

Abstract
The recent emergence of the novel coronavirus (SARS-CoV-2) has resulted in a devastating pandemic with global concern. However, to date, there are no regimens to prevent and treat SARS-CoV-2 virus. There is an urgent need to identify novel leads with anti-viral properties that impede viral pathogenesis in the host system. Esculentoside A (EsA), a saponin isolated from the root of Phytolacca esculenta, is known to exhibit diverse pharmacological properties, especially anti-inflammatory activity. To our knowledge, SARS-CoV-2 uses angiotensin converting enzyme 2 (ACE2) to enter host cells. This is mediated through the proteins of SARS-CoV-2, especially the spike glycoprotein receptor binding domain. Thus, our primary goal is to prevent virus replication and binding to the host, which allows us to explore the efficiency of EsA on key surface drug target proteins using the computational biology paradigm approach. Here, the anti-coronavirus activity of EsA in vitro and its potential mode of inhibitory action on the S-protein of SARS-CoV-2 were investigated. We found that EsA inhibited the HCoV-OC43 coronavirus during the attachment and penetration stage. Molecular docking results showed that EsA had a strong binding affinity with the spike glycoprotein from SARS-CoV-2. The results of the molecular dynamics simulation revealed that EsA had higher stable binding with the spike protein. These results demonstrated that Esculentoside A can act as a spike protein blocker to inhibit SARS-CoV-2. Considering the poor bioavailability and low toxicity of EsA, it is suitable as novel lead for the inhibitor against binding interactions of SARS-CoV-2 of S-protein and ACE2.
Keywords: 2019-nCoV, HCoV-OC43 coronavirus, Spike protein, Esculentoside A, Molecular simulation
1. Introduction
The coronavirus disease 2019 (COVID-19) pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2, 2019-nCoV) is an urgent public health emergency and has rapidly spread throughout the world [1]. However, there is currently no regimen to treat COVID-19. In view of the high mortality rate due to 2019-nCoV, the development of effective drugs against 2019- nCoV is essential [2,3]. 2019-nCoV consists of a spike protein (S), membrane protein (M), envelope protein (E), and nucleocapsid (N) [4]. 2019-nCoV enters the host cell by mediating viral attachment to angiotensin converting enzyme 2 (ACE2) receptor by the S glycoprotein [5]. The S-protein is made up of two subunits, the S1 subunit is involved in host cell receptor recognition and is the receptor-binding domain (RBD) and S2 subunit is responsible for fusion of the viral membrane and the host cellular membrane [6]. Due to the key role of S-protein in the process of SARS-CoV-2 infection, this structural component can be used as a drug target mediated by small molecule compounds, thus guiding the design and development of new therapeutic molecules [7]. It is important to conduct detailed molecular analyses and identification of the interaction between the ACE2 receptor and S-RBD of SARS-CoV-2, which is essential for the development of vaccines or therapeutic drugs for the prevention and treatment of SARS-CoV-2 infection [4]. In this study, the crystal structure of ACE2 and S-protein complex and the whole genome sequence of SARS-CoV-2 and human ACE2 were used to identify the virus binding residues on the ACE2 receptor using an in silico structure-based molecular docking and simulation approach. These residues of the SARS-CoV-2 S-protein were targeted to identify small molecules for the prevention and treatment of SARS-CoV-2 infection.
In addition, we focused on Esculentoside A (EsA, C42H66O16), a saponin isolated from the root of Phytolacca esculenta, commonly known as a herb for the treatment of bacteria, viruses and inflammation, which is widely used in China [8,9]. Studies have shown that it has strong anti-inflammatory, anti-tumor, anti-viral, and immunomodulatory effects [8,[10], [11], [12]].
We carried out a virtual screening simulation and the results demonstrated the therapeutic potential of EsA as an inhibitory agent targeting the interaction between S-protein and ACE-2. Thus, the main purpose of this study was to assess the anti-viral effect of EsA on the binding interactions between the S-protein and ACE2 receptor. The pharmacokinetics and pharmacodynamics of EsA were also studied. A classical molecular dynamics (MD) simulation study was performed and root-mean-square deviation (RMSD), hydrogen bond analysis, root mean square fluctuation (RMSF) calculation, radius of gyration (Rg), solvent accessible surface area (SASA), principal component analysis (PCA), and secondary structure of the protein were calculated to determine the stability of the protein–ligand complex. The free energy binding using molecular mechanics Poisson−Boltzmann surface area (MM-PBSA) and binding free energy, and energy decomposition calculation demonstrated the stability of EsA complexed with the S-protein [7]. Finally, the anti-viral activity of EsA in vitro on human coronavirus CoV-OC43 was investigated.
2. Materials and methods
2.1. Virus strain and cell culture
Human colon adenocarcinoma cell -8 (HCT-8, ATCC®CCL-244) was grown in Dulbecco's Modified Eagle's Medium (DMEM) containing 2% fetal bovine serum (FBS) and incubated in a humidified atmosphere of 5% CO2 at 37 °C. Human coronavirus OC43 (ATCC® VR-1558) was originally obtained from the American Type Culture Collection (ATCC, Manassas, VA 20108, USA).
2.2. Reagents and compounds
Esculentoside A [purity ≥98% by high-performance liquid chromatography (HPLC)] was obtained from Shanghai Yuanye Biotechnology Company (LOT: P09M10S87832, Shanghai, China), were diluted with saline to a final concentration of 0.5% Tween 80 and to the required concentrations for in vivo and in vitro experiments.
2.3. Antiviral activity assay
HCT-8 cells (2.5 × 104/well) were cultured in 96-well plates and in serum-free DMEM for 3 days at 37 °C. Cells were washed with serum-free Medium and infected with 100 μl of 100 mean tissue culture infective dose (TCID50) HCoV-OC43 for 2 h at 37 °C. After removing the virus-containing medium, the cells were treated with various doses of EsA (5, 10, or 20 μg/ml) solution in DMEM. All cultures were incubated at 37 °C for 72 h. All wells were then observed under a light microscope to determine cytopathic effect (CPE). To investigate the preventive and therapeutic effects of EsA at different stages of HCoV-OC43 replication, the effects on HCoV-OC43 infection were determined by three administration scheme: pre-incubation treatment, simultaneous treatment and post treatment.
2.4. Binding interaction analysis of the SARA-CoV-2 S-protein and ACE2
To study the binding interaction analysis of the SARA-CoV-2 S-protein and ACE2, the crystal structures of SARA-CoV-2 S-protein receptor binding domain bound to the ACE2 receptors (PDB ID:6LZG, 6M0J, 6VW1, and 7A94) were used [[13], [14], [15], [16]]. The crystal structure of spike glycoprotein of SARS-CoV-2 (PDB ID: 6XR8) and its receptor ACE2 (PDB ID: 1R42) were obtained from the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank [17,18]. The protein docking of the S-protein and ACE2 were performed using ZDOCK server (http://zdock.umassmed.edu/) [19]. The docking results of ZDOCK were optimized using the protein-protein docking in Rosetta software [20]. The docked structures of lowest interface score (I_sc) was selected for further analyzed for ACE2 and S-protein interaction using the Ligplot software [21].
2.5. Molecular docking simulations of EsA and S-protein
The molecular docking simulations of EsA and S-protein were performed using AutoDock Vina software [22]. The ligands were processed by autodock tools and transformed into pdbqt format. PyMOL software [23] was used to remove the water molecules, phosphate and redundant inactive ligands in the target protein. The target protein was hydrogenated and charged in autodock tools software. Autogrid was constructed around the active site defined in the crystal structure and the box of size x = 36.8 Å, y = 44.8 Å, z = 36.9 Å were placed over the S-protein internal region belonging to the ACE2, centering around residues (Phe490,Tyr489, Arg403, Asn487, Phe486, Gly485, Gly476, Gly502, Ser477, Thr478, Gln498, Tyr505, Gly496, Asn501, Tyr449, Asn450, Ser494, Leu452, Tyr453, Leu455, Gln493) with a lattice point spacing of 0.0375 nm. The semi-empirical potential function was used as the energy scoring function to search for the conformation and position of small molecules and ten independent docking experiments were carried out. The docking results were evaluated by scoring the binding affinity (kcal/mol). The best-scoring pose was judged and selected by the Vina docking score and visually analyzed using the LigPlot+1.4 software [21] and AutoDockTools 1.5.6 software (http://autodock.scripps.edu/).
