

Abstract
Advanced pancreatic cancer is characterized by few treatment options and poor outcomes. Oncolytic virotherapy and chemotherapy involve complementary pharmacodynamics and could synergize to improve therapeutic efficacy. Likewise, multimodality treatment may cause additional toxicity, and new agents have to be safe. Balancing both aims, we generated an oncolytic measles virus for 5-fluorouracil-based chemovirotherapy of pancreatic cancer with enhanced tumor specificity through microRNA-regulated vector tropism. The resulting vector encodes a bacterial prodrug convertase, cytosine deaminase-uracil phosphoribosyl transferase, and carries synthetic miR-148a target sites in the viral F gene. Combination of the armed and targeted virus with 5-fluorocytosine, a prodrug of 5-fluorouracil, resulted in cytotoxicity toward both infected and bystander pancreatic cancer cells. In pancreatic cancer xenografts, a single intratumoral injection of the virus induced robust in vivo expression of prodrug convertase. Based on intratumoral transgene expression kinetics, we devised a chemovirotherapy regimen to assess treatment efficacy. Concerted multimodality treatment with intratumoral virus and systemic prodrug administration delayed tumor growth and prolonged survival of xenograft-bearing mice. Our results demonstrate that 5-fluorouracil-based chemovirotherapy with microRNA-sensitive measles virus is an effective strategy against pancreatic cancer at a favorable therapeutic index that warrants future clinical translation.
Keywords: chemovirotherapy, cytosine deaminase, fluorouracil, measles virus, microRNAs, miR-148a, oncolytic viruses, pancreatic cancer, prodrugs, virotherapy
Graphical abstract
This study describes a novel, virus-based therapeutic approach for treatment of pancreatic cancer. Our results demonstrate that 5-fluorouracil-based chemovirotherapy with microRNA-sensitive and prodrug convertase-encoding measles virus is an effective strategy against pancreatic cancer at a favorable therapeutic index.
Introduction
Patients with pancreatic ductal adenocarcinoma (PDAC) frequently present with unresectable tumors or develop metastases despite primary resection.1 Treatment options for these patients are limited to palliative chemotherapy and best supportive care. Fluorinated pyrimidines were the first substances with clinical benefit in PDAC, leading to approval of gemcitabine (2,2-difluorodeoxycytidine) in 1996.2 Outcomes have been improved with subsequent combination chemotherapies, several of which incorporate 5-fluorouracil (2,4-dioxo-5-fluoropyrimidine [5-FU]).3 Today, systemic 5-FU with irinotecan and oxaliplatin (FOLFIRINOX) is the first-line regimen for advanced PDAC in patients with good performance status.4 Fluoropyrimidine-based adjuvant chemotherapy includes dose-modified FOLFIRINOX or gemcitabine plus capecitabine (5-fluoro-N-[(pentyloxy)carbonyl]-deoxycytidine), a prodrug of 5-FU.5,6 However, 5-year overall survival rates of ~9% underline that in most cases PDAC recurs and remains treatment-refractory despite multidisciplinary management.7
Oncolytic viruses (OVs) constitute a class of cancer therapeutics based on replication-competent viral vectors. Using multiple mechanisms of action, OVs may be capable of overcoming resistance to standard cancer therapies.8, 9, 10 Clinical observations of measles virus (MeV) inducing anti-tumor effects in naturally infected hosts motivated the generation of oncolytic MeV from attenuated vaccine strains.11 The MeV platform has since been developed toward clinical applicability in several tumor entities.12, 13, 14, 15 To this end, we and others have engineered MeV to express a wide range of therapeutic transgenes.16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26
Prodrug convertases catalyze the bioactivation of drugs from nontoxic precursors and can be used to enrich tumor tissue for active antineoplastics.27 Well-studied examples include a fusion protein of two Escherichia coli enzymes, cytosine deaminase and uracil phosphoribosyl transferase (CD-UPRT), which converts 5-fluorocytosine (5-FC) into 5-FU and its active metabolite 5-fluorouridine monophosphate (5-FUMP).28,29 We reasoned that the combination of oncolytic MeV and the CD-UPRT/5-FC system might be a promising approach in PDAC, based on the most effective chemotherapy regimens currently in clinical practice.
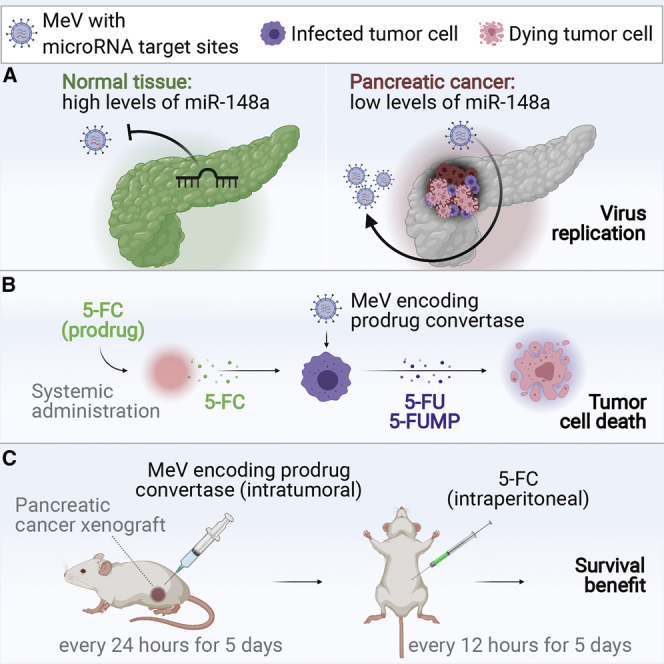
Besides therapeutic efficacy, patient safety determines the successful clinical translation of OVs. To avoid side effects, tumor-specific virus replication is critical. Cells acquiring malignant properties frequently develop altered expression levels of cellular small non-coding RNA, including microRNA (miRNA).30 In PDAC, microRNA expression profiling has revealed downregulation of miR-148a relative to normal parenchyma as a recurrent feature.31,32 Cancer-specific microRNA profiles can be exploited to generate post-entry targeted OVs with tumor-directed tropism.33 This is achieved by the introduction of microRNA target sites (miRTSs), which allow the cellular RNA interference machinery to act on viral genomes or transcripts.34, 35, 36 We have previously shown that physiologic microRNA expression in non-tumor tissue confines the propagation of microRNA-sensitive MeV and protects virus-exposed organs from adverse toxicity.37,38 In contrast, downregulation of the respective microRNA within tumors allows microRNA-sensitive MeV to replicate and to exert oncolytic effects.
In this study, we generated and characterized a miR-148a-sensitive oncolytic MeV expressing CD-UPRT as a therapeutic transgene. We show that 5-FU-based chemovirotherapy with miR-148a-sensitive MeV is feasible and effective against PDAC in vitro and in vivo. This is the first study linking microRNA-based vector reprogramming for tumor-specific replication with prodrug-based MeV chemovirotherapy.
Results
Generation of miR-148a-sensitive CD-UPRT-armed MeV
Recombinant vectors were based on the attenuated Edmonston tag laboratory strain of MeV. To produce transgene-armed MeV (MeV-CD) for 5-FU-based chemovirotherapy, an additional transcription unit encoding E. coli CD-UPRT was inserted upstream of the viral N gene (Figure 1A). MeV encoding enhanced green fluorescent protein (EGFP) instead of CD-UPRT was used for experiments involving fluorescence microscopy. To engineer microRNA-sensitive MeV, a miRTS box comprising three identical target sequences fully complementary to miR-148a-3p was designed as previously described (Figure 1B).38 The miRTS box was cloned into the 3′ untranslated region (UTR) of the MeV F gene resulting in MeV-CD-FmiRTS148a and MeV-EGFP-FmiRTS148a. MeV F encodes the viral fusion glycoprotein (F), which mediates membrane fusion at cell entry and lateral spread of MeV through formation of multicellular syncytia.39,40 MeV vectors harboring non-functional miRTS (either reverse complementary or reverse of the original target sequence) were generated as controls (MeV-CD-FmiRTS148aRC, MeV-EGFP-FmiRTS148aRC, and MeV-EGFP-FmiRTS148aREV). Due to co-transcriptional encapsidation of the MeV genome, cellular microRNAs can act on viral mRNA but not on MeV genomic RNA.37
Figure 1.

Generation of microRNA-sensitive CD-UPRT-armed MeV
(A) Genome representation of modified MeV. Generated vectors encode an additional transcription unit for CD-UPRT or EGFP upstream of the MeV N gene and a microRNA target site (miRTS) box in the 3′ UTR of the MeV F gene. Control vectors harbor non-functional reverse or reverse complementary miRTS. (B) Nucleotide sequence of the functional miRTS box. The orientation corresponds to viral messenger RNA. Three identical target sites complementary to mature miR-148a (underlined indicates seed sequence) are separated by non-coding heptanucleotide spacers.
Attenuation of microRNA-sensitive MeV in the presence of miR-148a
To model the exposure of non-tumor tissue to microRNA-sensitive MeV, we first used the virus production cell line Vero (African green monkey kidney cells). Vero cells were transfected with miR-148a (or subjected to mock transfection) prior to infection with miR-148a-sensitive MeV. Virus spread, progeny virus titers, and post-infection cell viability were determined. miR-148a reduced the spread of MeV-EGFP-FmiRTS148a across the cell layer as indicated by limited syncytia formation (Figure 2A). Compared to mock transfection, this resulted in a significant reduction of EGFP-positive cells from ~76% to ~34% at 40 h post-infection (p = 0.006, analysis of variance [ANOVA] and Tukey’s test, Figure 2B). In line with limited virus dissemination, progeny titers of MeV-CD-FmiRTS148a at 48 h post-infection were reduced ~7-fold in the presence of miR-148a (p < 0.001, Figure 2C). Finally, inhibition of syncytia formation and virus replication attenuated cell lysis by microRNA-sensitive MeV (Figure 2D). This preserved >60% viability among miR-148a-treated cells compared to ~16% in the absence of miR-148a (p = 0.001). On the contrary, miR-148a treatment had no effect on virus spread, progeny titers, or cytotoxicity of control MeV harboring non-functional miRTS (MeV-EGFP-FmiRTS148aRC).
Figure 2.

Regulation of microRNA-sensitive MeV in the presence of miR-148a
(A–D) Vero cells were transfected with miR-148a or subjected to mock transfection prior to infection with microRNA-sensitive MeV at a multiplicity of infection (MOI) of 0.03. (A) Syncytia formation and EGFP expression at 36 h post-infection. Fluorescence microscopy images were acquired using a ×5 objective. (B) Virus dissemination assay. The proportion of infected cells was determined by computational segmentation of fluorescence microscopy images obtained at 40 h post-infection (n = 3). (C) Progeny virus titers at 48 h post-infection expressed as cell infectious units (CIU) per mL (n = 4). (D) Cell viability at 64 h post-infection (n = 3). Each dataset in (B)–(D) represents pooled replicates from independent experiments. Median, first and third quartiles, and range are plotted. (E–G) IMIM-PC2 cells transduced to stably express miR-148a (IMIM-PC2-148a) were infected with microRNA-sensitive MeV or control viruses as indicated. Outcome measures were normalized to those in control (empty lentiviral vector-transduced) IMIM-PC2-LV cells treated in parallel (dotted lines). (E) Syncytia formation and EGFP expression at 36 h post-infection of IMIM-PC2-148a with MeV-EGFP-FmiRTS148a (MOI of 0.3) determined by computational segmentation of fluorescence microscopy images. Two biological replicates per group and mean values (gray) are shown. (F) Progeny virus titers at 48 h post-infection of IMIM-PC2-148a (MOI of 0.3). The dotted line corresponds to reference titers on IMIM-PC2-LV. Data represent biological triplicates; median, first and third quartiles, and range are plotted. (G) Viability of IMIM-PC-148a cells after treatment with CD-UPRT-armed MeV in the presence and absence of prodrug. Cells were infected with MeV-CD-FmiRTS148a or MeV-CD (MOI of 1), followed by 5-FC addition (1 mM) or mock treatment at 24 h post-infection. An XTT assay was performed at 48 h post-treatment, and cell viability was calculated relative to IMIM-PC2-LV. Data represent biological quadruplicates; median, first and third quartiles, and range are plotted.
