2017;45(W1):W229–W235. doi: 10.1093/nar/gkx439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Frappier V., Chartier M., Najmanovich R.J. ENCoM server: exploring protein conformational space and the effect of mutations on protein function and stability. Nucleic Acids Res. 2015;43(W1):W395–W400. doi: 10.1093/nar/gkv343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Portelli S., Phelan J.E., Ascher D.B., Clark T.G., Furnham N. Understanding molecular consequences of putative drug resistant mutations in Mycobacterium tuberculosis. Sci Rep. 2018;8(1):15356. doi: 10.1038/s41598-018-33370-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Welch B.L. The generalisation of student's problems when several different population variances are involved. Biometrika. 1947;34(1–2):28–35. doi: 10.1093/biomet/34.1-2.28. [DOI] [PubMed] [Google Scholar]
- 91.Team R. C. R A language and environment for statistical computing., R Foundation for Statistical Computing 2019 Vienna, Austria.
- 92.Wold S., Esbensen K., Geladi P. Principal component analysis. Chemomet Intell Lab Syst. 1987;2(1):37–52. [Google Scholar]
- 93.McInnes, L., John Healy, and James Melville, Umap: Uniform manifold approximation and projection for dimension reduction. arXiv preprint arXiv:1802.03426 (2018). 2018.
- 94.Pedregosa F., Varoquaux I., Gramfort A., Michel V., Thirion B. Machine Learning in Python. J. Mach. Learn. Res. 2011;12:2825–2830. [Google Scholar]
- 95.Guo X., Yin Y., Dong C., Yang G., Zhou G. In On the class imbalance problem, Fourth international conference on natural computation. IEEE. 2008;2008:192–201. [Google Scholar]
- 96.da Silveira C.H., Pires D.E., Minardi R.C., Ribeiro C., Veloso C.J., Lopes J.C. Protein cutoff scanning: A comparative analysis of cutoff dependent and cutoff free methods for prospecting contacts in proteins. Proteins. 2009;74(3):727–743. doi: 10.1002/prot.22187. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.