

Abstract
Analyzing every cell in a diverse sample provides insight into population-level heterogeneity, but abundant cell types dominate the analysis and rarer populations are scarcely represented in the data. To focus on specific cell types, the current paradigm is to physically isolate subsets of interest prior to analysis; however, it remains difficult to isolate and then single-cell sequence such populations because of compounding losses. Here, we describe an alternative approach that selectively merges cells with reagents to achieve enzymatic reactions without having to physically isolate cells. We apply this technique to perform single-cell transcriptome and genome sequencing of specific cell subsets. Our method for analyzing heterogeneous populations obviates the need for pre- or post-enrichment and simplifies single-cell workflows, making it useful for other applications in single-cell biology, combinatorial chemical synthesis, and drug screening.
Graphical Abstract
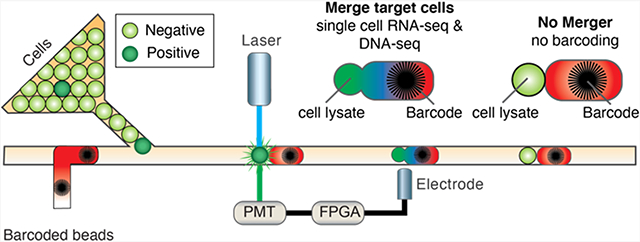
INTRODUCTION
A high-value product of droplet microfluidics has been scalable and cost-effective single-cell sequencing. This approach encapsulates individual cells in droplets with barcodes that uniquely label the genome,1,2 transcriptome3,4 or proteome.5,6 After barcoding, all material can be pooled, efficiently read by DNA sequencing, and separated in silico. Droplet microfluidic barcoding provides the throughput and precision necessary to characterize thousands of single cells and understand trajectories during cellular differentiation,7 heterogeneity in disease,8,9 transcriptional changes associated with genetic perturbations,10,11 and numerous other biological measurements. Indeed, the approach has heralded a new era in systems biology and enabled myriad systems to be decomposed into their most essential component, the single cell.
Existing technologies for high-throughput droplet-based single-cell sequencing analyze all cells in a sample. Often, however, the cells of interest comprise a small subset in a mixed population, necessitating sorting prior to barcoding. In addition to adding cumbersome steps, presorting of rare targets by fluorescence-activated cell sorting may not yield enough cells to run existing single-cell droplet workflows. One solution would be to integrate sorting directly into the barcoding chip, streamlining workflows for the user. However, droplet sorting is one of the most challenging and user-input intensive operations in droplet microfluidics.12-16 This has limited the integration of droplet sorting into commercial instruments that have been designed for simplicity and engineering reliability. In addition, on-chip sorting of cells during encapsulation is not compatible with diverse droplet detection modalities, including the polymerase chain reaction (PCR)-based detection of nucleic acids and assays on secreted molecules,16-19 which require a separate cell encapsulation and incubation step. A new paradigm is needed that allows droplet-based detection methods to be coupled directly to single-cell analysis workflows.
In this paper, we describe a technique to target cell subsets for molecular analysis without the need for physical separation. Our approach exploits the ability to selectively coalesce target droplets with essential reagents. Coalescence, via a droplet merger, combines all components needed to catalyze the desired reaction, yielding products that can be recovered. By contrast, negative droplets are not coalesced, yielding no reaction products. The droplet merger is triggered on only droplets of interest by integrating droplet fluorescence detection, gating, and merger electrode activation. In this way, selective coalescence achieves subset analysis without the need to separate the input or output of the microfluidic device, allowing bulk purification to be performed downstream of the microfluidic steps. We demonstrate the power of the selective merger by applying it to single-cell RNA-sequence and DNA-sequence specific cells from mixed populations. Our method comprises a simple and flexible strategy to integrate subset analysis into droplet-based single-cell workflows.
EXPERIMENTAL SECTION
Microfluidic Fabrication and Device Construction.
Three-inch silicon wafers are spin-coated with SU-8 2025 photoresist (MicroChem, Westborough, MA) and UV-patterned using a mask aligner (SUSS MJB3). Poly-(dimethylsiloxane) (PDMS) prepolymer and curing agent (Momentive, Waterford, NY; RTV 615) are mixed vigorously at a 10:1 ratio, degassed in a vacuum chamber, and poured onto the master mold. The mold is degassed and baked at 65 °C overnight before being removed and punched with a 0.75 mm biopsy punch (Ted Pella, Inc., Redding, CA; Harris Uni-Core 0.75). The PDMS replica and a glass slide (75 mm × 50 mm × 1.0 mm, 12-550C, Fisher Scientific) are plasma-treated (Technics Plasma etcher 500-II) and bonded. The complete device is placed at 150 °C to strengthen the bonds and further baked overnight. The device is treated with Aquapel for 5 min, purged with air, flushed with oil (Fluorinert FC-40), purged with air again, and baked for 30 min before use.
Electronics and Optical System.