2.6. MD simulation of complex with EsA and S-protein
MD simulation study is one of the effective tools to check the stability of the protein-ligand complex. The ligand with highest binding affinity scorer among the docking results was selected for further MD simulation studies.
Classical MD simulations of the systems were performed using two MD protocols, implemented in the GROMACS (version 5.0) [24] and AMBER software packages (http://ambermd.org/index.php) [25] as previously described [26]. All simulation work is done on the computer workstation: Intel®Xeon ®E5-2680 2.7 GHz CPU, 128 GB memory, 500GB hard disk, NVIDIA GeForce RTX 2080ti GPU graphics card.
-
(i)
AMBER: MD simulations were performed using the sander and pmemd modules in the AMBER 18 package, and the receptor and ligand proteins used the ff14SB force field. The complex was placed in a water cube with the center of mass in the center of the box. The system was solvated by the TIP3P water model. Sufficient numbers of Na + ad Cl− ions were added to neutralize the simulation system and further minimized the system by employing the steepest descent algorithm of 10,000 steps. NVT and NPT equilibration phases were carried for 1000 ps with 2.0 fs integration time step at 300 K temperature. After the system was in equilibrium, 50 ns MD simulation was carried out. The binding energy (ΔG binding) of the inhibitor with protein and the energy contribution during the MD simulation were analyzed by the AMBER in-built molecular mechanics/Poisson Boltzmann surface area (MM-PBSA) procedure during 50 ns simulation. The free energy of binding of either substrates/inhibitors with the protein was obtained by computing the difference between bound and unbound states: ∆ G0 bind,solv = ∆G0solv,complex − (∆G0 solv,ligand + ∆G0 solv,receptor) (http://ambermd.org/tutorials/advanced/tutorial3/index.php). The energy decomposition is calculated by GBSA (generalized Born/surface area) method [27]. The formula for the interaction energy of S-protein residues is as follows: △G = △GVDW + △GELE + △GGB + △GGbsur, where, △GVDW and △GELE are the van der Waals interaction energy and electrostatic interaction energy of each residue in vacuum, respectively; △GGB is the polar solvation energy of each residue, which is calculated by the generalized Bonn model; and △GGbsur is the nonpolar solvation energy, which is calculated by LCPO model. Each energy is divided into the main chain energy contribution and the side chain energy contribution of the residue.
-
(ii)
GROMACS. MD simulations for all systems were also repeated using the GROMACS MD package.
CHARMM36 force field of the inhibitors and proteins were selected from CHARMM General Force Field server(https://cgenff.umaryland.edu/). The Avogadro program(https://sourceforge.net/projects/avogadro/) was used to add the hydrogens to the heavy atoms. The prepared systems were first vacuum minimized for 1500 steps using the steepest descent algorithm. Then the structures were solvated in a cubic periodic box with water extending 2 Å outside the protein in all sides using TIP3P water model [28]. The system was then maintained at an appropriate salt concentration of 0.15 M by adding an appropriate amount of Na+ and Cl− counter ions. Then, the systems use the steepest descent algorithm to minimize the energy by 50,000 steps under solvation conditions. Subsequently, the V-type heavy-scale thermostat with coupling constant of 0.1 ps was used to gradually heat the system to reach 310 K temperature to balance in NVT ensemble. Then, a Parrinello Rahman constant with a coupling constant of 0.1 ps was used to maintain the solvent density at 1 bar and 310 K, and the equilibrium was carried out in the NPT ensemble. Finally, 100 ns unconstrained molecular dynamics simulation was carried out for the equilibrium system. The data generated from the simulation studies were analyzed using GROMACS simulation package (a package for molecular simulation and trajectory analysis).RMSD(backbone), Hydrogen bond analysis, RMSF calculation (Cα), Radius of gyration (Rg), SASA, and secondary structure of the protein were calculated using GROMACS utilities such as “gmx rms”, “gmx hbond”, “gmx rmsf”, “gmx gyrate”, “gmx sasa”, and “gmx do_dssp” respectively. In the RMSD plot, whole trajectory was considered for the analysis (for backbone atoms). While for RMSF, Rg and SASA the simulation window with an appreciable stability index was considered. PCA was performed to investigate the collective motion of proteins [29] and the gmx covar tool was used to generate the covariance matrix of backbone atoms for complex. The structural figures were visualized using PyMol software. The graphs and plots were generated using the Xmgrace tool (Boston, MA,USA).
2.7. Absorption, distribution, metabolism, elimination and toxicity(ADMET) and toxicity prediction by komputer assisted technology (TOPKAT) predictions
Physicochemical properties and pharmacokinetic properties play an important role in the discovery of new anti-viral drugs. The EsA was submitted to the ADMET and TOPKAT tools of small molecule protocol in Discovery Studio software (Acceltrs Software Inc. V2020,USA) for the in silico pharmacokinetics and pharmacodynamic studies. The ADMET study included parameters such as human intestinal absorption, aqueous solubility, blood–brain-barrier penetration (BBB), cytochrome CYP2D6 inhibition, plasma protein binding (PPB) and hepatotoxicity. The TOPKAT study included parameters such as animal models such as rat oral LD50, ocular irritation, skin irritancy, skin sensitization, weight of evidence carcinogenicity(WOE_prediction), rat inhalational LC50 and Ames mutagenicity. All these models were developed and validated based on a quantitative-structure toxicity relationship (QSTR) [30]. In addition, the chemical informatics analysis of EsA was carried out by SwissADME program (http://www.swissadme.ch/index.php) [31] and FAF-Drugs4 software (https://fafdrugs4.rpbs.univ-paris-diderot.fr/).
3. Result
3.1. Assessment of EsA anti-coronavirus ability in vitro
We examined the inhibitory effects of EsA on HCoV-OC43 coronavirus infection and progeny virus production in vitro in three different scheme, as shown in Table 1 . HCT-8 cells infected with HCoV-OC43 coronavirus microscopically showed cytopathic effect (CPE), including cell rounding, detachment and death. 5–20 μg/ml EsA significantly reduced CPE caused by infected HCT-8 cells. Treatment with EsA (5–20 μg/ml) before inoculation of coronavirus (HCoV-OC43) to HCT-8 cells resulted in a 10% less CPE (Table 1). The results showed that EsA could protect HCT-8 cells from virus infection.
Table 1.
Inhibitory effects of EsA on coronavirus(HCoV-OC43) infection and progeny virus production in vitro.
| Administration scheme | EsA (μg/ml) |
Virus-control | |||
|---|---|---|---|---|---|
| 20 | 10 | 5 | 2.5 | ||
| Pre-incubation treatment | − | − | − | + | ++++ |
| Simultaneous treatment | − | − | + | ++ | ++++ |
| Post treatment | − | ++ | +++ | +++ | ++++ |
HCT-8 cells (2.5 × 104/well) were treated with various amounts of EsA (2.5–20 μg/ml) before, at the same time as, and after inoculation with HCoV-OC43 coronavirus suspension (100TCID50/well). After incubation for 72 h at 37 °C, 5%CO2, the cell viability was determined by the cytopathic effect (CPE). The CPE occurrence was scored using an inverted microscope. “−”: < 10% CPE; “+”: < 25% CPE; “++: < 25% ~50% CPE; “+++”: < 50% ~75% CPE; “++++”: < 75% ~100% CPE.
3.2. Interaction of SARS-Cov-2 spike protein with human ACE2 receptor
The crystal structures of SARS-Cov-2 S-protein and its receptor ACE2 complex have been solved, such as 6LZG, 6M0J, 6VW1, and 7A94. By performing protein-protein docking, we obtained an interactive complex structure for S-protein of 2019-nCoV with ACE2. We further analyzed the interactions to examine the binding ability of S-protein to ACE2.
