

SUMMARY
The main limitation on axon regeneration in the peripheral nervous system (PNS) is the slow rate of regrowth. We recently reported that nerve regeneration can be accelerated by axonal G3BP1 granule disassembly, releasing axonal mRNAs for local translation to support axon growth. Here, we show that G3BP1 phosphorylation by casein kinase 2α (CK2α) triggers G3BP1 granule disassembly in injured axons. CK2α activity is temporally and spatially regulated by local translation of Csnk2a1 mRNA in axons after injury, but this requires local translation of mTor mRNA and buffering of the elevated axonal Ca2+ that occurs after axotomy. CK2α’s appearance in axons after PNS nerve injury correlates with disassembly of axonal G3BP1 granules as well as increased phospho-G3BP1 and axon growth, although depletion of Csnk2a1 mRNA from PNS axons decreases regeneration and increases G3BP1 granules. Phosphomimetic G3BP1 shows remarkably decreased RNA binding in dorsal root ganglion (DRG) neurons compared with wild-type and non-phosphorylatable G3BP1; combined with other studies, this suggests that CK2α-dependent G3BP1 phosphorylation on Ser 149 after axotomy releases axonal mRNAs for translation. Translation of axonal mRNAs encoding some injury-associated proteins is known to be increased with Ca2+ elevations, and using a dual fluorescence recovery after photobleaching (FRAP) reporter assay for axonal translation, we see that translational specificity switches from injury-associated protein mRNA translation to CK2α translation with endoplasmic reticulum (ER) Ca2+ release versus cytoplasmic Ca2+ chelation. Our results point to axoplasmic Ca2+ concentrations as a determinant for the temporal specificity of sequential translational activation of different axonal mRNAs as severed axons transition from injury to regenerative growth.
In Brief
Sahoo et al. find that axon injury triggers local translation of the mRNA encoding casein kinase 2α. CK2α phosphorylates axonal G3BP1 and causes RNA-protein granule disassembly. Phospho-G3BP1 has decreased binding to mRNAs needed for axon growth; thus, G3BP1 phosphorylation releases axonal mRNAs for translation to support nerve regeneration.
Graphical Abstract
INTRODUCTION
Peripheral nerve injury can activate protein synthesis in axons, where the new proteins support axon regrowth locally and retrogradely signal to activate regeneration-associated gene expression [1, 2]. Axonal mRNA populations are surprisingly complex, with 1,000’s of different axonal mRNAs in different neuron types [3]. mRNAs and translational machinery localize into axons in vivo [4–6], and mRNA translation in peripheral axons contributes to regeneration after axotomy [7, 8]. The distances separating distal axons from the soma as well as the rapidity of the translational response following axotomy indicate that mRNAs are present in axons before injury, with axotomy recruiting some mRNAs into translation [8–11]. G3BP1 is a stress granule protein [12] that we have recently shown is used to store mRNAs in adult peripheral nerves [13]. Axonal G3PB1 granules attenuate translation of their bound mRNAs, and disassembly of axonal G3BP1 granules increases intra-axonal protein synthesis and axon growth [13]. TIAR2, the mammalian ortholog of TIA1, is another protein linked to stress granule formation like G3BP1, and axonal TIAR-2 granules similarly slow axon regeneration in C. elegans [14].
G3BP1 self-assembles into granules through liquid-liquid phase separation (LLPS) driven by its intrinsically disordered regions (IDRs). G3BP1’s propensity for LLPS is increased by RNA binding and decreased by its phosphorylation on serine 149 (S149) [15, 16]. Axonal G3BP1 granules rapidly increase and then decrease after nerve injury, indicating that axons have endogenous mechanisms to disassemble G3BP1-mRNA complexes [13]. Consistent with this, phospho-G3BP1 levels increase as axonal G3BP1 granules decrease [13], suggesting that activity of a G3BP1 kinase is temporally regulated by axotomy. Work in other cellular systems indicate that casein kinase 2 (CK2) and Akt pathways are involved in G3BP1 S149 phosphorylation (G3BP1PS149) [17, 18]. Here, we show that CK2α expression increases axon growth through G3BP1 S149 phosphorylation. The CK2α catalytic subunit can be constitutively active [19]; we find that temporal specificity for CK2 activity is achieved by axotomy-induced increase in local translation of CK2α’s mRNA in axons. This axonal translation of Csnk2a1 mRNA requires post-injury translation of axonal mTor mRNA, and changes in axoplasmic Ca2+ levels allow intra-axonal translation to switch from mRNAs encoding injury-associated proteins to the growth-promoting CK2α.
RESULTS
Axonal CK2α Levels Rapidly Increase following Peripheral Nervous System Axotomy
We previously showed that axonal G3BP1PS149 levels are higher in regenerating than in uninjured sciatic nerve axons with a commensurate reduction in axonal G3BP1 granules [13]. CK2α was shown to phosphorylate G3BP1 S149 in U2OS cells [17], and we find that axonal CK2α immunoreactivity increases in sciatic nerve axons after axotomy (Figures 1B and 1C). Interestingly, the increase in axonal CK2α levels temporally correlates with a decrease in the number of axonal-stress-granule-like structures visualized by colocalization of G3BP1 and TIA1, where we used immunofluorescence (IF) signal excitation and acquisition parameters to only capture granular G3BP1 and TIA1 signals (Figures 1A–1C). These data show that axonal-stress-granule-like structures decrease in prevalence as axonal CK2α levels increase.
Figure 1. Axotomy Increases Axonal CK2α Protein.

(A) Exposure-matched confocal images for G3BP1 and TIA1 in naive and post-injury sciatic nerve. Left column shows granular G3BP1 and TIA1 signals merged with neurofilament (NF) in single XY planes; right column shows XYZ projections of colocalized G3BP1 + TIA1 (arrows) that overlap with NF (“axonal G3BP1 + TIA1”) across individual XY planes.
(B) Exposure-matched confocal images for CK2α in post-injury sciatic nerve. Left column shows CK2α merged with NF in single XY planes, and right column shows XYZ projections of CK2α signals that overlap with NF (“axonal CK2α”) across individual XY planes.
(C) Quantitation of axonal CK2α and colocalized G3BP1 + TIA1 from sciatic nerve axons imaged as in (A) and (B) shown as mean ± SEM (N = 6 animals; *p ≤ 0.05,**p ≤ 0.01, and ****p ≤ 0.0001 by Student’s t test versus t = 0 h; scale bars represent 5 μm in A and 10 μm in B).
CK2α Phosphorylates Axonal G3BP1
The catalytic CK2α subunit of the CK2 holoenzyme is encoded by two different genes, Csnk2A1 and Csnk2A2 for the CK2α and CK2α’ subunits, respectively [20]. CK2α and CK2α’ reported to serve the same cellular functions [21], and each can function as a kinase independent of the CK2b regulatory subunit [19]. The CK2α antibody used above did not distinguish between CK2α and CK2α’; thus, we used small interfering RNAs (siRNAs) to simultaneously deplete both Csnk2a1 and Csnk2a2 mRNAs for both from cultured DRG neurons (Figures 2A–2D and S1A–S1C). Using increased excitation and acquisition parameters for G3BP1 IF so that we detect total G3BP1 levels, Csnk2a1/2 knockdown did not affect axonal G3BP1 levels but decreased axonal and cell body G3BP1PS149 signals (Figures 2A–2D and S1A–S1C). Adjusting the imaging parameters for G3BP1 IF to detect total versus granular G3BP1 overexposed the G3BP1 granules, particularly for axons, and still likely underexposed the diffuse G3BP1 population. Thus, we used immunoblotting to determine whether Csnk2a1/2 mRNA knockdown affects G3BP1 levels. Consistent with CK2α functioning as a G3BP1 kinase, there was no significant effect on G3BP1 levels (Figures 2C and 2D), but the density of axonal G3BP1 granules increased upon Csnk2a1/2 mRNA depletion (Figures 2E and 2F). Conversely, overexpression of CK2α significantly decreased axonal G3BP1 granules and increased G3BP1PS149 levels but had no effect on total G3BP1 levels (Figures 2G, 2H, S1D, and S1E). These data indicate that neuronal CK2α expression regulates disassembly of axonal G3BP1 granules likely through G3BP1 phosphorylation.
Figure 2. CK2α Phosphorylates G3BP1 in DRG Neurons.

(A) Exposure-matched epifluorescent images for CK2α, G3BP1, and G3BP1PS149 in DRGs transfected with Cnsk2a1/2 (siCsnk2a1/2) and control (siCon) siRNAs.
(B) Quantitations of axonal CK2α, G3BP1, and G3BP1PS149 in DRG cultures transfected with indicated siRNAs shown as mean ± SEM. The G3BP1PS149 signals normalized to total G3BP1 (N ≥ 72 axons from 4 replicates; ***p ≤ 0.001 and ****p ≤ 0.0001 by one-way ANOVA versus siCon with Tukey honestly significant difference [HSD] post hoc).
(C and D) Representative immunoblots of DRGs transfected with siCon and siCsnk2a1/2 (C). Quantification of immunoblots for CK2α, G3BP1PS149, and G3BP1 levels (normalized to GAPDH loading control) are shown in (D) as mean ± SEM relative to siCon (N = 3; *p ≤ 0.05 by Student’s t test versus siCon).
(E and F) Representative exposure-matched XY confocal images for CK2α and G3BP1 granules in axons of siCon and siCsnk2a1/2 transfected DRG cultures (E). Quantitation of axonal G3BP1 granules in (F) as mean ± SEM is shown (N ≥ 16 axons from 3 replicates; *p ≤ 0.05 by Student’s t test).
(G and H) Exposure-matched single XY images for G3BP1 granules in axons of DRG cultures overexpressing GFP versus HA-tagged CK2α shown in (G). Quantitation of axonal G3BP1 granules in (H) as mean ± SEM is shown (N ≥ 28 axons from 3 replicates; ***p ≤ 0.001 by Student’s t test; scale bars represent 5 μm). Related to Figure S1.
CK2α mRNA Localizes into Axons and Is Locally Translated in Response to Injury
With activity of CK2α and CK2α’ able to phosphorylate substrates in the absence of the regulatory subunit [19], a localized increase in CK2α or α’ protein in axons could enable temporal and spatial regulation of CK2 activity. RNA profiling data from embryonic murine DRG and motor axons [22, 23], as well as our own axonal RNA profiles from adult mouse DRGs, show that both Csnk2a1 and Csnk2a2 mRNAs localize into axons (Figure S2A). By reverse transcriptase coupled droplet digital PCR (RTddPCR) using cultured adult rat lumbar 4–6 (L4–6) DRGs, both Csnk2a1 and Csnk2a2 mRNAs were detected in axonal preparations, but neither significantly changed after in vivo injury conditioning (Figures S2B and S2C). This suggests that the in vivo increase in axonal CK2α levels is not accounted for by altered mRNA levels. Because Csnk2a1 mRNA was more abundant in the axons, we focused on Csnk2a1 mRNA in subsequent experiments. Using single-molecule fluorescent in situ hybridization (smFISH) coupled with IF, we detected Csnk2a1 mRNA in distal axons of cultured DRG neurons and in sciatic nerves (Figures 3A–3C). There was no change in axonal Csnk2a1 mRNA levels comparing naive and regenerating sciatic nerve (Figure 3C), indicating that altered mRNA transport does not explain the in vivo increase in axonal CK2α protein levels.
