

Abstract
Lung cancer is the leading cause of cancer mortality worldwide. The treatment of lung cancer patients harboring mutant EGFR with orally administered EGFR tyrosine kinase inhibitors (TKIs) has been a paradigm shift. Osimertinib and rociletinib are third-generation irreversible EGFR TKIs targeting the EGFR T790M mutation. Osimertinib is the current standard of care for patients with EGFR mutations due to increased efficacy, lower side effects, and enhanced brain penetrance. Unfortunately, all patients develop resistance. Genomic approaches have primarily been used to interrogate resistance mechanisms. Here we characterized the proteome and phosphoproteome of a series of isogenic EGFR mutant lung adenocarcinoma cell lines that are either sensitive or resistant to these drugs, comprising the most comprehensive proteomic dataset resource to date to investigate third-generation EGFR TKI resistance in lung adenocarcinoma. Unbiased global quantitative mass spectrometry uncovered alterations in signaling pathways, revealed a proteomic signature of epithelial mesenchymal transition, and identified kinases and phosphatases with altered expression and phosphorylation in TKI-resistant cells. Decreased tyrosine phosphorylation of key sites in the phosphatase SHP2 suggests its inhibition, resulting in subsequent inhibition of RAS/MAPK and activation of PI3K/AKT pathways. Anticorrelation analyses of this phosphoproteomic dataset with published drug-induced P100 phosphoproteomic datasets from the Library of Integrated Network-Based Cellular Signatures program predicted drugs with the potential to overcome EGFR TKI resistance. The PI3K/MTOR inhibitor dactolisib in combination with osimertinib overcame resistance both in vitro and in vivo. Taken together, this study reveals global proteomic alterations upon third-generation EGFR TKI resistance and highlights potential novel approaches to overcome resistance.
Keywords: Phosphoproteomics, EGFR, Lung cancer, Osimertinib, Resistance
Introduction
Lung cancer continues to be the leading cause of cancer mortality in the world (1). Many lung adenocarcinoma patients with activating epidermal growth factor receptor (EGFR) mutations initially respond dramatically to the first- or second-generation EGFR tyrosine kinase inhibitors (TKIs). However, they eventually develop resistance. The most common mechanism of acquired resistance is the EGFR T790M gatekeeper site residue mutation (2). Osimertinib, a third generation irreversible EGFR TKI has been approved by the FDA to treat patients harboring the EGFR T790M mutation who have developed resistance to first- and second- generation EGFR TKIs (3). Recently, osimertinib was also approved for the front-line treatment of patients harboring EGFR mutations (4). Rociletinib is another irreversible inhibitor targeting the EGFR T790M mutation, which has minimal activity against wild-type EGFR. Both drugs have therapeutic benefits and have demonstrated activity in tumors with T790M-mediated resistance to other EGFR tyrosine kinase inhibitors (5,6). Further development of rociletinib was ceased in 2016 due to less than expected efficacy, poor brain penetration leading to tumor progression in brain tissues and off-target effects on IGFR activation leading to hyperglycemia (7,8).
Although 3rd-generation TKIs provide clinical benefit to most patients with EGFR mutations, some patients, demonstrating primary resistance, still do not respond to these inhibitors. Complete responses are rare, and all patients eventually develop resistance, suggesting primary and acquired resistance mechanisms decrease the efficacy of the drugs (9,10). Genomic approaches have been used primarily to interrogate osimertinib resistance mechanisms (9,11–15). Several mechanisms of osimertinib resistance have been identified (16), including novel second site EGFR mutations, activated bypass pathways such as MET amplification, HER2 amplification, RAS mutations, BRAF mutations, PIK3CA mutations, and novel fusion events (17). However, the resistance mechanism is complex and still not fully understood.
Previously, we have used SILAC-based quantitative phosphoproteomics to identify the global dynamic modification which occur upon treatment of TKI-sensitive and -resistant lung adenocarcinoma cells with the 1st and 2nd generation EGFR TKIs, erlotinib and afatinib. Utilizing this strategy, we identified the targets of mutant EGFR signaling pathways responsible for TKI resistance, and possible off-target effects of the drugs (18,19). In this study, we employed SILAC-based quantitative mass spectrometry to characterize alterations in the proteome and phosphoproteome which occur upon acquired resistance and sought to identify novel mechanisms of resistance to the third generation EGFR TKIs, osimertinib and rociletinib. To our knowledge, this is the most comprehensive 3rd generation EGFR TKI resistant proteome and phosphoproteome analysis resource available to date.
Materials and Methods
Cell culture, SILAC labeling, and drug treatment
H1975 parental cell line was obtained from ATCC. The resistant cell lines AZR3/AZR4 (resistant to osimertinib) and COR1/COR10 (resistant to rociletinib) have been described before (3,6) and were obtained from AstraZeneca and Clovis Oncology, respectively. All cell lines used in this study were tested for mycoplasma contamination using Universal Mycoplasma Detection Kit (ATCC) and were maintained in plasmosin (InvivoGen) for two weeks prior to performing experiments. Cells were authenticated by short tandem repeat (STR) profiling using the AmpFℓSTR Identifiler Direct PCR Amplification Kit at the Protein Expression Laboratory (NCI, Frederick, MD). Three-state SILAC labeling was performed as described before (18, 19). Further details of cell culture, SILAC labeling and drug treatment before harvesting are provided in Supplementary Materials and Methods.
Generation of osimertinib-resistant cell lines
Generation of osimertinib-resistant cells (PC9-OsiR-NCI1, and HCC827-OsiR-NCI1) is described in Supplementary Materials and Method section.
Experimental Mice
All animal experiments were reviewed and approved prior to animal use under the guidance of the IACUC of NCI/NIH. Mice were maintained in a pathogen-free facility approved by the NCI. Athymic nu/nu mice (6–12 weeks) were obtained from the Center for Cancer Research Mouse Facility at FNRL, Frederick, MD. Mice were injected in the flank with 2 × 106 cells using a 25G needle. Tumors were measured using a digital caliper twice a week. Tumor volume was calculated using the formula (W2 x L)/2. The mice were assigned to random groups before drug treatments and no approaches to blinding were applied. Tumors were harvested from the flank after euthanasia according to the NIH IACUC protocols.
Protein extraction and sample processing
See Supplementary Materials and Methods for details of protein extraction and sample processing for mass spectrometry analysis.
Basic reversed phase liquid chromatography (RPLC) fractionation
Basic-RPLC separation of resuspended peptides before phosphopeptide enrichment was performed as described before (18) and described in detail in Supplementary Materials and Methods.
Enrichment of Phosphopeptides
Enrichment of phosphopeptides was performed as described before (18, 19), and described in detail in Supplementary Materials and Methods.
LC-MS/MS Analysis
Peptides were analyzed on a LTQ-Orbitrap Elite mass spectrometer or a Q Exactive HF interfaced with an Ultimate™ 3000 RSLCnano System (Thermo Fisher Scientific, San Jose, CA). The dried peptides and the enriched phosphopeptides were loaded onto a nano-trap column (Acclaim PepMap100 Nano Trap Column, C18, 5 μm, 100 Å, 100 μm i.d. x 2 cm) and separated on an Easy-spray™ C18 LC column (Acclaim PepMap100, C18, 2 μm, 100 Å, 75 μm i.d. x 25 cm). Mobile phases A and B consisted of 0.1% formic acid in water and 0.1% formic acid in 90% ACN, respectively. Peptides were eluted from the column at 300 nL/min using the following linear gradient: from 4 to 35% B in 60 min, from 35 to 45% B in 5 min, from 45 to 90% B in 5 min, and held at 90% B for an additional 5 min. The heated capillary temperature and spray voltage were 275 °C and 1.7 kV, respectively. Full spectra were collected from 350 to 1800 m/z in the Orbitrap analyzer at a resolution of 120,000, followed by data dependent HCD MS/MS scans of the fifteen most abundant ions at a resolution of 30,000, using 40% collision energy and dynamic exclusion time of 30s.
Mass Spectrometry Data Analysis
Peptides and proteins were identified and quantified using the MaxQuant software package (version 1.5.7.4) with the Andromeda search engine (20,21). MS/MS spectra were searched against the Uniprot human protein database (Feb 2017, 70952 entries) and quantification was performed using default parameters for 3 state SILAC in MaxQuant. Combined normalized SILAC ratio of the proteins and the phosphopeptides and individual ratios of each experiment were obtained from the MaxQuant search. Perseus (version 1.5.5.3) was used to view and further analyze the data (22). Further details of the mass spectrometry data analysis and quantification of peptides are provided in Supplementary Materials and Methods.
Kinase Enrichment Analysis
The goal of the enrichment analysis was to identify kinases whose targets were significantly over-represented among the phosphorylation sites that were significantly hyper- or hypo-phosphorylated in drug resistant vs. sensitive cells when matched with the kinase-target site data in iPTMnet v.4.1 (23). To improve statistical power, the enrichment analysis was performed at the level of kinase families rather than individual kinases. Human kinases in iPTMnet were classified into families using a mapping table provided by KinBase (24)(http://kinase.com/web/current/human/; Dec 07 update). Further details of the kinase enrichment analysis are provided in Supplementary Materials and Methods.
Drug Signature Comparison
Drug signature comparisons were performed by querying Touchstone-P, a library of phosphoproteomic signatures from several cell lines treated with a panel of small molecule drugs (25) and the Proteomic Query tool, available through the ConnectivityMap web interface (http://clue.io/proteomics-query). Each signature in the library consists of the relative abundances of approximately 100 phosphorylation sites (P100). The Proteomic Query tool compares an input P100 phosphorylation signature to each signature in the library and reports a connectivity score ranging from −1 (strong negative connection/most “opposite” profile) to 1 (strong positive connection/most similar profile). Further details of the Drug Signature Comparison are provided in Supplementary Materials and Methods.