The contact between the ACE2 and the S-protein involve the interface region of RBG of S-protein. In these complexes, there are 14 (6LZG),17(6LZG),16(6LZG) and 12(6LZG) amino acid residues in 2019-nCoV RBD that are directly in contact with ACE2. The interface region between the S-protein (6XR8) and ACE2(1R42) had the maximum number of residues in direct contact with ACE2 in the 2019-nCoV RBD (Table 2 ).
Table 2.
Contact residues of the SARS-CoV-2 S-proteine-ACE2 interface.
| Complexes (PDB:ID) | S-proteina | Number of interactive residues in S-protein | ACE2a | Number of interactive residues in ACE2 |
|---|---|---|---|---|
| 6LZG | Gly496,Gly502,Tyr476,Asn501,Thr500,Ala475,Asn487,Lys417,Gln498,Phe456,Tyr489,Tyr453,Leu455,Gln493 | 14 | Gly354,Lys353,Arg357,Tyr41,Met82,Asp355,Asn330,Ser19,Gln24,Tyr83,Thr27,Asp30,Leu45,Phe28,Asp38,Gln42,Glu35,His34 | 18 |
| 6M0J | Asn501,Gly502,Tyr505,Thr500,Lys417,Tyr449,Gly446,Gln493,Gln498,Tyr453,Phe456,Leu455,Tyr489,Phe486,Asn487,Gly496,Ala475 | 17 | Asn330,Lys353,Asp355,Tyr41,Asp30,Gly354,Asp38,Leu79,Gln42,Lys31,His34,Arg357,Thr27,Glu37,Met82,Phe28,Thr83,Glu24 | 18 |
| 6VW1 | Tyr489,Ala475,Phe456,Tyr453,Gln498,Gln493,Asn501,Thr500,Leu455,Gly476,Tyr449,Asn487,Tyr505,Gly502,Gly496,Phe486 | 16 | Thr27,Phe28,Leu45,Ser19,Gln42,Asn330,His34,Glu35,Lys31,Tyr41,Arg357,Asp355,Leu79,Glu24,Glu37,Asp38,Tyr83,Lys353,Met82 | 19 |
| 7A94 | Phe486,Phe456,Asn487,Gly502,Gly476,Ala475,Tyr505,Asn501,Leu455,Gln498,Gln493,Thr500 | 12 | Gln24,Met82,Tyr79,Phe28,Ser19,Glu23,Thr27,Lys353,Lys31,His34,Tyr41,Arg357,Glu35 | 13 |
| 6XR8-1R42 | Phe490,Tyr489,Arg403,Asn487,Phe486,Gly485,Gly476,Gly502,Ser477,Thr478,Gln498,Tyr505,Gly496,Asn501,Tyr449,Asn450,Ser494,Leu452,Tyr453,Leu455,Gln493, | 21 | Pro135,Val49,Gln175,Leu162,Glu166,Pro490,Asn159,Gly130,Tyr158,Ser170,Glu171,Tyr613,Thr125,Pro612,Ala614,Asp615,Leu143,Lys131,Thr129,Cys141,Gln139,Pro138,Cys133,Val132 | 24 |
Analyzed in the Ligplot program.
A total of 21 residues of the RBD of 2019-nCoV were in stable and efficient contact with 24 residues of the ACE2 receptor (Fig. 1, Fig. 2 , Table 2). An important feature of the S-RBD-CoV-2/ACE2 interface is the number of hydrophilic interactions and hydrogen bonds (HBs) detected in the relevant crystal structures (Fig. 1). The hydrogen bonds (HBs) and 3 salt bridges (SBs) stably populate the S-protein CoV-2/ACE2 interface (Fig. 1). As atomistic structural and energetical information would greatly improve our understanding of interaction between the viral S-protein RBD and its cellular ACE2 receptor, providing fundamental indications about important protein targets for the molecular docking . A receptor grid-box was generated by centering around residues in 2019-nCoV S-protein that are directly in contact with ACE2(Fig. 2).
Fig. 1.

The S-protein interacts with the ACE2 receptor.
Fig. 2.

The molecular interactions of S-protein and ACE2, and molecular binding site interactions of EsA docked to 2019-nCoV.
3.3. Molecular docking of EsA with 2019-nCoV S-protein
The molecular docking was performed to investigate the potential binding between S-protein and EsA. In this study, we selected the centering around residues in 2019-nCoV RBD that are directly in contact with ACE2 as the molecular binding site (Fig. 3 ).
Fig. 3.

The details of the binding between EsA and S-protein (A), and the structures of complex of S-protein (B) and EsA(C).
The binding energy of EsA was summarized in Table 3 . The docking energy of EsA was −13.1 kcal/mol. The contributions of different energies were analyzed to further dissect the docking energy. In the interaction between S-protein and EsA, van der Waals's interaction, hydrogen bond (H-bond), desolvation energy and electrostatic energy were the main parts. The vdW + Hbond+desolv energy of EsA was −24.19 kcal/mol and electrostatic energy was −5.485 kcal/mol in the docking results (Table 3). All results showed that EsA had better binding ability to S-protein.
Table 3.
The energies for the binding between EsA and S-protein.
| Molecules | vdW + Hbond+desolv energy (kcal/mol) | Electrostatic energy (kcal/mol) | Total binding energy (kcal/mol) | The best docking energy (kcal/mol) | Binding free energy (kcal/mol) |
|---|---|---|---|---|---|
| EsA | −24.19 | −5.485 | −42.899 | −13.1 | −24.81 |
The binding pocket of EsA was located in the RBD site, which has been observed to be an acceptor for ACE2. EsA fitted well with the shape of the pocket, as shown in Fig. 3A–B. To further understand the details of S-protein binding to EsA, the interaction of S-protein with EsA was visualized in a two-dimensional plot (Fig. 4 ), which formed a total of six hydrogen bonds. The abundant H-bonds indicated that H-bonds play an important role in the binding of EsA to S-protein. The residues of the S-protein receptor, namely Arg-594 (bond length: 3.21 Å), Pro-595 (bond length: 3.26 Å), Tyr-141 (bond length: 2.69 Å), Asn-142(bond length: 3.15 Å), and Arg-238 (bond length: 2.80 and 2.63 Å), formed five hydrogen bonds with EsA. These results showed that EsA bound to the receptor in the active pocket, forming a stable complex that inhibited the activity of the S-protein.
Fig. 4.

The interaction between EsA and S-protein receptors.
3.4. The dynamics stability MD simulation of EsA with S-protein
EsA showed good binding ability to S-protein in molecular docking test. During the MD simulation, EsA and S-protein also showed stable binding in the binding pocket during the simulation period (Fig. 5 ).
Fig. 5.

Molecular dynamics structures at 5 ns (A), 10 ns (B), 30 ns (C) and 50 ns (D), and superimposed structures of EsA with S-protein, showing the position of the binding pocket over the stimulation period.
3.4.1. MD trajectory stability analysis
The RMSD of MD simulation trajectory is an important basis to measure whether the system is stable or not. It was found that the larger the RMSD value, the greater the molecular position variation; the smaller the RMSD value, the greater the molecular position variation and the more stable the system. Therefore, it is an important basis to measure whether the system is stable or not [32]. To test the stability of the MD trajectory, a molecular dynamics simulation of 100 ns was performed on the complex with EsA and S-protein. Taking the initial protein conformation of the crystal as a reference, the variation trend of the RMSD of the backbone atoms in the protein was analyzed with the simulation time, and the stability of the protein conformation of the complex system was investigated. There is a small change in the position of the ligand relative to the protein(Fig. 6 ). The binding pose was well maintained during the simulation, suggesting that the presence of EsA changed the conformation of the protein and that the overall range of change was small (Fig. 6A). This is mainly because EsA stabilized the overall conformation of the S protein, thus better maintaining the original conformation of the protein. The RMSD value reached the maximum at about 30 ns and then gradually balanced (Fig. 6B). After equilibration, the RMSD value ranged from 0.218 nm to 0.461 nm and the average RMSD value was 0.325 nm. Compared with the initial protein crystal conformation, complex system of EsA-S-protein changed very little (Fig. 6C). Both time series show the RMSD levels off to ~0.1 nm, indicating that the structure is very stable. Subtle differences between the plots indicate that the structure at t = 0 ns is slightly different from this crystal structure. The RMSD level of the two time series almost overlaps, indicating that the structure is very stable.