Figure 3. CK2α Is Locally Translated in PNS Axons.

(A) Images of smFISH/IF for Csnk2a1 mRNA and scrambled probe plus NF protein for cell body and axons of naive DRG neurons (scale bar represents 10 μm).
(B and C) Exposure-matched confocal images Csnk2a1 mRNA versus scrambled probe plus NF protein in naive sciatic nerves shown in (B). Upper panels of image pairs show Csnk2a1 mRNA merged with NF protein signals in XY planes, with orthogonal projections to right (arrows indicate location for YZ projections); lower panel of each pair shows XYZ projection of RNA signals that overlap with NF (“axonal RNA”) across individual XY planes. (C) shows quantitation of axonal Csnk2a1 mRNA from naive (Na) and crushed (Cr) sciatic nerve axons as mean ± SEM (N = 6 animals; *p ≤ 0.05 and **p ≤ 0.01 by Student’s t test for indicated groups versus scrambled control; scale bar represents 5 μm).
(D) Schematic of nerve ligation experiment used in (E) and (F) and Figure S2D.
(E and F) Exposure-matched confocal images for CK2α in post-injury sciatic nerve from proximal and distal to ligation site and proximal to crush site in (E). Upper rows of image pairs show CK2α merged with NF in single XY planes; bottom rows show XYZ projections of CK2α signals that overlap with NF (axonal CK2α) across individual XY planes. (F) shows quantitation of axonal CK2α from ligation and crush sites in sciatic nerve as mean ± SEM (N = 5 animals; **p ≤ 0.01; NS, non-significant by Student’s t test for indicated time points versus 3 h; scale bar represents 10 μm).
(G) Schematic of ex vivo sciatic nerve injury and puromycinylation experiment used in (H) and (I) and Figure S2E.
(H and I) Representative immunoblots of input and puromycinylated protein from axoplasm of Na and ex vivo Cr sciatic nerve shown in (H). Quantitation of puromycinylated CK2α versus total CK2α in sciatic nerve axoplasm is shown in (I) as mean ± SEM (N = 3; *p ≤ 0.05 and **p ≤ 0.01 by Student’s t test for indicated conditions).
Related to Figure S2.
We used a nerve ligation assay to determine whether the injury-induced increase in axonal CK2α protein is intrinsic to the nerve. For this, rat sciatic nerve was ligated, to restrict anterograde and retrograde transport [24], and then crushed 1 cm distal to the ligation (Figure 3D). Successful attenuation of axonal transport was indicated by accumulation of amyloid precursor protein (APP) proximal and Stat3 distal to the ligation (Figure S2D). CK2α was selectively increased after injury at the crush site, but not at the ligation (Figures 3E and 3F), pointing to a nerve-intrinsic increase in CK2α protein after injury. We next used an ex vivo puromycinylation assay to determine whether the increase in axonal CK2α was from mRNA translation. For this, excised segments of naive rat sciatic nerve were incubated in culture medium supplemented with cyclosporin A to delay Wallerian degeneration [25] and the ClickIt puromycin analog O-propargyl-puromycin (OPP) to label nascently synthesized proteins [26]. There was a significant increase in OPP-labeled axoplasmic CK2α protein after in vitro nerve crush, and this was completely attenuated by protein synthesis inhibition (Figures 3G–3I and S2E). Thus, CK2α protein is locally synthesized in injured peripheral nerve axons.
Csnk2a1 mRNA 3′ UTR Drives Axonal Localization, and Its 5′ UTR Regulates Axonal Translation
To address mechanisms of axonal CK2α synthesis, we first asked how its mRNA localizes into axons. Axonal localization motifs are largely restricted to untranslated regions (UTRs) of mRNAs [3]. Thus, we fused Csnk2a1 5′ and/or 3′ UTRs to the coding sequence of a diffusion-limited GFP cDNA (GFPMYR) to test for axonal localizing activity (Figure 4A). DRG neurons expressing GFPMYR with 5′ and 3′ UTRs of Csnk2a1 mRNA (GFPMYR5′/3′Csnk2a1) showed axonal Gfp mRNA signals (Figures 4B, S3A, and S3B). Axonal Gfp mRNA localization was still seen when the Csnk2a1 5′ UTR was replaced with the non-localizing Camk2α 5′ UTR (GFPMYR5′camk2α/3′Csnk2a1; Figures 4B, S3A, and S3B). However, GFPMYR mRNA with the Csnk2a1 5′ UTR plus a segment of Kpnb1 3′ UTR that lacks Kpnb1’s axonal localization motif [27] (GFPMYR5′Csnk2a1/3′kpnb1_1–134) showed no axonal Gfp mRNA signals above the negative control (Figures 4B, S3A, and S3B). Thus, Csnk2a1’s 3′ TR is sufficient for localizing mRNA into axons of cultured DRG neurons.
Figure 4. Intracellular Ca2+ Blocks Axonal Csnk2a1 mRNA Translation and Increases Axonal Kpbn1 mRNA Translation.

(A) Schematic of experimental pipeline used to test for 5′ and 3′ UTR activities used in (B)–(H) and Figure S3.
(B) Quantitation of axonal Gfp mRNA normalized to GFP protein levels in soma of DRGs transfected with indicated constructs as mean ± SEM. See Figures S3A and S3B for representative axonal smFISH/IF images and somal mRNA quantitation (N ≥ 78 axons from 3 replicates; ****p ≤ 0.0001 by one-way ANOVA with Tukey HSD post hoc from indicated transfection group versus control).
(C and D) Representative FRAP sequences for distal axons of DRG cultures expressing GFPMYR5′/3′Csnk2a1 are shown in (C) (top shows GFPMYR5′/3′Csnk2a1 schematic). Representative videos for these FRAP sequences are included as Videos S1 and S2. Quantitation of FRAP assays (D) for distal axons of DRGs expressing GFPMYR5′/3′Csnk2a1 treated with anisomycin or neurotrophin cocktail as normalized is shown, average % recovery ± SEM (N ≥ 12 neurons over 3 repetitions; *p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001 versus control by two-way ANOVA with Tukey HSD post hoc).
(E) FRAP quantitation for distal axons of DRGs expressing GFPMYR5′camk2α/3′Csnk2a1 treated with anisomycin or neurotrophin cocktail as in (D) (N ≥ 12 neurons over 3 repetitions; *p ≤ 0.05, ***p ≤ 0.001, and ****p ≤ 0.0001 versus control by two-way ANOVA with Tukey HSD post hoc; there was no significant difference with neurotrophin treatment).
(F–H) Representative FRAP sequences for basal recovery of distal axons of DRGs co-expressing GFPMYR5′/3′Csnk2a1 (left) plus mChMYR5′/3′kpnb1 (right) shown in (F) (schematics for GFPMYR5′/3′Csnk2a1 and mChMYR5′/3′kpnb1 shown above image sequences). (G) and (H) show FRAP for distal axons of DRGs co-expressing GFPMYR5′/3′Csnk2a1 (G) and mChMYR5′/3′kpnb1 (H) for basal (control) and anisomycin, thapsigargin, and EGTA treatment shown as in (D). Representative videos for these FRAP sequences are included as Videos S3, S4, S5, and S6 (N ≥ 10 neurons over 3 repetitions; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001 versus control by two-way ANOVA with Tukey HSD post hoc; significance symbols/lines are matched for the respective treatment conditions; scale bars represent 10 μm).
Fluorescence recovery after photobleaching (FRAP) assays showed translation-dependent fluorescent recovery in distal axons for both the GFPMYR5′camk2α/3′Csnk2a1 and GFPMYR5′/3′Csnk2a1 transfected DRGs (Figures 4C–4E and S3C; Videos S1 and S2). Interestingly, GFPMYR5′camk2α/3′Csnk2a1 showed higher basal recovery than GFPMYR5′/3′Csnk2a1, which suggests translational regulation from the Csnk2a1 5′ UTR. Consistent with this, stimulation with neurotrophins significantly increased recovery of GFPMYR5′/3′Csnk2a1 fluorescence in axons but had no effect on GFPMYR5′camk2α/3′Csnk2a1 (Figures 4C–4E and S3C). Upregulation of neurotrophins by glial cells is well established in injured peripheral nervous system (PNS) nerves [28], so this may provide a route for the injury-induced increase in axonal CK2α. Regardless of mechanism, these data indicate that axonal transport of Csnk2a1 mRNA is driven by its 3′ UTR and translation by its 5′ UTR.
Cytoplasmic Ca2+ Provides a Switch for Regulation of Cskn2α1 mRNA Translation in Axons
Axotomy induces a near-immediate increase in axoplasmic Ca2+ [29, 30], and previous work has linked this to translation of injury-associated mRNAs in PNS axons [9–11, 31]. Thus, we asked whether Csnk2a1 mRNA is translationally regulated by Ca2+. Acute treatment with thapsigargin, to block Ca2+ uptake by endoplasmic reticulum (ER), significantly decreased recovery of photobleached axonal GFPMYR5′/3′Csnk2a1 (Figure S3D). In contrast, axonal GFPMYR5′/3′Csnk2a1 recovery rose significantly above basal recovery levels when intracellular Ca2+ was chelated using BAPTA-AM (Figure S3D). In Aplysia, in vitro axotomy increases in axoplasmic Ca2+ that needs to be buffered for subsequent axon growth [32]. Thus, Csnk2a1 mRNA translation may be activated in axons only after the Ca2+ increase seen after injury is resolved. Increased Ca2+ can block generalized translation by increasing phosphorylation of the translation initiation factor eIF2α [33], but translation of several axonal mRNAs, including Kpnb1, Vim, RanBP1, and Stat3α, is increased by elevated axoplasmic Ca2+ [9–11, 31]. These mRNAs are translated within 3–6 h post-axotomy, which is earlier than the increase in axonal CK2α seen above. This suggests that translation of different axonal mRNA cohorts might be determined by changes in axoplasmic Ca2+ levels after PNS injury. To test this possibility, we used dual reporter FRAP where DRG neurons were co-transfected with GFPMYR5′/3′Csnk2a1 and a myristoylated mCherry (mChMYR) reporter with the 5′ and 3′ UTRs of Kpnb1 mRNA (mChMYR5′/3′kpnb1; Figure 4F). This long Kpnb1 3′ UTR localizes reporter mRNAs into axons [7, 27], and we included Kpnb1’s 5′ UTR for potential translational control motifs needed for axonal importin β1 synthesis. As expected, translational inhibitors significantly attenuated recovery of both reporters in axons (Figures 4G and 4H; Videos S3 and S4). Thapsigargin treatment attenuated recovery of axonal GFPMYR5′/3′Csnk2a1 but increased recovery of axonal mChMYR5′/3′kpnb1 (Figures 4G and 4H; Video S5). BAPTA-AM had only modest effects on axonal mChMYR5′/3′kpnb1 recovery (Figure S3F), but Ca2+ chelation using EGTA significantly decreased recovery of mChMYR5′/3′kpnb1 and both BAPTA-AM and EGTA significantly increased recovery of axonal GFPMYR5′/3′Csnk2a1 (Figures 4G, 4H, and S3E; Video S6). The inhibition of mChMYR5′/3′kpnb1 translation by EGTA, but not BAPTA-AM, may reflect distinct roles for different Ca2+ sources on Kpnb1 mRNA translation, as BAPTA-AM does not chelate extracellular Ca2+. Regardless, these data are consistent with changes in axoplasmic Ca2+ as a mechanism to switch specificity of translation between different axonal mRNAs.
