

Abstract
Amyotrophic lateral sclerosis (ALS) is an orphan neurodegenerative disease currently without a cure. The arylsulfanyl pyrazolone (ASP) scaffold was one of the active scaffolds identified in a cell-based high throughput screening assay targeting mutant Cu/Zn superoxide dismutase 1 (SOD1)-induced toxicity and aggregation as a marker for ALS. The initial ASP hit compounds were potent and had favorable ADME properties, but had poor microsomal and plasma stability. Here, we identify the microsomal metabolite and describe synthesized analogs of these ASP compounds to address the rapid metabolism. Both in vitro potency and pharmacological properties of the ASP scaffold have been dramatically improved via chemical modification to the corresponding sulfone and ether derivatives. One of the ether analogs (13), with superior potency and in vitro pharmacokinetic properties, was tested in vivo for its pharmacokinetic profile, brain penetration, and efficacy in an ALS mouse model. The analog showed sustained blood and brain levels in vivo and improved activity in the mouse model of ALS, thus validating the new, aryloxanyl pyrazolone scaffold as an important novel therapeutic lead for the treatment of this neurodegenerative disorder.
Graphical Abstract
Introduction
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig’s disease, is a fatal neurodegenerative disease resulting in progressive muscle loss and paralysis.1 The incidence of ALS is 1–2/100,000 per year,2,3 with approximately 2–5 years from diagnosis to death. There is a disproportionate incidence of ALS among military personnel, particularly among Gulf War veterans.4,5,6 No effective treatment for the disease is currently known.7 Riluzole, the only FDA approved drug for treating ALS, extends patient lives by 2–3 months.8
The sporadic disease (SALS) accounts for approximately 90% of all cases and familial disease (FALS) the remaining 10%. Over 100 mutations in Cu/Zn superoxide dismutase 1 (SOD1) have been identified since the pioneering work of Brown and colleagues in identifying this FALS gene,9 and studies on SOD1 have advanced the understanding of the molecular mechanisms and underlying pathogenesis of this disease.10 Mutations in SOD1 are believed to be responsible for about 20% of all FALS cases.11,12 The latter findings resulted in the development of transgenic mouse models, which have further elucidated pathophysiological mechanisms as well as provided a model for preclinical drug trials.13 The clinical and pathological features of FALS and sporadic ALS are similar, which supports the strategy of using laboratory models of FALS mutations for elucidating disease pathogenesis and for identifying potential therapeutic targets for both forms of the disease.14,15 Protein aggregation is a pathological hallmark of ALS13,16 the appearance of intracellular inclusions containing detergent-insoluble SOD1 protein aggregates represents a common early event associated with SOD1 cellular toxicity.17,1
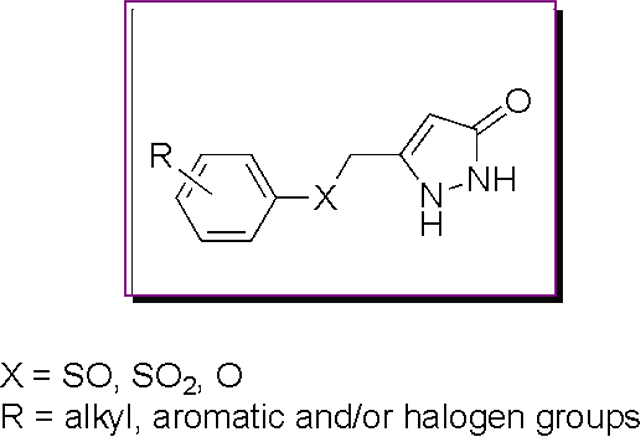
In previous studies, PC12 cells expressing mutant G93A SOD1 were utilized in a cell-based high throughput screening (HTS) assay.18,19 The arylsulfanyl pyrazolone (ASP) scaffold was identified as one of the active scaffolds showing protection against cytotoxicity from protein aggregation from a 50,000-compound library screened in the HTS assay (Figure 1; two general structures were active, where Type II was, in general, more potent than Type I).20 Structural optimization of this scaffold led via 1 (EC50 1.93 μM) and 2 (EC50 0.71 μM) to a more potent analog (3) than the original screening hits, with an EC50 value of 170 nM.
Figure 1.

ASP HTS hits. R1, alkyl or substituted aromatic groups; R2, proton or substituted aromatic groups; R, various alkyl and/or halogen groups.
In vitro pharmacokinetic (PK) and in vivo brain/plasma ratio studies were also performed at an early stage of this drug development project. Other than having low microsomal stability and plasma stability, the ASP scaffold generally showed good PK properties and permeated the blood brain barrier.21 Identifying and modifying the metabolic liability in the ASP scaffold, therefore, became a priority. The results of the metabolism studies, SAR investigations of the new scaffold, PK and toxicity properties, the ability to cross the blood-brain barrier, and effects on an ALS mouse model are described herein.
Results and Discussion
Chemistry
ASP analogs were synthesized as described earlier.21 The syntheses of Type II ASP analogs with sulfoxide and sulfone linkers in place of the sulfide are shown in Scheme 1. The sulfide linker β-ketoester intermediates (20–22) were treated with VO(acac)2-TBHP22,23 to give the sulfoxide and sulfone intermediates (23–26) or with H2O224 to give sulfone 27, which were treated with hydrazine to give the corresponding sulfoxide (4,5) and sulfone (6-8) products. The corresponding Type II aryloxanyl pyrazolone derivatives (9-14) were prepared from phenol and ethyl 4-chloroacetoacetate to generate the β-hydroxyester intermediates (28-33),25 followed by treatment with hydrazine (Scheme 2). Although this two-step synthetic method is simple and direct, it is not efficient, mainly because of the low stability of the enolate derivatives from both the ethyl 4-chloroacetoacetate and the β-hydroxyester intermediates. Improved strategies to prepare the β-hydroxyester intermediates are described in Scheme 3. Compounds 42–47 were synthesized from the corresponding phenols and 2-bromo-N-methoxy-N-methylacetamide, which was obtained from 2-bromoacetyl bromide and N,O-dimethylhydroxylamine hydrochloride.26 The corresponding β-hydroxyester intermediates were prepared by condensation of 42–47 with the enolate of ethyl acetate. The aryloxanyl pyrazolones (13-19) were obtained by treatment of the β-hydroxyester intermediates with hydrazine. Intermediates 39 and 41 were obtained via Suzuki coupling of aryl bromides 38 and 40, respectively, with phenylboronic acid using a palladium catalyst.27
Scheme 1a.

aReagents and conditions: (a) TBHP, VO(acac)2, DCM, room temperature, overnight; (b) H2O2, AcOH, EtOAc, room temperature; (c) NH2NH2, EtOH, room temperature, overnight.
Scheme 2a.

aReagents and conditions: (a) ethyl 4-chloroacetoacetate, NaH, THF, DMF, −20 °C to 70 °C; (b) NH2NH2, EtOH, room temperature, overnight.
Scheme 3a.

aReagents and conditions: (a) ethyl 2-bromoacetate, NaOEt, EtOH, reflux, overnight; (b) NaOH, H2O, 80 °C, overnight; (c) oxalyl chloride, DCM, DMF, room temperature, 4 h; (d) N,O-dimethylhydroxylamine hydrochloride, DIEA, DCM, room temperature, 1 h; (e) N,O-dimethylhydroxylamine hydrochloride, K2CO3, Et2O, H2O, room temperature, 30 min; (f) phenylboronic acid, PdCl2(PPh3)2, K2CO3, dioxane, H2O, 100 °C, 16 h; (g) NaOEt, EtOH, 70 °C, overnight; (h) EtOAc, LiHMDS, THF, −78 °C, overnight; (i) NH2NH2, EtOH, room temperature, overnight.
To avoid hydrolysis of Weinreb amide 36 under concentrated and strong basic conditions,28,29 3,5-dichlorophenol (34) was allowed to react with ethyl 2-bromoacetate then treated with NaOH/H2O to afford 35. This intermediate was transformed to 42 with oxalyl chloride and N,O-dimethylhydroxylamine hydrochloride, and the Weinreb amide was converted to 13 as described above.
Microsomal Stability
ASP analogs, like 5-((2,4-dichloro-5-methylphenylthio)methyl)-1H-pyrazol-3(2H)-one (48, Figure 2) and 5-((4-chloro-2,5-dimethylphenylthio)methyl)-1H-pyrazol-3(2H)-one (49), were found to have low mouse liver microsomal stability. One possible site of metabolism by cytochrome P450 (CYP) isozymes30 is the aromatic methyl group. Therefore, an analog lacking an aromatic methyl group (1) was tested for microsomal stability in the presence of NADPH, and the results were compared to minaprine (50), an antidepressant drug known to undergo rapid microsomal metabolism, as a positive control.31 After incubation for 20 minutes with rat liver microsomes, minaprine and 1 were metabolized by 41% and 88%, respectively (Figure 3). Therefore, the aromatic methyl group is not responsible for the metabolic instability of 1. Based on the above result, the most likely site of CYP metabolism is the sulfide sulfur atom, which is known to undergo oxidative metabolism.32
Figure 2.

Compounds tested in metabolic studies.
Figure 3.

Microsomal stability of minaprine (50) and 1. Rat liver microsomes were incubated at 37 °C for 20 minutes in the presence of 15 μM compound.
Metabolite Profiling of 1
Metabolic profiling was carried out on 1 despite its relatively low in vitro potency and because it contained a chlorine atom, which has a natural isotope abundance (35Cl:37Cl = 3:1) that can simplify the mass spectral analysis. The two likely metabolites are the corresponding sulfoxide (4) and sulfone (6), which were synthesized as shown in Scheme 1.
Compound 1 (15 μM) was incubated with rat liver microsomes at 37 °C for 0, 5, 10, 20, and 60 min in the presence of NADPH. HPLC analysis showed a new peak (5.90 min) that was not present prior to the addition of microsomes, and which was identified as the corresponding sulfoxide by comparison with those of synthetic standards of sulfoxide 4 (5.95 min) and sulfone 6 (10.8 min) (Figure 4).
Figure 4.

