

Abstract
Individuals exhibiting an intermediate alcohol drinking pattern in conjunction with signs of metabolic risk present clinical features of both alcohol‐associated and metabolic‐associated fatty liver diseases. However, such combination remains an unexplored area of great interest, given the increasing number of patients affected. In the present study, we aimed to develop a preclinical DUAL (alcohol‐associated liver disease plus metabolic‐associated fatty liver disease) model in mice. C57BL/6 mice received 10% vol/vol alcohol in sweetened drinking water in combination with a Western diet for 10, 23, and 52 weeks (DUAL model). Animals fed with DUAL diet elicited a significant increase in body mass index accompanied by a pronounced hypertrophy of adipocytes, hypercholesterolemia, and hyperglycemia. Significant liver damage was characterized by elevated plasma alanine aminotransferase and lactate dehydrogenase levels, extensive hepatomegaly, hepatocyte enlargement, ballooning, steatosis, hepatic cell death, and compensatory proliferation. Notably, DUAL animals developed lobular inflammation and advanced hepatic fibrosis. Sequentially, bridging cirrhotic changes were frequently observed after 12 months. Bulk RNA‐sequencing analysis indicated that dysregulated molecular pathways in DUAL mice were similar to those of patients with steatohepatitis. Conclusion: Our DUAL model is characterized by obesity, glucose intolerance, liver damage, prominent steatohepatitis and fibrosis, as well as inflammation and fibrosis in white adipose tissue. Altogether, the DUAL model mimics all histological, metabolic, and transcriptomic gene signatures of human advanced steatohepatitis, and therefore serves as a preclinical tool for the development of therapeutic targets.
DUAL model perfectly mimics all histological, metabolic and transcriptomic gene signatures of human advanced steatohepatitis, and thus serve as a preclinical tool for the development of therapeutic targets.

Abbreviations
- ALD
alcohol‐associated liver disease
- ALT
alanine aminotransferase
- ANOVA
analysis of variance
- AST
aspartate aminotransferase
- Bcl2
B cell lymphoma 2
- BMI
body mass index
- BW
body weight
- CPT‐1c
carnitine palmitoyltransferase 1c
- DEN
diethylnitrosamine
- DUAL
ALD plus MAFLD
- ECM
extracellular matrix
- EtOH
ethanol
- FFA
free fatty acid
- H&E
hematoxylin and eosin
- HSC
hepatic stellate cell
- IF
immunofluorescence
- IHC
immunohistochemistry
- LDH
lactate dehydrogenase
- MAFLD
metabolic associated fatty liver disease
- mRNA
messenger RNA
- MS
metabolic syndrome
- NAFLD
nonalcoholic fatty liver disease
- NF‐κB
nuclear factor kappa B
- ORO
Oil Red O
- PCNA
proliferating cell nuclear antigen
- Pi3K
phosphoinositide 3‐kinase
- RNA‐seq
RNA sequencing
- SR
sirius red
- TEM
transmission electron microscopy
- TG
triglycerides
- TLR
toll‐like receptor
- TNF‐α
tumor necrosis factor‐α
- WAT
white adipose tissue
- WD
Western diet
- α‐SMA
α‐smooth muscle actin
Excessive alcohol drinking is a leading cause of chronic liver disease and accounts for up to 60%‐80% of liver‐related mortality in Europe.( 1 ) These data become even more relevant considering that alcohol‐associated liver disease (ALD) receives only about 5% of the attention in the field of hepatology.( 2 )
The principal fact that only about 6%‐30% of heavy drinkers develop cirrhosis indicates that additional factors modulate the risk of ALD progression.( 1 ) Clinical observations commonly suggest a wide individual susceptibility, and indicate several risk factors for ALD including drinking patterns, female gender, genetic background, cigarette smoking, occupational hazards, and hepatotropic viruses. Obesity and metabolic syndrome (MS) represent another important group of risk factors that accelerate fibrosis progression, hepatic carcinogenesis, and mortality in ALD.( 3 ) Epidemiological studies using a large cohort of patients( 4 , 5 , 6 ) clearly showed that obese patients with alcoholism have 2‐3‐times higher risk of developing steatohepatitis and progression to fibrosis or cirrhosis. Hence, obese individuals consuming 15 or more drinks per week have an adjusted relative rate of liver‐related death of 18.9 compared with 3.16 in their lean counterparts.( 5 )
However, the patients with intermediate levels of ethanol use plus the presence of metabolic risks (i.e., dual clinical features of ALD and metabolic associated fatty liver disease [MAFLD]) represent a large understudied area in hepatology with a huge unmet need in preclinical and clinical studies.( 7 )
Herein we report a physiological, innovative experimental DUAL (ALD plus MAFLD) model that synergistically combines the effects of alcohol and Western diet (WD). The taste of alcohol was camouflaged by adding glucose to the drinking water, consequently increasing the daily ethanol (EtOH) consumption and remarkably intensifying liver damage. Within 23 weeks, this preclinical DUAL model reproduced all of the key metabolic and histological features of human steatohepatitis, with consistent development of hepatic fibrosis, enhanced obesity, glucose intolerance, as well as inflammation of white adipose tissue (WAT). Altogether, this preclinical model might be well‐considered as a useful experimental model to study the dangerous combination of ALD plus MAFLD in human, and therefore be further used for the development of therapeutic options.
Materials And Methods
Animal Husbandry
All animal procedures were carried out according to Spanish legal requirements and animal protection law, and approved by the authority of environment conservation and consumer protection of the Regional Government of Madrid (PROEX210/18). All animals were maintained in the Animal Facility at the Faculty of Biology, Complutense University Madrid, in a temperature‐controlled room with 12‐hour light/dark cycle with free access to food and water according to the guidelines of the Federation for Laboratory Animal Science Associations.
Animal Experimentation
Female 10‐week‐old C57BL6/J mice were randomly assigned to four different experimental groups. The total number of mice per group was five to seven. Mice were treated with a DUAL diet consisting of WD (D09100301; Research Diets, Inc., New Brunswick, NJ) and 10% vol/vol EtOH absolute in sweetened drinking water containing 6.75% D‐glucose (Sigma‐Aldrich, St. Louis, MO). Controls were fed with either normal diet only (Altomin, Lage, Germany), WD only, or EtOH in sweetened drinking water only (Supporting Tables S1‐S3).
Statistical Analysis
Data are expressed as mean ± SD. GraphPad Prism version 8.0 (https://www.graphpad.com/scientific‐software/prism/) (San Diego, CA) was used for statistical analysis and graph design. Statistical significance was determined by one‐way analysis of variance ANOVA followed by a Tukey post hoc test. One‐way paired ANOVA followed by Bonferroni’s post hoc test was used to evaluate the differences between 10‐week and 23‐week feeding groups. Values with different superscripts are significantly different from each other (P < 0.05), as assessed by one‐way ANOVA. Differences (P < 0.05) between time points (10 weeks vs. 23 weeks) for each pairing group were assessed by pairing one‐way ANOVA and denoted by “#.”
Results
Characterization of a Preclinical DUAL Model
Because low EtOH intake due to natural aversion in mice is the main limiting factor for ALD development,( 8 ) we masked the taste of alcohol by adding 6.75% D‐glucose to the drinking water and gradually increased EtOH concentration from 1% to 10% vol/vol. Mice usually consume alcohol up to 25% vol/vol.( 9 ) Thus, initially we performed a pilot study to assess the optimal concentration of EtOH in the drinking water. Surprisingly we found that the concentration of EtOH significantly affected the volume, and, consequently the quantity of the daily consumed ethanol. As the concentration of EtOH increased (20%), the mice drank less volume, and intake significantly decreased. At a 5% concentration, the quantity of consumed EtOH remained relatively low (Supporting Fig. S1A). Only a concentration of 10% vol/vol of EtOH resulted in severe pathophysiologic changes (Supporting Fig. S1B,C). Consequently, for all of the following experiments, animals were randomly assigned to four groups: (1) control group; (2) WD plus sweetened water group; (3) 10% vol/vol EtOH in sweetened water plus chow diet group; and (4) DUAL group (Supporting Fig. S2A).
Obesity, Dyslipidemia, and Glycemia: Features of the DUAL Diet
Body weight (BW) increased steadily in all treated groups throughout the experimental period (Fig. 1A [left] and Supporting Table S4). Previous studies in both humans and rodents showed significantly elevated EtOH intake in subjects consuming fat.( 10 ) Consistently, our model nicely reflected this positive correlation, as DUAL‐fed animals demonstrated greater EtOH intake compared with the EtOH‐fed group (Supporting Fig. S3A). In turn, alcohol stimulated the ingestion of a fat‐rich diet, thus creating a positive feedback loop (Supporting Fig. S3B). This finally resulted in higher daily caloric consumption (Fig. 1A [middle panel]) and robust increase in the body mass index (BMI) in DUAL‐fed mice (Fig. 1A [right panel]).