Custom optics, electronics, and software are used to selectively merge droplets. Custom software that monitors droplet fluorescence and controls the selective merger is written using LabView. Custom hardware is similar to the previously described systems16,20,21 and is diagrammed in Figure S1A. The optical system consists of two lasers (OptoEngine MLL-FN-473, MRL-III-640) aligned through dichroic mirrors (Semrock). Aligned lasers enter an inverted microscope (Motic AE31) and are focused onto a spot on the microfluidic device using the objective. Fluorescent signals emitted by drops are collected by the objective and diverted through filters to photomultiplier tubes (PMTs) (Thorlabs PMM01 and PMM02). Signals from the PMTs are processed using LabView FPGA software, which detects droplet fluorescence and triggers a high-voltage power amplifier (Trek Model 609E-6) to generate an on-chip dielectrophoretic force via a 2 M NaCl salt-water electrode. Once gates have been set, the FPGA operates without communication with the host computer such that detection, gating, and electrode activation occur before the drop can traverse a drop length in the channel.
Dye-Based Validation of the Selective Merger.
A FAM-labeled oligonucleotide in phosphate-buffered saline (PBS) is used to generate 55 μm droplets using a bubble-triggered droplet generator running 2% ionic Krytox, prepared as previously described.22 Two concentrations (1 and 0.1 μM) of the FAM droplets are produced and mixed. The drops are reinjected and paired with 75 μm droplets generated on-chip that contain BSA-CY5 (1 μM) in PBS. The selective merger of paired droplets is achieved by triggering a salt-water electrode (2 M NaCl) (Figures 2A and 4A) or by triggering direct electrification of the aqueous reagent stream (Figure 3A) in response to fluorescence. Analysis of the droplet fluorescence is performed on the droplet cytometer, as described above. The system contains three lasers (473, 532, 638 nm) for excitation and filter sets to direct fluorescence to three photomultiplier tubes (PMM01, Thorlabs). Droplet fluorescence values are recorded, exported, and analyzed in FlowJo.
Figure 2.

Efficient reagent addition using droplet coalescence. (A) Microfluidic device used for dye-based validation of the selective merger consists of an upstream reagent T-junction drop maker and downstream drop reinjector. Reinjected drops (colored green) and reagent drops (colored red) enter the microfluidic device, are paired due to their size mismatch, and selectively merged based on fluorescence. (B) Microscope images of droplets when the merger is not triggered. (C) Microscope images of droplets when the merger is triggered on the FAM+ drops. (D) Drop size distributions before and after the selective merger. (E) Dropometry plot showing the average fluorescence of drops before the selective merger. (F) Dropometry plot showing the average fluorescence of drops after the selective merger. The percentage of FAM− and FAM+ drops with respect to the total number of small drops is shown to assess the precision and recall of our technique.
Figure 4.

Single-cell DNA seq of cancer-related genomic loci from a leukemia cell subpopulation using the fluorescence-activated droplet merger. (A) Microscope image of the microfluidic device for the selective merger of barcoded beads with cell lysate drops. Left: Operation of the merger device showing bead reinjection, reagent drop formation, and cell lysate drop reinjection. The drops are colored red (negative, CEM) and green (positive, K562) to aid in the figure interpretation. Middle: drop pairing and merger. Right: Higher-magnification images showing bead-drop:cell-drop pairing and drop merger. The PCR mix is colored blue, oil is colored brown, positive cell drops are colored green, negative cell drops are colored red, barcoded beads are colored gray, and electrode is colored red-orange. (B) Bioinformatic analysis of single-cell genome sequencing. The clustering of genomic mutations identifies the cell of origin. The cells are classified accordingly. (C) Distribution of the classified cells in different samples. Merging all drops with the barcoded beads results in equal sequencing of both cancer cell types, while selectively merging with the K562 cells results in targeted scDNA seq of the correct subpopulation.
Figure 3.

Single-cell RNA seq of an immune subpopulation using the fluorescence-activated droplet merger. (A) Cytometer plot of droplet fluorescence. Four quadrants (Q1–Q4) are defined, and the percentage of drops in each quadrant is displayed. Q1 contains calcein green positive drops, Q2 contains calcein green and CellTrace Far red positive drops, Q3 contains CellTrace Far red positive drops, and Q4 contains empty drops. The Q1 gate (shaded green) is used to selectively merge T-cells. (B) Cells are merged with a stream of barcoded beads for mRNA capture and reverse transcription. Only merged cells are co-encapsulated with beads and reagents for cDNA synthesis. The stream immediately forms droplets at a T-junction downstream of the merging event. The cell drops are colored red (negative) and green (positive, merge), PCR reagents are colored blue, barcoded beads are colored gray, and oil is colored brown. (C) Single-cell RNA-seq and t-SNE clustering confirms that the desired subpopulation is targeted for sequencing. Merging all of the cells with barcoded beads performs scRNAseq on both B-cell and T-cells, while merging onlyT-cells targets that population.
