

Abstract
The prevalence of congenital hydrocephalus has been estimated at 1.1 per 1000 infants when including cases diagnosed before 1 year of age after exclusion of neural tube defects. Classification criteria are based either on CSF dynamics, pathophysiological mechanisms or associated lesions. Whereas inherited syndromic hydrocephalus has been associated with more than 100 disease-causing genes, only four genes are currently known to be linked to congenital hydrocephalus either isolated or as a major clinical feature: L1CAM, AP1S2, MPDZ and CCDC88C. In the past 10 years, pathogenic variants in CCDC88C have been documented but the neuropathology remains virtually unknown. We report the neuropathology of two foetuses from one family harbouring two novel compound heterozygous pathogenic variants in the CCDC88C gene: a maternally inherited indel in exon 22, c.3807_3809delinsACCT;p.(Gly1270Profs*53) and a paternally inherited deletion of exon 23, c.3967-?_c.4112-?;p.(Leu1323Argfs*10). Medical termination of pregnancy was performed at 18 and 23 weeks of gestation for severe bilateral ventriculomegaly. In both fetuses, brain lesions consisted of multifocal atresia-forking along the aqueduct of Sylvius and the central canal of the medulla, periventricular neuronal heterotopias and choroid plexus hydrops. The second fetus also presented lumbar myelomeningocele, left diaphragmatic hernia and bilateral renal agenesis. CCDC88C encodes the protein DAPLE which contributes to ependymal cell planar polarity by inhibiting the non-canonical Wnt signaling pathway and interacts with MPDZ and PARD3. Interestingly, heterozygous variants in PARD3 result in neural tube defects by defective tight junction formation and polarization process of the neuroepithelium. Besides, during organ formation Wnt signalling is a prerequisite for planar cell polarity pathway activation, and mutations in planar cell polarity genes lead to heart, lung and kidney malformations. Hence, candidate variants in CCDC88C should be carefully considered whether brain lesions are isolated or associated with malformations suspected to result from disorders of planar cell polarity.
Supplementary Information
The online version contains supplementary material available at 10.1186/s40478-021-01207-5.
Keywords: CCDC88C pathogenic variants, Autosomal recessive inheritance, Foetal hydrocephalus, Neuropathology, Multiple ependymal malformations, Choroid plexus hydrops, Neural tube defect, Renal agenesis, Diaphragmatic defect, Planar cell polarity
Introduction
Congenital hydrocephalus affects 4.65 per 10,000 births and its prevalence has been estimated at 1.1 per 1000 infants when including cases diagnosed before 1 year of age in the absence of other extrinsic causes and after exclusion of neural tube defects [16, 20]. Several classifications are recognized based on CSF dynamics (communicating vs. non-communicating; non-obstructive vs. obstructive), pathophysiological mechanisms (developmental vs. acquired) or associated lesions (syndromic vs. non-syndromic; associated with other major abnormalities vs. with brain lesions only) [15]. Whereas syndromic hydrocephalus has been associated with more than 100 disease-causing genes, only four genes are currently known to be linked to congenital hydrocephalus as isolated or as the major clinical feature: L1CAM, AP1S2, MPDZ and CCDC88C. Pathogenic L1CAM variants are responsible for a wide phenotypic spectrum, X-linked hydrocephalus with stenosis of the aqueduct of Sylvius (AS) being the most common genetic form, with a prevalence of 1:30,000 and accounting for approximately 5–10% of males with non-syndromic congenital hydrocephalus. L1CAM is a neuronal adhesion molecule of the immunoglobulin superfamily which plays major roles in intercellular adhesion, neuronal cell survival and migration, growth cone morphology and neurite outgrowth, axonal guidance and fasciculation, synaptogenesis and myelination [1]. Pathogenic AP1S2 variants have been linked to Pettigrew syndrome characterized by intellectual disability with prominent basal ganglia iron deposition or calcification and variable severity of hydrocephalus. AP1S2 is a subunit of the AP1 adaptin protein complex, one of the major regulators of lysosomal protein sorting involved in clathrin-coated vesicle assembly and transport of proteins between the trans-Golgi network and lysosomes [14]. More recently, deleterious variants in MPDZ and CCDC88C genes were shown to be responsible for non-syndromic autosomal recessive congenital hydrocephalus. MPDZ, named alternatively MUPP1, is a modular scaffold protein consisting of 13 PDZ and one L27 domains localized to apical junction complexes [17]. The pathophysiology of MPDZ-linked hydrocephalus has been attributed to hyperpermeability of the choroid plexus epithelial cells in mice [21]. CCDC88C encodes the segment polarity protein disheveled homolog (Dvl)-binding protein DAPLE, a guanine exchange factor trimeric G proteins [2]. DAPLE contains a PDZ-binding motif at its C-terminus which contributes to ependymal cell planar polarity by inhibiting the non-canonical Wnt signaling pathway through its interaction with Dishevelled, leading to Rho-ROCK and Rac-JNK activation [10, 19]. In the past 10 years, pathogenic variants in CCDC88C have been documented but the neuropathology of the disease remains virtually unknown [4, 6, 13, 20]. We report herein the prenatal phenotype and neuropathological lesions in two fetuses from one family, which harboured two novel compound heterozygous variants in the CCDC88C gene.