FIG. 1.
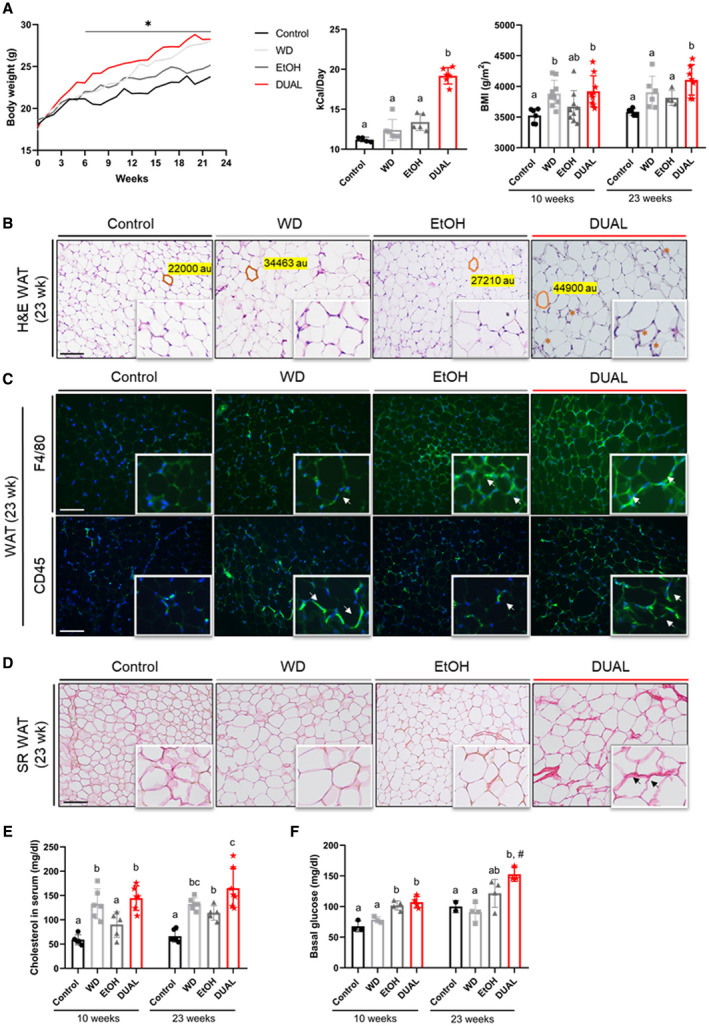
Metabolic profile of mice treated with DUAL diet and control groups. (A) Left: BW curve during the feeding period. Statistical differences between DUAL and control groups are shown (n = 5‐7). Differences (P < 0.05) between control and DUAL group are denoted by “*.” Middle: Calorie intake per day including calories in food and in drinking water (D‐glucose and/or EtOH) (n = 4‐6). Right: BMI was calculated after 10 and 23 weeks of feeding (body surface area [m2/BW]; n = 5‐9). (B) Representative WAT H&E. Scale bar = 100 µm (n = 3). (C) Representative CD45 and F4/80 IF staining of WAT. Positive immune cells are stained in green. Nuclei are stained in blue using DAPI as a counterstain. Scale bar = 100 µm (n = 3). (D) SR staining performed in WAT. Scale bar = 100 µm (n = 3). (E) Levels of cholesterol in serum (n = 5‐7). (F) Basal glucose levels in blood after 12 hours of fasting (n = 3). Values with different superscripts are significantly different from each other (P < 0.05), assessed by one‐way ANOVA. Differences (P < 0.05) between time points (10 weeks vs. 23 weeks) for each pairing group were assessed by pairing one‐way ANOVA and denoted as “#.” Abbreviation: DAPI, 4′,6‐diamidino‐2‐phenylindole.
Obesity and ALD are associated with profound changes in the function of adipose tissue, which has important systemic and hepatic consequences.( 11 ) The morphometric evaluation of adipocytes in WAT from visceral fat pad showed that DUAL diet led to an increase in adipocyte size by 93.2% after 23 weeks of feeding (Fig. 1B and Supporting Fig. S4A). Additionally, many crown‐like structures, formed by macrophages aggregated around dying adipocytes, were observed in the DUAL group (Fig. 1B). These results were confirmed by a trend toward increased F4/80 and CD45 positive cell infiltration of WAT in DUAL‐fed animals (Fig. 1C).
Excessive amounts of extracellular matrix (ECM) and fibrosis in WAT accelerates adipose tissue dysfunction in obese individuals.( 12 ) Consistently, sirius red (SR) staining revealed that DUAL diet dramatically increased collagen deposition in WAT compared with the rest of the experimental groups (Fig. 1D and Supporting Fig. S4B).
Obesity is strongly associated with other features of MS, including glycemia and dyslipidemia.( 13 ) In fact, the circulating levels of total cholesterol were constantly higher in DUAL‐fed animals (10 weeks and 23 weeks) compared with control groups (Fig. 1E). Additionally, DUAL‐fed mice developed hyperglycemia at 10 weeks that was sustained up to 23 weeks of treatment (Fig. 1F). Hence, no significant difference between control animals and treated ones was detected by glucose tolerance test. Insulin tolerance test (ITT) after 6 hours of fasting showed the impaired insulin sensitivity only in WD and EtOH groups (Supporting Fig. S5A,B).
DUAL Feeding Triggers Hepatomegaly and Fatty Liver Disease
Obesity and MS predispose to the development of fatty liver disease.( 14 ) After 23 weeks of DUAL diet, animals exhibited enlarged livers, which were pale and yellowish in color, indicating lipid accumulation (Fig. 2A [left]). Accordingly, the hepatic mass was increased, and the hepatosomatic ratio of DUAL mice reached almost 8.5% (Fig. 2A [middle and right panels]). Hepatocytes of DUAL‐fed animals lost their typical hexagonal shape and became enlarged round cells, as demonstrated by phalloidin staining (Fig. 2B).
FIG. 2.
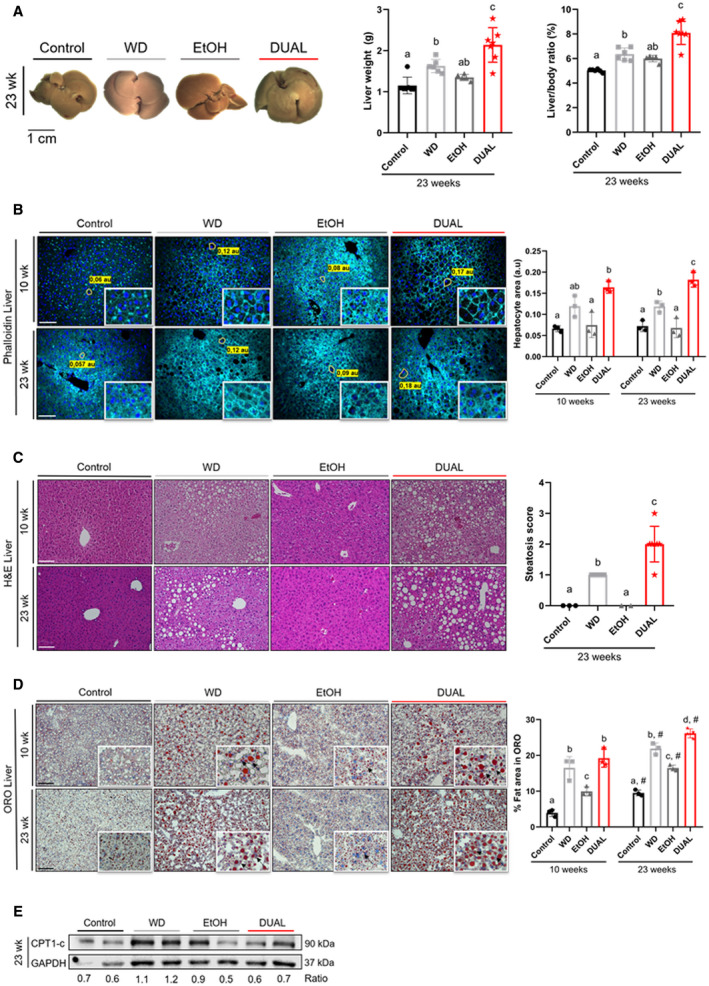
DUAL mice develop hepatomegaly and advanced steatosis. (A) Left: Liver macroscopic images after 23 weeks of feeding. Middle: Liver weight (g) (n = 5‐7). Right: Liver weight–to‐BW ratio (%) (n = 5‐7). (B) Representative phalloidin‐stained liver images and size of hepatocytes in phalloidin‐stained liver pictures quantified by ImageJ software. Scale bar = 100 µm (n = 3). (C) H&E representative images after 10 weeks or 23 weeks of feeding. Scale bar = 100 µm. Steatosis score assigned after 23 weeks of treatment (n = 3‐7). (D) Illustrative ORO‐stained liver sections from each group and time‐point feeding. Scale bar = 100 µm. Quantification of ORO‐stained area (n = 3). (E) CPT‐1c immunoblot using GAPDH as loading control. Ratio between CPT‐1c and GAPDH was calculated. Values with different superscripts are significantly different from each other (P < 0.05), assessed by one‐way ANOVA. Differences (P < 0.05) between time points (10 weeks vs. 23 weeks) for each pairing group were assessed by pairing one‐way ANOVA and denoted as “#.” Abbreviation: GAPDH, glyceraldehyde 3‐phosphate dehydrogenase.
