

Abstract
Acetaminophen (N‐acetyl‐para‐aminophenol [APAP]) overdose is the most common cause of drug‐induced liver injury in the Western world and has limited therapeutic options. As an important dietary component intake, fructose is mainly metabolized in liver, but its impact on APAP‐induced liver injury is not well established. We aimed to examine whether fructose supplementation could protect against APAP‐induced hepatotoxicity and to determine potential fructose‐sensitive intracellular mediators. We found that both high‐fructose diet feeding before APAP injection and fructose gavage after APAP injection reduced APAP‐induced liver injury with a concomitant induction of the hepatic carbohydrate‐response element‐binding protein α (ChREBPα)–fibroblast growth factor 21 (FGF21) pathway. In contrast, Chrebpα liver‐specific‐knockout (Chrebpα‐LKO) mice failed to respond to fructose following APAP overdose, suggesting that ChREBPα is the essential intracellular mediator of fructose‐induced hepatoprotective action. Primary mouse hepatocytes with deletion of Fgf21 also failed to show fructose protection against APAP hepatotoxicity. Furthermore, overexpression of FGF21 in the liver was sufficient to reverse liver toxicity in APAP‐injected Chrebpα‐LKO mice. Conclusion: Fructose protects against APAP‐induced hepatotoxicity likely through its ability to activate the hepatocyte ChREBPα–FGF21 axis.
Abbreviations
- AAV‐TBG‐Cre
adeno‐associated viral‐thyroxine binding globulin promoter‐Cre
- Ad
adenovirus
- AKT
protein kinase B
- ALT
alanine transaminase
- AMPK
adenosine monophosphate–activated protein kinase
- ANOVA
analysis of variance
- APAP
N‐acetyl‐para‐aminophenol; acetaminophen
- ChREBP
carbohydrate‐response element‐binding protein
- CYP
cytochrome P450
- FGF21
fibroblast growth factor 21
- GFP
green fluorescent protein
- GSH
glutathione
- Gst‐π
glutathione S transferase pi
- H&E
hematoxylin and eosin
- HFrD
high‐fructose diet
- Ho1
heme oxygenase 1
- JNK
c‐Jun N‐terminal kinase
- LDH
lactate dehydrogenase
- LKO
liver‐specific knockout
- L‐pk
L‐type pyruvate kinase
- mRNA
messenger RNA
- mTORC1
mammalian target of rapamycin complex 1
- NAPQI
N‐acetyl‐p benzoquinone imine
- NRF2
nuclear factor erythroid 2
- PGC1α
peroxisome proliferator‐activated receptor gamma coactivator 1 alpha
- ROS
reactive oxygen species
- RT‐qPCR
quantitative reverse‐transcription polymerase chain reaction
- TUNEL
terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick‐end labeling
- WT
wild type
Acetaminophen (N‐acetyl‐para‐aminophenol [APAP]) is one of the key ingredients in the most commonly used over‐the‐counter painkillers and cold medicines in the United States. However, overdose of APAP (>4 g/day for adults) has become the most common cause of drug‐related liver injury or death, resulting in 26,000 hospitalizations in the United States each year.( 1 ) APAP at the therapeutic dose is metabolized mainly through conjugation with glucuronic acid and sulfate before excretion. A minor portion of APAP is oxidized by cytochrome P450 (CYP)2E1 and CYP1A2 to generate N‐acetyl‐p benzoquinone imine (NAPQI), the reactive intermediate of APAP. NAPQI saturates hepatocyte conjugation pathways, resulting in accumulation of NAPQI‐protein adducts, sustained c‐Jun N‐terminal kinase (JNK) activation, and eventually massive reactive oxygen species (ROS) accumulation and hepatocyte death.( 1 ) N‐acetylcysteine (NAC) is by far the mainstay of therapy for APAP toxicity by enhancing NAPQI sulfate conjugation and NAPQI clearance. However, the efficacy of NAC is highly time dependent and significantly dwindles at the late stage of APAP overdose when most patients seek medical attention.( 2 ) Therefore, there is an urgent need to identify novel factors as targets for novel preventive or therapeutic measures for APAP‐induced hepatotoxicity.
Factors affecting severity of APAP hepatotoxicity include age, nutritional status, preexisting liver disease, use of alcohol and other liver‐metabolized medications, as well as genetic factors.( 3 ) Among these factors, nutritional status is a potential target that can be manipulated for the relief of APAP liver toxicity. The influence of nutritional states, such as alcohol consumption and malnutrition, has been extensively studied.( 4 , 5 , 6 ) However, as an important component of daily caloric intake, the impact of fructose consumption on drug‐induced liver injury remains largely unknown. For example, fructose has been found to impact APAP‐induced hepatotoxicity in rodents.( 7 , 8 ) Rats fed with 25% (weight/volume) fructose for 5 weeks showed resistance to APAP hepatotoxicity,.( 7 ) and 8‐week fructose feeding reduced liver toxicity in mice after APAP in spite of elevated liver lipid content.( 8 ) Although this latter study showed that chronic fructose feeding increased basal glutathione (GSH) content and modified intestinal microbiota composition,( 8 ) the exact mechanisms by which fructose feeding reduces APAP liver toxicity remains undefined. More importantly, whether fructose can be used as a therapeutic approach to reverse APAP liver injury has never been explored.
Hepatocytes are the major cell type that metabolizes fructose. In hepatocytes, fructose undergoes fructolysis to generate intermediate metabolic products that are readily incorporated into de novo lipogenesis.( 9 ) Fructose also activates the transcription of key enzymes for de novo lipogenesis, largely through carbohydrate‐response element‐binding protein (ChREBP).( 9 ) We and others have reported that a fructose‐rich diet potently activates de novo lipogenesis and induces liver steatosis in wild‐type (WT) mice, whereas in Chrebp‐knockout mice, fructose feeding results in nonalcoholic steatohepatitis‐like liver injury without triggering liver steatosis.( 10 , 11 ) These findings point to an unrecognized hepatoprotective role of ChREBP against diet‐induced liver injury. As of now, the role of the hepatic ChREBPα pathway in APAP hepatotoxicity remains unknown.
Fibroblast growth factor 21 (FGF21) is one of the main hepatokines produced within hepatocytes in response to fasting, stress, and dietary stimulation. FGF21 is reportedly induced during APAP‐induced liver injury, and supplementation of recombinant FGF21 reduced liver injury in APAP‐treated mice.( 12 ) Ye et al. showed that FGF21 functions as an autocrine or paracrine signal to induce the nuclear factor erythroid 2 (NRF2)– peroxisome proliferator‐activated receptor gamma coactivator 1 alpha (PGC1α)‐antioxidant pathway and protect liver against APAP toxicity. Interestingly, hepatocyte Fgf21 messenger RNA (mRNA) is potently induced by fructose in a ChREBP‐dependent manner. Fisher et al.( 13 ) reported that ChREBPα, the dominant isoform of ChREBP in hepatocytes, directly binds to the promoter of the Fgf21 gene following fructose‐diet feeding. Based on these data, it is tempting to speculate that fructose‐mediated protection against APAP toxicity may depend on the hepatic ChREBPα–FGF21 axis.
In the current study, we demonstrate that fructose not only prevents but also has a therapeutic effect against APAP‐induced liver injury in a mouse model. Mechanistic characterization revealed that the ChREBPα–FGF21 axis mediates a major hepatoprotective effect of fructose in APAP‐treated mice. Mice lacking hepatocyte ChREBPα developed more severe liver injury following APAP injection in spite of prefeeding with fructose, but such a condition could be reversed by restoring liver FGF21 expression. Thus, our findings highlight the ChREBPα–FGF21 axis as a potential critical intracellular mediator that links nutritional status and drug‐induced liver injury.