Case presentation
A 31-year-old woman, gravida I, para 0, underwent routine ultrasonography (US) at 22 weeks of gestation (WG), which revealed macrocephaly (head circumference >> 97th percentile) with severe bilateral ventriculomegaly, but with no other associated brain, visceral or growth parameter abnormalities. Based on these findings, a medical termination of the pregnancy (TOP) was achieved at 23 WG in accordance with the French law. Nine months later, TOP was performed at 18 WG after the discovery of similar brain lesions on US, associated with growth restriction and myelomeningocele. Chromosomal analysis revealed a normal karyotype, 46, XX and 46, XY respectively. CGH analysis was normal. The unrelated parents were in good health and there was no particular medical family history. At autopsy (Additional file 1), both fetuses displayed similar facial particularities consisting of macrocephaly, malar hypoplasia, small nose with anteverted nostrils and microretrognathism. Neither skeletal nor visceral anomalies were identified in the first fetus. Conversely, the second fetus presented bilateral clubfoot and severe amyotrophy of the lower limbs secondary to lumbar myelomeningocele. Associated visceral malformations consisted of left diaphragmatic hernia and bilateral renal agenesis. On CNS examination, brain weights were in accordance with the term despite hydrocephalus. No primary fissures were present with the exception of a dimple-shaped sylvian fissure. Olfactory bulbs and optic chiasm were present. External examination of the cerebellum and brainstem was normal. On sections passing through the mesencephalon, the AS was indiscernible. On coronal sections, the lateral ventricles were severely dilated. The third and fourth ventricles appeared to be collapsed in the second case. Histologically, the two brains had identical lesions. In the mesencephalon, the subcommissural organ was normal. The inferior colliculi were fused in the second case. AS atresia consisted of few rosettes lined by ependymal cells (Fig. 1a, b). Similar lesions were noted in the central canal of the medulla from the level of the area postrema to the level of decussation of the pyramids (Fig. 1c, d). At the supratentorial level, the internal capsule was normal and callosal fibres were present. Subependymal gray matter heterotopias were observed in both cases (Fig. 1e, f) and were made up with several cell types comprising an admixture of SOX2, nestin, vimentin, MAP2, GABA and GFAP immunoreactive cells. One of the most striking features was the abnormal morphology of the choroid plexuses which were voluminous due to hydrops of the connective tissue core (Fig. 2a, b) covered by intact basal lamina (Fig. 2c) and epithelial cells (Fig. 2d). No other lesions were observed in any of the different infra- and supratentorial brain structures analyzed. A targeted capture-sequencing panel including L1CAM, MPDZ and CCDC88C was performed in fetus 1-analysis in solo (Additional file 1). Two CCDC88C (NM_001080414.2) compound heterozygous variations were found: a indel in exon 22, c.3807_3809delinsACCT;p.(Gly1270Profs*53) and a deletion of exon 23, c.3967-?_c.4112-?;p.(Leu1323Argfs*10) (Fig. 3). Segregation analysis by Sanger sequencing demonstrated that the indel variation in exon 22 was maternally inherited, whereas the deletion of exon 23 was paternally inherited. These variations are not reported in gnomAD. According to ACMG classification, both variants are classified as pathogenic (class 5). Due to the presence of associated visceral malformations in the second case, whole exome sequencing (WES) was performed in the second fetus and his parents (trio analysis) but no additional causal variation for a second disorder was identified.