Blinded quantitative analysis performed by an experienced pathologist revealed that animals treated with DUAL diet exhibited microvesicular and macrovesicular steatosis grade 2 associated with hepatocyte ballooning, reaching in most of the animals a S2A4 NAFLD score (moderate steatosis, severe activity according to Bedossa system( 15 )) (Fig. 2C and Supporting Fig. S6A‐C).
All experimental groups after 23 weeks of treatment displayed positive Oil Red O (ORO) staining compared with control mice. However, numerous macrolipid and microlipid droplets were more profuse in DUAL‐fed mice (Fig. 2D). Consistently, hepatic triglycerides (TG) were increased in all mice, with the highest levels found in those treated with DUAL diet (Supporting Fig S6D).
Dietary free fatty acids (FFAs) are the main source of TG in the liver. Interestingly, while the level of nonesterified fatty acids (NEFAs) in the serum of WD‐fed mice was significantly increased, the NEFAs in the DUAL animals were not up‐regulated (Supporting Fig. S6E). This indicates the massive FFA flux into the liver. Consistently we found significant up‐regulation of CD36 expression (FA translocase) in the livers of DUAL animals (Supporting Fig. S6F). At the same time, the level of TG in serum of DUAL mice was not proportionally increased, reflecting the possible impairment in hepatic secretion of very low‐density lipoproteins( 16 ) (Supporting Fig. S6G).
Expectedly, the lipid load induced an increase in lipid oxidation, as observed by raised carnitine palmitoyltransferase 1c (CPT‐1c) levels in WD. However, protein expression of CPT‐1c in the DUAL group was not as high as in WD mice. Altogether, high FFA flux into the liver, poor TG secretion, in combination with reduced lipid oxidation contributed to accumulation of a remarkable amount of fat in the liver parenchyma of DUAL mice (Fig. 2E).
Oxidative Stress and Hepatocyte Cell Death in DUAL‐Fed Mice
Extensive fat accumulation in the liver tissue together with alcohol consumption generate oxidative stress.( 17 ) Immunoblotting showed increased expression of cytochrome P450 in the DUAL group after 23 weeks feeding (Fig. 3A). Consequently, we performed 4‐hydroxinonenal staining in all groups of mice. Overall, DUAL diet led to significant induction of lipid peroxides in the liver (Fig. 3B).
FIG. 3.
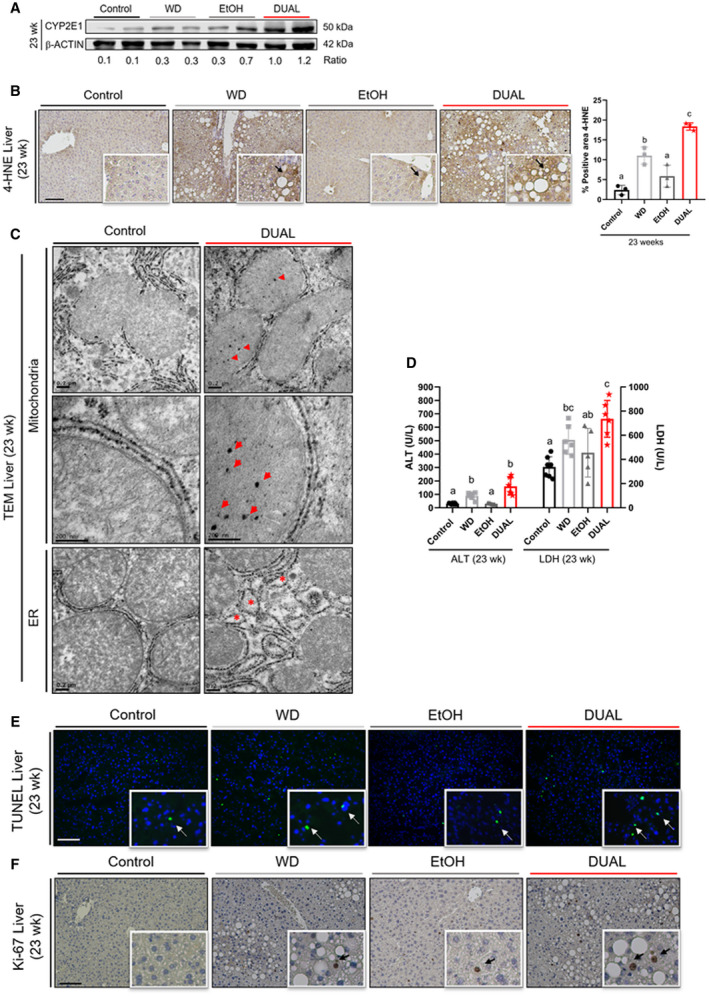
DUAL diet stimulates early oxidative stress and hepatocyte cell death. (A) CYP2E1 western blot, β‐actin used as a loading control. Ratio between CYP2E1 and β‐actin was calculated. (B) Illustrative 4‐HNE‐stained liver sections from each group after 23 weeks of feeding and 4‐HNE quantification (n = 3). Scale bar = 100 µm. (C) Representative TEM pictures of control and DUAL groups. Mitochondria and endoplasmic reticulum are shown. Arrows mark cristae inclusions in mitochondrial matrix. (D) ALT and LDH measurements in serum after 12 hours of fasting (n = 5‐7). (E) Representative TUNEL‐stained photomicrographs at 23 weeks. Scale bar = 100 µm. (F) Ki‐67 liver IHC staining after 23 weeks of feeding. Scale bar = 100 µm. Values with different superscripts are significantly different from each other (P < 0.05), assessed by one‐way ANOVA. Abbreviations: 4‐HNE, 4‐hydroxinonenal; CYP2E1, cytochrome P450; ER, endoplasmic reticulum; TUNEL, terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick‐end labeling.
Oxidative stress led to significant ultrastructural morphological changes in DUAL animals identified by transmission electron microscopy (TEM).( 18 ) After 23 weeks, DUAL diet promoted important morphological changes in mitochondria, which exhibited irregular circled shapes, accumulation of cristae, and multiple electron dense particles (Fig. 3C). Importantly, enlargement of the rough endoplasmic reticulum and cisternae dilation were specifically found in DUAL mice (Fig. 3C and Supporting Fig. S7A,B).
Oxidative and mitochondrial stress likely contributed to liver damage and caused modest but significant increases of the plasma levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), lactate dehydrogenase (LDH), all major clinical indicators of cellular liver injury (Fig. 3D and Supporting Fig. S7C). Consistently, the cell death by TUNEL (terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick‐end labelling) staining in DUAL‐fed livers was significantly higher compared with other experimental groups (Fig. 3E and Supporting Fig. S7D).
Death of hepatocytes may trigger compensatory proliferation in surrounding cells to maintain tissue homeostasis. Accordingly, Ki‐67 staining revealed that cellular proliferation was higher in DUAL mice compared with the rest of experimental groups (Fig. 3F and Supporting Fig. S7E).
DUAL Feeding Induces Extensive Hepatic Inflammation
Fat accumulation and cell death in the liver further caused immune cell infiltration and hepatic inflammation.( 19 ) All treated groups showed an increased accumulation of CD45 and F4/80 positive Kupffer cells/macrophages, as assessed by immunofluorescence (IF) staining, but such infiltration was clearly more pronounced in DUAL animals (Fig. 4A‐C and Supporting Fig. S8A,B).
FIG. 4.