HPLC traces of the incubation of 1 (15 μM) with rat liver microsomes at 37 °C
Because of the limit of HPLC detection, the metabolite profiling of 1 was also carried out by LC/MS/MS. To assist in this analysis, an analog of 1 with 15N substituted for both pyrazole nitrogens was synthesized by the route in Scheme 1 using 15NH2-15NH2 in place of hydrazine. Both 1 and [15N2]-1 (5 μM) were incubated with rat liver microsomes in the presence and absence of NADPH. After 0, 10, 20, 40, and 60 min, the samples were processed and analyzed by LC/MS/MS. Both compounds, in the presence of NADPH but not in its absence, produced one new peak not detected in the controls without microsomes or compound. The mass spectrum of the new peak corresponded to the sulfoxide (4). No sulfone was detected. The rate of formation of sulfoxide corresponded well with the loss of the sulfide (Figure 5).
Figure 5.

Rate of formation of sulfoxide 4 and rate of metabolism of 1
Metabolism Guided Design and Synthesis of ASP Analogs
Because of the rapid metabolism of 1, the sulfide was replaced by sulfoxide, sulfone, and ether functional groups, synthesized as shown in Schemes 1-3. These compounds were screened in the toxicity protection assay, and compounds that retained activity in this assay were tested for in vitro microsomal stability.
Mutant SOD1-induced Cytotoxicity Protection Assay of Modified Compounds
All of the ASP analogs exhibited 100% viability in the cytotoxicity protection assay, except for 4 (maximun viability ~ 35%). The EC50 values of these analogs in the protection assay are summarized in Table 1. In general, the potencies of the analogs were greater with the ether linkage, when compared with the sulfide, sulfone, and sulfoxide. The low sulfoxide potency indicated that metabolism of the ASPs to the corresponding sulfoxide results in deactivation of the compounds and that pursuing a prodrug strategy with the thioether linked compounds would not be possible. Therefore, a series of ether analogs, aryloxanyl pyrazolones, was synthesized (Table 1). Ether 13 (CMB-087229) was the most potent (67 nM) of these compounds. Analogs of 13, having F, CF3, Br, and Ph in place of the Cl atoms, indicated that size and electronics were important features at the meta-positions; the potency decreased in the following order: Cl > CF3 > F > Br > Ph.
Table 1.
SAR of ASP analogs with -S-, -SO-, -SO2-, and -O- linkersa
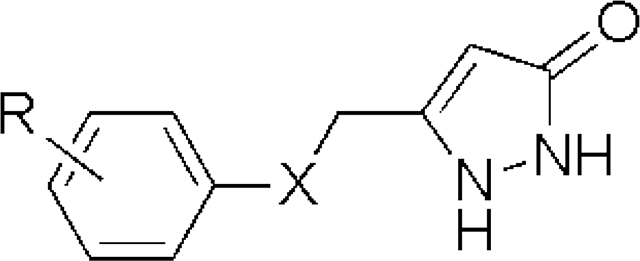 | |||
|---|---|---|---|
| X | Compound | R | EC50 (μM) |
| S | 1 | 4-Cl | 1.93 |
| 2 | 2.6-diCl | 0.71 | |
| 3 | 3,5-diCl | 0.17 | |
| SO | 4 | 4-Cl | > 32 |
| 5 | 4-Cl-2,5-diMe | 2.88 | |
| SO2 | 6 | 4-Cl | 4.7 |
| 7 | 3,5-diCl | 1.41 | |
| 8 | 4-Cl-2,5-diMe | 1.47 | |
| O | 9 | 4-Cl | 0.79 |
| 10 | 4-Et | 1.96 | |
| 11 | 3-Et | 0.72 | |
| 12 | 3-t-Bu | 0.87 | |
| 13 | 3,5-diCl | 0.067 | |
| 14 | 3,5-diCF3 | 0.58 | |
| 15 | 3,5-diF | 1.07 | |
| 16 | 3,5-diBr | 0.51 | |
| 17 | 3-Br | 1.02 | |
| 18 | 3-Ph | 2.84 | |
| 19 | 3,5-diPh | 3.70 | |
Average Z’ factor value = 0.5
In Vitro ADME Assays
Microsomal Stability.
Human and mouse microsomal stabilities of sulfone 7 and ether analogs 9 and 13 were tested at 5 μM concentration at 37 °C for 1 h in the presence and absence of NADPH (Table 2). All three compounds showed good stability.
Table 2.
In Vitro Microsomal Stability of 7, 9, and 13.
| Compd | Human | Mouse | ||||
|---|---|---|---|---|---|---|
| CL’inta (mL/min/kg) | T1/2b (min) | CL’int NADPH-free (mL/min/kg) | CL’int (mL/min/kg) | T1/2 (min) | CL’int NADPH-free (mL/min/kg) | |
| 7 | 4.5 | > 60 | 1.3 | 15.3 | > 60 | 0 |
| 9 | 15.3 | 139 | 0 | 24.5 | 143 | 6.1 |
| 13 | 25 | 93 | 13 | 64 | 36 | 21 |
Microsomal intrinsic clearance.
Half-life.
Experimental procedures and data analysis are described in reference 29. Data were obtained from Apredica and Biogen Idec.
Aqueous Solubility.
The aqueous solubility of 7, 9, and 13 was evaluated by diluting them from a stock solution in DMSO to a final concentration of 1% DMSO in PBS (Table 3). The maximum solubility was considered to be the highest concentration that showed no precipitation. All three compounds showed good aqueous solubility.
Table 3.
In Vitro Aqueous Solubility of Compounds 7, 9, and 13a
| Compound | Maximum Solubility (μM) | |
|---|---|---|
| 45 min | 16 h | |
| 7 | ≥500 | ≥500 |
| 9 | 250 | 250 |
| 13 | 250 | 250 |
Data were obtained at Apredica Inc. Experimental procedures and data analysis are described in reference 21.
Caco-2 Permeability.
Compounds 7, 9, and 13 all had high33 permeability in the Caco-2 permeability assay (Table 4).
Table 4.
In Vitro Caco-2 Permeability of 7, 9, and 13a
| Compd | Pappb (A→B) (10−6 cm/s) | Papp (B→A) (10−6 cm/s) | Ratio (B→A / A→B) |
|---|---|---|---|
| 7 | 27.0 | 4.4 | 0.2 |
| 9 | 43.0 | 28.0 | 0.6 |
| 13 | 36.7 | 14.1 | 0.4 |
Data were obtained from Apredica Inc. Experimental procedures and data analysis are described in reference 21.
Apparent permeability.
Protection of Primary Cortical Neurons.
It is not uncommon to observe that compounds that are active in tissue culture cells will not be active in differentiated neurons. Therefore sulfide 2, sulfone 7, and ether linker analog 13 were tested in a primary cortical neuron cell protection assay that showed all of these compounds were active. This issue was of particular concern because compounds from another scaffold, cyclohexane 1,3-diones, identified in the original high-throughput screen had poor activity in this assay, potentially due to poor cell permeability.34
PK Profiling of 13
Sulfone 7 and ether linker analogs 9 and 13 all showed good or adequate in vitro ADME properties. Compound 13 was selected based on its potency and in vitro pharmacological profile for in vivo PK profiling and efficacy in a mouse model of ALS.
Mouse In Vivo Steady-state Level and Blood-Brain Barrier Penetration for 13.
The in vivo steady-state level of ether analog 13 from plasma was determined in mice. The animals were administered 300 mg/kg 13 and sacrificed at progressive time points, (0, 3, 6, 12, and 24 h, Table 5), Blood and brain samples were harvested and analyzed by mass spectrometry using ESI ionization in the MRM mode.
Table 5.
In Vivo Plasma Concentration of Compound 13a
| Time (h) | Plasma Concentration (μg/mL; [μM]} |
|---|---|
| 0 | 0 |
| 3 | 88.8 [342] |
| 6 | 90 [347] |
| 12 | 64.4 [248] |
| 24 | 1.7 [6.5] |
Brain and plasma samples were analyzed at Apredica, Inc.
In Vivo Rat PK Profiling of 13.
Compound 13 was administered to Sprague-Dawley rats both intravenously and orally at 1 mg/kg in a single bolus dose. The results are summarized in Table 6.
Table 6.
Rat Plasma PK Profile of Compound 13a
| AUC Last (ng × h/mL) | AUC/Dose (ng × h/mL/dose) | T1/2 (h) | CL (mL/min/kg) | Vss (L/kg) | Cmax (ng/mL) | Tmax (h) | F (%) | |
|---|---|---|---|---|---|---|---|---|
| i.v. | 179 | 184 | 2.1 | 92 | 5.7 | - | - | - |
| p.o. | 42 | 50 | 3.6 | - | - | 39 | 0.25 | 27 |
Single bolus administration of 1 mg/kg i.v. and p.o. to SD rats; data were obtained from Biogen Idec.
Effect of 13 on the Potassium Channel.
The in vitro effect of 13 (10 μM) on the hERG potassium channel current expressed in human embryonic kidney cells (HEK293) was evaluated using a patch-clamp technique, the most definitive in vitro hERG inhibition assay.35 A positive control (E-4031) was used to confirm the sensitivity of this test system, and the TurboSol evaluation system was performed on 13 to confirm that its insolubility was not a reason for low hERG inhibition observed in this test system (see Supporting Information for results). The mean % hERG antagonism (two experiments) was 0.6 ± 1.5%. These results indicate that 13 does not affect the hERG channel.
Effect of 13 on Cytochrome P450 Isozymes.
Compound 13 was tested for its inhibition CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4, present in human liver microsomes. IC50 values of 13 to CYP isozymes inhibition were evaluated using single drug substrates except for CYP3A4 (testosterone and midazolam tested). IC50 values were > 50 μM except for CYP2C19, which was 20 μM.
Effect of 13 on Enzymes and Receptors.
Compound 13 was screened at 10 μM concentration in duplicate by MDS Pharma Services (Taipei, Taiwan) with LeadProfilingScreen, a suite of 68 in vitro enzyme and receptor assays of the most commonly observed adverse CNS, cardiovascular, pulmonary, and genotoxic activities. No significant activity was detected in any of the 68 assays. See Supporting Information for results.
The Effect of 13 administration on SOD1 G93A ALS Mice.