Fig. 1.
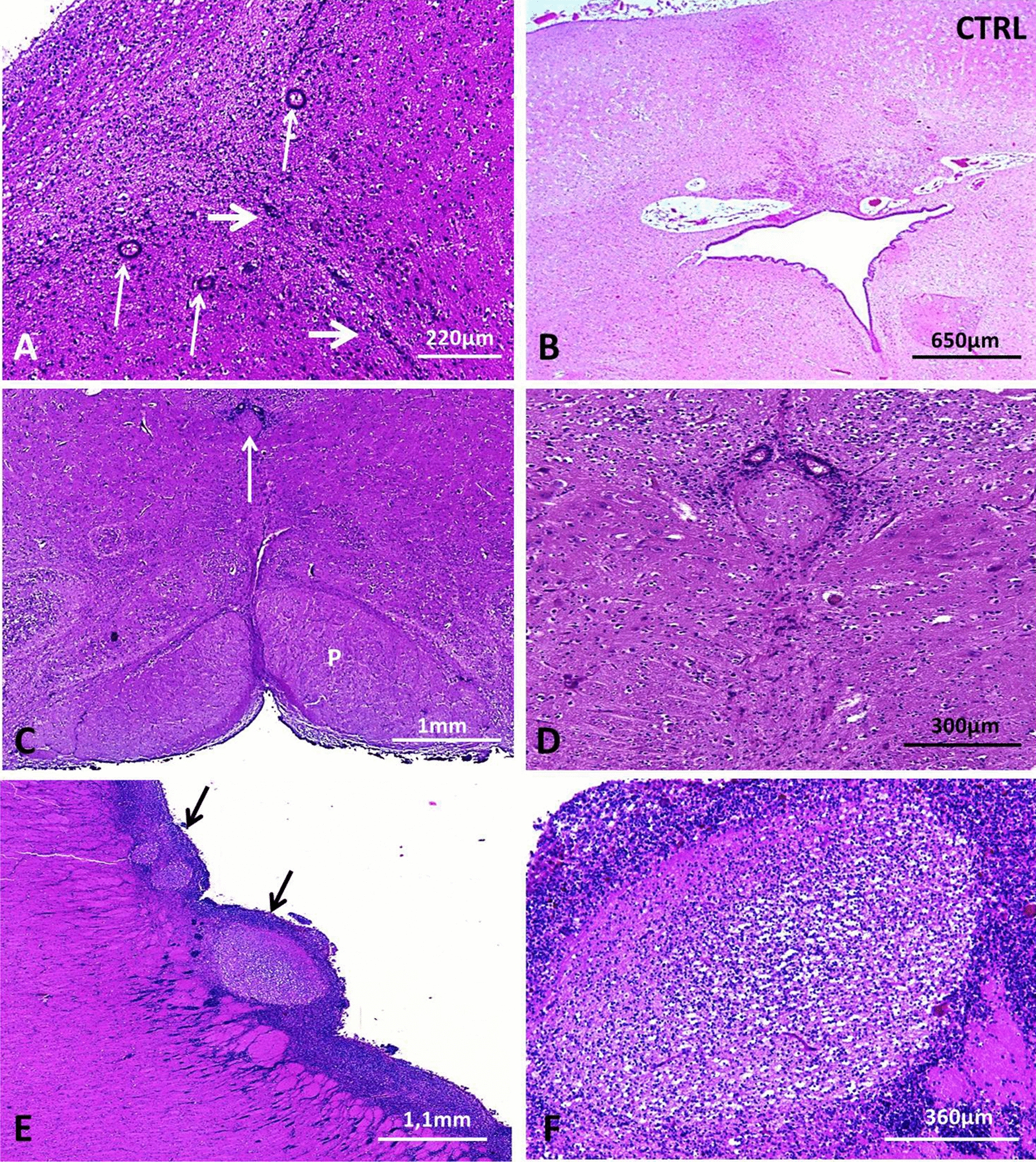
Main neuropathological hallmarks of hydrocephalus linked to CCDC88C pathogenic variants. a Coronal section through the mesencephalon reveals AS atresia in the first case (thick arrows) associated with small- and medium-sized channels lined by ependymal cells (thin arrows) [H&E, OM × 100)]. b By comparison with a normal aqueduct of Sylvius [H&E, OM × 100)]. c Identical lesions (arrow) observed in the second case at the level of the central canal of the medulla the AS [H&E, OM × 25]. d With, at higher magnification, presence of several small ependymal channels [H&E, OM × 100]. e Periventricular nodular heterotopias of various size (thick arrows) [H&E, OM × 25]. f With at higher magnification, a lower cell density [H&E, OM × 100)]. H&E: Hematoxylin and eosin stain; OM: original magnification; P: pyramids
Fig. 2.
