

Abstract
“Low-dose” acute intermittent hypoxia (AIH; 3-15 episodes/day) is emerging as a promising therapeutic strategy to improve motor function after incomplete cervical spinal cord injury (cSCI). Conversely, chronic “high-dose” intermittent hypoxia (CIH; > 80–100 episodes/day) elicits multi-system pathology and is a hallmark of sleep apnea, a condition highly prevalent in individuals with cSCI. Whereas daily AIH (dAIH) enhances phrenic motor plasticity in intact rats, it is abolished by CIH. However, there have been no direct comparisons of prolonged dAIH versus CIH on phrenic motor outcomes after chronic cSCI. Thus, phrenic nerve activity and AIH-induced phrenic long-term facilitation (pLTF) were assessed in anesthetized rats. Experimental groups included: 1) intact rats exposed to 28 days of normoxia (Nx28; 21% O2; 8 h/day), and three groups with chronic C2 hemisection (C2Hx) exposed to either: 2) Nx28; 3) dAIH (dAIH28; 10, 5-min episodes of 10.5% O2/day; 5-min intervals); or 4) CIH (IH28-2/2; 2-min episodes; 2-min intervals; 8 h/day). Baseline ipsilateral phrenic nerve activity was reduced in injured versus intact rats but unaffected by dAIH28 or IH28-2/2. There were no group differences in contralateral phrenic activity. pLTF was enhanced bilaterally by dAIH28 versus Nx28 but unaffected by IH28-2/2. Whereas dAIH28 enhanced pLTF after cSCI, it did not improve baseline phrenic output. In contrast, unlike shorter protocols in intact rats, CIH28-2/2 did not abolish pLTF in chronic C2Hx. Mechanisms of differential responses to dAIH versus CIH are not yet known, particularly in the context of cSCI. Further, it remains unclear whether enhanced phrenic motor plasticity can improve breathing after cSCI.
Keywords: acute intermittent hypoxia, cervical spinal cord injury, chronic intermittent hypoxia, phrenic long-term facilitation, respiratory rehabilitation, sleep apnea
Introduction
Insufficient motor drive to respiratory muscles is a major cause of morbidity and mortality in individuals with cervical spinal cord injury (cSCI).1–4 Although limited spontaneous recovery of lost breathing function is observed shortly after injury, prevailing damage to bulbospinal pathways and/or respiratory motor neurons often leads to life-long respiratory compromise. Since the mainstay of respiratory management following cSCI is mechanical ventilation, there is pressing need for new strategies to restore independent breathing.5 One emerging strategy to restore respiratory (and non-respiratory) motor function is to invoke respiratory motor plasticity via low-dose acute intermittent hypoxia (AIH).6,7
AIH is characterized by brief intermittent reductions in inspired oxygen (3-15 episodes per day), with normoxic intervals. AIH triggers spinal respiratory neuroplasticity, leading to sustained increases in respiratory motor output.8-10 Repetitive/daily AIH (rAIH/dAIH) partially restores breathing capacity in rats with incomplete cSCI,9,11-13 and upregulates key pro-plasticity molecules in the spinal cord.9,14,15 Ongoing efforts are exploring rAIH/dAIH as a therapeutic modality to improve motor function after SCI.6,16-18 Indeed, a growing body of evidence from studies in animal models and humans with chronic SCI highlights the promise of rAIH to improve both respiratory7,9,11-13 and non-respiratory16,17,19–24 motor function following incomplete cSCI.
On the other end of the spectrum, chronic intermittent hypoxia (CIH; > 80-100 episodes per day) elicits multi-system pathology and is a hallmark of sleep apnea, a condition highly prevalent in individuals with cSCI.5,25-27 Indeed, sleep-disordered breathing is experienced by nearly 80% of individuals with cSCI28,29 and evidence from experimental models further reinforces an apparent association between sleep-disordered breathing and cSCI.30-32 Although most studies exploring CIH highlight its detrimental effects (e.g., cardiovascular, metabolic, cognitive impairment), there are suggestions that mild to moderate CIH has beneficial effects.33–37 We hypothesize that the balance of beneficial versus pathogenic effects of intermittent hypoxia relates to the “dose.”38,39 Indeed, 7 days of low-dose dAIH pre-conditioning (< 15 episodes per day) enhances phrenic motor plasticity,15 whereas it is abolished by 1-7 days of moderate CIH (15 episodes per hour, 8 h per day; 120 episodes/day).40 In contrast, mild CIH (∼six episodes per hour, 12 h per day, 72 episodes/day) enhances phrenic motor plasticity similar to dAIH.35,36 Thus, mild CIH associated with sleep apnea could in fact serve as a form of “self-medication,” contributing to spontaneous recovery of motor function. However, the comparative effects of IH protocols that differ in intensity (“dose”) on respiratory outcomes are unclear, particularly in the context of chronic cSCI.
Here, we compare prolonged (28 days), low-dose dAIH (10 episodes/day) versus high-dose CIH simulating moderate sleep apnea (120 episodes/day) on phrenic motor outcomes in rats with chronic, incomplete cSCI. Specifically, we explored the impact of IH pre-conditioning on baseline phrenic nerve activity, phrenic responses to chemoreflex activation, and moderate AIH (mAIH)-induced phrenic long-term facilitation (pLTF) in an anesthetized preparation. We hypothesized that dAIH pre-conditioning would enhance phrenic motor output and AIH-induced plasticity, whereas moderate CIH would not. These data provide the first direct comparison of different IH protocols in a rat model of cSCI. We demonstrate protocol-specific IH effects on phrenic motor plasticity; however, these effects did not translate to improvement in spontaneous or chemoreflex-driven phrenic motor output.
Methods
Animals
We used adult, male, Sprague-Dawley rats (n = 42; 11-12 weeks old and 375 ± 24 g at study enrollment; Colony 208A; ENVIGO Laboratories, Indianapolis, IN), housed in pairs in a controlled environment (12 h light/dark cycles, 20-30% relative humidity, ∼22°C) with food and water ad libitum. All experimental protocols were approved by the University of Florida Institutional Animal Care and Use Committee, using standards set forth in the National Institutes of Health Guide for Care and Use of Laboratory Animals. Rats were assigned to one of four groups: 1) spinal intact, exposed to 28 days of normoxia (Intact Nx28; n = 11); 2) chronic cervical hemisection (C2Hx) exposed to 28 days of normoxia (C2Hx Nx28; n = 8); 3) chronic C2Hx exposed to 28 days of daily acute intermittent hypoxia (C2Hx dAIH28, n = 8); or 4) C2Hx exposed to 28 days of CIH (C2Hx IH28-2/2, n = 15).
Surgical procedures
Anesthesia and spinal cord injury methods have been described previously.13,41,42 Briefly, rats were anesthetized with 2.5% isoflurane (in 100% O2) and transferred to a heated surgical station. The adequacy of surgical anesthesia was confirmed by the absence of toe pinch and palpebral responses. Meloxicam was administered (2 mg/kg, subcutaneously [s.q.]) to minimize post-surgical pain and inflammation. The incision site was shaved and disinfected with alternating chlorhexidine and ethanol (70%) scrubs. A 1-inch dorsal midline incision was made from the base of the skull to the fifth cervical segment. After C2 laminectomy and durotomy just caudal to the C2 dorsal rootlets, complete transection of the left spinal cord (midline to lateral border) was performed at C2 (Fig. 1A) using a microscalpel, followed by gentle aspiration to ensure a complete lesion. The dura was closed using interrupted 9-0 sutures, overlying muscle with interrupted 4-0 sutures, and the skin closed with stainless steel surgical clips.
FIG. 1.

C2 Hemi-section model and intermittent hypoxia protocols. (A) Schema depicting C2 spinal cord hemi-section injury model. Briefly, the left side of the spinal cord was cut at C2, severing descending projections to ipsilateral phrenic motoneurons, paralyzing the ipsilateral hemidiaphragm. (B) Schema depicting 28 days of normoxia (Nx28), daily acute intermittent hypoxia (dAIH28), and intermittent hypoxia simulating moderate sleep apnea (IH28-2/2) pre-conditioning protocols. Pre-conditioning was conducted daily for 4 weeks, beginning 8 weeks post-injury (or the equivalent timepoint in intact rats). Nx28 consisted of 8 h of continuous normoxia (21% O2) per day. dAIH consisted of ten 5-min episodes of 10.5% O2, with 5-min normoxic intervals. IH28-2/2 consisted of 8 h of 2-min hypoxic episodes (10.5% O2), with 2-min normoxic intervals.
After surgery, buprenorphine (0.03 mg/kg, s.q.) and sterile lactated Ringer's solution (5 mL s.q.) were administered. Rats recovered on a heated pad while breathing 100% O2 via nose cone until anesthesia was reversed. Post-surgical supportive care included buprenorphine (0.03 mg/kg, s.q.) for the initial 72 h post-injury, lactated Ringer's solution (5 mL s.q., 2 times/day), and oral DietGel recovery supplement (1-3 mL, 1-2 times/day, Clear H2O, Westbrook, ME) until volitional drinking and eating had resumed. Manual bladder expression was performed at least twice per day until spontaneous voiding returned (typically <1 week post-injury). Randomly selected, age-matched rats in intact control groups did not undergo surgical procedures but were maintained in the same environmental conditions.
Intermittent hypoxia protocols
The basic Nx, dAIH, and CIH protocols have been described in prior reports.9,13,40 Briefly, daily exposures were administered during the light phase for 28 consecutive days, beginning 8 weeks post-injury (or analogous time-point in intact rats). During the 28-day exposure, rats were housed in pairs in custom Plexiglas cages, with access to food and water ad libitum. Gas delivery was controlled by programmable mass flow controllers attached to a specialized cage top equipped to deliver protocol-specific gas mixtures (O2, balance N2; Fig. 1B; Therapeutiq, Kansas City, MO; cage volume = 4.5 L; total gas flow rate = 16 L/min per cage). Normoxic and hypoxic gases were created by mixing room air with medical grade nitrogen. Gases were filtered for microparticles and bacteria using a high purity compressed air-filter (Parker-Hannifin, Cleveland, OH) and humidified to ∼30-50% relative humidity using a custom-built system comprised of a Nafion Fuel Cell Humidifier (PermaPure, Lakewood, NJ) connected to a circulating water bath. For normoxia protocols, cages were flushed with filtered, humidified air (21% O2) for 8 h/day; for intermittent hypoxia protocols,10.5% and 21% O2 were alternated in 5-min episodes with 5-min intervals for a total of 10 episodes (dAIH28), or at 2-min episodes with 2-min intervals for 8 h/day (IH28-2/2), for a total of 120 episodes.