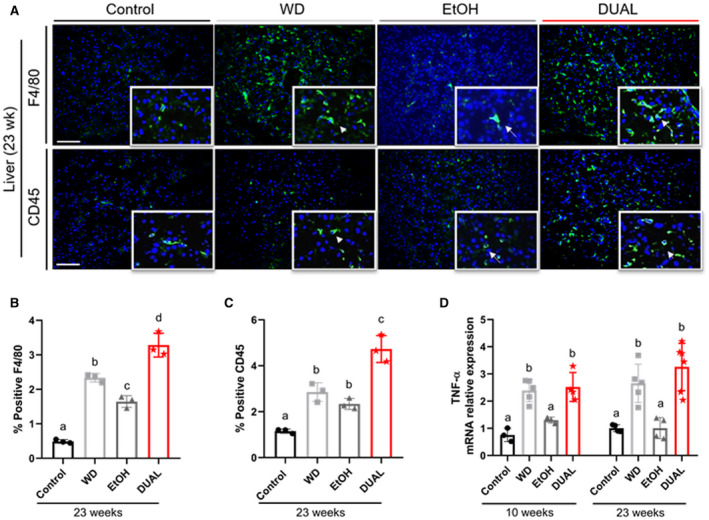
DUAL mice manifest enhanced hepatic inflammation. (A) Illustrative CD45 and F4/80 IF staining in liver sections of mice fed for 23 weeks. Positive immune cells are stained in green. Nuclei are stained in blue using DAPI as a counterstain. Arrows indicate CD45 or F4/80 positive cells, respectively. Scale = 100 µm. (B,C) Quantification of %CD45 and F4/80 positive cells, respectively, using ImageJ software (n = 3). (D) TNF‐α mRNA relative expression to GAPDH after 10 weeks and 23 weeks on diet (n = 3‐6). Values with different superscripts are significantly different from each other (P < 0.05), assessed by one‐way ANOVA. Abbreviations: DAPI, 4′,6‐diamidino‐2‐phenylindole; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase.
Neutrophil infiltration is an important hallmark of alcoholic hepatitis and correlates with the severity of disease.( 20 ) Immunohistochemistry (IHC) staining revealed that neutrophil infiltration in the liver was significantly higher in DUAL mice (Supporting Fig. S8C,D). Infiltrating cells actively produce different cytokines and further contribute to create a pro‐inflammatory microenvironment. Consistently, messenger RNA (mRNA) expression of tumor necrosis factor‐α (TNF‐α) was significantly increased in mice fed with DUAL diet, especially after 23 weeks of feeding (Fig. 4D).
DUAL Diet Leads to Increased Hepatic Stellate Cell Activation and Hepatic Fibrogenesis
TNF‐α overproduction induces activation of hepatic stellate cells (HSCs) in the liver.( 21 ) We found strong expression of α‐smooth muscle actin (α‐SMA), a marker of HSCs activation, using western blot analysis and IHC staining in DUAL‐fed animals (Fig. 5A,B [upper panel]). Activated HSCs are the major source of ECM during progression of fibrosis.( 22 ) Hence, SR staining clearly demonstrated that feeding a WD or EtOH alone induced only minor collagen expression in the liver, whereas rapid and severe progression of fibrosis was a notable histological feature of DUAL‐fed mice. The first signs of fibrosis were detectable as soon as 10 weeks of treatment (Supporting Fig. S9A,B), with remarkable escalation of fibrogenesis at 23 weeks (Fig. 5B,C [middle panel]). These findings were additionally confirmed by IF staining for collagen I (Fig. 5B,D [lower panel]). Histopathological evaluation of fibrosis revealed F1a stage after 10 weeks and F1b in all DUAL animals after 23 weeks of feeding (Fig. 5E). Additionally, collagen fibers were further identified though TEM analysis in DUAL mice (Supporting Fig. S10). Finally, quantitative polymerase chain reaction confirmed that DUAL treatment significantly up‐regulated pro‐fibrogenic genes such as collagen1α1 and MCP1 (monocyte chemoattractant protein 1) (Fig. 5F).
FIG. 5.
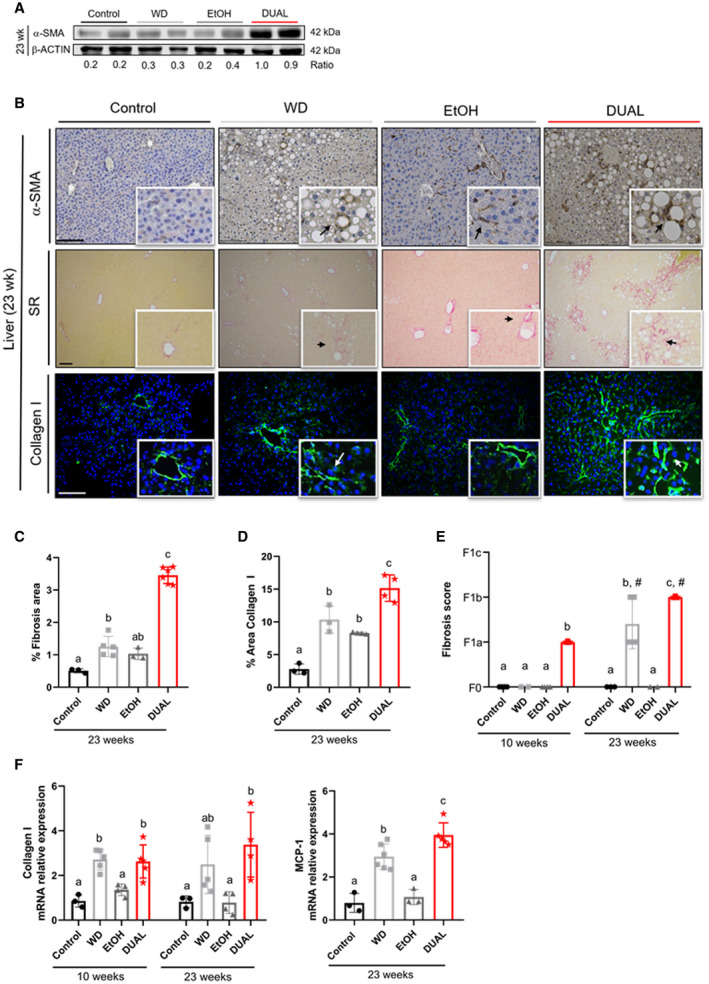
DUAL feeding leads to prominent hepatic fibrosis. (A) α‐SMA western blot using β‐actin as a loading control. Ratio between α‐SMA and β‐actin is calculated. (B) HSCs and fibrosis‐related stainings in liver. Representative liver images stained with α‐SMA (IHC), SR, and collagen I (IF). Scale bar = 100 µm. (C,D) Quantification of positive SR‐stained and collagen I–stained areas after 23 weeks of treatment, respectively (SR, n = 3‐6; collagen I, n = 3‐4). (E). Fibrosis score assigned after 10 weeks and 23 weeks of feeding in all groups (n = 3‐7) (F) Respective collagen I (10 weeks and 23 weeks) and MCP1 (23 weeks) mRNA expression relative to GAPDH (n = 3‐6). Values with different superscripts are significantly different from each other (P < 0.05), assessed by one‐way ANOVA. Differences (P < 0.05) between time points (10 weeks vs. 23 weeks) for each pairing group were assessed by pairing one‐way ANOVA and denoted as “#.” Abbreviation: MCP1, monocyte chemoattractant protein 1.
DUAL Diet Drives the Activation of Characteristic Disease‐Associated Signaling Pathways
To better define the relevance of the DUAL model and to unveil potential modulation of cell regulatory processes, the livers of 10‐week‐old DUAL mice were subjected to bulk RNA sequencing (RNA‐seq). A total of 1,563 genes were significantly (adjusted P value < 0.05 and fold change > 2) altered in liver after DUAL feeding compared with the corresponding controls, of which 1,259 were up‐regulated and 304 down‐regulated (Fig. 6A). Differentially expressed mRNAs were predominantly enriched in inflammation, fibrosis, and metabolism‐related pathways. Affected inflammatory pathways ranged from inflammatory response, neutrophil‐mediated immunity and activation, chemokine and cytokine biosynthesis and activity, and phagocytic and platelet activation. The fibrotic pathways included ECM organization, collagen fibril organization, and collagen binding (Fig. 6B). Furthermore, pathway analysis was performed with reference to the Kyoto Encyclopedia of Genes and Genomes database (Supporting Fig. S11), and, consistently, DUAL animals showed an up‐regulation of important metabolic/inflammatory/fibrogenic pathways including phosphoinositide 3‐kinase (PI3K)/AKT, TNF, nuclear factor kappa B NF‐κB, CAMs, ECM interaction, focal adhesion, chemokine, and toll‐like receptor (TLR) signaling pathways. Additionally, protein–protein interaction networks were built using the differentially expressed genes (Fig. 6C). The results indicate the presence of central regulatory genes coordinating the expression of many others. Most of these genes are involved in inflammatory response (Btk [Bruton’s tyrosine kinase], Csf1r [colony stimulating factor 1 receptor], Ifr8 [interferon regulatory factor 8], Syk, and Nlrp3 [NLR family pyrin domain containing 3]), apoptosis and cell cycle regulation (Bcl2 [B cell lymphoma 2], Plcg2 [phospholipase C gamma 2], and Fyn), and metabolism (Pik3 [phosphoinositide 3‐kinase] and Foxo3 [forkhead box protein O3]).
FIG. 6.