Intra-axonal Synthesis of CK2α Requires Local Translation of mTOR mRNA
We recently showed that mTor mRNA localizes into PNS axons, where it is translated in response to axotomy [8]. The kinetics of mTor mRNA translation in injured axons is intermediate between that reported for Kpnb1 [9] and Csnk2a1. Thus, we asked whether axonal Csnk2a1 mRNA translation requires mTOR activity. By FRAP, axonal recovery of GFPMYR5′/3′Csnk2a1 was blocked by the mTOR inhibitor torin similar to complete translational inhibition with anisomycin (data not shown). We next tested the possibility that axonally synthesized mTOR is needed for translation of Csnk2a1 using mTOR 3′ UTR deletion mice (mTOR-3′ UTR−/−) that show decreased axonal mTor mRNA levels and markedly decreased axonal protein synthesis after axotomy [8]. Strikingly, the injury-induced appearance of axonal CK2α after sciatic nerve crush was not observed in mTOR-3′ UTR−/− axons, although wild-type mice showed a robust increase in axonal CK2α (Figures 5A and 5B).
Figure 5. Axonally Synthesized mTOR Regulates Axonal CK2α Synthesis.

(A and B) Exposure-matched confocal images for CK2α in naive and 24 h post-injured sciatic nerve from wild-type versus mTOR 3′ UTR−/− mice shown in (A). Upper rows of image pairs show CK2α merged with NF signals in single XY planes; lower rows show XYZ projections of CK2α signals that overlap with NF (axonal CK2α) across individual XY planes. (B) shows quantitation of axonal CK2α from sciatic nerve axons as mean ± SEM (N = 3 animals; **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001 between the indicated groups by one-way ANOVA with Tukey HSD post hoc; scale bar represents 10 μm).
(C and D) FRAP quantifications of FRAP for distal axons of DRGs expressing mCHMYR5′/3′calr (C) versus mCHMYR5′/3′mTOR (D) shown for control, anisomycin, thapsigargin, and BAPTA-AM as normalized, average % recovery ± SEM (N ≥ 15 neurons over 3 repetitions; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001 versus control by two-way ANOVA with Tukey HSD post hoc; significance symbols/lines are matched for the respective treatment conditions).
Based on the above observations, we hypothesized that differential regulation by Ca2+ levels after injury could provide a molecular mechanism for switching from mTor to Csnk2a1 mRNA translation. We tested that possibility using intracellular Ca2+ release versus buffering to mimic the axonal environment after injury. Similar to Kpnb1 reporters above, we have previously shown that axonal Calr mRNA translation is increased by release of Ca2+ from the ER [34]. As anticipated, axonal translation of mChMYR reporter with 5′ and 3′ UTRs of Calr (mChMYR5′/3′calr) showed translation-dependent FRAP recovery that was increased by thapsigargin (Figure 5C). This recovery was completely blocked by Ca2+ chelation with BAPTA-AM (Figure 5C). Interestingly, mChMYR reporter with the 5′ and 3′ UTRs of mTor mRNA (mChMYR5′/3′mtor) showed similar response, with increased recovery with thapsigargin treatment and BAPTA-AM-treated cultures showing decreased recovery (Figure 5D). Together with Figures 4G and 4H, these data emphasize the role of Ca2+ levels in switching mRNA preference of axonal translational machinery from mRNAs encoding injury-associated proteins (Kpnb1, Calr, and mTor) to Csnk2a1, whose encoded protein could support axon growth.
Increasing CK2α Protein Increases Axonal Growth through Phosphorylation of G3BP1
Because we found that the injury-induced synthesis of axonal CK2α disassembles G3BP1 granules through G3BP1 phosphorylation, we asked whether CK2α activity impacts axon growth using a combination of mRNA depletion, pharmacological inhibitors, and expression of G3BP1 mutants (Figure 6A). siRNA-mediated depletion of Csnk2a1/2 significantly decreased axon growth in adult DRG cultures, and this was reversed by simultaneous depletion of G3bp1 mRNA (Figures 6B and 6C). DRG cultures treated with the CK2 inhibitor CX-4945 similarly showed reduced axon growth that was reversed by simultaneous G3bp1 mRNA depletion (Figures S4A and S4B). These data indicate that G3BP1 lies downstream of CK2α.
Figure 6. Axonally Synthesized CK2α Regulates Axon Growth via G3BP1.

(A) Schematic for experimental approach to test for CK2α effects on signaling to G3BP1 for axon growth.
(B and C) Representative images for siRNA-transfected DRG neurons at 60 h post-transfection in (B). Axon growth in siRNA-transfected DRGs is shown as mean ± SEM in (C) (N ≥ 200 neurons over 3 repetitions; ****p ≤ 0.0001 versus siCon and ####p ≤ 0.0001 for siCsnk2a1/2 versus siCsnk2a1/2 + siG3b1 by one-way ANOVA with Tukey HSD post hoc; scale bar represents 100 mm).
(D) Axon length for DRG cultures transfected with indicated siRNAs ± G3BP1S149E as mean ± SEM (N ≥ 96 neurons over 3 repetitions; ***p ≤ 0.001 versus siCon and ##p ≤ 0.01 for siCsnk2a1/2 versus siCsnk2a1/2 + G3BP1S149E by one-way ANOVA with Tukey HSD post hoc; siCsnk2a1/2 + G3BP1S149E is not statistically different than siCon).
(E) Axon length for DRG neurons transfected with indicated constructs as mean ± SEM (N ≥ 101 neurons over 3 repetitions; ***p ≤ 0.001 and ****p ≤ 0.0001 for indicated transfection group versus GFP and ####p ≤ 0.0001 for CK2α-HA versus CK2α-HA + G3BP1S149A by one-way ANOVA with Tukey HSD post hoc).
(F) Schematic for the in vivo depletion of Csnk2a1/2 mRNAs from sciatic nerve by injection of penetratin-tagged (“pen”) siRNAs that is used in (G)–(J) and Figures S5A–S5D, with timing of nerve crush injury and siRNA injection indicated. Nerves were harvested 7 days post-crush injury.
(G) Representative immunoblots of axoplasm from animals injected pen-siCon and pen-siCsnk2a1/2.
(H and I) Exposure-matched confocal images for CK2α, G3BP1 granules, and TIA1 granules in 168 h post-injury sciatic nerve injected with pen-siCon versus pen-siCsnk2a1/2 in (H). CK2α, G3BP1 granules, and TIA1 granules merged with NF in single XY planes are shown in upper left image pairs; remaining image pairs show CK2α (upper right), G3BP1 granular (lower left), and TIA1 granular (lower right) signals that overlap with NF (axonal) across individual XY planes projected as a separate channels in XYZ. (I) shows quantitation for the axonal CK2α intensity and G3BP1 and TIA1 granule intensity in sciatic nerve axons as mean Log2 fold-change ± SEM relative to siCon (N = 4; *p ≤ 0.05 and **p ≤ 0.01 by Student’s t test versus respective siCon; scale bar represents 10 μm).
(J) Quantitation of NF-positive axon profiles relative to the crush site for the pen-siCon and pen-siCsnk2a1/2 injected nerves as mean ± SEM (N = 4; *p ≤ 0.05 by two-way ANOVA with Bonferroni multiple comparisons test post hoc).
Related to Figures S4 and S5.
Notably, siG3bp1 did not restore the siCsnk2a1/2- or CX-4945-treated DRG cultures to the axon growth levels that siG3bp1 did by itself, so we asked whether expression of G3BP1 S149 phosphomimetic (G3BP1S149E) or non-phosphorylatable (G3BP1S149A) mutants might affect CK2α’s axon growth promotion. A recent report questioned the role of G3BP1 S149 phosphorylation based on additional mutations detected in the NTF2-like domain of G3BP1 constructs used in several publications [35], including ours [13]. We thus generated G3BP1S419A and G3BP1S149E constructs with intact NTF2-like domains (and no other mutations). Transfecting Csnk2a1/2-depleted DRG neurons with G3BP1S149E reversed the axon growth deficit seen with CK2α depletion alone (Figure 6D). In contrast to our previous work [13], transfecting only the G3BP1S149E increased axon outgrowth similar to G3BP1 depletion (Figure 6E). Considering that overexpression of CK2α in DRG neurons decreased axonal G3BP1 granules (Figures 2G and 2H), we asked whether this effect requires phosphorylation of G3BP1. Co-transfection of CK2α-hemagglutinin (HA) with G3BP1S149A completely blocked the growth promotion seen with CK2α overexpression (Figures 6E and S4C). The intra-molecular interactions that drive LLPS for G3BP1 granule formation strongly correlate with charge of the acidic domain and require RNA interaction [15, 16]. The G3BP1S149E mutation also strongly inhibited LLPS by G3BP1 by shifting phase separation threshold to significantly higher G3BP1 and RNA concentrations [15, 16]. We tested the new S149 G3BP1 mutants for mRNA co-immunoprecipitation from transected DRG neurons and find significantly lower mRNA binding by G3BP1S149E compared to wild-type G3BP1 and G3BP1S149A (Figures S4D and S4E). This extends the previous in vitro RNA binding data by showing that S149 phosphorylation attenuates G3BP1’s interactions with mRNAs within neurons and, together with the growth assessments above, indicates that CK2α-dependent phosphorylation of G3BP1 likely regulates axon growth by affecting G3BP1’s LLPS and RNA binding activity [15].
To determine whether CK2α-dependent phosphorylation of G3BP1 can promote axon growth in vivo, we used the vector peptide penetratin [36] covalently attached to siCsnk2a1/2 versus scrambled (siCon) siRNA to deplete Csnk2a1/2 mRNA from distal nerve. These penetratin-tagged siRNAs were rapidly taken up by cultured cells (Figure S5A). Immediately after crush injury, the siRNAs were injected into sciatic nerves proximal to the crush site and then nerves were harvested 7 days later (Figure 6F). Csnk2a1 and Csnk2a2 mRNAs as well as CK2α protein were significantly depleted from siCsnk2a1/2-injected nerves (Figures 6G–6I, S5C, and S5D). By IF, there was a significant increase in axonal G3BP1, but not TIA1, granules and significant decrease in G3BP1PS149 comparing siCsnk2a1/2 versus siCon-injected nerves (Figures 6H and 6I); axonal granules also appeared to be larger for both G3BP1 and TIA1 after siCsnk2a1/2 treatment (Figure 6H). Immunoblotting confirmed the reduction in G3BP1PS149 in sciatic nerve axoplasm but also showed no change in total G3BP1 (Figures 6G and S5D). Finally, in vivo depletion of axonal Csnk2a1/2 mRNAs significantly attenuated nerve regeneration (Figures 6J and S5B). Taken together, these data indicate that injury-induced synthesis of axonal CK2α promotes axon regeneration by phosphorylating G3BP1 and likely promoting intra-axonal protein synthesis by releasing mRNAs from G3BP1 granules.