Neurophysiology
The basic neurophysiology preparation has been described previously40,42–45; neurophysiological assessments were performed by a single experimenter blinded to experimental group. Briefly,1 day after the final exposure, rats were anesthetized with 3% isoflurane (in 100% O2), and transferred to a heated surgical station, where core body temperature was recorded via rectal probe (Kent Scientific) and maintained at ∼37.5 ± 1°C. A tail vein catheter was placed for intravenous delivery of urethane anesthesia and supplemental fluids. Using a ventral approach, the trachea was cannulated, rats were pump-ventilated (Rodent Ventilator, Harvard Apparatus, Holliston, MA) and mid-cervical vagotomy was performed bilaterally. Tracheal pressure was continuously monitored, and rats were maintained on 2.5-3.5% isoflurane (50-60% O2) with inspired CO2 added to maintain end-tidal CO2 between 45-50 mm Hg (Capnogard; Respironics, Inc., Andover, MA). A femoral arterial catheter was placed for real-time assessment of arterial blood pressure and sampling arterial blood for analysis of arterial blood gases, pH and base excess (ABL90 FLEX, Radiometer, Copenhagen, Denmark). Rats were then slowly converted to urethane anesthesia (2.1 g/kg, intravenously [i.v.], 6 mL/h) and inhaled isoflurane was simultaneously withdrawn. Adequate anesthetic depth was verified by assessing blood pressure responses to toe-pinch. Urethane supplements (e.g., 0.2 mL bolus) were given if blood pressure increased significantly in response to toe pinch. Following urethane conversion, rats received the paralytic drug, pancuronium bromide (2.5 mg/kg, i.v., Hospira, Inc.), to minimize artifact from respiratory efforts out of phase with the ventilator. Continuous infusion (1-3 mL/h) of a 1:4 solution (8.4% sodium bicarbonate + lactated Ringer's, i.v.) was maintained throughout the experiment; the infusion rate adjusted to maintain blood pressure and acid-base balance (SBE: ±3 mEq/L).
During a 1-h stabilization period following conversion to urethane, the left and right phrenic nerves were isolated with a dorsal approach, cut distally, and desheathed. The proximal nerve stump was bathed in 0.9% saline and recorded via custom bipolar silver wire suction electrodes. Nerve activity was amplified (gain: 10k), band pass-filtered (100 Hz to 10 kHz; A-M Systems, Carlsberg, WA), and integrated (50 msec time constant; Power 1401, Cambridge Electronic Design Limited, Cambridge, UK). The signal was digitized, recorded continuously and analyzed with Spike2.v8 software (CED). Baseline nerve activity was established at an FIO2 ∼0.5-0.6 (PaO2 > 180 mm Hg). End-tidal CO2 was monitored and maintained throughout the experiment using a flow-through capnograph (Respironics, Andover, MA). The apneic threshold of each rat was determined by progressively lowering inspired CO2 until phrenic nerve discharge ceased. Inspired CO2 was then slowly increased until phrenic nerve activity resumed, marking the recruitment threshold. End tidal PCO2 was set 2-3 mm Hg above the recruitment threshold for the remainder of the experiment. After a stable period of phrenic nerve activity (> 15 min), an arterial blood sample (∼70 mL per sample) was drawn to establish baseline blood gases, and an AIH protocol was administered, consisting of three 5-min hypoxic episodes (10.5% O2; target PaO2: 40-50 mm Hg) separated by 5-min intervals of normoxia.40,45 Blood samples were drawn during hypoxic episodes, and at 15, 30, 60, and 90 min post-AIH to document and adjust physiological conditions throughout the recording period. At 90 min post-hypoxia, a chemoreflex stimulation challenge was administered by increasing inspired CO2 to reach an end-tidal PCO2 of 90 mm Hg; typically, this required an FICO0 of ∼10-11%. On completion of experimental procedures, the proximal stump of the phrenic nerve was bathed in lidocaine to establish a “zero” value. Rats were then humanely overdosed with urethane.
Statistical analysis
Electrophysiological data were collected using Spike2.v8 software (Cambridge Electronic Design, CED, Cambridge, UK). Data were included in analysis only if they met all of the following previously established criteria40,41: 1) PaO2 during baseline and recovery was >180 mm Hg; 2) PaO2 during hypoxic episodes was 40 to 50 mm Hg; and 3) PaCO2 remained within 1.5 mm Hg of baseline post-AIH. Average phrenic nerve amplitude was evaluated for 1 min prior to each blood sample (baseline; first hypoxic episode; and 15, 30, 60, and 90 min post-AIH; for simplicity, phrenic burst amplitude is reported only during baseline, hypoxic episodes, and 90 min post-AIH. Phrenic burst amplitude during each hypoxic response was determined from Minute 3 of each hypoxic episode (H1, H2, and H3). In addition to average amplitudes expressed as a raw value (volts), integrated nerve burst amplitude was normalized as a percent change from baseline (e.g., pLTF). Group data were averaged and expressed as means ±1 standard error of the mean. Groups were compared using one-way analysis of variance (ANOVA) with Tukey honest significant difference post hoc tests. For comparisons of phrenic output, cardiovascular variables and blood gas values across experimental protocols, groups were compared using a two-way ANOVA with a repeated measures design and Tukey honest significant difference post hoc tests. Statistical analyses were performed in GraphPad Prism 8 (GraphPad Software Inc., San Diego, CA) and SigmaPlot version 14 (Systat software, San Jose, CA). Differences were considered statistically significant if p < 0.05.
Results
Phrenic nerve burst amplitude
Consistent with prior reports,46,47 C2Hx resulted in persistent reductions in phrenic nerve burst amplitude ipsilateral to injury, under both baseline and chemoreflex stimulated conditions (Fig. 2; Table 1). Indeed, baseline ipsilateral phrenic output was reduced by 67% (C2Hx Nx28), 72% (C2Hx dAIH28) and 73% (C2Hx IH28-2/2) post-injury versus intact normoxia-treated rats (p < 0.001 for all; Fig. 2A, 2B). No protocol-specific differences in ipsilateral baseline burst amplitude were observed among injured groups (p > 0.9 for all). Similarly, ipsilateral phrenic burst amplitude was reduced during hypercapnic chemoreflex challenge in rats with chronic C2Hx by 55%, 53% and 64%, respectively (p < 0.003 for all vs. intact; Fig. 2C, 2D), with no protocol-specific differences observed (p > 0.84 across injured groups). Contralateral phrenic burst amplitude was not different between groups under baseline or chemoreflex stimulated conditions, regardless of injury or intermittent hypoxia exposure (Fig. 3; Table 1; p = 0.494 and p = 0.177, respectively).
FIG. 2.

Left phrenic nerve activity (ipsilateral to C2 hemisection) during baseline and chemoreflex stimulated conditions. (A) Representative compressed traces depict raw (bottom) and integrated (top) left phrenic (i.e., ipsilateral to injury) neurograms under baseline conditions from intact rats, and rats with chronic C2 hemisection (C2Hx), exposed to 28 days of normoxia (Nx28), daily acute intermittent hypoxia (dAIH28), or chronic intermittent hypoxia simulating moderate sleep apnea (IH28-2/2). (B) Under baseline conditions, average ipsilateral/left integrated phrenic burst amplitude (volts) was reduced in all C2Hx rats vs. intact rats exposed to Nx28. In injured rat groups, no protocol-induced differences could be detected. (C) Representative traces depict raw and integrated left phrenic neurograms under chemoreflex stimulated conditions. (D) During chemoreflex stimulation, average ipsilateral/left integrated phrenic burst amplitude was reduced in C2Hx vs. intact rats. No IH protocol-specific differences were observed in injured rats. *Significantly different from intact Nx28; p < 0.05.
Table 1.
Phrenic Nerve Burst Amplitude
| Intact Nx28 | C2Hx Nx28 | C2Hx dAIH28 | C2Hx IH28-2/2 | |
|---|---|---|---|---|
| Ipsilateral phrenic (raw amplitude - volts) | ||||
| Baseline | 0.063 ± 0.007 | 0.021 ± 0.002* | 0.017 ± 0.005* | 0.017 ± 0.002* |
| H1 | 0.117 ± 0.012 | 0.057 ± 0.004* | 0.055 ± 0.010* | 0.047 ± 0.005* |
| H2 | 0.103 ± 0.013 | 0.041 ± 0.003* | 0.045 ± 0.011* | 0.043 ± 0.006* |
| H3 | 0.103 ± 0.013 | 0.044 ± 0.005* | 0.046 ± 0.011* | 0.043 ± 0.005* |
| 90 min | 0.078 ± 0.010 | 0.035 ± 0.005* | 0.037 ± 0.009* | 0.028 ± 0.004* |
| Chemoreflex stimulation | 0.139 ± 0.016 | 0.084 ± 0.008 | 0.084 ± 0.017 | 0.078 ± 0.009* |
| Contralateral phrenic (raw amplitude - volts) | ||||
| Baseline | 0.088 ± 0.015 | 0.094 ± 0.017 | 0.084 ± 0.012 | 0.112 ± 0.013 |
| H1 | 0.147 ± 0.021 | 0.161 ± 0.030 | 0.149 ± 0.020 | 0.216 ± 0.030 |
| H2 | 0.137 ± 0.019 | 0.142 ± 0.027 | 0.152 ± 0.022 | 0.208 ± 0.029 |
| H3 | 0.139 ± 0.017 | 0.148 ± 0.030 | 0.163 ± 0.024 | 0.208 ± 0.026 |
| 90 min | 0.116 ± 0.016 | 0.130 ± 0.032 | 0.153 ± 0.023 | 0.167 ± 0.019 |
| Chemoreflex stimulation | 0.209 ± 0.033 | 0.219 ± 0.048 | 0.278 ± 0.041 | 0.326 ± 0.042 |
| Ipsilateral phrenic (% change relative to baseline) | ||||
| H1 | 90.9 ± 10.0 | 180.4 ± 23.4 | 274.4 ± 34.4* | 194.8 ± 20.6* |
| H2 | 63.4 ± 7.6 | 102.7 ± 21.1 | 183.4 ± 17.7* | 163.0 ± 25.2* |
| H3 | 63.1 ± 7.1 | 118.1 ± 29.7 | 185.8 ± 19.5* | 162.2 ± 19.8* |
| 90 min | 24.6 ± 8.6 | 79.2 ± 29.1 | 142.2 ± 35.5* | 72.4 ± 18.2 |
| Chemoreflex stimulation | 123.5 ± 13.3 | 330.5 ± 50.1* | 456.1 ± 54.6* | 383.0 ± 39.4* |
| Contralateral phrenic (% change relative to baseline) | ||||
| H1 | 75.2 ± 6.7 | 74.2 ± 2.7 | 86.4 ± 17.0 | 88.4 ± 9.2 |
| H2 | 63.3 ± 6.8 | 52.7 ± 5.0 | 83.2 ± 11.7 | 80.4 ± 9.1 |
| H3 | 68.6 ± 8.2 | 59.7 ± 7.5 | 96.9 ± 15.3 | 83.7 ± 8.2 |
| 90 min | 39.9 ± 8.3 | 27.8 ± 10.8 | 85.4 ± 16.0 | 49.7 ± 10.6 |
| Chemoreflex stimulation | 150.1 ± 15.7 | 124.1 ± 12.7 | 239.6 ± 31.0# | 190.5 ± 22.1 |
Ipsilateral (left) and contralateral (right) phrenic nerve burst amplitude (raw and % change relative to baseline) at baseline, first, second, and third hypoxic episodes (H1, H2, & H3), 90 min after the final hypoxia, and during hypercapnic chemoreflex stimulated conditions.