Single-Cell Transcriptomics.
Raji (ATCC CCL-86) and Jurkat (ATCC CRL-2901) cells are cultured with Gibco RPMI Media 1640 (ThermoFisher 11875093) containing 10% (w/v) fetal bovine serum with penicillin and streptomycin (ThermoFisher 15140122). The cells are stained with CellTrace Far Red (ThermoFisher C34564) and CellTrace Calcein green (ThermoFisher C34852), respectively. The cells are counted, mixed at a ratio of 10:1 Raji:Jurkat, and diluted to a starting concentration of 80 000 cells/mL in phosphate-buffered saline at 1× with 18% (vol/vol) OptiPrep (Iodixanol solution, ThermoFisher D1556). Single-cell RNA barcoding is performed using a custom microfluidic device for the selective merger (Figures 3 and S1B). Cell droplets, generated on-chip, are selectively merged with a liquid stream containing reagents for reverse transcription and barcoded beads (inDrops v3, Harvard Single Cell Core), according to the established recipes and protocols.3,23 The microfluidic device is run with the following flow rates: 250 μL/h cells, 150 μL/h barcoded beads, 600 μL/h 1.3× RT premix,21 and 1300 μL/h droplet generation oil (Biorad, 1864005). This corresponds to an approximate dropmaking frequency of 275 Hz. To selectively barcode 1000 cells at a rarity of 1% requires approximately 1 h. Sequencing reads are processed using the inDrops pipeline available on github (https://github.com/indrops/indrops) to generate a table of counts per genes per cell. t-Distributed stochastic neighbor embedding (t-SNE) is used to cluster and visualize single-cell RNA-seq data.24
Single-Cell Genomics.
Acute lymphoblastic leukemia cells (CEM/C1 ATCC CRL-2265) are cultured with Gibco RPMI Media 1640 (ThermoFisher 11875093) containing 10% fetal bovine serum with penicillin and streptomycin (ThermoFisher 15140122). Chronic myelogenous leukemia cells (K562 ATCC CCL-243) are cultured in Iscove’s modified Dulbecco’s medium (ThermoFisher 31980097) containing 10% fetal bovine serum with penicillin and streptomycin. The CEM cells are stained with 25 μM calcein red-orange (ThermoFisher C34851) and the K562 cells are stained with 25 μM calcein green AM (ThermoFisher C34852) by incubating on ice for 30 min in 1× PBS. After staining, the cells are washed twice (HBSS, no calcium, no magnesium, ThermoFisher 14170112) and resuspended in HBSS containing 18% OptiPrep density gradient medium (Sigma-Aldrich). The cells are counted, resuspended to 3 M cells/mL, mixed at a ratio of 1:1, and co-flowed with Mission Bio lysis buffer to generate 45 μm droplets. The drops are incubated at 50 °C for 60 min, 80 °C for 20 min, and kept at 4 °C for no more than 2 h while preparing the droplet merging setup. Cascade Blue is included as a drop dye to enable the merging of all drops as an experimental control. Custom barcode beads are generated as recently described,25 targeting 16 amplicons of the Mission Bio Acute Myeloid Leukemia panel (Table S1). These amplicons are chosen because K562 and CEM have different single-nucleotide polymorphisms (SNPs) in those genomic locations that allow for their subsequent identification. To generate beads with barcoded primers, acrylamide precursors are combined microfluidically and polymerized in water-in-oil droplets with an acrydited primer. Polyacrylamide beads containing the bound primer are removed from oil using 1H,1H,2H,2H-Perfluoro-1-octanol (Sigma-Aldrich), and barcodes are added using a split-pool ligation-based approach.25
The droplets containing the cell lysate are prepared on the Mission Bio Tapestri instrument, according to the manufacturer’s specifications. Cell lysate droplets are merged with barcoded beads and PCR reagents on a custom microfluidic chip (Figure 4A). First, Mission Bio Barcoding PCR Mix is diluted to 0.7× and supplemented with 20 U/mL User II enzyme mix (NEB) (final concentration; for barcode release), Mission Bio barcoding additives, and 16 reverse primers (Table S1), each at 0.1 μM final concentration. This solution is co-flowed at 150 μL/h with close-packed barcoding beads at 75 y«L/h and HFE-7500 supplemented with 2% (w/v) poly(ethylene glycol) (PEG)–perfluoropolyether (PFPE) amphiphilic block copolymer at 450 μL/h to generate bead-containing droplets. The cell lysate droplets are reinjected at 35 μL/h, spaced with 200 μL/h HFE (without surfactant), paired with barcode bead droplets, and selectively merged using a salt-water electrode. The droplets are collected, the excess is oil removed, and the emulsion is washed in FC-40 with 5% (w/v) PEG–PFPE amphiphilic block copolymer in HFE-7500. Emulsion PCR is performed with the following cycling conditions: 95 °C 10 min, [95 °C 30 s, 72 °C 10 s, 61 °C 4 min, 72 °C 30 s] × 20, 72 °C 2 min, 4 °C hold. Library preparation and sequencing is performed according to Mission Bio’s protocol.