Materials and Methods
Animal Experiments
All animal experiments were approved by the Institutional Animal Care and Research Advisory Committee at the University of Michigan. All animal care and use were in accordance with guidelines of the University of Michigan Institutional Animal Care and Use Committee. C57BL/6 mice were maintained on 12‐hour/12‐hour light/dark cycles with access to food and water ad libitum. As a preventative model, mice were fed a high‐fructose diet (HFrD) (70 kcal%, D08040107; Research Diets) for 2 weeks with regular chow (Purina LabDiet #5008) as control and injected with APAP. In a second model, mice were injected with APAP; 45 minutes later, these mice were gavaged with fructose or a 1‐M glucose solution at a dose of 4 g/kg body weight. APAP (A7085; Sigma) was dissolved in warm water at 15.1 mg/mL and injected by intraperitoneal injection at 500 mg/kg body weight. Adult‐onset Chrebpα liver‐specific‐knockout (Chrebpα‐LKO) mice were generated by injecting ChrebpαFlox/Flox mice with adeno‐associated viral‐thyroxine binding globulin promoter‐Cre (AAV‐TBG‐Cre) by tail vein. For liver‐specific Fgf21 overexpression, 2 weeks after AAV‐TBG‐Cre injection into Chrebpαflox/flox mice, adenovirus (Ad)‐Fgf21 or Ad‐green fluorescent protein (GFP) was delivered by tail vein injection at a dose of 1 × 1012 plaque‐forming units.
Liver Injury and Cytotoxicity Assessment
Terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick‐end labeling (TUNEL) staining was performed with the In Situ Cell Death Detection Kit (cat. #11684795910; Roche). The lactate dehydrogenase (LDH) assay was performed with the LDH Cytotoxicity Detection Kit (cat. #MK401; Takara). ROS in liver tissue were determined with 2',7'‐dichlorodihydrofluorescein diacetate (H2DCFDA) (D6883; Sigma). The alanine aminotransferase (ALT) assay kit was from POINTE. The Mouse/Rat FGF‐21 Immunoassay Kit (MF2100) was from R&D Systems. Liver GSH levels were determined with the GSH/GSH Disulfide Ratio Detection Assay Kit (cat. #ab138881; Abcam) after trichloroacetic acid deproteinization with the liver lysates.
Microarray
Total RNA was extracted from WT or Chrebp−/− primary mouse hepatocytes treated with/without 25 mM fructose for 16 hours. A microarray assay was performed in the DNA Sequencing Core of the University of Michigan with the Mouse Gene 2.1 ST Strip from Affymetrix. Gene expression levels were compared between WT‐fructose and WT‐control and between Chrebp−/− fructose and WT‐fructose; a heat map was generated with Excel. Microarray data were submitted to the Gene Expression Omnibus of the National Center for Biotechnology Information (accession no. GSE164321)
Statistics
Statistical analysis was performed using Prism version 6.0 (GraphPad Software, San Diego, CA). Statistical significance was determined either by the unpaired two‐tailed Student t test for comparison between two groups or by one‐way analysis of variance (ANOVA) with Tukey’s test for multiple group comparison. All results are presented as mean ± SEM. Differences were considered statistically significant with P < 0.05.
Other detailed methods, including adenoviral production, liver histology, complementary DNA synthesis, and quantitative reverse‐transcription polymerase chain reaction (RT‐qPCR), are presented in the Supporting Experimental Procedures.
Results
Short‐Term HFrD Feeding Before APAP Overdose Ameliorates APAP Hepatotoxicity
Chronic overconsumption of a fructose‐rich diet has been linked to obesity, insulin resistance, and diabetes in both animal and human studies. Unexpectedly, it has been reported that mice fed with high fructose by drinking water for up to 8 weeks are resistant to APAP hepatotoxicity( 7 , 8 ) despite developing obesity and the metabolic syndrome. Whether a short duration (3 weeks or shorter) of fructose feeding also impacts the susceptibility to APAP overdose has not been previously tested. To answer this question, we fed WT male mice (age 8‐10 weeks) with regular chow versus an HfrD (70 kcal% fructose) for 2 weeks before giving a single dose of intraperitoneal injection of APAP (450 mg/kg body weight). We then assessed liver injury at 1 hour, 2 hours, 6 hours, and 24 hours after APAP injection by serum ALT and LDH and by hematoxylin and eosin (H&E) staining for liver histology. In the chow‐fed group, APAP overdose injection induced severe liver injury, which was featured by high levels of serum ALT and LDH peaking at 6 hours after APAP injection as well as necrotic regions (highlighted within the black‐dotted line) in the liver (Fig. 1A‐C). In contrast, no apparent necrotic areas were found in the liver from mice on the HFrD. Moreover, serum ALT and LDH levels were 90% lower than those in mice on the HFrD at the 6‐hour and 24‐hour time points (Fig. 1B,C). It has been established that mitochondrial leakage and ROS accumulation drive APAP‐induced hepatocyte injury. Indeed, ROS were highly induced in the liver of mice on regular chow 6 hours after APAP injection. However, ROS level was more than 70% lower in HFrD‐fed mice (Fig. 1D). It has also been reported that APAP overdose depletes hepatic GSH and that the recovery rate of reduced GSH significantly influences liver injury.( 14 ) In the regular chow group, we found that liver GSH was nearly depleted within 1 hour after APAP injection but gradually returned to the basal level 24 hours later. In comparison, liver GSH levels in the HFrD‐fed group, although lower than the basal level in the saline‐injected and regular chow‐fed group, were significantly higher 1 hour and 2 hours after APAP injection (Fig. 1E).
FIG. 1.
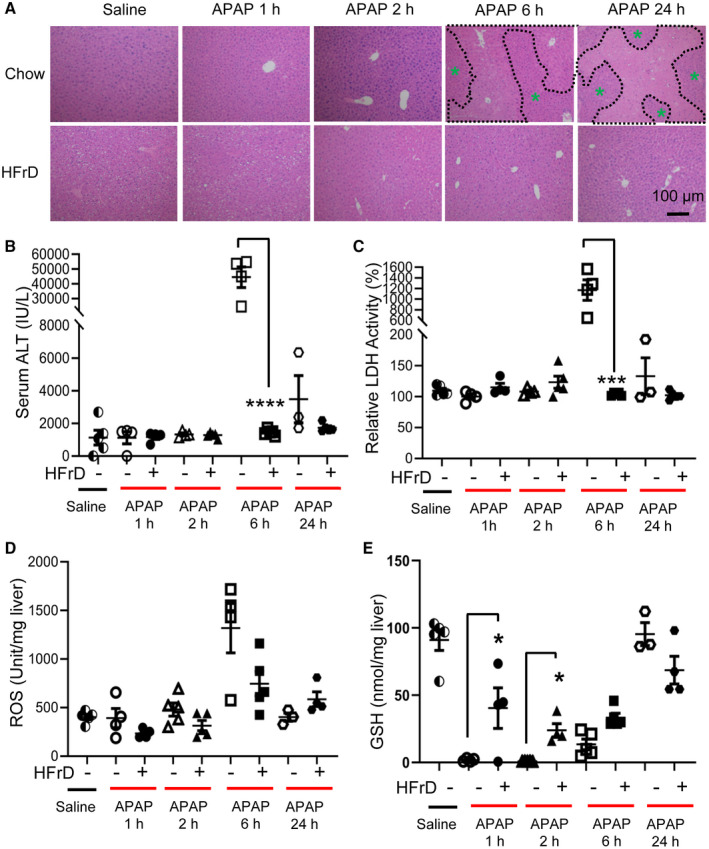
Short‐term HFrD feeding prevents APAP‐induced hepatotoxicity. Male mice (age 8 weeks) were fed a 70% HFrD for 2 weeks, injected with APAP (450 mg/kg), and killed 1 hour, 2 hours, 6 hours, or 24 hours later for tissue collecting (n = 4‐5 for each group). (A‐C) Liver H&E staining (magnification ×100; necrotic areas are outlined in black, green asterisks indicate healthy areas) and serum ALT and LDH assays were used to assess liver injury. (D) ROS levels in the liver were examined by the 2', 7'‐dichlorofluorescein assay. (E) Liver GSH level was determined with a commercial kit. *P < 0.05, ***P < 0.001, ****P < 0.0001 by one‐way ANOVA with Tukey’s test. Data are presented as mean ± SEM; scale bar, 100 µm. Abbreviation: h, hours.