Different from Intact Nx28.
Different from C2Hx Nx28.
Nx28, 28 days of normoxia; C2Hx, chronic C2 hemisection; dAIH, daily acute intermittent hypoxia; IH28-2/2, intermittent hypoxia simulating moderate sleep apnea; H, hypoxic episode.
FIG. 3.

Right phrenic nerve activity (contralateral to C2 hemisection) during baseline and chemoreflex stimulated conditions. (A) Representative compressed traces depict raw (bottom) and integrated (top) right phrenic (i.e., contralateral to injury) neurograms under baseline conditions from intact rats, and rats with chronic C2 hemisection (C2Hx) exposed to 28 days of normoxia (Nx28), daily acute intermittent hypoxia (dAIH28), or chronic intermittent hypoxia simulating moderate sleep apnea (IH28-2/2). (B) Under baseline conditions, average contralateral/right integrated phrenic burst amplitude (volts) in C2Hx rats was not different from intact rats exposed to Nx28. No protocol-specific differences were observed across groups. (C) Representative traces depict raw and integrated right phrenic neurograms under hypercapnic chemoreflex stimulated conditions. (D) During chemoreflex stimulation, average contralateral/right integrated phrenic burst amplitude was not different between intact vs. injured rats. No protocol-specific differences were observed across groups.
Phrenic nerve burst frequency
Consistent with literature,48,49 baseline phrenic nerve burst frequency was increased with chronic C2Hx (p = 0.002; Fig. 4; Table 2). Indeed, baseline frequency was higher in chronically injured rats versus intact rats (p < 0.044 for all), with no protocol-specific differences observed (p > 0.821 for all). No evidence of frequency LTF was observed in any group (p = 0.584). Further, phrenic nerve burst frequency during chemoreflex activation was not different between groups (p = 0.326).
FIG. 4.
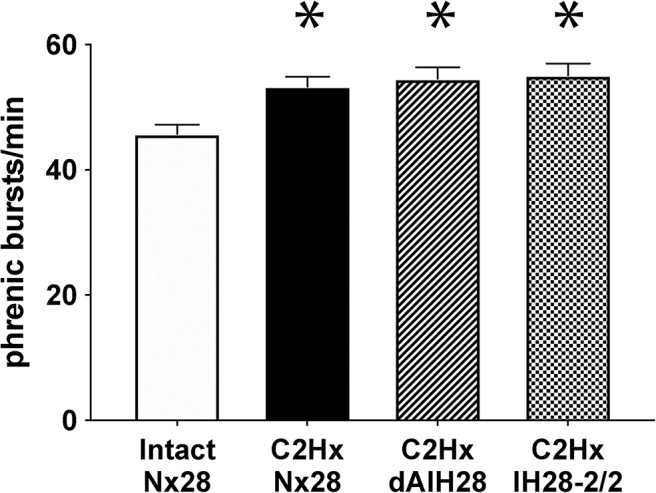
Baseline phrenic nerve burst frequency. Average phrenic nerve burst frequency during baseline conditions. Baseline frequency was increased following C2 hemisection (C2Hx); no intermittent hypoxia (IH) protocol-specific differences were observed. *Significantly different from intact 28 days of normoxia (Nx28), p < 0.05.
Table 2.
Phrenic Nerve Burst Frequency
| Intact Nx28 | C2Hx Nx28 | C2Hx dAIH28 | C2Hx IH28-2/2 | |
|---|---|---|---|---|
| Baseline | 46 ± 1 | *53 ± 2 | *55 ± 2 | *55 ± 2 |
| H1 | 61 ± 2 | 66 ± 2 | 62 ± 2 | 63 ± 2 |
| H2 | 63 ± 2 | 71 ± 2 | 68 ± 2 | 65 ± 2 |
| H3 | 64 ± 2 | 71 ± 3 | 68 ± 2 | 66 ± 2 |
| 90 min | 47 ± 1 | 57 ± 2 | 56 ± 1 | 55 ± 2 |
| Chemoreflex stimulation | 53 ± 2 | 56 ± 2 | 57 ± 2 | 57 ± 1 |
Phrenic nerve burst frequency at baseline, H1, H2, H3, 90 min after moderate acute intermittent hypoxia, and during hypercapnic chemoreflex stimulation.
Different from intact Nx28.
Nx28, 28 days of normoxia; C2Hx, chronic C2 hemisection; dAIH, daily acute intermittent hypoxia; IH28-2/2, intermittent hypoxia simulating moderate sleep apnea; H, hypoxic episode.
Hypoxic phrenic response
Phrenic burst amplitude was increased bilaterally during hypoxic episodes (relative to baseline) in all groups (Fig. 5; Table 1; ipsilateral, p < 0.001; contralateral, p < 0.001). As expected, raw ipsilateral phrenic burst amplitude during hypoxic episodes was reduced in all injured groups versus intact rats (p < 0.0003 for all); no protocol-specific differences in ipsilateral hypoxic responses were observed within injured groups (all p > 0.47). Contralateral phrenic nerve burst amplitude during hypoxic episodes was not different among groups when expressed as raw amplitude (p = 0.194) or percent change from baseline (p = 0.225), and no protocol-specific differences observed (all p > 0.05). We found no evidence of progressive augmentation (e.g., increasing hypoxic phrenic response with successive hypoxic episodes)50 in ipsilateral or contralateral phrenic nerves. Phrenic nerve burst frequency increased during hypoxic episodes in all groups (all p < 0.001), but no differences were observed between treatment groups (all p > 0.07).
FIG. 5.

Short-term hypoxic phrenic responses. (A) Average ipsilateral integrated phrenic nerve burst amplitude (volts) during the first (H1), second (H2), and third (H3) hypoxic episodes in each rat. Average phrenic nerve amplitude was reduced ipsilateral to injury during hypoxic episodes vs. intact rats, despite similar levels of arterial PaO2. No intermittent hypoxia (IH) protocol-specific differences were observed, nor was there evidence for progressive augmentation in any group. (B) Average contralateral integrated phrenic nerve burst amplitude during the first, second, and third hypoxic episodes. Average phrenic nerve amplitude was not different between groups, nor was there evidence for progressive augmentation in successive hypoxic episodes. *Significantly different from intact 28 days of normoxia (Nx28); p < 0.05.
Expression of mAIH-induced phrenic motor plasticity
Representative traces of raw and integrated phrenic nerve activity at baseline (black) and 90 min post-AIH (red) are overlain in Figure 6 to illustrate progressive increases in phrenic burst amplitude (i.e., pLTF). Although ipsilateral phrenic nerve amplitude was reduced versus intact rats at baseline and 90 min post-AIH (p < 0.0036), pLTF was evident in all groups. Nx28-treated rats (intact and C2Hx) exhibited moderate pLTF in both ipsilateral and contralateral phrenic nerves (Fig. 6B and 6D). pLTF was enhanced bilaterally in injured rats following dAIH28 (ipsilateral, p = 0.01; contralateral, p = 0.041) versus intact rats. Contralateral pLTF was also greater in injured rats following dAIH28 versus injured rats treated with normoxia (p = 0.015). Injured rats exposed to IH28-2/2 did express pLTF bilaterally; however, the magnitude of pLTF expressed in these rats was comparable to that expressed in intact (ipsilateral, p = 0.426; contralateral, p = 0.927) and injured rats treated with Nx28 (ipsilateral, p = 0.997; contralateral, p = 0.606). A trend for lower pLTF in injured rats exposed to IH28-2/2 versus dAIH28 was not significant (ipsilateral, p = 0.168; contralateral, p = 0.100).
FIG. 6.

Phrenic long-term facilitation (pLTF) following moderate acute intermittent hypoxia (mAIH). (A) Representative traces depict integrated ipsilateral (left) phrenic neurograms at baseline (black) overlaid on neurogram traces 90 min post-hypoxia (red). (B) Average ipsilateral pLTF (% increase from baseline) was enhanced in rats with chronic C2 hemisection (C2Hx) exposed to daily acute intermittent hypoxia (dAIH28) vs. intact rats. In C2Hx rats exposed to intermittent hypoxia simulating moderate sleep apnea (IH28-2/2), pLTF was not different from intact 28 days of normoxia (Nx28) rats; an apparent difference from dAIH28 rats did not reach statistical significance (p = 0.168). (C) Representative traces depict integrated contralateral (right) phrenic neurograms at baseline (gray) vs. 90 min post-mAIH (black). (D) Average contralateral pLTF was enhanced in dAIH-treated rats with chronic C2Hx. Although C2Hx rats exposed to IH28-2/2 tended to have blunted pLTF (vs. dAIH-treated), this difference did not reach statistical significance (p = 0.1). *Significantly different from intact Nx28; p < 0.05. #Significantly different from C2Hx Nx28; p < 0.05.