We then studied microRNA-based MeV attenuation in miR-148a-expressing cells of pancreatic origin, as a representative compartment exposed to potential off-target effects of MeV. To this end, microRNA-sensitive MeV was used to infect a PDAC cell line stably transduced to express miR-148a (IMIM-PC2-148a).41 Outcomes in IMIM-PC2-148a cells were normalized to those in the control-transduced cell line IMIM-PC2-LV to analyze virus-specific differences related to miR-148a presence. MeV spread, progeny virus titers, and cell viability upon infection of IMIM-PC2-148a were assessed (Figures 2E–2G). We observed miR-148a-specific attenuation of MeV-EGFP-FmiRTS148a both in terms of virus spread and progeny virus titers. Relative to IMIM-PC2-LV controls, miR-148a expression led to a reduction in EGFP-positive cells of ~75% and a reduction in progeny virus titers of ~1 log-fold after infection with MeV-EGFP-FmiRTS148a (Figures 2E and 2F). Cell viability of IMIM-PC-148a was assessed after exposition to CD-UPRT-armed MeV with and without subsequent prodrug addition (Figure 2G). Infection with MeV-CD-FmiRTS148a alone resulted in reduced cytotoxicity toward IMIM-PC2-148a compared to IMIM-PC2-LV controls, with ~30% excess viability of miR-148a-expressing cells. Conversely, we did not observe a net effect of miR-148a-specific attenuation in IMIM-PC2-148a cells when MeV-CD-FmiRTS148a was combined with 5-FC. Interestingly, IMIM-PC2-148a cells were less susceptible than the more rapidly proliferating IMIM-PC2-LV in terms of virus replication and cytotoxicity also of control MeV.41 While these nonspecific effects were less pronounced than the additional specific attenuation of microRNA-sensitive MeV, they may point toward cellular miR-148a functions that support MeV tumor specificity in general.
Infection of non-tumor tissue with MeV-EGFP-FmiRTS148a was further assessed in live primary human liver specimens (Figure S5). Fluorescence microscopy at 24–144 h after ex vivo infection suggested formation of fewer EGFP-positive syncytia with MeV-EGFP-FmiRTS148a compared to control MeV. In principle, these observations are in line with the specific attenuation of microRNA-sensitive MeV at physiologic levels of hepatic miR-148a. However, the small sample size precluded an additional quantitative analysis to statistically confirm this notion.
Taken together, our evaluation of microRNA de-targeting in several models of non-target tissue indicates that miR-148a-mediated regulation of MeV F protein expression is suitable to protect susceptible cells from potential off-target effects of MeV-CD-FmiRTS148a.
Endogenous expression of miR-148a in PDAC cells
Before moving on to infection of target PDAC cells, we characterized the endogenous expression of miR-148a in our panel of PDAC cell lines. Absolute quantification revealed mean copy numbers of miR-148a ranging from 9 to 5,131 molecules per cell (Figure 3). We found very low levels of miR-148a in IMIM-PC2 and MIA PaCa-2 cells (~9 and ~43 molecules per cell), followed by T3M4 and BxPC-3 cells (~1,359 and ~1,489 molecules per cell). Endogenous expression of miR-148a was highest in Capan-1 cells (~5,131 molecules per cell). PDAC cells stably transduced to express miR-148a (IMIM-PC2-148a) were included as a positive control (22,816 molecules per cell).41 We also confirmed low expression of miR-148a by the virus production cell line Vero (~337 molecules per cell).
Figure 3.

Abundance of microRNA-148a in PDAC cells
Total RNA was isolated from 3 × 105 cells per cell line. The average number of miR-148a molecules per cell was determined by quantitative real-time PCR using a standard curve. The stably transduced cell line IMIM-PC2-148a served as a positive control. Data represent mean values and SD of three technical replicates, respectively.
Replication of MeV-CD-FmiRTS148a in PDAC cells
MeV infection of target cells and replication characteristics were assessed in PDAC cell lines representing subtypes with epithelial (BxPC-3) or mesenchymal (MIA PaCa-2, T3M4) differentiation.42,43 Syncytia formation and EGFP expression were similar with MeV-EGFP-FmiRTS148a and MeV-EGFP-FmiRTS148aRC but differed between individual cell lines (Figure 4A). Virus growth curves of MeV-CD-FmiRTS148a also indicated varying replication of MeV in different PDAC cells (Figure 4B). MeV-CD-FmiRTS148a yielded maximum titers of 8.4 × 102, 6.2 × 103, and 3.1 × 104 cell infectious units (CIU) per mL in BxPC-3, MIA PaCa-2, and T3M4 cells, respectively. Overall, titers of MeV expressing the CD-UPRT transgene did not differ substantially from those of MeV-EGFP on PDAC cells. Likewise, replication efficiency was in the same range with both MeV-CD-FmiRTS148a and control MeV-CD harboring the unmodified F gene.
Figure 4.

MeV replication in PDAC cells
(A) Fluorescence microscopy. PDAC cells were infected with MeV-EGFP-FmiRTS148a or MeV-EGFP-FmiRTS148aRC (MOI of 0.3), and images were acquired at 64 h post-infection. Scale bars represent 50 μm. (B) Virus growth curves. Progeny virus titers were determined at the indicated time points after infection of PDAC cells with MeV-EGFP, MeV-CD, or MeV-CD-FmiRTS14a (MOI of 0.03). Data represent mean values ± SD of three biological replicates.
Prodrug activation upon MeV-CD-FmiRTS148a infection
To examine the transgene expression kinetics and functionality of CD-UPRT in PDAC cells upon infection with MeV-CD-FmiRTS148a, we used a photometric approach to determine prodrug turnover. BxPC-3 cells were infected with MeV-CD-FmiRTS148a or unmodified control MeV at a multiplicity of infection (MOI) of 0.1. Repeated sampling of cell lysates every 12 h revealed a marked increase in CD-UPRT enzymatic activity between 24 and 48 h after infection. This was followed by constant, sustained 5-FC turnover throughout the observation period of 72 h (Figure 5). As expected, infection with control MeV lacking the therapeutic transgene had no effect on prodrug concentration. Taken together, this indicated protein expression, stability, and sustained activity of CD-UPRT in PDAC cells after infection with MeV-CD-FmiRTS148a.
Figure 5.

Transgene activity in MeV-infected PDAC cells
To determine prodrug turnover in target cells, BxPC-3 cells were infected with MeV-CD-FmiRTS148a or parental MeV (MOI of 0.1). At designated time points after infection, cell lysates were collected, heat-inactivated, and incubated with 5-FC (1.5 mM) until readout. (A) Spectrophotometry. The dotted line corresponds to 1.5 mM 5-FC. (B) CD-UPRT activity over time. Absorbance data shown in (A) were normalized to MeV-treated controls and maximum activity was set to 100%. Mean values ± SD of three biological replicates are shown.
Combined cytotoxicity of MeV-CD-FmiRTS148a and prodrug in PDAC cells
The cytotoxicity of miR-148a-sensitive, CD-UPRT-armed MeV in combination with 5-FC was studied both in infected and in non-infected (bystander) PDAC cells. To evaluate the effects on infected cells, BxPC-3, Capan-1, MIA PaCa-2, and T3M4 cells were inoculated with MeV-CD-FmiRTS148a or control MeV (MOI of 0.1). 5-FC was added at 36 h post-infection, and cell viability was determined repeatedly after combination treatment. The resulting time course up to 72 h post-treatment is summarized in Figure 6.
Figure 6.

Cytotoxicity of MeV-CD-FmiRTS148a in combination with prodrug
Cell viability of PDAC cells was studied after MeV infection and 5-FC treatment. Cells were infected with MeV-EGFP, MeV-CD, MeV-CD-FmiRTS148a, or MeV-CD-FmiRTS148aRC (MOI of 0.1) followed by addition of 1 mM 5-FC at 36 h post-infection. An XTT assay was performed every 24 h after combination treatment, and cell viability was calculated relative to uninfected controls. (Left) Cell viability over time. (Right) Endpoint cell viability at 72 h post-treatment. Data represent mean ± SEM from two experiments each performed in biological quadruplicates.
We found variable degrees of cytolysis among PDAC cells that were treated with MeV alone. MeV-CD-FmiRTS148a infection had the most pronounced effect on MIA PaCa-2 cells (~32% mean viability at 72 h relative to mock-treated cells). On the contrary, Capan-1 cells were largely refractory to MeV cytolysis (~92% mean viability at 72 h). Importantly, in the absence of prodrug the cytotoxicity of MeV-CD-FmiRTS148a and MeV-EGFP across all cell lines was not significantly different (p > 0.99, ANOVA and Tukey’s test). This indicated that MeV cytolysis was unaffected by the insertion of the larger CD-UPRT transgene.
Irrespective of the variable susceptibility of PDAC cells to MeV infection, the addition of 5-FC to CD-armed MeV consistently reinforced cytotoxicity. Averaged across all cell types, this further reduced mean cell viability by ~29% (95% confidence interval [CI], 29% ± 7%, p < 0.001) compared to MeV alone. In pairwise comparisons of MeV-CD-FmiRTS148a plus 5-FC and MeV-CD-FmiRTS148a, the effect varied between cell lines and ranged from ~22% to ~38% of additional toxicity after combination treatment (p < 0.05 for each comparison). Addition of 5-FC exerted the highest toxicity in T3M4 cells (~26% versus ~63% mean viability at 72 h) and in Capan-1 cells (~54% versus ~92% mean viability at 72 h). The detected differences in cell-specific susceptibility to MeV and 5-FC did not indicate any cross-resistance between the respective treatment components. Overall, these findings were in line with the observed sensitivity of the tested PDAC cell lines toward 5-FU (Figure S1A).
As expected, addition of 5-FC did not significantly alter the viability of MeV-EGFP-infected cells (p = 0.81). This confirmed that interactions of 5-FC and MeV depended on CD-UPRT expression. We also noted that the simultaneous insertion of CD-UPRT and miRTS148a did not impair the effects of MeV on PDAC cells, as combination treatment with MeV-CD-FmiRTS148a or MeV-CD was similarly effective. Likewise, there was no indication of a correlation between cellular miR-148a expression levels and 5-FU sensitivity in our panel of PDAC cells (Figure S1B). Taken together, we did not observe any interference of miRTS148a or CD-UPRT with the therapeutic efficacy of MeV in target cells.
In a post hoc analysis of the results with MeV-CD-FmiRTS148a chemovirotherapy, we analyzed the observed efficacy after stratification by molecular PDAC subtype, following a transcriptomic classification of cell lines based on the PDAC subtypes defined by Moffitt et al.44,45 The analysis was restricted to cell lines within our panel with available conclusive classifications. We found more pronounced cytotoxicity in cell lines of the “basal” (BxPC-3 and T3M4) versus “classical” (Capan-1) subtype of PDAC (Figure S2). This relationship was consistent both for treatment with MeV-CD-FmiRTS148a alone and for MeV-CD-FmiRTS148a plus prodrug (p < 0.05 for each comparison, Mann-Whitney U test).