Immunoblot Analysis
Immunoblot analysis of cell extracts with different antibodies is described in Supplementary Materials and Methods section.
Drug Treatment and Cell Viability Assay
See Supplementary Materials and Methods section for drug treatment and cell survival analysis.
Knock-down of AXL in osimertinib sensitive and resistant cells by siRNA treatment
Knock-down of AXL in H1975, H1975_AZR3, and H1975_AZR4 cells by AXL siRNAs is described in Supplementary Materials and Methods section.
Quantitative RT-PCR
See Supplementary Materials and Methods section for isolation of total RNA from cell lines and quantitative RT-PCR.
Experimental Design and Statistical Rationale
Three and five biological replicates were processed for H1975/AZR3/AZR4 and H1975/COR1/COR10 SILAC Mass spectrometry experiments, respectively. Both proteome and phosphoproteome data were acquired and the quantitation was done by 3-state SILAC labeling. Volcano plots were generated for the Log 2 ratio of M/L and H/L. Proteins or phosphosites with 1.5-fold change and p value greater than 0.05 were considered as significant.
Data Access
The MS proteomics data in this study have been deposited in the ProteomeXchange Consortium (http://proteomecentral-.proteomexchange.org) via the PRIDE partner repository (26,27) with the dataset identifier PXD020108. Username: reviewer14844@ebi.ac.uk Password: npddP7.
Results
Quantitative mass spectrometry to identify and quantify the global proteome and phosphoproteome
H1975, an EGFR-L858R/T790M mutant 3rd generation EGFR TKI-sensitive cell line, and the corresponding isogenic osimertinib (AZR3 and AZR4) or rociletinib (COR1 and COR10) resistant cell lines were used for 3-state SILAC experiments to characterize the proteome and phosphoproteome by quantitative mass spectrometry. Labelled cells were treated with either DMSO or the 3rd generation EGFR TKIs, osimertinib or rociletinib (Fig. 1A, left panel). Both proteome and phosphoproteome (including TiO2 enriched phospho-serine / threonine and anti-phophotyrosine antibody-enriched phosphotyrosine peptides) analyses were performed. Overall, we identified and quantified thousands of proteins and phosphosites (Fig. 1A, right panel and Table S1A–D, S2A–D). Approximately 66–69% of the phosphosites identified were Class I sites. These are phosphosites whose localization probability of phosphorylation is greater than or equal to 0.75. More proteins and phosphosites were identified from the H1975/AZR3/AZR4 experiment than the H1975/COR1/COR10 experiment. 77% of the proteins and 39% of the phosphosites identified were common to both the osimertinib and rociletinib experiments. More unique proteins and phosphosites were identified in the osimertinib experiment than the rociletinib experiment (Fig. 1B). This could be due to the use of the QE HF mass spectrometer for the osimertinib experiments, compared to the slower, less sensitive orbitrap elite for the rociletinib experiments. In addition, more biological replicates were performed for the osimertinib experiments, especially for the phosphotyrosine enrichment. The hierarchical clustering of proteins and phosphosites (Fig. 1C) clearly showed that the two drug resistant cell lines (osimertinib and rociletinib) were grouped into two separate clusters. The osimertinib resistant cell lines (AZR3 and AZR4), with or without drug treatment, were clustered together as were the rociletinib resistant cell lines (COR1 and COR10). Overall, more proteins and phosphopeptides were less abundant in the resistant cells as evidenced by the distribution of the log2 ratio of the resistant and sensitive cells (Supplementary Fig. 1A, B) and the actual numbers of significantly altered proteins and phosphopeptides (Supplementary Fig. 2A). COR1 and COR10 cell lines were similar to each other but there was greater difference between AZR3 and AZR4 cell lines (Supplementary Fig. 2A).
Fig. 1.

Summary of SILAC-based quantitative proteome and phosphoproteome analyses of isogenic 3rd generation EGFR TKI-sensitive and resistant lung adenocarcinoma cells. (A) Experimental workflow showing treatment of SILAC-labelled cells, enrichment of phosphopeptides, and detection by tandem mass spectrometry. TKI-sensitive H1975, osimertinib-resistant AZR3/AZR4 cells, and rociletinib-resistant COR1/COR10 cells were treated with DMSO or the corresponding TKI (Left panel). Summary table showing the number of proteins and phosphosites identified in each experiment (Right panel). (B) Venn diagrams of proteins (left panel) and phosphosites (right panel) identified in osimertinib and rociletinib experiments. (C) Hierarchical clustering of proteins (left panel) and phosphosites (right panel) based on SILAC ratios. Columns represent different cell lines treated as indicated. Rows represent quantified proteins or phosphopeptides identified in all experimental conditions.
Next, one sample t-tests were performed on the protein and phosphopeptide SILAC ratios to determine significant differences in abundance between the resistant and sensitive cells. Hundreds of proteins and phosphosites were identified whose abundances were significantly different in resistant and sensitive cell lines (P < 0.05, 1.5-fold change) (Supplementary Fig. 2A, B, Table S2A–D). We generated volcano plots using the log10 (p value) and log2 (fold change) of the proteins and phosphosites (Supplementary Fig. 3A, B). The abundance of 113 proteins and 79 phosphosites were significantly altered in AZR3 and AZR4 cells treated with either DMSO or osimertinib. 463 proteins and 122 phosphosites were significantly altered in COR1 and COR10 cells treated with either DMSO or rociletinib. Among the proteins differentially expressed in the resistant cell lines were kinases, phosphatases, transcription regulators, transporters, and enzymes (Supplementary Fig. 2B). Examination of protein localization and function for all significantly altered proteins by Gene Ontology (GO) classification analyses showed that the differentially regulated proteins localized to the cytoplasm, nucleus, plasma membrane and the extracellular space (Fig. 2A). Many translation regulator proteins were significantly more altered in rociletinib compared to osimertinib-resistant cells (Table S3). The heatmap of selected translation regulators illustrates the altered expression of proteins including several EIF proteins (Fig. 2B).
Fig. 2.

GO classification analyses for localization and function of proteins with altered abundance, and heatmaps of protein SILAC ratios (TKI-resistant/sensitive) of translation regulators and EMT proteins and ratio of transcripts of EMT genes (TKI-resistant/sensitive). (A) Percentage of differentially expressed proteins in TKI-resistant cells with specific subcellular localization (up) and function (bottom). (B) Heatmap of SILAC ratios of protein abundance (TKI-resistant/sensitive) of selected translation regulators across all experiments demonstrate significantly more alterations in rociletinib-resistant cells. (C) Hierarchical clustering of quantified EMT proteins in three state SILAC experiments based on SILAC ratios of protein abundance in presence and absence of corresponding TKI. (D) Hierarchical clustering of transcript ratios of EMT genes quantified by qPCR.
Epithelial-mesenchymal transition (EMT) is a biological program observed in several types of epithelial cancers including NSCLC. It has been associated with metastatic spread and EGFR inhibitor resistance (28–30). We asked whether the osimertinib and rociletinib resistant cells underwent EMT and whether we can identify an EMT protein signature from the mass spectrometry data. We evaluated the expression of EMT proteins across all the resistant cell lines. In addition, we examined the expression of a set of EMT transcripts by quantitative RT-PCR (Table S4). The protein SILAC ratios of COR1 and COR10 in presence or absence of rociletinib clustered together based on EMT protein signature, while those of AZR3 and AZR4 in presence or absence of osimertinib clustered together (Fig. 2C). Similarly, based on transcript signature, COR1 and COR10 clustered together while AZR3 and AZR4 clustered together. EMT associated proteins, VIM, VCAN, SERPINE1, TFPI2, FN1 and others had increased protein expression in COR1 and COR10 cells compared to the AZR3 and AZR4 cells (Fig. 2C). Transcripts of various EMT genes such as ZEB1, MSN, MAP1B, VIM, TFPI2, VCAN, and FN1 were also expressed more in COR1 and COR10 cells compared to AZR3 and AZR4 cells (Fig. 2D). This data shows that both transcripts and protein expression of targets associated with EMT were higher in rociletinib resistant cells. E-cadherin protein (CDH1) expression, associated with the epithelial state, has been associated with longer time to progression and a trend toward longer overall survival following combination erlotinib/chemotherapy (31). Consistent with this, we observed lower protein and transcript expression of CDH1 in resistant cells. Taken together, our results suggest that EMT is associated with TKI drug resistance in both rociletinib and osimertinib resistant cells, although EMT signature is more pronounced in rociletinib resistant cells, despite the heterogeneity.
Changes in the abundance and phosphorylation of protein kinases and phosphatases in osimertinib and rociletinib resistant cells
To further examine the altered protein expression of kinases and phosphatases identified (Fig. 2A), the significantly altered SILAC ratios for kinases (Supplementary Fig. 4A) and phosphatases (Supplementary Fig. 4B) in the resistant cell lines, both DMSO and drug treated, were visualized in a heatmap. The significantly altered proteins (p<0.05) with a fold change cut-off of 1.5 were chosen. There were many kinases and phosphatases with altered expression in the TKI-resistant cells. There were more proteins dysregulated in the rociletinib resistant cells than osimertinib resistant cells, especially those expressed at lower levels. Expression of protein tyrosine kinase 7 (PTK7), an inactive tyrosine kinase involved in Wnt signaling (32–35), and two phosphatases, translocase of inner mitochondrial membrane 50 (TIMM50) and protein tyrosine phosphatase receptor type F (PTPRF), was increased in all resistant lines. Abundance of several kinases, such as thymidine kinase 1 (TK1), P21 activated kinase 1 (PAK1), phosphatidylinositol-5-phosphate 4-kinase type 2 gamma (PIP4K2C), and phosphatases, RNA guanylyltransferase and 5’-phosphatase (RNGTT), protein phosphatase, Mg2+/Mn2+ dependent 1B (PPM1B) and protein phosphatase 1 regulatory inhibitor subunit 14B (PPP1R14B) was decreased in all resistant lines. Interestingly, several proteins displayed opposite patterns in the TKI-resistant cell lines, such as 5’-Nucleotidase Ecto (NT5E), was more abundant in osimertinib-resistant, but less abundant in rociletinib-resistant cells, in comparison to their sensitive counterparts.