Arterial pressure and heart rate
Arterial blood pressure and heart rate measurements are presented in Figure 7 and Table 3. Blood pressure exhibited no significant protocol-specific differences among treatment groups in systolic (Intact Nx28 = 162 ± 4 mm Hg; C2Hx Nx28 = 147 ± 7 mm Hg; C2Hx dAIH28 = 153 ± 6 mm Hg; C2Hx IH28-2/2 = 151 ± 4 mm Hg; p = 0.2628), diastolic (Intact Nx7 = 100 ± 4 mm Hg; C2Hx Nx7 = 94 ± 6 mm Hg; C2Hx dAIH7 = 99 ± 4 mm Hg; C2Hx IH7-2/2 = 99 ± 3 mm Hg; p = 0.7485) or mean arterial pressures (Intact Nx28 = 121 ± 4; C2Hx Nx28: 112 ± 6; C2Hx dAIH28 = 117 ± 5; and C2Hx IH28-2/2 = 116 ± 3 mm Hg; p = 0.518). Mean arterial pressure was reduced during hypoxic episodes (H1: p = 0.116; H2: p < 0.001; H3: p < 0.001), as is characteristic of this experimental preparation,51 but returned to baseline levels on return to control conditions; mean arterial pressure was not different from baseline 90 min post-AIH (all p = 0.538), nor were there differences in 90-min systolic, diastolic, or mean arterial pressures from baseline or between groups (all p > 0.123).
FIG. 7.

Mean arterial pressure and heart rate. (A) Representative traces depict instantaneous heart rate (top) and arterial blood pressure (bottom) under baseline conditions. (B) Average baseline mean arterial pressure (mm Hg) was not different between groups. (C) Average baseline heart rate (beats/min) was not different between groups.
Table 3.
SBP, DBP, MAP, and HR at Baseline, H1, H2, H3 and 90 Min after Moderate Acute Intermittent Hypoxia
| Intact Nx28 | C2Hx Nx28 | C2Hx dAIH28 | C2Hx IH28-2/2 | |
|---|---|---|---|---|
| Systolic blood pressure | ||||
| Baseline | 162 ± 4 | 147 ± 7 | 153 ± 6 | 151 ± 4 |
| H1 | 163 ± 6 | 149 ± 9 | 154 ± 7 | 143 ± 7 |
| H2*#% | 154 ± 7 | 129 ± 10 | 145 ± 8 | 128 ± 6 |
| H3*#% | 155 ± 8 | 131 ± 11 | 148 ± 8 | 127 ± 5 |
| 90 min | 167 ± 6 | 145 ± 11 | 146 ± 7 | 146 ± 6 |
| Diastolic blood pressure | ||||
| Baseline | 100 ± 4 | 94 ± 5 | 99 ± 4 | 99 ± 3 |
| H1 | 94 ± 5 | 89 ± 7 | 92 ± 7 | 88 ± 7 |
| H2*#% | 81 ± 5 | 72 ± 7 | 79 ± 6 | 71 ± 6 |
| H3*#% | 84 ± 5 | 75 ± 7 | 80 ± 6 | 67 ± 6 |
| 90 min | 108 ± 4 | 96 ± 9 | 90 ± 6 | 100 ± 5 |
| Mean arterial pressure | ||||
| Baseline | 121 ± 4 | 112 ± 6 | 117 ± 4 | 116 ± 3 |
| H1 | 117 ± 5 | 109 ± 7 | 112 ± 7 | 103 ± 7 |
| H2*# | 106 ± 6 | 91 ± 8 | 101 ± 7 | 90 ± 6 |
| H3*# | 108 ± 6 | 94 ± 8 | 102 ± 6 | 87 ± 5 |
| 90 min | 128 ± 5 | 112 ± 10 | 108 ± 6 | 111 ± 5 |
| Heart rate | ||||
| Baseline | 403 ± 6 | 422 ± 8 | 419 ± 8 | 412 ± 5 |
| H1*# | 421 ± 7 | 449 ± 6 | 442 ± 9 | 412 ± 14 |
| H2# | 413 ± 7 | 437 ± 7 | 429 ± 8 | 417 ± 6 |
| H3# | 401 ± 9 | 434 ± 7 | 426 ± 8 | 407 ± 7 |
| 90 min* | 395 ± 7 | 409 ± 11 | 401 ± 9 | 397 ± 4 |
Different from baseline.
Different from 90 min.
Different from H1.
SBP, systolic blood pressure; DBP, diastolic blood pressure; MAP, mean arterial pressure; HR, heart rate; Nx28, 28 days of normoxia; C2Hx, chronic C2 hemisection; dAIH, daily acute intermittent hypoxia; IH28-2/2, intermittent hypoxia simulating moderate sleep apnea; H, hypoxic episode.
Baseline heart rate was unaffected by treatment group (Intact Nx28: 402 ± 6; C2Hx Nx28: 422 ± 8; C2Hx dAIH28: 419 ± 8; and C2Hx IH28-2/2: 412 ± 6 mm Hg; p = 0.952); 90 min post-AIH, heart rate was reduced versus baseline (p < 0.001), but this decrease was less than 5%, and average heart rate remained within published norms.52
Body mass, apneic and recruitment thresholds, and arterial blood gases
Body mass was reduced by ∼11% in rats with chronic C2Hx exposed to IH28-2/2 versus intact rats exposed to Nx28 (p = 0.014; Table 4). No other group differences in body mass were observed. No differences in apneic or recruitment thresholds were observed between groups (p = 0.081 and 0.196, respectively; Table 5). Baseline arterial blood samples confirmed that all rats were well oxygenated and within the acceptable PaCO2 range, with no differences between groups (Table 6; p = 0.33 and 0.22, respectively). As expected, PaO2 decreased during all hypoxic episodes versus baseline (all p < 0.001) and 90 min post-AIH (all p < 0.001). At 90 min post-AIH, PaO2 was also reduced versus baseline (p < 0.001), although average PaO2 remained above 300 mm Hg in all groups. No differences in PaO2 were observed across experimental groups at any time-point (p = 0.223). PaCO2 tended to increase slightly in all groups at 90 min post-AIH versus baseline; however, this apparent increase was <1 mm Hg, and was not significant (p = 0.062). No PaCO2 differences were observed during hypoxic episodes, and no group differences were observed at any time (p = 0.265). pH was similar across groups at baseline (p = 0.097), although some time-dependent differences were apparent; nevertheless, pH was maintained within 0.05 units of baseline at all times. No significant differences in arterial pH were observed across groups (p = 0.150).
Table 4.
Body Mass (g)
| Intact Nx28 | C2Hx Nx28 | C2Hx dAIH28 | C2Hx IH28-2/2 | |
|---|---|---|---|---|
| Body mass | 469.1 ± 10.7 | 452.9.1 ± 13.3 | 443.8 ± 11.2 | *422.0 ± 10.1 |
Different from Intact Nx28; p = 0.0142.
Nx28, 28 days of normoxia; C2Hx, chronic C2 hemisection; dAIH, daily acute intermittent hypoxia; IH28-2/2, intermittent hypoxia simulating moderate sleep apnea; H, hypoxic episode.
Table 5.
Apneic and Recruitment Thresholds in Anesthetized, Paralyzed, Mechanically Ventilated Rats
| Intact Nx28 | C2Hx Nx28 | C2Hx dAIH28 | C2Hx IH28-2/2 | |
|---|---|---|---|---|
| Apneic threshold | 39.3 ± 0.6 | 38.0 ± 0.7 | 38.8 ± 0.7 | 37.7 ± 0.6 |
| Recruitment threshold | 45.5 ± 0.8 | 43.5 ± 0.4 | 44.6 ± 0.6 | 43.3 ± 0.5 |
Nx28, 28 days of normoxia; C2Hx, chronic C2 hemisection; dAIH, daily acute intermittent hypoxia; IH28-2/2, intermittent hypoxia simulating moderate sleep apnea.
Table 6.
PAO2, PACO2, and pH at Baseline, H1, H2, H3 and 90 Min after Moderate Acute Intermittent Hypoxia
| Intact Nx28 | C2Hx Nx28 | C2Hx dAIH28 | C2Hx IH28-2/2 | |
|---|---|---|---|---|
| PAO2 | ||||
| Baseline | 345.2 ± 5.1 | 344.6 ± 4.9 | 343.2 ± 4.4 | 334.1 ± 5.4 |
| H1*# | 43.8 ± 2.1 | 46.2 ± 2.1 | 44.3 ± 1.7 | 47.9 ± 1.3 |
| H2*# | 43.1 ± 0.8 | 44.3 ± 0.9 | 43.1 ± 1.1 | 43.9 ± 1.2 |
| H3*# | 43.3 ± 0.6 | 43.7 ± 0.5 | 42.4 ± 0.7 | 43.1 ± 0.7 |
| 90 min* | 335.9 ± 3.2 | 335.6 ± 7.1 | 322.8 ± 7.6 | 315.1 ± 8.8 |
| PACO2 | ||||
| Baseline | 44.5 ± 1.1 | 41.9 ± 0.6 | 44.5 ± 1.0 | 43.9 ± 0.7 |
| H1 | 45.7 ± 1.3 | 42.4 ± 0.7 | 45.0 ± 1.1 | 44.1 ± 0.9 |
| H2 | 44.8 ± 1.4 | 42.6 ± 0.9 | 44.6 ± 0.9 | 44.2 ± 0.8 |
| H3 | 45.5 ± 1.1 | 41.5 ± 0.9 | 45.2 ± 1.3 | 44.3 ± 0.8 |
| 90 min | 45.6 ± 1.1 | 42.8 ± 0.8 | 44.9 ± 1.0 | 44.1 ± 0.7 |
| pH | ||||
| Baseline | 7.38 ± 0.01 | 7.40 ± 0.01 | 7.39 ± 0.00 | 7.39 ± 0.00 |
| H1# | 7.37 ± 0.01 | 7.40 ± 0.01 | 7.38 ± 0.00 | 7.38 ± 0.00 |
| H2*# | 7.37 ± 0.01 | 7.40 ± 0.01 | 7.37 ± 0.01 | 7.38 ± 0.00 |
| H3# | 7.36 ± 0.01 | 7.40 ± 0.01 | 7.37 ± 0.01 | 7.38 ± 0.00 |
| 90 min* | 7.35 ± 0.01 | 7.36 ± 0.01 | 7.36 ± 0.01 | 7.37 ± 0.01 |
Different from baseline.