We went on to assess whether the release of activated prodrug by infected cells allowed for an expansion of cytotoxicity to non-infected (bystander) PDAC cells. To this end, PDAC cells were exposed to culture medium collected from donor cells previously treated with MeV-CD-FmiRTS148a plus 5-FC. Different dilutions (1:2 to 1:1,000) of transferred medium were used in parallel and cell viability was assessed after 72 h. Medium derived from combination-treated cells induced a concentration-dependent toxicity on naive PDAC cells, which resulted in a minimal mean cell viability of ~10% (BxPC-3 and MIA PaCa-2), ~16% (T3M4), and ~20% (IMIM-PC2) after 72 h (Figure 7). Cytotoxic effects were reproducible on all PDAC cell lines at dilutions of activated medium ranging from 1:2 to 1:100 (p < 0.05 for pairwise comparisons to mock-treated controls). All cell lines except MIA PaCa-2 also responded to medium from combination-treated cells at a dilution of 1:1,000. Medium from MeV only-treated controls had no effect on PDAC cell viability. Medium derived from 5-FC only-treated cells did affect the viability of T3M4 cells when applied at a high concentration (1:2). This is in line with T3M4 cell responses to prodrug concentrations exceeding 0.5 mM in a separate dose escalation experiment (data not shown). However, medium derived from combination treatment at 1:2 dilution was still associated with superior cytotoxicity toward T3M4 cells compared to medium containing 5-FC only (mean difference and 95% CI, 43% ± 18%, p < 0.0001).
Figure 7.

Bystander effect of 5-FC activation on PDAC cells
Cell viability of bystander PDAC cells after chemovirotherapy. PDAC cells were exposed to four different dilutions of heat-inactivated medium from either combination-treated (MeV-CD-FmiRTS148a + 1 mM 5-FC) or control-treated Vero cells. After 72 h of incubation with donor cell medium, cell viability was determined by an XTT assay and calculated relative to untreated controls. Mean values ± SD of four biological replicates are shown.
In addition to cell viability, we studied the expression of apoptosis and cell proliferation markers in bystander PDAC cells. Cells were treated with activated medium at 1:2 or 1:10 dilution as described, and immunofluorescence staining was performed after 48–72 h to quantify the proportion of cells expressing cleaved caspase-3 or Ki-67. Fluorescence imaging results with quantitative analysis are summarized in Figure S3. We observed a significant increase in the fraction of cleaved caspase-3-positive cells across all cell lines (p < 0.05 for all comparisons, Pearson’s chi-square test) after exposition to activated medium from combination-treated donor cells (Figures S3A and S3B). This effect was pronounced in Capan-1 and MIA PaCa-2 cells, and modest in BxPC-3 and T3M4 cells. With regard to Ki-67 expression, immunofluorescence results were less consistent, and highly variable fractions of positive cells were found both across different cell lines and between treatment groups (Figure S3B). In line with the previously documented cytotoxicity of activated culture medium, treatment at both dilutions markedly reduced the density of the cell layer indicated by the total number of DAPI (4',6-diamidino-2-phenylindole)-positive cells per field of view.
Collectively, these data demonstrate the oncolytic activity of MeV-CD-FmiRTS148a in PDAC cells and show complementary effects with the addition of 5-FC. Furthermore, activated prodrug released from MeV-CD-FmiRTS148a-treated cells expanded the toxicity to non-infected bystander PDAC cells.
Dynamics of transgene expression in PDAC xenografts
The successful translation of chemovirotherapy in vivo requires a coordinated application of complementary treatment modalities. To develop a treatment schedule of intratumoral MeV-CD-FmiRTS148a and systemic prodrug, we explored the temporal pattern of CD-UPRT expression in infected PDAC tumors. Non-obese diabetic (NOD)/severe combined immunodeficiency (SCID)/gamma (NSG) mice with subcutaneous (s.c.) T3M4 tumors received one intratumoral injection with 5 × 106 CIU of MeV-CD-FmiRTS148a. Whole tumors were obtained for protein isolation and immunodetection of CD-UPRT up to 7 days after MeV administration. We detected intratumoral CD-UPRT protein at all time points studied. As revealed by semiquantitative immunoblot of whole tumor lysates, CD-UPRT levels peaked at 48 h after virus injection (Figure S4). While at 12 h the relative CD-UPRT level tended to be very low, it increased markedly by 24 h post-infection, indicating de novo transgene expression in infected tumors. CD-UPRT protein persisted at detectable quantities for at least 7 days following a single virus injection.
Therapeutic efficacy of chemovirotherapy in vivo
We then explored the therapeutic efficacy of MeV-CD-FmiRTS148a chemovirotherapy in vivo. NSG mice bearing subcutaneous T3M4 tumors were treated with daily intratumoral injections of 6.9 × 106 CIU of MeV-CD-FmiRTS148a on 5 consecutive days. In order to synchronize CD-UPRT expression and 5-FC bioavailability, intraperitoneal 5-FC treatment was initiated 48 h after the first MeV injection and continued at 200 mg per kg twice daily for 5 days. Treatment effects on tumor growth and overall survival were assessed.
Tumor growth kinetics for individual animals and between-group comparisons are shown in Figures 8A and 8B. Mock-treated tumors reached a mean volume of 505 mm3 on day 16 after implantation, the last day all animals were alive (Figure 8C). Compared to mock-treated controls, both MeV-CD-FmiRTS148a alone and MeV-CD-FmiRTS148a plus 5-FC significantly reduced mean tumor volume (p = 0.005 and p = 0.0001, ANOVA and Tukey’s test). Mean tumor volume on day 16 was lower after treatment with MeV-CD-FmiRTS148a plus 5-FC (158 mm3) than after MeV-CD-FmiRTS148a alone (253 mm3), but the difference was not statistically significant (p = 0.52). Systemic 5-FC alone had no significant effect on mean tumor volume compared to mock treatment (p = 0.99). Survival outcomes are summarized in Figure 8D. Chemovirotherapy with MeV-CD-FmiRTS148a plus 5-FC significantly prolonged animal survival compared to MeV-CD-FmiRTS148a alone (p = 0.039, log-rank test). Mice treated with MeV-CD-FmiRTS148a plus 5-FC survived a median of 28 days after tumor implantation, in contrast to 18 days with mock treatment. While local administration of MeV-CD-FmiRTS148a alone also prolonged survival compared to mock treatment (p = 0.002), systemic 5-FC alone had no significant effect (p = 0.19). There were no events indicating 5-FC-associated toxicity. In summary, combination treatment with intratumoral virus and systemic pro-drug administration effected the most prolonged survival of mice bearing PDAC xenografts.
Figure 8.

Efficacy of MeV-CD-FmiRTS148a chemovirotherapy in vivo
Subcutaneous T3M4 tumors were implanted into the right flank of NOD/SCID/gamma mice. On day 9 post-implantation, mice were treated with daily intratumoral injections of MeV-CD-FmiRTS148a (6.9 × 106 CIU) for 5 days. Starting 48 h after the first virus injection, intraperitoneal 5-FC (200 mg/kg) was applied twice daily for 5 days. Control-treated mice received either MeV-CD-FmiRTS148a, 5-FC, or mock treatment. A total number of 36 animals were treated in the study (n = 9 per group). (A) Tumor growth dynamics of individual mice. Arrows indicate intratumoral injections, and gray areas mark the duration of intraperitoneal treatments. (B) Tumor growth trajectories aggregated by treatment group. Regression lines (dashed) based on the data in (A) with 95% confidence bands (colored) are shown. (C) Tumor volume of individual mice (gray) on day 16 after tumor implantation (the last day all animals were alive). Horizontal bars indicate mean values. (D) Kaplan-Meier survival analysis; p-value was obtained by log-rank test comparing MeV-CD-FmiRTS148a + 5-FC and MeV-CD-FmiRTS148a alone.
Discussion
This study introduces microRNA-sensitive CD-UPRT-armed oncolytic MeV with systemic 5-FC as a novel chemovirotherapeutic approach for advanced PDAC.
To limit adverse effects of chemovirotherapy in patients, spatial control of virus replication is important. The viral hemagglutinin protein and its cellular receptors, CD46, SLAMF1, and nectin-4, mediate the cell entry of MeV vaccine strains.46,47 Overexpression of these receptors in epithelial cancers, including PDAC, facilitates MeV cytolysis.48, 49, 50 Other nectin-4-directed agents have been evaluated in clinical trials which underscore its use as a molecular target for cancer therapy.51 Nonetheless, the physiologic expression of both CD46 and nectin-4 in non-tumor tissues limits the tumor selectivity of MeV at cell entry.
Our study applies the regulation of MeV by miR-148a as a safety strategy to direct MeV replication toward PDAC. Comprehensive work has defined the role of miR-148a in cancer.52 In PDAC, miR-148a expression is frequently suppressed, a finding we could reproduce in our panel of PDAC cell lines. In this study, we evaluated miRTSs introduced in the 3′ UTR of MeV F, conferring intermediate regulation compared to alternative locations within the MeV genome. We have recently shown that positional effects of miRTS insertion are useful to fine-tune vector control, and introduced microRNA targeting of several MeV genes in parallel.38,53 Clearly, individual clinical applications and tumor characteristics will guide the optimal selection of both miRTS species and insertion sites when moving toward clinical trials.
Importantly, the insertion of miRTS did not affect the therapeutic efficacy of MeV. This is in line with the overall low expression levels of miR-148a in PDAC. In a previous study in PDAC xenografts, a related multi-microRNA-sensitive MeV also retained the same in vivo efficacy as the respective control vector.38 However, we still observed variable sensitivity to MeV infection among different cell lines. Our findings reflect the molecular, phenotypic, and clinical heterogeneity of PDAC tumors. Based on genomic and transcriptomic profiling, several classifications of PDAC subtypes have been introduced and linked to clinical outcomes.42,44,54,55 We analyzed the relationship between cellular PDAC subtypes and the efficacy of MeV chemovirotherapy based on our in vitro cytotoxicity data. This analysis follows a recent transcriptomic classification of PDAC cell lines into subtypes defined in datasets from The Cancer Genome Atlas (TCGA).44,45 Of note, both MeV-CD-FmiRTS148a alone and chemovirotherapy with 5-FC appeared to have more pronounced effects on cells of the basal subtype previously associated with inferior survival. We also found pronounced MeV cytolysis in MIA PaCa-2 cells, a cell line that could not be conclusively classified as classical or basal, but also has been assigned to a PDAC subtype with inferior prognosis.42,45,55
In the selection of patients for future chemovirotherapy trials, inter-tumor heterogeneity may pose important challenges. Improvements in subclassification of PDAC are therefore expected to enable optimal stratification of patients. Matching molecular profiling of patient-derived materials with results of preclinical chemovirotherapy in each subtype could represent one way of identifying the patients with the greatest potential benefit. In a previously published study involving patient-derived xenografts of other cancer types, the authors established a classification model based on gene signatures to predict clinical responses to MeV therapy.56 Further research is needed to delineate the influence of tumor characteristics concerning interferon response, receptor status, and transcriptional networks on MeV efficacy in PDAC.
Although MeV infection induced variable responses, 5-FC added to its efficacy in all PDAC cell lines tested. We also demonstrated complementary cytotoxicity to non-infected bystander tumor cells after prodrug conversion. This allows for therapeutic effects beyond the fraction of infected tumor cells. These results are further corroborated by the observed relevant sensitivity of all cell lines toward 5-FU, which in turn was independent of the respective cellular miR-148a expression levels. Taken together, these findings suggest the absence of cross-resistance between the different treatment components of the 5-FU-based chemovirotherapy regimen. Fluorouracil was shown previously to synergize with MeV in several tumor entities.57, 58, 59, 60 In one study, PDAC cells were transduced ex vivo with a CD-expressing vector prior to engraftment in mice.61 Subsequent 5-FC treatment induced complete remission of subcutaneous tumors, emphasizing the efficacy of the CD/5-FC system upon optimal transgene expression.