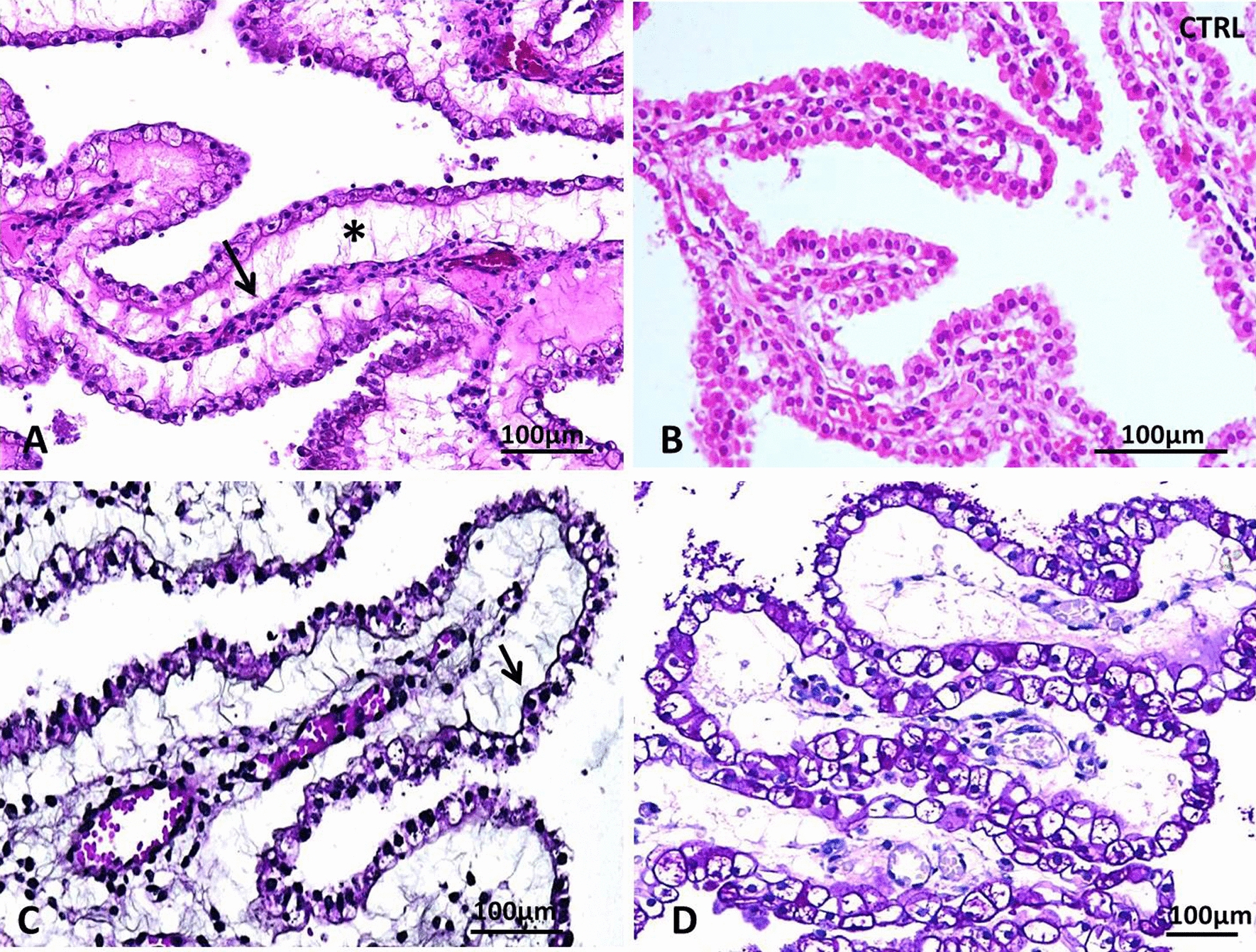
Main neuropathological hallmarks of hydrocephalus linked to CCDC88C pathogenic variants. a Accumulation of fluid (asterisk) under the choroid epithelium causing collapse of the villus mesenchymatous core which contains collapsed vessels (arrow) [H&E, OM × 200]. b Compared with the normal choroid villus morphology in an age- matched control [H&E, OM × 200]. c Without disruption of the basal lamina as evidenced by Jones’ silver impregnation method [H&E, OM × 200]. d Covered by epithelial cells still containing glycogen [PAS, OM × 200]. H&E: Hematoxylin and eosin stain; OM: original magnification; PAS: periodic Schiff staining
Fig. 3.