After sequencing on a Miseq V2 300 (Illumina), the cells are genotyped by demultiplexing the sequencing reads by cell barcodes and performing variant calling of all amplicons. The barcodes are extracted from the sequencing reads by a custom script and the valid cells are selected based on the inflection point on a plot of cells versus reads per cell. Amplicon sequences are aligned to the hg19 reference with bowtie2 (v2.3.4.1), filtered, sorted, and indexed with samtools (v1.8). GVCF files are produced with HaplotypeCaller from the GATK suite (v.4.1.3.0). The 1100 called variants in the cell pool are reduced to the most informative polymorphisms, which display a high fraction of cells with alternate homozygous or heterozygous calls and a low drop-out rate, giving rise to a short list (Table S2). Using these polymorphisms, we construct a cell–cell similarity matrix by performing the dot product between the one-hot encoded feature vectors of each cell pair. This is, in principle, a non-normalized variant of the Jaccard index.
RESULTS
Efficient Targeting of Droplet Subpopulations Using Selective Droplet Merger.
Droplet merger is an important component of many droplet microfluidic workflows because it allows the composition of each droplet to be precisely modified. This enables two-step workflows that utilize off chip temperature regulation, incompatible chemicals,26-28 or that require precise temporal control of assay components.29 Droplet merger has been used in numerous studies with microfluidics, including single-cell genome sequencing,1,2 single-molecule haplotyping,30 and high-throughput screening.31 We exploit this technique to target subpopulations by selectively adding reagents only to the desired drops. To perform the selective merger, target drops are selectively merged with reagent drops based on their fluorescence (Figure 1). Cell- or analyte-containing droplets are reinjected and interdigitated with reagent droplets; the two drops pair due to their different speeds, and the small drop is optically probed when passing through the detection region, which sits just upstream of the merger device. Merging is triggered on small droplets that fall within user-defined optical gates, and all droplets, whether merged or not, are collected in the output container. The processed emulsion is incubated to allow reactions to occur in the merged droplets. The emulsion is ruptured and the reaction products are recovered for analysis. Since these products are specific to the merged droplet pairs, no physical separation is required.
Figure 1.

Schematic of the experimental system used for the selective merger. Reinjected droplets (shown in green) enter the microfluidic device downstream of reagent droplets and pair in the long channel before the merger junction. Target drops (shown in dark green) are merged with reagent droplets (shown in red) based on their fluorescence, while negative drops (shown in light green) remain unmerged. Lasers are focused with an objective lens onto flowing droplets to excite the fluorescent molecules (Step 1, Excite). Fluorescence is collected through the same objective and measured using photomultiplier tubes (PMTs) (Step 2, Detect). Integrating under time trace data results in single-fluorescence values for each droplet, which are plotted and used to assign gates (Step 3, Gate). Droplets that fall within the desired gate are merged with reagents by applying a high-voltage pulse to the salt-water electrode embedded on the microfluidic device (Step 4, Merge). Drops that fall outside of the gates remain unmerged and are collected in the same tube. Drops that fall inside the gates are merged and molecular reactions commence. All drops are collected in the same tube (Step 5, Collect).
To characterize the efficiency of our microfluidic approach for selectively targeting droplet subpopulations, we introduce droplets containing two concentrations of a fluorescent dye (1 μM FAM+, 0.1 μM FAM−) and target the positive population for merger. Reagent droplets (CY5+) are formed in a T-junction just upstream of the reinjected droplets (Figure 2A); larger reagent drops plug the channel, allowing smaller reinjected drops to catch up and efficiently pair in the leading channel (Figure 2A). The paired droplets enter the merger junction in contact, allowing efficient coalescence when the electrode is on (Figure 2A). The selective merger is triggered with a salt-water electrode, merging the small and large droplets. Additional oil can be added to space droplets after the point of the merger to ensure that no off-target coalescence occurs. With the electrode off, the paired droplets do not merge and thus instances of double-positive (FAM+ CY5+) are rare (Figure 2B). In this case, three drop populations are observed: large CY5+ reagent droplets and small reinjected droplets containing two distinct fluorophore concentrations (FAM− and FAM+). With the electrode triggered on the high fluorescent population, FAM+ drops are merged with CY5+ drops, resulting in three main populations: unmerged reagent drops (CY5+), unmerged low fluorescent drops (FAM−), and merged high fluorescent drops (CY5+ FAM+). In this condition, the number of double positives (FAM+ CY5+) increases significantly (Figure 2C), and the diameters of the droplets shift from a population of two sizes (55 μm targets, 75 μm reagents) to a population of three sizes (55 μm targets, 75 μm reagents, 85 μm merged) (Figure 2D). To quantitatively assess selective merger efficiency, we use dropometry to measure droplet fluorescence for a large population of drops (Figure 2E,F). We estimate that the precision of the selective merger is 99.8%, while the estimated recall is 96.1% (Figure 2F). These results demonstrate the highly specific ability of fluorescence-activated droplet merger to add reagents and target droplet subpopulations.