Control and transgenic mice of the same age (± 3 days) and from the same “f” generation were selected from multiple litters to form experimental cohorts. The tolerable dose range for 13 was determined in wild type mice by increasing the dose b.i.d. one-fold each i.p. injection; the maximum tolerated dose was 75 mg/kg. Based on the studies performed to determine tolerance and blood brain levels, the dose levels of 1.0, 10, and 20 mg/kg once a day were administered, starting at 6 weeks of age, throughout the lives of the G93A mice. Administration of 13 resulted in a significant extension in survival in the G93A ALS mice in a dose dependent manner in comparison to untreated G93A mice (Figure 5). The most efficacious dose (20 mg/kg) resulted in a life extension of 13.3%.
Discussion
We had previously found that ASP compounds are metabolically unstable.29 In vitro metabolic studies were carried out to determine the basis of microsomal instability. The early analogs had aromatic methyl groups (48 and 49) and a sulfide sulfur atom. Metabolic degradation of an ASP without an aromatic methyl (1) was more rapid than that of minaprine (50), a CNS drug known to undergo rapid metabolism, indicating that the instability was not caused by the aromatic methyl (Figure 3). Following microsome incubation, a new metabolite of 1 was identified by HPLC (Figure 4), which comigrated with the corresponding sulfoxide (4); no sulfone (6) was detected. The formation of sulfoxide 4 quantitatively followed the degradation of 1 (Figure 5). The structure of the NADPH-dependent metabolite was further confirmed by LC/MS/MS; the NADPH dependence suggests that metabolism is catalyzed by a cytochrome P450.
Identification of the metabolite of 1 as the corresponding sulfoxide, which had poor activity in the cytotoxicity protection assay, guided our redesign of the arylsulfanyl pyrazolones to the corresponding sulfones and ethers (aryloxanyl pyrazolones). In general, the potency of compounds with the various linker groups increases (EC50 decreases) in the order ether > sulfide > sulfone >> sulfoxide.
A series of ether analogs was synthesized (Table 1) and all of the compounds were assayed in the cytotoxicity protection assay, which uses PC12 cells that express mutant G93A SOD1 as a YFP fusion protein.20 In general, the most potent analogs were the 3,5-disubstituted phenyl analogs, where dichloro > dibromo > ditrifluoromethyl > difluoro > diphenyl. The dichloro analog (13) has an EC50 value of 67 nM.
Sulfone 7, ether 9, and ether 13 were much more metabolically stable than arylsulfanyl pyrazolone 1,29 with the ether being considerably more stable than the sulfone (Table 2). Because of the stability of the ethers, further ADME testing was carried out with the most potent analog (13).
The aqueous solubility of ether 13 was good (250 μM) as compared with that of 48 (56 μM21); although the solubility of sulfone 7 was greater than twice that of 13. Permeability through Caco-2 monolayer cells correlates with in vivo intestinal permeability. The efflux of compounds in the opposite direction also can be estimated by this method. The high permeability values (Papp 27–43 × 10−6 cm/s) and low efflux ratios (Papp (B→A)/Papp (A→B); 0.2–0.6) of 7, 9, and 13 suggest that these compounds have high membrane permeability36 and are likely poor substrates for efflux transport proteins, such as p-glycoprotein.37
To determine if these compounds are active in other types of cells, including neurons, a screen of selected compounds was carried out with four other cell types; SHSY-5Y, HeLa, HEK293, and primary cortical neuronal cells. Ether 13 was active in all four cell types, which is a positive indication of utility for the treatment of ALS, a non cell autonomous disease.38
Compound 13 was evaluated for its metabolic stability and blood-brain barrier penetration in mice. Mouse plasma stability was good, peaking at 3–6 hours; after 4 hours 13 was detected at 194 μM concentration in brain tissue. The loss of 13 does not follow first-order kinetics following intraperitoneal administration. Pharmacokinetics could be influenced by the high plasma protein binding by 13 (98% in rat and human), which can limit compound bioavailability in the plasma compartment, thereby prolonging the half-life.39,40 However, high plasma protein binding alone does not determine the effect on the pharmacokinetic properties; the binding affinity (on/off rate) also is a major factor.72 Pharmacokinetics in Sprague-Dawley rats was investigated both by i.v. and p.o. administration. The compound appears to be reasonably stable and is orally active; half-lives were 2.1 and 3.6 hours, respectively, for these two routes of administration with oral bioavailability (%F) of 27%, which is above the typical cutoff for continued development of drug candidates41 (Table 6). The difference in plasma stability from the mouse and rat studies could be the result of a combination of factors, including, different species (mouse vs. rat), different amount of compound used (5 mg/kg vs. 1 mg/kg), and route of administration (i.p. vs. i.v.).
Toxicity is an important cause for attrition of drug candidates at later stages of drug development. To reduce the time and effort of drug discovery, various toxicological criteria need to be met early on in the development of drug candidates. One is the effect of the compound on the human Ether-à-go-go Related Gene (hERG), a gene that encodes a cardiac potassium ion channel to regulate cardiac repolarization, which determines cardiac rhythmicity. When a compound inhibits the hERG ion channel, it can produce a disorder called long QT syndrome, resulting in torsade de pointes, heart arrhythmias and potentially sudden death. Therefore, it is essential that drug candidates do not affect hERG. Compound 13 did not antagonize hERG at a concentration of 10 μM.
The family of cytochrome P450 isozymes are also associated with drug toxicity.42 Molecules that either inhibit or upregulate these enzymes can cause drug-drug interactions. If they inhibit the isozymes, metabolism of other drugs is blocked; if they upregulate the isozymes, it leads to rapid metabolism of drugs. The IC50 of compound 13 was > 50 μM for human liver CYP1A2, CYP2C19, CYP2D6, and CYP3A4 and 20 μM for CYP2C9, indicating that 13 should not cause drug-drug interactions.
To determine if there are other receptors or enzymes that might be potential off-targets, 13 was screened by MDS Pharma Services (Taipei, Taiwan) using its LeadProfilingScreen, a suite of 68 in vitro enzyme and receptor assays of the most commonly observed adverse CNS, cardiovascular, pulmonary, and genotoxic activities. Compound 13 showed no significant binding to any of these targets at 10 μM, suggesting that it is highly target selective.
Expression of genes with ALS-associated G93A SOD1 mutations produces a striking ALS phenotype motor neuron degeneration and paralysis in rats.43,44 Transgenic mice that express human G93A mutant SOD1 develop hind limb weakness, muscle wasting, and neuropathological sequelae similar to those observed in both familial and sporadic ALS patients.45 Candidate compounds are commonly tested in the ALS mouse model to evaluate their potential for the treatment of ALS.13 The in vivo efficacy of riluzole, the only FDA-approved drug for ALS, was tested in G93A SOD1 mice and showed a lifespan extension of 7.5% when tested at 16 mg/kg46 and of 11% at 22 mg/kg.47 The in vivo efficacy of 13 in the mutant SOD1 G93A ALS mouse model was assessed to validate the aryloxanyl pyrazolone scaffold as a potential therapy for ALS,. Compound 13 produced a life extension of 13.3% at 20 mg/kg (Figure 6), comparable to or superior to riluzole.
Figure 6.

Kaplan-Meier plot of 13-treated SOD1 G93A ALS mice. Untreated Group: 125.7+/−4.9 d; Group 1 (1 mg/kg): 129.2+/−5.3 d; Group 2 (10 mg/kg): 135+/−5.5 d; Group 3 (20 mg/kg): 142.5+/−8.2 d; p < 0.05.
Conclusions
We had previously identified arylsulfanyl pyrazolones (ASP) as a class of compounds exhibiting good potency against toxicity from protein aggregation by mutant SOD1.21 These compounds could not be developed further, however, because of poor metabolic stability. Here we identify the cause for metabolic instability as the sulfide sulfur atom, which is oxygenated to the corresponding sulfoxide and show that the resulting sulfoxide compounds have significantly lower activity than the sulfides. Conversion to the corresponding sulfones and ethers, the aryloxanyl pyrazolones, resulted in much greater metabolic stability. The most potent analog was aryloxanyl pyrazolone 13, with an IC50 of 67 nM, having good aqueous solubility, excellent Caco-2 permeability with low efflux potential, a rat plasma half-life of 3.6 h, rat oral bioavailability of 27%, neuron permeability, good mouse blood-brain barrier penetration (194 μM after 4 h), no effect on the hERG channel or 68 off-target proteins, and a life extension of G93A ALS mice of 13.3% at 20 mg/kg, as good or better than that previously reported for riluzole, the only FDA-approved therapeutic for ALS. These results support 13 as a novel drug candidate for the treatment of ALS.
Experimental Section
Chemistry. General methods, reagents and materials.
All reagents were purchased from Aldrich Chemical Co. (Milwaukee, WI) or Alfa Aesar (Ward Hill, MA) and were used without further purification, unless stated otherwise. Tetrahydrofuran was distilled under nitrogen from sodium/benzophenone. Dichloromethane was redistilled from CaH2 under nitrogen. Other dry solvents were directly purchased. Thin-layer chromatography was carried out on E. Merck precoated silica gel 60 F254 plates. Compounds were visualized with ferric chloride reagent or a UV lamp. Column chromatography was performed with E. Merck silica gel 60 (230–400 mesh). Proton nuclear magnetic resonances (1H NMR) were recorded in deuterated solvents on a Varian Inova 400 (400 MHz), a Varian Inova 500 (500 MHz) or a Bruker 500 (500 MHz) spectrometer. Chemical shifts are reported in parts per million (ppm, δ) using various solvents as internal standards (CDCl3, δ 7.26 ppm; DMSO-d6, δ 2.50 ppm). 1H NMR splitting patterns are designated as singlet (s), doublet (d), triplet (t), or quartet (q). Splitting patterns that could not be interpreted or easily visualized were recorded as multiplet (m) or broad (br). Coupling constants are reported in Hertz (Hz). Proton-decoupled carbon (13C NMR) spectra were recorded on a Varian Inova 500, a Varian Inova 400, or a Bruker 500 (125.7, 100.6, and 125.7 MHz, respectively) spectrometer and are reported in ppm using various solvents as internal standards (CDCl3, δ 77.23 ppm; DMSO-d6, δ 39.52 ppm). Electrospray mass spectra (ESMS) were obtained using an LCQ-Advantage spectrometer with methanol as the solvent in the positive ion mode. High-resolution mass spectra were carried out using a VG70–250SE mass spectrometer. Chemical ionization (CI) or electron impact (EI) was used as the ion source. Elemental analyses were performed by Atlantic Microlab Inc., Norcross, GA. All final compounds were analyzed for purity by HPLC using a Luna C18 (2) column (4.6 × 150, 5 μm; Phenomenex, Torrance, CA) at a flow rate of 1 mL/min with multiple HPLC conditions. Sample elution was detected by absorbance at 254 nm. HPLC was performed on a Beckman System Gold chromatograph (Model 125P solvent module and Model 166 detector). All tested compounds had a purity of at least 95% as demonstrated by HPLC (see Supporting Information).