Identification of the two compound heterozygous variations in the CCDC88C gene. a Pedigree structure of the family. Targeted NGS sequencing of a panel including L1CAM, MPDZ and CCDC88C was performed in fetus II.1 (red star). Whole exome sequencing (WES) was performed in fetus II.2 and his parents (I.1 and I.2; black stars). b Targeted NGS sequencing and WES identified a heterozygous frameshift variant in the CCDC88C gene, c.3807_3809delinsACCT; p.(Gly1270Profs*53), which was shown to be maternally inherited by Sanger sequencing of the fetuses and their parents. c Targeted NGS sequencing and WES also identified a heterozygous deletion of CCDC88C exon 23 (c.3967-?_c.4112-?; p.Leu1323Argfs*10), which was shown to be paternally inherited and was confirmed by a relative quantification ddPCR assay of fetus II.1 and her parents. Top: Representative result of the ddPCR assay, using of the CCDC88C gene (primers located in the exon 23, blue droplets) compared to a reference housekeeping gene (HMBS gene, green droplets). Bottom: Quantification of copy number in a control DNA (target, 1358 copies/μL; reference, 1426 copies/μL), fetus II.1 (target, 551 copies/μL; reference, 1057 copies/μL), parent I.1 (target, 1011 copies/μL; reference, 1019 copies/μL), and parent II.2 (target, 447 copies/μL; reference, 894 copies/μL). d Schematic representation of DAPLE protein organization. DAPLE contains a Hook domain, a Gα binding and activating domain (GBA), a coiled coil region, a frizzled binding domain (FBD) and a carboxy-terminal PDZ binding motif (PBM). The compound heterozygous variants identified in this study (in red) were localized in the coiled-coil domain. Published homozygous loss-of-function variants are depicted in black whereas heterozygous gain-of-function variations are represented in green (2,8–11). Nt: amino-terminal; Ct: carboxy-terminal
Discussion
When excluding X-linked hydrocephalus which encompasses approximately 5–15% of congenital hydrocephalus cases, the frequency of non-syndromic forms in humans is very low, with an empiric recurrence risk rate ranging from < 1% to 4% [20], indicative of the rarity of autosomal recessive congenital hydrocephalus [19]. Using WES, variants in other genes have been recently reported as disease-causing. In the work by Shaheen et al. exploring 27 families with recurrent hydrocephalus, the most common etiologies were dystroglycanopathies (26%) and ciliopathies (15%) indicating that non-syndromic hydrocephalus is rare [16]. Using the same technical approaches, Jin et al. explored a series of 381 undiagnosed living patients with sporadic hydrocephalus and found that damaging de novo variations accounted for more than 17% of the cases. Five genes (TRIM71, SMARCC1, PTEN, PIK3C, MTOR) were found to be involved in syndromic forms of hydrocephalus, and three other genes (FMN2, FOXJ1 and PTCH1) to be responsible for severe hydrocephalus with AS stenosis [9]. Similarly to families with recurrent hydrocephalus, non-syndromic hydrocephalus is rare in patients with sporadic hydrocephalus. To date, only 6 CCDC88C homozygous pathogenic variants have been identified in 14 patients with non-syndromic hydrocephalus from 6 unrelated families, including three frameshift, one splice site, and two nonsense mutations [4, 6, 13, 20]. Conversely, CCDC88C heterozygous gain-of-function variations have been reported in patients without hydrocephalus who suffered from adult-onset spinocerebellar ataxia SCA40 [10, 19].