Fig. 6.

MD simulation trajectory analysis. (A) RMSD change of LIG_Heavy atoms in MD simulation. (B) RMSD change of S-protein backbone atoms in MD simulation. (C) Change of backbone RMSD of complex system relative to crystal structure position with simulation time. (D) RMSF of residues in the protein during dynamics simulations.
To study the fluctuation of amino acid residues in S-protein, the RMSF of Cα atoms for each residue of S-protein was calculated during molecular dynamics simulation (Fig. 6D). The average RMSF value of amino acid residues in the complex was 0.146 nm and most of the residues fluctuated at lower values. In general, the RMSF profile shows fluctuations at the loop regions with ligand. The RMSF of EsA showed a fluctuation with a high peak at regions 29–54, 119–123, 321–325 and 408–412 of S-protein. Apart from these, no obvious conformational fluctuations were observed in the active site and the binding residues with EsA were very stable.
3.4.2. Radius of gyration (Rg)
To understand the influence of inhibitors on protein compactness, we calculated radius of gyration (Rg) of the protein. The radius of gyration of a protein is a measure of its compactness which is greatly influenced by the combination of secondary structures (alpha and beta proteins). If a protein is stably folded, it will likely maintain a relatively steady value of Rg. If a protein unfolds, its Rg will change over time. As can be seen from the plot of Rg values over time, the protein maintains a very stable compact (folded) form over the course of 100 ns, indicating that the Rg of the system is very stable after EsA binding to the protein (Fig. 7 ). The value of Rg in the complex is between 0.2484 and 2.659 nm, with an average value of 2.590 nm. It reaches the maximum value at about 10 ns and then gradually balances (Fig. 7).
Fig. 7.
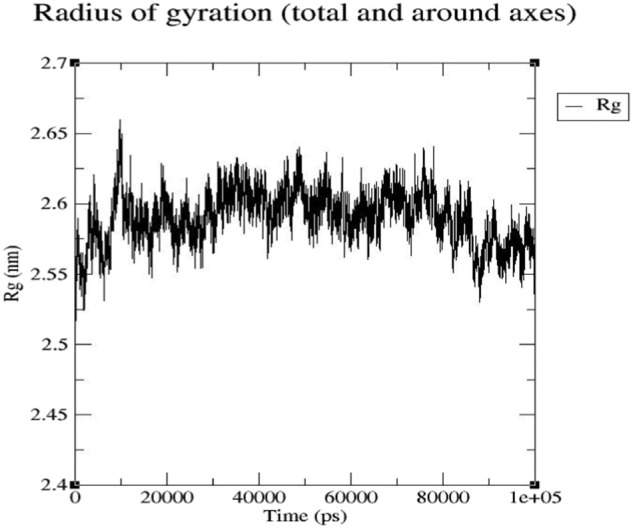
Plot of radius of gyration varies with time.
3.4.3. Hydrogen bond analysis
Hydrogen bonding is an important force in substrate recognition and in maintaining the stability of proteins and ligands. The hydrogen bond formed between S-protein and EsA was analyzed by GROMACS program. The geometric criterion for hydrogen bond is that the angle between hydrogen donor receptor is not more than 30° and the distance between donor receptor is not more than 0.3500 nm. In order to analyze the hydrogen bonding during the simulation more visually, we analyzed the number, length and angle of the hydrogen bonds. During the simulation, the hydrogen bond numbers varied between the EsA and S-protein were recorded and plotted in Fig. 8 A. EsA showed stable hydrogen bond interactions with S-protein during the simulation period, with 1–3 hydrogen bonds, and the highest number reached 5 hydrogen bonds. These results show that the hydrogen bond interaction between EsA and S-protein was very stable and continuous.
Fig. 8.

Inter-molecular H-bond for complex of EsA-S-protein. (A) Plot of hydrogen bond number varies with time. (B) Plot of hydrogen bond angle varies with time.(C) Plot of hydrogen bond distance varies with time.
The angle of hydrogen bond fluctuates in the range of 0.5°–29.5° and the average hydrogen bond angle was 15° (Fig. 8B). The bond length of hydrogen bond fluctuated in the distance of 0.242–0.347 nm, and the average length of hydrogen bond was 0.295 nm (Fig. 8C). The above results showed that the hydrogen bonds between S-protein and EsA were relatively stable and strong during the simulation, which played an important role in the stability of the complex.
3.4.4. Secondary structure analysis
Time evolutions of secondary structural units were also analyzed using DSSP, GROMACS result analysis module, as shown in Fig. 9 . It was observed that the secondary structure of amino acid residues existed mainly in the form of α-helix during the period of 50–100 ns. With the progress of simulation, the secondary structure mainly transformed between coil, β-sheet, bend and turn. Finally, the secondary structure was stable in the form of bend. It can be seen that the presence of ligands stabilized the secondary structure of S-protein, but also caused changes in the secondary structure of the residues.
Fig. 9.

Secondary structure changes of complex.
3.4.5. Solvent accessible surface area (SASA) analysis
The solvent exposed surface area of a protein was analyzed using SASA, which the interaction of proteins with various solvents and ligands depends primarily on their surface properties. The total solvent accessible surface area of the complexes ranged from 263.125–301.527 nm with an average of 287.17 nm and was relatively stable during the simulation (Fig. 10 ). These results indicated that the interaction between EsA and S-protein has little effect on the solvent accessible area of protein structure.
Fig. 10.
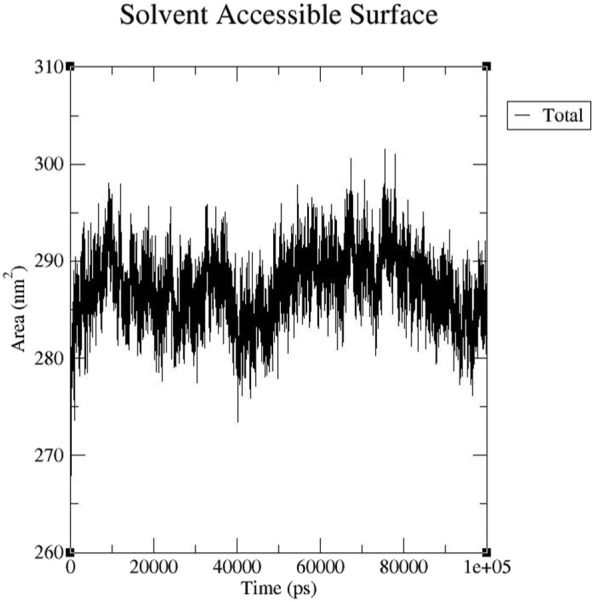
Plot of solvent accessible surface area (SASA) of complex.
3.4.6. Binding energy and energy decomposition analysis
The binding energy and the energy contribution were analyzed by the MM-PASA program during the 50 ns simulation (Fig. 11 ). The binding energy of S-protein with EsA was −13.12 kcal/mol. The residue TPR503 in the complex of EsA-S-protein contributed the most to the binding energy (<−4.0 kcal/mol) and PRO-241 and Arg-238 were also the main primary contributors (Fig. 11). Most of the residues involved in the contribution to the binding energy were consistent with the major residues in the above docking results.
Fig. 11.

Residue binding energy contribution of EsA-S-protein complex. The marked residues contribute to the binding energy values ≦−4.0 kcal/mol.
A molecular simulation approach was used to study the binding properties of EsA to S protein to explain its antiviral molecular mechanism. Binding free energy of S-protein with EsA was over −24.81 kcal/mol (Table 2). According to the trajectories, the RMSF of EsA binding with S-protein is shown in Fig. 6B. In general, the RMSF profile shows fluctuations at the loop regions with ligand. The RMSF of EsA showed a fluctuation with a high peak at regions 29–54, 85–99, 109–147, 270–290 and 475–482 of S-protein. Apart from these, no obvious conformational fluctuations were observed in the active site and the binding residues with EsA was very stable.