5-(4-Chlorophenylthio)-1H-[15N2]pyrazol-3(2H)-one (3).
4-Chlorothiophenol (1.1 g, 7.61 mmol) was mixed with ethyl 4-chloroacetoacetate (0.95 mL, 7.00 mmol) in CH2Cl2 (100 mL) at 0 °C. Triethylamine (1.5 mL, 10.8 mmol) was then added dropwise. After the resulting suspension was stirred at 0 °C for another 30 min, the reaction mixture was poured into water, and the aqueous layer was extracted with EtOAc. The combined organic layer was washed with saturated NaHCO3, HCl (0.25 N), brine, concentrated in vacuo, and purified by flash column chromatography (ethyl acetate/hexanes = 1/9) to afford 20 (1.82 g) as a light yellow oil, which was not very stable. Therefore, 20 was used directly in the next step immediately after flash column chromatography purification. A proton NMR spectrum was taken immediately after the flash column purification. 1H NMR (400 MHz, CDCl3, δ): 7.37 (dd, J = 18.4, 8.4 Hz, 4H), 4.12 (d, J = 14.0, 7.2 Hz, 2H), 4.06 (s, 2H), 3.72 (s, 2H), 1.21 (t, J = 7.2 Hz, 3H). Compound 20 (0.52 g, 1.89 mmol) was stirred in EtOH (15 mL) and H2O (6 mL), then 15NH215NH2·H2SO4 (0.25 g, 1.89 mmol) and NaHCO3 (0.32, 3.79 mmol) were added. The resulting solution was stirred overnight at room temperature, during which time a precipitate formed. The precipitate was filtered, washed with cold EtOH, and dried in vacuo to afford 3 (0.21 g, 44%, two steps) as a white solid. 1H NMR (500 MHz, DMSO-d6, δ): 11.48 (br s, 1H), 9.48 (br s, 1H), 7.36 (s, 4H), 5.30 (s, 1H), 4.08 (s, 2H); 13C NMR (125 MHz, DMSO-d6, δ): 161.3, 139.3, 135.0, 130.5, 129.8, 128.8, 89.6, 27.7; HRMS (m/z): [M + H]+ calcd for C10H9Cl15N2OS, 243.0142; found 243.0192.
5-(4-Chlorophenylsulfinyl)-1H-pyrazol-3(2H)-one (4).
Compound 20 was obtained from 4-chlorothiophenol and ethyl 4-chloroacetoacetate and used directly after flash column as described above. Compound 20 (1.27 g, 4.66 mmol) was mixed with t-butyl hydrogen peroxide (70 wt% in water, 1.1 mL, 7.69 mmol) in CH2Cl2 (50 mL) at room temperature, and vanadyl acetylacetonate (0.1% mol) was added slowly. Additional t-butyl hydrogen peroxide (0.5 mL, 3.50 mmol) was added to the reaction mixture after 2 h. The resulting suspension was stirred overnight at room temperature. The reaction mixture was then concentrated under vacuum and purified by flash column chromatography (ethyl acetate/hexanes = 1/4) to afford compound 23 (0.98 g) as a light yellow solid, which was not very stable. A proton NMR spectrum was taken immediately after the flash column purification. 1H NMR (500 MHz, CDCl3, δ): 7.46–7.34 (m, 4H), 4.20 (q, J = 7.5 Hz, 2H), 4.13 (s, 2H), 3.80 (s, 2H), 1.28 (t, J = 7.5 Hz, 3H). Therefore, 23 was used directly in the next step immediately after the flash column chromatography purification. Compound 23 (0.50 g, 1.73 mmol) was stirred in EtOH (6 mL), and an ethanolic solution of NH2NH2 (2 N, 0.87 mL, 1.74 mmol) was added. The resulting solution was stirred overnight at room temperature, during which time a precipitate formed. The precipitate was filtered, washed with cold EtOH, and dried in vacuo to afford 4 (0.10 g, 17%, three steps) as a white solid. 1H NMR (500 MHz, DMSO-d6, δ): 11.49 (br s, 1H), 9.75 (br s, 1H), 7.62 (d, J = 8.0 Hz, 2H), 7.54 (d, J = 9.0 Hz, 2H), 5.17 (s, 1H), 4.13–3.99 (m, 2H); 13C NMR (125 MHz, DMSO-d6, δ): 160.9, 142.5, 135.7, 132.1, 129.1, 126.3, 90.2, 53.3; HRMS (m/z): [M + H]+ calcd for C10H9ClN2O2S, 256.00733; found 256.00732.
5-(4-Chloro-2,5-dimethylphenylsulfinyl)-1H-pyrazol-3(2H)-one (5).
Analogous to 4, compound 22 was prepared from 4-chloro-2,5-dimethylbenzenethiol (9 g, 52.1 mmol) to afford 22 (13.11 g). Immediately after flash chromatography, 22 (4.69 g, 14.2 mmol) was converted to 24 (3.20 g). Immediately after flash column chromatography, 24 (1.39 g, 4.00 mmol) was converted to 5 as a white solid (0.42 g, 19%, three steps). 1H NMR (500 MHz, DMSO-d6, δ): 11.52 (br s, 1H), 9.50 (br s, 1H), 7.58 (s, 1H), 7.37 (s, 1H), 5.22 (s, 1H), 4.03–3.93 (m, 2H), 2.35 (s, 3H), 2.15 (s, 3H); 13C NMR (125 MHz, DMSO-d6, δ): 161.1, 140.8, 135.7, 134.7, 134.1, 132.5, 130.3, 126.1, 90.9, 52.3, 19.3, 16.8.
5-(4-Chlorophenylsulfonyl)-1H-pyrazol-3(2H)-one (6).
Compound 20 was obtained from 4-chlorothiophenol and ethyl 4-chloroacetoacetate and used directly after flash column as described above. Compound 20 (3.0 g, 11.0 mmol) was mixed with t-butyl hydrogen peroxide (70 wt% in water, 1.5 mL, 10.5 mmol) in CH2Cl2 (50 mL) at room temperature, and vanadyl acetylacetonate (0.1% mol) was added slowly. Additional t-butyl hydrogen peroxide (6 mL, 41. 94 mmol) was added to the reaction mixture gradually until all starting material was consumed, as determined by TLC. The resulting suspension was stirred overnight at room temperature. The reaction mixture was then concentrated under vacuum and purified by flash column chromatography (ethyl acetate/hexanes = 1/4) to afford 25 (1.61 g) as a light yellow oil, which was not very stable. Therefore, 25 was used directly in the next step immediately after flash column chromatography purification. Compound 25 (1.60 g, 5.25 mmol) was stirred in EtOH (6 mL), and an ethanolic solution of NH2NH2 (2 N, 2.64 mL, 5.28 mmol) was added. The resulting solution was stirred overnight at room temperature, during which time a precipitate formed. The precipitate was filtered, washed with cold EtOH, and dried under vacuum to afford 6 (0.46 g, 12%, three steps) as a white solid. 1H NMR (500 MHz, DMSO-d6, δ): 11.24 (br s, 1H), 7.75 (d, J = 9.0 Hz, 2H), 7.67 (d, J = 8.5 Hz, 2H), 5.21 (s, 1H), 4.56 (s, 2H); 13C NMR (125 MHz, DMSO-d6, δ): 159.1, 139.0, 137.2, 130.1, 129.4, 89.9, 54.1; HRMS (m/z): [M + H]+ calcd for C10H9ClN2O3S, 272.00224; found 272.00164.
5-(3,5-Dichlorophenylsulfonyl)-1H-pyrazol-3(2H)-one (7).
Analogous to 6, compound 21 (4.20 g) was prepared from 3,5-dichlorobenzenethiol (2.5 g, 14.0 mmol). Immediately after flash chromatography, 21 (4.10 g, 13.3 mmol) was converted to 26 (2.24 g). Immediately after flash chromatography, 26 (2.14 g, 6.31 mmol) was converted to 7 as a white solid (0.38 g, 10%, three steps). 1H NMR (500 MHz, DMSO-d6, δ): 11.62 (br s, 1H), 9.55 (br s, 1H), 7.77–7.36 (m, 3H), 5.26 (s, 1H), 4.70 (s, 2H); 13C NMR (125 MHz, DMSO-d6, δ): 161.3, 141.2, 135.0, 133.8, 129.5, 126.7, 92.1, 52.5; HRMS (m/z): [M + H]+ calcd for C10H8Cl2N2O3S, 301.0408; found 301.0415.
5-(4-Chloro-2,5-dimethylphenylsulfonyl)-1H-pyrazol-3(2H)-one (8).