Targeted Single-Cell RNA Sequencing of Immune Cell Subpopulations.
Single-cell RNA sequencing is one of the broadest and most important contributions of droplet microfluidics to biology. It allows massive, heterogeneous populations of cells to be characterized at the single-cell level rapidly and cost efficiently. However, existing methods cannot focus analysis on interesting subpopulations, resulting in a significant waste of reagents and sequencing on uninteresting cells. Single-cell sequencing approaches employ bead-based reactions to amplify and label cellular mRNAs with unique barcodes that enable in silico assignment of sequencing data to single cells. In such workflows, the cells are paired with barcode beads, irrespective of identity, and the whole population is sequenced. The selective merger provides a simple way to sequence a subpopulation without having to presort cells. To illustrate this, we apply the approach to a mixed population of B-cells (Raji) and T-cells (Jurkat), stained separately so they can be identified by their fluorescence (Figure 3A). The B-cells are loaded at 10 times the T-cell concentration and the resultant emulsion is processed in a selective droplet merger device. For this device, the bead solution is sufficiently conductive to induce droplet merger by direct electrification of the liquid stream (Figure 3B). We induce merger with a barcoded bead only when a droplet containing a T-cell (green) is detected. When a B-cell (red) passes through the detection window, the stream is unelectrified, resulting in no merging. All droplets are collected, but reverse transcription with barcoded primers contained on the bead only occurs in merged drops. As a control, we also collect a sample with electrification always on, resulting in pairing and merging of all cells with the beads. When we sequence the result, we find that, as expected, merging all droplets results in single cells from both input populations: ~90% B-cells (red, n = 1191) and ~10% T-cells (green, n = 157), as determined by single-cell gene expression clustering (Figure 3C, left). By contrast, when bead pairing is triggered only for the T-cells, the vast majority (99.6%, 697 in 700) classify correctly (Figure 3C, right). Misclassified cells (Figure 3C, right, red), resulting from incorrect merger, are a small percentage (0.43%, n = 2) and are still sequenced at single-cell resolution. These results illustrate that droplet merger can selectively target specific cell populations for single-cell RNA sequencing.
Targeted Single-Cell DNA Sequencing of Mutation Hotspots in Leukemia Cells.
Droplet microfluidic technologies are enabling population-scale single-cell DNA sequencing with unprecedented throughput and cost efficiency.28,32,33 These technologies allow generation of rich and detailed genomic maps that relate clonal lineages and track the genomic reprogramming of resistance to cancer therapy. Droplet merger is an essential step in these workflows because it allows cellular lysis and targeted PCR, two incompatible steps, to be performed sequentially.26,27,30,34-36 By separating genomic lysis from PCR amplification of single-cell DNA, these workflows achieve efficient completion of both steps. Because the cell lysate is merged with beads to enable target gene amplification and barcoding, methods for single-cell DNA seq can utilize selective merger to analyze specific cell subsets. To illustrate this, we target leukemia cells for single-cell genome sequencing of a tumor hotspot panel containing 16 loci. We stain and mix at a 1:1 ratio two cancer cell lines—CEM (acute lymphoblastic leukemia) cells and K562 (bone marrow-derived lymphoblast) cells. We co-flow the mixed cell suspension with lysis buffer and Cascade Blue dye; this dye acts as a droplet tag that allows us to detect all droplets, including ones devoid of cells, which aids in population gating. After lysis incubation, the cell droplets are introduced into the selective merger device, which merges the amplification mastermix, barcoded beads, and genomic hotspot PCR primers with cells of interest (Figure 4A).
We perform three experiments with different merger conditions. K562 cells (merge on high green), K562 and CEM cells (merge all), and no cells (merge none, electrode off) are collected and libraries are prepared in individual tubes with identical library amplification conditions. We load equimolar amounts from each library on the sequencer and sequence approximately 1600 cells per merger condition. DNA sequencing of the 16 genomic loci is used to genotype the single cells. We cluster the resulting similarity matrix with Ward’s minimum variance method (Figure 4B). The two top tier clusters represent the two cell lines K562 and CEM, each containing known genomic mutations. Next, we split the cells into their assigned clusters and plot with respect to their merger condition (Figure 4C). As expected, when all cells are merged with reagents, we recover roughly equal proportions of each cell type, and when no cells are merged, we recover only a single cell (Figure 4C), presumably a result of off-target droplet coalescence after collection. The selective merger enriches the K562 population from 42 to 97%—only 39 of the 1603 cells sequenced are CEM in origin, and these are still sequenced with single-cell resolution. This result illustrates how cell subsets can be targeted for single-cell DNA sequencing. Focusing sequencing power on cancer subpopulations of most interest allows deeper and more cost-efficient recovery of important and actionable information.