DISCUSSION
The main limitation on axon regeneration in the PNS is the slow rate of regrowth, typically limited to 1 to 2 mm per day [37]. Hence, there is keen interest to understand the mechanisms underlying our recent demonstration of accelerated nerve regeneration upon axonal G3BP1 granule disassembly [13]. CK2 phosphorylates G3BP1 S149 during recovery from stress to trigger stress granule disassembly [17]. Our data link CK2α to axon growth promotion through axonal G3BP1 S149 phosphorylation. CK2α overexpression increases axon growth in cultured neurons and CK2α knockdown or inhibition decreases axon growth, effects that are mitigated by co-transfection with non-phosphorylatable and phosphomimetic G3BP1 S149 mutants. CK2 is well known to have constitutive kinase activity and is highly promiscuous in terms of substrates [19]. We find that localized translation of axonal Csnk2a1 mRNA determines when and where CK2α is introduced, thereby bringing both temporal and spatial specificity to the kinase in axons. This specificity is driven by altered axoplasmic Ca2+, with elevated Ca2+ sequentially triggering axonal synthesis of mTOR and then CK2α. However, it should be noted that our data do not exclude effects of Ca2+ on stability of these and other axonal mRNAs.
Our data for axonal CK2α synthesis fit with many previous publications showing that intra-axonal mRNA translation supports PNS axon regeneration [1]. The regulation of axonal Csnk2a1 mRNA translation uncovered here also provides new insight into how specificity of intra-axonal translation facilitates transition from an injured to a regenerating axon. Hundreds of mRNAs localize into axons, but there is only a rudimentary understanding of the mechanisms underlying how the axon determines which mRNAs to translate when [3]. We find that Ca2+ elevation blocks intra-axonal translation of Csnk2a1 mRNA, but this requires local mTor mRNA translation that is activated by Ca2+ increases. Ca2+ elevations are well known to block protein synthesis through phosphorylation of eIF2α, but translation of some mRNAs, particularly mRNAs encoding proteins linked to cellular stress responses, is paradoxically increased by phospho-eIF2α [38]. PNS axotomy triggers a rapid increase in axoplasmic Ca2+ levels, and translation of endogenous Kpnb1, Ranbp1, Stat3α, and Vim mRNAs is blocked by chelating Ca2+ [9, 10, 31, 39]. Like mTor mRNA, we find that axonal Kpnb1 mRNA translation is increased by Ca2+ elevation. Axons have functional equivalents of rough ER and Golgi apparatus [40–42], and Ca2+ release from the ER can trigger ER stress, referred to as the “unfolded protein response” (UPR) [43]. Axotomy-induced UPR in axons promotes PNS nerve regeneration [44] and translation of axonal Creb3 (also known as Luman), Calr, and Grp78 (encodes Hspa8) mRNAs can be activated by UPR [34, 45]. The mRNAs that are translationally activated with Ca2+ and axonal UPR constitute an injury response by the axon. Interestingly, Terenzio et al. [8] found that axonal synthesis of importin β1 (Kpnb1), Stat3, Calr, and Hspa8 required local mTOR activity by ex vivo translation assays as used herein. Combined with our finding that axonal synthesis of CK2α requires mTOR and lower Ca2+ levels, these observations indicate that changes in axoplasmic Ca2+ levels provide a mechanism to switch mTOR translational specificity from injury- to regeneration-associated mRNAs in axons (Figure 7).
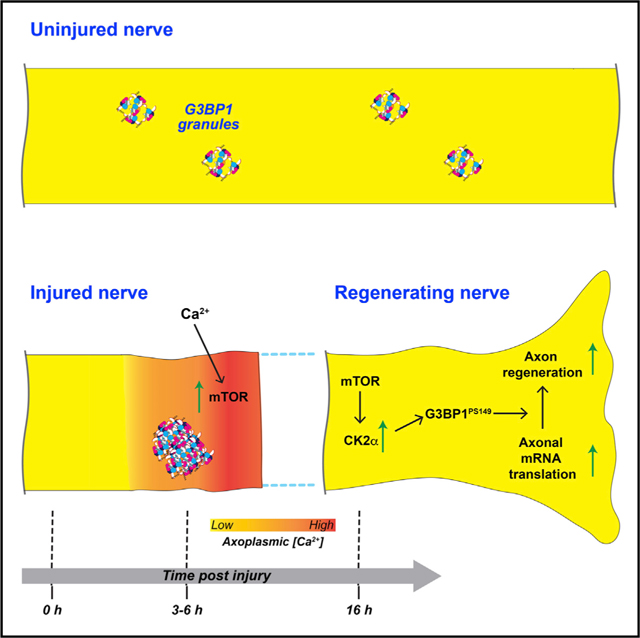
Figure 7. Axoplasmic Ca2+ Alterations Can Drive mRNA Translation Specificity after Axonal Injury.

(A) G3BP1 granules in uninjured PNS axons serve as a storage depot for axonal mRNAs.
(B) Nerve crush injury rapidly increases axonal G3BP1 granules. Injury to the axoplasmic membrane allows extracellular Ca2+ to enter the axon and likely also results in release of Ca2+ stores in the ER. This elevated Ca2+ triggers translation of axonal injury-response mRNAs, including mTor.
(C) Upregulation of mTOR enables translation of other axonal mRNAs. However, for translation of Csnk2a1 mRNA and likely other growth-associated mRNAs, Ca2+ must be buffered back to normal levels.
It is curious that there was no increase in CK2α protein at the ligation site, as this also represents an injury to the axons. Two possibilities may explain the selective activation of Csnk2a1 mRNA at the crush site. First, the injury resulting from ligation may not allow for the full sequence of translational events needed to synthesize axonal CK2α. This could reflect insufficient [Ca2+] to start the sequence or lack of Ca2+ buffering to allow translation of Csnk2a1 mRNA. There is no Wallerian degeneration distal to the ligation, although axon degeneration clearly occurs distal to the crush site, indicating that ligation is less severe than crush injury. Work in cultured mammalian sensory neurons showed that growth cone formation after in vitro axotomy requires intra-axonal mRNA translation [46]. Translation of axonal Calr mRNA, which is activated by phospho-eIF2α, is needed for axon extension after in vitro axotomy [47], and regeneration does initiate from the ligation site. Second, we see that axonal GFPMYR5′/3′Csnk2a1 reporter translation is increased by neurotrophins. Axotomized Schwann cells show increased synthesis and release of neurotrophins and other trophic factors distal to crush injured PNS nerves [28], so the severed nerve may receive a boost in trophic signaling that does not occur at the ligation site. Additional experiments will be needed to distinguish these possibilities, but the differential increase in Csnk2a1 mRNA translation upon nerve crush versus ligation suggests that the nature of the injury can impact translation in axons.
The axon-growth-promoting activity of CK2α seen here correlates with G3BP1 S149 phosphorylation. This phosphorylation was initially reported to trigger disassembly of stress granules [48], and our previous work showed an inverse correlation between axonal G3BP1 granules and G3BP1PS149 levels [13]. However, Panas et al. [35] recently refuted that G3BP1 S149 phosphorylation influences stress granule assembly from observations in U2OS cells treated with sodium arsenite, a potent SG inducer. These authors concluded that S99P mutation in the previously published G3BP1S149E plasmid accounted for the effects of what was presumed to be a simple phosphomimetic mutant, questioning the relevance of G3BP1 phosphorylation. Here, we have used S149E and S149A G3BP1 plasmids, free of any additional mutations, to show that expression of G3BP1S149E and G3BP1S149A mitigates the growth-regulating effects seen with Csnk2a1 mRNA knockdown and CK2α overexpression, respectively. S149 lies in the first of G3BP1’s three IDRs [15]. We had previously shown that exogenous introduction of this conserved IDR can trigger G3BP1 granule disassembly and accelerate axon growth [13]. Very recent work showed that G3BP1 is a core stress granule protein, with G3BP1-RNA binding lowering its biophysical threshold for LLPS [15, 16]. Moreover, S149E mutation was shown to disrupt G3BP1’s RNA binding affinity and LLPS [15, 16]. Our data are consistent with these in vitro analyses and add that G3BP1S149E has remarkably decreased binding for endogenous mRNAs in primary neurons. Both G3BP1 and TIA1 granules increase in prevalence shortly after axon injury, and axonal translation of Csnk2a1 mRNA with subsequent phosphorylation of the nascent protein allows the axon to disassemble G3BP1 granules, thereby releasing mRNAs to promote regeneration. Serine phosphorylation of C. elegans TIAR2 similarly blocks LLPS and promotes axon regeneration [14]. It is not clear which kinase phosphorylates TIAR2, and based on sequence alignments, the S338 and S339 that were mutated to generate phosphomimetic C. elegans TIAR2 are not conserved in human or rodent TIA1.
In summary, we show that axoplasmic Ca2+ levels control the translational specificity for different mRNAs in injured PNS axons, with elevated cytoplasmic Ca2+ increasing translation of axonal Kpnb1, Calr, and mTor mRNAs and Ca2+ chelation increasing translation of axonal Csnk2a1 mRNA. Axonally synthesized mTOR is required for the post-axotomy increase in axonal CK2α that subsequently drives phosphorylation of G3BP1 to support regeneration. Liao et al. [49] recently implicated G3BP1 granules in vesicle-linked transport of mRNAs into axons. It is intriguing to speculate that these G3BP1 transport granules mature into a storage structure like the granules studied here. Increased cytoplasmic Ca2+ supports membrane sealing, retrograde signaling, and cytoskeletal instability needed for growth cone formation [29, 32, 50, 51] but also activates a translation sequence needed for regeneration. Despite that increased axoplasmic Ca2+ inhibits translation of axonal Csnk2a1 mRNA, the initial Ca2+ elevation is essential to introduce CK2α into the injured axon by activating translation of axonal mTor mRNA. Together, our findings provide evidence for a sequential translation of different axonal mRNAs after nerve injury that is driven by axonal Ca2+ dynamics and converges on axonal G3BP1 granules that are used to store axonal mRNAs.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jeffery L. Twiss (twiss@mailbox.sc.edu).
Materials Availability
All unique reagents generated in this study (Csnk2a1, Kpnb1, and mTor translation reporter constructs, G3BP1 S149-phospho mutants) are available from the Lead Contact upon completion of Materials Transfer Agreement.
Data and Code Availability
Raw FASTQ files of GSE51572, and GSE66230 were downloaded from NCBI GEO database.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal use and survival surgery –
Institutional Animal Care and Use Committees of University of South Carolina and Weizmann Institute of Science approved all animal procedures. Male Sprague Dawley rats (175–250 g) and wild-type or mTOR−/− ICR mice [8] were used for all sciatic nerve injury experiments.. Isoflurane was used for anesthesia for peripheral nerve injuries.
For peripheral nerve injury, anesthetized rats or mice were subjected to a sciatic nerve crush at mid-thigh as described [54]. Rat sciatic nerve ligation experiments were done in rats as described previously [55]. Sciatic nerves were ligated close to the sciatic notch, immediately followed by crush injury as above at ~1 cm distal to the ligation site for the 3, 6, and 16 h time points. For the 72 h time point, nerve crush injury was done first followed by nerve ligation 6 h later. For in vivo siRNA injections, 2.5 nmoles of penetratin-tagged siRNAs (diluted in 10 μl phosphate-buffered saline [PBS]) were injected into sciatic nerves proximal to the crush site immediately after injury. Axoplasm extruded from sciatic nerve segments as previously described [13].