Magnetic resonance imaging performed in seven out of the 12 patients with CCDC88C homozygous pathogenic variants revealed associated schizencephaly/porencephaly in three patients, along with either arachnoid cyst, small vermis or biparietal polymicrogyria in three other patients. Neuropathological examination performed in a single fetus interrupted at 21 WG revealed neither AS stenosis or atresia, nor ependymal rosettes or denudation [4]. Yet, neuropathological examination of the two fetuses allowed the detection of three major signs in favour of maldevelopment and malfunction of neuroepithelial/ependymal cells. First, the loss of ependyma in the AS with rosette formation leads to fusion of its dorsal and ventral walls and consequently to severe hydrocephalus. Second, early loss of ventricular zone integrity results in abnormal neurogenesis and migration. In the disrupted areas, radial glial cells disappear, depriving the neuroepithelial cells of scaffolds to migrate on. Consequently, the cells stay along the ventricular wall and form nodular periventricular heterotopias that are source of seizures in infants notwithstanding surgical CSF shunting. Third, late developmental disruption of the ventricular zone results in loss of multiciliated cells leading to alterations of the laminar CSF flow and hydrocephalus [8, 12]. Moreover, orientation of beating of cilia is defined by ependymal planar cell polarity (PCP) which involves translational and rotational movements of basal bodies. Basal body translational position movement depends on the primary cilium in connection with microtubules and apical junctions. Alterations in these junctions disrupt the natural barrier between the parenchyma and the CSF, leading to hydrocephalus, as observed in MPDZ-mutated patients [8]. MPDZ has been shown to bind directly DAPLE and to act as a scaffold which gathers DAPLE and other proteins involved in planar and apicobasal cell polarity pathways [11]. Therefore, the neuropathological similarities between the phenotypes linked to MPDZ and CCDC88C variants are not surprising [15]. DAPLE functions as a cellular “compass” for establishing and maintaining contact-triggered planar polarity through interaction with the third PDZ domain of MPDZ and the third PDZ domain of PARD3 [5, 18]. Interestingly, in a recent study including 138 patients with neural tube defect (NTD), rare heterozygous variants in the PARD3 gene were identified and were shown to be significantly enriched in the aPKC-binding region resulting in defective tight junction formation via disrupted aPKC binding, suggesting that these deleterious variants contribute to human NTDs possibly by preventing apical tight junction formation and subsequent polarization process of the neuroepithelium [3]. Besides, during organ formation, proper Wnt signalling is a prerequisite for the activation of the PCP pathway, and it has also been shown that mutations in PCP genes may lead to congenital abnormalities other than NTD, notably heart, lung and kidney developmental defects. Noteworthy, in human foetuses, DAPLE expression is high in the brain and in the kidneys [6, 7]. Moreover, incomplete development of the diaphragm in the second fetus could also result also from Wnt signalling disruption, which could lead to defects in proliferation/survival of myogenic progenitor cells and defects in muscle morphogenesis by affecting extensive convergent migration of myogenic precursors during early embryogenesis. At last, our findings are in line with what already observed in MPDZ syndromic forms in which eye anomalies (chorioretinal coloboma), thoracic malformations (atrial septal defect and congenital diaphragmatic hernia) have been described and also very likely result from PCP disturbances.
Conclusion
Multifocal atresia-forking along the AS and of the central canal of the medulla associated with periventricular neuronal heterotopias and disruption of the choroid plexus epithelium are the key hallmarks of variations in the CCDC88C gene, whether these brain lesions are isolated or associated with malformations suspected to result from PCP disorders. Variations in the CCDC88C gene together with those previously identified in the MPDZ gene represent a new group of emerging diseases causing congenital hydrocephalus.
Supplementary Information
Additional file 1: Pathological and molecular methods.
Acknowledgements
This work was co-supported by European Union and Région Normandie in the context of Recherche Innovation Normandie (RIN 2018). Europe gets involved in Normandie with European Regional Development Fund (ERDF). This work was generated within the European Reference Network for Developmental abnormalities and Intellectual Disability.The authors of this publication are members of the European Reference Network for Developmental Anomalies and Intellectual Disability (ERN-ITHACA).