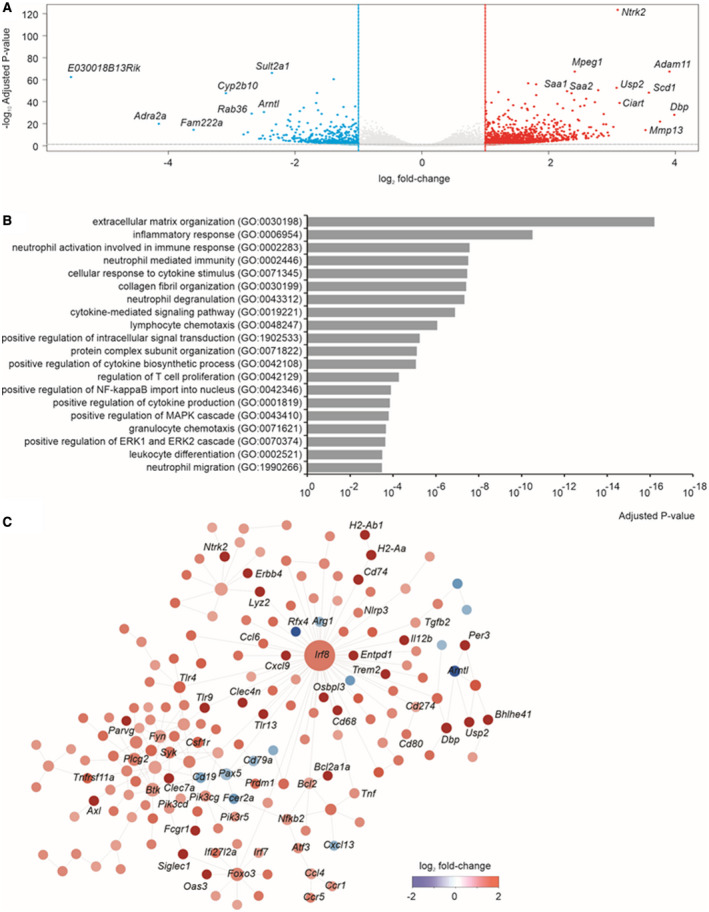
Single‐cell RNA‐seq of liver tissue after 10 weeks of DUAL feeding. (A) The volcano plot was constructed using fold‐change values and adjusted P values. The vertical blue lines corresponds to 2‐fold up and down, and horizontal green line represents adjusted P value of 0.05. The red points indicate differentially expressed genes with statistical significance, and nonsignificant genes are in black. (B) Gene ontology enrichment analysis performed for up‐regulated and down‐regulated genes between C57BL/6 control diet and DUAL diet. The top 10 enriched the biological process, molecular function, and cellular component. The x‐axis represents the P‐value ranking, and the y‐axis represents the gene ontology term. Red and blue bars indicate up‐regulated and down‐regulated gene ontology terms, respectively. (C) Overview of the protein–protein interaction network. The network was generated using the NetworkAnalyst. Color represents the expression of nodes. Specifically, red and green represent up‐regulated and down‐regulated nodes, respectively. The color gradient indicates the expression level. The size of the nodes indicates the degrees that the nodes connect to others. Abbreviations: Bcl2, B‐cell lymphoma 2; Btk, Bruton tyrosine kinase; Csf1, Macrophage colony‐stimulating factor 1; ERK, extracellular signal‐regulated kinase; Foxo1, Forkhead Box O1; Fyn, Proto‐oncogene tyrosine‐protein kinase Fyn; GO, gene ontology; Irf8, Interferon Regulatory Factor 8; LogFC, fold change; MAPK, mitogen‐activated protein kinase; NF‐κB, Nuclear factor kappa B; Pi3k, Phosphoinositide 3‐kinases; Plcg2, Phospholipase C Gamma 2; Ptgs2, Prostaglandin‐Endoperoxide Synthase 2; Syk, Spleen Associated Tyrosine Kinase; TNF, tumor necrosis factor.
Importantly, most up‐regulated pathways as well as overexpressed genes such as Tnf,( 23 ) Nf‐κB,( 24 ) Bcl2,( 25 ) Tlr4/9,( 26 ) and pi3K ( 27 ) have been previously reported in relation to the progression of clinical and preclinical steatohepatitis and liver fibrosis (Supporting Fig. S11B).
DUAL Diet Drives Fibrogenesis and Tumorigenesis
Epidemiological data suggest that the combination of alcohol and metabolic factors is associated with an increased risk of cirrhosis and cancer.( 3 ) Therefore, we challenged mice to long‐term DUAL feeding. Within 52 weeks of DUAL diet, livers became extraordinarily enlarged, whitish‐yellow in color, with pronounced scar tissue on the surface and multiple nodules in some, but not in all mice (Fig. 7A,B). Moreover, animals developed splenic enlargement (Supporting Fig. S12A), and significant increases of plasma AST, ALT, LDH, and serum cholesterol (Supporting Fig. S12B‐E). Hematoxylin and eosin (H&E) and SR stainings demonstrated intense immune infiltration, significant fatty changes, extensive collagen deposition, and well‐differentiated micronodules, surrounded by fibrotic connective tissue extending between portal regions, which overall indicated the stage of hepatic cirrhosis (Fig. 7C). Notably, we detected HSP70/HSP72 and CK19 positive staining, a broadly used marker of hepatocellular carcinoma (HCC), inside the nodules (Fig. 7D). Nodules exhibit positive proliferating cell nuclear antigen (PCNA) nuclei staining, an indicator of proliferation and cell‐cycle up‐regulation. Consistently, cyclin E mRNA expression was increased in the liver tissue of DUAL mice (Supporting Fig. S13A,B).
FIG. 7.
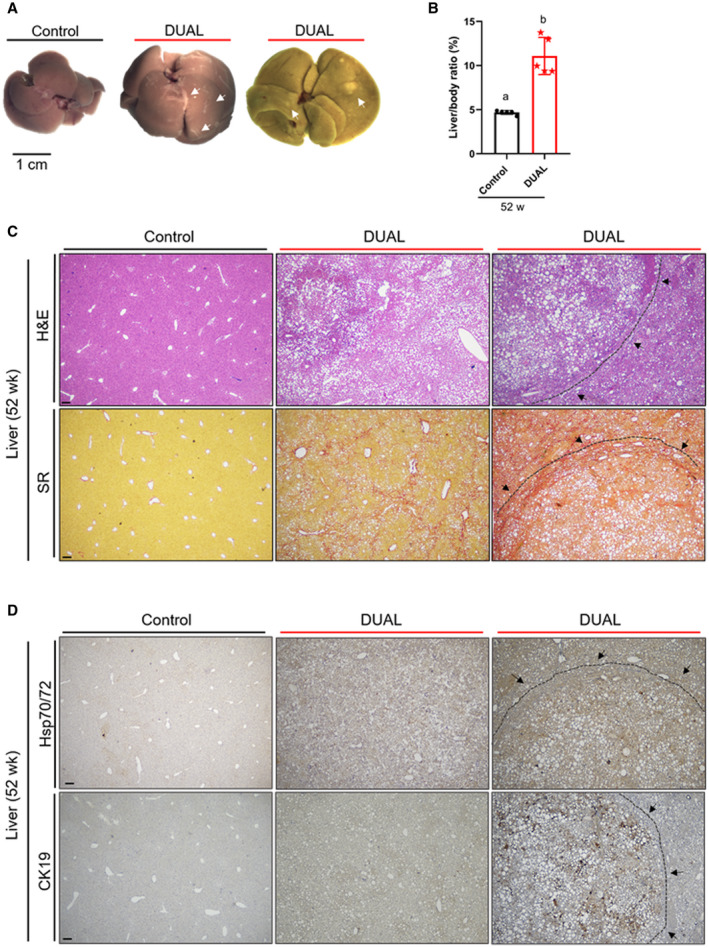
Advanced fibrosis and tumor development in DUAL diet as an end‐point of metabolic disease. (A) Liver pictures after 52 weeks of feeding: control, DUAL, and DUAL with regenerative nodule. Fibrotic scars and nodules on the surface are marked with arrows. (B) Liver/BW ratio (%) (n = 5). (C) Representative pictures of H&E and SR stainings after 52 weeks: control, DUAL, and DUAL with regenerative nodule. Scale = 100 µm. (D) Representative pictures of Hsp70/72 and CK19 after 52 weeks treatment. Scale = 100 µm. Values with different superscripts are significantly different from each other (P < 0.05), assessed by one‐way ANOVA.