There were also differences in expression of certain kinases and phosphatases between the two different isogenic cell lines resistant to either osimertinib (AZR3 and 4) and rociletinib (COR1 and 10). For example, we saw contrasting expression patterns for the SRC proto-oncogene, an important non-receptor tyrosine kinase activated downstream of RTK, which plays a role in the activation of other protein tyrosine kinase (PTK) families. Whereas expression of SRC was increased in the COR1, COR10 and AZR3 cells, but decreased in the AZR4 cells. Such a difference in individual protein expression in isogenic TKI-resistant cells, such as AZR3 and 4 may represent their differential modulation during resistance. The phosphatase, Inositol Polyphosphate-5-Phosphatase (OCRL) involved in regulating membrane trafficking was less abundant in the AZR4 cells only. N-Myc Downstream Regulated 1 (NDRG1) had lower expression in the resistant cell lines, AZR4 and COR1. Overall, the differences in the modulation of the expression of specific kinases and phosphatases among isogenic versions of TKI-resistant cells underscore the heterogeneity in the development of resistance to the EGFR TKIs, osimertinib and rociletinib.
Since phosphorylation is regulated by both kinases and phosphatases, which are themselves regulated by phosphorylation, we evaluated the altered phosphorylation of specific phosphosites on kinases and phosphatases in the resistant cells. Heatmaps of the SILAC phosphorylation ratios (resistant/sensitive) highlight significant alterations in the phosphorylation of individual sites in kinases (Supplementary Fig. 5A) and phosphatases (Supplementary Fig. 5B) across the TKI-resistant cell lines with/without TKI treatment. Phosphorylation of many important sites on kinases, including EGFR-Y1172, MET-Y1003/Y1234, GAB1-Y659/Y406, MAPK1-Y187 was significantly reduced in resistant cells, independent of TKI treatment. On the other hand, phosphorylation of many sites, such as CDK1-Y15, CDK2-T14/Y15, CHEK2-S379, ROCK2-S1374, was significantly increased in resistant cells. Interestingly, we identified phosphorylation sites within several kinases which were differentially modulated with and without TKI treatment. In osimertinib-resistant cells, reduced phosphorylation of MAPK3K2-S154, EGFR-Y1172, HIPK-Y359, YES1-Y194, EPHB4-Y590, CDK5-Y4, GAB1-Y405/Y659, LYN-Y32 was more pronounced in untreated as compared to osimertinib-treated resistant cells. Similarly, in rociletinib-resistant cells, reduced phosphorylation of MARK3-S469, TJP2-S986, MAST4-S1373, SRP72-S621, MTOR-T1162, BAIAP2-T360, TNK2-Y859, STK10-S450, AAK1-S624, compared to rociletinib treated resistant cells. These dynamic phosphorylation changes suggest that, in TKI-resistant cells, phosphorylation at these sites is less dependent on EGFR signaling.
We also investigated changes in phosphorylation on phosphatases (Supplementary Fig. 5B). The phosphatase, PTPN11, located downstream of EGFR, regulates the RAS/MAPK signaling pathway. Phosphorylation of PTPN11-Y62 was reduced in both osimertinib and rociletinib resistant cells, regardless of TKI treatment. In rociletinib resistant cells phosphorylation of PTPN11-Y584 was also reduced. We also identified another differentially modulated phosphatase, PTPN3. Interestingly, PTPN3 has been shown to be upregulated in both cisplatin and doxorubicin-resistant ovarian cancer cells, suggesting its role in tumorigenesis, stemness and drug resistance in ovarian cancer and potential its use as a therapeutic target for ovarian cancer (36). Phosphorylation of PTPN3-S359 was significantly reduced in COR1 and COR10 cells but increased in AZR3 and AZR4 cells. Integrin-linked Kinase-Associated Serine Threonine Phosphatase (ILKAP) also known as protein phosphatase 2C selectively associates with Integrin Linked kinase (ILK) and modulates cell adhesion and growth factor signaling (37). ILKAP inhibits S9 phosphorylation of GSK3β. ILKAP-S13 phosphorylation was reduced significantly in AZR3 and AZR4 cells. Taken together, we identified altered phosphorylation of key phophosites on kinases and phosphatases implying that resistance is accompanied by dysregulation of kinase and phosphatase activity and in turn the relevant signaling pathways.
Protein and phosphorylation abundance comparisons between osimertinib and rociletinib-resistant cells in presence of the respective TKI
Osimertinib and rociletinib are both 3rd generation EGFR TKIs with similar mechanisms of action. They are both irreversible inhibitors which covalently bind C797 of mutant EGFR, and do not inhibit wild type EGFRs. However, small molecule inhibitors such as TKIs often have different off target effects that have implications for targeted therapy and resistance. In addition, there is significant heterogeneity of resistance mechanisms to osimertinib even in individual patients (11). We sought to specifically analyze the proteins and phosphosites whose modulation is similar or different upon resistance to both TKIs. We examined proteins with significantly altered expression in all four TKI-resistant cell lines; the osimertinib resistant cell lines AZR3 and AZR4 as well as the rociletinib resistant cell lines, COR1 and COR10. There were 60 proteins with altered expression in all four TKI resistant cell lines treated with their corresponding TKIs, osimertinib and rociletinib (Fig. 3A and Table S5A). The heatmap and hierarchical clustering of the 60 common differentially expressed proteins shows that the protein expression is either increased or decreased in both COR1 and COR10 cells or AZR3 and AZR4 cells, suggesting that these proteins are likely involved in the resistance mechanisms to the individual EGFR TKIs. Of these 60 proteins, 35 were less abundant in all resistant lines, while nine proteins were expressed at significantly higher levels in all resistant lines. Among the 9 proteins that had increased abundance across the osimertinib and rociletinib treatment conditions, were TAGLN (Transgelin), a TGFβ-inducible gene, that has been shown to be a poor prognostic factor in colorectal cancer and promotes motility and metastasis that are associated with EMT (38–40) and EDIL3 (EGF-like repeat & Discoidin 1-like domain), that is a secreted glycoprotein and integrin ligand that has been implicated in EMT and microvessel density in lung adenocarcinoma (41,42). Among the common proteins with reduced abundance across the rociletinib and osimertinib resistant cells in presence of the respective TKI, was PIP4K2C, a PI3K that results in hyperactivation of the immune system, and XRCC1, a well-known DNA repair protein that is involved in DNA single strand break repair induced by ionizing radiation and alkylating agents.
Fig. 3.

Comparison of protein abundance differences between osimertinib-resistant and rociletinib-resistant cells treated with respective TKI. (A) Differentially expressed proteins in TKI-resistant cells compared to the sensitive cells. 60 differentially expressed proteins were common to all four resistant cell lines. (B) Heatmap of SILAC ratios of protein abundance (TKI-resistant/sensitive) and hierarchical clustering of the 60 differentially expressed proteins common to all TKI-resistant cell lines. (C) Protein network of the 60 common differentially expressed proteins together with EGFR by STRING analysis. Many of these proteins are direct and indirect downstream targets of EGFR. (D) Heatmap of SILAC ratios of phosphorylation abundance (TKI-resistant/sensitive) and hierarchical clustering of the 49 differentially phosphorylated phosphosites common to all TKI-resistant cell lines.
Interestingly, expression of 16 proteins were differentially modulated in the osimertinib and rociletinib resistant cells. Of these proteins, expression of 15 increased in rociletinib resistant cells and decreased in osimertinib resistant cells compared to the isogenic TKI sensitive cells (Fig. 3B). Included in these group is fibronectin 1 (FN1), Transglutaminase 2 (TGM2), Complement C3 (C3), HLA-A and Inosine Triphosphatase (ITPA). Only one protein, ALCAM, a member of a subfamily of immunoglobulin receptors, was down-regulated in rociletinib resistant cells but up-regulated in osimertinib resistant cells. STRING analysis of the 60 common differentially expressed proteins revealed a network of proteins in which 27 of the 60 are direct or indirect downstream targets of EGFR (Fig. 3C), indicating EGFR downstream targets are altered in the EGFR TKI-resistant cells. We also visualized the SILAC ratios of phosphorylation of TKI-resistant/sensitive cells in a heatmap for the phosphosites identified in AZR3 and AZR4 cells in presence of osimertinib and COR1 and COR10 cells in the presence of rociletinib (Fig. 3D, Table S5B). Among the sites in proteins that were hyperphosphorylated in all TKI-resistant cells was BAD-S118. BAD is a BH3 member of the BCL2 family that regulates apoptosis. Site-specific phosphorylation of BAD results in the pro- or anti-apoptotic function of BAD (43). Platinum resistant ovarian cancer cell lines and patient samples have increased phosphorylation at S118, as well as S75 and S99. Serine to Alanine mutations of either S99 or S118 or pharmacological inhibition of upstream kinase PKA restored cisplatin-induced apoptosis (44). Among the phosphosites that showed reduced phosphorylation in all 4 TKI resistant cell lines was PTPN11-Y62. There was significant reduction of phosphorylation at this site in all 4 resistant cell lines in presence of the respective TKIs (Fig. 3D, Table S5B). Taken together, despite the heterogeneity of resistance mechanisms, potentially different off target effects of similar EGFR TKIs, and technical limitations of mass spectrometry, the comparisons of identified proteins and phosphorylation sites and their SILAC ratios across osimertinib and rociletinib resistant isogenic cell lines identified key differentially expressed proteins and phosphorylation sites that were common across all 4 resistant cell lines.