Cell culture –
For primary neuronal cultures, DRGs were harvested in Hybernate-A medium (BrainBits) and then dissociated as described [54]. Rats with sciatic nerve injury (7 d) were used to isolate injury conditioned DRG neurons. After centrifugation and washing in DMEM/F12 (Life Technologies), dissociated ganglia were resuspended in DMEM/F12, 1 x N1 supplement (Sigma), 10% fetal bovine serum (Hyclone), and 10 μM cytosine arabinoside (Sigma). Dissociated DRGs were plated immediately on poly-L-lysine (Sigma) + laminin (Millipore) coated surfaces [54]. Cells were plated onto glass coverslips for immunostaining, polyethylene-tetrathalate (PET) membrane inserts (1 μm pores; Corning) for isolation of axons from cell bodies, or tissue culture plates for isolation of protein. Axon and cell body preparations were isolated from neurons grown on PET membranes as described [56] for preparation of RNA (see below).
For DRG neuron transfection, dissociated ganglia were pelleted by centrifugation at 100 x g for 5 min and resuspended in ‘nucleofector solution’ (Rat Neuron Nucleofector kit; Lonza). 5–7 μg plasmid was electroporated using an AMAXA Nucleofector apparatus (program Neurons Rat DRG, G-013; Lonza). For siRNA transfection, 100 nM siRNAs (Dharmacon) were used with DharmaFECT 3 reagent and incubated for 60 h. Dharmacon On-target plus-SMART pool siRNA against G3bp1 or Csnk2a1/2 mRNAs were used in G3BP1PS149 level determination and axon growth assays. Non-targeting siRNAs were as control. CK2 inhibitor (CX-4945; Selleckchem) was used at 10 μM.
NIH 3T3 cells were used for testing cell permeability of pentratin-tagged siRNAs (see below). These cells were maintained in DMEM (Life Technologies) supplemented with 10% FBS (GIBCO) and 100 U/ml of Penicillin-Streptomycin (Life Technologies). For penetratin-tagged siRNA, siRNA dosage was determined by applying 10–100 nM to NIH 3T3 cells at 12 h after plating. Cells were incubated with FITC-tagged siRNAs for 4 h, cultures were fixed, and analyzed by confocal microscopy. Non-tagged siRNAs were used as controls.
METHOD DETAILS
Penetratin conjugation of siRNAs –
We used pentratin-tagged, fluorescent siRNAs for depletion of Csnk2a1/2 mRNA from sciatic nerve in vivo. For this, the four siRNAs from the Dharmacon On-target plus-SMART pool siRNA against Csnk2a1/2 mRNA were tested individually for depletion of Csnk2a1 and Csnk2a2 in primary rat DRG cultures transfected as above. The siRNA targeting nt 299–317, and 722–738 of rat Csnk2a1 and Csnk2a2 mRNAs, respectively, provided highest efficiency; this siRNA plus scrambled sequence siRNA (siCon) were used to generate siRNA duplexes with a 5′ thiol (sense strand) and 3′ FITC (anti-sense strand); labeled, modified siRNAs were HPLC purified (Dharmacon, Lafayette, CO and Xeragon, Germantown, MD) [36, 57]. Csnk2a1/2-specific and scrambled control sequences are as follows: siCon – sense, 5′ S-S.ggucucacguccuaauaaauu 3′ and antisense, 5′ Fl.uuuauuaggacgugagaccuu 3′; siCsnk2a1/2 – sense, 5′ S-S.cuaccagcuuguucgaaaauu 3′ and antisense, 5′ Fl.uuuucgaacaagcugguaguu 3′. Activated penetratin (MP Biol) was used to conjugate siRNAs. For this, annealed siRNA duplexes were resuspended in siRNA buffer (Dharmacon) and treated with an equimolar mixture of activated penetratin solution (MP Bio). The mixture was heated to 65°C for 15 min and then incubated at 37°C for 1 h.
Plasmids –
All fluorescent reporter constructs for analyses of RNA translation were based on eGFP with myristoylation element (GFPMYR; originally provided by Dr. Erin Schuman, Max-Plank Inst., Frankfurt) [58] or mCherry plasmid with myristoylation element (mChMYR) [34]. Reporter constructs containing 5′ and 3′UTRs of rat Kpnb1 mRNAs with GFPMYR have been published [13]. For generation of mChMYR5′/3′kpnb1 construct, GFPMYR was replaced with mCherryMYR (mChMYR) using BamHI plus NotI sites.
For generation of GFPMYR5′camk2α/3Csnk2a1 construct, 3′ UTR of Csnk2a1 (XM_006235220.3) was PCR amplified from rat genomic DNA using gene specific primers with terminal NotI and XhoI sites. PCR amplified products were digested with NotI plus XhoI and cloned into the same sites of GFPMYR5′camk2α/3′nrn1 [59] construct, replacing the Nrn1 mRNA 3′UTR. The 5′UTR of Csnk2a1 mRNA was custom synthesized by IDT, Inc. and cloned into NheI and BamHI sites of GFPMYR5′camk2α/3′Csnk2a1 to generate the GFPMYR5′/3′Csnk2a1 construct. 5′ UTR of GFPMYR5′camk2α/3′kpnb1–1-134 [8] was replaced with 5′ UTR of Csnk2a1 mRNA to generate GFPMYR5′Csnk2a1/3′kpnb1–1-134. For generation of mCHMYR5′/3′mtor construct, 5′ and 3′UTRs of mouse mTor mRNA (NM_020009.2) were used to replace the Kpnb1 mRNA 5′ and 5′UTRs in mCHMYR5′/3′kpnb1 construct using NheI/BamHI and NotI/XhoI sites, respectively. The mChMYR5′/3′calr construct has been published [34]. The G3BP1-WT, G3BP1S149A and G3BP1S149E clones were generously provided by Prof. Jamal Tazi (Institut de Génétique Moléculaire de Montpellier, France). Like Anderson’s group [35], we also detected NTF2 domain mutations consisting of A54T and S99P in G3BP1S149A and G3BP1S149E, respectively (hereafter referred to as G3BP1A54T/S149A and G3BP1S99P/S149A; G3BP1 plasmid was found to be free of any mutation). To correct these domain mutations in the G3BP1A54T/S149A and G3BP1S99P/S149A, the NTF2 domain of each was replaced with that from G3BP1 using BglII and EcoRV sites. Human CK2α-HA construct was from Addgene (plasmid # 27086; RRID:Addgene_27086; originally generated by David Litchfield). All the clones used for transfections were validated by bidirectional sequencing.
Analysis of RNA-Seq data –
Raw FASTQ files of GSE51572, and GSE6623′ were downloaded from NCBI GEO database and adaptor sequences were trimmed as necessary [22, 23]. Unpublished axonal RNA-seq data from adult C57BL/6 mouse DRGs were similarly analyzed using a standard analysis pipeline (https://github.com/icnn/RNAseq-PIPELINE.git). Briefly, reads were then aligned to mm10 genome using STAR with default parameters. Mapped reads for mRNAs were counted by HTSeq and then normalized by trimmed mean of M values using EdgeR. Normalized counts were used to quantify levels of Csnk2a1 and Csnk2a2 mRNA levels in axons or cell bodies.
Immunofluorescent staining –
All procedures were performed at room temperature (RT) unless specified otherwise. Cultured neurons were fixed in 4% paraformaldehyde (PFA) in PBS and processed as described [59]. Primary antibodies consisted of: RT97 mouse anti-neurofilament (NF; 1:500, Devel. Studies Hybridoma Bank), mouse anti-CK2α (1:200, Abcam), and rabbit anti-HA (1:500, Abcam). FITC-conjugated donkey anti-rabbit and Cy3-conjugated donkey anti-mouse (both at 1:200, Jackson ImmunoRes.) were used as secondary antibodies.
For effect of CK2α knockdown on G3BP1 granularity and G3BP1PS149 level, Zenon antibody-labeling kit (Life Technologies) was used to directly label antibodies with fluorophores. To label G3BP1 granules, rabbit anti-G3BP1 (Sigma) + Alexa 488, mouse anti-CK2α (Abcam) or rabbit anti-HA (Abcam) + Alexa-555, and mouse anti-neurofilament (DSHB) + Alexa-633 antibodies were used. To detect G3BP1PS149 levels, rabbit anti-G3BP1 (Sigma) + Alexa 488, rabbit anti-G3BP1PS149 (Sigma) + Alexa-405, rabbit anti-CK2α (Abcam) + Alexa-555, and mouse anti-neurofilament (DSHB) + Alexa-633 antibodies were used. All the primary antibodies were used at 1:50 dilution. Equal amounts of rabbit-IgG or mouse-IgG labeled with Alexa-405, −488, −555 and −633 were used as control.
For quantifying axonal content of G3BP1, TIA1, and CK2α in peripheral nerve, sciatic nerve segments were fixed for 4 h in 4% PFA and then cryoprotected overnight in 30% sucrose, PBS at 4°C. 10 μm cryostat sections were processed for immunostaining as previously described [59]. Primary antibodies consisted of rabbit anti-G3BP1 (1:100, Sigma), goat anti-TIA1 (1:75, Santacruz), mouse anti-CK2α (1:100, Abcam) and RT97 mouse anti-NF (1:300). Secondary antibodies were FITC-conjugated donkey anti-goat, Cy3-conjugated donkey anti-rabbit, Cy3-conjugated donkey anti-rabbit, and Cy5-conjugated donkey anti-mouse (all at 1:200, Jackson ImmunoRes.).
All samples were mounted with Prolong Gold Antifade (Invitrogen) and analyzed by epifluorescent or confocal microscopy. Leica DMI6000 epifluorescent microscope with ORCA Flash ER CCD camera (Hamamatsu), Leica SP8X confocal microscope, or Image-Express Micro XLS high content imaging system (Molecular Dynamics) were used for imaging. For quantitation between samples, imaging parameters were matched for exposure, gain, offset and post-processing.
Fluorescence in situ hybridization (FISH) –
For FISH, DRG cultures were fixed for 15 min in 2% PFA in PBS and then processed for FISH as described [13]. RNA-FISH was performed using custom 5′ Cy3-labeled ‘Stellaris’ probes against rat Csnk2a1 mRNA (probe sequences available upon request; Bio-Search Tech). Scrambled probes were used as control for specificity; samples processed without addition of primary antibody were used as control for antibody specificity. Primary antibody consisted of RT97 mouse anti-NF (1:200). FITC-conjugated donkey anti-mouse (1:200) was used as secondary. Samples were mounted as above and analyzed using a Leica DMI6000 epifluorescent microscope with ORCA Flash ER CCD camera (Hamamatsu).
For the detection of Csnk2a1 mRNA in tissue, rat sciatic nerve segments were fixed for 4 h in 2% PFA and then cryoprotected overnight in in 30% buffered sucrose at 4°C. 10 μm thick sections were processed for FISH; slides were stored at −20°C until used. Slides were brought to room temperature and all steps were performed at room temperature unless indicated otherwise. FISH was performed as described [8], using the probes from above with minor modifications. Briefly, warmed tissue sections were washed three times in PBS (5 min each), followed by three times in 20 mM Glycine (10 min each), and 0.25 M NaBH4 three times (5 min each). Samples were rinsed with 0.1 M triethanolamine (TEA) followed by 0.1M TEA plus 0.25% acetic anhydride (10 min) and then dehydrated in 70, 95, and 100% ethanol (3 min each). Dehydrated tissues were delipidated in chloroform (5 min) and rehydrated through 100 and 95% ethanol (3 min each) followed by washing in 2X SSC. Sections were the incubated overnight at 37°C in a humidified chamber with Stellaris probes (250 nM) and RT97 anti-NF (1:200) in hybridization buffer [27]. Hybridization washes consisted of 2X SSC + 10% formamide at 37°C (30 min), followed by twice in 2X SSC (5 min each). Sections were then equilibrated in PBS + 1% Triton X-100 (5 min) and incubated in donkey anti-mouse FITC antibody (1:200) diluted in 0.3% Triton X-100 + 10X blocking buffer (1:100; Roche) for 1 h. After 5 min PBS wash, post-fixing in 2% PFA for 15 min [60], and 5 min PBS wash followed by rince in DEPC-treated water, samples were mounted with Prolong Gold Antifade (Invitrogen).