DISCUSSION
Droplet microfluidics has significantly expanded the throughput and repertoire of single-cell analysis by allowing soluble molecular biology reagents to be co-incubated with individual cells. In this paper, we develop a microfluidic system to target cell subsets with greatly reduced engineering complexity compared to droplet sorting. Instead of physically separating the cells, we tag them by selective reagent addition using fluorescence-activated droplet merger. Only droplets of interest receive reagents and therefore only droplets of interest are analyzed in downstream processing steps. Chemicals, small molecules, proteins, DNA, RNA, oligonucleotides, buffers, enzymes, beads, cells, or viruses can be selectively added to droplets of interest so that reactions (chemical synthesis, polymerase chain reactions, reverse transcription, transfection, transduction, transformation, etc.) occur only in those droplets.
There are several important advantages of selective reagent addition for the isolation of sample subpopulations. First, the challenges of handling small cell numbers are avoided. Second, the complexity of droplet sorting is replaced with a significantly simpler device. Droplet sorting requires substantial expertise, and as a result, commercial systems are not currently available. Third, fluorescent signals resulting from soluble assays or two-step droplet workflows can be selectively triggered, which expands the type of subpopulations that can be targeted. Workflows that require preincubation in droplets, lysis using proteases, or droplet culturing are now amenable to enrichment without sorting. Finally, the technique can be implemented via any method that rapidly combines separate streams in an oil-based carrier phase, including droplet merger (Figures 2A and 4A),37-39 picoinjection,40,41 or stream merger (Figure 3B). Selective merger is therefore compatible with many existing commercial and academic droplet workflows. A valuable attribute of our method is thus that it can be integrated into existing merger devices without modification of microfluidic chips, which already have all of the fluidic components necessary. This includes existing instruments for single-cell DNA sequencing that already incorporate the droplet merger as an essential step in the barcoding workflow (Mission Bio). To operate such devices, the electrode is switched on and off to merge selected drops, instead of being always on to merge all drops.
An important application of our approach, which we demonstrate here, is single-cell sequencing. Many droplet microfluidic systems use barcoded beads to obtain single-cell resolution, but these devices barcode and sequence every cell loaded in a droplet. An important but rare subpopulation representing 1% of the total would mean 99% of the information generated is uninformative. The cost of sequencing is therefore distributed over a large population, instead of the cells of interest. Although sorting subpopulations prior to encapsulation is feasible, this becomes prohibitive as the number of cells decreases due to difficulties in handling limited inputs. Integrating enrichment into the barcoding step avoids these problems. Selective merger also offers an attractive alternative to droplet sorting in microfluidic workflows that implement advanced detection assays, such as those that measure soluble molecules released from the cells or growth phenotypes. In addition, it reduces the complexity of many single-cell workflows by combining, sorting, and barcoding into a single operation, which means that it is easier to integrate into commercial systems that require robust and operator-free usage.
Selective droplet merger has numerous additional applications. It can be used to recover materials without the need to physically sort by merging with probes or beads that can be later purified or by triggering the formation of hydrogels that are easily recovered by filtration. Therefore, selective merger can replace sorting in many cases but also adds the additional ability to perform reactions that aid downstream processing. Selectively targeting the cells of interest can also be used to balance or shift the relative proportion of the sequenced cells from the samples with diverse cell types. For example, the selective merger can predefine the desired proportions of T-cells, B-cells, natural killer cells, and monocytes from peripheral blood mononuclear cell (PBMC) samples that are barcoded and sequenced. Or, the approach can be applied to selectively barcode the subsets of interest sequentially. In this manner, and without sorting, it is possible to serially barcode and divert each subpopulation into a different collection tube with a simple valve.
CONCLUSIONS
In this study, we describe a novel method for the enrichment of cells or molecules based on high-throughput droplet microfluidic merger. Instead of sorting, our technique selectively triggers coalescence of the target droplets with molecular reagents, thereby chemically modifying their contents. In contrast, nontarget droplets are not coalesced, yielding no reaction products. Thus, enrichment is achieved without physical separation and at reduced engineering complexity. We focus on single-cell sequencing applications of this approach and demonstrate that fluorescently labeled cell subsets can be single-cell DNA or RNA sequenced to high purity. Our approach can be integrated into existing academic and commercial single-cell barcoding workflows and has numerous applications related to combinatorial chemical synthesis, drug screening, protein engineering, and cell-based assays.
Supplementary Material
ACKNOWLEDGMENTS
Research reported in this publication was supported by NIAID of the National Institutes of Health under Award Numbers 1U01AI129206-01 and UM1AI126611-01 and by the Chan Zuckerberg Biohub.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.0c03059.