Abbreviations
- AS
Aqueduct of Sylvius
- CGH
Comparative genomic hybridization
- CSF
Cerebrospinal fluid
- CNS
Central nervous system
- NGS
Next generation sequencing
- NTD
Neural tube defects
- PCP
Planar cell polarity
- TOP
Medical termination of the pregnancy
- WES
Whole exome sequencing
- WG
Weeks of gestation
Authors' contributions
SAB followed the family and PM performed autopsies. AL, FM and PM were responsible for interpretation of neuropathological studies and contributed to the diagnosis. KC, ND, PC and PSV carried out genetic studies and MV and PSV were responsible for exome sequencing. AL, FM, and PSV designed the study and wrote the manuscript. AH and BG contributed to the design of the study and critically reviewed the manuscript. All authors read and approved the final version of the manuscript.
Funding
This work was supported by the Rouen University, the Normandy University, the Institut National de la Santé et de la Recherche Médicale (INSERM; UMR1245).
Availability of data and materials
In the departments of Pathology and Genetics, Rouen University Hospital F76000, Rouen, France; in the department of Pathology at Brest University Hospital F29609, Brest, France.
Declarations
Ethics approval and consent to participate
Medical terminations of the pregnancy were performed after approval by our local ethical committee. Autopsies were performed in the two foetuses with the informed written consent of the parents in accordance with the French law. The parents provided written informed consent for targeted sequencing and Whole Exome Sequencing.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflicts of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Annie Laquerriere and Pascale Saugier-Veber contributed equally to this work
References
- 1.Adle-Biassette H, Saugier-Veber P, Fallet-Bianco C, Delezoide AL, Razavi F, Drouot N, et al. Neuropathological review of 138 cases genetically tested for X-linked hydrocephalus: evidence for closely related clinical entities of unknown molecular bases. Acta Neuropathol. 2013 doi: 10.1007/s00401-013-1146-1. [DOI] [PubMed] [Google Scholar]
- 2.Aznar N, Midde KK, Dunkel Y, Lopez-Sanchez I, Pavlova Y, Marivin A, et al. Daple is a novel non-receptor GEF required for trimeric G protein activation in Wnt signaling. Elife. 2015 doi: 10.7554/eLife.07091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen X, An Y, Gao Y, Guo L, Rui L, Xie H, et al. Rare Deleterious PARD3 Variants in the aPKC-binding region are implicated in the pathogenesis of human cranial neural tube defects via disrupting apical tight junction formation. Hum Mutat. 2017 doi: 10.1002/humu.23153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Drielsma A, Jalas C, Simonis N, Désir J, Simanovsky N, Pirson I, et al. Two novel CCDC88C mutations confirm the role of DAPLE in autosomal recessive congenital hydrocephalus. J Med Genet. 2012 doi: 10.1136/jmedgenet-2012-101190. [DOI] [PubMed] [Google Scholar]
- 5.Ear J, Saklecha A, Rajapakse N, Choi J, Ghassemian M, Kufareva I, Ghosh P. Tyrosine-based signals regulate the assembly of Daple⋅PARD3 complex at cell-cell junctions. iScience. 2020 doi: 10.1016/j.isci.2020.100859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ekici AB, Hilfinger D, Jatzwauk M, Thiel CT, Wenzel D, Lorenz I, et al. Disturbed Wnt signalling due to a mutation in CCDC88C causes an autosomal recessive non-syndromic hydrocephalus with medial diverticulum. Mol Syndromol. 2010 doi: 10.1159/000319859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Henderson DJ, Long DA, Dean CH. Planar cell polarity in organ formation. Curr Opin Cell Biol. 2018 doi: 10.1016/j.ceb.2018.06.011. [DOI] [PubMed] [Google Scholar]