In order to study the binding mechanism of S-protein and EsA, the GBSA module of AMBER 18 software was used to calculate the interaction between EsA and the residues of S-protein. The energies decomposition of the main residues was shown in Table 4 .
Table 4.
Energy contribution of the key residues computed by GB model (kcal/mol).
| Location | ∆GVDW |
∆GELE |
∆GGB |
∆GGbsur |
Total energy | ||||
|---|---|---|---|---|---|---|---|---|---|
| Contribution of side chain | Contribution of main chain | Contribution of side chain | Contribution of main chain | Contribution of side chain | Contribution of main chain | Contribution of side chain | Contribution of main chain | ||
| R-TRP593 | −2.664 | −0.167 | −2.559 | −0.328 | 2.586 | 0.222 | −0.358 | −0.013 | −3.280 |
| R-PRO241 | −1.532 | −0.382 | 0.467 | −0.193 | −0.435 | 0.338 | −0.285 | −0.000 | −2.023 |
| R-SER240 | −1.323 | −0.591 | −0.378 | 0.306 | 1.093 | −0.497 | −0.088 | −0.040 | −1.517 |
| R-ARG238 | −0.920 | −0.600 | −1.649 | −0.705 | 2.208 | 0.385 | −0.074 | −0.024 | −1.380 |
| R-PHE586 | −0.904 | −0.128 | −0.074 | 0.003 | 0.421 | 0.013 | −0.207 | −0.017 | −0.893 |
| R-ILE239 | −0.259 | −0.639 | 0.034 | −0.621 | −0.005 | 0.851 | 0.000 | −0.046 | −0.685 |
| R-PRO595 | −0.519 | −0.645 | −0.514 | 0.306 | 0.549 | 0.279 | −0.007 | −0.122 | −0.673 |
| R-TYR596 | −0.323 | −0.340 | −0.055 | 0.020 | 0.115 | 0.134 | −0.022 | −0.040 | −0.511 |
| L-LIG | −18.707 | 0.000 | −2.743 | 0.000 | 11.060 | 0.000 | 2.734 | −0.000 | −13.123 |
Note: △GVDW: van der Waals interaction energy; GELE: electrostatic interaction energy; △GGB: Polar Solvation energy; △GGbsur: Non-polar Solvation energy.
It can be seen from Table 4 that the binding free energy of EsA with the residues of TPR593, PRO241, SER240 and ARG238 were greater. The contribution of each residue on the receptor to the binding energy was smaller than that on the ligand. Although PRO241 had smaller van der Waals interaction energy (−1.664 kcal/mol) and the smallest polar solar energy (−0.435 kcal/mol), the contribution of PRO241 (−2.023 kcal/mol) ranked second in the contribution of receptor residues due to the larger electronic interaction energy (0.467 kcal/mol) on the side chain. TPR593 (−3.280 kcal/mol) contributed the most to the binding free energy of the receptor with smaller van der Waals interaction energy (−2.664 kcal/mol) and the smallest non polar solar energy (−0.358 kcal/mol) on its side chain, which was offset by its larger polar solar energy (2.586 kcal/mol). The ligand makes the largest contribution to the binding free energy (−13.123 kcal/mol), mainly from the van der Waals interaction energy (−18.707 kcal/mol) and the electrochemical interaction energy on the side chain (−2.743 kcal/mol). As found in the molecular docking structure, the whole ligand was trapped like a key in the cavity composed of TRP593, SER240, PRO241, ARG238, etc. (Fig. 3). The above energy decomposition results well validated the binding mode of S-protein and EsA. These results provided clues for the rational design of inhibitors based on the S-protein structure.
3.4.7. Principal component analysis (PCA)
The PCA was performed to analyze the atomic motions of S-protein alone and in the presence of compounds using Cartesian coordinates of the Cα atoms. The principal motion was used to get insight into the correlated motion of a protein characterized by eigenvector and eigenvalues.
We calculated the cross-correlation matrix describing the fluctuation of protein Cα atoms to further explore the effect of EsA binding on the internal dynamics of S-protein, as shown in Fig. 12 . The matrix showed the covariance between atoms. Red indicated that two atoms moved in the same direction, while blue indicated that they moved in opposite directions. The depth of red indicated the amplitude of the wave. The combination of EsA had a significant effect on the motion mode of S-protein (Fig. 12A). The first two eigenvectors accounted for the significant amount of overall motion of complex, thus first two eigenvectors were selected for the analysis. The PCA highlight that complex covered a larger phase space along PC1 and PC2 (Fig. 12B). Fig. 12C showed the relationship between the eigenvalue and the eigenvector index after diagonalization of the covariance matrix constructed by atomic coordinates. The results showed that the complex of S- protein and EsA decreased in exercise intensity and maintained a relatively stable structure.
Fig. 12.
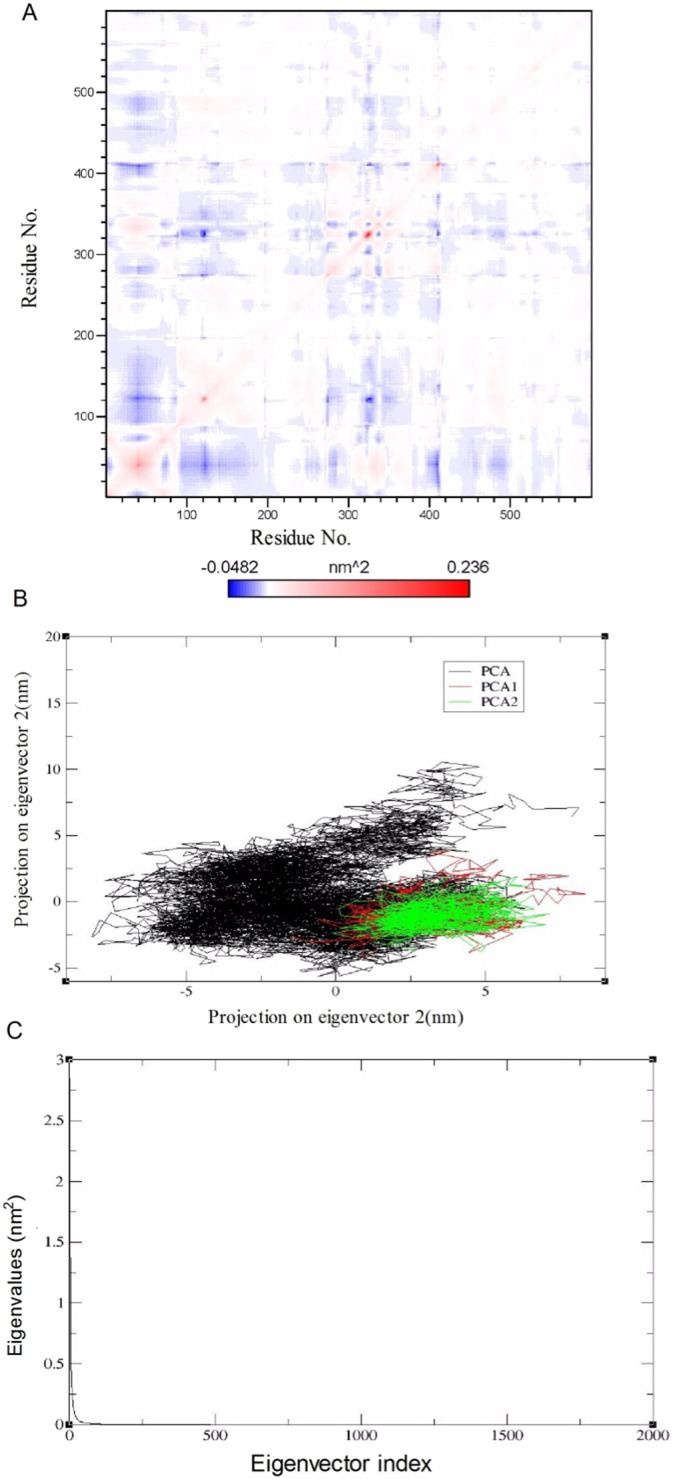
Principal components analysis (PCA) of MD trajectory. (A) Cross-correlation matrix of Cα atom fluctuation near its corresponding average position after equilibrium. The matrix shows the covariance between atoms. Red indicated that two atoms moved in the same direction, while blue indicated that they moved in opposite directions. The depth of red indicated the amplitude of the wave. (B) 2D-projection of the atomic motion of complex in phase space along the first two principal eigenvectors. (C) Eigenvector index obtained from the Cα covariance matrix constructed in the equilibrium phase of MD simulation. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.5. Computational pharmacoinformatic study on EsA
Estimation of ADMET properties for EsA using Discovery Studio software are given in Table 5 . We evaluated EsA absorption, distribution, metabolism and excretion in five aspects: water solubility, BBB, PPB, hepatotoxicity and human intestinal absorption. The absorption profile is directly related to the availability of drug in systemic circulation. The ADMET results revealed that EsA exhibited poor intestinal absorption and water solubility. Distribution of drugs to the various tissue and organs guaranteed its better treatment efficiency. EsA has low blood–brain barrier penetration and Plasma-Protein Binding. The drug metabolism analysis suggested that EsA was predicted as either non-inhibitors of CYP2D6 enzymes or non-toxicity for hepatocytes. Overall, EsA is non-toxic to hepatic cells, non-inhibitors of a metabolic enzyme (CPYD26), and exhibit very high to medium penetration across the BBB and PPB (Table 5).