Different from 90 min.
Nx28, 28 days of normoxia; C2Hx, chronic C2 hemisection; dAIH, daily acute intermittent hypoxia; IH28-2/2, intermittent hypoxia simulating moderate sleep apnea; H, hypoxic episode.
Discussion
To our knowledge, this is the first report directly comparing the effects of different intermittent hypoxia protocols (therapeutic vs. pathogenic) on phrenic motor output and plasticity in rats with chronic, incomplete cSCI. Contrary to expectations, one essential finding of this study is that dAIH28 did not improve and IH28-2/2 did not impair baseline or maximal phrenic motor output after chronic C2 hemisection. In contrast, we had originally predicted that prolonged repetitive (low dose) AIH would improve phrenic motor output ipsilateral to chronic, as it does with acute (1-2 weeks), cervical spinal hemisection in rats.9,13 On the other hand, our findings are consistent with results from chronically injured rats exposed to 1 week of dAIH (beginning 8-9 weeks post-injury).12 We originally predicted that longer repetitive AIH exposure may be more effective with chronic injury, but this does not appear to be the case. Based on this and other studies, we now propose that unknown factor(s) undermine the therapeutic efficacy of repetitive AIH in rats with chronic (vs. acute) spinal hemisection, such as adenosine 2A receptor activation,12 age,53 or chronic inflammation.40,43,54,55
Although dAIH28 did not improve phrenic motor output in rats with chronic spinal injury, it did enhance the capacity for mAIH-induced phrenic motor plasticity (i.e., pLTF) post-injury, just as with intact rats.15 The ability for intermittent hypoxia pre-conditioning to enhance the capacity for subsequent pLTF represents a form of metaplasticity.8,56,57 Although the mechanisms of repetitive AIH-enhanced pLTF and metaplasticity are not known, it may result from increased expression of pro-plasticity molecules, and/or the addition of new mechanisms.36,57 Our finding that this form of meta plasticity persists following chronic cSCI is important, since it suggests the potential to accumulate functional benefits of therapeutic AIH once the relevant constraints are removed.
To our surprise, we did not detect any adverse consequences of IH28-2/2 on either: 1) spontaneous or chemoreflex-driven phrenic motor output; 2) expression of mAIH-induced pLTF; or 3) mean arterial pressure. Although a CIH protocol simulating that experienced during mild sleep apnea (5-min episodes, 5-min intervals, 8-12 h per day, 7 consecutive days) enhances pLTF36 and strengthens spinal synaptic inputs to phrenic motor neurons,35 more intense protocols are prone to generate neuroinflammation,38,40 a condition that undermines mAIH-induced pLTF.40,43,55 Thus, we were surprised to see expression of mAIH-induced pLTF in chronic C2Hx rats exposed to IH28-2/2. The mechanistic basis for spared mAIH-induced pLTF in these rats warrants further investigation, as it may reveal a previously unrecognized form of neuroplasticity that occurs with chronic spinal cord injury. From a different perspective, IH28-2/2 did blunt pLTF metaplasticity since pLTF in rats exposed to IH28-2/2 was similar to Nx28-treated rats; in this case, CIH-induced inflammation may still have exerted detrimental effects on phrenic motor plasticity, though to a lesser degree than in prior reports.40
“Low-dose” dAIH with chronic cSCI
One of the most extensively described models of neuroplasticity in respiratory motor control is mAIH-induced pLTF.56 In intact rats, mAIH (PaO2 > 35 mm Hg) elicits pLTF by a serotonin-dependent mechanism.10,39,58 Advances in our understanding of mAIH-induced pLTF have guided development of new, experimental therapeutic approaches to improve breathing ability after cervical spinal cord injury.6,59 The present study represents a single step in that translational process, although our unexpected findings were negative.
One recent realization is that mAIH-induced pLTF is complex, consisting of a dominant spinal serotonin receptor-induced cellular cascade to phrenic motor facilitation,45,60,61 that is undermined by a competing spinal adenosine-dependent mechanism.8,10,57 The potential for spinal adenosine accumulation to undermine serotonin-induced phrenic motor plasticity may be an especially important consideration following chronic SCI.
In the intact spinal cord, mAIH elicits modest adenosine accumulation, partially constraining phrenic62 and diaphragm LTF.63 With spinal injury, there is accumulating evidence that adenosine plays a greater relative role. For example, with acute cervical injury, serotonergic innervation of the phrenic motor nucleus is reduced,64 transiently diminishing the potential for serotonin-dependent mechanisms of functional recovery. Indeed, dAIH-induced functional recovery early post-injury (1-2 weeks) is serotonin-independent,11 and adenosine-dependent.13 With time post-injury, serotonergic innervation of the phrenic motor nucleus partially recovers,64 restoring marginal, serotonin-dominant,11 and adenosine-constrained functional recovery by 8-9 weeks post-injury.12 The power of this constraint is illustrated by the observation that greater functional recovery results when each AIH exposure is preceded by an adenosine 2A receptor inhibitor.12 The net impact of the adenosine 2A receptor constraint may increase post-injury since: 1) serotonergic innervation of the phrenic motor nucleus is still not fully restored 8 weeks post-C2Hx; and/or 2) another factor increases adenosine accumulation during moderate hypoxic episodes.
The failure of prolonged dAIH to improve phrenic motor output in chronically injured rats may be due to changes in spinal oxygen delivery and greater tissue hypoxia at the same level of arterial PO2. A recent study described injury-induced vascular pericyte constriction, limiting capillary blood flow below spinal injury. Restricted blood flow would decrease spinal tissue PO2,65 increasing extracellular adenosine at the same arterial PO2 during moderate hypoxic episodes, thereby increasing the constraint to serotonin-dominant pLTF. The findings of Navarette-Opazo and colleagues,12 that adenosine receptor inhibition increases functional benefits of dAIH, are consistent with this hypothesis.
An alternative mechanism that has been proposed to constrain serotonin-dependent dAIH28-induced functional recovery in chronically injured rats is neuroinflammation. Chronic SCI elicits persistent, low-grade systemic inflammation,66-68 and individuals with chronic SCI are highly susceptible to lung, bladder, and skin infections,69 increasing circulating levels of pro-inflammatory molecules that cross the blood–brain barrier and trigger neuro-inflammation.70 Even mild inflammation undermines serotonin-dependent phrenic motor facilitation.40,43,55 In contrast, inflammation has minimal impact on adenosine-dependent phrenic motor facilitation.71 Thus, although serotonin reinnervation of the ipsilateral phrenic motor nucleus is extensive 12 weeks post-injury,72 persistent inflammation may undermine its ability to elicit functional recovery. Anti-inflammatory drugs (targeting NF-κB vs. COX-2 enzymatic activity)73 may improve the efficacy of dAIH as a therapeutic modality.
Finally, it is likely that, as in non-respiratory motor systems,16,21 dAIH may be most effective when paired with task specific training, in this case, repeated activation of the phrenic motor circuit.74
“High-dose” pathogenic IH and cSCI
The reason pre-conditioning with prolonged IH28-2/2 blunts, but does not abolish, pLTF in rats with chronic cSCI is also unclear. In intact rats, 1 day40 or 1 week of IH-2/275 abolishes pLTF secondary to inflammation. Thus, either the extended duration of the IH28-2/2 exposures or some feature unique to chronic spinal cord injury prevents IH28-2/2 from blocking mAIH-induced pLTF. One possibility is that serotonin-dependent recovery is completely suppressed by inflammation,40,43,55,76 leaving residual, adenosine-dependent mechanisms of phrenic motor facilitation intact post-injury.77 Exploring the mechanism by which pLTF is spared in CIH-treated rats with chronic spinal injury may provide important clinical insights regarding the application of therapeutic intermittent hypoxia in cSCI, since sleep-disordered breathing is so prevalent in this population.
Similarly, unlike prior studies in spinal intact preparations,78–81 CIH did not induce hypertension in rats with chronic C2Hx, although this finding is not entirely unexpected. Indeed, Fuller and colleagues demonstrated a similar finding using a milder CIH protocol in rats with C2Hx.35 The proposed mechanism by which CIH elicits hypertension in intact rats involves sustained sympathoexcitation with reduced parasympathetic influences.82–86 Injury to the cervical spinal cord often leads to hemodynamic dysfunction due to complex interactions between factors including dysregulation of sympathetic outflow, altered baroreflex function, and the loss of skeletal muscle pumping action.87,88 Indeed, hypotension is common with cervical injury, owing to an overall reduction in sympathetic tone caudal to injury and uncontrolled parasympathetic outflow via the vagus nerves.89–92 Thus, the impact of CIH on cardiovascular parameters may differ after cSCI since many of the descending inputs to sympathetic neurons that may mediate CIH-induced hypertension have been severed by injury. Alternatively, the apparent lack of hypertension in IH28-2/2-treated rats may relate to factors associated with the experimental preparation itself, including vagotomy,93,94 anesthesia,95 and maintenance of blood volumes with supplemental fluids.
Finally, an important consideration is that the model of CIH employed in the present study does not faithfully mimic the lived experience of a person with sleep-disordered breathing. Specifically, individuals with sleep apnea rarely experience episodes of pure hypoxia. Indeed, episodic hypercapnia and sleep fragmentation also occur with sleep-disordered breathing,79,85,86,96–99 although the individual and combined effects of these factors have not been extensively explored. Nevertheless, CIH has been used extensively in the literature and models a key component of sleep-disordered breathing that is known to elicit systemic pathology similar to that observed in individuals with sleep apnea.79 Future work should consider the impact of concurrent hypercapnia and/or sleep fragmentation with CIH to more accurately reflect the mechanisms whereby sleep-disordered breathing affects baseline physiology and the potential to express plasticity.