Canonical pathways dysregulated in osimertinib and rociletinib resistant cells
Next, we used Ingenuity Pathway Analysis (IPA) to evaluate the activated or inhibited canonical pathways enriched among the proteins with significantly altered phosphorylation (Table S6). Interestingly, a number of signaling pathways enriched in both the osimertinib resistant cells (Supplementary Fig. 6A) and rociletinib resistant cells (Supplementary Fig. 6B) had a negative Z-score, which is indicative of pathway inhibition. In both TKI-resistant lines, ERBB signaling, EGF signaling, regulation of eIF4 and p70S6K signaling, ILK signaling, JAK/STAT signaling, RAC signaling, and PAK signaling were inhibited. Many of the pathways enriched in the resistant cells had positive Z-scores, suggesting activation, including PTEN, PPARγ, RHOA, RhoGDI, p53, and PPARα/RXRα signaling. There were a few signaling pathways, in the osimertinib and rociletinib resistant cells, which were dysregulated in an opposing manner. For example, CDC42 signaling and regulation of Actin-based motility by RHO pathway signaling were activated in the osimertinib resistant cells and inhibited in the rociletinib resistant cells. Overall, we found that, more signaling pathways were inhibited in the resistant cells, correlating with the overall significant reduction of phosphorylation in the resistant cells.
Validation of differentially phosphorylated proteins and phosphosites in resistant cells
To further validate and supplement our global quantitative mass spectrometry data, we performed Western blots of select proteins (Fig. 4). Changes in protein expression and phosphorylation at specific phosphosites in TKI-sensitive cells, H1975 and the TKI-resistant cells, COR1, COR10, AZR3, and AZR4 with or without TKI treatment, were examined (Fig. 4A). Relative quantification of phosphorylated proteins normalized to total protein expression was performed on the Western blots. Phosphorylation of EGFR, ERK, AKT-T308, SRC-Y416, MEK-S217/221 and p70S6K-T389 was reduced in at least one of the resistant cell lines. Phosphorylation of MET, an RTK with known activation of parallel signaling pathway upon EGFR TKI resistance, was increased in COR10 and both AZR3 and AZR4. AXL expression was upregulated in both the osimertinib and rociletinib resistant cells; however, AXL-Y779 phosphorylation was reduced in AZR3 and AZR4 cells without osimertinib treatment, but not significantly in COR1 and COR10 cells. Among the epithelial-mesenchymal transition (EMT) proteins, E-cadherin expression consistently decreased in both the osimertinib and rociletinib resistant cell lines, again confirming EMT associated with the resistant cells (Fig. 4A, B).
Fig. 4.

Validation of differentially phosphorylated phosphosites and differentially expressed proteins in osimertinib- or rociletinib-resistant cells. (A-B) Western blots showing changes in phosphorylation and total protein expression without TKI treatment and upon 1 hour of rociletinib (100 nM) or osimertinib (50 nM) treatment in H1975, COR1, COR10, AZR3, and AZR4 cells. Bar graphs show the relative quantification of phosphorylated proteins normalized to total protein expression in each cell line.
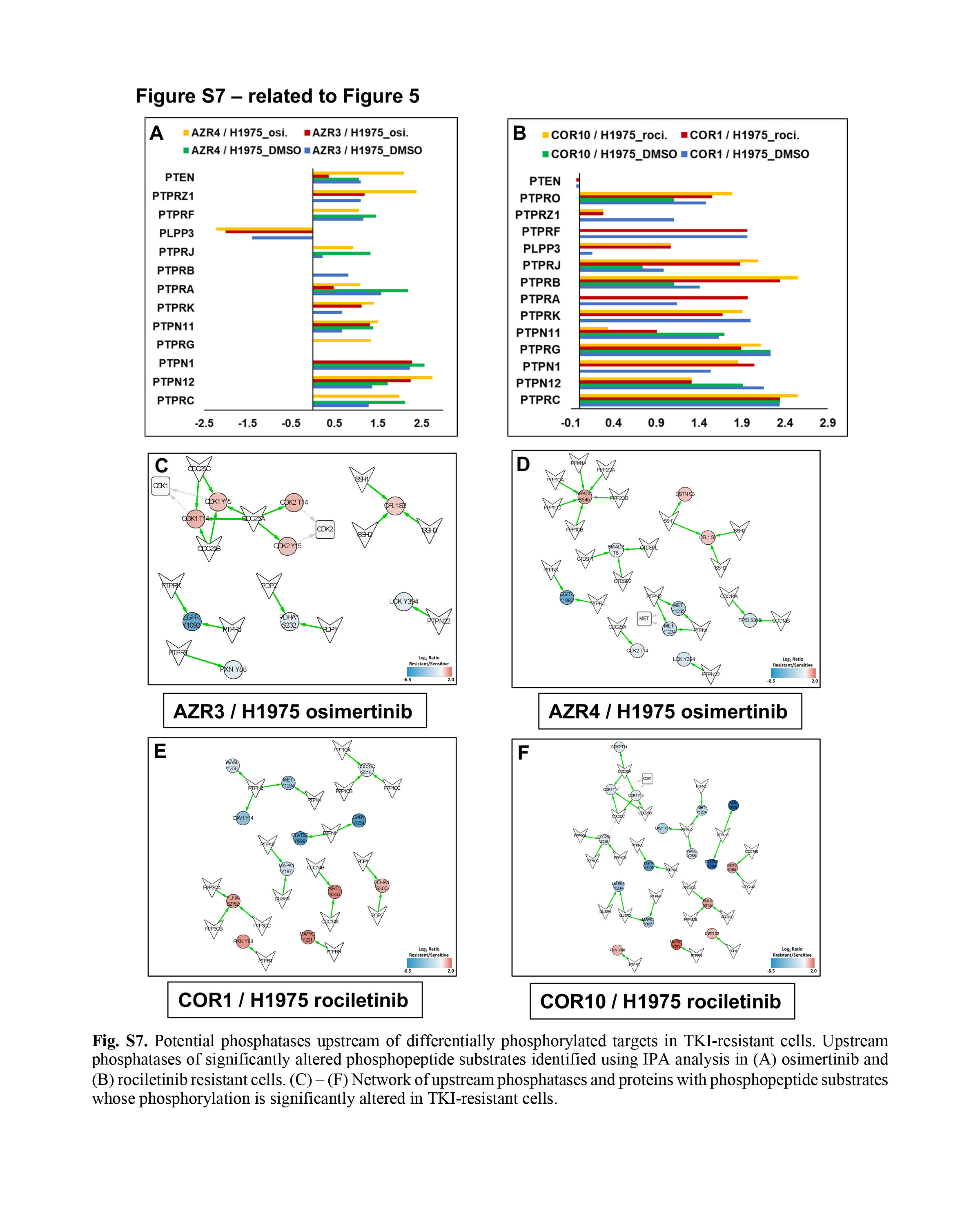
Upstream phosphatase analysis of significantly altered phosphopeptide substrates in TKI-resistant cells
Phosphorylation of downstream targets is regulated by upstream kinases and phosphatases. To identify the upstream regulatory phosphatases, we performed IPA analysis on the phosphorylation sites that were significantly hyper- or hypo-phosphorylated in the resistant cells. We identified 13 osimertinib-specific (Supplementary Fig. 7A) and 14 rociletinib-specific (Supplementary Fig. 7B) upstream phosphatases in the resistant cells. Almost all had positive Z-scores, indicating they are active in the TKI-resistant cell lines.
Next, we made networks showing connections between altered phosphosites and their experimentally validated phosphatases from the human DEPhOsphorylation Database (DEPOD) (45) (Supplementary Fig. 7C–F). Many of these phosphatases, such as PTPN11, PTPN1, PTPRK, and PTPRJ, overlapped with those identified by IPA analysis. Phosphorylation of downstream phosphorylation targets of these phosphatases was reduced in the resistant cells, suggesting activation of these phosphatases.
Validation of altered phosphorylation sites of the phosphatase PTPN11
The phosphatase, PTPN11 (more commonly known as SHP2) can activate the RAS/MAPK pathway and inhibit the PI3K/AKT cascade through different mechanisms. Once SHP2 is associated with adaptor protein GAB1, it inhibits PI3K by dephosphorylating GAB1-PI3K binding sites. We asked whether phosphorylation changes in SHP2 are associated with PI3K/AKT and MAPK signaling in TKI resistant cells. Multiple phosphorylation sites of PTPN11 were identified and quantified in both osimertinib and rociletinib resistant cells (Fig. 5A, upper panel). Most of the sites identified, including S580, Y546, Y584, and Y62, were significantly hypo-phosphorylated in all resistant cells. MS and MS/MS spectra of the PTPN11 phosphopeptides VpYENVGLMQQQK (Y584) (Fig. 5A, middle panel) and IQNTGDpYYDLYGGEK (Y62) (Fig. 5A, lower panel) demonstrate that phosphorylation at these sites was significantly lower in all resistant cells, AZR3 and AZR4, as well as COR1 and COR10. Western blots confirmed the PTPN11 phosphorylation changes in sensitive and resistant cells with or without drug treatment. PTPN11-Y584 phosphorylation was significantly reduced in both the osimertinib and rociletinib resistant cells (Fig. 5B). Taken together, reduced phosphorylation of key PTPN11 sites in resistant cells, which is consistent with inactive phosphatase activity, is associated with increased activation of PI3K/AKT pathway and decreased activation of the RAS/MAPK signaling (Fig. 5C).
Fig. 5.