Compound 22 was obtained from 4-chloro-2,5-dimethylbenzenethiol and ethyl 4-chloroacetoacetate and used directly after flash column chromatography as described above. Compound 22 (4.68 g, 14.1 mmol) was mixed with AcOH (5 mL) in EtOAc (10 mL), and H2O2 (30 % in water, 10 mL, 84.6 mmol) was added. The resulting solution was left stirring at room temperature overnight after which additional H2O2 (30 % in water, 5 mL, 42.3 mmol) was added. The reaction mixture was then evaporated under vacuum and purified by flash column chromatography (ethyl acetate/hexanes = 1/3) to afford 27 (4.34 g) as a yellowish oil, which was not very stable. Therefore, 27 was used directly in the next step immediately after flash column chromatography purification. Compound 27 (4.33 g, 11.9 mmol) was stirred in EtOH (20 mL), and an ethanolic solution of NH2NH2 (2 N, 5.98 mL, 11.9 mmol) was added. The resulting solution was stirred overnight at room temperature, during which time a precipitate formed. The precipitate was filtered, washed with cold EtOH, and dried under vacuum to afford 8 (0.71 g, 17%, three steps) as a white solid. 1H NMR (500 MHz, DMSO-d6, δ): 11.62 (br s, 1H), 9.59 (br s, 1H), 7.67 (s, 1H), 7.54 (s, 1H), 5.24 (s, 1H), 4.49 (s, 2H), 2.47 (s, 3H), 2.33 (s, 3H); 13C NMR (125 MHz, DMSO-d6, δ): 160.3, 138.8, 137.7, 135.2, 133.9, 132.5,132.4, 131.0, 91.8, 53.1, 43.3, 19.1; HRMS (m/z): [M + H]+ calcd for C12H13ClN2O3S, 306.9705; found 306.9713.
Synthesis of ether analogs (9–13) via method A.
5-((4-Chlorophenoxy)methyl)-1H-pyrazol-3(2H)-one (9).
A solution of 4-chlorophenol (6.4 g, 50 mmol) in THF (25 mL) was treated with NaH (60% in mineral oil, 2 g, 50 mmol) at 0 °C. In another flask, a solution of ethyl 4-chloroacetoacetate (10.21 mL, 75 mmol) in THF (25 mL) was treated with NaH (60% in mineral oil, 3.5 g, 75 mmol) at −20 °C. The resulting yellowish suspension was slowly added to the solution of sodium 4-chlorophenoxide, which was kept at 0 °C. After the addition of DMF (10 mL), the reaction temperature was slowly raised to 70 °C. After the reaction mixture was stirred at 70 °C overnight, it was cooled and evaporated to dryness. The residue was purified by flash column chromatography (ethyl acetate/hexanes = 1/9) to afford 28 (10.7 g, contains 30% 4-chloroacetonacetate) as a yellowish oil. The obtained mixture was used directly in the next step. To the mixture of 28 and 4-chlorophenol (10.7 g, assumed 41.7 mmol) was added an ethanolic solution of NH2NH2 (2 N, 14.5 mL, 29.0 mmol). The resulting solution was stirred overnight at room temperature, during which time a precipitate formed. The precipitate was filtered, washed with cold EtOH, and dried under vacuum to afford 9 (1.38 g, 13%, two steps) as a white solid. 1H NMR (500 MHz, DMSO-d6, δ): 11.79 (br s, 1H), 9.56 (br s, 1H), 7.33 (d, J = 9.0 Hz, 2H), 7.02 (d, J = 9.0 Hz, 2H), 5.52 (s, 1H), 4.92 (s, 2H); 13C NMR (125 MHz, DMSO-d6, δ): 160.2, 157.0, 139.2, 129.3, 124.6, 116.6,89.5, 62.1.
5-((4-Ethylphenoxy)methyl)-1H-pyrazol-3(2H)-one (10).
Analogous to 9, compound 29 was prepared from 4-ethylphenol (3.0 g, 24.6 mmol). The mixture of 29 and 4-ethylphenol (4.05 g, assumed 16.2 mmol) was converted to 10 (1.62 g, 30%, two steps) as a white solid. 1H NMR (500 MHz, DMSO-d6, δ): 11.75 (br s, 1H), 9.50 (br s, 1H), 7.10 (d, J = 8.0 Hz, 2H), 6.89 (d, J = 8.5 Hz, 2H), 5.50 (s, 1H), 4.87 (s, 2H), 2.54–2.51 (m, 2H), 1.13 (t, J = 7.5 Hz, 3H); 13C NMR (125 MHz, DMSO-d6, δ): 160.0, 156.2, 140.3, 136.1, 128.7, 114.6, 89.4, 61.6, 27.4, 16.0.
5-((3-Ethylphenoxy)methyl)-1H-pyrazol-3(2H)-one (11).
Analogous to 9, compound 30 was prepared via method A from 3-ethylphenol (3.0 g, 24.6 mmol). The mixture of 30 and 3-ethylphenol (1.81 g, assumed 7.23 mmol) was converted to 10 (0.30 g, 6%, two steps) as a white solid. 1H NMR (500 MHz, DMSO-d6, δ): 11.79 (br s, 1H), 9.55 (br s, 1H), 7.18 (t, J = 7.8 Hz, 1H), 6.83–6.79 (m, 3H), 5.52 (s, 1H), 4.89 (s, 2H), 2.56 (q, J = 7.5 Hz, 2H), 1.16 (t, J = 7.5 Hz, 3H); 13C NMR (125 MHz, DMSO-d6, δ): 160.7, 158.2, 145.4, 139.3, 129.3,120.4, 114.3, 111.8, 89.8, 61.0, 28.3, 15.6.
5-((3-tert-Butylphenoxy)methyl)-1H-pyrazol-3(2H)-one (12).
Analogous to 9, compound 31 was prepared via method A from 3-tert-butylphenol (3.0 g, 20.0 mmol). The mixture of 31 and 3-tert-butylphenol (2.47 g, assumed 8.86 mmol) was treated with NH2NH2 (2 N, 4.40 mL, 8.80 mmol) to give 10 (1.52 g, 10%, two steps) as a white solid. 1H NMR (500 MHz, DMSO-d6, δ): 11.72 (br s, 1H), 9.50 (br s, 1H), 7.20 (t, J = 8.0 Hz, 1H), 6.97–6.80 (m, 3H), 5.52 (s, 1H), 4.91 (s, 2H); 13C NMR (125 MHz, DMSO-d6, δ): 160.7, 157.9, 152.4, 138.7, 129.0, 117.8, 112.3, 111.1, 89.7, 61.0, 34.5, 31.3.
5-((3,5-Dichlorophenoxy)methyl)-1H-pyrazol-3(2H)-one (13).
Analogous to 9, compound 32 was prepared via method A from 3,5-dichlorophenol (2.0 g, 12.3 mmol). The mixture of 32 and 3,5-dichlorophenol (1.10 g, assumed 3.78 mmol) was treated with NH2NH2 (2 N, 1.90 mL, 3.80 mmol) to give 13 (66.1 mg, 2%, two steps) as a white solid. See below (Method B) for characterization of 13.
5-((3,5-Bis(trifluoromethyl)phenoxy)methyl)-1H-pyrazol-3(2H)-one (14).
Analogous to 9, compound 33 was prepared via method A from 3,5-bis(trifluoromethyl)phenol (1.00 mL, 5.93 mmol). The mixture of 33 and 3,5-bis(trifluoromethyl)phenol (0.72 g, assumed 2.00 mmol) was treated with NH2NH2 (2 N, 1.00 mL, 2.00 mmol) to give 14 (0.40 g, 20%, two steps) as a white solid. 1H NMR (500 MHz, DMSO-d6, δ): 11.62 (br s, 1H), 9.50 (br s, 1H), 7.99 (s, 2H), 7.86 (s, 1H), 5.35 (s, 1H), 4.31 (s, 2H); 13C NMR (125 MHz, DMSO-d6, δ): 159.8, 140.9, 131.2, 130.9, 130.5, 130.2, 127.6, 127.2, 124.5, 121.8, 119.1, 118.8, 89.0, 27.6.
Synthesis of ether analogs (13, 15–19) via method B.
Detailed experimental procedures and characterization of 35, 36, 39, 41, and 42–47 can be found in the Supporting Information.
5-((3,5-Dichlorophenoxy)methyl)-1H-pyrazol-3(2H)-one (13).
EtOAc (3.20 mL. 32.7 mmol) was added to a solution of LiHMDS (1 N in THF, 75.0 mL, 75.0 mmol) at 0 °C and stirred. After 60 min, a THF solution of 42 (8.55 g, 32.4 mmol) was added dropwise at −78 °C. After the resulting solution was stirred at −78 °C for another 8 h, the reaction mixture was quenched with diluted HCl (0.25 N), the pH was adjusted to 3–5, and the aqueous layer was extracted with Et2O. The combined organic layer was dried over Na2SO4 and concentrated under vacuum. The residue was purified by flash column chromatography (ethyl acetate/hexanes = 1/9) to give ethyl 4-(3,5-dichlorophenoxy)-3-oxobutanoate (3.28 g) as a white solid. The proton NMR spectrum was taken immediately after recrystallization (ethyl acetate/hexanes). 1H NMR (500 MHz, CDCl3, δ): 7.02 (t, J = 1.7 Hz, 1H), 6.81 (d, J = 1.7 Hz, 2H), 4.68 (s, 2H), 4.21 (q, J = 7 Hz, 2H), 3.61 (s, 2H), 1.28 (t, J = 7 Hz). Ethyl 4-(3,5-dichlorophenoxy)-3-oxobutanoate was used directly in the next step immediately after the flash column chromatography purification. Ethanolic hydrazine (2 N, 5.60 mL, 11.2 mmol) was added to a solution of ethyl 4-(3,5-dichlorophenoxy)-3-oxobutanoate (3.28 g, 11.3 mmol) in EtOH (25 mL). The resulting solution was stirred at room temperature overnight. The reaction mixture was then evaporated under vacuum, purified by flash column chromatography (ethyl acetate/hexanes = 1/2) and recrystallized in ethyl acetate/hexanes to give 13 (0.88 g, 11%, two steps) as white crystals. 1H NMR (500 MHz, DMSO-d6, δ): 11.82 (br s, 1H), 9.54 (br s, 1H), 7.16 (s, 1H), 7.12 (d, J = 1.5 Hz, 2H), 5.53 (s, 1H), 4.99 (s, 2H); 13C NMR (125 MHz, DMSO-d6, δ): 161.0, 159.4, 137.8, 134.6, 120.6, 114.2, 90.7, 61.5. Anal. Calcd for C10H8Cl2N2O2: C, 46.36; H, 3.11; Cl, 27.37; N, 10.81. Found: C, 46.40; H, 3.08; Cl, 27.24; N, 10.65. All values are given as percentages.
5-((3,5-Difluorophenoxy)methyl)-1H-pyrazol-3(2H)-one (15).