Several factors, particularly APAP metabolism and the regeneration capacity of the remaining hepatocytes, determine the degree of liver injury following APAP overdose.( 3 ) Whether fructose feeding can impact these pathways simultaneously remains unknown. CYP2E1‐mediated APAP metabolism and the formation of APAP‐protein adducts are critical factors that promote hepatocyte oxidative stress and mitochondrial damage.( 15 , 16 , 17 ) We therefore measured APAP‐protein adducts in the livers 1 hour and 2 hours after APAP injection as well as the expression levels of CYP2E1. In the livers of chow‐fed mice, levels of APAP‐protein adduct were elevated at 1 hour and further increased at 2 hours after APAP injection. In contrast, the accumulation of APAP‐protein adducts was dramatically blunted at both time points in the liver of HFrD‐fed mice (Supporting Fig. S1A). Next, we examined both mRNA and protein expression of CYP2E1. When compared with the chow‐fed group, HFrD‐fed mice showed lowered levels of Cyp2e1 mRNA and protein 1 hour after APAP injection (Supporting Fig. S1B,C).
Liver regeneration is a critical step for recovery from acute liver injury. To examine whether HFrD feeding also impacts liver regeneration after APAP injection, we specifically examined liver regeneration 24 hours after APAP injection when liver regeneration normally occurs. Signal transducer and activator of transcription (STAT)3 and its downstream targets are required for this process.( 18 ) Our results showed that the classical makers for liver regeneration, including STAT3 phosphorylation and proliferating cell nuclear antigen, were modestly up‐regulated whereas p27 was suppressed by HFrD feeding (Supporting Fig. S1). We previously showed that short‐term HFrD feeding alters the protein kinase B (AKT) signaling pathway in liver, which could impact liver regeneration.( 10 ) In contrast, the prolonged activation of stress kinase JNK plays a central role in hepatocyte death during APAP overdose.( 19 , 20 , 21 ) We therefore examined the activities of the JNK pathway and survival kinases, such as AKT and adenosine monophosphate–activated protein kinase (AMPK), in APAP‐treated liver tissues from both chow and HFrD‐fed mice. In the livers of chow‐fed mice, the phosphorylation level of JNK was elevated 2 hours and 6 hours after APAP injection whereas it remained unchanged in the HFrD‐fed group (Supporting Fig. S1D). Consistent with the liver‐protective effect of fructose intake, the activities of AKT and AMPK activity in the liver were increased by HFrD feeding (Supporting Fig. S1D).
Transcription factor NRF2 plays an essential role in the mammalian response to oxidative stress. An overdose of APAP activates NRF2,( 22 ) whereas Nrf2‐knockout mice are more susceptible to APAP‐induced hepatotoxicity.( 23 ) NRF2 activation is responsible for the protective function of several compounds against APAP‐induced liver injury.( 24 , 25 , 26 ) To our surprise, HFrD feeding before APAP injection selectively modulated the mRNA levels of glutamate cysteine ligase catalytic (Gclc), nicotinamide adenine dinucleotide phosphate (reduced form) quinone dehydrogenase 1 (Nqo1), and heme oxygenase 1 (Ho1) (Supporting Fig. S2), indicative of an altered oxidative stress in the livers of HFrD‐fed mice. Taken together, our data demonstrated that short‐term HFrD feeding protects against APAP hepatotoxicity potentially through suppression of CYP2E1 and formation of APAP‐protein adducts, stimulation of liver regeneration, and activation of survival kinases, such as AKT.
Acute Fructose Intake Shortly After APAP Overdose Reduces Liver Injury
The preventative effect of fructose against hepatotoxicity of APAP promoted us to speculate that fructose gavage after APAP overdose may reverse liver injury. If this is true, fructose supplementation may be used as a therapy strategy for patients who have overdosed on APAP. To access the therapeutic potential of acute fructose intake shortly after APAP injection, we gave mice a single dose of APAP injection (450 mg/kg body weight) and gavaged them with fructose solution (4 g/kg body weight) or the same volume of saline after 45 minutes, 2 hours, or 6 hours. Liver histology showed almost no sign of liver necrosis in mice gavaged with fructose at 45 minutes after APAP injection (Fig. 2A). Compared with mice gavaged with saline 45 minutes after APAP injection, mice gavaged with fructose showed less liver necrosis at both 2 hours and 6 hours after APAP (Fig. 2A). Consistent with liver histology, we found that serum ALT and LDH levels were down more than 90% in mice gavaged with fructose 45 minutes after APAP injection and were about 70% reduced in mice gavaged with fructose at 2 hours and 6 hours after APAP injection (Fig. 2B,C). We also observed similar changes in liver ROS, which showed the lowest levels at 45 minutes after APAP injection (Fig. 2D). Liver GSH levels were almost completely depleted in mice gavaged with saline but were significantly elevated in mice gavaged with fructose at all time points after APAP injection (Fig. 2E). Taken together, our data demonstrated that fructose supplementation shortly after APAP overdose has therapeutic effects on reducing liver toxicity, possibly by enhancing GSH levels. Furthermore, our data showed that the timing of fructose treatment determines its efficacy against APAP live injury.
FIG. 2.
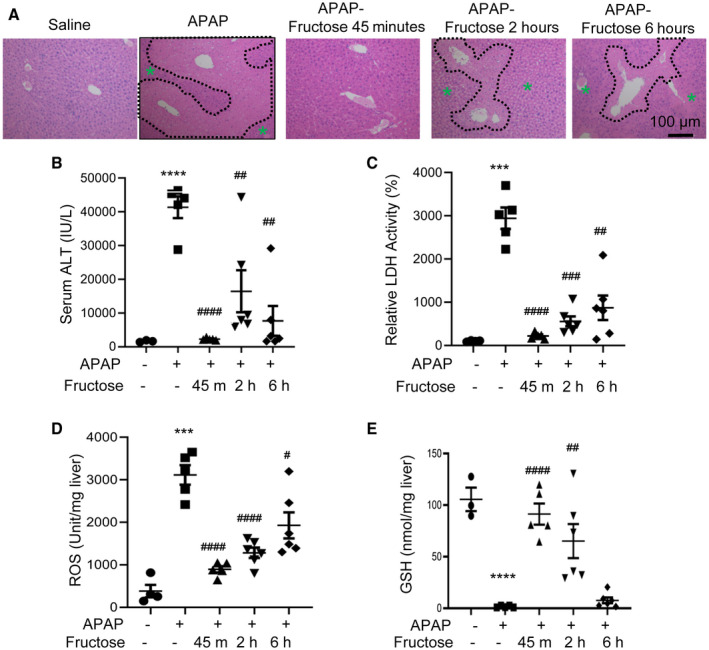
Fructose gavage rescues APAP‐induced hepatotoxicity. Male mice (age 8 weeks) were injected with APAP; gavaged with saline or fructose 45 minutes, 2 hours, or 6 hours later; and killed 24 hours later for tissue collecting (n = 4‐5 for each group). (A‐C) Liver H&E staining (magnification ×100; necrotic areas are outlined in black, green asterisks indicate healthy areas) and serum ALT and LDH assays were used to assess liver injury. (D) ROS levels in the liver were examined by the 2', 7'‐dichlorofluorescein assay. (E) Liver GSH level was determined with a commercial kit. * indicates APAP+saline versus saline+saline; # indicates APAP+fructose versus APAP+saline of the same time point. # P < 0.05, ## P < 0.01, *** or ### P < 0.001, **** or #### P < 0.0001 by one‐way ANOVA with Tukey’s test. Data are presented as mean ± SEM; scale bar, 100 µm. Abbreviations: h, hours; m, minutes.