Phosphorylation alteration of different phosphosites of the phosphatases PTPN11. (A) Multiple phosphorylation sites of PTPN11 identified and quantified in both osimertinib and rociletinib resistant cells. The SILAC ratios indicate the relative abundance between the resistant and H1975 sensitive cells (upper panel). Annotated MS/MS spectra (left) and MS spectra of their parent ions (middle and right) of the phosphopeptides VpYENVGLMQQQK (Y584) (middle panel) and IQNTGDpYYDLYGGEK (Y62) (lower panel) of PTPN11 showing the relative abundance changes between the sensitive and resistant cells in osimertinib and rociletinib experiment. (B) Western blots showing changes in phosphorylation and total protein expression of PTPN11 and EGFR without TKI treatment and upon 1 hour of rociletinib (100 nM) or osimertinib (50 nM) treatment in H1975, COR1, COR10, AZR3, and AZR4 cells. (C) Schematic showing the role of SHP2 in the activation of RAS/MAPK and inhibition of PI3K/AKT signaling pathways downstream from EGFR.
Upstream kinase analysis of significantly altered phosphopeptide substrates in TKI-resistant cells
To identify kinases whose targets were significantly over-represented among the significantly hyper- or hypo-phosphorylated sites in drug-resistant vs. sensitive cells, we matched the significantly altered phosphorylation sites with the kinase-target site data in iPTMnet v.4.1 (23). To improve statistical power, the enrichment analysis was performed at the level of kinase families rather than individual kinases (Tables S7, S8). Networks were generated showing the upstream kinase families and the downstream phosphorylated targets that were either significantly hyper- or hypo- phosphorylated in AZR3, AZR4, COR1 and COR10 cell lines in presence of the corresponding EGFR TKIs (Fig. 6). We determined that many downstream CDK kinase family phosphosites in the CMGC group were hypo-phosphorylated; while many downstream AKT kinase family and STE20 kinase family phosphosites were hyper-phosphorylated in all four resistant cell lines (Fig. 6A–D). We also found phosphosites in proteins downstream of the PKA kinase family were hyper-phosphorylated in the two rociletinib resistant cell lines (Fig. 6C, D). There was more similarity between networks identified for the rociletinib-resistant lines, COR1 vs COR10, than between the osimertinib resistant cell lines, AZR3 vs AZR4. The CK2, SGK and WEE kinase family’s downstream proteins were hyper-phosphorylated in the AZR3 cell line (Fig. 6A). Phosphosites downstream of several CAMK group (CAMK1, RAD53, and MAPKAPK) and the TK group (ACK and MET) kinase families were either hyper- or hypo-phosphorylated in the other osimertinib resistant cell, AZR4 (Fig. 6B). In summary, global quantitative phosphoproteomic analyses have identified key regulatory networks consisting of the upstream kinase families and the downstream phosphorylation targets that are potentially activated or inhibited in EGFR TKI resistant cells.
Fig. 6.

Networks generated by upstream kinase analysis of significantly altered phosphopeptide substrates in TKI-resistant cells using iPTMNet. (A) AZR3/H1975 in the presence of osimertinib; (B) AZR4/H1975 in the presence of osimertinib; (C) COR1/H1975 in the presence of rociletinib; (D) COR10/H1975 in the presence of rociletinib. The scale bars show the log2 SILAC ratio of phosphorylation (resistant/sensitive). Phosphosites in red are hyperphosphorylated and blue are hypophosphorylated in the resistant cells.
Prediction of drugs to circumvent resistance and in vitro / in vivo efficacy testing in osimertinib resistant cells
Next, we used the P100 phosphoproteomic data generated from cancer cell lines with and without drug treatment by the NIH Library of Integrated Network-Based Cellular Signatures (LINCS) program (http://www.lincsproject.org/) (46) to identify drugs that might reverse the phosphoproteomic signature from the TKI resistant cells. We looked for anti-correlation of the phosphoproteomic signatures from osimertinib and rociletinib resistant cells with specific P100 phosphoproteomic signatures from cell lines treated with specific drugs. We postulated that the drugs whose phosphorylation signatures are anticorrelated with those from the TKI resistant cell lines will have the potential to circumvent resistance to the EGFR TKIs, osimertinib and rociletinib. Using this approach, several inhibitors of multiple signaling pathways were identified for both osimertinib and rociletinib resistant cells (Supplementary Fig. 8A, Table S9). We selected dactolisib, a dual PI3K/AKT and MTOR inhibitor and VX-970, an ATR kinase inhibitor for further validation. The osimertinib and rociletinib resistant cells used for the large-scale SILAC quantitative mass spectrometry in this study had EC50s to their respective TKIs in the micromolar range. The osimertinib resistant AZR3 and AZR4 cells were 162-fold and 136-fold resistant to osimertinib-sensitive H1975 cells (Fig. 7A). Similarly, the COR1 and COR10 cells were 45 and 38-fold more resistant to rociletinib compared to H1975 cells, respectively (Fig. 7B). We generated two more EGFR mutant osimertinib-sensitive and -resistant isogenic pairs of lung adenocarcinoma cell lines by stepwise increase in concentration of osimertinib treatment for these validation experiments. These include PC9 and PC9-OsiR-NCI1 cells and HCC827 and HCC827-OsiR-NCI1 cells. These cells harbor the EGFR 746–750 Del mutant. The fold increase of EC50 was in the range of 34 for PC9-OsiR-NCI1 cells to 336 for the HCC827-OsiR-NCI1 cells (Fig. 7C–D). Dactolisib could overcome TKI resistance in osimertinib resistant cell lines, AZR3, AZR4, PC9-OsiR-NCI1, and HCC827-OsiR-NCI1 (Fig. 7A, C, D), as well as rociletinib resistant cell lines, COR1 and COR10 (Fig. 7B), either as single drug treatment or in the presence of 2μM osimertinib. Similarly, VX970 could also overcome osimertinib resistance in AZR3 and AZR4 cells either as single drug treatment or in the presence of 2μM osimertinib (Fig. 7E). We interrogated phosphorylated and total EGFR, AKT, ERK, AXL and PTPN11 by Western blot analyses in the isogenic H1975, AZR3, and AZR4 cells (Fig. 7F) and the isogenic PC9/PC9-OsiR-NCI1 and HCC827/HCC827-OsiR-NCI1 cells (Fig. 7G). There was reduced EGFR and ERK phosphorylation in the osimertinib-resistant cells upon treatment with 50nM Osimertinib. However, while osimertinib was unable to reduce phosphorylation of AKT, 50nM dactolisib treatment reduced AKT phosphorylation in AZR3, AZR4, PC9-OsiR-NCI1 and HCC827-OsiR-NCI1 cells. Similar reduction in phosphorylation of AKT was observed upon 1 hour of dactolisib (50nM) treatment or combination of dactolisib (50nM) and osimertinib (50nM) treatment in all osimertinib resistant cells (Fig. 7F–G). While AXL protein expression was increased in all osimertinib resistant cells compared to their isogenic counterparts, pAXL-Y779 was reduced in all osimertinib resistant cells. Osimertinib could inhibit pAXL expression in both sensitive and resistant cells while dactolisib treatment had no effect on AXL or pAXL expression (Fig. 7F–G). We further investigated whether dactolisib could inhibit xenograft tumor growth in vivo in mice. Mice were injected with AZR3 cells to develop tumors in flank. Dactolisib in combination with osimertinib was able to inhibit tumor growth of AZR3 xenografts in vivo. There was significant reduction of the size of flank tumors upon combination osimertinib and dactolisib treatment compared to the other treatment groups by Day 20 of treatment (Fig. 7H, I). We followed these tumors for 36 days of treatment, by which time mice from the other treatment groups were starting to get euthanized for reaching threshold tumor size (Supplementary Fig. S8B). There was significant decrease in size of tumors on Day 36 of combination treatment (Fig. 7J). The results show the feasibility of predicting drugs that overcome TKI resistance by leveraging the phosphoproteomic signature of drug treatment in cancer cells from the LINCS program.
Fig. 7.

In vitro and in vivo sensitivity of osimertinib and rociletinib resistant cells to dactolisib, a PI3K/mTOR inhibitor, and in vitro sensitivity of osimertinib resistant cells to VX-970, an ATR inhibitor. (A) Cell viability curves of H1975, H1975-AZR 3 and 4 cells showing the effect of osimertinib, dactolisib, and dactolisib in presence of 2 μM of osimertinib treatment. The EC50 values and fold resistant of the AZR3 and AZR4 cells compared to H1975 sensitive cells are shown. (B) Cell viability curves of H1975, H1975-COR 1 and 10 cells showing the effect of rociletinib, dactolisib, and dactolisib in presence of 2 μM of rociletinib treatment. The EC50 values and fold resistance of COR1 and COR10 cells compared to H1975 sensitive cells are shown. (C-D) Cell viability curves of PC9, PC9-OsiR-NCI1 (C) and HCC827, HCC827-OsiR-NCI1 (D) cell lines showing the effect of osimertinib, dactolisib, and dactolisib in presence of 2 μM of osimertinib treatment. The corresponding EC50 of each drug is shown in the right panels. (E) Cell viability curves of H1975, H1975-AZR 3 and 4 cell lines showing the effect of VX-970, and VX-970 in presence of 2 μM of osimertinib treatment. The EC50 of VX970 alone or in presence of 2μM osimertinib are shown in the right panel. (F) Western blots showing changes in phosphorylation and total protein expression without TKI treatment and upon 1 hour of dactosilib (50nM), osimertinib (50nM) or a combination of dactolisib (50nM) and osimertinib (50nM) treatment in H1975, H1975-AZR3, and H1975-AZR4 cells (G) Western blots showing changes in phosphorylation and total protein expression without TKI treatment and upon 1 hour of dactosilib (50nM) or osimertinib (50nM) or a combination of dactolisib (50nM) and osimertinib (50nM) treatment treatment in PC9, PC9-OsiR-NCI1, HCC827, and HCC827-OsiR-NCI1 cells. (H-J) Dactolisib in combination with osimertinib inhibits tumor growth of H1975-AZR3 xenografts in vivo. The in vivo growth of xenograft tumors is shown for 20 days of treatment (H) and the percent tumor growth at Day 20 (I) and Day 36 (J) of treatment is shown.