Table 5.
Computational pharmacokinetic parameters (ADMET) properties of EsA.
| Compounds | MW | Aqueous solubility level | Blood-brain barrier (BBB) | Cytochrome (CYP-2D6) binding | Hepato-toxicity | Intestinal absorption level | Plasma-protein binding (PPB) prediction |
|---|---|---|---|---|---|---|---|
| EsA | 826.9 | 2 (Yes, low) | 4 (very low) | False (non-inhibitor) | FALSE (non-Toxic) | 3 (very poor) | False (poorly bounded) |
Further, EsA was also evaluated for its carcinogenicity and hepatotoxicity (Table 6 ). Quantitative-structure toxicity relationship (QSTR)-based toxicity parameters such as rat (TD50), rat_oral (LD50), rat_inhalation_ fathead minnow (LC50), Skin sensitization, Skin irritancy, and Ocular irritancy are summarized in Table 6. The pharmacodynamic models of EsA are free from carcinogens and mutagens in silico. EsA showed weak skin sensitization but moderate-to-severe irritation to the skin and eyes (Table 6). In a short, EsA possess poor bioavailability and low toxicity and carcinogenicity.
Table 6.
Computational toxicity risk assessment parameters (DS TOPKAT, Accelrys, USA) OF EsA.
| Compounds | Rat oral LD50 (g/kg) | TOPKAT_Rat_inhalational_LC50 (mg/m3/h) | TOPKAT_Ames mutagenicity | TOPKAT_WOE_prediction | Skin sensitization | Skin irritancy | Ocular irritancy |
|---|---|---|---|---|---|---|---|
| EsA | 3.15164 | 1.35905 | Non-mutagen | Non-carcinogen | Weak | Moderate | Severe |
SwissADME and FAF-Drugs were employed to perform chemical informatic analyses to further evaluate the drug abilities, and Lipophilicity of EsA. The results were showed in Table 7 and Fig. 13 . The logarithm of the partition coefficient between water and 1-octanol (known as log P) of EsA was 1.72, less than five, and the values of Log P o/w, Log P o/w, Log P o/w, Log P o/w, Log P o/w, Consensus Log P o/w were 3.92, 0.83, 0.62, −0.62, 0.21 and 0.99 respectively, which indicated that EsA was a liposolubility drug with good absorption. These results indicated that EsA had low gastrointestinal tract absorption and no probability of penetration into the CNS via the BBB.
Table 7.
The chemical informatics analyses of EsA.
| Molecules | LogP | Log Po/w (iLOGP) |
Log Po/w (XLOGP3) |
Log Po/w (WLOGP) |
Log Po/w (MLOGP) |
Log Po/w (SILICOS-IT) |
Consensus Log Po/w |
|---|---|---|---|---|---|---|---|
| EsA | 1.72 | 3.92 | 0.83 | 0.62 | −0.62 | 0.21 | 0.99 |
Fig. 13.
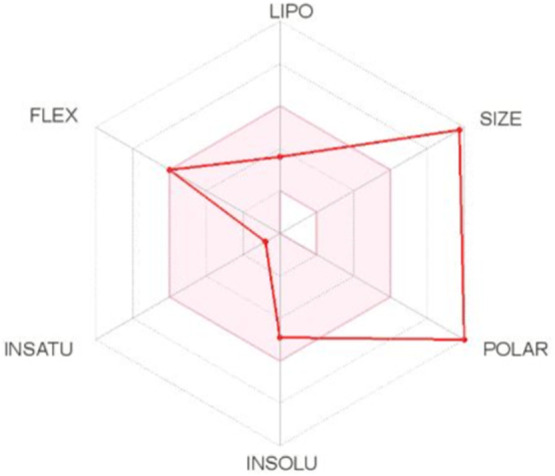
Bioavailability radar of EsA. The colored zone is the suitable physicochemical space for oral bioavailability. LIPO(Lipophilicity):−0.7 < ALOGP3 < +5.0; SIZE:150 g/mol < MV < 500 g/mol; POLAR(Polarity):20 Å2 < TPSA <130 Å2; INSOLU(Insolubility):0 < Log S (ESOL) < 6; INSATU (Insaturation):0.25 < Fraction Csp3 < 1; FLEX(Flexibility): 0 < Num.rotable bonds <9. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
4. Discussion
2019-nCoV results in a wide spectrum of life-threatening symptoms in patients, mainly affecting the respiratory and gastrointestinal systems, which is based on the S-protein protruding from the viral envelope [[33], [34], [35]]. There are no approved vaccines or drugs that are effective against this deadly strain of the virus. Thus, there is an urgent need to manage the situation by identifying potential inhibitors of 2019-nCoV. This preliminary study was carried out to identify effective inhibitors of the 2019-nCoV spike glycoprotein.
Esculentoside A, a triterpene saponin, was isolated from the root of the perennial herb, Phytolacca esculenta [36]. It was previously found that EsA has activity in regulating immune response, inhibiting cell proliferation and apoptosis, and anti-inflammation [12,37,38]. In particular, EsA has strong anti-inflammatory activity, and inhibits pro-inflammatory factors and inflammatory cytokine production in vitro and in vivo [[39], [40], [41]]. This in silico and in vitro study also suggested that EsA is a bioactive compound against the S-protein of 2019-nCoV.
We found that EsA had an inhibitory effect on the human CoV-OC43 coronavirus during the attachment and penetration stage. The inhibitory effect of EsA on coronavirus was high both before and upon viral inoculation, but was low after viral inoculation. EsA inhibited viral replication when added before viral adsorption.
SARS-CoV-2, which causes COVID-19 disease, uses the fusion spike glycoprotein to penetrate into the host cell via ACE2, a transmembrane protein expressed on the surface of lung alveolar cells. This results in a major immune inflammatory response. Deaths are due to respiratory failure, which has been linked to a cytokine storm with high serum levels of pro-inflammatory cytokines and chemokines [42]. As this interaction is essential for SARS-CoV-2 entry into the host cell and infection, drugs targeting S-protein–ACE2 interface protein-protein interactions could potentially inhibit virus entry into host cells and thus, may provide a solution for controlling SARS-CoV-2 infections [2].