- 8.Jiménez AJ, Domínguez-Pinos MD, Guerra MM, Fernández-Llebrez P, Pérez-Fígares JM. Structure and function of the ependymal barrier and diseases associated with ependyma disruption. Tissue Barriers. 2014 doi: 10.4161/tisb.28426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jin SC, Dong W, Kundishora AJ, Panchagnula S, Moreno-De-Luca A, Furey CG, et al. Exome sequencing implicates genetic disruption of prenatal neuro-gliogenesis in sporadic congenital hydrocephalus. Nat Med. 2020 doi: 10.1038/s41591-020-1090-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leńska-Mieciek M, Charzewska A, Królicki L, Hoffman-Zacharska D, Chen ZS, Lau KF, Chan HYE, Gambin T, Fiszer U. Familial ataxia, tremor, and dementia in a polish family with a novel mutation in the CCDC88C gene. Mov Disord. 2019 doi: 10.1002/mds.27536. [DOI] [PubMed] [Google Scholar]
- 11.Marivin A, Garcia-Marcos M. DAPLE and MPDZ bind to each other and cooperate to promote apical cell constriction. Mol Biol Cell. 2019 doi: 10.1091/mbc.E19-02-0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rodríguez EM, Guerra MM, Ortega E. Physiopathology of fetal onset hydrocephalus. In: Limbrick D, Leonard J, editors. Cerebrospinal Fluid Disorders. Cham: Springer International Publishing; 2019. pp. 3–30. [Google Scholar]
- 13.Ruggeri G, Timms AE, Cheng C, Weiss A, Kollros P, Chapman T, et al. Bi-allelic mutations of CCDC88C are a rare cause of severe congenital hydrocephalus. Am J Med Genet A. 2018 doi: 10.1002/ajmg.a.38592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saillour Y, Zanni G, Des Portes V, Heron D, Guibaud L, Iba-Zizen MT, et al. Mutations in the AP1S2 gene encoding the sigma 2 subunit of the adaptor protein 1 complex are associated with syndromic X-linked mental retardation with hydrocephalus and calcifications in basal ganglia. J Med Genet. 2007 doi: 10.1136/jmg.2007.051334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saugier-Veber P, Marguet F, Lecoquierre F, Adle-Biassette H, Guimiot F, Cipriani S, et al. Hydrocephalus due to multiple ependymal malformations is caused by mutations in the MPDZ gene. Acta Neuropathol Commun. 2017 doi: 10.1186/s40478-017-0438-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shaheen R, Sebai MA, Patel N, Ewida N, Kurdi W, Altweijri I, et al. The genetic landscape of familial congenital hydrocephalus. Ann Neurol. 2017 doi: 10.1002/ana.24964. [DOI] [PubMed] [Google Scholar]
- 17.Simpson EH, Suffolk R, Jackson IJ. Identification, sequence, and mapping of the mouse multiple PDZ domain protein gene, Mpdz. Genomics. 1999 doi: 10.1006/geno.1999.5853. [DOI] [PubMed] [Google Scholar]
- 18.Takagishi M, Sawada M, Ohata S, Asai N, Enomoto A, Takahashi K, et al. Daple coordinates planar polarized microtubule dynamics in ependymal cells and contributes to hydrocephalus. Cell Rep. 2017 doi: 10.1016/j.celrep.2017.06.089. [DOI] [PubMed] [Google Scholar]
- 19.Tsoi H, Yu AC, Chen ZS, Ng NK, Chan AY, Yuen LY, et al. A novel missense mutation in CCDC88C activates the JNK pathway and causes a dominant form of spinocerebellar ataxia. J Med Genet. 2014 doi: 10.1136/jmedgenet-2014-102333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wallis M, Baumer A, Smaili W, Jaouad IC, Sefiani A, Jacobson E, et al. Surprisingly good outcome in antenatal diagnosis of severe hydrocephalus related to CCDC88C deficiency. Eur J Med Genet. 2018 doi: 10.1016/j.ejmg.2017.12.002. [DOI] [PubMed] [Google Scholar]
- 21.Yang J, Simonneau C, Kilker R, Oakley L, Byrne MD, Nichtova Z, et al. Murine MPDZ-linked hydrocephalus is caused by hyperpermeability of the choroid plexus. EMBO Mol Med. 2019 doi: 10.15252/emmm.201809540. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Pathological and molecular methods.
Data Availability Statement
In the departments of Pathology and Genetics, Rouen University Hospital F76000, Rouen, France; in the department of Pathology at Brest University Hospital F29609, Brest, France.