Discussion
The use of oral EGFR TKIs for the treatment of lung cancer patients with tumors harboring mutant EGFR has been a paradigm shift. Osimertinib, the 3rd generation EGFR TKI, is the current standard of care for patients with EGFR mutations due to its increased efficacy, lower side effects, and increased brain penetrance. Unfortunately, all patients treated with this drug develop resistance. Genomic approaches have primarily been used to interrogate resistance mechanisms to EGFR TKIs, including the 3rd generation EGFR TKIs. Here, we have characterized the proteome and the phosphoproteome of a series of isogenic EGFR mutant lung adenocarcinoma cell lines that are either sensitive or resistant to the 3rd generation EGFR TKIs, osimertinib and rociletinib. To our knowledge, this is the most comprehensive proteomic dataset resource to date used to investigate the 3rd generation EGFR TKI resistance in lung adenocarcinoma. We have utilized this unbiased global quantitative proteomic and phosphoproteomic dataset to uncover alterations in signaling pathways in the TKI-resistant lines. In addition, we used this resource to reveal a proteomic EMT signature as well as kinases and phosphatases with altered protein expression and phosphorylation in the TKI resistant cells. Furthermore, we used anticorrelation analysis of this phosphoproteomic dataset with the published drug-induced P100 phosphoproteomic datasets from the LINCS analysis to postulate drugs with potential to overcome EGFR TKI resistance. We validated one such drug, dactolisib, a PI3K/mTOR inhibitor, which was able to overcome osimertinib and rociletinib resistance in vitro and in combination with osimertinib, was able to overcome osimertinib resistance in vivo.
Four distinct osimertinib and rociletinib resistant isogenic cell lines were independently developed from H1975 cells which harbor the EGFR-L858R/T790M mutation, by stepwise increases in drug dosage over several months (3,47). In addition, in this study we describe two additional osimertinib resistant EGFR mutant cell lines, PC9-OsiR NCI1 and HCC827-OsiR-NCI1, harboring the EGFR 746–750 Del mutation that were generated by the same approach from their sensitive counterparts, PC9 and HCC827. The H1975 sensitive cells phenocopy approximately 60% of patients who develop resistance to the 1st and 2nd generation EGFR TKIs via acquisition of the EGFR-T790M mutation. The PC9 and HCC827 cells phenocopy EGFR TKI-sensitive frontline patients who harbor the TKI sensitive, but not any resistant EGFR mutation and are now treated mostly with osimertinib following FDA approval for frontline patients. The EGFR TKI resistant cells serve as a valid model system for patients who undergo osimertinib treatment, and eventually develop secondary resistance. We and others have primarily used genomic strategies to investigate mechanisms of osimertinib resistance (9,11–15). To our knowledge, this is the first study to identify and compare resistance mechanism of these 3rd generation EGFR TKIs by utilizing unbiased global proteome and phosphoproteome modification data. This approach provides a powerful tool to examine activated signaling pathways without bias and identify vulnerable nodes to potentially overcome resistance.
The experimental approach used in this study employed 3–5 global SILAC quantitative mass spectrometry biological replicates. We identified thousands of proteins and phosphosites in both experiments, hundreds of which had statistically significant alterations in the resistant cells. Surprisingly, we observed more proteins and phosphosites with decreased abundance in both osimertinib and rociletinib resistant cells with/without drug treatment. Moreover, many translation regulator proteins were significantly more altered in rociletinib-resistant cells compared to osimertinib-resistant cells, such as several of the EIF proteins (EIF3J, EIF1AX, EIF6, EIF2S3, EIF2S2, EIF4A2, EIF1A2, EIF4G1, EIF5, EIF4H and EIF4B), RPS27L, EEFSEC, IGF2BP2, PAIP1 (Fig. 2B). The results suggest that the regulation of translation may have been altered in rociletinib resistant cells in contrast to the osimertinib resistant cells. Global alteration of protein translation would result in changes in protein expression contributing to TKI resistance. Furthermore, translation could be used as a therapeutic target (48).
EMT has been associated with resistance to targeted therapies, including EGFR TKIs (28,29). A transcriptomic EMT signature has been validated in clinical studies (29). We investigated quantitative transcript expression of EMT genes using qPCR and further estimated the protein expression changes in TKI resistant cells from the quantitative mass spectrometry data. Many EMT signature proteins and transcripts were altered significantly in the resistant cells, especially in the rociletinib resistant cells. For example, vimentin (VIM) and fibronectin 1 (FN1) were more abundant and E-cadherin protein (CDH1) expression was reduced in TKI-resistant cells. Our results suggest that EMT is associated with TKI drug resistance for both rociletinib and osimertinib-resistant cells. We propose an EMT protein signature that can be identified and analyzed using mass spectrometry. This EMT protein signature needs to be further validated with large datasets employing targeted mass spectrometry assays, such as multiple reaction monitoring (MRM) in a QQQ mass spectrometer (49–51).
We observed more differences between the two isogenic osimertinib resistant cells, AZR3 and AZR4 compared to the two rociletinib resistance cells, COR1 and COR10. The iPTMnet of upstream kinase analysis showed that downstream protein targets of the CK2, SGK and WEE kinase families were hyper-phosphorylated in the resistant cell line, AZR3 (Fig. 6A). Phosphosites downstream of several kinase families in the CAMK group (CAMK1, RAD53, and MAPKAPK) and the TK group (ACK and MET) were either hyper- or hypo-phosphorylated in the other osimertinib resistant cell, AZR4 (Fig. 6B). To the contrary, the two rociletinib resistant cells, COR1 and COR10 were more similar to each other with respect to the upstream kinases and the downstream targets. These results obtained from isogenic TKI-resistant cells underscore the proteomic heterogeneity that is likely to impact treatment response and resistance.
We have identified and quantified many kinases and phosphatases with altered expression and phosphorylation in the osimertinib and rociletinib resistant cells. Moreover, many important phosphorylation sites on kinases were hyper- or hypo-phosphorylated significantly in the resistant cells, both with and without drug treatment. Many of them are important downstream EGFR signaling pathway proteins, including EGFR-Y1172, MET-Y1003/Y1234, GAB1-Y659/Y406, MAPK1-Y187, CDK1-Y15, CDK2-T14/Y15, CHEK2-S379, ROCK2-S1374.
The activation of AXL kinase is a known mechanism of intrinsic and acquired resistance to EGFR TKIs, including osimertinib (28,29,52,53). In our study AXL expression was upregulated in AZR¾, COR1/10, PC9-OsiR-NCI1, and HCC827-OsiR-NCI cells (Fig. 4A, Fig. 7D–E). However, phosphorylated AXL (pY779) was reduced in the TKI resistant cells used in this study. Treatment of the AZR3 and AZR4 osimertinib resistant cells with osimertinib further reduced pAXL, demonstrating the known crosstalk between EGFR and AXL. In the PC9-OsiR-NCI1 and HCC827-OsiR-NCI1 cells, pAXL was significantly reduced in the resistant cells and there was no further alteration upon drug treatment. We also interrogated whether AXL inhibition by siRNA knockdown or AXL inhibitor, R428 (54) could overcome osimertinib resistance in AZR3 and AZR4 cells. Both these approaches showed that AXL inhibition was unable to overcome osimertinib resistance (Supplementary Fig. 9A–E). The EC50 of osimertinib after AXL knockdown or of R428 in the presence of 2μM osimerttinib were in the high micromolar ranges, suggesting AXL inhibition could not overcome osimertinib resistance. This suggests that in the osimertinib resistant cells used in this study, although there was increased AXL protein expression, activated AXL was not increased and the resistant cells were not dependent on AXL for survival. This underscores the need to develop appropriate biomarkers for clinical studies, including for phosphorylated targets.
PTPN11, commonly known as SHP2, is a ubiquitously expressed well characterized PTP oncogene. SHP2 activates the RAS/MAPK signaling pathway and inhibits the PI3K/AKT cascade downstream of RTKs by various mechanisms (55–57). The SHP2 protein contains two SH2 domains (N-SH2/C-SH2), a catalytic PTP domain, and a C-terminal tail. SHP2 exists in the closed inactive conformation whereby the N-SH2 is wedged into the PTP domain, which is relieved upon pTyr binding on substrate proteins upon RTK signaling resulting in enzyme activity. Active SHP2 is marked by phosphorylation of Y546 and Y584 in the C-terminal tail (mostly referred to as Y542 and Y580 in literature based on the short isoform). We identified various phosphorylated tyrosine sites in SHP2, including Y62 (Fig. 3D), Y546 and Y584 (Fig. 5). Phosphorylation at all of these sites was reduced in TKI-resistant cells, indicating SHP2 exists in the closed inactive state in the resistant cells. This may partly explain the paradoxically lower activation of RAS/MAPK signaling as evidenced by reduced phosphorylation of ERK and increased activation of the PI3K/AKT pathway as evidenced by increased phosphorylation of AKT in resistant cells (Fig. 5D). Reduced SHP2 phosphorylation and activation may be partly a result of overall reduced activation of mutant EGFRs (Figs. 4 and 5). However, further upstream inhibitors of SHP2 activity in TKI resistant cells remains to be elucidated. PDGFRB, ALK and ABL have been demonstrated to be kinases phosphorylating Y584 as well as the other key tyrosine site in the tail, Y542 (58–60). We speculate any of these upstream tyrosine kinases could be phosphorylating PTPN11 in the EGFR TKI-resistant cells that may be inhibited in the resistant cells. This can be further followed by CRISPR mediated introduction of PTPN11-Y584F mutant that inhibit PTPN11 and conversely a phosphomimetic mutant (Y584E) will activate PTPN11. Introduction of nonhydrolyzable phosphotyrosine analogues at Y542 and Y584 by expressed protein ligation have been shown to activate PTPN11 by interaction with its N- and C-terminal SH2 domains, respectively (61). If this phosphorylation event is responsible for TKI resistance, then the Y584E mutant may overcome resistance. Conversely, Y584F (in addition to Y542F) is likely to promote resistance.