To further study the effect of our DUAL model on tumor progression, male mice were administered a single dose of diethylnitrosamine (DEN) at the age of 14 days. Eight weeks later, DEN‐treated mice were placed on a DUAL diet or control chow diet for 16 weeks (Supporting Fig. S14A). Macroscopic evaluation identified that all mice fed with DUAL diet exhibited single or multiple tumor nodules larger than 0.5 cm (Fig. 8A,B). In contrast, only one out of nine mice developed DEN‐induced nodule on chow treatment. Additionally, the DUAL diet also increased the hepatosomatic ratio, ALT, AST, and LDH in DEN‐treated mice (Supporting Fig. S14B‐E). Pathological examination (Fig. 8C) revealed well‐circumscribed lesions with compressed adjacent parenchyma, loss of lobular architecture, and moderate fatty changes. All tumor nodules in DEN + DUAL mice showed strong nuclear expression of PCNA (Fig. 8D). Next, we assessed the characteristic tumor markers and found strong overexpression of HSP70/72 (Fig. 8E) and CK19 stainings (Supporting Fig. S14F) in the nodules of DEN + DUAL treated mice.
FIG. 8.
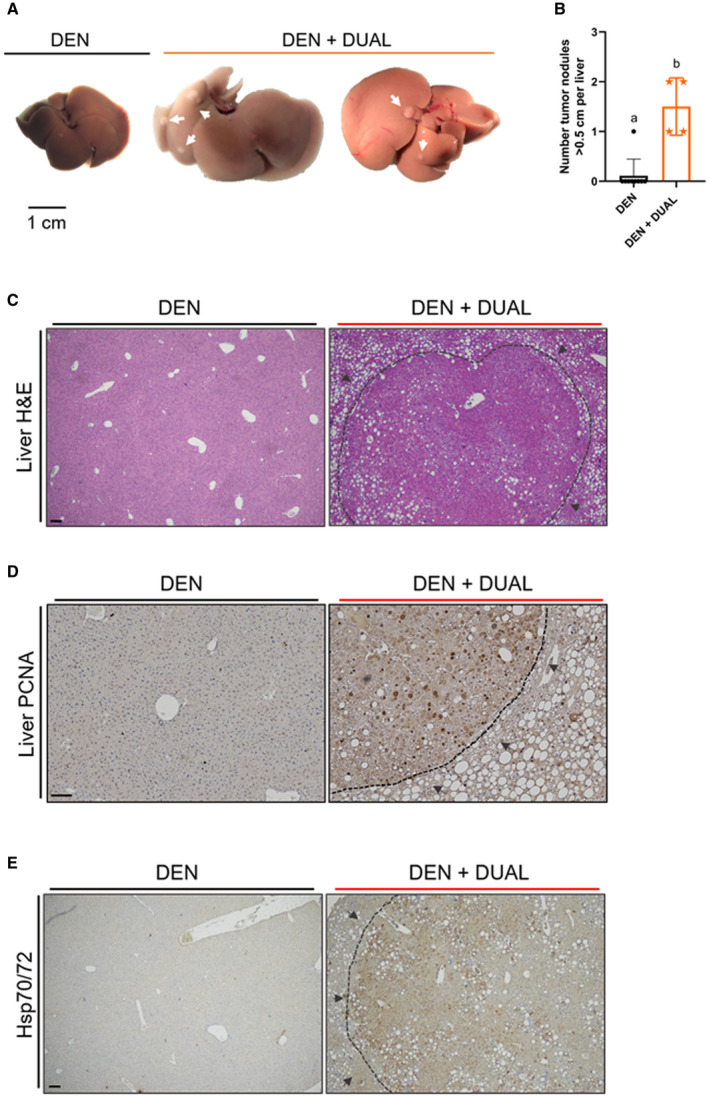
DUAL diet + DEN injection as a model of rapid tumor progression. (A) Liver pictures of DEN and DEN + DUAL model. Tumor nodules are marked with arrows. (B) Number of tumor nodules > 0.5 cm per liver (n = 4‐9). (C‐E) Representative pictures of H&E, Hsp70/72, and PCNA, respectively. Scale = 100 µm. Values with different superscripts are significantly different from each other (P < 0.05), assessed by one‐way ANOVA.
Taken together, our experimental model showed hepatic hyperplasia confirmed following histological examination and significant up‐regulation of HSP70/72, CK19, and cell proliferation (PCNA), providing evidence for the synergistic and tumor promoting action of DUAL diet in DEN‐induced early HCC.
Discussion
Over 2 billion people worldwide consume alcohol, and up to 75 million are diagnosed with alcohol‐use disorders and are at high risk of ALD.( 1 ) Moreover, modern societies have acquired unhealthy fast food habits that tremendously promote weight gain and, consequently, the development of MAFLD.( 28 , 29 )
The latest figures indicate that overweight affects from 30%‐70% of the population.( 30 ) Thus, very likely, both dietary habits, alcohol drinking pattern, and high caloric consumption would overlap in one part of the population. Moreover, high caloric meals are often combined with mild and regular alcohol intake.( 31 ) In fact, for patients with BMI over 35 kg/m2, the hepatotoxicity of alcohol doubles.( 1 )
Recent epidemiological and experimental evidence has highlighted the dangerous synergism of alcohol, obesity, and MS in the progression of chronic liver disease.( 4 , 5 , 6 ) One of these conditions is often predominant, with the other acting as a cofactor of morbi‐mortality.( 3 )
Therefore, an appropriate animal model that faithfully reproduces a combined type of liver damage would definitely improve the understanding of the synergistic mechanism of alcohol and MS. In the current study, we developed a physiological experimental murine model that mimics the human dietary habits by combining high fat, high fructose, high cholesterol, Western‐style diet with alcohol administration.
Although several hybrid models have been suggested,( 32 , 33 ) most of them have relevant limitations and do not fully mirror all physiological, metabolic, histologic, and common clinical features of human steatohepatitis (Supporting Table S7), including hepatic inflammation and advanced fibrosis.( 8 ) The main reason is that animals naturally display strong aversion to alcohol. Therefore, if alcohol is incorporated in the drinking water, mice drink much less than expected, which is sufficient to induce moderate clear steatosis but simply not high enough to cause significant liver damage.( 9 )
In our innovative model, we combined WD with EtOH in the drinking water sweetened by D‐glucose. We used WD from the Amylin liver NASH study( 16 ) as a combination of 40% fat, 22% fructose, and 2% cholesterol, resulting in relatively strong steatohepatitis. Moreover, glucose in the drinking water potentiates absorption of fructose from the diet, whereas fructose catalyzes glucose uptake and storage in the liver, leading together to stronger harm.( 34 )
More importantly, by incorporating D‐glucose in the water we overcome mouse aversion to alcohol. Sweetened water successfully masked the taste of alcohol, thus increasing alcohol intake. DUAL‐fed animals consume 32.2 g/kg of alcohol daily. Hence, the relative risk of alcohol‐associated cirrhosis increases in humans who drink more than 25 g/day.( 35 )
Additionally, the consumption of fatty WD further elevated EtOH intake. Previous reports suggested the existence of a positive correlation between EtOH and fat, whereby each nutrient stimulates the intake of the other. Within this synergistic vicious cycle, calorie intake can dramatically increase.( 10 )
Consequently, following initiation of the DUAL diet, mice started to gain weight and developed obesity, characterized by an increase in BMI. Over a period of 23 weeks, the DUAL group became 19% heavier than the control group. Abdominal obesity is a predominant underlying risk factor for MS.( 13 ) Simultaneously with obesity, DUAL animals developed dyslipidemia and hyperglycemia: altogether three important medical conditions of MS.( 36 ) Hence, it is essential to mention that impaired fasting glycemia was not accompanied either by glucose intolerance or by insulin resistance in DUAL mice. In fact, the ITT area under the curve was significantly lower in animals treated with DUAL diet compared with controls. Nevertheless, the explanation for this phenomenon by enhanced insulin action is an obvious misinterpretation of the results. We believe that DUAL mice exhibited a defect in the counter‐regulatory response to insulin in—particularly—gluconeogenesis.( 37 ) In fact, RNA‐seq data demonstrated that the level of glucose‐6 phosphatase was remarkably lower in the DUAL group.
Another key feature of the DUAL obesity is the remarkable damage of WAT: Hypertrophic adipocytes develop an inflammatory phenotype and crown‐like structures, consisting of necrotic adipocytes. Adipocyte death and subsequent inflammation increase lipolysis, flux of lipids to the liver, production of pro‐inflammatory cytokines, and significantly contribute to disease progression.( 3 , 11 ) Previous animal studies showed that high‐fat diet in combination with EtOH binge feeding synergistically induce liver injury by stimulating hepatocytes to produce chemokine (C‐X‐C motif) ligand 1 (CXCL1), which subsequently promoted hepatic neutrophil recruitment.( 38 ) RNA‐seq data showed up‐regulation of CXCL1 (up to 30 times) and concomitant pathways related to neutrophil‐mediated immunity in the liver of DUAL animals.
Hence, hepatic steatosis induced by DUAL diet takes a short period of time. Due to the massive flux of FFAs into the liver, relatively poor ß‐oxidation and reduced TG secretion, lipids accumulate in large amounts already after 10 weeks of feeding and gradually increase over time. DUAL mice developed microvesicular and macrovesicular steatosis with hepatocytes ballooning. Consequently, the increased accumulation of FFAs in the liver led to metabolism deregulation and the generation of oxidative stress, which triggered cell death, inflammation, and immune cell infiltration into the hepatic parenchyma.