Analogous to 13, compound 15 was prepared via method B from 43 (1.24 g, 5.36 mmol) to afford initially ethyl 4-(3,5-difluorophenoxy)-3-oxobutanoate (0.89 g, 3.44 mmol). Because of instability, immediately after flash column chromatography, ethyl 4-(3,5-difluorophenoxy)-3-oxobutanoate (0.89 g, 3.44 mmol) was treated with NH2NH2 (2 N in EtOH, 1.70 mL, 3.40 mmol) to give 15 as a white solid (50.9 mg, 5%, two steps). 1H NMR (500 MHz, DMSO-d6, δ): 11.82 (br s, 1H), 9.53 (br s, 1H), 6.80–6.79 (m, 3H), 5.56 (s, 1H), 4.95 (s, 2H); 13C NMR (125 MHz, DMSO-d6, δ): .1, 164.0, 162.1, 162.0, 160.0, 146.4, 137.8, 99.1, 98.9, 96.4, 90.7, 61.8.
5-((3,5-Dibromophenoxy)methyl)-1H-pyrazol-3(2H)-one (16).
Analogous to 13, compound 16 was prepared via method B from 44 (1.07 g, 3.03 mmol) to afford initially ethyl 4-(3,5-dibromophenoxy)-3-oxobutanoate (0.35 g). Immediately after flash column, ethyl 4-(3,5-dibromophenoxy)-3-oxobutanoate (0.35 g, 0.92 mmol) was treated with NH2NH2 (2 N in EtOH, 0.46 mL, 0.92 mmol) to give 16 as a white solid (23.7 mg, 2%, two steps). 1H NMR (500 MHz, DMSO-d6, δ): 11.80 (br s, 1H), 9.52 (br s, 1H), 7.39 (s, 1H), 7.28 (m, 2H), 5.55 (s, 1H), 4.99 (s, 1H); 13C NMR (125 MHz, DMSO-d6, δ): 161.0, 159.5, 137.7, 125.9, 122.8, 117.4, 90.6, 61.5.
5-((3-Bromophenoxy)methyl)-1H-pyrazol-3(2H)-one (17).
Analogous to 13, compound 17 was prepared via method B from 45 (2.43 g, 8.87 mmol) to afford initially ethyl 4-(3-bromophenoxy)-3-oxobutanoate (1.07 g). Immediately after flash column, ethyl 4-(3-bromophenoxy)-3-oxobutanoate (1.07 g, 3.55 mmol) was treated with NH2NH2 (2 N in EtOH, 1.70 mL, 3.40 mmol) to give 17 as a white solid (0.42 g, 18%, two steps). 1H NMR (500 MHz, DMSO-d6, δ): 11.78 (br s, 1H), 9.50 (br s, 1H), 7.26–7.23 (m, 1H), 7.14–7.13 (m, 1H), 7.02–7.00 (m, 2H), 5.58 (s, 1H), 4.96 (s, 2H); 13C NMR (125 MHz, DMSO-d6, δ): 161.6, 159.1, 138.4, 131.2, 123.8, 122.1, 117.6, 114.3, 90.5, 61.3.
5-((Biphenyl-3-yloxy)methyl)-1H-pyrazol-3(2H)-one (18).
Analogous to 13, compound 18 was prepared via method B from 46 (0–.74 g, 2.74 mmol) to afford initially ethyl 4-(biphenyl-3-yloxy)-3-oxobutanoate (0.50 g). Immediately after flash column, ethyl 4-(biphenyl-3-yloxy)-3-oxobutanoate (0.50 g, 1.68 mmol) was treated with NH2NH2 (2 N in EtOH, 0.84 mL, 1.68 mmol) to give 18 as a white solid (0.19 g, 27%, two steps). 1H NMR (500 MHz, DMSO-d6, δ): 11.80 (br s, 1H), 9.50 (br s, 1H), 7.67–6.99 (m, 9H), 5.55 (s, 1H), 5.02 (s, 2H); 13C NMR (125 MHz, DMSO-d6, δ): 161.3, 158.6, 138.7, 141.7, 140.0, 130.0, 128.9, 127.7, 126.8, 119.4, 113.9, 113.0, 90.3, 60.8.
5-((5-Phenylbiphenyl-3-yloxy)methyl)-1H-pyrazol-3(2H)-one (19).
Analogous to 13, compound 19 as prepared via method B from 47 (0.61 g, 1.75 mmol) to afford initially ethyl 4-(5-phenylbiphenyl-3-yloxy)-3-oxobutanoate (0.45 g). Immediately after flash column, ethyl 4-(5-phenylbiphenyl-3-yloxy)-3-oxobutanoate (0.45 g, 1.21 mmol) was treated with NH2NH2 (2 N in EtOH, 0.60 mL, 1.20 mmol) to give 19 as a white solid (0.19 g, 27%, two steps). 1H NMR (500 MHz, DMSO-d6, δ): 11.82 (br s, 1H), 9.50 (br s, 1H), 7.76 (d, J = 8.0 Hz, 4H), 7.50–7.38 (m, 7H), 7.27 (s, 2H), 5.60 (s, 1H), 5.13 (s, 2H); 13C NMR (125 MHz, DMSO-d6, δ): 161.3, 159.1, 142.3, 140.0, 139.0, 129.0, 127.8, 127.0, 118.0, 112.3, 90.4, 61.2.
In Vitro Microsomal Stability of Compound 1 and Minaprine. General methods, reagents and materials.
The microsome assay was measured on a Beckman HPLC system using Luna C18 (2) column (4.6 × 150, 5 μm; Phenomenex, Torrance, CA) at a flow rate of 1 mL/min at 254 nm for minaprine and 260 nm for 1. Solid phase extraction was carried out using Waters Sep-Pak®Vac C18 1 cc cartridges (Waters Chromatography, Milford, MA). NADPH regeneration solutions and rat liver microsomes were purchased from Bectin-Dickinson.
To PBS (10 mL) was added NADPH regeneration buffer A (500 μL) and NADPH regeneration buffer B (100 μL) to make the PBS+ solution. To the PBS+ solution (255 μL) was added the compound of interest (30 μL of 150 μM stock in PBS+) in an Eppendorf tube. Each reaction mixture was carried out in PBS (pH 7.4), which contained 1 mg/mL microsomal protein with 1.3 mM NADP+, 3.3 mM glucose-6-phosphate, 0.4 U/mL glucose-6-phosphate dehydrogenase, and 3.3 mM magnesium chloride. The tubes were vortexed and equilibrated at 37 °C. The samples were incubated at 37 °C for 0 and 20 min after the rat liver microsomes (15 μM) were added. Acetonitrile (40 μL) and haloperidol (internal standard; 100 μL) were added as a quench solution. The reaction mixture was vortexed and left in a −78 °C ice bath immediately after it was quenched. The samples were centrifuged (13.4 krpm, 12 min), and the supernatant was loaded onto a Waters Sep-Pak®Vac C18 1 cc SPE cartridge. The loaded SPE column was washed with H2O (2 × 1 mL), and the compounds were eluted with acetonitrile/H2O = 1/1 (2 × 1 mL). The solvent was removed under vacuum. The samples were reconstituted in DMSO/H2O = 1/9 (25 μL), and an aliquot (20 μL) was analyzed by HPLC (isocratic; acetonitrile/H2O = 25/75, 60 min; 0.1% TFA). The response ratio (RR) was calculated by dividing the peak area of the analyte by the peak area of the internal standard. Peak integrals were normalized by dividing by the area for the peak at t = 0 min. Sample HPLC spectrum and RR data can be found in the Supporting Information.
Metabolite Profiling of Compound 1.
Compound 1 was incubated with rat liver microsomes and NADPH regeneration buffers as described above for the in vitro microsomal stability study of 1. To the PBS+ solution (85 μL) was added 1 (10 μL of 150 μM stock in PBS+). The sample was incubated at 37 °C for 0, 5, 10, 20, 40, and 60 min after the rat liver microsomes (15 μM) were added. Acetonitrile (50 μL) was added as a stop solution. The reaction was vortexed and left on a −78 °C ice bath immediately after it was quenched. The samples were centrifuged (13.4 krpm, 12 min), and the supernatant (20 μL) was directly analyzed by HPLC (isocratic; acetonitrile/H2O = 25/75, 30 min; 0.1% TFA). Compounds 4 (20 μL, 10 μM) and 6 (20 μL, 10 μM) were analyzed by the same HPLC program. Spectra of the microsomal incubation residue of 1, standard solution of 4, and standard solution of 6 were compared in parallel.
Further metabolite profiling of 1 and 3.
Metabolite studies were also performed at Apredica, Inc. (Watertown, MA). Samples were analyzed by LC/MS/MS using either an Agilent 6410 mass spectrometer coupled with an Agilent 1200 HPLC and a CTC PAL chilled autosampler or an ABI2000 mass spectrometer coupled with an Agilent 1100 HPLC and a CTC PAL chilled autosampler. After separation of compounds on a C18 reverse phase HPLC column using an acetonitrile-water gradient system, peaks were analyzed by mass spectrometry using ESI ionization in MRM mode.
Each test agent (1 and 3) was incubated in duplicate at 5 μM with or without rat liver microsomes in the presence and absence of NADPH. After 0, 10, 20, 40, and 60 min incubation, an aliquot was removed from each reaction, the protein was precipitated with acetonitrile containing internal standard. The supernatant was analyzed by LC/MS/MS. Detailed experimental data, HPLC, and MS spectra can be found in the Supporting Information.
Mutant SOD1-induced Cytotoxicity Protection Assay.
Viability and EC50 values of 1-19 were determined according to the previously reported assay procedure.20 PC12 cells expressing mutant G93A SOD1 were seeded at 15000 cells/well in 96-well plates and incubated fro 24 h prior to compound addition. Compounds were assayed in 12-point dose-response experiments to determine potency and efficacy. The highest compound concentration tested was 32 μM, which was decreased by one-half with each subsequent dose. After 24 h incubation with the compounds, MG132 was added at a final concentration of 100 nM. MG132 is a well-characterized proteasome inhibitor, which enhances the appearance of protein aggregation by blocking the proteosomal clearance of aggregated proteins. Cell viability was measured 48 h later using a fluorescent viability probe, Calcein-AM (Molecular Probes). Briefly, cells were washed twice with PBS, Calcein-AM was added at a final concentration of 1 μM for 20 min at room temperature, and fluorescence intensity was read in a POLARstar fluorescence plate reader (BMG). Fluorescence data were coupled with compound structural data, then stored, and analyzed using the CambridgeSoft Chemoffice Enterprise Ultra software package.