The P100 assay includes a set of 100 phosphopeptide probes representing key cancer signaling pathways developed by the LINCS program (62). Phosphorylation of these sites from cell lines treated with a large set of therapeutic molecules was quantitated using a targeted MS-based P100 assay screen (46). Our anticorrelation analysis of quantitated phosphopeptides in our dataset that overlapped with the P100 assay peptides revealed dactolisib targeting PI3K/AKT and mTOR pathways as having potential to reverse TKI resistance. Dactolisib alone was very effective in inhibiting osimertinib- or rociletinib-resistant cell growth in vitro with EC50 in the nanomolar range. The presence of 2μM rociletinib or osimertinib did not significantly alter the EC50 of dactolisib in vitro. Importantly, while osimertinib could not inhibit phospho-AKT in resistant cells, rociletinib could by itself reduce pAKT levels in resistant cells. In contrast while dactolisib by itself had no effect on reducing pERK levels, as expected, osimertinib could still inhibit pERK in resistant cells (Fig. 7D–E). Either dactolisib or osimertinib had no effect on pPTPN11/PTPN11, indicating further that pAKT is downstream of PTPN11 (Fig. 5D). Although there was no sensitization from in vitro data, greater combination response was observed from in vivo data in Fig. 7E. The osimertinib/dactolisib combination inhibited tumor growth significantly more than dactolisib or osimertinib alone. Although these isogenic TKI-resistant cells are resistant to the respective EGFR TKIs (growth in presence of 2μM TKI), there is some sensitivity to the respective TKIs that was apparent in vivo (Fig. 7F–H). In addition, tumor heterogeneity is well established in the development of osimertinib resistance in patients (11). There are tumors in osimertinib resistant patients that continue to have stable disease or continued response when a distinct other tumor is resistant to osimeritnib. Hence, we propose combination therapy of osimertinib and dactolisib rather than monotherapy with dactolisib.
We were able to identify several other inhibitors of multiple signaling pathways that can potentially be used to treat TKI-resistant lung cancer that needs further validation using our anticorrelation analysis with the P100 assay (Fig. 7A). One such target is the ATR inhibitor, VX970. The potential vulnerability of ATR inhibition to overcome resistance is indicated from additional evidence from our study. XRCC1, involved in DNA single strand break repair, was among the proteins whose expression was significantly reduced in all osimertinib and rociletinib resistant cell lines (Fig. 3B). ATR inhibition by small molecule ATR inhibitors was synthetically lethal in XRCC1 deficient ovarian cancer cells (63). Similar to dactolisib, the ATR inhibitor, VX-970 could circumvent osimertinib resistance in vitro (Fig. 7C). Hence, the decrease in XRCC1 protein expression in osimertinib and rociletinib resistant cells may be responsible for the vulnerability of the resistant cells to the ATR inhibitor, VX970. Further in vivo validation studies are needed in osimertinib resistant models, including patient derived xenografts (PDX) to ascertain the suitability of this combination to overcome resistance in vivo. Our study underscored the possibility that the assay involving global or targeted MS-based phosphoproteomics can be used to identify drug targets in the treatment of cancer.
In conclusion, this study identified and quantified, in an unbiased manner, the global proteome and phosphoproteome changes in 3rd generation TKI resistant lung cancer cells in presence and absence of the respective TKIs. This study provides a new dimension and complements the existing studies involving genomics to identify resistance mechanisms and drug targets to overcome resistance. Our study identified TKI resistant mechanisms and also an actionable target based on cellular signaling perturbations in resistant cells.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Statement of Significance:
Global quantitative proteomics reveals changes in the proteome and phosphoproteome in lung cancer cells resistant to third-generation EGFR TKIs, identifying the PI3K/MTOR inhibitor dactolisib as a potential approach to overcome resistance.
Acknowledgments
We thank AstraZeneca and Clovis Oncology for providing TKI resistant cell lines. We also thank Daren E. Cross (AstraZeneca) and Thomas Harding (Clovis Oncology) for helpful discussions. This research was supported by the NIH Intramural Research Program, Center of Cancer Research, National Cancer Institute, NIH U54HL127624 and NIH U01GM120953, and research funds from AstraZeneca. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Declaration of Interests
U.G. has a clinical trial agreement (CTA) with AstraZeneca and had received research funding from AstraZeneca, Esanex and Aurigene. U.G. is currently an employee of Bristol Myers Squibb. The other authors have no conflicts of interest to report.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin 2013;63:11–30 [DOI] [PubMed] [Google Scholar]
- 2.Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2005;2:e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov 2014;4:1046–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N Engl J Med 2018;378:113–25 [DOI] [PubMed] [Google Scholar]
- 5.Janne PA, Yang JC, Kim DW, Planchard D, Ohe Y, Ramalingam SS, et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med 2015;372:1689–99 [DOI] [PubMed] [Google Scholar]
- 6.Sequist LV, Soria JC, Goldman JW, Wakelee HA, Gadgeel SM, Varga A, et al. Rociletinib in EGFR-mutated non-small-cell lung cancer. N Engl J Med 2015;372:1700–9 [DOI] [PubMed] [Google Scholar]
- 7.Sequist LV, Soria JC, Camidge DR. Update to Rociletinib Data with the RECIST Confirmed Response Rate. N Engl J Med 2016;374:2296–7 [DOI] [PubMed] [Google Scholar]
- 8.Sequist LV, Piotrowska Z, Niederst MJ, Heist RS, Digumarthy S, Shaw AT, et al. Osimertinib Responses After Disease Progression in Patients Who Had Been Receiving Rociletinib. JAMA Oncol 2016;2:541–3 [DOI] [PubMed] [Google Scholar]
- 9.Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med 2015;21:560–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Piotrowska Z, Niederst MJ, Karlovich CA, Wakelee HA, Neal JW, Mino-Kenudson M, et al. Heterogeneity Underlies the Emergence of EGFRT790 Wild-Type Clones Following Treatment of T790M-Positive Cancers with a Third-Generation EGFR Inhibitor. Cancer Discov 2015;5:713–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roper N, Brown AL, Wei JS, Pack S, Trindade C, Kim C, et al. Clonal Evolution and Heterogeneity of Osimertinib Acquired Resistance Mechanisms in EGFR Mutant Lung Cancer. Cell Rep Med 2020;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oxnard GR, Hu Y, Mileham KF, Husain H, Costa DB, Tracy P, et al. Assessment of Resistance Mechanisms and Clinical Implications in Patients With EGFR T790M-Positive Lung Cancer and Acquired Resistance to Osimertinib. JAMA Oncol 2018;4:1527–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Le X, Puri S, Negrao MV, Nilsson MB, Robichaux J, Boyle T, et al. Landscape of EGFR-Dependent and -Independent Resistance Mechanisms to Osimertinib and Continuation Therapy Beyond Progression in EGFR-Mutant NSCLC. Clin Cancer Res 2018;24:6195–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang Z, Yang N, Ou Q, Xiang Y, Jiang T, Wu X, et al. Investigating Novel Resistance Mechanisms to Third-Generation EGFR Tyrosine Kinase Inhibitor Osimertinib in Non-Small Cell Lung Cancer Patients. Clin Cancer Res 2018;24:3097–107 [DOI] [PubMed] [Google Scholar]
- 15.Piotrowska Z, Isozaki H, Lennerz JK, Gainor JF, Lennes IT, Zhu VW, et al. Landscape of Acquired Resistance to Osimertinib in EGFR-Mutant NSCLC and Clinical Validation of Combined EGFR and RET Inhibition with Osimertinib and BLU-667 for Acquired RET Fusion. Cancer Discov 2018;8:1529–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang ZH, Lu JJ. Osimertinib resistance in non-small cell lung cancer: Mechanisms and therapeutic strategies. Cancer Lett 2018;420:242–6 [DOI] [PubMed] [Google Scholar]
- 17.Murtuza A, Bulbul A, Shen JP, Keshavarzian P, Woodward BD, Lopez-Diaz FJ, et al. Novel Third-Generation EGFR Tyrosine Kinase Inhibitors and Strategies to Overcome Therapeutic Resistance in Lung Cancer. Cancer Res 2019;79:689–98 [DOI] [PubMed] [Google Scholar]
- 18.Zhang X, Belkina N, Jacob HK, Maity T, Biswas R, Venugopalan A, et al. Identifying novel targets of oncogenic EGF receptor signaling in lung cancer through global phosphoproteomics. Proteomics 2015;15:340–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang X, Maity T, Kashyap MK, Bansal M, Venugopalan A, Singh S, et al. Quantitative Tyrosine Phosphoproteomics of Epidermal Growth Factor Receptor (EGFR) Tyrosine Kinase Inhibitor-treated Lung Adenocarcinoma Cells Reveals Potential Novel Biomarkers of Therapeutic Response. Mol Cell Proteomics 2017;16:891–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 2008;26:1367–72 [DOI] [PubMed] [Google Scholar]
- 21.Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M. Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res 2011;10:1794–805 [DOI] [PubMed] [Google Scholar]