Inflammatory cytokines, produced by immune cells (e.g., TNF‐α), further activated HSCs and stimulated the production of collagen fibers and ECM deposition in the liver, leading to fibrogenesis.( 39 ) Mice fed a DUAL diet exhibited steatohepatitis already after 10 weeks, whereas feeding for 23 weeks resulted in more fibrotic stage, particularly affecting the portal and bridging areas.
Notably, transcriptomic changes relevant to chronic liver diseases in humans were also demonstrated in DUAL mice, including (1) Btk, Csf1, Sfpi1, Irf8 and Syk, which play critical roles in the development and function of myeloid and lymphoid cells, associated with chronic inflammation( 40 , 41 , 42 , 43 ); (2) Bcl2, Ptgs2, Plcg2 and Fyn, which promote apoptosis and proliferation( 25 , 44 , 45 ); and (3) Pi3k and Foxo1, which are master regulators of glucose/lipid homeostasis.( 46 , 47 ) The up‐regulation of TNF and NF‐κB( 24 ) in DUAL animals entirely recapitulated the pathogenesis of human‐like steatohepatitis and correlated with disease progression to advanced fibrosis. Importantly, the activation of TLR4/9 indicated that besides the direct toxic effect of alcohol and WD on the liver, the abnormal microbiome and the loss of intestinal barrier function may potentially contribute to the pathogenesis in DUAL mice.( 26 , 48 )
DUAL mice were similar to human disease, not only in terms of initiation of steatohepatitis, but also in development of cirrhosis and tumorigenesis. Thus, after 1 year of DUAL diet, animals demonstrated extensive collagen accumulation and micronodular cirrhotic changes.
The reported DUAL diet additionally functions as an excellent tumor promoter for DEN‐induced liver tumorigenesis in mice. With this diet, preneoplastic nodules develop within only 26 weeks following DEN injection. This observation supports the importance of identifying patients with excess alcohol consumption and MS, as they are at a higher risk of liver‐related cancer.
Altogether, our preclinical model induces the development of liver damage in the context of key risk factors for the human condition (e.g., alcohol consumption, obesity, MS), naturally mimicking human pathology and the progression to advanced liver fibrosis, cirrhosis, and end‐stage tumorigenesis. Importantly, it is an easy, affordable, highly reproducible, time‐efficient diet that does not require any special skill or expensive equipment. Moreover, it is associated with no mortality even using long‐term feeding, and as a mild procedure, it should be easily approved by ethical committees.
However, there are some limitations and methodological difficulties in our DUAL model. First, according to our observations, as typical nocturnal animals, mice consumed significant amounts of EtOH in the drinking water primarily during the dark cycle. Hence, given the fast metabolic capacity of mice, on the following morning their blood alcohol level levels drop significantly. Therefore, we calculated the daily amount of consumed alcohol based on the volume of the water drunk from the bottle. Second, we tested our model on male and female mice. Independently of gender, all mice developed steatohepatitis and fibrosis, and advanced cirrhotic changes. Nonetheless, the phenotype in male mice was slightly stronger, including a more elevated BMI, more pronounced steatosis, and moderately increased ALT (data not showed). However, we dealt with some difficulties with the housing of male mice. Food intake was negatively affected by the subordination, raising concerns about welfare, alcohol consumption, and negatively impacted research validity. Hence, according to our experience, the only possibility for male mice is individual housing (for DUAL diet and the corresponding control groups).
Finally, lack of clinical studies with tissue of patients with DUAL alcoholic and metabolic‐associated steatohepatitis was a limitation of our study. Moreover, there were major methodological concerns about the existing studies, as many reports failed to consider the pattern and type of alcohol use and/or did not separate between lifetime and current abstainers. In addition, underreporting of alcohol use is a major concern, particularly when assessing patients who are aware of their liver disease.( 49 , 50 )
Altogether, DUAL alcoholic and MAFLD remains as not well‐explored area of great interest, despite the increased number of affected patients. Our innovative preclinical DUAL model can be a valuable toolbox, as it mimics all histological, metabolic, transcriptomic gene signatures of human disease, thereby contributing to the development of very much needed therapeutic targets (Supporting Fig. S13).
Supporting information
Fig S1
Fig S2
Fig S3
Fig S4
Fig S5
Fig S6
Fig S7
Fig S8
Fig S9
Fig S10
Fig S11
Fig S12
Fig S13
Fig S14
Fig S15
Supplementary Material
Supported by EXOHEP‐CM (S2017/BMD‐3727), Ramón y Cajal (RYC‐2014‐15242 and RYC‐2015‐17438), NanoLiver‐CM (Y2018/NMT‐4949), COST Action (CA17112), AMMF (2018/117), ERAB (EA 18/14), MINECO Retos (SAF2016‐78711 and SAF2017‐87919‐R), and German Research Foundation (DFG NE 2128/2‐1, SFB 1382‐403224013/A02, and SFB/TRR57/P04). FJC is a Gilead Research Liver Scholar. The research group belongs to the validated Research group Ref. 970935 “Liver Pathophysiology”, 920631 “Lymphocyte immunology”, 920361 “Immunogenética e inmunología de las mucosas” and IBL‐6 (imas12‐associated). FG and KZ are Chinese Scholarship Council (CSC) fellows. O.E.‐V is supported by Beca FPI (associated to MINECO SAF2017‐87919R) and R.B.‐U. by Contratos predoctorales de personal investigador en formación UCM‐Banco Santander (CT63/19).
Potential conflict of interest: Dr. Bruns consults for Intercept. He received grants from Gilead, Falk, Abbvie, Norgine, and Merck.
References
Author names in bold designate shared co‐first authorship.
- 1. Pimpin L, Cortez‐Pinto H, Negro F, Corbould E, Lazarus JV, Webber L, et al. Burden of liver disease in Europe: epidemiology and analysis of risk factors to identify prevention policies. J Hepatol 2018;69:718‐735. [DOI] [PubMed] [Google Scholar]
- 2. Ndugga N, Lightbourne TG, Javaherian K, Cabezas J, Verma N, Barritt ASt, Bataller R. Disparities between research attention and burden in liver diseases: implications on uneven advances in pharmacological therapies in Europe and the USA. BMJ Open 2017;7:e01362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ntandja Wandji LC, Gnemmi V, Mathurin P, Louvet A. Combined alcoholic and non‐alcoholic steatohepatitis. JHEP Rep 2020;2:100101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Naveau S, Cassard‐Doulcier AM, Njike‐Nakseu M, Bouchet‐Delbos L, Barri‐Ova N, Boujedidi H, et al. Harmful effect of adipose tissue on liver lesions in patients with alcoholic liver disease. J Hepatol 2010;52:895‐902. [DOI] [PubMed] [Google Scholar]
- 5. Hart CL, Morrison DS, Batty GD, Mitchell RJ, Davey SG. Effect of body mass index and alcohol consumption on liver disease: analysis of data from two prospective cohort studies. BMJ 2010;340:c1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chang Y, Cho YK, Kim Y, Sung E, Ahn J, Jung HS, et al. Nonheavy drinking and worsening of noninvasive fibrosis markers in nonalcoholic fatty liver disease: a cohort study. Hepatology 2019;69:64‐75. [DOI] [PubMed] [Google Scholar]
- 7. Bellentani S, Tiribelli C. Is it time to change NAFLD and NASH nomenclature? Lancet Gastroenterol Hepatol 2017;2:547‐548. [DOI] [PubMed] [Google Scholar]
- 8. Nevzorova YA, Boyer‐Diaz Z, Cubero FJ, Gracia‐Sancho J. Animal models for liver disease—a practical approach for translational research. J Hepatol 2020;73:423‐440. [DOI] [PubMed] [Google Scholar]
- 9. D'Souza El‐Guindy NB, Kovacs EJ, De Witte P, Spies C, Littleton JM, de Villiers WJ, et al. Laboratory models available to study alcohol‐induced organ damage and immune variations: choosing the appropriate model. Alcohol Clin Exp Res 2010;34:1489‐1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Barson JR, Karatayev O, Chang GQ, Johnson DF, Bocarsly ME, Hoebel BG, et al. Positive relationship between dietary fat, ethanol intake, triglycerides, and hypothalamic peptides: counteraction by lipid‐lowering drugs. Alcohol 2009;43:433‐441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Parker R, Kim SJ, Gao B. Alcohol, adipose tissue and liver disease: mechanistic links and clinical considerations. Nat Rev Gastroenterol Hepatol 2018;15:50‐59. [DOI] [PubMed] [Google Scholar]