The treatment of pre-established tumors in vivo resembles clinical applications more accurately than in vitro infection. In pre-established PDAC xenografts in mice, we demonstrated intratumoral expression of prodrug convertase after a single administration of MeV-CD-FmiRTS148a, indicating effective virus replication and transgene expression in vivo. However, further potential challenges exist in the clinical translation of MeV chemovirotherapy for PDAC. The stromal microenvironment of PDAC has been identified as a prognostic factor and is characterized by a dense extracellular matrix that can pose barriers to the distribution of conventional drugs.62, 63, 64 This has implications for PDAC chemovirotherapy, as intratumoral virus spread contributes to efficacy, and sufficient intratumoral uptake and diffusion of prodrug is a prerequisite for therapeutic bystander effects. Interestingly, cytokine signaling between stromal and malignant cells in PDAC was shown to enforce rather than hinder the replication of several OVs in patient-derived xenografts.65 Moreover, different strategies have been explored to improve the distribution of OVs in stroma-rich tumors.66,67 With regards to MeV therapy, targeting both neoplastic cells and cells of the stromal compartment has been shown to result in superior efficacy.68 It is conceivable that through similar mechanisms, MeV chemovirotherapy may lead to remodeling of the extracellular matrix in PDAC with positive therapeutic effects. However, in light of the functional heterogeneity among non-neoplastic cell types in PDAC tumors, outcomes of stromal targeting will need careful assessment.69
Our results with chemovirotherapy in infected and bystander PDAC cells suggest that the respective effects of MeV-CD-FmiRTS148a and activated prodrug on tumor cells are not fully additive. Instead, our findings point toward a more complex interaction of both treatment components. Previous studies have identified a possible interference of prodrug activation with MeV-CD replication, especially at early time points after infection.57 Prodrug application schedules therefore need to incorporate data on virus replication kinetics and on the temporal dynamics of transgene expression applicable to the specific setting. We therefore repeatedly assessed intratumoral prodrug convertase expression in vivo up to 7 days after MeV-CD-FmiRTS148a infection of PDAC xenografts and detected peak expression at 48 h. This is in line with other studies of intratumoral MeV replication and time to maximum transgene expression.70,71
Based on our analysis of prodrug convertase expression in PDAC xenografts, we devised a chemovirotherapy schedule aiming at optimal prodrug activation in vivo. Our regimen of MeV-CD-FmiRTS148a plus 5-FC significantly prolonged animal survival compared to MeV alone. We found no toxicity of systemic 5-FC in mice at 200 mg per kg twice daily, and similar doses were safely applied by others.72 Overall, the efficacy we observed goes beyond previous results of PDAC chemovirotherapy with MeV and a fludarabine-based regimen.73 While the absolute survival benefit was still modest, it may indicate a meaningful improvement in light of the highly refractory nature of PDAC toward standard therapies. However, there were no complete or long-term remissions. As expected, the outcomes in our xenograft model differ from results in immunocompetent tumor models, where MeV therapy induces both complete and durable remissions and tumor vaccination effects.23,24
The immunostimulatory potential of virotherapy has become an overarching theme in the field with widespread therapeutic implications.74 In a subset of cancer patients, immunotherapies have already surpassed other modalities in long-term tumor control.75,76 However, even in immunogenic tumors, clinical responses often manifest with delay. Therefore, both rapid proliferation and poor immunogenicity of PDAC counteract the induction of effective antitumor immunity.77 Importantly, studies in preclinical models suggest that fluoropyrimidines, including 5-FU, can induce the selective depletion of intratumoral immune cell populations with immunosuppressive functions, such as myeloid-derived suppressor cells.78 Alternatively, various mechanisms have been characterized by which MeV oncolysis induces and sustains antitumor immunity.79 Combining conventional antineoplastics and MeV virotherapy could therefore open a window of opportunity for the formation of immune-mediated therapeutic effects in PDAC. This concept warrants the evaluation of multimodal approaches that incorporate elements of chemotherapy, immunotherapy, and virotherapy. So far, most trials in PDAC have focused only on combinations of OVs and chemotherapy.80,81
From a clinical perspective, 5-FU-based MeV chemovirotherapy could add to the management of PDAC in several settings. In locally advanced unresectable PDAC, activation of 5-FC upon intratumoral virus injection may serve as a radio-sensitizing strategy.82 In disseminated stages, testing of alternative virus administration routes should be encouraged.80 This may include intravenous or intracavitary treatment of manifestations often refractory to standard treatment, such as peritoneal carcinomatosis.83, 84, 85 MeV-CD-FmiRTS148a chemovirotherapy could also be evaluated in other tumor entities with matching microRNA profiles and clinical sensitivity to 5-FU, including gastric and colorectal cancer.86, 87, 88
The interplay of OVs and the tumor microenvironment allows for anti-tumor effects along various mechanisms of action. Genetic engineering of OVs expands this pharmacodynamic range and realizes new combination therapies. In this study, we generated and characterized an oncolytic MeV for 5-FU-based chemovirotherapy of PDAC with an improved safety profile. Further research is needed to elucidate and circumvent resistance mechanisms, and to align virotherapy and other modalities in optimal synergism. Hopefully, this will advance the treatment of entities such as PDAC and enable new ways of tumor control for the benefit of patients.
Materials and methods
Cloning of recombinant MeV
Cloning of recombinant viruses was based on the p(+)MV-NSe cDNA antigenome of MeV Edmonston tag laboratory strain.89,90 To generate microRNA-sensitive MeV, a miRTS box encoding three copies of either the nucleotide sequence complementary to mature hsa-miR-148a-3p (as per http://www.mirbase.org) or the corresponding reverse complementary control sequence was designed as described previously.38 For experiments involving IMIM-PC2-148a cells, a control miRTS box containing three copies of the target site in reverse orientation separated by 14-nt spacer sequences was designed. The following DNA oligonucleotides including full-length miRTS boxes with PvuI-PacI compatible ends for cloning were synthesized by Eurofins Genomics (Ebersberg, Germany): 5′-CGACAAAGTTCTGTAGTGCACTGAGAGCTTGACAAAGTTCTGTAGTGCACTGAGAGCTTGACAAAGTTCTGTAGTGCACTGAGAGTTAAT-3′ (miRTS148a forward), 5′-TAACTCTCAGTGCACTACAGAACTTTGTCAAGCTCTCAGTGCACTACAGAACTTTGTCAAGCTCTCAGTGCACTACAGAACTTTGTCGAT-3′ (miRTS148a reverse); 5′-CGTCAGTGCACTACAGAACTTTGTGAGCTTGTCAGTGCACTACAGAACTTTGTGAGCTTGTCAGTGCACTACAGAACTTTGTGAGTTAAT-3′ (miRTS148aRC forward), 5′-TAACTCACAAAGTTCTGTAGTGCACTGACAAGCTCACAAAGTTCTGTAGTGCACTGACAAGCTCACAAAGTTCTGTAGTGCACTGACGAT-3′ (miRTS148aRC reverse); 5′-CGAGTCACGTGATGTCTTGAAACAAATAATACCCAATGAGTCACGTGATGTCTTGAAACATTCGATCCCTTCTTAGTCACGTGATGTCTTGAAACAGTTAAT-3′ (miRTS148aREV forward), 5′-TAACTGTTTCAAGACATCACGTGACTAAGAAGGGATCGAATGTTTCAAGACATCACGTGACTCATTGGGTATTATTTGTTTCAAGACATCACGTGACTCGAT-3′ (miRTS148aREV reverse).
Oligonucleotides encoding miRTS boxes were inserted into the 3′ UTR of MeV F encoded by the plasmid pCG-F, followed by cloning of NarI-PacI fragments of pCG-FmiRTS into pMV-EGFP genomic cDNA as described previously.37,38 Plasmid DNA encoding the E. coli CD-UPRT fusion gene in an open reading frame (pORF-codA::upp) was purchased from InvivoGen (San Diego, CA, USA). To yield microRNA-sensitive MeV encoding CD-UPRT, an exchange of the additional transcription unit upstream of the MeV N gene was performed by cloning the MluI-SdaI fragment of pMV-CD57 into pMV-EGFP-FmiRTS.
Cell culture
Vero (African green monkey kidney), Capan-1, and BxPC-3 cells were obtained from ATCC (Manassas, VA, USA). IMIM-PC2, IMIM-PC2-148a, and IMIM-PC2-LV cells were kindly provided by Stephan A. Hahn (Ruhr University, Bochum, Germany) and MIA PaCa-2 and T3M4 cells by Zahari Raykov (DKFZ, Heidelberg, Germany). Cell culture media were supplemented with 10% fetal bovine serum (Biosera, Nuaille, France) unless indicated otherwise. Vero and MIA PaCa-2 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Life Technologies, Darmstadt, Germany). BxPC-3, IMIM-PC2, and T3M4 cells were maintained in Roswell Park Memorial Institute medium (Life Technologies). The medium of IMIM-PC2-148a and IMIM-PC2-LV cells was supplemented with 2 μg/mL puromycin (Life Technologies). Capan-1 cells were maintained in Iscove’s modified Dulbecco’s medium (Life Technologies) supplemented with 20% fetal bovine serum and 1% ultraglutamine (Lonza, Basel, Switzerland). Cultured cell lines were incubated at 37°C in a humidified atmosphere with 5% CO2 and routinely tested to exclude mycoplasma contamination using the Venor GeM kit (Minerva Biolabs, Berlin, Germany) according to the manufacturer’s instructions.
Virus generation and propagation
Rescue of recombinant MeV was based on the RNA polymerase II-dependent method.91,92 Virus generation and propagation were carried out in Vero cells. For propagation, cells seeded on 15-cm culture dishes (Greiner, Frickenhausen, Germany) were infected at an MOI of 0.03 and incubated at 32°C, 5% CO2. After aspiration of culture medium at 36–72 h post-infection, infected cell layers were harvested using a sterile cell scraper. Harvested cells were subjected to one freeze-thaw cycle followed by centrifugation at 5,000 × g, 4°C for 5 min. The supernatant was aliquoted and stored at −80°C.
In vitro virus infection
Cells were seeded on multi-well plates at indicated densities. Virus suspended in Opti-MEM (Life Technologies) was applied at the indicated MOI in exchange of, or in addition to, cell culture media, depending on the experiment. Cells were incubated with virus at 37°C, 5% CO2. Plates were gently shaken four times per hour to allow for even distribution of virus. Where indicated, medium was changed to fresh culture medium after 2 h of virus adsorption.
Multi-step virus growth curves
Pancreatic cancer cells were seeded on 12-well plates at 1 × 105 to 1.5 × 105 cells per well depending on the cell line and were infected with MeV variants at an MOI of 0.03 as described. At the designated time points post-infection, cells were harvested, vortexed, subjected to one freeze-thaw cycle, and centrifuged at 5,000 × g, 4°C for 5 min. Supernatants were stored at −80°C until progeny virus titration.
MicroRNA precursor transfection
miR-148a-3p microRNA mimics were purchased from Life Technologies (mirVana microRNA mimics). Cell transfection with microRNA mimics was performed using Lipofectamine 2000 (Life Technologies) according to the manufacturer’s instructions. Transfection complexes were prepared in Opti-MEM (Life Technologies) and added to cells seeded on 12- or 24-well plates, resulting in a microRNA mimic concentration of 60 nmol/L. Mock transfection was performed in parallel without the addition of microRNA mimics. Following microRNA transfection, cells were incubated at 37°C, 5% CO2 until MeV infection was carried out as described. At the indicated time points post-infection, the respective readout was performed by fluorescence microscopy, progeny virus titration, or a cell viability assay.