- 22.Tyanova S, Cox J. Perseus: A Bioinformatics Platform for Integrative Analysis of Proteomics Data in Cancer Research. Methods Mol Biol 2018;1711:133–48 [DOI] [PubMed] [Google Scholar]
- 23.Huang H, Arighi CN, Ross KE, Ren J, Li G, Chen SC, et al. iPTMnet: an integrated resource for protein post-translational modification network discovery. Nucleic Acids Res 2018;46:D542–D50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science 2002;298:1912–34 [DOI] [PubMed] [Google Scholar]
- 25.Litichevskiy L, Peckner R, Abelin JG, Asiedu JK, Creech AL, Davis JF, et al. A Library of Phosphoproteomic and Chromatin Signatures for Characterizing Cellular Responses to Drug Perturbations. Cell Syst 2018;6:424–43 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vizcaino JA, Cote RG, Csordas A, Dianes JA, Fabregat A, Foster JM, et al. The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res 2013;41:D1063–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vizcaino JA, Csordas A, del-Toro N, Dianes JA, Griss J, Lavidas I, et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res 2016;44:D447–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet 2012;44:852–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Byers LA, Diao L, Wang J, Saintigny P, Girard L, Peyton M, et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res 2013;19:279–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Izumchenko E, Chang X, Michailidi C, Kagohara L, Ravi R, Paz K, et al. The TGFbeta-miR200-MIG6 pathway orchestrates the EMT-associated kinase switch that induces resistance to EGFR inhibitors. Cancer Res 2014;74:3995–4005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yauch RL, Januario T, Eberhard DA, Cavet G, Zhu W, Fu L, et al. Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin Cancer Res 2005;11:8686–98 [DOI] [PubMed] [Google Scholar]
- 32.Bin-Nun N, Lichtig H, Malyarova A, Levy M, Elias S, Frank D. PTK7 modulates Wnt signaling activity via LRP6. Development 2014;141:410–21 [DOI] [PubMed] [Google Scholar]
- 33.Dunn NR, Tolwinski NS. Ptk7 and Mcc, Unfancied Components in Non-Canonical Wnt Signaling and Cancer. Cancers (Basel) 2016;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hayes M, Naito M, Daulat A, Angers S, Ciruna B. Ptk7 promotes non-canonical Wnt/PCP-mediated morphogenesis and inhibits Wnt/beta-catenin-dependent cell fate decisions during vertebrate development. Development 2013;140:1807–18 [DOI] [PubMed] [Google Scholar]
- 35.Peradziryi H, Kaplan NA, Podleschny M, Liu X, Wehner P, Borchers A, et al. PTK7/Otk interacts with Wnts and inhibits canonical Wnt signalling. EMBO J 2011;30:3729–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li S, Cao J, Zhang W, Zhang F, Ni G, Luo Q, et al. Protein tyrosine phosphatase PTPN3 promotes drug resistance and stem cell-like characteristics in ovarian cancer. Sci Rep 2016;6:36873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kumar AS, Naruszewicz I, Wang P, Leung-Hagesteijn C, Hannigan GE. ILKAP regulates ILK signaling and inhibits anchorage-independent growth. Oncogene 2004;23:3454–61 [DOI] [PubMed] [Google Scholar]
- 38.Kim IG, Lee JH, Kim SY, Hwang HM, Kim TR, Cho EW. Hypoxia-inducible transgelin 2 selects epithelial-to-mesenchymal transition and gamma-radiation-resistant subtypes by focal adhesion kinase-associated insulin-like growth factor 1 receptor activation in non-small-cell lung cancer cells. Cancer Sci 2018;109:3519–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Elsafadi M, Manikandan M, Dawud RA, Alajez NM, Hamam R, Alfayez M, et al. Transgelin is a TGFbeta-inducible gene that regulates osteoblastic and adipogenic differentiation of human skeletal stem cells through actin cytoskeleston organization. Cell Death Dis 2016;7:e2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou HM, Fang YY, Weinberger PM, Ding LL, Cowell JK, Hudson FZ, et al. Transgelin increases metastatic potential of colorectal cancer cells in vivo and alters expression of genes involved in cell motility. BMC Cancer 2016;16:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jeong D, Ban S, Oh S, Jin Lee S, Yong Park S, Koh YW. Prognostic Significance of EDIL3 Expression and Correlation with Mesenchymal Phenotype and Microvessel Density in Lung Adenocarcinoma. Sci Rep 2017;7:8649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gasca J, Flores ML, Jimenez-Guerrero R, Saez ME, Barragan I, Ruiz-Borrego M, et al. EDIL3 promotes epithelial-mesenchymal transition and paclitaxel resistance through its interaction with integrin alphaVbeta3 in cancer cells. Cell Death Discov 2020;6:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bui NL, Pandey V, Zhu T, Ma L, Basappa, Lobie PE. Bad phosphorylation as a target of inhibition in oncology. Cancer Lett 2018;415:177–86 [DOI] [PubMed] [Google Scholar]
- 44.Marchion DC, Cottrill HM, Xiong Y, Chen N, Bicaku E, Fulp WJ, et al. BAD phosphorylation determines ovarian cancer chemosensitivity and patient survival. Clin Cancer Res 2011;17:6356–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Damle NP, Kohn M. The human DEPhOsphorylation Database DEPOD: 2019 update. Database (Oxford) 2019;2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abelin JG, Patel J, Lu X, Feeney CM, Fagbami L, Creech AL, et al. Reduced-representation Phosphosignatures Measured by Quantitative Targeted MS Capture Cellular States and Enable Large-scale Comparison of Drug-induced Phenotypes. Mol Cell Proteomics 2016;15:1622–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walter AO, Sjin RT, Haringsma HJ, Ohashi K, Sun J, Lee K, et al. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov 2013;3:1404–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grzmil M, Hemmings BA. Translation regulation as a therapeutic target in cancer. Cancer Res 2012;72:3891–900 [DOI] [PubMed] [Google Scholar]
- 49.Zhao L, Whiteaker JR, Pope ME, Kuhn E, Jackson A, Anderson NL, et al. Quantification of proteins using peptide immunoaffinity enrichment coupled with mass spectrometry. J Vis Exp 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Whiteaker JR, Zhao L, Abbatiello SE, Burgess M, Kuhn E, Lin C, et al. Evaluation of large scale quantitative proteomic assay development using peptide affinity-based mass spectrometry. Mol Cell Proteomics 2011;10:M110 005645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Whiteaker JR, Zhao L, Anderson L, Paulovich AG. An automated and multiplexed method for high throughput peptide immunoaffinity enrichment and multiple reaction monitoring mass spectrometry-based quantification of protein biomarkers. Mol Cell Proteomics 2010;9:184–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Taniguchi H, Yamada T, Wang R, Tanimura K, Adachi Y, Nishiyama A, et al. AXL confers intrinsic resistance to osimertinib and advances the emergence of tolerant cells. Nat Commun 2019;10:259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang F, Liu XW, Bartholdy BA, Cheng HY, Halmos B. Blockade of AXL activation overcomes acquired resistance to EGFR tyrosine kinase inhibition in non-small cell lung cancer. Transl Cancer Res 2019;8:2425-+ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Holland SJ, Pan A, Franci C, Hu Y, Chang B, Li W, et al. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res 2010;70:1544–54 [DOI] [PubMed] [Google Scholar]
- 55.Tajan M, de Rocca Serra A, Valet P, Edouard T, Yart A. SHP2 sails from physiology to pathology. Eur J Med Genet 2015;58:509–25 [DOI] [PubMed] [Google Scholar]
- 56.Zhang SQ, Tsiaras WG, Araki T, Wen G, Minichiello L, Klein R, et al. Receptor-specific regulation of phosphatidylinositol 3’-kinase activation by the protein tyrosine phosphatase Shp2. Mol Cell Biol 2002;22:4062–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Montagner A, Yart A, Dance M, Perret B, Salles JP, Raynal P. A novel role for Gab1 and SHP2 in epidermal growth factor-induced Ras activation. J Biol Chem 2005;280:5350–60 [DOI] [PubMed] [Google Scholar]
- 58.Bennett AM, Tang TL, Sugimoto S, Walsh CT, Neel BG. Protein-tyrosine-phosphatase SHPTP2 couples platelet-derived growth factor receptor beta to Ras. Proc Natl Acad Sci U S A 1994;91:7335–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Voena C, Conte C, Ambrogio C, Boeri Erba E, Boccalatte F, Mohammed S, et al. The tyrosine phosphatase Shp2 interacts with NPM-ALK and regulates anaplastic lymphoma cell growth and migration. Cancer Res 2007;67:4278–86 [DOI] [PubMed] [Google Scholar]
- 60.Mitra S, Beach C, Feng GS, Plattner R. SHP-2 is a novel target of Abl kinases during cell proliferation. J Cell Sci 2008;121:3335–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lu W, Gong D, Bar-Sagi D, Cole PA. Site-specific incorporation of a phosphotyrosine mimetic reveals a role for tyrosine phosphorylation of SHP-2 in cell signaling. Mol Cell 2001;8:759–69 [DOI] [PubMed] [Google Scholar]
- 62.Stathias V, Turner J, Koleti A, Vidovic D, Cooper D, Fazel-Najafabadi M, et al. LINCS Data Portal 2.0: next generation access point for perturbation-response signatures. Nucleic Acids Res 2020;48:D431–D9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sultana R, Abdel-Fatah T, Perry C, Moseley P, Albarakti N, Mohan V, et al. Ataxia telangiectasia mutated and Rad3 related (ATR) protein kinase inhibition is synthetically lethal in XRCC1 deficient ovarian cancer cells. PLoS One 2013;8:e57098. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.