We also measured CYP2E1 levels and the accumulation of APAP‐protein adducts in the liver from APAP‐injected mice after gavage with saline versus fructose. We found that fructose gavage 45 minutes after APAP injection suppressed CYP2E1 protein level in the liver while slightly increasing its mRNA (Supporting Fig. S3A,B). However, to our surprise, fructose gavage did not appear to change hepatic APAP‐protein adducts (Supporting Fig. S3C). In terms of signaling pathways, we found that fructose intake greatly reduced the phosphorylation levels of JNK and AMPK but not AKT (Supporting Fig. S3D).
Given the potent effects of fructose on liver GSH (Figs. 1 and 2), we analyzed the mRNA expression of NRF2 targets in the livers of saline‐ and fructose‐gavaged mice. Our results showed that acute fructose intake differentially affected the mRNA expression of NRF2 pathways. Notably, the mRNA levels of catalase, glutathione S transferase pi (Gst‐π), and Nqo1 were higher in the group gavaged with fructose compared to the saline control (Supporting Fig. S4).
Both fructose and glucose are common monosaccharides in diets, prompting us to examine whether glucose supplementation after APAP overdose had a similar hepatoprotective effect. Unlike fructose, the same dose of glucose gavage did not block APAP‐induced serum ALT increase (Supporting Fig. S5), indicating that the liver protective effect is unique to fructose intake. Taken together, our data showed that fructose supplementation shortly after APAP overdose has therapeutic effects on reducing liver toxicity, likely through enhancing GSH levels.
Fructose Reverses APAP‐Induced Suppression on the ChREBPα Pathway in Liver
Our data demonstrated fructose intake either before or shortly after APAP overdose could greatly reduce liver injury in mice; however, the intracellular targets of fructose that confer hepatoprotection are unclear. This is an important issue because long‐term excessive fructose intake could impair metabolism and lead to metabolic syndrome.( 27 ) We previously reported that the ChREBP pathway is required for metabolic adaption to HFrD feeding, raising the possibility that the ChREBP pathway might play a role in fructose‐mediated protection against APAP liver toxicity. To date, the impact of APAP on the ChREBP pathway has not been reported. We found that the mRNA levels of ChREBPα and its transcriptional targets, including Chrebpβ and L‐type pyruvate kinase (L‐pk), were reduced significantly in chow‐fed APAP‐injected mice (Fig. 3A). In contrast, our short‐term HFrD feeding blocked the suppression of both Chrebpβ and L‐pk after APAP injection (Fig. 3A) while increasing nuclear ChREBPα abundance in the liver (Fig. 3B). Next, we examined whether fructose gavage could have a similar effect on APAP‐induced inhibition of the ChREBP pathway. Indeed, fructose gavage restored the expression of ChREBPα targets (Chrebpβ and L‐pk) (Fig. 3C) and ChREBPα protein (Fig. 3D). These findings for the first time demonstrate fructose intake counters the potent suppression of hepatic ChREBP pathways induced by APAP overdose.
FIG. 3.
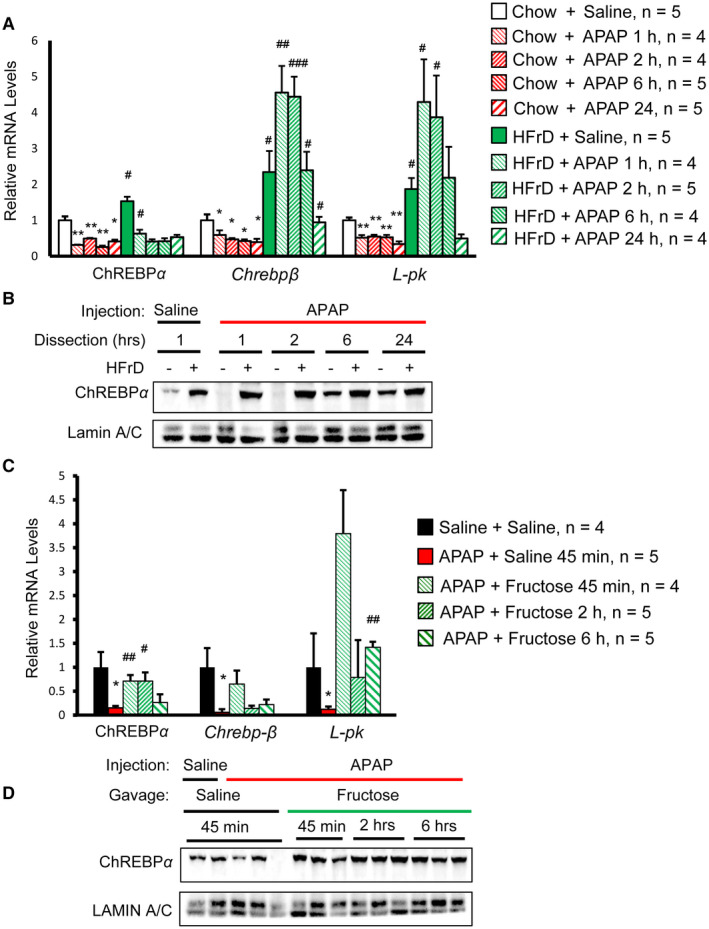
Fructose reverses hepatic ChREBP suppression by APAP. (A,B) High‐fructose feeding prevents suppression of the hepatic ChREBP pathway by APAP. (A) ChREBP transcription activity was determined for ChREBP target genes by RT‐qPCR. * indicates chow+APAP versus chow+saline; # indicates HFrD+APAP versus chow+APAP for the same time point. (B) Liver nuclear lysates of mice with the same treatment were pooled together to measure the abundance of ChREBPα by western blot. (C,D) Fructose gavage reverses suppression of the hepatic ChREBP pathway by APAP. (C) ChREBP transcription activity was determined by RT‐qPCR for ChREBP target genes. * indicates APAP+saline versus saline+saline; # indicates APAP+fructose versus APAP+saline. (D) Liver nuclear lysates of representative mice were used to measure the abundance of nuclear ChREBPα by western blot. * or # P < 0.05, ** or ## P < 0.01, ### P < 0.001 by one‐way ANOVA with Tukey’s test. Data are presented as mean ± SEM. Abbreviations: h, hrs, hours; min, minutes.
Chrebp‐Deficient Hepatocytes Lose Fructose Protection Against APAP Toxicity
Our study has demonstrated that fructose could protect against APAP‐induced hepatotoxicity while activating the hepatic ChREBP pathway, suggesting that ChREBP may be a critical mediator between fructose intake and reduced APAP hepatotoxicity. To test this possibility, we isolated primary hepatocytes from WT and Chrebp−/− mice treated with 10 mM APAP in the presence or absence of 25 mM fructose and evaluated hepatocyte injury by measuring LDH activity in the culture medium. In WT primary mouse hepatocytes, fructose treatment effectively reduced LDH activity in the medium. In contrast, Chrebp deficiency not only sensitized hepatocytes to APAP‐induced toxicity but also reduced the efficacy of fructose‐induced protection (Fig. 4A). Next, we used TUNEL staining to measure hepatocyte death following APAP treatment. Similar to the LDH assay data, TUNEL staining showed reduced hepatocyte death (from 16% to 3%) in fructose‐treated WT hepatocytes. However, fructose treatment offered minimal protection in Chrebp−/− hepatocytes (Fig. 4B). Together, these results suggest that fructose protects hepatocytes from APAP toxicity through ChREBPα in a cell‐autonomous manner.
FIG. 4.