Quantitative microRNA PCR
Total RNA was isolated from 3 × 105 cells per cell line using the miRNeasy mini kit (QIAGEN, Hilden, Germany). RNA oligonucleotides encoding mature hsa-miR-148a-3p (Eurofins, Ebersberg, Germany) were used to prepare a standard dilution series of 101–108 molecules in 300 ng each of E. coli carrier RNA (Sigma-Aldrich). RNA samples and standards were reverse transcribed using the miScript II RT kit (QIAGEN). Real-time PCR was performed on a LightCycler 480 with software version 1.5 (Roche) using miScript primer assays and the miScript SYBR Green PCR kit (both from QIAGEN). Crossing point (Cp) values were defined by the second derivative maximum method.93 Cp values were normalized to RNU6-2 as an endogenous reference, and absolute microRNA copy numbers were calculated using the standard curve.
Progeny virus titration
Cells were pretreated according to the experiment, infected, and harvested as described. Virus samples were briefly thawed in a water bath at 37°C and serial 1:10 dilutions in DMEM were prepared in octuplicates. Vero cells seeded on 96-well plates (104 per well) were incubated with diluted samples at 37°C, 5% CO2. After 48 h, formation of individual syncytia was determined and CIU per mL was calculated.
Fluorescence microscopy of infected cells
An Axiovert 200 microscope, AxioCam MR, and Axiovision software (all from Carl Zeiss, Jena, Germany) were used for microscopy and image acquisition. Fluorescence-based analysis of cell layer infection was performed by automated segmentation with ImageJ 2.1.0 software (General Public License)94 with uniform threshold settings across all samples.
Spectrophotometry
To study prodrug activation in vitro, a photometric assay was established based on the method by Nishiyama et al.27 CD-UPRT activity was estimated as the relative change in substrate concentration based on the optical absorbance spectrum of 5-FC (maximum at 285 nm) and the Beer-Lambert-Bouguer law. At designated time points post-infection (MOI of 0.1), 1.5 × 105 BxPC-3 cells cultured in 12-well plates were scraped in their medium and lysed by one freeze-thaw cycle. 5-FC (Sigma-Aldrich, Darmstadt, Germany) stock solution was prepared using 0.9% NaCl (B. Braun, Melsungen, Germany). Whole-cell lysates were incubated at 37°C with 5-FC at a final concentration of 1.5 mmol/L for 48 h. After centrifugation at 6,000 × g for 5 min at 4°C, absorbance at 290 nm was determined using a NanoDrop 2000 spectrophotometer (Thermo Scientific, Waltham, MA, USA).
Cell viability assay
PDAC cells were seeded on 96-well plates at 104 cells per well and infected as described (MOI of 0.1). 5-FC stock solution was diluted in Opti-MEM and added to cells at 36 h post-infection, resulting in a final concentration of 1 mmol/L. Each experiment was carried out in biological quadruplicates per condition. At the designated time points after prodrug treatment, XTT reagent was applied to cells using the Colorimetric Cell Viability Kit III (PromoKine, Heidelberg, Germany) following the manufacturer’s instructions. Absorbance at 450 nm of each sample was measured in technical triplicates on a GENios plate reader using XFLUOR4 v4.51 software (Tecan, Maennedorf, Switzerland). Cell viability was calculated relative to mock-treated or prodrug-treated references.
Bystander cell viability assay
Vero cells seeded on six-well plates (4 × 105 per well) were inoculated with MeV-CD-FmiRTS148a in Opti-MEM at an MOI of 0.1 or mock-infected as described before, including an exchange of medium to fresh DMEM after 2 h. At 36 h post-infection, cells were treated with 5-FC at a concentration of 1 mmol/L or with carrier DMEM (mock treatment). After incubation for 12 h, cell culture supernatants were collected and centrifuged at 15,000 × g, 22°C for 5 min to remove cell debris. Cleared medium samples were heated to 60°C for 30 min in order to inactivate remaining virus particles and stored at −20°C. PDAC cells seeded on 96-well plates (104 per well) were treated with the respective conditioned supernatant at indicated dilutions. After 72 h of incubation at 37°C, 5% CO2, an XTT (2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)2H-tetrazolium-5-carboxanilide) cell viability assay was carried out as described above.
Infection of primary liver tissue
Patient’s informed consent was obtained according to the research proposals approved by the Institutional Review Boards at Heidelberg Medical Faculty. Primary human liver tissue was provided by the surgical department at Heidelberg University Hospital. Sections of 300 μm thickness were prepared using a vibratome as described previously.38 Tissue slices were maintained on culture inserts for six-well plates (BD Biosciences, Heidelberg, Germany) in William’s E medium (Life Technologies) supplemented with 434 mg/L l-alanyl-l-glutamine, 25 mM glucose, and 50 μg/mL gentamicin. Immediately after preparation, specimens were infected with 5 × 106 CIU of the respective virus by inoculation into the culture medium and incubated at 37°C, 5% CO2 for 24 h. Medium was renewed daily using fresh supplemented William’s E medium. Multichannel fluorescence microscopy images were acquired as overlapping tiles at 24–144 h post-infection. Composite images were assembled, and individual channels were used to correct for tissue autofluorescence by background subtraction.
Immunofluorescence staining
Cells were seeded on six-well plates (3.5 × 105 per well) containing glass coverslips (thickness no. 1.5, VWR, Darmstadt, Germany). At 6–24 h after seeding, cells were treated with conditioned supernatant as described above. After 48–72 h of incubation, cells were washed with PBS, fixed with 4% paraformaldehyde (ROTI Histofix, Carl Roth, Karlsruhe, Germany), and permeabilized in PBS containing 0.2% Triton X-100 (Sigma-Aldrich). Cells were incubated with blocking buffer (2% bovine serum albumin [Sigma-Aldrich] and 0.1% Triton X-100 in PBS) for 60 min at room temperature (RT). Primary antibodies were diluted in PBS containing 1% bovine serum albumin and 0.1% Triton X-100 and incubated at 4°C overnight. The monoclonal rabbit anti-Ki-67 (#M7240, Agilent Technologies, Waldbronn, Germany) was diluted at 1:30, and the polyclonal rabbit anti-cleaved caspase-3 (#9661, Cell Signaling Technology, Frankfurt, Germany) was diluted at 1:300. After washing, secondary antibodies (goat anti-rabbit Cy3, 111-165-003, Dianova, Hamburg, Germany and goat anti-mouse Alexa Fluor 488, 715-545-150, Dianova) were diluted at 1:200 along with DAPI (AppliChem, Darmstadt, Germany) in PBS and incubated for 60–90 min at RT. Cells were washed and the coverslips were mounted onto microscope slides (Thermo Scientific) containing mounting medium (Dako, Hamburg, Germany). Fluorescence images were acquired on an AxioCam MR R3 (Zeiss) connected to an Axio Observer Z1 (Zeiss) with ZEN 2.5 blue edition software (Zeiss). Analysis of immunofluorescence images was performed by automated segmentation and particle analysis with ImageJ 2.1.0 software using uniform settings across samples from the same cell line and acquisition channel.
Dose-response assay
PDAC cells were seeded on 96-well plates at 104 cells per well. Dilutions of 5-FU (Sigma-Aldrich) stock solution in 0.9% NaCl (B. Braun) were prepared using cell-specific medium and added to cells at 6–8 h after seeding, resulting in final concentrations from 0.05 to 2 mM. Each experiment was carried out in biological quadruplicates per condition. At 72 h post-treatment, an XTT assay was performed as described and cell viability was calculated relative to mock-treated references.
In vivo experiments
Animal experiments were approved by the local regulatory authority (Regierungspraesidium Karlsruhe, Germany) and conducted in line with the German Animal Protection Law. NOD.Cg-Prkdcscid Il2rgtm1WjI (NOD/SCID/gamma) mice were bred and housed in the DKFZ Central Animal Laboratory (DKFZ, Heidelberg, Germany). For evaluation of intratumoral prodrug convertase expression, 106 T3M4 cells were implanted subcutaneously into the right flank of NOD/SCID/gamma mice. Treatment was initiated when mean tumor volume reached ≥100 mm3. MeV-CD-FmiRTS148a was administered as one intratumoral injection of 5 × 106 CIU. At the designated time points after virus application, animals were sacrificed, whole tumors were excised, and tissue was stored at −80°C until protein isolation. For assessment of in vivo chemovirotherapy efficacy, 6 × 106 T3M4 cells suspended in 100 mm3 PBS were implanted subcutaneously into the right flank of NOD/SCID/gamma mice. Treatment was initiated once mean tumor volume reached ≥80 mm3. MeV-CD-FmiRTS148a was injected intratumorally at 6.9 × 106 CIU in 100 mm3 of Opti-MEM per day on 5 consecutive days. Starting 48 h after the first virus application, 5-FC was administered intraperitoneally at 200 mg per kg in 800 mm3 of sterile NaCl 0.9% (B. Braun) every 12 h for a total of 5 days. Control-treated animals received sham treatment using the respective carrier substances: intratumoral MeV-CD-FmiRTS148a + intraperitoneal NaCl 0.9% (MeV only), intratumoral Opti-MEM + intraperitoneal 5-FC (5-FC only), or intratumoral Opti-MEM + intraperitoneal NaCl 0.9% (mock). Tumor volume was determined using a caliper and the following formula: tumor volume (mm3) = 0.5 × largest diameter (mm) × (smallest diameter [mm])2. Animals were sacrificed when one of the following criteria was met: tumor volume ≥ 1500 mm3, tumor ulceration, or deterioration indicating pain or distress, such as ruffed fur, apathy, or cachexia.
Protein isolation and immunoblot
Frozen T3M4 xenograft tumors were weighed, kept on dry ice, and minced using a scalpel. Tissue samples were homogenized in 8 μL of radioimmunoprecipitation assay (RIPA) buffer (Thermo Scientific, Waltham, MA, USA) per mg using a tissue grinder (Wheaton, Millville, NJ, USA) and subjected to three freeze-thaw cycles. Tumor lysates were centrifuged at 16,000 × g for 10 minutes (4°C). The protein concentration of supernatants was determined by a bicinchoninic acid (BCA) protein assay (Merck, Darmstadt, Germany) and was adjusted across samples. After denaturation in modified Laemmli buffer (Carl Roth), equal amounts of protein from each sample were loaded on a Mini-PROTEAN TGX gel (Bio-Rad, Feldkirchen, Germany) and electrophoresis was performed using a Mini-PROTEAN Tetra cell chamber (Bio-Rad). Wet transfer to an Immobilon-P membrane (Merck) was performed in Tris-glycine buffer (Life Technologies) using a Mini Trans-Blot insert (Bio-Rad). Membranes were washed in Tris-buffered saline with polysorbate 20 (TBS-T; Carl Roth), and binding sites were blocked with 5% milk powder in TBS-T. For immunodetection, membranes were incubated for 3 h at RT with monoclonal mouse anti-CD antibody (mAb 16D8F2, provided by Johannes Gebert, DKFZ, Heidelberg, Germany) diluted at 1:1,000 in TBS-T plus 5% milk powder.95 After incubation with anti-mouse immunoglobulin G (IgG)-horseradish peroxidase (HRP) (Bethyl Laboratories, Montgomery, TX, USA), activated peroxidase substrate (SuperSignal West Pico, Fisher Scientific, Schwerte, Germany) was applied according to the manufacturer’s instructions, and chemiluminescence was recorded on a ChemiDoc XRS+ system with Image Lab software 3.0 (both by Bio-Rad). ImageJ 2.0.0 software was used to analyze the lane profiles of digital scans. To determine the abundance of intratumoral CD-UPRT protein independent from individual tumor growth kinetics, each calculated band density was normalized to the mass of the corresponding tumor by the following formula: normalized expression = relative signal intensity × initial tumor weight.