An overview of the hardware and microfluidics devices used (Figure S1); a list of the primers used for single-cell DNA sequencing (Table S1); and a list of the polymorphisms used for cell identification (Table S2) (PDF)
Operation of the microfluidic device used in Figure 2 (Movie S1) (MP4)
Operation of the microfluidic device used in Figure 3 (Movie S2) (MP4)
Operation of the microfluidic device used in Figure 4 (Movie S3) (MP4)
The authors declare the following competing financial interest(s): I.C.C. and A.R.A. have submitted a patent application related to this work.
Contributor Information
Iain C. Clark, Department of Bioengineering and Therapeutic Sciences, University of California, San Francisco, San Francisco, California 94158, United States; Ann Romney Center for Neurologic Diseases, Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts 02115, United States.
Cyrille L. Delley, Department of Bioengineering and Therapeutic Sciences, University of California, San Francisco, San Francisco, California 94158, United States
Chen Sun, Department of Bioengineering and Therapeutic Sciences, University of California, San Francisco, San Francisco, California 94158, United States.
Rohan Thakur, Center for Engineering in Medicine, Massachusetts General Hospital, Harvard Medical School, Boston, Massachusetts 02114, United States.
Shannon L. Stott, Center for Engineering in Medicine, Massachusetts General Hospital, Harvard Medical School, Boston, Massachusetts 02114, United States
Shravan Thaploo, Ann Romney Center for Neurologic Diseases, Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts 02115, United States.
Zhaorong Li, Ann Romney Center for Neurologic Diseases, Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts 02115, United States.
Francisco J. Quintana, Ann Romney Center for Neurologic Diseases, Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts 02115, United States; Broad Institute of MIT and Harvard, Cambridge, Massachusetts 02142, United States
Adam R. Abate, Department of Bioengineering and Therapeutic Sciences, University of California, San Francisco, San Francisco, California 94158, United States; Chan Zuckerberg Biohub, San Francisco, California 94158, United States
REFERENCES
- (1).Demaree B; Weisgerber D; Lan F; Abate AR J. Vis. Exp 2018, No. e57598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Lan F; Demaree B; Ahmed N; Abate AR Nat. Biotechnol 2017, 35, 640–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Klein AM; Mazutis L; Akartuna I; Tallapragada N; Veres A; Li V; Peshkin L; Weitz DA; Kirschner MW Cell 2015, 161, 1187–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Macosko EZ; Basu A; Satija R; Nemesh J; Shekhar K; Goldman M; Tirosh I; Bialas AR; Kamitaki N; Martersteck EM; Trombetta JJ; Weitz DA; Sanes JR; Shalek AK; Regev A; McCarroll SA Cell 2015, 161, 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Shahi P; Kim SC; Haliburton JR; Gartner ZJ; Abate AR Sci. Rep 2017, 7, No. 44447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Stoeckius M; Hafemeister C; Stephenson W; Houck-Loomis B; Chattopadhyay PK; Swerdlow H; Satija R; Smibert P Nat. Methods 2017, 14, 865–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Wagner DE; Weinreb C; Collins ZM; Briggs JA; Megason SG; Klein AM Science 2018, 360, 981–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Wheeler MA; Clark IC; Tjon EC; Li Z; Zandee SEJ; Couturier CP; Watson BR; Scalisi G; Alkwai S; Rothhammer V; Rotem A; Heyman JA; Thaploo S; Sanmarco LM; Ragoussis J; Weitz DA; Petrecca K; Moffitt JR; Becher B; Antel JP; et al. Nature 2020, 578, 593–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Zilionis R; Engblom C; Pfirschke C; Savova V; Zemmour D; Saatcioglu HD; Krishnan I; Maroni G; Meyerovitz CV; Kerwin CM; Choi S; Richards WG; De Rienzo A; Tenen DG; Bueno R; Levantini E; Pittet MJ; Klein AM Immunity 2019, 50, 1317–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Adamson B; Norman TM; Jost M; Cho MY; Nunez JK; Chen Y; Villalta JE; Gilbert LA; Horlbeck MA; Hein MY; Pak RA; Gray AN; Gross CA; Dixit A; Parnas O; Regev A; Weissman JS Cell 2016, 167, 1867–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Dixit A; Parnas O; Li B; Chen J; Fulco CP; Jerby-Arnon L; Marjanovic ND; Dionne D; Burks T; Raychowdhury R; Adamson B; Norman TM; Lander ES; Weissman JS; Friedman N; Regev A Cell 2016, 167, 1853–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Ahn K; Kerbage C; Hunt TP; Westervelt RM; Link DR; Weitz DA Appl. Phys. Lett 2006, 88, No. 024104. [Google Scholar]