In this study, the interaction between EsA and the S-protein was studied using a molecular docking and MD simulation. The docking results of EsA with S-protein showed that the ligand and receptor formed a stable complex. The MD simulation of this complex system was carried out using GROMACS. Then, RMSD, hydrogen bond, RMSF, Rg, SASA, secondary structure of the protein, PCA and binding free energy and energy decomposition calculation were used to analyze the complex system after the dynamic simulation. The results showed that the complex system of EsA and S-protein demonstrated less fluctuation and formed a low-energy dynamic equilibrium state during the MD simulation. The ligand EsA can limit the conformation of S-protein, and play a key role in controlling the changes in protein secondary structure, core distance of the key residual matrix, etc., resulting in a stable EsA-S-protein complex. The results of PCA showed that motion of the complex system was not obvious during the simulation process. Following simulation, EsA forms stable hydrogen bonds with some important residues at the active site of S-protein. EsA can not only form hydrogen bonds with Arg594, Pro595, Tyr141, Asn142, and Arg238, but also form key hydrophobic interactions with Asp592, Trp593, Tyr596, Ser237, Ala597, Arg238, Leu145, Val474, Tyr596, and Ser237 in the active pocket, which makes it stable in the active pocket. It was found that van der Waals energy, electrostatic interaction energy and nonpolar solvation energy are favorable for the binding of EsA with S-protein. During the MD simulation, the main residues Trp593, Ser240, Pro241, Arg238, Tyr505, Asn501, Gln498, and Thr500 near the active pocket play an important role in binding the inhibitor with the S-protein. The results of the bioactivity test, molecular docking and binding energy calculation of MM-PBSA were consistent. These results provide an important theoretical basis for further understanding of the anti-viral mechanism of EsA on the S-protein.
Our docking results demonstrated that EsA has an inhibitory effect on the S-protein. EsA is a triterpene saponin derived from the root of Phytolacca esculenta, which has been traditionally used in China for the management of various disease such as inflammation, fever, and treatment of infections, and autoimmune disorders such as arthritis [36]. EsA is also a well-known regulator of various interleukins, chemokines, and cytokines. Recent studies have shown that the levels of tumor necrosis factor-α, interleukin-1, interleukin-6, nitric oxide, and interferons were elevated in patients with SARS-CoV-2 [43]. Therefore, EsA can be used as an alternative therapy for the management of various signs and symptoms associated with SARS-CoV-2 infection.
We further analyzed the ADMET and TOPKAT of EsA. According to the Lipinski rule for drug-likeness, any drug molecule with MW ≥ 500 g/mol, hydrogen-bond-donating atoms ≥5, and hydrogen-bond-accepting atoms ≥10 is not considered a good pharmaceutical agent in terms of oral activity. As EsA did not meet 3 of the 5 criteria of Lipinski's rule, EsA was considered not to have drug-likeness and would have problems with oral bioavailability. EsA is a liposoluble drug with good absorption, and is a safe compound. Therefore, designing an attachment inhibitor from EsA may provide a safe and effective method for combating SARS-CoV-2 infection. The findings in the present study could facilitate the use of EsA as a useful lead for drug structure transformation of potent anti-viral agents for the prevention and treatment of SARS-CoV-2. In the future, we aim to perform clinical, in vitro, and in vivo experiments to confirm the efficacy of EsA against SARS-CoV-2.
5. Conclusion
In conclusion, EsA could act as a spike protein blocker to inhibit SARS-CoV-2. Considering the pharmacokinetic and toxicity of EsA, It is suitable for EsA as novel lead for the drug structure transformation of inhibitor against the S-protein of SARS-CoV-2.
CRediT authorship contribution statement
All authors have contributed substantially to the design and execution of this study and approve the final version of this manuscript. Miss Maosen Zeng performed the presented experiments and wrote the manuscript. Miss Wendi Yu and Huixian Wang conducted experiments and analyzed data. Dr. Jinyuan Liu conducted checking and reviewing of manuscript. Prof. Peiping Xu provided financial support and designed the presented experiments and revised the manuscript. All authors read and approved the manuscript.
Declaration of competing interest
We wish to confirm that there are no known conflicts of interest associated with this publication and there has been no significant financial support for this work that could have influenced its outcome.
Acknowledgements
We thank International Science Editing (Co. Clare, Ireland) for the language editorial assistance in the preparation of the manuscript.
References
- 1.Wang C., Horby P.W., Hayden F.G., Gao G.F. A novel coronavirus outbreak of global health concern. Lancet. 2020;395(10223):470–473. doi: 10.1016/S0140-6736(20)30185-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pillaiyar T., Manickam M., Namasivayam V., Hayashi Y., Jung S.H. An overview of severe acute respiratory syndrome-coronavirus (SARS-CoV) 3CL protease inhibitors: peptidomimetics and small molecule chemotherapy. J. Med. Chem. 2016;59(14):6595–6628. doi: 10.1021/acs.jmedchem.5b01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vijayaraj R., Altaff K., Rosita A.S., Ramadevi S., Revathy J. Bioactive compounds from marine resources against novel corona virus (2019-nCoV):in silicostudy for corona viral drug. Nat. Prod. Res. 2020;1:1–5. doi: 10.1080/14786419.2020.1791115. [DOI] [PubMed] [Google Scholar]
- 4.Wu F., Zhao S., Yu B., Chen Y.M., Wang W., Song Z.G., Hu Y., Tao Z.W., Tian J.H., Pei Y.Y., et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Starr T.N., Greaney A.J., Hilton S.K., Ellis D., Crawford K.H.D., Dingens A.S., Navarro M.J., Bowen J.E., Tortorici M.A., Walls A.C., et al. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell. 2020;182(5):1295–1310. doi: 10.1016/j.cell.2020.08.012. (1295-+) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonavia A., Zelus B.D., Wentworth D.E., Talbot P.J., Holmes K.V. Identification of a receptor-binding domain of the spike glycoprotein of human coronavirus HCoV-229E. J. Virol. 2003;77(4):2530–2538. doi: 10.1128/JVI.77.4.2530-2538.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Romeo A., Iacovelli F., Falconi M. Targeting the SARS-CoV-2 spike glycoprotein prefusion conformation: virtual screening and molecular dynamics simulations applied to the identification of potential fusion inhibitors. Virus Res. 2020;286:198068. doi: 10.1016/j.virusres.2020.198068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang W.W., Hong L., Xu X.X., Wang Q., Huang J.L., Jiang L. Study on the inhibitory effects of Esculentoside A on cervical cancer through PI3K/AKT/mTOR signaling pathway in vitro and in vivo experiments. J. Biomater. Tissue Eng. 2018;8(4):568–573. doi: 10.1166/jbt.2018.1778. [DOI] [Google Scholar]
- 9.Xiao Z.Y., Zheng Q.Y., Jiang Y.Y., Zhou B., Yin M., Wang H.B., Zhang J.P. Effects of Esculentoside A on production of interleukin-1,2 and prostaglandin E-2. Acta Pharmacol. Sin. 2004;25(6):817–821. [PubMed] [Google Scholar]
- 10.Zhang F., Wang X.T., Qiu X.C., Wang J.J., Fang H., Wang Z.H., Sun Y., Xia Z.F. The protective effect of Esculentoside A on experimental acute liver injury in mice. PLoS One. 2014;9(11):e113107. doi: 10.1371/journal.pone.0113107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang H.B., Fang J., Zheng Q.Y. Inhibitory effect of Esculentoside A on prostaglandin E(2) production from murine peritoneal macrophages and rabbit synovial cells in vitro. Mediat. Inflamm. 1997;6(1):22–24. doi: 10.1080/09629359791884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu Z.L., Qiu L., Xiao Z.Y., Wang J., Yu Q., Li J.Z., Feng H., Guo C., Zhang J.P. Effects of Esculentoside A on autoimmune syndrome induced by Campylobacter jejuni in mice and its modulation on T-lymphocyte proliferation and apoptosis. Int. Immunopharmacol. 2010;10(1):65–71. doi: 10.1016/j.intimp.2009.09.022. [DOI] [PubMed] [Google Scholar]