Fluorescence-based immunoblot was performed on an Amersham WB analyzer system (GE Healthcare Bio-Sciences, Uppsala, Sweden) with Amersham ECL Plex system consumables (GE Healthcare). For Cy5-based total protein normalization, tumor lysates in RIPA buffer were prelabeled with Amersham QuickStain (GE Healthcare) according to the manufacturer’s instructions. Samples adjusted to 30 μg of total protein each were loaded on an Amersham ECL precast gel (8%–16%) (GE Healthcare). Electrophoresis was carried out at 250–600 V for 50 min. Proteins were transferred to an Amersham WB polyvinylidene fluoride (PVDF) card (GE Healthcare) in Tris-glycine plus 20% ethanol at 100 V for 30 min. After blocking with ECL Prime blocking reagent (GE Healthcare), the membrane was incubated with primary mAb 16D8F2 (1:1,000) overnight at 4°C. Probing was performed with Amersham ECL Plex goat-anti-mouse IgG-Cy3 (PA43009, GE Healthcare) for 30 min. Fluorescence signals were detected on an Amersham WB analyzer scan unit and analyzed with Amersham WB software version 1.0.33 (GE Healthcare).
Statistical analysis
Statistical analysis was performed using the R framework software versions 3.4.2 and 4.0.0 (R Foundation for Statistical Computing, Vienna, Austria).96 p values were adjusted for multiple comparisons and were interpreted as significant when <0.05. Unless indicated otherwise, experiments involving independent observations of continuous variables from more than two groups were analyzed by ANOVA, and pairwise comparisons were performed by a Tukey’s test with a family-wise confidence level of 95%. Count data were analyzed by Pearson’s chi-square test for individual comparisons and by a Cochran-Mantel-Haenszel test for effects across composite datasets. Longitudinal tumor growth data were analyzed by means of a nonlinear mixed-effects model with parameter estimation based on the method by Lindstrom and Bates.97 Survival data were analyzed by the Kaplan-Meier method. Survival distributions were compared by a log-rank test with a Benjamini-Hochberg correction to control the false discovery rate.
Acknowledgments
We thank Jessica Albert, Birgit Hoyler, Stefanie Sawall, and Alexandra Just for excellent technical assistance, and Elena Busch and Johannes Heidbuechel for support with animal experiments. We thank Johannes Gebert for kindly providing anti-CD antibody, and Stephan Hahn and Zahari Raykov for IMIM-PC2 cells and MIA PaCa-2 and T3M4 cells, respectively. We thank Jürgen Weitz and Hubertus Schmitz-Winnenthal for provision and Frank Bergmann and Marc-Andrea Baertsch for processing of primary tissue material. We thank Eva Besemfelder for support with immunoblotting, and Assia Angelova for expertise in setting up the immunofluorescence microscopy. The graphical abstract was created with BioRender.com. This work was supported by Deutsche Krebshilfe (German Cancer Aid), Max Eder Program no. 108307 (to G.U.), a Helmholtz Association/DKFZ PhD Fellowship and a Physician-Scientist Fellowship of the Medical Faculty, Heidelberg University (both to M.F.L.), and by a DKFZ Heinrich F.C. Behr Scholarship (to H.M.S.). H.M.S. is a Fellow of the MD/PhD Program at Heidelberg University, Germany.
Author contributions
Conceptualization, H.M.S., M.F.L., and G.U.; methodology, H.M.S., M.F.L., C.E.E., K.Z., and S.B.; investigation, H.M.S., M.F.L., C.E.E., C.G., and J.D.; writing – original draft, H.M.S.; writing – review & editing, M.F.L., C.E.E., and G.U.; funding acquisition, H.M.S., J.C.B., C.v.K., D.J., and G.U.; resources, K.Z.; supervision, M.F.L., S.B., J.C.B., D.J., C.v.K., and G.U.
Declaration of interests
G.U. is co-founder, stakeholder, and CMO/CSO of CanVirex AG, a company investigating oncolytic viruses as novel cancer immunotherapeutics. J.C.B. is co-founder, stakeholder, and co-director of Turnstone Biologics, a company developing viral immunotherapeutics against cancer. The remaining authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2021.04.015.
Supplemental information
References
- 1.Ryan D.P., Hong T.S., Bardeesy N. Pancreatic adenocarcinoma. N. Engl. J. Med. 2014;371:2140–2141. doi: 10.1056/NEJMc1412266. [DOI] [PubMed] [Google Scholar]
- 2.Burris H.A., 3rd, Moore M.J., Andersen J., Green M.R., Rothenberg M.L., Modiano M.R., Cripps M.C., Portenoy R.K., Storniolo A.M., Tarassoff P. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997;15:2403–2413. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- 3.Conroy T., Desseigne F., Ychou M., Bouché O., Guimbaud R., Bécouarn Y., Adenis A., Raoul J.L., Gourgou-Bourgade S., de la Fouchardière C., Groupe Tumeurs Digestives of Unicancer. PRODIGE Intergroup FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011;364:1817–1825. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 4.Sohal D.P., Mangu P.B., Khorana A.A., Shah M.A., Philip P.A., O’Reilly E.M., Uronis H.E., Ramanathan R.K., Crane C.H., Engebretson A. Metastatic pancreatic cancer: American Society of Clinical Oncology clinical practice guideline. J. Clin. Oncol. 2016;34:2784–2796. doi: 10.1200/JCO.2016.67.1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neoptolemos J.P., Palmer D.H., Ghaneh P., Psarelli E.E., Valle J.W., Halloran C.M., Faluyi O., O’Reilly D.A., Cunningham D., Wadsley J., European Study Group for Pancreatic Cancer Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): A multicentre, open-label, randomised, phase 3 trial. Lancet. 2017;389:1011–1024. doi: 10.1016/S0140-6736(16)32409-6. [DOI] [PubMed] [Google Scholar]
- 6.Conroy T., Hammel P., Hebbar M., Ben Abdelghani M., Wei A.C., Raoul J.L., Choné L., Francois E., Artru P., Biagi J.J., Canadian Cancer Trials Group and the Unicancer-GI–PRODIGE Group FOLFIRINOX or gemcitabine as adjuvant therapy for pancreatic cancer. N. Engl. J. Med. 2018;379:2395–2406. doi: 10.1056/NEJMoa1809775. [DOI] [PubMed] [Google Scholar]
- 7.Howlader, N.N.A., Krapcho, M., Miller, D., Brest, A., Yu, M., Ruhl, J., Tatalovich, Z., Mariotto, A., Lewis, D.R., Chen, H.S., et al., eds. (2019). SEER Cancer Statistics Review, 1975–2016, National Cancer Institute, Bethesda, MD. https://seer.cancer.gov/csr/1975_2016/, based on November 2018 SEER data submission.
- 8.Breitbach C.J., Burke J., Jonker D., Stephenson J., Haas A.R., Chow L.Q., Nieva J., Hwang T.H., Moon A., Patt R. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature. 2011;477:99–102. doi: 10.1038/nature10358. [DOI] [PubMed] [Google Scholar]
- 9.Andtbacka R.H., Kaufman H.L., Collichio F., Amatruda T., Senzer N., Chesney J., Delman K.A., Spitler L.E., Puzanov I., Agarwala S.S. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 2015;33:2780–2788. doi: 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 10.Desjardins A., Gromeier M., Herndon J.E., 2nd, Beaubier N., Bolognesi D.P., Friedman A.H., Friedman H.S., McSherry F., Muscat A.M., Nair S. Recurrent glioblastoma treated with recombinant poliovirus. N. Engl. J. Med. 2018;379:150–161. doi: 10.1056/NEJMoa1716435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cattaneo R., Miest T., Shashkova E.V., Barry M.A. Reprogrammed viruses as cancer therapeutics: Targeted, armed and shielded. Nat. Rev. Microbiol. 2008;6:529–540. doi: 10.1038/nrmicro1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miest T.S., Cattaneo R. New viruses for cancer therapy: Meeting clinical needs. Nat. Rev. Microbiol. 2014;12:23–34. doi: 10.1038/nrmicro3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matveeva O.V., Guo Z.S., Senin V.M., Senina A.V., Shabalina S.A., Chumakov P.M. Oncolysis by paramyxoviruses: Preclinical and clinical studies. Mol. Ther. Oncolytics. 2015;2:150017. doi: 10.1038/mto.2015.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pol J.G., Lévesque S., Workenhe S.T., Gujar S., Le Boeuf F., Clements D.R., Fahrner J.E., Fend L., Bell J.C., Mossman K.L. Trial watch: Oncolytic viro-immunotherapy of hematologic and solid tumors. OncoImmunology. 2018;7:e1503032. doi: 10.1080/2162402X.2018.1503032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Msaouel P., Opyrchal M., Dispenzieri A., Peng K.W., Federspiel M.J., Russell S.J., Galanis E. Clinical trials with oncolytic measles virus: Current status and future prospects. Curr. Cancer Drug Targets. 2018;18:177–187. doi: 10.2174/1568009617666170222125035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dingli D., Peng K.W., Harvey M.E., Greipp P.R., O’Connor M.K., Cattaneo R., Morris J.C., Russell S.J. Image-guided radiovirotherapy for multiple myeloma using a recombinant measles virus expressing the thyroidal sodium iodide symporter. Blood. 2004;103:1641–1646. doi: 10.1182/blood-2003-07-2233. [DOI] [PubMed] [Google Scholar]
- 17.Springfeld C., von Messling V., Frenzke M., Ungerechts G., Buchholz C.J., Cattaneo R. Oncolytic efficacy and enhanced safety of measles virus activated by tumor-secreted matrix metalloproteinases. Cancer Res. 2006;66:7694–7700. doi: 10.1158/0008-5472.CAN-06-0538. [DOI] [PubMed] [Google Scholar]
- 18.Ungerechts G., Springfeld C., Frenzke M.E., Lampe J., Johnston P.B., Parker W.B., Sorscher E.J., Cattaneo R. Lymphoma chemovirotherapy: CD20-targeted and convertase-armed measles virus can synergize with fludarabine. Cancer Res. 2007;67:10939–10947. doi: 10.1158/0008-5472.CAN-07-1252. [DOI] [PubMed] [Google Scholar]
- 19.Liu C., Erlichman C., McDonald C.J., Ingle J.N., Zollman P., Iankov I., Russell S.J., Galanis E. Heat shock protein inhibitors increase the efficacy of measles virotherapy. Gene Ther. 2008;15:1024–1034. doi: 10.1038/gt.2008.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grossardt C., Engeland C.E., Bossow S., Halama N., Zaoui K., Leber M.F., Springfeld C., Jaeger D., von Kalle C., Ungerechts G. Granulocyte-macrophage colony-stimulating factor-armed oncolytic measles virus is an effective therapeutic cancer vaccine. Hum. Gene Ther. 2013;24:644–654. doi: 10.1089/hum.2012.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iankov I.D., Federspiel M.J., Galanis E. Measles virus expressed Helicobacter pylori neutrophil-activating protein significantly enhances the immunogenicity of poor immunogens. Vaccine. 2013;31:4795–4801. doi: 10.1016/j.vaccine.2013.07.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Engeland C.E., Grossardt C., Veinalde R., Bossow S., Lutz D., Kaufmann J.K., Shevchenko I., Umansky V., Nettelbeck D.M., Weichert W. CTLA-4 and PD-L1 checkpoint blockade enhances oncolytic measles virus therapy. Mol. Ther. 2014;22:1949–1959. doi: 10.1038/mt.2014.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Veinalde R., Grossardt C., Hartmann L., Bourgeois-Daigneault M.C., Bell J.C., Jäger D., von Kalle C., Ungerechts G., Engeland C.E. Oncolytic measles virus encoding interleukin-12 mediates potent antitumor effects through T cell activation. OncoImmunology. 2017;6:e1285992. doi: 10.1080/2162402X.2017.1285992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Speck T., Heidbuechel J.P.W., Veinalde R., Jaeger D., von Kalle C., Ball C.R., Ungerechts G., Engeland C.E. Targeted BiTE expression by an oncolytic vector augments therapeutic efficacy against solid tumors. Clin. Cancer Res. 2018;24:2128–2137. doi: 10.1158/1078-0432.CCR-17-2651. [DOI] [PubMed] [Google Scholar]