Responses to intermittent hypoxia after spinal cord injury
Mechanisms underlying cumulative IH exposure effects are not well understood, particularly in the context of chronic spinal cord injury. The injured spinal cord has been referred to as a “new” spinal cord,100 meaning that biological responses after spinal injury may be quite distinct from intact animals. Thus, physiological mechanisms in the intact spinal cord are not necessarily expected to pertain to the injured cord. Responses to dAIH and CIH in chronically injured rats may exemplify this caveat since some of the expected functional changes were not observed in this study. These differences between prediction and outcome warrant further exploration, since an improved understanding of differences between intact and injured spinal cords may speed the development and optimization of therapeutic strategies to improve functional recovery in persons with SCI.
Implications
The results of these experiments have profound implications for the clinical management of individuals with chronic spinal cord injury, since dAIH is actively being explored in clinical trials as a therapeutic strategy for improving breathing and somatic motor function following chronic spinal cord injury (www.clinicaltrials.gov; keywords: spinal cord injury, intermittent hypoxia). Although we confirm the ability to induce phrenic motor plasticity and metaplasticity with prolonged dAIH pre-conditioning after chronic cSCI, these studies highlight the need for additional protocol optimization before dAIH can be harnessed for therapeutic benefit. Indeed, dAIH pre-conditioning alone may not elicit sufficient plasticity to trigger meaningful functional improvements in phrenic motor output after chronic cSCI. Although sleep-disordered breathing is highly prevalent in a number of neurological conditions, including SCI,25,28,29 we found little evidence that moderate CIH had a significant impact on phrenic motor plasticity post-injury, though we do not account for other important aspects of sleep-disordered breathing, including concomitant chronic intermittent hypercapnia and/or sleep fragmentation.
Ironically, studies of intermittent hypoxia-induced respiratory motor plasticity guided translation to non-respiratory motor systems after chronic spinal injury.9,16,19,21,24 Indeed, the same AIH protocols appear to elicit greater benefits in limb versus respiratory function.12,18 However, it is important to note that AIH alone is of only modest benefit to limb function, and that the greatest functional benefits occur when AIH is combined with task specific training.74 The same may be true for breathing, although this idea has yet to be formally tested.
The data presented here represent an important step in our deepening understanding that respiratory plasticity and functional recovery after prolonged IH are dose-dependent. In addition to the intensity (frequency and number of IH episodes), outcomes may be affected by exposure duration, diurnal variations, inflammation, and/or background expression of pro-plasticity molecules (e.g., growth/trophic factors).37,38 We are working towards identifying optimal AIH protocols for specific circumstances, such as chronic spinal cord injury.
Acknowledgments
The authors wish to express their appreciation to Zachary A. Asa, Kristin N. Smith, Ashley Holland, Juliet V. Santiago, Kelsey Stefan, Ashley Ross and Yasin B. Seven for their outstanding technical contributions.
Authors' Contributions
EGR, LLA, AT and GSM: conception & design of research; EGR and LLA: project administration and oversight; EGR, AT, LLA, MCC and ME: performed experiments & data collection; EGR and GSM: analysis & interpretation of data; EGR: initial draft and revisions of manuscript; EGR, AT and GSM: final approval of manuscript.
Funding Information
This work was supported by funding from The National Institutes of Health (1OT2OD0023854); and the University of Florida McKnight Brain Institute. E.J. Gonzalez-Rothi was supported by K12 HD055929. L.L. Allen was supported by T32 HD043730.
Author Disclosure Statement
No competing financial interests exist.
References
- 1. van den Berg, M.E., Castellote, J.M., de Pedro-Cuesta, J., and Mahillo-Fernandez, I. (2010). Survival after spinal cord injury: a systematic review. J. Neurotrauma 27, 1517–1528 [DOI] [PubMed] [Google Scholar]
- 2. Waddimba, A.C., Jain, N.B., Stolzmann, K., Gagnon, D.R., Burgess, J.F., Kazis, L.E., and Garshick, E. (2009). Predictors of cardiopulmonary hospitalization in chronic spinal cord injury. Arch. Phys. Med. Rehabil. 90, 193–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Winslow, C. and Rozovsky, J. (2003). Effect of spinal cord injury on the respiratory system. Am. J. Phys. Med. Rehabil. 82, 803–814 [DOI] [PubMed] [Google Scholar]
- 4. Winslow, C., Bode, R.K., Felton, D., Chen, D., and Meyer, P.R. (2002). Impact of respiratory complications on length of stay and hospital costs in acute cervical spine injury. Chest 121, 1548–1554 [DOI] [PubMed] [Google Scholar]
- 5. Berlowitz, D.J., Wadsworth, B., and Ross, J. (2016). Respiratory problems and management in people with spinal cord injury. Breathe (Sheff.) 12, 328–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gonzalez-Rothi, E.J., Lee, K.Z., Dale, E.A., Reier, P.J., Mitchell, G.S., and Fuller, D.D. (2015). Intermittent hypoxia and neurorehabilitation. J. Appl. Physiol. (1985) 119, 1455–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dale-Nagle, E.A., Hoffman, M.S., MacFarlane, P.M., Satriotomo, I., Lovett-Barr, M.R., Vinit, S., and Mitchell, G.S. (2010). Spinal plasticity following intermittent hypoxia: implications for spinal injury. Ann. N. Y. Acad. Sci. 1198, 252–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Devinney, M.J., Huxtable, A.G., Nichols, N.L., and Mitchell, G.S. (2013). Hypoxia-induced phrenic long-term facilitation: emergent properties. Ann. N. Y. Acad. Sci. 1279, 143–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lovett-Barr, M.R., Satriotomo, I., Muir, G.D., Wilkerson, J.E., Hoffman, M.S., Vinit, S., and Mitchell, G.S. (2012). Repetitive intermittent hypoxia induces respiratory and somatic motor recovery after chronic cervical spinal injury. J. Neurosci. 32, 3591–3600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fuller, D.D. and Mitchell, G.S. (2017). Respiratory neuroplasticity—overview, significance and future directions. Exp. Neurol. 287, 144–152 [DOI] [PubMed] [Google Scholar]
- 11. Dougherty, B.J., Terada, J., Springborn, S.R., Vinit, S., MacFarlane, P.M., and Mitchell, G.S. (2017). Daily acute intermittent hypoxia improves breathing function with acute and chronic spinal injury via distinct mechanisms. Respir. Physiol. Neurobiol. 256, 50–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Navarrete-Opazo, A., Dougherty, B.J., and Mitchell, G.S. (2017). Enhanced recovery of breathing capacity from combined adenosine 2A receptor inhibition and daily acute intermittent hypoxia after chronic cervical spinal injury. Exp. Neurol. 287, 93–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Navarrete-Opazo, A., Vinit, S., Dougherty, B.J., and Mitchell, G.S. (2015). Daily acute intermittent hypoxia elicits functional recovery of diaphragm and inspiratory intercostal muscle activity after acute cervical spinal injury. Exp. Neurol. 266, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Satriotomo, I., Dale, E.A., Dahlberg, J.M., and Mitchell, G.S. (2012). Repetitive acute intermittent hypoxia increases expression of proteins associated with plasticity in the phrenic motor nucleus. Exp. Neurol. 237, 103–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wilkerson, J.E. and Mitchell, G.S. (2009). Daily intermittent hypoxia augments spinal BDNF levels, ERK phosphorylation and respiratory long-term facilitation. Exp. Neurol. 217, 116–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hayes, H.B., Jayaraman, A., Herrmann, M., Mitchell, G.S., Rymer, W.Z., and Trumbower, R.D. (2014). Daily intermittent hypoxia enhances walking after chronic spinal cord injury: a randomized trial. Neurology 82, 104–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Trumbower, R.D., Jayaraman, A., Mitchell, G.S., and Rymer, W.Z. (2012). Exposure to acute intermittent hypoxia augments somatic motor function in humans with incomplete spinal cord injury. Neurorehabil. Neural Repair 26, 163–172 [DOI] [PubMed] [Google Scholar]
- 18. Tester, N.J., Fuller, D.D., Fromm, J.S., Spiess, M.R., Behrman, A.L., and Mateika, J.H. (2014). Long-term facilitation of ventilation in humans with chronic spinal cord injury. Am. J. Resp. Crit. Care Med. 189, 57–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Trumbower, R.D., Hayes, H.B., Mitchell, G.S., Wolf, S.L., and Stahl, V.A. (2017). Effects of acute intermittent hypoxia on hand use after spinal cord trauma: a preliminary study. Neurology 89, 1904–1907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hassan, A., Arnold, B.M., Caine, S., Toosi, B.M., Verge, V.M.K., and Muir, G.D. (2018). Acute intermittent hypoxia and rehabilitative training following cervical spinal injury alters neuronal hypoxia- and plasticity-associated protein expression. PLoS One 13, e0197486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Prosser-Loose, E.J., Hassan, A., Mitchell, G.S., and Muir, G.D. (2015). Delayed intervention with intermittent hypoxia and task training improves forelimb function in a rat model of cervical spinal injury. J. Neurotrauma 32, 1403–1412 [DOI] [PubMed] [Google Scholar]