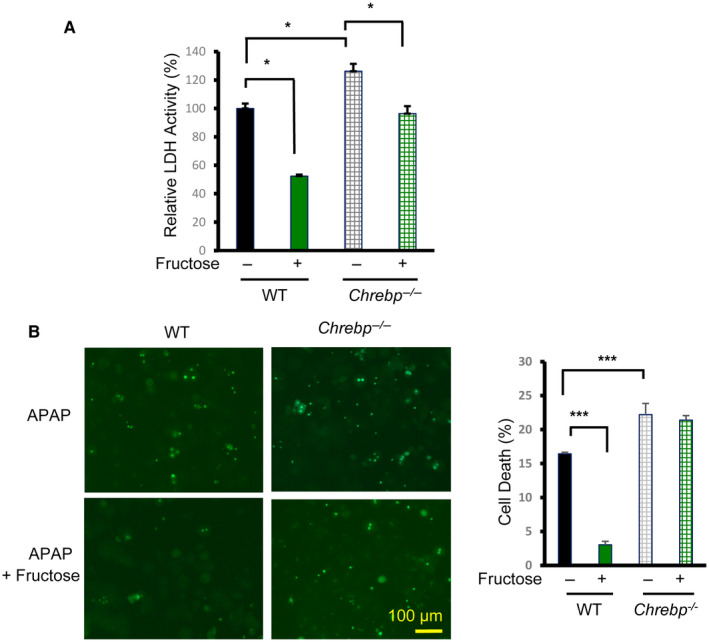
ChREBP is required for fructose protection against APAP cytotoxicity in hepatocytes. Primary mouse hepatocytes were isolated from the liver of WT and Chrebp−/− mice and cultured in medium containing 10 mM APAP in the presence or absence of 25 mM fructose for 16 hours. Cytotoxicity was determined by (A) LDH assay and (B) TUNEL staining (bright green dots). *P < 0.05, ***P < 0.001 by one‐way ANOVA with Tukey’s test. Data are presented as mean ± SEM.
Hepatocyte ChREBPα is Required For Fructose Protection Against APAP Liver Toxicity
We next tested whether hepatic ChREBPα is required for the protective effect of fructose against APAP liver toxicity in vivo. We generated Chrebpα‐LKO by injecting Chrebpαflox/flox with AAV‐TBG‐Cre by tail vein. The control group was injected with the same dose of AAV‐TBG‐GFP. Deletion of Chrebpα in the liver was confirmed by immunoblotting (Fig. 5A). As expected, hepatic Chrebpα deletion resulted in significantly reduced expression of ChREBPα target gene L‐pk but had no impact on the Chrebpβ isoform (Supporting Fig. S7A). We then fed hepatic Chrebpα‐deleted mice the HFrD for 3 weeks and injected them with saline or an overdose of APAP (450 mg/kg body weight) and collected serum and liver samples 6 hours later for liver injury assessment. H&E staining showed that there was no difference between the livers of control and Chrebpα‐LKO mice following saline injection. However, HFrD feeding protected most hepatocytes from necrosis by APAP in control mice but failed in Chrebpα‐LKO mice (Fig. 5B). We also observed increased serum ALT and LDH as well as increased liver ROS and reduced liver GSH levels in mice with hepatic Chrebpα deletion (Fig. 5C‐F). Unexpectedly, the levels of APAP‐protein adduct at 2 hours after APAP injection were comparable between control and Chrebpα‐LKO mice (Supporting Fig. S7B). Our results support that ChREBPα is required for fructose‐induced protection against APAP‐induced liver injury, but such protective effects are largely independent of the accumulation of APAP‐protein adduct.
FIG. 5.
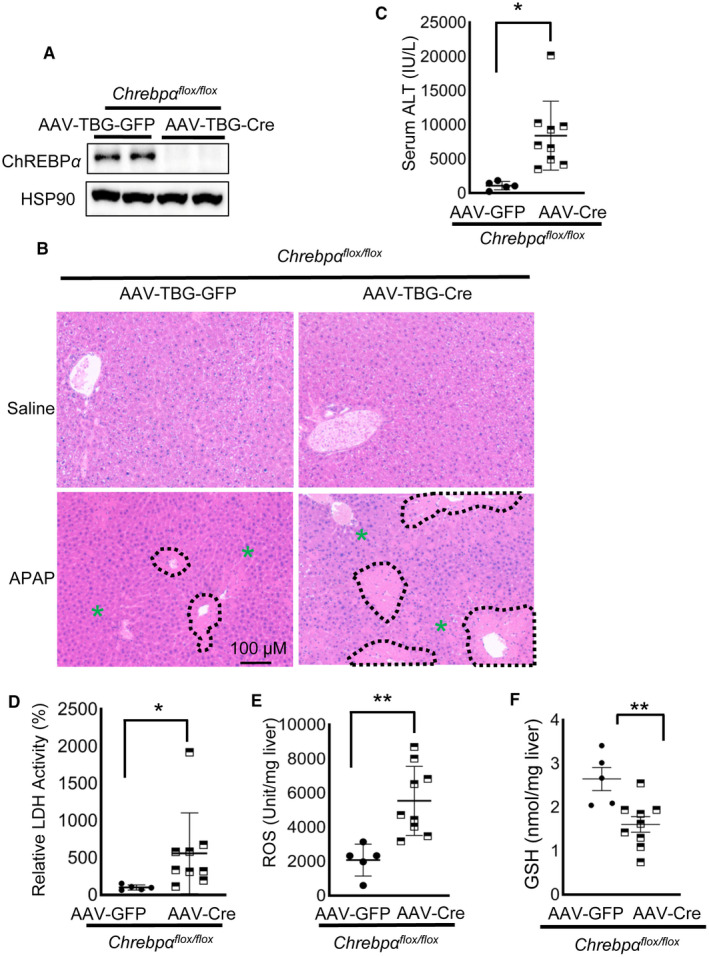
ChREBPα is required for fructose protection against APAP liver toxicity. One week after injection with AAV‐TBG‐Cre or AAV‐TBG‐GFP control by tail vein, Chrebpαflox/flox mice were fed an HFrD for 2 weeks. At the end of the HFrD feeding, mice were injected with APAP and killed 6 hours later for tissue collecting (n = 5 for chow, n = 9 for HFrD). (A) Hepatic ChREBPα deletion was confirmed by western blot against ChREBP. (B‐F) Liver Chrebpα deficiency abolishes the hepatoprotective effect of fructose, resulting in increased liver necrosis in (B) H&E staining (necrotic areas are outlined in black, green asterisks indicate healthy areas), (C) serum ALT, (D) LDH, and (E) liver tissue ROS as well as (F) decreased GSH. *P < 0.05, **P < 0.01 by two‐tailed Student t test (C,E,F) and Mann Whitney test (D). Data are presented as mean ± SEM; scale bar, 100 µm. Abbreviation: HSP90, heat shock protein 90.
Restoring FGF21 Expression Reverses Hepatotoxicity in APAP‐Treated Chrebp−/− Hepatocytes
We and others have observed that ChREBPα is indeed required for fructose‐stimulated de novo lipogenesis in the liver.( 10 , 28 ) To identify hepatic genes specifically regulated by fructose‐activated ChREBPα, we performed an unbiased gene expression array analysis in WT and Chrebp−/− hepatocytes treated with or without fructose. The genes up‐regulated or down‐regulated by fructose in WT but not Chrebp−/− hepatocytes are listed in (Supporting Fig. S8A). Among those top fructose‐regulated genes, Fgf21 has been shown to be highly relevant to APAP liver toxicity. Several groups showed that Fgf21‐knockout mice are more sensitive to APAP treatment and that FGF21 supplementation reduces liver injury.( 12 , 29 , 30 ) Interestingly, fructose was known to induce hepatic Fgf21 in a ChREBP‐dependent manner,( 13 , 31 ) raising the possibility that fructose may activate the ChREBPα–FGF21 pathway to protect against APAP‐induced toxicity.
We next validated the microarray data by RT‐qPCR in primary mouse hepatocytes. Indeed, fructose potently induced Fgf21 mRNA in WT mouse hepatocytes (about 15‐fold) but not in Chrebp−/− hepatocytes (Supporting Fig. S8B). We also confirmed the expression of other genes identified by microarray (Supporting Fig. S8C‐E). Next, we measured serum levels of FGF21 in previously described mouse models. Both HFrD feeding and fructose gavage slightly increased the mRNA level of Fgf21 (Supporting Fig. S9) and significantly elevated serum FGF21 in the liver (Fig. 6A,B). In contrast, the hepatic mRNA level of Fgf21 was reduced in the livers of HFrD‐fed and APAP‐injected Chrebpα‐LKO mice, although it did not reach statistical significance (Supporting Fig. S9C). Serum FGF21 was significantly lower (about 50%) in liver‐specific Chrebpα‐deleted mice treated with the HFrD and APAP (Fig. 6C). These data support that the fructose‐activated ChREBPα–FGF21 axis may be responsible for protection against APAP‐induced liver injury.