- 25.Backhaus P.S., Veinalde R., Hartmann L., Dunder J.E., Jeworowski L.M., Albert J., Hoyler B., Poth T., Jäger D., Ungerechts G., Engeland C.E. Immunological effects and viral gene expression determine the efficacy of oncolytic measles vaccines encoding IL-12 or IL-15 agonists. Viruses. 2019;11:914. doi: 10.3390/v11100914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Busch E., Kubon K.D., Mayer J.K.M., Pidelaserra-Martí G., Albert J., Hoyler B., Heidbuechel J.P.W., Stephenson K.B., Lichty B.D., Osen W. Measles vaccines designed for enhanced CD8+ T cell activation. Viruses. 2020;12:242. doi: 10.3390/v12020242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nishiyama T., Kawamura Y., Kawamoto K., Matsumura H., Yamamoto N., Ito T., Ohyama A., Katsuragi T., Sakai T. Antineoplastic effects in rats of 5-fluorocytosine in combination with cytosine deaminase capsules. Cancer Res. 1985;45:1753–1761. [PubMed] [Google Scholar]
- 28.Tiraby M., Cazaux C., Baron M., Drocourt D., Reynes J.P., Tiraby G. Concomitant expression of E. coli cytosine deaminase and uracil phosphoribosyltransferase improves the cytotoxicity of 5-fluorocytosine. FEMS Microbiol. Lett. 1998;167:41–49. doi: 10.1111/j.1574-6968.1998.tb13205.x. [DOI] [PubMed] [Google Scholar]
- 29.Porosnicu M., Mian A., Barber G.N. The oncolytic effect of recombinant vesicular stomatitis virus is enhanced by expression of the fusion cytosine deaminase/uracil phosphoribosyltransferase suicide gene. Cancer Res. 2003;63:8366–8376. [PubMed] [Google Scholar]
- 30.Lu J., Getz G., Miska E.A., Alvarez-Saavedra E., Lamb J., Peck D., Sweet-Cordero A., Ebert B.L., Mak R.H., Ferrando A.A. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 31.Szafranska A.E., Davison T.S., John J., Cannon T., Sipos B., Maghnouj A., Labourier E., Hahn S.A. MicroRNA expression alterations are linked to tumorigenesis and non-neoplastic processes in pancreatic ductal adenocarcinoma. Oncogene. 2007;26:4442–4452. doi: 10.1038/sj.onc.1210228. [DOI] [PubMed] [Google Scholar]
- 32.Hanoun N., Delpu Y., Suriawinata A.A., Bournet B., Bureau C., Selves J., Tsongalis G.J., Dufresne M., Buscail L., Cordelier P., Torrisani J. The silencing of microRNA 148a production by DNA hypermethylation is an early event in pancreatic carcinogenesis. Clin. Chem. 2010;56:1107–1118. doi: 10.1373/clinchem.2010.144709. [DOI] [PubMed] [Google Scholar]
- 33.Bell J.C., Kirn D. MicroRNAs fine-tune oncolytic viruses. Nat. Biotechnol. 2008;26:1346–1348. doi: 10.1038/nbt1208-1346. [DOI] [PubMed] [Google Scholar]
- 34.Edge R.E., Falls T.J., Brown C.W., Lichty B.D., Atkins H., Bell J.C. A let-7 microRNA-sensitive vesicular stomatitis virus demonstrates tumor-specific replication. Mol. Ther. 2008;16:1437–1443. doi: 10.1038/mt.2008.130. [DOI] [PubMed] [Google Scholar]
- 35.Ylösmäki E., Hakkarainen T., Hemminki A., Visakorpi T., Andino R., Saksela K. Generation of a conditionally replicating adenovirus based on targeted destruction of E1A mRNA by a cell type-specific microRNA. J. Virol. 2008;82:11009–11015. doi: 10.1128/JVI.01608-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kelly E.J., Hadac E.M., Greiner S., Russell S.J. Engineering microRNA responsiveness to decrease virus pathogenicity. Nat. Med. 2008;14:1278–1283. doi: 10.1038/nm.1776. [DOI] [PubMed] [Google Scholar]
- 37.Leber M.F., Bossow S., Leonard V.H., Zaoui K., Grossardt C., Frenzke M., Miest T., Sawall S., Cattaneo R., von Kalle C., Ungerechts G. MicroRNA-sensitive oncolytic measles viruses for cancer-specific vector tropism. Mol. Ther. 2011;19:1097–1106. doi: 10.1038/mt.2011.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baertsch M.A., Leber M.F., Bossow S., Singh M., Engeland C.E., Albert J., Grossardt C., Jäger D., von Kalle C., Ungerechts G. MicroRNA-mediated multi-tissue detargeting of oncolytic measles virus. Cancer Gene Ther. 2014;21:373–380. doi: 10.1038/cgt.2014.40. [DOI] [PubMed] [Google Scholar]
- 39.Buckland R., Gerald C., Barker R., Wild T.F. Fusion glycoprotein of measles virus: nucleotide sequence of the gene and comparison with other paramyxoviruses. J. Gen. Virol. 1987;68:1695–1703. doi: 10.1099/0022-1317-68-6-1695. [DOI] [PubMed] [Google Scholar]
- 40.Harrison S.C. Viral membrane fusion. Nat. Struct. Mol. Biol. 2008;15:690–698. doi: 10.1038/nsmb.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liffers S.T., Munding J.B., Vogt M., Kuhlmann J.D., Verdoodt B., Nambiar S., Maghnouj A., Mirmohammadsadegh A., Hahn S.A., Tannapfel A. MicroRNA-148a is down-regulated in human pancreatic ductal adenocarcinomas and regulates cell survival by targeting CDC25B. Lab. Invest. 2011;91:1472–1479. doi: 10.1038/labinvest.2011.99. [DOI] [PubMed] [Google Scholar]
- 42.Collisson E.A., Sadanandam A., Olson P., Gibb W.J., Truitt M., Gu S., Cooc J., Weinkle J., Kim G.E., Jakkula L. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011;17:500–503. doi: 10.1038/nm.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dijk F., Veenstra V.L., Soer E.C., Dings M.P.G., Zhao L., Halfwerk J.B., Hooijer G.K., Damhofer H., Marzano M., Steins A. Unsupervised class discovery in pancreatic ductal adenocarcinoma reveals cell-intrinsic mesenchymal features and high concordance between existing classification systems. Sci. Rep. 2020;10:337. doi: 10.1038/s41598-019-56826-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moffitt R.A., Marayati R., Flate E.L., Volmar K.E., Loeza S.G., Hoadley K.A., Rashid N.U., Williams L.A., Eaton S.C., Chung A.H. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015;47:1168–1178. doi: 10.1038/ng.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu K., Chen B., Aran D., Charalel J., Yau C., Wolf D.M., van ’t Veer L.J., Butte A.J., Goldstein T., Sirota M. Comprehensive transcriptomic analysis of cell lines as models of primary tumors across 22 tumor types. Nat. Commun. 2019;10:3574. doi: 10.1038/s41467-019-11415-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Naniche D., Varior-Krishnan G., Cervoni F., Wild T.F., Rossi B., Rabourdin-Combe C., Gerlier D. Human membrane cofactor protein (CD46) acts as a cellular receptor for measles virus. J. Virol. 1993;67:6025–6032. doi: 10.1128/jvi.67.10.6025-6032.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mühlebach M.D., Mateo M., Sinn P.L., Prüfer S., Uhlig K.M., Leonard V.H., Navaratnarajah C.K., Frenzke M., Wong X.X., Sawatsky B. Adherens junction protein nectin-4 is the epithelial receptor for measles virus. Nature. 2011;480:530–533. doi: 10.1038/nature10639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fishelson Z., Donin N., Zell S., Schultz S., Kirschfink M. Obstacles to cancer immunotherapy: Expression of membrane complement regulatory proteins (mCRPs) in tumors. Mol. Immunol. 2003;40:109–123. doi: 10.1016/s0161-5890(03)00112-3. [DOI] [PubMed] [Google Scholar]
- 49.Anderson B.D., Nakamura T., Russell S.J., Peng K.W. High CD46 receptor density determines preferential killing of tumor cells by oncolytic measles virus. Cancer Res. 2004;64:4919–4926. doi: 10.1158/0008-5472.CAN-04-0884. [DOI] [PubMed] [Google Scholar]
- 50.Awano M., Fujiyuki T., Shoji K., Amagai Y., Murakami Y., Furukawa Y., Sato H., Yoneda M., Kai C. Measles virus selectively blind to signaling lymphocyte activity molecule has oncolytic efficacy against nectin-4-expressing pancreatic cancer cells. Cancer Sci. 2016;107:1647–1652. doi: 10.1111/cas.13064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rosenberg J.E., O’Donnell P.H., Balar A.V., McGregor B.A., Heath E.I., Yu E.Y., Galsky M.D., Hahn N.M., Gartner E.M., Pinelli J.M. Pivotal trial of enfortumab vedotin in urothelial carcinoma after platinum and anti-programmed death 1/programmed death ligand 1 therapy. J. Clin. Oncol. 2019;37:2592–2600. doi: 10.1200/JCO.19.01140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Friedrich M., Pracht K., Mashreghi M.F., Jäck H.M., Radbruch A., Seliger B. The role of the miR-148/-152 family in physiology and disease. Eur. J. Immunol. 2017;47:2026–2038. doi: 10.1002/eji.201747132. [DOI] [PubMed] [Google Scholar]
- 53.Leber M.F., Baertsch M.A., Anker S.C., Henkel L., Singh H.M., Bossow S., Engeland C.E., Barkley R., Hoyler B., Albert J. Enhanced control of oncolytic measles virus using microRNA target sites. Mol. Ther. Oncolytics. 2018;9:30–40. doi: 10.1016/j.omto.2018.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Waddell N., Pajic M., Patch A.M., Chang D.K., Kassahn K.S., Bailey P., Johns A.L., Miller D., Nones K., Quek K., Australian Pancreatic Cancer Genome Initiative Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495–501. doi: 10.1038/nature14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bailey P., Chang D.K., Nones K., Johns A.L., Patch A.M., Gingras M.C., Miller D.K., Christ A.N., Bruxner T.J., Quinn M.C., Australian Pancreatic Cancer Genome Initiative Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531:47–52. doi: 10.1038/nature16965. [DOI] [PubMed] [Google Scholar]
- 56.Kurokawa C., Iankov I.D., Anderson S.K., Aderca I., Leontovich A.A., Maurer M.J., Oberg A.L., Schroeder M.A., Giannini C., Greiner S.M. Constitutive interferon pathway activation in tumors as an efficacy determinant following oncolytic virotherapy. J. Natl. Cancer Inst. 2018;110:1123–1132. doi: 10.1093/jnci/djy033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zaoui K., Bossow S., Grossardt C., Leber M.F., Springfeld C., Plinkert P.K., Kalle Cv., Ungerechts G. Chemovirotherapy for head and neck squamous cell carcinoma with EGFR-targeted and CD/UPRT-armed oncolytic measles virus. Cancer Gene Ther. 2012;19:181–191. doi: 10.1038/cgt.2011.75. [DOI] [PubMed] [Google Scholar]