- 22. Lynch, M., Duffell, L., Sandhu, M., Srivatsan, S., Deatsch, K., Kessler, A., Mitchell, G.S., Jayaraman, A., and Rymer, W.Z. (2016). Effect of acute intermittent hypoxia on motor function in individuals with chronic spinal cord injury following ibuprofen pretreatment: a pilot study. J. Spinal Cord Med. 40, 295–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sandhu, M.S., Gray, E., Kocherginsky, M., Jayaraman, A., Mitchell, G.S., and Rymer, W.Z. (2019). Prednisolone pretreatment enhances intermittent hypoxia-induced plasticity in persons with chronic incomplete spinal vord injury. Neurorehabil. Neural Repair 33, 911–921 [DOI] [PubMed] [Google Scholar]
- 24. Navarrete-Opazo, A., Alcayaga, J., Sepúlveda, O., Rojas, E., and Astudillo, C. (2017). Repetitive intermittent hypoxia and locomotor training enhances walking cunction in incomplete spinal cord injury subjects: a randomized, triple-blind, placebo-controlled clinical trial. J. Neurotrauma 34, 1803–1812 [DOI] [PubMed] [Google Scholar]
- 25. Chiodo, A.E., Sitrin, R.G., and Bauman, K.A. (2016). Sleep disordered breathing in spinal cord injury: s systematic review. J. Spinal Cord Med. 39, 374–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fuller, D.D., Lee, K.Z., and Tester, N.J. (2013). The impact of spinal cord injury on breathing during sleep. Respir. Physiol. Neurobiol. 188, 344–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Berlowitz, D.J., Brown, D.J., Campbell, D.A., and Pierce, R.J. (2005). A longitudinal evaluation of sleep and breathing in the first year after cervical spinal cord injury. Arch. Phys. Med. Rehabil. 86, 1193–1199 [DOI] [PubMed] [Google Scholar]
- 28. Sankari, A., Martin, J.L., Bascom, A.T., Mitchell, M.N., and Badr, M.S. (2015). Identification and treatment of sleep-disordered breathing in chronic spinal cord injury. Spinal Cord 53, 145–149 [DOI] [PubMed] [Google Scholar]
- 29. Sankari, A. and Badr, M.S. (2016). Diagnosis of sleep disordered breathing in patients with chronic spinal cord injury. Arch. Phys. Med. Rehabil. 97, 176–177 [DOI] [PubMed] [Google Scholar]
- 30. Lee, K.Z. (2019). Impact of cervical spinal cord contusion on the breathing pattern across the sleep-wake cycle in the rat. J. Appl. Physiol. (1985) 126, 111–123 [DOI] [PubMed] [Google Scholar]
- 31. Esteves, A.M., Mello, M.T., Squarcini, C.F., Lancellotti, C.L., Comparoni, A., and Tufik, S. (2007). Sleep patterns over 15-day period in rats with spinal cord injury. Spinal Cord 45, 360–366 [DOI] [PubMed] [Google Scholar]
- 32. Sankari, A., Minic, Z., Farshi, P., Shanidze, M., Mansour, W., Liu, F., Mao, G., and Goshgarian, H.G. (2019). Sleep disordered breathing induced by cervical spinal cord injury and effect of adenosine A1 receptors modulation in rats. J. Appl. Physiol. (1985) 127, 1668–1676 [DOI] [PubMed] [Google Scholar]
- 33. Avezov, K., Aizenbud, D., and Lavie, L. (2018). Intermittent hypoxia induced formation of “endothelial cell-colony forming units (EC-CFUs)” is affected by ROS and oxidative stress. Front. Neurol. 9, 447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lavie, L. (2015). Oxidative stress in obstructive sleep apnea and intermittent hypoxia—revisited—the bad ugly and good: implications to the heart and brain. Sleep Med. Rev. 20, 27–45 [DOI] [PubMed] [Google Scholar]
- 35. Fuller, D.D., Johnson, S.M., Olson, E.B., and Mitchell, G.S. (2003). Synaptic pathways to phrenic motoneurons are enhanced by chronic intermittent hypoxia after cervical spinal cord injury. J. Neurosci. 23, 2993–3000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ling, L., Fuller, D.D., Bach, K.B., Kinkead, R., Olson, E.B., and Mitchell, G.S. (2001). Chronic intermittent hypoxia elicits serotonin-dependent plasticity in the central neural control of breathing. J. Neurosci. 21, 5381–5388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mateika, J.H. and Narwani, G. (2009). Intermittent hypoxia and respiratory plasticity in humans and other animals: does exposure to intermittent hypoxia promote or mitigate sleep apnoea? Exp. Physiol. 94, 279–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Navarrete-Opazo, A. and Mitchell, G.S. (2014). Therapeutic potential of intermittent hypoxia: a matter of dose. Am. J. Physiol. Regul. Integr. Comp. Physiol. 307, R1181–R1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dale-Nagle, E.A., Hoffman, M.S., MacFarlane, P.M., and Mitchell, G.S. (2010). Multiple pathways to long-lasting phrenic motor facilitation. Adv. Exp. Med. Biol. 669, 225–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Huxtable, A.G., Smith, S.M., Peterson, T.J., Watters, J.J., and Mitchell, G.S. (2015). Intermittent hypoxia-induced spinal inflammation impairs respiratory motor plasticity by a spinal p38 MAP kinase-dependent mechanism. J. Neurosci. 35, 6871–6880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dougherty, B.J., Fields, D.P., and Mitchell, G.S. (2015). Mammalian target of rapamycin is required for phrenic long-term facilitation following severe but not moderate acute intermittent hypoxia. J. Neurophysiol. 114, 1784–1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gonzalez-Rothi, E.J., Streeter, K.A., Hanna, M.H., Stamas, A.C., Reier, P.J., Baekey, D.M., and Fuller, D.D. (2017). High-frequency epidural stimulation across the respiratory cycle evokes phrenic short-term potentiation after incomplete cervical spinal cord injury. J. Neurophysiol. 118, 2344–2357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Huxtable, A.G., Smith, S.M., Vinit, S., Watters, J.J., and Mitchell, G.S. (2013). Systemic LPS induces spinal inflammatory gene expression and impairs phrenic long-term facilitation following acute intermittent hypoxia. J. Appl. Physiol. (1985) 114, 879–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Perim, R.R., Fields, D.P., and Mitchell, G.S. (2020). Spinal AMP kinase activity differentially regulates phrenic motor plasticity. J. Appl. Physiol. (1985) 128, 523–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tadjalli, A. and Mitchell, G.S. (2019). Cervical spinal 5-HT2A and 5-HT2B receptors are both necessary for moderate acute intermittent hypoxia-induced phrenic long-term facilitation. J. Appl. Physiol. (1985) 127, 432–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lee, K.Z., Dougherty, B.J., Sandhu, M.S., Lane, M.A., Reier, P.J., and Fuller, D.D. (2013). Phrenic motoneuron discharge patterns following chronic cervical spinal cord injury. Exp. Neurol. 249, 20–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fuller, D.D., Doperalski, N.J., Dougherty, B.J., Sandhu, M.S., Bolser, D.C., and Reier, P.J. (2008). Modest spontaneous recovery of ventilation following chronic high cervical hemisection in rats. Exp. Neurol. 211, 97–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fuller, D.D., Golder, F.J., Olson, E.B., and Mitchell, G.S. (2006). Recovery of phrenic activity and ventilation after cervical spinal hemisection in rats. J. Appl. Physiol. (1985) 100, 800–806 [DOI] [PubMed] [Google Scholar]
- 49. Minic, Z., Wilson, S., Liu, F., Sankari, A., Mao, G., and Goshgarian, H. (2017). Nanoconjugate-bound adenosine A. Exp. Neurol. 292, 56–62 [DOI] [PubMed] [Google Scholar]
- 50. Powell, F.L., Milsom, W.K., and Mitchell, G.S. (1998). Time domains of the hypoxic ventilatory response. Respir. Physiol. 112, 123–134 [DOI] [PubMed] [Google Scholar]
- 51. Bavis, R.W. and Mitchell, G.S. (2003). Intermittent hypoxia induces phrenic long-term facilitation in carotid-denervated rats. J. Appl. Physiol. (1985) 94, 399–409 [DOI] [PubMed] [Google Scholar]
- 52. Praman, S., Mulvany, M.J., Allenbach, Y., Marston, A., Hostettmann, K., Sirirugsa, P., and Jansakul, C. (2011). Effects of an n-butanol extract from the stem of Tinospora crispa on blood pressure and heart rate in anesthetized rats. J. Ethnopharmacol. 133, 675–686 [DOI] [PubMed] [Google Scholar]
- 53. McGuire, M. and Ling, L. (2005). Ventilatory long-term facilitation is greater in 1- vs. 2-mo-old awake rats. J. Appl. Physiol. (1985) 98, 1195–1201 [DOI] [PubMed] [Google Scholar]
- 54. Huxtable, A.G., Vinit, S., Windelborn, J.A., Crader, S.M., Guenther, C.H., Watters, J.J., and Mitchell, G.S. (2011). Systemic inflammation impairs respiratory chemoreflexes and plasticity. Respir. Physiol. Neurobiol. 178, 482–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Agosto-Marlin, I.M., Nichols, N.L., and Mitchell, G.S. (2018). Systemic inflammation inhibits serotonin receptor 2-induced phrenic motor facilitation upstream from BDNF/TrkB signaling. J. Neurophysiol .119, 2176–2185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mitchell, G.S. and Johnson, S.M. (2003). Neuroplasticity in respiratory motor control. J. Appl. Physiol. (1985) 94, 358–374 [DOI] [PubMed] [Google Scholar]
- 57. Fields, D.P. and Mitchell, G.S. (2015). Spinal metaplasticity in respiratory motor control. Front. Neural Circuits 9, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mitchell, G.S., Baker, T.L., Nanda, S.A., Fuller, D.D., Zabka, A.G., Hodgeman, B.A., Bavis, R.W., Mack, K.J., and Olson, E.B. (2001). Invited review: intermittent hypoxia and respiratory plasticity. J. Appl. Physiol. (1985) 90, 2466–2475 [DOI] [PubMed] [Google Scholar]