FIG. 6.
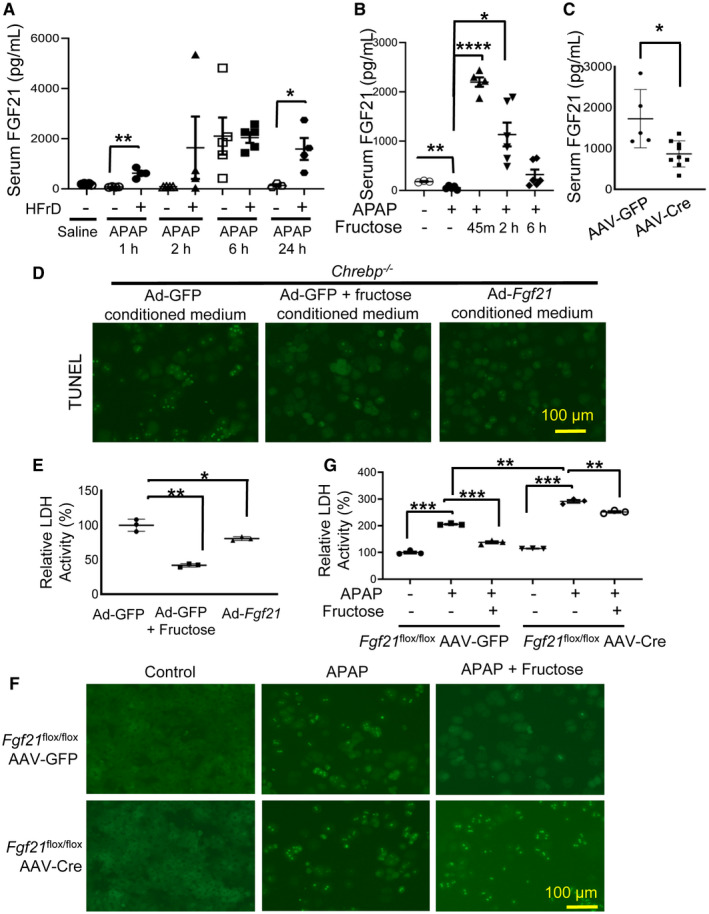
Fructose protects against APAP‐induced hepatotoxicity by activating the ChREBPα–FGF21 axis. (A) HFrD feeding and (B) fructose gavage elevated serum FGF21, whereas (C) acute Chrebpα deficiency abolished HFrD‐induced circulating FGF21. (D,E) FGF21 rescues APAP‐induced cell death in Chrebp−/− hepatocytes. Six hours after transduction with Ad‐GFP or Ad‐Fgf21, WT primary hepatocytes were switched to serum‐free medium supplemented with or without 25 mM fructose; 24 hours later, culture medium was collected to incubate primary Chrebp−/− hepatocytes treated with 10 mM APAP. Cytotoxicity was determined 24 hours after incubation by (D) TUNEL staining and (E) LDH assay. (F,G) FGF21 deficiency impairs fructose protection against APAP cytotoxicity. Primary hepatocytes were isolated from Fgf21 flox/flox mice injected with either AAV‐TBG‐GFP or AAV‐TBG‐Cre and treated with APAP and fructose. Cytotoxicity was determined by (F) TUNEL staining and (G) LDH assay. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by one‐way ANOVA with Tukey’s test (A,B,E,G) and two‐tailed Student t test (C). Data are presented as mean ± SEM; scale bar, 100 µm. Abbreviations: h, hours; m, minutes.
To test whether FGF21‐containing culture medium could reduce APAP‐induced cell death in Chrebp‐deficient hepatocytes, we first collected medium from WT hepatocytes transduced with Ad‐GFP control, Ad‐GFP plus fructose treatment, or Ad‐Fgf21. Next, we incubated Chrebp‐deficient hepatocytes in Ad‐GFP, Ad‐GFP plus fructose treatment, or Ad‐Fgf21 medium, respectively, overnight and added in APAP before analysis of cell toxicity. The percentage of APAP‐induced hepatocyte death was ~45% in Ad‐GFP‐conditioned medium, whereas it decreased to <20% in both Ad‐GFP plus fructose treatment and Ad‐Fgf21‐conditioned medium (Fig. 6D; Supporting Fig. S10). LDH analysis showed a similar trend (Fig. 6E), supporting that supplementation of hepatocytes with FGF21 protects Chrebp−/− hepatocytes from APAP toxicity.
Furthermore, we tested the impact of hepatic Fgf21 deficiency on fructose protection against APAP hepatotoxicity. We isolated primary mouse hepatocytes from Fgf21flox/flox mice injected with either AAV‐TBG‐Cre or AAV‐TBG‐GFP and then treated the cells with APAP alone or APAP plus fructose. We found that AAV‐TBG‐Cre transduction almost completely abolished the expression of Fgf21 in Fgf21flox/flox primary hepatocytes (Supporting Fig. S11A). As expected, fructose was able to reduce hepatocyte death in AAV‐TBG‐GFP‐transduced Fgf21flox/flox hepatocytes as well as LDH enzymatic activity in the medium collected from those cells after APAP treatment. However, the protective effects of fructose were lost in AAV‐TBG‐Cre‐transduced Fgf21flox/flox hepatocytes (Fig. 6F,G; Supporting Fig. S11B), confirming the crucial role of Fgf21 in fructose protection against APAP hepatotoxicity. Taken together, all the evidence demonstrates that the hepatic ChREBPα–FGF21 axis mediates the protective effect of fructose against APAP‐induced hepatotoxicity.
Ectopic Expression of Fgf21 Mitigates APAP Hepatotoxicity in Chrebpα‐LKO Mice
To further test whether restoring FGF21 expression in the liver is sufficient to ameliorate APAP‐induced liver injury in mice with liver‐specific deletion of Chrebpα, we injected Chrebpαflox/flox mice with AAV‐TBG‐Cre to generate Chrebpα‐LKO mice. Two weeks later, mice were divided into two groups before injection with either Ad‐GFP or Ad‐Fgf21, and 10 days later, mice were subjected to APAP injection before dissection. Serum FGF21 level in Ad‐Fgf21‐injected mice rose to about 1,500 pg/mL (Fig. 7A), a level comparable to what was observed in the HFrD‐fed WT mice (Fig. 6C). Compared with the Ad‐GFP control group, the Ad‐Fgf21 group showed reduced regions of liver necrosis, lowered levels of serum ALT and LDH, and decreased ROS but increased GSH levels in the liver (Fig. 7B‐F). This supports the essential role of FGF21 in mediating the protective effect against APAP. Furthermore, JNK phosphorylation was markedly reduced in the liver with Fgf21 overexpression, indicative of reduced liver stress responses (Supporting Fig. S12A).
FIG. 7.