- 58.Kaufmann J.K., Bossow S., Grossardt C., Sawall S., Kupsch J., Erbs P., Hassel J.C., von Kalle C., Enk A.H., Nettelbeck D.M., Ungerechts G. Chemovirotherapy of malignant melanoma with a targeted and armed oncolytic measles virus. J. Invest. Dermatol. 2013;133:1034–1042. doi: 10.1038/jid.2012.459. [DOI] [PubMed] [Google Scholar]
- 59.Hartkopf A.D., Bossow S., Lampe J., Zimmermann M., Taran F.A., Wallwiener D., Fehm T., Bitzer M., Lauer U.M. Enhanced killing of ovarian carcinoma using oncolytic measles vaccine virus armed with a yeast cytosine deaminase and uracil phosphoribosyltransferase. Gynecol. Oncol. 2013;130:362–368. doi: 10.1016/j.ygyno.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 60.Maurer S., Salih H.R., Smirnow I., Lauer U.M., Berchtold S. Suicide gene-armed measles vaccine virus for the treatment of AML. Int. J. Oncol. 2019;55:347–358. doi: 10.3892/ijo.2019.4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Inoko K., Hiraoka K., Inagaki A., Takahashi M., Kushibiki T., Hontani K., Takano H., Sato S., Takeuchi S., Nakamura T. Therapeutic activity of retroviral replicating vector-mediated prodrug activator gene therapy for pancreatic cancer. Cancer Gene Ther. 2018;25:184–195. doi: 10.1038/s41417-018-0020-7. [DOI] [PubMed] [Google Scholar]
- 62.Mahadevan D., Von Hoff D.D. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol. Cancer Ther. 2007;6:1186–1197. doi: 10.1158/1535-7163.MCT-06-0686. [DOI] [PubMed] [Google Scholar]
- 63.Olive K.P., Jacobetz M.A., Davidson C.J., Gopinathan A., McIntyre D., Honess D., Madhu B., Goldgraben M.A., Caldwell M.E., Allard D. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maurer C., Holmstrom S.R., He J., Laise P., Su T., Ahmed A., Hibshoosh H., Chabot J.A., Oberstein P.E., Sepulveda A.R. Experimental microdissection enables functional harmonisation of pancreatic cancer subtypes. Gut. 2019;68:1034–1043. doi: 10.1136/gutjnl-2018-317706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ilkow C.S., Marguerie M., Batenchuk C., Mayer J., Ben Neriah D., Cousineau S., Falls T., Jennings V.A., Boileau M., Bellamy D. Reciprocal cellular cross-talk within the tumor microenvironment promotes oncolytic virus activity. Nat. Med. 2015;21:530–536. doi: 10.1038/nm.3848. [DOI] [PubMed] [Google Scholar]
- 66.Diop-Frimpong B., Chauhan V.P., Krane S., Boucher Y., Jain R.K. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc. Natl. Acad. Sci. USA. 2011;108:2909–2914. doi: 10.1073/pnas.1018892108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.LaRocca C.J., Warner S.G. A new role for vitamin D: The enhancement of oncolytic viral therapy in pancreatic cancer. Biomedicines. 2018;6:104. doi: 10.3390/biomedicines6040104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jing Y., Chavez V., Khatwani N., Ban Y., Espejo A.P., Chen X., Merchan J.R. In vivo antitumor activity by dual stromal and tumor-targeted oncolytic measles viruses. Cancer Gene Ther. 2020;27:910–922. doi: 10.1038/s41417-020-0171-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hosein A.N., Brekken R.A., Maitra A. Pancreatic cancer stroma: An update on therapeutic targeting strategies. Nat. Rev. Gastroenterol. Hepatol. 2020;17:487–505. doi: 10.1038/s41575-020-0300-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jung M.Y., Offord C.P., Ennis M.K., Kemler I., Neuhauser C., Dingli D. In vivo estimation of oncolytic virus populations within tumors. Cancer Res. 2018;78:5992–6000. doi: 10.1158/0008-5472.CAN-18-0447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kemler I., Ennis M.K., Neuhauser C.M., Dingli D. In vivo imaging of oncolytic measles virus propagation with single-cell resolution. Mol. Ther. Oncolytics. 2018;12:68–78. doi: 10.1016/j.omto.2018.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Leveille S., Samuel S., Goulet M.L., Hiscott J. Enhancing VSV oncolytic activity with an improved cytosine deaminase suicide gene strategy. Cancer Gene Ther. 2011;18:435–443. doi: 10.1038/cgt.2011.14. [DOI] [PubMed] [Google Scholar]
- 73.Bossow S., Grossardt C., Temme A., Leber M.F., Sawall S., Rieber E.P., Cattaneo R., von Kalle C., Ungerechts G. Armed and targeted measles virus for chemovirotherapy of pancreatic cancer. Cancer Gene Ther. 2011;18:598–608. doi: 10.1038/cgt.2011.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Twumasi-Boateng K., Pettigrew J.L., Kwok Y.Y.E., Bell J.C., Nelson B.H. Oncolytic viruses as engineering platforms for combination immunotherapy. Nat. Rev. Cancer. 2018;18:419–432. doi: 10.1038/s41568-018-0009-4. [DOI] [PubMed] [Google Scholar]
- 75.Rosenberg S.A. Decade in review-cancer immunotherapy: Entering the mainstream of cancer treatment. Nat. Rev. Clin. Oncol. 2014;11:630–632. doi: 10.1038/nrclinonc.2014.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kruger S., Ilmer M., Kobold S., Cadilha B.L., Endres S., Ormanns S., Schuebbe G., Renz B.W., D’Haese J.G., Schloesser H. Advances in cancer immunotherapy 2019—latest trends. J. Exp. Clin. Cancer Res. 2019;38:268. doi: 10.1186/s13046-019-1266-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kalbasi A., Ribas A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 2020;20:25–39. doi: 10.1038/s41577-019-0218-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vincent J., Mignot G., Chalmin F., Ladoire S., Bruchard M., Chevriaux A., Martin F., Apetoh L., Rébé C., Ghiringhelli F. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010;70:3052–3061. doi: 10.1158/0008-5472.CAN-09-3690. [DOI] [PubMed] [Google Scholar]
- 79.Pidelaserra-Martí G., Engeland C.E. Mechanisms of measles virus oncolytic immunotherapy. Cytokine Growth Factor Rev. 2020;56:28–38. doi: 10.1016/j.cytogfr.2020.07.009. [DOI] [PubMed] [Google Scholar]
- 80.Hajda J., Lehmann M., Krebs O., Kieser M., Geletneky K., Jäger D., Dahm M., Huber B., Schöning T., Sedlaczek O. A non-controlled, single arm, open label, phase II study of intravenous and intratumoral administration of ParvOryx in patients with metastatic, inoperable pancreatic cancer: ParvOryx02 protocol. BMC Cancer. 2017;17:576. doi: 10.1186/s12885-017-3604-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Eissa I.R., Bustos-Villalobos I., Ichinose T., Matsumura S., Naoe Y., Miyajima N., Morimoto D., Mukoyama N., Zhiwen W., Tanaka M. The current status and future prospects of oncolytic viruses in clinical trials against melanoma, glioma, pancreatic, and breast cancers. Cancers (Basel) 2018;10:356. doi: 10.3390/cancers10100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Freytag S.O., Barton K.N., Brown S.L., Narra V., Zhang Y., Tyson D., Nall C., Lu M., Ajlouni M., Movsas B., Kim J.H. Replication-competent adenovirus-mediated suicide gene therapy with radiation in a preclinical model of pancreatic cancer. Mol. Ther. 2007;15:1600–1606. doi: 10.1038/sj.mt.6300212. [DOI] [PubMed] [Google Scholar]
- 83.Angelova A.L., Aprahamian M., Grekova S.P., Hajri A., Leuchs B., Giese N.A., Dinsart C., Herrmann A., Balboni G., Rommelaere J., Raykov Z. Improvement of gemcitabine-based therapy of pancreatic carcinoma by means of oncolytic parvovirus H-1PV. Clin. Cancer Res. 2009;15:511–519. doi: 10.1158/1078-0432.CCR-08-1088. [DOI] [PubMed] [Google Scholar]
- 84.Réjiba S., Bigand C., Parmentier C., Masmoudi A., Hajri A. Oncosuppressive suicide gene virotherapy “PVH1-yCD/5-FC” for pancreatic peritoneal carcinomatosis treatment: NFκB and Akt/PI3K involvement. PLoS ONE. 2013;8:e70594. doi: 10.1371/journal.pone.0070594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang P., Li X., Wang J., Gao D., Li Y., Li H., Chu Y., Zhang Z., Liu H., Jiang G. Re-designing Interleukin-12 to enhance its safety and potential as an anti-tumor immunotherapeutic agent. Nat. Commun. 2017;8:1395. doi: 10.1038/s41467-017-01385-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li Y., Deng X., Zeng X., Peng X. The role of Mir-148a in cancer. J. Cancer. 2016;7:1233–1241. doi: 10.7150/jca.14616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Meyerhardt J.A., Mayer R.J. Systemic therapy for colorectal cancer. N. Engl. J. Med. 2005;352:476–487. doi: 10.1056/NEJMra040958. [DOI] [PubMed] [Google Scholar]
- 88.Van Cutsem E., Sagaert X., Topal B., Haustermans K., Prenen H. Gastric cancer. Lancet. 2016;388:2654–2664. doi: 10.1016/S0140-6736(16)30354-3. [DOI] [PubMed] [Google Scholar]
- 89.Duprex W.P., McQuaid S., Hangartner L., Billeter M.A., Rima B.K. Observation of measles virus cell-to-cell spread in astrocytoma cells by using a green fluorescent protein-expressing recombinant virus. J. Virol. 1999;73:9568–9575. doi: 10.1128/jvi.73.11.9568-9575.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Singh M., Cattaneo R., Billeter M.A. A recombinant measles virus expressing hepatitis B virus surface antigen induces humoral immune responses in genetically modified mice. J. Virol. 1999;73:4823–4828. doi: 10.1128/jvi.73.6.4823-4828.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Radecke F., Spielhofer P., Schneider H., Kaelin K., Huber M., Dötsch C., Christiansen G., Billeter M.A. Rescue of measles viruses from cloned DNA. EMBO J. 1995;14:5773–5784. doi: 10.1002/j.1460-2075.1995.tb00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Martin A., Staeheli P., Schneider U. RNA polymerase II-controlled expression of antigenomic RNA enhances the rescue efficacies of two different members of the Mononegavirales independently of the site of viral genome replication. J. Virol. 2006;80:5708–5715. doi: 10.1128/JVI.02389-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Luu-The V., Paquet N., Calvo E., Cumps J. Improved real-time RT-PCR method for high-throughput measurements using second derivative calculation and double correction. Biotechniques. 2005;38:287–293. doi: 10.2144/05382RR05. [DOI] [PubMed] [Google Scholar]
- 94.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. Fiji: An open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Haack K., Moebius U., Knebel Doeberitz M.V., Herfarth C., Schackert H.K., Gebert J.F. Detection of cytosine deaminase in genetically modified tumor cells by specific antibodies. Hum. Gene Ther. 1997;8:1395–1401. doi: 10.1089/hum.1997.8.11-1395. [DOI] [PubMed] [Google Scholar]
- 96.R Core Team . R Foundation for Statistical Computing; 2017. R: A Language and Environment for Statistical Computing.https://www.R-project.org/ [Google Scholar]
- 97.Lindstrom M.L., Bates D.M. Nonlinear mixed effects models for repeated measures data. Biometrics. 1990;46:673–687. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.