In Vitro ADME Assays.
In vitro microsomal stability of 7 and 9, aqueous solubility, and Caco-2 permeability of 7, 9, and 13 were determined according to the previously reported procedure.21 In vitro microsomal stability of 13 was obtained from Biogen Idec.
Preliminary Cortical Neuron Permeability.
Primary rat cortical tissue was purchased from Neuromics Inc., Edina, MN and used to initiate primary cortical neuron cultures. The tissue was isolated from micro-surgically dissected E18 embryonic Sprague-Dawley or Fischer 344 rat brain and shipped in a nutrient rich medium under refrigeration.
To isolate neurons, the tissue was incubated with papain at a concentration of 2 mg/mL in Hibernate without calcium for 30 min at 37 °C. The enzymatic solution was then removed and 1 mL of culture media (Neurobasal, B27, 0.5 mM glutamine) was added. A sterile Pasteur pipette was used to gently disperse the cells, which were then washed, re-suspended and counted. The cells were plated on poly-D-lysine coated 96-well plates at a density of 20,000 cells/well and incubated at 37 °C in a 5% CO2-humidified atmosphere for 5 days prior to use in compound testing. By microscopic inspection, the resulting culture consisted of ~90% neurons.
Test compounds were assayed in six-point dose response experiments. The highest compound concentration tested was 100 μM, which was then diluted by approximately one-third with each subsequent dose. After 24 h incubation with the compounds, MG132 was added at a final concentration of 100 nM, a dose that produces an approximately 70% loss of viability. Cell viability was measured 48 h later using the fluorescent viability probe, Calcein-AM (Molecular Probes, Invitrogen, Carlesbad, CA). Briefly, cells were washed twice with PBS, Calcein-AM was added at a final concentration of 1 μM for 20 min at room temperature, and fluorescence intensity was then read in a POLARstar fluorescence plate reader (BMG). Compounds that restored viability at any dose to a level equal or higher than 5 standard deviations from MG132 controls were considered active.
In Vivo Mouse Drug Steady-State Level Determination and in Vivo Brain Penetration.
In Vivo PK Profiling
In vivo PK profiling was performed by administering a 300 mg/kg bolus intraperitoneal injection of 13 into B6SJL mice. Blood and brain samples were collected at 0, 1, 3, 6, 12, and 24 h time points and flash frozen in liquid nitrogen, stored at −80 °C, and shipped to Apredica, Inc. for analysis. The analysis was carried out as for for in vivo mouse drug steady-state level determination and in vivo brain penetration described in Supporting Information.
Effect of Compound 13 on Potassium Channels
FASTPatch hERG screen assay, using cloned hERG potassium channels expressed in human embryonic kidney cells (HEK293), was carried out by ChanTest Corporation (Cleveland, OH).
In Vivo SOD1 G93A ALS Mice Study of 13.
G93A SOD1 mice and littermate SOD1 mice were mated with B6SJL females, and the offspring were genotyped by PCR using tails. A 12-h light-dark cycle was maintained, and animals were given free access to food and water. Control and transgenic mice of the same age (± 3 days) and from the same “f” generation were selected from multiple litters to form experimental cohorts (n = 20 per group). Standardized criteria for age, weight, and parentage were used for placing individual mice into experimental groups/cohorts. Wild type mice were used for initial toxicity, tolerability, and pharmacokinetics studies. The tolerable dose range and LD50 for 13 was determined in the wild type mice by increasing the dose b.i.d. one-fold each injection and observed at 75 mg/kg. Drug steady-state level was determined in animals that had been dosed for one week prior to sacrifice. The dosing levels of 1.0, 10, and 20 mg/kg once a day were administered throughout the levels of the G93A mice.
Efficacy was measured using endpoints that clearly indicate neuroprotective function. These include amelioration of degenerative changes in the spinal cord, improved motor function, and prolonged survival. The mice cohorts were sacrificed at end stage disease using criteria for euthanasia, followed temporally for survival analyses. Mice were observed three times daily (morning, noon, and later afternoon) throughout the experiment. Mice were euthanized when disease progression was sufficiently severe that they were unable to initiate movement and right themselves after gentle prodding for 30 seconds.
Data sets were generated and analyzed for each clinical and neuropathological measure. Effects on behavior and neuropathology were compared in treatment and control groups. Dose-dependent effects were assessed in each treatment group using multiple two-sided ANOVA testes. Multiple comparisons in the same subject groups were dealt with post hoc. Kaplan-Meier analysis was used for survival function. All other statistical analyses were performed using Student’s t-test. Data are expressed as the mean ± SEM. Statistical comparisons of histological data were made by ANOVA.
Supplementary Material
Acknowledgements.
We thank the National Institutes of Health [1R43NS057849], the ALS Association (TREAT program), the Department of Defense [AL093052] and the Center for ALS Research at the Univesity of Pittsburgh for their generous support of the research project. The authors are grateful to Biogen Idec Inc. for carrying out the in vivo rat PK profiling tests, in vitro P450 reversible inhibition study, the plasma binding affinity assay, the hERG inhibition assays; and MDS Pharma Service (now Ricerca Biosciences, LLC) for carrying out the LeadProfilingScreen of 13.
a Abbreviations:
- ADME
absorption, distribution, metabolism, and excretion
- ALS
amyotrophic lateral sclerosis
- ASP
arylsulfanyl pyrazolone
- CYP
cytochrome P450
- FALS
familial amyotrophic lateral sclerosis
- HEK
human embryonic kidney
- hERG
human Ether-à-go-go-Related Gene
- HTS
high-throughput screen
- MRM
multiple reaction monitoring
- PK
pharmacokinetics
- SOD1
Cu/Zn superoxide dismutase 1
- SALS
sporadic amyotrophic lateral sclerosis
Footnotes
Supporting Information Available: Experimental details and data for 35, 36, 39, 41 and 42–47, HPLC data and spectra for 1-19, HPLC spectra and data of microsomal stability of minaprine and 1, data and spectra of metabolite profiling of 1 and 3, bioanalysis data of in vivo mouse steady-state level study and in vivo blood brain barrier study of 13, data for the effect of 13 on hERG potassium channel and data for the effect of 13 on enzymes and receptors. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Bruijn LI; Miller TM; Cleveland DW Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu. Rev. Neurosci 2004, 27, 723–749. [DOI] [PubMed] [Google Scholar]
- 2.Hirtz D; Thurman DJ; Gwinn-Hardy K; Mohamed M; Chaudhuri AR; Zalutsky R How common are the “common” neurologic disorders. Neurology 2007, 68, 326–337. [DOI] [PubMed] [Google Scholar]
- 3.Cronin S; Hardiman O; Traynor BJ Ethnic variation in the incidence of ALS. Neurology 2007, 68, 1002–1007. [DOI] [PubMed] [Google Scholar]
- 4.Haley RW Excess incidence of ALS in young Gulf War veterans, Neurology 2003, 61, 750–756. [DOI] [PubMed] [Google Scholar]
- 5.Horner RD; Kamins KG; Feussner JR; Grambow SC; Hoff-Lindquist J; Harati Y; Mitsumoto H; Pascuzzi R; Spencer PS; Tim R; Howard D; Smith TC; Ryan MAK; Coffman CJ; Kasarskis EJ Occurrence of amyotrophic lateral sclerosis among Gulf War veterans. Neurology 2003, 61, 742–749. [DOI] [PubMed] [Google Scholar]
- 6.Weisskopf MG; O’Reilly EJ; McCullough ML; Calle EE; Thun MJ; Cudkowicz M; Ascherio A Prospective study of military service and mortality from ALS. Neurology 2005, 64, 32–37. [DOI] [PubMed] [Google Scholar]
- 7.Rowland LP; Shneider NA Amyotrophic lateral sclerosis. N. Engl. J. Med 2001, 344, 1688–1700. [DOI] [PubMed] [Google Scholar]
- 8.Miller RG; Mitchell JD; Moore DH Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Amyotroph. Lat. Scler. Other Motor Neuron Disord 2003, 4, 191–206. [PubMed] [Google Scholar]
- 9.Rosen DR; Siddique T; Patterson D; Figlewicz DA; Sapp P; Hentati A; Donaldson D; Goto J; O’Regan JP; Deng H; Rahmani Z; Krizus A; Mckenna-Yasek D; Cayabyab A; Gaston SM; Berger R; Tanzi RE; Halperin JJ; Herzfeldt B; Van den Bergh R; Hung W; Bird T; Deng G; Mulder DW; smyth C; Laing NG; Soriano E; Pericak-Vance MA; Haines J; Rouleau GA; Gusella J; Horvitz HR; Brown RH Jr. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [DOI] [PubMed] [Google Scholar]
- 10.Pasinelli P; Brown RH Molecular biology of amyotrophic lateral sclerosis: insight from genetics Nat. Rev. Neurosci 2006, 7, 710–723. [DOI] [PubMed] [Google Scholar]
- 11.Mulder DW; Kurland LT; Offord KP; Beard CM Familial adult motor neuron disease: amyotrophic lateral sclerosis. Neurology 1986, 36, 511–517. [DOI] [PubMed] [Google Scholar]