- 13.Wang Q.H., Zhang Y.F., Wu L.L., Niu S., Song C.L., Zhang Z.Y., Lu G.W., Qiao C.P., Hu Y., Yuen K.Y., et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell. 2020;181(4):894–904. doi: 10.1016/j.cell.2020.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lan J., Ge J.W., Yu J.F., Shan S.S., Zhou H., Fan S.L., Zhang Q., Shi X.L., Wang Q.S., Zhang L.Q., et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581(7807):215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- 15.Shang J., Ye G., Shi K., Wan Y.H., Luo C.M., Aihara H., Geng Q.B., Auerbach A., Li F. Structural basis of receptor recognition by SARS-CoV-2. Nature. 2020;581(7807):221–224. doi: 10.1038/s41586-020-2179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benton D.J., Wrobel A.G., Xu P.Q., Roustan C., Martin S.R., Rosenthal P.B., Skehel J.J., Gamblin S.J. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature. 2020;588(7837):327–330. doi: 10.1038/s41586-020-2772-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cai Y.F., Zhang J., Xiao T.S., Peng H.Q., Sterling S.M., Walsh R.M., Rawson S., Rits-Volloch S., Chen B. Distinct conformational states of SARS-CoV-2 spike protein. Science. 2020;369(6511):1586–1592. doi: 10.1126/science.abd4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Towler P., Staker B., Prasad S.G., Menon S., Tang J., Parsons T., Ryan D., Fisher M., Williams D., Dales N.A., et al. ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J. Biol. Chem. 2004;279(17):17996–18007. doi: 10.1074/jbc.M311191200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pierce B.G., Wiehe K., Hwang H., Kim B.H., Vreven T., Weng Z. ZDOCK server: interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics. 2014;30(12):1771–1773. doi: 10.1093/bioinformatics/btu097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schaap M.G., Leij F.J., van Genuchten M.T. ROSETTA: a computer program for estimating soil hydraulic parameters with hierarchical pedotransfer functions. J. Hydrol. 2001;251(3–4):163–176. doi: 10.1016/S0022-1694(01)00466-8. [DOI] [Google Scholar]
- 21.Laskowski R.A., Swindells M.B. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011;51(10):2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- 22.Trott O., Olson A.J. Software news and update AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31(2):455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seeliger D., de Groot B.L. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput. Aid. Mol. Des. 2010;24(5):417–422. doi: 10.1007/s10822-010-9352-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pronk S., Pall S., Schulz R., Larsson P., Bjelkmar P., Apostolov R., Shirts M.R., Smith J.C., Kasson P.M., van der Spoel D., et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29(7):845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Case D.A., Cheatham T.E., Darden T., Gohlke H., Luo R., Merz K.M., Onufriev A., Simmerling C., Wang B., Woods R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005;26(16):1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang H.X., Zeng M.S., Ye Y., Liu J.Y., Xu P.P. Antiviral activity of puerarin as potent inhibitor of influenza virus neuraminidase. Phytother. Res. 2020;35(1):324–336. doi: 10.1002/ptr.6803. [DOI] [PubMed] [Google Scholar]
- 27.Cai Y.F., Schiffer C.A. Decomposing the energetic impact of drug resistant mutations in HIV-1 protease on binding DRV. J. Chem. Theory Comput. 2010;6(4):1358–1368. doi: 10.1021/ct9004678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bjelkmar P., Larsson P., Cuendet M.A., Hess B., Lindahl E. Implementation of the CHARMM force field in GROMACS: analysis of protein stability effects from correction maps, virtual interaction sites, and water models. J. Chem. Theory Comput. 2010;6(2):459–466. doi: 10.1021/ct900549r. [DOI] [PubMed] [Google Scholar]
- 29.DMF VanAalten, BL DeGroot, JBC Findlay, HJC Berendsen, Amadei A. A comparison of techniques for calculating protein essential dynamics. J. Comput. Chem. 1997;18(2):169–181. doi: 10.1002/(SICI)1096-987X(19970130)18:2<169::AID-JCC3>3.0.CO;2-T. [DOI] [Google Scholar]
- 30.Shanmugarajan D., Prabitha P., BRP Kumar, Suresh B. Curcumin to inhibit binding of spike glycoprotein to ACE2 receptors: computational modelling, simulations, and ADMET studies to explore curcuminoids against novel SARS-CoV-2 targets. RSC Adv. 2020;10(52):31385–31399. doi: 10.1039/D0RA03167D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Daina A., Michielin O., Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang S., He H.Q., Shen L., Wan H. Understanding peptide competitive inhibition of botulinum neurotoxin a binding to SV2 protein via molecular dynamics simulations. Biopolymers. 2015;103(10):597–608. doi: 10.1002/bip.22682. [DOI] [PubMed] [Google Scholar]
- 33.Zhang G.Q., Lu W.H., Chen Y.S., Zhu L., Wei Z.Y., Li Z.L., Sun B.L., Xie Q.M., Bi Y.Z., Ma J.Y. Complete genome sequence of two variant porcine reproductive and respiratory syndrome viruses isolated from vaccinated piglets. J. Virol. 2012;86(20):11396–11397. doi: 10.1128/JVI.01843-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang C.X., Zheng W., Huang X.Q., Bell E.W., Zhou X.G., Zhang Y. Protein structure and sequence reanalysis of 2019-nCoV genome refutes snakes as its intermediate host and the unique similarity between its spike protein insertions and HIV-1. J. Proteome Res. 2020;19(4):1351–1360. doi: 10.1021/acs.jproteome.0c00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Malik Y.S., Sircar S., Bhat S., Sharun K., Dhama K., Dadar M., Tiwari R., Chaicumpa W. Emerging novel coronavirus (2019-nCoV)-current scenario, evolutionary perspective based on genome analysis and recent developments. Vet. Q. 2020;40(1):68–76. doi: 10.1080/01652176.2020.1727993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu C.L., Dong L.H., Sun Z., Wang L., Wang Q.P., Li H.Y., Zhang J., Wang X.J. Esculentoside A suppresses breast cancer stem cell growth through stemness attenuation and apoptosis induction by blocking IL-6/STAT3 signaling pathway. Phytother. Res. 2018;32(11):2299–2311. doi: 10.1002/ptr.6172. [DOI] [PubMed] [Google Scholar]
- 37.Wang L.D., Zhang S.L., Cheng H., Lv H.M., Cheng G.H., Ci X.X. Nrf2-mediated liver protection by Esculentoside A against acetaminophen toxicity through the AMPK/Akt/GSK3 beta pathway. Free Radical Bio. Med. 2016;101:401–412. doi: 10.1016/j.freeradbiomed.2016.11.009. [DOI] [PubMed] [Google Scholar]
- 38.Zhong W.T., Jiang L.X., Wei J.Y., Qiao A.N., Wei M.M., Soromou L.W., Xie X.X., Zhou X., Ci X.X., Wang D.C. Protective effect of Esculentoside A on lipopolysaccharide-induced acute lung injury in mice. J. Surg. Res. 2013;185(1):364–372. doi: 10.1016/j.jss.2013.05.018. [DOI] [PubMed] [Google Scholar]
- 39.Dong L.H., Li Y., Wang S.J., Wang G., Sheng L.L., Pan X.F., Sun P.D. Esculentoside A inhibits LPS-induced BV2 microglia activation through activating PPAR-gamma. Eur. J. Pharmacol. 2017;813:61–65. doi: 10.1016/j.ejphar.2017.07.029. [DOI] [PubMed] [Google Scholar]
- 40.Chen D.Z., Chen L.Q., Lin M.X., Gong Y.Q., Ying B.Y., Wei D.Z. Esculentoside A inhibits LPS-induced acute kidney injury by activating PPAR-gamma. Microb. Pathog. 2017;110:208–213. doi: 10.1016/j.micpath.2017.06.037. [DOI] [PubMed] [Google Scholar]
- 41.Ju D.W., Zheng Q.Y., Cao X.T., Fang J., Wang H.B. Esculentoside A inhibits tumor necrosis factor, interleukin-1, and interleukin-6 production induced by lipopolysaccharide in mice. Pharmacology. 1998;56(4):187–195. doi: 10.1159/000028197. [DOI] [PubMed] [Google Scholar]
- 42.Feldmann M., Maini R.N., Woody J.N., Holgate S.T., Winter G., Rowland M., Richards D., Hussell T. Trials of anti-tumour necrosis factor therapy for COVID-19 are urgently needed. Lancet. 2020;395(10234):1407–1409. doi: 10.1016/S0140-6736(20)30858-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fatma B., Kumar R., Singh V.A., Nehul S., Sharma R., Kesari P., Kuhn R.J., Tomar S. Alphavirus capsid protease inhibitors as potential antiviral agents for Chikungunya infection. Antivir. Res. 2020;179:104808. doi: 10.1016/j.antiviral.2020.104808. [DOI] [PubMed] [Google Scholar]