To detect if the Csnk2a1 mRNA 5′ or 3′UTR have axonal localizing activity, DRG neurons were transfected with construct expressing GFPMYR reporter constructs containing various 5′ and 3′UTRs (see Figure 4B) using Amaxa Nucleofector (Lonza). 36 h post transfection FISH for Gfp mRNA was performed as described using digoxigenin labeled oligonucleotide probes [8]. Primary antibodies were mouse anti-DIG (Jackson ImmunoRes., 1:200) and Chicken anti-NF (1:500; Aves). Secondary antibodies were Cy5-conjugated donkey anti-mouse and Cy3-conjugated donkey anti-chicken (both at 1:200, Jackson ImmunoRes.).
For imaging FISH signals, scrambled probes were used to assign image acquisition parameters that would not acquire or would minimize any nonspecific signal from the scrambled probe in each experiment. FISH/IF for culture preparations was analyzed by epifluorescent microscropy using a Leica DMI6000 epifluorescent microscope with ORCA Flash ER CCD camera (Hamamatsu). NIH ImageJ was used to determine mRNA levels in axons and cell bodies. To quantify axonal mRNA levels in cultured neurons, an ROI of 150 μm was assessed that was separated from the cell body by at least 200 μm. For cell body GFP mRNA level, soma was traced from DIC images and signal intensity was calculated. FISH/IF on tissue sections was imaged by confocal microscopy using a Leica SP8X confocal microscope with HyD detectors and post-processing measures to distinguish axonal signals from non-neuronal signals. For each sample, a 144 × 144 μm nerve segment was scanned by taking xyz image stacks with 63x oil-immersion objective (1.4 NA). This xyz tile scan sequence was captured at two locations along each nerve section. The Colocalization Plug-in (https://imagej.nih.gov/ij/plugins/colocalization.html) for NIH ImageJ was used to extract RNA signals from FISH probes in each optical plane overlapping with axonal markers (NF) in the xyz tile [8]. All FISH signal quantifications for axonal mRNA signals from tissue culture sections were generated by analyses of pixel intensity across each xy plane of the extracted ‘axon only’ channels for the image sequences using ImageJ. These FISH signal intensities across the individual xy planes were then normalized to the area of NF immunoreactivity in each XY plane and averaged across the image Z stack of the tile images [6]. The relative mRNA signal intensity was averaged for all tiles in each biological replicate.
Fluorescence recovery after photobleaching (FRAP) –
FRAP was used to test for axonal mRNA protein synthesis using diffusion-limited GFPMYR and mCherryMYR reporters as described with minor modifications [13]. In each case, DRG neurons were transfected with GFPMYR or GFPMYR plus mChMYR reporter constructs. Cells were maintained at 37°C, 5% CO2 during imaging sequences. 488 nm and 514 nm laser lines on Leica SP8X confocal microscope were used to bleach GFP and mCh signals, respectively (Argon laser at 70% power, pulsed every 0.82 s for 80 frames). Pinhole was set to 3 Airy units to ensure full thickness bleaching and acquisition (63X/1.4 NA oil immersion objective) [11]. Prior to photobleaching, neurons were imaged every 60 s for 2 min to acquire baseline fluorescence the region of interest (ROI; 15% laser power, 498–530 nm for GFP and 565–597 nm for mCh emissions, respectively). The same excitation and emission parameters were used to assess recovery over 15 min post-bleach with images acquired at 30 s intervals. Cultures were preteated with a neutrophin cocktail (10 ng/ml each of NGF [Harlan Labs], BDNF [Alomone Labs], and NT3 [Alomone Labs]) for 3 h to test for trophic effects on fluorescence recovery.To determine if fluorescence recovery in axons was from translation, cultures were treated with 100 μm anisomycin (Sigma) for 30 min prior to photobleaching. Cultures were 1 μM Thapsigargin (Sigma), 3 μM BAPTA-AM (Sigma), or 50 μM EGTA (Sigma) for 1 h to test for effects of Ca2+ in FRAP experiments.
Fluorescent intensities in the ROIs were calculated by the Leica LASX software. For normalizing across experiments, fluorescence intensity value at t = 0 min post-bleach from each image sequence was set as 0%. The percentage of fluorescence recovery at each time point after photobleaching was then calculated by normalizing relative to the pre-bleach fluorescence intensity (set at 100%) [11].
Puromycin labeling –
Rat sciatic nerves from two animals per condition were left naive or 50% of the nerve was crushed and incubated in medium (DMEM + 10% FBS + CyclosporinA (20 μM) + 1% penicillin/streptomycin). 36 h later nerves were treated with 200 μg/ml anisomycin or vehicle (0.1% DMSO) for 2 h at 37°C, followed by adding 100 μg/ml O-propargyl-puromycin (OPP) for 2 h at 37°C. Axoplasm was extracted in 1 mL of modified transport buffer (20 mM HEPES, 100 mM NaAc, 5 mM MgAc, pH 7.4), after extraction SDS added to 1%. Protein concentration was measured by BCA assay and 300 μg of total protein was processed for biotin conjugation by Click-It chemistry (100 μM biotin-PEG3-azide) according to manufacturer’s instruction (Life Tech) by incubating the reaction mix for 2 h at room temperature on a rotator. Proteins were precipitated using 5 volumes ice-cold acetone. Protein pellets were resuspended in PBS containing 1% SDS. 1/20th of protein used for pulldown was reserved as input. Biotinylated proteins were isolated by overnight 4°C incubation with in 1 mL in PBS containing 60 μL of streptavidin magnetic beads (Invitrogen), 1% NP40, 0.1% SDS, and 1x protease inhibitor cocktail (Complete EDTA-free, Roche). Beads were washed three times (10 min each) with PBS plus 1% NP40, 0.1% SDS at room temperature. Proteins were eluted by boiling for 10 min in 2x Laemmli sample buffer (LSB); LSB concentration was adjusted to 1x with PBS and precipitated proteins plus input samples were fractionated by SDS/PAGE.
Immunoblotting –
For immunoblotting, gels were transferred to nitrocellulose membranes, blocked for 1 h at room temperature with 5% BSA in Tris-buffered saline with 0.1% Tween 20 (TBST) for anti-G3BP1PS149 and Streptavidin-HRP or with 5% milk in TBST for anti-CK2α, anti-G3BP1, anti-HA, and anti-GAPDH antibodies. Primary antibodies diluted in appropriate blocking buffer and incubated on membranes overnight incubation at 4°C with rocking. Primary antibodies consisted of: mouse anti-CK2α (1:1,000; Abcam), rabbit anti-G3BP1 (Sigma, 1:2,000), rabbit anti-G3BP1PS149 (Sigma, 1:1,000), rabbit anti-HA (1:5,000; Abcam), and rabbit anti-GAPDH (1:5,000; Cell Signaling Tech). After washing in TBST, blots were incubated HRP-conjugated anti-rabbit IgG antibodies (1:5,000; Jackson ImmunoRes) diluted in blocking buffer for 1 h at room temperature. For Biotinylated proteins Streptavidin-HRP (Abcam, 1:10,000) was used. After liberally washing in TBST, immunocomplexes were detected using ECL PrimeTM (GE Healthcare).
RNA immunoprecipitation (RIP) –
DRG neurons were lysed in 100 mM KCl, 5 mM MgCl2, 10 mM HEPES [pH 7.4], 1 mM DTT, and 0.5% NP-40 (RIP buffer) supplemented with 1 × protease inhibitor cocktail (Roche) and RNasin Plus (Invitrogen). Cells were passed through 25 Ga needle 5–7 times and cleared by centrifugation at 12,000 x g, 4°C for 20 min. Cleared lysates were pre-absorbed with Protein A-Dynabeads (Invitrogen) for 3′ min. Supernatants were then incubated with rabbit anti-GFP antibody (5 μg, Abcam) for 3 h and then immunocomplexes precipitated with Protein G-Dynabeads (Invitrogen) for additional 2 h at 4°C with rotation. Beads were washed six times with cold RIP buffer and then processed for isolation of RNA as below.
RNA isolation and PCR analyses –
RNA was isolated from dissociated DRG neurons, cell body/neurite fractionations, and RIP assays using RNeasy microisolation kit (QIAGEN). Fluorimetry with Ribogreen (Life Technologies) was used for RNA quantification in the DRG and cell body/neurite preparations; 10–5′ ng of RNA were used for reverse transcription (RT) with SensiFAST cDNA synthesis kit (Bioline) according to the manufacturer’s protocol. For RIP assays, an equal proportion of each RIP was used for RT with Sensifast. To assess the purity of axona RNA, RT-PCR was performed with primers designed to detect cell body-restricted (cJun and Map2) and glial cell-specific mRNAs (Gfap). ddPCR was performed using custom transcript-specific primer sets (IDT; sequences available on request) and detected with Evagreen using a QX200™ droplet reader (Biorad).
Image analyses and processing –
For analyses of protein and RNA levels in tissues, z planes of the xyz scans from 2 locations along each nerve section were analyzed using ImageJ. Colocalization Plug-in was used to extract protein or RNA signals that overlap with axonal marker (NF) in each plane, with the extracted ‘axon-only’ signal projected as a separate channel [6]. For calculating axonal protein or mRNA signal intensities, absolute signal intensity was quantified in each xy plane of the ‘Colocalization’ extracted images for axonal only protein and mRNA using ImageJ. Protein or mRNA signal intensities across the individual xy planes were then normalized to NF immunoreactivity area. The relative protein or mRNA signal intensity was averaged for all image locations in each biological replicate.
For quantitation of G3BP1 granules in in vitro culture DRG neurons, a 100 μm of the axon shaft was considered (≥ 200 μm from cell body). In both in vitro culture DRG neuron axons, and sciatic nerve axons in vivo, confocal imaging was used to image the G3BP1 granules. Exposure parameters were set to only capture the granular G3BP1 pool. Further ImageJ particle analyzer was used for analysis. G3BP1 granules with area ≥ 1 μm2 were considered as SG-like structures.
For neurite outgrowth, images were analyzed for neurite length and branching using WIS-Neuromath [61] with NF immunostained cultures.