- 59. Dale, E.A., Ben Mabrouk, F., and Mitchell, G.S. (2014). Unexpected benefits of intermittent hypoxia: enhanced respiratory and nonrespiratory motor function. Physiology (Bethesda) 29, 39–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Baker-Herman, T.L. and Mitchell, G.S. (2002). Phrenic long-term facilitation requires spinal serotonin receptor activation and protein synthesis. J. Neurosci. 22, 6239–6246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. MacFarlane, P.M., Vinit, S., and Mitchell, G.S. (2011). Serotonin 2A and 2B receptor-induced phrenic motor facilitation: differential requirement for spinal NADPH oxidase activity. Neuroscience 178, 45–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hoffman, M.S., Golder, F.J., Mahamed, S., and Mitchell, G.S. (2010). Spinal adenosine A2(A) receptor inhibition enhances phrenic long term facilitation following acute intermittent hypoxia. J. Physiol. 588, 255–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Navarrete-Opazo, A.A., Vinit, S., and Mitchell, G.S. (2014). Adenosine 2A receptor inhibition enhances intermittent hypoxia-induced diaphragm but not intercostal long-term facilitation. J. Neurotrauma 31, 1975–1984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Golder, F.J. and Mitchell, G.S. (2005). Spinal synaptic enhancement with acute intermittent hypoxia improves respiratory function after chronic cervical spinal cord injury. J. Neurosci. 25, 2925–2932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Li, Y., Lucas-Osma, A.M., Black, S., Bandet, M.V., Stephens, M.J., Vavrek, R., Sanelli, L., Fenrich, K.K., Di Narzo, A.F., Dracheva, S., Winship, I.R., Fouad, K., and Bennett, D.J. (2017). Pericytes impair capillary blood flow and motor function after chronic spinal cord injury. Nat. Med. 23, 733–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gensel, J.C. and Zhang, B. (2015). Macrophage activation and its role in repair and pathology after spinal cord injury. Brain Res. 1619, 1–11 [DOI] [PubMed] [Google Scholar]
- 67. Schwab, J.M., Zhang, Y., Kopp, M.A., Brommer, B., and Popovich, P.G. (2014). The paradox of chronic neuroinflammation, systemic immune suppression, autoimmunity after traumatic chronic spinal cord injury. Exp. Neurol. 258, 121–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Windelborn, J.A. and Mitchell, G.S. (2012). Glial activation in the spinal ventral horn caudal to cervical injury. Respir. Physiol. Neurobiol. 180, 61–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Stillman, M.D., Barber, J., Burns, S., Williams, S., and Hoffman, J.M. (2017). Complications of spinal cord injury over the first year after discharge from inpatient rehabilitation. Arch. Phys. Med. Rehabil. 98, 1800–1805 [DOI] [PubMed] [Google Scholar]
- 70. Jaerve, A. and Müller, H.W. (2012). Chemokines in CNS injury and repair. Cell Tissue Res. 349, 229–248 [DOI] [PubMed] [Google Scholar]
- 71. Agosto-Marlin, I.M., Nichols, N.L., and Mitchell, G.S. (2017). Adenosine-dependent phrenic motor facilitation is inflammation resistant. J. Neurophysiol. 117, 836–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ciesa, M.C., Seven, Y.B., Allen, L.L., Smith, K., Asa, Z., Simon, A., Holland, A., Santiago, J., Stefan, K., Ross, A., Gonzalez-Rothi, E.J., and Mitchel, G.S. (2018). Recovery of serotonergic innervation in spinal motor nuclei below a cervical spinal injury: effects of intermittent hypoxia. FASEB J. 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Huxtable, A.G., Kopp, E., Dougherty, B.J., Watters, J.J., and Mitchell, G.S. (2018). Cyclooxygenase enzyme activity does not impair respiratory motor plasticity after one night of intermittent hypoxia. Respir. Physiol. Neurobiol. 256, 21–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Welch, J.F., Sutor, T., Vose, A.K., Perim, R.R., Fox, E.J., and Mitchell, G.S. (2020). Synergy between acute intermittent hypoxia and task-specific training. Exerc. Sport Sci. Rev. 48, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. El Chami, M., Mitchel, G.S., and Gonzalez-Rothi, E.J. (2020). Chronic intermittent hypoxia blunts phrenic motor plasticity: role of inflammation. FASEB J. 34 [Google Scholar]
- 76. Vinit, S., Windelborn, J.A., and Mitchell, G.S. (2011). Lipopolysaccharide attenuates phrenic long-term facilitation following acute intermittent hypoxia. Respir. Physiol. Neurobiol. 176, 130–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Nichols, N.L., Dale, E.A., and Mitchell, G.S. (2012). Severe acute intermittent hypoxia elicits phrenic long-term facilitation by a novel adenosine-dependent mechanism. J. Appl. Physiol. (1985) 112, 1678–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Tamisier, R., Pepin, J.L., Remy, J., Baguet, J.P., Taylor, J.A., Weiss, J.W., and Levy, P. (2011). 14 nights of intermittent hypoxia elevate daytime blood pressure and sympathetic activity in healthy humans. Eur. Respir. J 37, 119–128 [DOI] [PubMed] [Google Scholar]
- 79. Levy, P., Pepin, J.L., Arnaud, C., Tamisier, R., Borel, J.C., Dematteis, M., Godin-Ribuot, D., and Ribuot, C. (2008). Intermittent hypoxia and sleep-disordered breathing: current concepts and perspectives. Eur. Respir. J. 32, 1082–1095 [DOI] [PubMed] [Google Scholar]
- 80. Lesske, J., Fletcher, E.C., Bao, G., and Unger, T. (1997). Hypertension caused by chronic intermittent hypoxia–influence of chemoreceptors and sympathetic nervous system. J. Hypertens. 15, 1593–1603 [DOI] [PubMed] [Google Scholar]
- 81. Fletcher, E.C. (2001). Invited review: physiological consequences of intermittent hypoxia: systemic blood pressure. J. Appl. Physiol. (1985) 90, 1600–1605 [DOI] [PubMed] [Google Scholar]
- 82. Shell, B., Faulk, K., and Cunningham, J.T. (2016). Neural control of blood pressure in chronic intermittent hypoxia. Curr. Hypertens. Rep. 18, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Leuenberger, U.A., Brubaker, D., Quraishi, S.A., Hogeman, C.S., Imadojemu, V.A., and Gray, K.S. (2005). Effects of intermittent hypoxia on sympathetic activity and blood pressure in humans. Auton. Neurosci. 121, 87–93 [DOI] [PubMed] [Google Scholar]
- 84. Sharpe, A.L., Calderon, A.S., Andrade, M.A., Cunningham, J.T., Mifflin, S.W., and Toney, G.M. (2013). Chronic intermittent hypoxia increases sympathetic control of blood pressure: role of neuronal activity in the hypothalamic paraventricular nucleus. Am. J. Physiol. Heart Circ. Physiol. 305, H1772–H1780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Dergacheva, O., Dyavanapalli, J., Pinol, R.A., and Mendelowitz, D. (2014). Chronic intermittent hypoxia and hypercapnia inhibit the hypothalamic paraventricular nucleus neurotransmission to parasympathetic cardiac neurons in the brain stem. Hypertension 64, 597–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Dyavanapalli, J., Jameson, H., Dergacheva, O., Jain, V., Alhusayyen, M., and Mendelowitz, D. (2014). Chronic intermittent hypoxia-hypercapnia blunts heart rate responses and alters neurotransmission to cardiac vagal neurons. J. Physiol. 592, 2799–2811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hou, S. and Rabchevsky, A.G. (2014). Autonomic consequences of spinal cord injury. Compr. Physiol. 4, 1419–1453 [DOI] [PubMed] [Google Scholar]
- 88. Bloch, R.F. (1986). Autonomic dysfunction in management of spinal cord injuries. In: Management of spinal cord injuries. Williams and Wilkins: Baltimore, MD, pps. 149–163 [Google Scholar]
- 89. Grigorean, V.T., Sandu, A.M., Popescu, M., Iacobini, M.A., Stoian, R., Neascu, C., Strambu, V., and Popa, F. (2009). Cardiac dysfunctions following spinal cord injury. J. Med. Life 2, 133–145 [PMC free article] [PubMed] [Google Scholar]
- 90. Teasell, R.W., Arnold, J.M., Krassioukov, A., and Delaney, G.A. (2000). Cardiovascular consequences of loss of supraspinal control of the sympathetic nervous system after spinal cord injury. Arch. Phys. Med. Rehabil. 81, 506–516 [DOI] [PubMed] [Google Scholar]
- 91. Mathias, C.J. and Frankel, H.L. (1992). Autonomic disturbances in spinal cord lesions, in: Autonomic Failure. Oxford University Press: Oxford, U.K., pps. 839–881 [Google Scholar]
- 92. Mathias, C.J. and Frankel, H.L. (1992). The cardiovascular system in tetraplegia and paraplegia. In: Spinal Cord Trauma. Handbook of Clinical Neurology. Elsevier Science Publishers: Amsterdam, the Netherlands, pps. 435–456 [Google Scholar]
- 93. Ter Horst, G.J., Hautvast, R.W., De Jongste, M.J., and Korf, J. (1996). Neuroanatomy of cardiac activity-regulating circuitry: a transneuronal retrograde viral labelling study in the rat. Eur. J. Neurosci. 8, 2029–2041 [DOI] [PubMed] [Google Scholar]
- 94. Schrier, R.W. and Berl, T. (1972). Mechanism of the antidiuretic effect associated with interruption of parasympathetic pathways. J. Clin. Invest. 51, 2613–2620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Janssen, B.J., De Celle, T., Debets, J.J., Brouns, A.E., Callahan, M.F., and Smith, T.L. (2004). Effects of anesthetics on systemic hemodynamics in mice. Am. J. Physiol. Heart Circ. Physiol. 287, H1618–H1624 [DOI] [PubMed] [Google Scholar]
- 96. Morrell, M.J., Finn, L., Kim, H., Peppard, P.E., Badr, M.S., and Young, T. (2000). Sleep fragmentation, awake blood pressure, and sleep-disordered breathing in a population-based study. Am. J. Respir. Crit. Care Med. 162, 2091–2096 [DOI] [PubMed] [Google Scholar]
- 97. McGuire, M., Tartar, J.L., Cao, Y., McCarley, R.W., White, D.P., Strecker, R.E., and Ling, L. (2008). Sleep fragmentation impairs ventilatory long-term facilitation via adenosine A1 receptors. J. Physiol. 586, 5215–5229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Smith, R.P., Veale, D., Pepin, J.L., and Levy, P.A. (1998). Obstructive sleep apnoea and the autonomic nervous system. Sleep Med Rev 2, 69–92 [DOI] [PubMed] [Google Scholar]
- 99. Hunyor, I. and Cook, K.M. (2018). Models of intermittent hypoxia and obstructive sleep apnea: molecular pathways and their contribution to cancer. Am. J. Physiol. Regul. Integr. Comp. Physiol. 315, R669–R687 [DOI] [PubMed] [Google Scholar]
- 100. Edgerton, V.R., Leon, R.D., Harkema, S.J., Hodgson, J.A., London, N., Reinkensmeyer, D.J., Roy, R.R., Talmadge, R.J., Tillakaratne, N.J., Timoszyk, W., and Tobin, A. (2001). Retraining the injured spinal cord. J. Physiol. 533, 15–22 [DOI] [PMC free article] [PubMed] [Google Scholar]