- (13).Baret J-C; Miller OJ; Taly V; Ryckelynck M; El Harrak A; Frenz L; Rick C; Samuels ML; Hutchison JB; Agresti JJ; Link DR; Weitz DA; Griffiths AD Lab Chip 2009, 9, 1850–1858. [DOI] [PubMed] [Google Scholar]
- (14).Franke T; Braunmüller S; Schmid L; Wixforth A; Weitz DA Lab Chip 2010, 10, 789–794. [DOI] [PubMed] [Google Scholar]
- (15).Franke T; Abate AR; Weitz DA; Wixforth A Lab Chip 2009, 9, 2625–2627. [DOI] [PubMed] [Google Scholar]
- (16).Mazutis L; Gilbert J; Ung WL; Weitz DA; Griffiths AD; Heyman JA Nat. Protoc 2013, 8, 870–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Konry T; Dominguez-Villar M; Baecher-Allan C; Hafler DA; Yarmush ML Biosens. Bioelectron 2011, 26, 2707–2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Konry T; Lerner A; Yarmush ML; Smolina IV Technology (Singapore World Sci) 2013, 01, 88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Mazutis L; Araghi AF; Miller OJ; Baret J-C; Frenz L; Janoshazi A; Taly V; Miller BJ; Hutchison JB; Link D; Griffiths AD; Ryckelynck M Anal. Chem 2009, 81, 4813–4821. [DOI] [PubMed] [Google Scholar]
- (20).Clark IC; Thakur R; Abate AR Lab Chip 2018, 18, 710–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Agresti JJ; Antipov E; Abate AR; Ahn K; Rowat AC; Baret J-C; Marquez M; Klibanov AM; Griffiths AD; Weitz DA Proc. Natl. Acad. Sci. U.S.A 2010, 107, 4004–4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Clark IC; Abate AR Lab Chip 2018, 18, 3598–3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Zilionis R; Nainys J; Veres A; Savova V; Zemmour D; Klein AM; Mazutis L Nat. Methods 2016, 12, 44–73. [DOI] [PubMed] [Google Scholar]
- (24).van der Maaten L; Hinton GE J. Mach. Learn. Res 2008, 9, 2579–2605. [Google Scholar]
- (25).Delley CL; Abate AR Modular barcode beads for microfluidic single cell genomics. bioRxiv 2020.09.10.292326. 2020, DOI: 10.1101/2020.09.10.292326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Eastburn DJ; Sciambi A; Abate AR Anal. Chem 2013, 85, 8016–8021. [DOI] [PubMed] [Google Scholar]
- (27).Eastburn DJ; Sciambi A; Abate AR Nucleic Acids Res. 2014, 42, No. e128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Pellegrino M; Sciambi A; Treusch S; Durruthy-Durruthy R; Gokhale K; Jacob J; Chen TX; Geis JA; Oldham W; Matthews J; Kantarjian H; Futreal PA; Patel K; Jones KW; Takahashi K; Eastburn DJ Genome Res. 2018, 28, 1345–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Kim SC; Clark IC; Shahi P; Abate AR Anal. Chem 2018, 90, 1273–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Lan F; Haliburton JR; Yuan A; Abate AR Nat. Commun 2016, 7, No. 11784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Brouzes E; Medkova M; Savenelli N; Marran D; Twardowski M; Hutchison JB; Rothberg JM; Link DR; Perrimon N; Samuels ML Proc. Natl. Acad. Sci. U.S.A 2009, 106, 14195–14200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).McMahon CM; Ferng T; Canaani J; Wang ES; Morrissette JJD; Eastburn DJ; Pellegrino M; Durruthy-Durruthy R; Watt CD; Asthana S; Lasater EA; DeFilippis R; Peretz CAC; McGary LHF; Deihimi S; Logan AC; Luger SM; Shah NP; Carroll M; Smith CC; et al. Cancer Discovery 2019, 9, 1050–1063. [DOI] [PubMed] [Google Scholar]
- (33).Xu L; Durruthy-Durruthy R; Eastburn DJ; Pellegrino M; Shah O; Meyer E; Zehnder J Sci. Rep 2019, 9, No. 11119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Lim SW; Lance ST; Stedman KM; Abate AR J. Virol. Methods 2017, 242, 14–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Lim SW; Tran TM; Abate AR PLoS One 2015, 10, No. e0113549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Pellegrino M; Sciambi A; Yates JL; Mast JD; Silver C; Eastburn DJ BMC Genomics 2016, 17, 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Mazutis L; Baret JC; Griffiths AD Lab Chip 2009, 9, 2665–2672. [DOI] [PubMed] [Google Scholar]
- (38).Niu X; Gulati S; Edel JB; deMello AJ Lab Chip 2008, 8, 1837–1841. [DOI] [PubMed] [Google Scholar]
- (39).Akartuna I; Aubrecht DM; Kodger TE; Weitz DA Lab Chip 2015, 15, 1140–1144. [DOI] [PubMed] [Google Scholar]
- (40).Abate AR; Hung T; Mary P; Agresti JJ; Weitz DA Proc. Natl. Acad. Sci. U.S.A 2010, 107, 19163–19166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Eastburn DJ; Sciambi A; Abate AR PLoS One 2013, 8, No. e62961. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.