- 12. Divoux A, Tordjman J, Lacasa D, Veyrie N, Hugol D, Aissat A, et al. Fibrosis in human adipose tissue: composition, distribution, and link with lipid metabolism and fat mass loss. Diabetes 2010;59:2817‐2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Paschos P, Paletas K. Non alcoholic fatty liver disease and metabolic syndrome. Hippokratia 2009;13:9‐19. [PMC free article] [PubMed] [Google Scholar]
- 14. Boyle M, Masson S, Anstee QM. The bidirectional impacts of alcohol consumption and the metabolic syndrome: cofactors for progressive fatty liver disease. J Hepatol 2018;68:251‐267. [DOI] [PubMed] [Google Scholar]
- 15. Bedossa P, Poitou C, Veyrie N, Bouillot J‐L, Basdevant A, Paradis V, et al. Histopathological algorithm and scoring system for evaluation of liver lesions in morbidly obese patients. Hepatology 2012;56:1751‐1759. [DOI] [PubMed] [Google Scholar]
- 16. Clapper JR, Hendricks MD, Gu G, Wittmer C, Dolman CS, Herich J, et al. Diet‐induced mouse model of fatty liver disease and nonalcoholic steatohepatitis reflecting clinical disease progression and methods of assessment. Am J Physiol Gastrointest Liver Physiol 2013;305:G483‐495. [DOI] [PubMed] [Google Scholar]
- 17. Mantena SK, King AL, Andringa KK, Eccleston HB, Bailey SM. Mitochondrial dysfunction and oxidative stress in the pathogenesis of alcohol‐ and obesity‐induced fatty liver diseases. Free Radic Biol Med 2008;44:1259‐1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jayakumar S, Guillot S, Argo C, Redick J, Caldwell S. Ultrastructural findings in human nonalcoholic steatohepatitis. Expert Rev Gastroenterol Hepatol 2011;5:141‐145. [DOI] [PubMed] [Google Scholar]
- 19. Wang K. Molecular mechanisms of hepatic apoptosis. Cell Death Dis 2014;5:e996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bertola A, Park O, Gao B. Chronic plus binge ethanol feeding synergistically induces neutrophil infiltration and liver injury in mice: a critical role for E‐selectin. Hepatology 2013;58:1814‐1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang YM, Seki E. TNFα in liver fibrosis. Curr Pathobiol Rep 2015;3:253‐261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, Dapito DH, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun 2013;4:2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wandrer F, Liebig S, Marhenke S, Vogel A, John K, Manns MP, et al. TNF‐receptor‐1 inhibition reduces liver steatosis, hepatocellular injury and fibrosis in NAFLD mice. Cell Death Dis 2020;11:212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Luedde T, Schwabe RF. NF‐kappaB in the liver–linking injury, fibrosis and hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol 2011;8:108‐118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ramalho RM, Cortez‐Pinto H, Castro RE, Sol?? S, Costa A, Moura MC, et al. Apoptosis and Bcl‐2 expression in the livers of patients with steatohepatitis. Eur J Gastroenterol Hepatol 2006;18:21‐29. [DOI] [PubMed] [Google Scholar]
- 26. Mencin A, Kluwe J, Schwabe RF. Toll‐like receptors as targets in chronic liver diseases. Gut 2009;58:704‐720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Matsuda S, Nakanishi A, Wada Y, Kitagishi Y. Roles of PI3K/AKT/PTEN pathway as a target for pharmaceutical therapy. Open Med Chem J 2013;7:23‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Eslam M, Sanyal AJ, George J, International Consensus P . MAFLD: a consensus‐driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology 2020;158:1999‐2014 e1991. [DOI] [PubMed] [Google Scholar]
- 29. Eslam M, Newsome PN, Sarin SK, Anstee QM, Targher G, Romero‐Gomez M, et al. A new definition for metabolic dysfunction‐associated fatty liver disease: an international expert consensus statement. J Hepatol 2020;73:202‐209. [DOI] [PubMed] [Google Scholar]
- 30. WHO . Obesity data and statistics. WHO [Internet]. https://www.euro.who.int/en/health‐topics/noncommunicable‐diseases/obesity/data‐and‐statistics. 2014. Accessed February 2020.
- 31. Mahli A, Hellerbrand C. Alcohol and obesity: a dangerous association for fatty liver disease. Dig Dis 2016;34(Suppl. 1):32‐39. [DOI] [PubMed] [Google Scholar]
- 32. Lazaro R, Wu R, Lee S, Zhu N‐L, Chen C‐L, French SW, et al. Osteopontin deficiency does not prevent but promotes alcoholic neutrophilic hepatitis in mice. Hepatology 2015;61:129‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gabele E, Dostert K, Dorn C, Patsenker E, Stickel F, Hellerbrand C. A new model of interactive effects of alcohol and high‐fat diet on hepatic fibrosis. Alcohol Clin Exp Res 2011;35:1361‐1367. [DOI] [PubMed] [Google Scholar]
- 34. Laughlin MR. Normal roles for dietary fructose in carbohydrate metabolism. Nutrients 2014;6:3117‐3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mathurin P, Bataller R. Trends in the management and burden of alcoholic liver disease. J Hepatol 2015;62:S38‐S46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lonardo A, Ballestri S, Marchesini G, Angulo P, Loria P. Nonalcoholic fatty liver disease: a precursor of the metabolic syndrome. Dig Liver Dis 2015;47:181‐190. [DOI] [PubMed] [Google Scholar]
- 37. Ayala JE, Samuel VT, Morton GJ, Obici S, Croniger CM, Shulman GI, et al. Standard operating procedures for describing and performing metabolic tests of glucose homeostasis in mice. Dis Model Mech 2010;3:525‐534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chang B, Xu M‐J, Zhou Z, Cai Y, Li M, Wang W, et al. Short‐ or long‐term high‐fat diet feeding plus acute ethanol binge synergistically induce acute liver injury in mice: an important role for CXCL1. Hepatology 2015;62:1070‐1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yang YM, Seki E. TNFalpha in liver fibrosis. Curr Pathobiol Rep 2015;3:253‐261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Giordano M, Roncagalli R, Bourdely P, Chasson L, Buferne M, Yamasaki S, et al. The tumor necrosis factor alpha‐induced protein 3 (TNFAIP3, A20) imposes a brake on antitumor activity of CD8 T cells. Proc Natl Acad Sci U S A 2014;111:11115‐11120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Palumbo T, Nakamura K, Lassman C, Kidani Y, Bensinger SJ, Busuttil R, et al. Bruton tyrosine kinase inhibition attenuates liver damage in a mouse warm ischemia and reperfusion model. Transplantation 2017;101:322‐331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Langlais D, Barreiro LB, Gros P. The macrophage IRF8/IRF1 regulome is required for protection against infections and is associated with chronic inflammation. J Exp Med 2016;213:585‐603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Qu C, Zheng D, Li S, Liu Y, Lidofsky A, Holmes JA, et al. Tyrosine kinase SYK is a potential therapeutic target for liver fibrosis. Hepatology 2018;68:1125‐1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Reinehr R, Sommerfeld A, Haussinger D. The Src family kinases: distinct functions of c‐Src, Yes, and Fyn in the liver. Biomol Concepts 2013;4:129‐142. [DOI] [PubMed] [Google Scholar]
- 45. Martin‐Sanz P, Casado M, Bosca L. Cyclooxygenase 2 in liver dysfunction and carcinogenesis: facts and perspectives. World J Gastroenterol 2017;23:3572‐3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tikhanovich I, Cox J, Weinman SA. Forkhead box class O transcription factors in liver function and disease. J Gastroenterol Hepatol 2013;28(Suppl. 1):125‐131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Taniguchi CM, Kondo T, Sajan M, Luo J, Bronson R, Asano T, et al. Divergent regulation of hepatic glucose and lipid metabolism by phosphoinositide 3‐kinase via Akt and PKClambda/zeta. Cell Metab 2006;3:343‐353. [DOI] [PubMed] [Google Scholar]
- 48. Albillos A, de Gottardi A, Rescigno M. The gut‐liver axis in liver disease: pathophysiological basis for therapy. J Hepatol 2020;72:558‐577. [DOI] [PubMed] [Google Scholar]
- 49. Aberg F, Farkkila M. Drinking and obesity: alcoholic liver disease/nonalcoholic fatty liver disease interactions. Semin Liver Dis 2020;40:154‐162. [DOI] [PubMed] [Google Scholar]
- 50. Sanchez‐Jimenez BA, Brizuela‐Alcantara D, Ramos‐Ostos MH, Alva‐Lopez LF, Uribe‐Esquivel M, Chavez‐Tapia NC. Both alcoholic and non‐alcoholic steatohepatitis association with cardiovascular risk and liver fibrosis. Alcohol 2018;69:63‐67. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Fig S4
Fig S5
Fig S6
Fig S7
Fig S8
Fig S9
Fig S10
Fig S11
Fig S12
Fig S13
Fig S14
Fig S15
Supplementary Material