To assess regeneration in vivo, tile scans of NF-stained nerve sections were post-processed by ImageJ Straighten Plug-in (http://imagej.nih.gov/ij/). NF positive axon profiles (> 30 μm long) were then counted in 30 μm bins at 0.25 mm intervals proceeding distally from the crush site. Crush site was identified by DAPI staining and DIC imaging. Axon profiles present in the proximal crush site was treated as the baseline, and values from the distal bins were normalized to this to calculate the percentage of regenerating axon profiles.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses –
Prism (GraphPad) and Excel (Microsoft) software packages were used for statistical analyses. One-way ANOVA or Two-way ANOVA was used to compare means of > 2 independent groups and Student’s t test was used to compare between 2 groups. p values of ≤ 0.05 were considered as statistically significant.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| RT97 mouse anti-neurofilament | Devel. Studies Hybridoma Bank | Cat # RT97; RRID: AB_528399 |
| mouse anti-CK2α | Abcam | Cat # ab70774; RRID: AB_1280818 |
| rabbit anti-HA | Abcam | Cat # ab9110; RRID: AB_307019 |
| rabbit anti-G3BP1 | Sigma | Cat # G6046; RRID: AB_1840864 |
| rabbit anti-G3BP1PS149 | Sigma | Cat # G8046; RRID: AB_1840867 |
| goat anti-TIA1 | Santacruz | Cat # sc-1751; RRID: AB_2201433 |
| mouse anti-Digoxigenin | Jackson ImmunoRes | Cat # 200-002-156; RRID: AB_2339005 |
| rabbit anti-GAPDH | Cell Sig. Tech | Cat # 2118S; RRID: AB_561053 |
| Chicken anti-Neurofilament (heavy) | Aves | Cat # NFH; RRID: AB_2313552 |
| Chicken anti-Neurofilament (medium) | Aves | Cat # NFM; RRID: AB_2313554 |
| Chicken anti-Neurofilament (light) | Aves | Cat # NFL; RRID: AB_2313553 |
| FITC-conjugated donkey anti-rabbit | Jackson ImmunoRes | Cat# 711-095-152; RRID: AB_2315776 |
| Cy3-conjugated donkey anti-mouse | Jackson ImmunoRes | Cat# 715-165-151; RRID: AB_2315777 |
| Cy3-conjugated donkey anti-goat | Jackson ImmunoRes | Cat# 705-165-147; RRID: AB_2307351 |
| Cy3-conjugated donkey anti-rabbit | Jackson ImmunoRes | Cat# 711-165-152; RRID: AB_2307443 |
| Cy5-conjugated donkey anti-mouse | Jackson ImmunoRes | Cat# 715-175-151; RRID: AB_2619678 |
| FITC-conjugated donkey anti-mouse | Jackson ImmunoRes | Cat# 715-095-151; RRID: AB_2335588 |
| Cy3-conjugated donkey anti-chicken | Jackson ImmunoRes | Cat # 703-166-155; RRID: AB_2340364 |
| Rabbit-IgG | Sigma | Cat# I5506; RRID: AB_1163593 |
| Mouse-IgG | Sigma | Cat# I5381; RRID: AB_1163670 |
| Bacterial and Virus Strains | ||
| N/A | N/A | N/A |
| Biological Samples | ||
| DRGs and Sciatic Nerve isolated from SD rats and | N/A | N/A |
| WT/mTOR−/− mouse | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| CK2 inhibitor (CX-4945) | Selleckchem | S2248 |
| Thapsigargin | Sigma | SML 1845 |
| BAPTA-AM | Sigma | A1076 |
| EGTA | Sigma | E3889 |
| Anisomycin | Sigma | A5862 |
| BDNF | Alomone Labs | B-250 |
| NGF | Harlan Labs | 5017 |
| NT3 | Alomone Labs | N-260 |
| CyclosporinA | TCI Chemicals | C2408 |
| Torin | Selleckchem | A8312 |
| O-propargyl-puromycin | Thermo Fisher Sc. | C10459 |
| Biotin Azide | Thermo Fisher Sc. | B10184 |
| Critical Commercial Assays | ||
| Click-IT Biotin Protein Analysis Detection Kit | Life Technologies | Cat# C33372 |
| Activated Penetratin | MP Bio | Cat# PENA0500 |
| Experimental Models: Cell Lines | ||
| NIH/3T3 | ATCC | ATCC-CRL-1658 |
| Experimental Models: Organisms/Strains | ||
| Rats | Sprague Dawley strain | N/A |
| WT, mTOR−/− mouse | ICR strain | N/A |
| Oligonucleotides | ||
| G3BP1 On-target plus-SMART pool siRNA | Dharmacon | Cat no. L-101659-02-0005 |
| CK2α On-target plus-SMART pool siRNA | Dharmacon | Cat no. L-096197-02-0005 |
| CK2α’ On-target plus-SMART pool siRNA | Dharmacon | Cat no. L-092756-02-0005 |
| Stellaris probes against rat csnk2a1 | Biosearch Tech | Cat# SS442599-01-38 |
| Probes for gfp mRNA FISH | IDT | [52] |
| siControl for penetratin conjugation | Dharmacon | Sense: 5’ S-S.ggucucacguccuaauaaauu 3’ |
| Antisense: 5’ Fl.uuuauuaggacgugagaccuu 3’ | ||
| siCsnk2a1/2 for penetratin conjugation | Dharmacon | Sense: 5’ S-S.cuaccagcuuguucgaaaauu 3’ |
| Antisense: 5’ Fl.uuuucgaacaagcugguaguu 3’ | ||
| Recombinant DNA | ||
| mChMYR5′/3′kpnb1 | This paper | N/A |
| GFPMYR5′camk2a/3′ csnk2a1 | This paper | N/A |
| GFPMYR5′/3′csnk2a1 | This paper | N/A |
| GFPMYR5′camk2a/3′ kpnb1-1-134 | [8] | N/A |
| mCHMYR5′/3′mtor | This paper | N/A |
| mCHMYR5′/3′calr | [34] | N/A |
| G3BP1S149A-GFP | This paper | N/A |
| G3BP1S149E-GFP | This paper | N/A |
| Human CK2α-HA | http://www.addgene.org/27086/ | [53] |
| Software and Algorithms | ||
| Graphpad Prism | https://www.graphpad.com | N/A |
| ImageJ | https://imagej.nih.gov/ij/ | N/A |
| ImageJ Colocalization Plugin | https://imagej.nih.gov/ij/plugins/colocalization.html | N/A |
Highlights.
Axonal CK2α phosphorylates G3BP1 to support axon regeneration
Axonal translation of CK2α mRNA after injury disassembles G3BP1 granules
CK2α translation requires initial translation of axonal mTOR mRNA
Specificity of axonal mRNA translation is driven by axoplasmic Ca2+ dynamics
ACKNOWLEDGMENTS
This work was supported by grants from NIH (R01-NS117821 and R01-NS089633 to J.L.T.), the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (to G.C., M.F., and J.L.T.), the South Carolina Spinal Cord Injury Research Fund (2019-PD-02 and 2018-PD-01 to P.K.S.), Wings for Life Foundation (WFL-US-09/18 to P.P.), and Magellan Fellowship and ASPIRE Award from the University of South Carolina Office of Research (to B.J. and A.N.K., respectively). Any opinions, findings, and conclusions or recommendations expressed are those of the author(s) and do not necessarily reflect those of the funding agencies. M.F. is the incumbent of the Chaya Professorial Chair in Molecular Neuroscience at the Weizmann Institute of Science. J.L.T. is the incumbent SmartState Chair in Childhood Neurotherapeutics at the University of South Carolina.
Footnotes
DECLARATIONS OF INTERESTS
The authors declare no competing interests. P.K.S. and J.L.T. hold US patent no. 10,668,128 for inhibition of G3BP1 function to accelerate nerve regeneration.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.cub.2020.09.043.
REFERENCES
- 1.Terenzio M, Schiavo G, and Fainzilber M. (2017). Compartmentalized signaling in neurons: from cell biology to neuroscience. Neuron 96, 667–679. [DOI] [PubMed] [Google Scholar]
- 2.Sahoo PK, Smith DS, Perrone-Bizzozero N, and Twiss JL (2018). Axonal mRNA transport and translation at a glance. J. Cell Sci 131, jcs196808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kar AN, Lee SJ, and Twiss JL (2018). Expanding axonal transcriptome brings new functions for axonally synthesized proteins in health and disease. Neuroscientist 24, 111–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hafner AS, Donlin-Asp PG, Leitch B, Herzog E, and Schuman EM (2019). Local protein synthesis is a ubiquitous feature of neuronal pre- and postsynaptic compartments. Science 364, eaau3644. [DOI] [PubMed] [Google Scholar]
- 5.Shigeoka T, Jung H, Jung J, Turner-Bridger B, Ohk J, Lin JQ, Amieux PS, and Holt CE (2016). Dynamic axonal translation in developing and mature visual circuits. Cell 166, 181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kalinski AL, Sachdeva R, Gomes C, Lee SJ, Shah Z, Houle JD, and Twiss JL (2015). mRNAs and protein synthetic machinery localize into regenerating spinal cord axons when they are provided a substrate that supports growth. J. Neurosci 35, 10357–10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perry RB, Doron-Mandel E, Iavnilovitch E, Rishal I, Dagan SY, Tsoory M, Coppola G, McDonald MK, Gomes C, Geschwind DH, et al. (2012). Subcellular knockout of importin β1 perturbs axonal retrograde signaling. Neuron 75, 294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Terenzio M, Koley S, Samra N, Rishal I, Zhao Q, Sahoo PK, Urisman A, Marvaldi L, Oses-Prieto JA, Forester C, et al. (2018). Locally translated mTOR controls axonal local translation in nerve injury. Science 359, 1416–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanz S, Perlson E, Willis D, Zheng JQ, Massarwa R, Huerta JJ, Koltzenburg M, Kohler M, van-Minnen J, Twiss JL, and Fainzilber M. (2003). Axoplasmic importins enable retrograde injury signaling in lesioned nerve. Neuron 40, 1095–1104. [DOI] [PubMed] [Google Scholar]
- 10.Perlson E, Hanz S, Ben-Yaakov K, Segal-Ruder Y, Seger R, and Fainzilber M. (2005). Vimentin-dependent spatial translocation of an activated MAP kinase in injured nerve. Neuron 45, 715–726. [DOI] [PubMed] [Google Scholar]
- 11.Yudin D, Hanz S, Yoo S, Iavnilovitch E, Willis D, Gradus T, Vuppalanchi D, Segal-Ruder Y, Ben-Yaakov K, Hieda M, et al. (2008). Localized regulation of axonal RanGTPase controls retrograde injury signaling in peripheral nerve. Neuron 59, 241–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anderson P, and Kedersha N. (2006). RNA granules. J. Cell Biol 172, 803–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sahoo PK, Lee SJ, Jaiswal PB, Alber S, Kar AN, Miller-Randolph S, Taylor EE, Smith T, Singh B, Ho TS, et al. (2018). Axonal G3BP1 stress granule protein limits axonal mRNA translation and nerve regeneration. Nat. Commun 9, 3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andrusiak MG, Sharifnia P, Lyu X, Wang Z, Dickey AM, Wu Z, Chisholm AD, and Jin Y. (2019). Inhibition of axon regeneration by liquid-like TIAR-2 granules. Neuron 104, 290–304.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang P, Mathieu C, Kolaitis R-M, Zhang P, Messing J, Yurtsever U, Yang Z, Wu J, Li Y, Pan Q, et al. (2020). G3BP1 is a tunable switch that triggers phase separation to assemble stress granules. Cell 181, 325–345.e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guillen-Boixet J, Kopach A, Holehouse AS, Wittmann S, Jahnel M, Schlüßler R, Kim K, Trussina IREA, Wang J, Mateju D, et al. (2020). RNA-induced conformational switching and clustering of G3BP drive stress granule assembly by condensation. Cell 181, 346–361.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reineke LC, Tsai WC, Jain A, Kaelber JT, Jung SY, and Lloyd RE (2017). Casein kinase 2 is linked to stress granule dynamics through phosphorylation of the stress granule nucleating protein G3BP1. Mol. Cell. Biol 37, e00596–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kwok HH, Poon PY, Mak KH, Zhang LY, Liu P, Zhang H, Mak NK, Yue PY, and Wong RN (2017). Role of G3BP1 in glucocorticoid receptor-mediated microRNA-15b and microRNA-23a biogenesis in endothelial cells. Cell. Mol. Life Sci 74, 3613–3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pinna LA (2003). The raison d’être of constitutively active protein kinases: the lesson of CK2. Acc. Chem. Res 36, 378–384. [DOI] [PubMed] [Google Scholar]