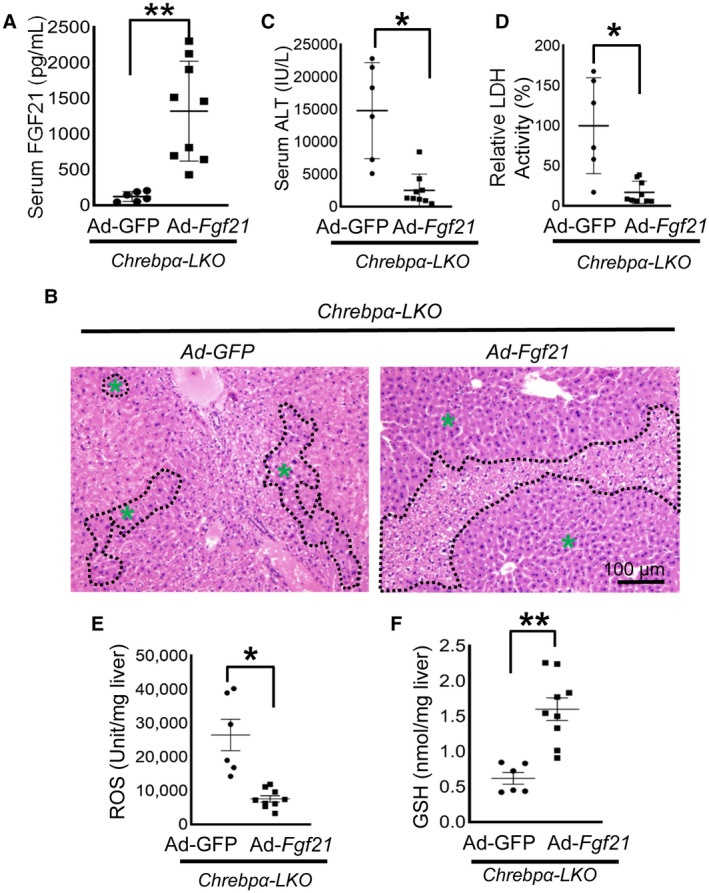
Restoring FGF21 alleviates APAP hepatotoxicity in hepatic Chrebpα‐deleted mice. Chrebpαflox/flox male mice were injected with AAV‐TBG‐Cre by tail vein to delete hepatic Chrebpα (Chrebpα‐LKO) and 2 weeks later were injected with Ad‐GFP or Ad‐Fgf21 to overexpress Fgf21 in the liver. Ten days later, the mice received an intraperitoneal injection with APAP and were killed 6 hours later for tissue collecting (n = 6 for Ad‐GFP, n = 9 for Ad‐Fgf21). (A) Adenoviral Fgf21 overexpression was confirmed by elevated serum FGF21. (B‐D) Liver H&E staining (magnification ×100; necrotic areas are outlined in black, green asterisks indicate healthy areas) and serum ALT and LDH assays were used to assess liver injury. (E) ROS levels in the liver were examined to assess oxidative stress. (F) Liver GSH level was determined with a commercial kit. *P < 0.05, **P < 0.01 by two‐tailed Student t test (A,E,F) and Mann Whitney test (C,D). Data are presented as mean ± SEM; scale bar, 100 µm.
Ye et al.( 12 ) reported that FGF21 protects against APAP liver injury by enhancing the NRF2‐PGC1a pathway and reducing oxidative stress. We therefore checked the expression of classical NRF2 targets in liver tissues and observed up‐regulation of catalase, Gst‐π, and Ho1 (Supporting Fig. S12B). Both the mRNA and protein levels of CYP2E1 were comparable between the two groups (Supporting Fig. S6A,B). Altogether, our data support that fructose protects against APAP overdose‐induced liver injury by activating the ChREBPα–FGF21 axis.
Discussion
In this study, we observed a potent protective effect of fructose intake against APAP overdose‐induced acute liver injury. Such a protective action of fructose can be achieved by either feeding mice a fructose‐rich diet before APAP injection or gavaging mice with fructose shortly after APAP injection. Our findings suggest that fructose might be considered for treating patients with acute APAP overdose. Moreover, we discovered that fructose‐induced hepatoprotection largely depends on the hepatic ChREBPα–FGF21 axis. Fructose fails to reduce APAP‐induced liver injury in liver‐specific Chrebpα‐knockout mice, which demonstrate lower levels of serum FGF21. In contrast, restoring hepatic Fgf21 expression reduces APAP toxicity in liver‐specific Chrebpα‐knockout mice. In summary, we uncovered a fructose‐based therapeutic pathway for APAP liver injury.
We and others reported that fructose feeding potently induces hepatic de novo lipogenesis by activating the ChREBPα pathway. However, the overall impact of fructose on hepatic transcriptome remains elusive. In this study, we compared the gene expression profile between Chrebp−/− and WT primary hepatocytes with or without fructose treatment. We identified a panel of novel genes that are controlled by fructose and ChREBP, including regulator of G protein signaling 16 (Rgs16) and thioredoxin interacting protein (Txnip). Rgs16 and Txnip were reported to be targets of ChREBP in the context of glucose stimulation,( 32 , 33 , 34 ) but their regulation by fructose had not been reported. Txnip was reported to be a ChREBP‐regulated gene involved in inflammation, oxidative stress, and apoptosis in pancreatic β cells.( 33 ) RGS16 is one of the guanosine triphosphatase‐activating proteins that control the intensity and duration of G protein‐coupled receptor signaling. ChREBP‐controlled RGS16 was reported to inhibit fatty acid oxidation in hepatocytes( 32 ) and promote the accumulation of lipid droplets in β cells.( 35 ) It would be of great interest to investigate whether the induction of either RGS16 or TXNIP contributes to fructose protection against APAP liver toxicity.
FGF21 is a hepatic hormone mainly produced by hepatocytes in response to nutritional and stress signals. Dushay et al.( 31 ) first reported that fructose potently induces the FGF21 expression in both rodents and humans whereas glucose only modestly increases FGF21 production. Our results confirm their findings and further demonstrate that the induction of FGF21 by fructose requires hepatic ChREBPα. What are the underlying mechanisms responsible for FGF21 hepatoprotection? Previous work has suggested that FGF21 may stimulate the PGC1α/NRF2 pathway to enhance the antioxidant pathway following APAP intoxication in hepatocytes.( 12 ) Our results also showed that FGF21 overexpression increases the expression of NRF2 targets, including catalase, Gst‐π, and Ho1 (Fig. 7H). However, fructose treatment increases hepatic FGF21 with very limited impact on the NRF2 pathway, suggesting that under the fructose condition, FGF21 may have other targets that could mediate hepatoprotective effects.
In adipose tissue, FGF21 binds to the receptor complex of fibroblast growth factor receptor 1c and β‐klotho and activates the extracellular signal‐regulated kinase (ERK)/mitogen‐activated protein kinase pathway.( 36 ) Minard et al.( 37 ) identified mammalian target of rapamycin complex 1 (mTORC1) as a major regulatory node in the FGF21 signaling network in adipocytes. They showed that blocking mTORC1 activity almost abrogated FGF21‐stimulated glucose uptake and improved insulin sensitivity. Whether hepatic FGF21 activates the ERK/mTORC1 axis in fructose‐induced protection against APAP toxicity in mouse models will be a focus of our future study.
During the time course of the HFrD study, we found that fructose feeding effectively abrogates the accumulation of APAP‐protein adducts while maintaining low levels of CYP2E1 in the liver after APAP injection. Cho et al.( 8 ) reported a similar finding that fructose intake by drinking water significantly reduces mRNA levels and enzymatic activities of CYP2E1 and CYP1A2, two critical enzymes responsible for converting APAP to the toxic metabolite NAPQI. The underlying mechanisms for reduced CYP2E1 protein expression in the liver of fructose‐fed mice remain unclear. Cho et al.( 8 ) attributed it to altered gut microbiota following fructose diet feeding. Because we found that fructose gavage shortly after APAP injection reduces CYP2E1 protein abundance in the liver, it is unlikely that gut microbiota is involved in this reduction due to the short duration of treatment. We speculate that it is more likely that CYP2E1 proteolysis might be involved in this process. It has been reported that the endoplasmic reticulum (ER)‐associated degradation (ERAD) system targets ER‐anchored P450 enzymes, including CYP2E1, for degradation through the ubiquitination‐proteasome or autophagy‐lysosome system.( 38 ) Beside the ERAD system, the ubiquitin E3 ligase gycoprotein 78 has been found to be a relevant E3 ligase for CYP2E1.( 39 , 40 ) Our future study will examine whether fructose can stimulate the CYP2E1 protein turnover by these degradation systems to block APAP‐protein adduct formation.