- 12.Pramatarova A; Figlewicz DA; Krizus A; Han FY; Ceballos-Picot I; Nicole A; Dib M; Meininger V; Brown RH; Rouleau GA Identification of new mutationsin the Cu/Zn superoxide dismutase gene of patients with familial amyotrophic lateral sclerosis. Am. J. Hum. Genet 1995, 56, 592–596. [PMC free article] [PubMed] [Google Scholar]
- 13.Ryu H; Ferrante RJ Translational therapeutic strategies in amyotrophic lateral sclerosis. Mini-Reviews Med. Chem 2007, 7, 141–150. [DOI] [PubMed] [Google Scholar]
- 14.Andersen PM; Sims KB; Xin WW; Kiely R; O’Neill G; Ravits J; Pioro E; Harati Y; Brower RD; Levine JS; Heinicke HU; Seltzer W; Boss M; Brown RH Jr. Sixteen novel mutations in the Cu/Zn superoxide dismutase gene in amyotrophic lateral sclerosis: a decade of discoveries, defects and diputes. Amyotroph. Lat. Scler. Other Motor Neuron Disord 2003, 4, 62–73. [DOI] [PubMed] [Google Scholar]
- 15.Brown RH Jr.; Robberecht W Amyotrophic lateral sclerosis: pathogenesis. Semin. Neurol 2001, 21, 131–139. [DOI] [PubMed] [Google Scholar]
- 16.Newbery HJ; Abbott CM Of mice, men and motor neurons. Trends Mol. Med 2002, 8, 88–92. [DOI] [PubMed] [Google Scholar]
- 17.Bruijn LI; Houseweart MK; Kato S; Anderson KL; Anderson SD; Ohama E; Reaume AG; Scott RW; Cleveland DW Aggregation and Motor Neuron Toxicity of an ALS-linked SOD1 Mutant Independent from Wild-type SOD1. Science 1998, 281, 1851–1854. [DOI] [PubMed] [Google Scholar]
- 18.Matsumoto G; Stojanovic A; Holmber CI; Kim S; Morimoto RI Structural Properties and Neuronal Toxicity of Amyotrophic Lateral Sclerosis-associated Cu/Zn Superoxide Dismutase 1 Aggregates. J. Cell Biol 2005, 171, 75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matsumoto G; Kim S; Morimoto RI Huntingtin and mutant SOD1 form aggregate structures with distinct molecular properties in human cells. J. Biol. Chem 2006, 281, 4477–4485. [DOI] [PubMed] [Google Scholar]
- 20.Benmohamed R; Arvanites AC; Silverman RB; Morimoto RI; Ferrante RJ; Kirsch DR Identification of compounds protective against G93A SOD1 toxicity for the treatment of amyotrophic lateral sclerosis. Amyotroph. Lat. Scler. Other Motor Neuron Disord 2011, 12, 87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen T; Benmohamed R; Arvanites AC; Ranaivo HR; Morimoto RI; Ferrante RJ; Watterson DM; Kirsch DR; Silverman RB Arylsulfanyl pyrazolones block mutant SOD1-G93A aggregation. Potential application for the treatment of amyotrophic lateral sclerosis. Bioorg. Med. Chem 2011, 19, 613–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Basak A; Barlan AU; Yamamoto H Catalytic enantioselective oxidation of sulfides and disulfides by a chiral complex of bis-hydroxamic acid and molybdenum. Tetrahedron: Asymmetry 2006, 17, 508–511. [Google Scholar]
- 23.Zhang W; Yamamoto H Vanadium-catalyzed asymmetric epoxidation of homoallylic alcohols. J. Am. Chem. Soc 2007, 129, 286–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sonda S; Kawahara T; Murozono T; Sato N; Asona K; Haga K Design and synthesis of orally active benzamide derivatives as potent serotonin 4 receptor agonist. Bioorg. Med. Chem 2003, 11, 4225–4234. [DOI] [PubMed] [Google Scholar]
- 25.Banfi L; Casicio G; Ghiron C; Guanti G; Manghisi E; Narisano E; Riva R Microbiological enatioselecrive synthesis of (S) and (R) 4-(p-anisyloxy)-3-hydroxybutyrates as new chiral building blocks for the synthesis of β-lactam antibiotics. Tetrahedron 1994, 50, 11983–11994. [Google Scholar]
- 26.Hirner S; Panknin O; Edefuhr M; Somfai P Synthesis of aryl glycines by the α arylation of weinreb amides. Angew. Chem. Int. Ed 2008, 47, 1907–1909. [DOI] [PubMed] [Google Scholar]
- 27.Gu R; Hameurlaine A; Dehaen W Facile one-pot synthesis of 6-monosubstituted and 6,12-disubstituted 5,11-dihydroindolo[3,2-b]carbazoles and preparation of various functionalized derivatives. J. Org. Chem 2007, 72, 7207–7213. [DOI] [PubMed] [Google Scholar]
- 28.Doherty EM; Fotsch M; Bo Y; Chakrabarti PP; Chen N; Gavva N; Han N; Kelly MG; Kincaid J; Klionsky L; Liu Q; Ognyanov VI; Tamir R; Wang X; Zhu J; Norman MH; Treanor JJS Discovery of potent, orally available vanilloid receptor-1 antagonists. structure−Activity relationship of N-aryl cinnamides. J. Med. Chem 2005, 48, 71–90. [DOI] [PubMed] [Google Scholar]
- 29.Hillier MC; Davidson JP; Martin SF Cyclopropane-derived peptidomimetics. design, synthesis, and evaluation of novel ras farnesyltransferase inhibitors. J. Org. Chem 2001, 66, 1657–1671. [DOI] [PubMed] [Google Scholar]
- 30.Nakajima T; Wang R; Elovaara E, Gonzalez F.J.; Gelboin H.V.; Raunio H.; Pelkonen O.; Vainio H.; Aoyama T. Toluene metabolism by cDNA-Expressed human hepatic cytochrome P450. Biochem. Pharmacol 1997, 53, 271–277. [DOI] [PubMed] [Google Scholar]
- 31.McNaney CA; Drexler DM; Hnatyshy SY; Zvyaga TA; Knipe JO; Belcastro JV; Sanders M An automated liquid chromatography-mass spectrometry process to determine metabolic stability half-life and intrinsic clearance of drug candidates by substrate depletion. Assay Drug Dev. Techol 2008, 6, 121–129. [DOI] [PubMed] [Google Scholar]
- 32.(a) Mitchell SC; Waring RH The early history of xenobiotic sulphoxidation. Drug Metab. Rev. 1985, 16, 255–284. [DOI] [PubMed] [Google Scholar]; (b) Fruetel J; Chang Y; Collins J; Loew G; Ortiz de Montellano PR Thioanisole sulfoxidation by cytochrome P450cam (CYP101): experimental and calculated absolute stereochemistries. J. Am. Chem. Soc 1994, 116, 11643–11648. [Google Scholar]
- 33.Kerns EH; Di L Drug-like Properties: Concepts, Structure, Design, and Methods; Elsevier, Inc.; Amsterdam, 2008; pp. 291. [Google Scholar]
- 34.Zhang W; Benmohamed R; Arvanites AC; Morimoto RI; Ferrante RJ; Kirsch DR; Silverman RB Cyclohexane 1,3-diones and their inhibition of mutant SOD1-dependent protein aggregation and toxicity in PC12 cells. Bioorg. Med. Chem, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kerns EH; Di L Drug-like Properties: Concepts, Structure, Design, and Methods; Elsevier, Inc.: Amsterdam, 2008; pp. 378. [Google Scholar]
- 36.Kerns EH; Di L Drug-like Properties: Concepts, Structure Design and Methods; Elsevier, Inc.: Amsterdam, 2008; pp. 288–291. [Google Scholar]
- 37.Press B; Di Grandi D Permeability for intestinal absorption: Caco - 2 assay and related issues. Curr. Drug Metab 2008, 9(9), 893–900. [DOI] [PubMed] [Google Scholar]
- 38.Ilieva H; Polymenidou M; Cleveland DW Non—Cell Autonomous Toxicity in Neurodegenerative Disorders: ALS and Beyond. J. Cell. Biol 2009, 187, 761–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Talber AM; Transter GE; Holmes E; Francis PL Determination of drug-plasma protein binding kinetics and equilibria by chromatographic profiling: exemplification of the method using L-tryptophan and albumin. Anal. Chem 2002, 74, 446–452. [DOI] [PubMed] [Google Scholar]
- 40.Weisiger RA Dissociation from albumin: a potentially rate-limiting step in the clearance of substances by the liver. Proc. Natl. Acad. Sci. USA. 1985, 82, 1563–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.(a) Kerns EH; Di L Drug-like Properties: Concepts, Structure, Design, and Methods; Elsevier, Inc.: Amsterdam, 2008; p. 234. [Google Scholar]; (b) Mei H; Korfmacher W; Morrison R Rapid in vivo oral screening in rats: reliability, acceptance criteria, and filtering efficiency. AAPS J 2006, 8, E493–E500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.(a) Bambal RB; Clarke SE Cytochrome P450: structure, function and application in drug discovery and development. Eval. Drug Cand. Preclin. Develop 2010, 55–107. [Google Scholar]; (b) Wang B; Zhou S-F Synthetic and natural compounds that interact with human cytochrome P450 1A2 and implications in drug development. Curr. Med. Chem 2009, 16(31), 4066–4218. [DOI] [PubMed] [Google Scholar]
- 43.Nagai M; Aoki M; Miyoshi I; Kato M; Pasinelli P; Kasai N; Brown RH Jr.; Itoyama Y Rats expressing human cytosolic copper-zinc superoxide dismutase transgenes with amyotrophic lateral sclerosis: associated mutations develop motor neuron disease. J. Neurosci 2001, 21, 9246–9254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aoki M; Kato S; Nagai M; Itoyama Y Development of a rat model of amyotrophic lateral sclerosis expressing a human SOD1 transgene. Neuropathology 2005, 25, 365–370. [DOI] [PubMed] [Google Scholar]
- 45.Gurney ME; Pu H; Chiu AY; Dal Canto MC; Polchow CY; Alexander DD; Caliendo J; Hentati A; Kown YW; Deng HX; Chen W; Zhai P; Sufit RL; Siddique T Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [DOI] [PubMed] [Google Scholar]
- 46.Del Signore SJ; Amante DJ; Km J; Stack EC; Goodrich S; Cormier K; Smith K; Cudkowicz ME; Ferrante RJ Combined riluzole and sodium phenylbutylrate therapy in transgenic amyotrophic lateral sclerosis mice. Amyotroph. Lat. Scler. Other Motor Neuron Disord 2009, 10, 85–94. [DOI] [PubMed] [Google Scholar]
- 47.Gurney ME; Cutting FB; Zhai P; Doble A; Taylor CP; Andrus PK; Hall ED Benefit of vitamin E, riluzole, and gababapentin in a transgenic model of familial amyotrophic lateral sclerosis. Ann. Neurol 1996, 39, 147–157. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.