- 20.Montenarh M, and Götz C. (2018). Ecto-protein kinase CK2, the neglected form of CK2. Biomed. Rep 8, 307–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Litchfield DW (2003). Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem. J 369, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Minis A, Dahary D, Manor O, Leshkowitz D, Pilpel Y, and Yaron A. (2014). Subcellular transcriptomics-dissection of the mRNA composition in the axonal compartment of sensory neurons. Dev. Neurobiol 74, 365–381. [DOI] [PubMed] [Google Scholar]
- 23.Briese M, Saal L, Appenzeller S, Moradi M, Baluapuri A, and Sendtner M. (2016). Whole transcriptome profiling reveals the RNA content of motor axons. Nucleic Acids Res. 44, e33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Michaelevski I, Segal-Ruder Y, Rozenbaum M, Medzihradszky KF, Shalem O, Coppola G, Horn-Saban S, Ben-Yaakov K, Dagan SY, Rishal I, et al. (2010). Signaling to transcription networks in the neuronal retrograde injury response. Sci. Signal 3, ra53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Villegas R, Martinez NW, Lillo J, Pihan P, Hernandez D, Twiss JL, and Court FA (2014). Calcium release from intra-axonal endoplasmic reticulum leads to axon degeneration through mitochondrial dysfunction. J. Neurosci 34, 7179–7189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koppel I, and Fainzilber M. (2018). Omics approaches for subcellular translation studies. Mol. Omics 14, 380–388. [DOI] [PubMed] [Google Scholar]
- 27.Perry RB, Rishal I, Doron-Mandel E, Kalinski AL, Medzihradszky KF, Terenzio M, Alber S, Koley S, Lin A, Rozenbaum M, et al. (2016). Nucleolin-mediated RNA localization regulates neuron growth and cycling cell size. Cell Rep. 16, 1664–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heumann R, Lindholm D, Bandtlow C, Meyer M, Radeke MJ, Misko TP, Shooter E, and Thoenen H. (1987). Differential regulation of mRNA encoding nerve growth factor and its receptor in rat sciatic nerve during development, degeneration, and regeneration: role of macrophages. Proc. Natl. Acad. Sci. USA 84, 8735–8739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cho Y, Sloutsky R, Naegle KM, and Cavalli V. (2013). Injury-induced HDAC5 nuclear export is essential for axon regeneration. Cell 155, 894–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ziv NE, and Spira ME (1997). Localized and transient elevations of intracellular Ca2+ induce the dedifferentiation of axonal segments into growth cones. J. Neurosci 17, 3568–3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ben-Yaakov K, Dagan SY, Segal-Ruder Y, Shalem O, Vuppalanchi D, Willis DE, Yudin D, Rishal I, Rother F, Bader M, et al. (2012). Axonal transcription factors signal retrogradely in lesioned peripheral nerve. EMBO J. 31, 1350–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gitler D, and Spira ME (1998). Real time imaging of calcium-induced localized proteolytic activity after axotomy and its relation to growth cone formation. Neuron 20, 1123–1135. [DOI] [PubMed] [Google Scholar]
- 33.Laitusis AL, Brostrom CO, Ryazanov AG, and Brostrom MA (1998). An examination of the role of increased cytosolic free Ca2+ concentrations in the inhibition of mRNA translation. Arch. Biochem. Biophys 354, 270–280. [DOI] [PubMed] [Google Scholar]
- 34.Vuppalanchi D, Merianda TT, Donnelly C, Pacheco A, Williams G, Yoo S, Ratan RR, Willis DE, and Twiss JL (2012). Lysophosphatidic acid differentially regulates axonal mRNA translation through 5’UTR elements. Mol. Cell. Neurosci 50, 136–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Panas MD, Kedersha N, Schulte T, Branca RM, Ivanov P, and Anderson P. (2019). Phosphorylation of G3BP1-S149 does not influence stress granule assembly. J. Cell Biol 218, 2425–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davidson TJ, Harel S, Arboleda VA, Prunell GF, Shelanski ML, Greene LA, and Troy CM (2004). Highly efficient small interfering RNA delivery to primary mammalian neurons induces MicroRNA-like effects before mRNA degradation. J. Neurosci 24, 10040–10046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Höke A. (2006). Mechanisms of Disease: what factors limit the success of peripheral nerve regeneration in humans? Nat. Clin. Pract. Neurol 2, 448–454. [DOI] [PubMed] [Google Scholar]
- 38.Brostrom CO, and Brostrom MA (1990). Calcium-dependent regulation of protein synthesis in intact mammalian cells. Annu. Rev. Physiol 52, 577–590. [DOI] [PubMed] [Google Scholar]
- 39.Yudin D, and Fainzilber M. (2009). Ran on tracks–cytoplasmic roles for a nuclear regulator. J. Cell Sci 122, 587–593. [DOI] [PubMed] [Google Scholar]
- 40.Merianda T, and Twiss J. (2013). Peripheral nerve axons contain machinery for co-translational secretion of axonally-generated proteins. Neurosci. Bull 29, 493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Merianda TT, Lin AC, Lam JS, Vuppalanchi D, Willis DE, Karin N, Holt CE, and Twiss JL (2009). A functional equivalent of endoplasmic reticulum and Golgi in axons for secretion of locally synthesized proteins. Mol. Cell. Neurosci 40, 128–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.González C, Cánovas J, Fresno J, Couve E, Court FA, and Couve A. (2016). Axons provide the secretory machinery for trafficking of voltagegated sodium channels in peripheral nerve. Proc. Natl. Acad. Sci. USA 113, 1823–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brostrom MA, and Brostrom CO (2003). Calcium dynamics and endoplasmic reticular function in the regulation of protein synthesis: implications for cell growth and adaptability. Cell Calcium 34, 345–363. [DOI] [PubMed] [Google Scholar]
- 44.Oñate M, Catenaccio A, Martínez G, Armentano D, Parsons G, Kerr B, Hetz C, and Court FA (2016). Activation of the unfolded protein response promotes axonal regeneration after peripheral nerve injury. Sci. Rep 6, 21709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ying Z, Zhai R, McLean NA, Johnston JM, Misra V, and Verge VM (2015). The unfolded protein response and cholesterol biosynthesis link Luman/CREB3 to regenerative axon growth in sensory neurons. J. Neurosci 35, 14557–14570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Verma P, Chierzi S, Codd AM, Campbell DS, Meyer RL, Holt CE, and Fawcett JW (2005). Axonal protein synthesis and degradation are necessary for efficient growth cone regeneration. J. Neurosci 25, 331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pacheco A, Merianda TT, Twiss JL, and Gallo G. (2020). Mechanism and role of the intra-axonal Calreticulin translation in response to axonal injury. Exp. Neurol 323, 113′72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tourrière H, Chebli K, Zekri L, Courselaud B, Blanchard JM, Bertrand E, and Tazi J. (2003). The RasGAP-associated endoribonuclease G3BP assembles stress granules. J. Cell Biol 160, 823–831. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 49.Liao Y-C, Fernandopulle MS, Wang G, Choi H, Hao L, Drerup CM, Patel R, Qamar S, Nixon-Abell J, Shen Y, et al. (2019). RNA granules hitchhike on lysosomes for long-distance transport, using annexin A11 as a molecular tether. Cell 179, 147–164.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Detrait ER, Yoo S, Eddleman CS, Fukuda M, Bittner GD, and Fishman HM (2000). Plasmalemmal repair of severed neurites of PC12 cells requires Ca(2+) and synaptotagmin. J. Neurosci. Res 62, 566–573. [DOI] [PubMed] [Google Scholar]
- 51.Gitler D, and Spira ME (2002). Short window of opportunity for calpain induced growth cone formation after axotomy of Aplysia neurons. J. Neurobiol 52, 267–279. [DOI] [PubMed] [Google Scholar]
- 52.Vuppalanchi D, Coleman J, Yoo S, Merianda TT, Yadhati AG, Hossain J, Blesch A, Willis DE, and Twiss JL (2010). Conserved 3′-untranslated region sequences direct subcellular localization of chaperone protein mRNAs in neurons. J. Biol. Chem 285, 18025–18038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Turowec JP, Duncan JS, French AC, Gyenis L, St Denis NA, Vilk G, and Litchfield DW (2010). Protein kinase CK2 is a constitutively active enzyme that promotes cell survival: strategies to identify CK2 substrates and manipulate its activity in mammalian cells. Methods Enzymol. 484, 471–493. [DOI] [PubMed] [Google Scholar]
- 54.Twiss JL, Smith DS, Chang B, and Shooter EM (2000). Translational control of ribosomal protein L4 mRNA is required for rapid neurite regeneration. Neurobiol. Dis 7, 416–428. [DOI] [PubMed] [Google Scholar]
- 55.Cavalli V, Kujala P, Klumperman J, and Goldstein LS (2005). Sunday Driver links axonal transport to damage signaling. J. Cell Biol 168, 775–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zheng JQ, Kelly TK, Chang B, Ryazantsev S, Rajasekaran AK, Martin KC, and Twiss JL (2001). A functional role for intra-axonal protein synthesis during axonal regeneration from adult sensory neurons. J. Neurosci 21, 9291–9303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rivieccio MA, Brochier C, Willis DE, Walker BA, D’Annibale MA, McLaughlin K, Siddiq A, Kozikowski AP, Jaffrey SR, Twiss JL, et al. (2009). HDAC6 is a target for protection and regeneration following injury in the nervous system. Proc. Natl. Acad. Sci. USA 106, 19599–19604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aakalu G, Smith WB, Nguyen N, Jiang C, and Schuman EM (2001). Dynamic visualization of local protein synthesis in hippocampal neurons. Neuron 30, 489–502. [DOI] [PubMed] [Google Scholar]
- 59.Merianda TT, Gomes C, Yoo S, Vuppalanchi D, and Twiss JL (2013). Axonal localization of neuritin/CPG15 mRNA in neuronal populations through distinct 5′ and 3′ UTR elements. J. Neurosci 33, 13735–13742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Spillane M, Ketschek A, Merianda TT, Twiss JL, and Gallo G. (2013). Mitochondria coordinate sites of axon branching through localized intra-axonal protein synthesis. Cell Rep. 5, 1564–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rishal I, Golani O, Rajman M, Costa B, Ben-Yaakov K, Schoenmann Z, Yaron A, Basri R, Fainzilber M, and Galun M. (2013). WIS-NeuroMath enables versatile high throughput analyses of neuronal processes. Dev. Neurobiol 73, 247–256. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw FASTQ files of GSE51572, and GSE66230 were downloaded from NCBI GEO database.