How nutrients regulate hepatic ChREBP activity has been well established. For instance, glucose stimulates ChREBP activity by phosphorylation.( 41 ) We recently observed that fructose enhances ChREBP protein stability by inhibiting its proteolysis.( 42 ) In contrast, very little is known about the effects of hepatocyte stress, such as APAP overdose, on the hepatic ChREBP pathway. APAP overdose causes cellular oxidative stress and mitochondrial impairment. In this context, APAP treatment within 6 hours potently reduces ChREBPα protein expression and expression of its target genes in the liver. Our findings also raise an important question about possible negative effects of hepatocyte stress on the ChREBP pathway. Whether this is a unique response to APAP remains to be addressed. We suspect that APAP‐induced down‐regulation of the ChREBP pathway could occur at both transcriptional and posttranslational levels. A number of transcription factors, including hepatocyte nuclear factor 1α,( 43 ) farnesoid X receptor,( 44 ) NRF2,( 23 ) and nuclear factor kappa B,( 45 ) that are known to be involved in cytotoxic response to APAP could regulate the transcription of ChREBP. Transcriptional activity and protein stability of ChREBP are also tightly regulated by posttranslational modifications, such as acetylation, phosphorylation, and ubiquitination.( 46 , 47 ) Does APAP suppress the ChREBP protein level and transcriptional activity through posttranslational modifications? Which pathway(s) mediate the suppression? These questions would be interesting to address.
In addition to liver, ChREBPα is also abundantly expressed in intestinal epithelial cells and participates in fructose absorption and metabolism.( 48 , 49 ) Cho et al.( 8 ) showed altered gut microbiota following 8 weeks of fructose diet feeding, which could contribute to fructose protective action against APAP toxicity. Whether enterocyte ChREBPα plays a role during this process warrants further investigation; however, it is not likely that fructose gavage after APAP intake reduces liver injury by modulating colon microbiota due to the relatively short time frame.
Supporting information
Fig S1‐S12
Supplementary Material
Acknowledgment
Chrebpαflox/flox mice were generously provided by Dr. Lawrence Chan, Dr. Pradip Saha, and Dr. Alli Artar at Baylor College of Medicine. Dr. Ling Qi (University of Michigan) provided Fgf21 flox/flox mice, and Dr. Jun Wu (University of Michigan) provided Ad‐Fgf21.
Supported by the National Institutes of Health (NIH) (R00 DK077449, R01 DK099593 to L.Y.; R01 DK121170 to X.T.; R01 DK107583 to J.W.; R01 DK114356 to P.S.), Michigan Nutrition Obesity Research Center (P30 DK089503 to L.Y.), Michigan Diabetes Research Training Center (P30 DK020570 to X.T.), Center for Gastrointestinal Research (P30 DK034933 to D.Z.), and Michigan Diabetes Research Center (NIH grant P30‐DK020572 to D.Z.).
Potential conflict of interest: Nothing to report.
Contributor Information
Xin Tong, Email: xintong@umich.edu.
Lei Yin, Email: leiyin@umich.edu.
References
Author names in bold designate shared co‐first authorship.
- 1. Ramachandran A, Jaeschke H. Acetaminophen hepatotoxicity. Semin Liver Dis 2019;39:221‐234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Smilkstein MJ, Knapp GL, Kulig KW, Rumack BH. Efficacy of oral N‐acetylcysteine in the treatment of acetaminophen overdose. Analysis of the national multicenter study (1976 to 1985). N Engl J Med 1988;319:1557‐1562. [DOI] [PubMed] [Google Scholar]
- 3. Yoon E, Babar A, Choudhary M, Kutner M, Pyrsopoulos N. Acetaminophen‐induced hepatotoxicity: a comprehensive update. J Clin Transl Hepatol 2016;4:131‐142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schmidt LE, Dalhoff K, Poulsen HE. Acute versus chronic alcohol consumption in acetaminophen‐induced hepatotoxicity. Hepatology 2002;35:876‐882. [DOI] [PubMed] [Google Scholar]
- 5. Waring WS, Stephen AF, Malkowska AM, Robinson OD. Acute ethanol coingestion confers a lower risk of hepatotoxicity after deliberate acetaminophen overdose. Acad Emerg Med 2008;15:54‐58. [DOI] [PubMed] [Google Scholar]
- 6. Whitcomb DC, Block GD. Association of acetaminophen hepatotoxicity with fasting and ethanol use. JAMA 1994;272:1845‐1850. [DOI] [PubMed] [Google Scholar]
- 7. Ishida K, Hanada T, Sakai T, Doi K. Effects of fructose‐induced hypertriglyceridemia on hepatorenal toxicity of acetaminophen in rats. Exp Toxicol Pathol 1995;47:509‐516. [DOI] [PubMed] [Google Scholar]
- 8. Cho S, Tripathi A, Chlipala G, Green S, Lee H, Chang EB, et al. Fructose diet alleviates acetaminophen‐induced hepatotoxicity in mice. PLoS One 2017;12:e0182977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Softic S, Cohen DE, Kahn CR. Role of dietary fructose and hepatic de novo lipogenesis in fatty liver disease. Dig Dis Sci 2016;61:1282‐1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang D, Tong X, VanDommelen K, Gupta N, Stamper K, Brady GF, et al. Lipogenic transcription factor ChREBP mediates fructose‐induced metabolic adaptations to prevent hepatotoxicity. J Clin Invest 2017;127:2855‐2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hannou SA, Haslam DE, McKeown NM, Herman MA. Fructose metabolism and metabolic disease. J Clin Invest 2018;128:545‐555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ye D, Wang Y, Li H, Jia W, Man K, Lo CM, et al. Fibroblast growth factor 21 protects against acetaminophen‐induced hepatotoxicity by potentiating peroxisome proliferator‐activated receptor coactivator protein‐1α‐mediated antioxidant capacity in mice. Hepatology 2014;60:977‐989. [DOI] [PubMed] [Google Scholar]
- 13. Fisher FM, Kim MiSung, Doridot L, Cunniff JC, Parker TS, Levine DM, et al. A critical role for ChREBP‐mediated FGF21 secretion in hepatic fructose metabolism. Mol Metab 2016;6:14‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ramachandran A, Jaeschke H. Mechanisms of acetaminophen hepatotoxicity and their translation to the human pathophysiology. J Clin Transl Res 2017;3(Suppl. 1):157‐169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Snawder JE, Roe AL, Benson RW, Roberts DW. Loss of CYP2E1 and CYP1A2 activity as a function of acetaminophen dose: relation to toxicity. Biochem Biophys Res Commun 1994;203:532‐539.Erratum in: Biochem Biophys Res Commun 1995;206:437. [DOI] [PubMed] [Google Scholar]
- 16. Zaher H, Buters JTM, Ward JM, Bruno MK, Lucas AM, Stern ST, et al. Protection against acetaminophen toxicity in CYP1A2 and CYP2E1 double‐null mice. Toxicol Appl Pharmacol 1998;152:193‐199. [DOI] [PubMed] [Google Scholar]
- 17. McGill MR, Jaeschke H. Metabolism and disposition of acetaminophen: recent advances in relation to hepatotoxicity and diagnosis. Pharm Res 2013;30:2174‐2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Taub R. Liver regeneration: from myth to mechanism. Nat Rev Mol Cell Biol 2004;5:836‐847. [DOI] [PubMed] [Google Scholar]
- 19. Hanawa N, Shinohara M, Saberi B, Gaarde WA, Han D, Kaplowitz N. Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen‐induced liver injury. J Biol Chem 2008;283:13565‐13577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Das J, Ghosh J, Manna P, Sil PC. Acetaminophen induced acute liver failure via oxidative stress and JNK activation: protective role of taurine by the suppression of cytochrome P450 2E1. Free Radic Res 2010;44:340‐355. [DOI] [PubMed] [Google Scholar]
- 21. Kim Y‐H, Hwang JH, Kim K‐S, Noh J‐R, Choi D‐H, Kim D‐K, et al. Metformin ameliorates acetaminophen hepatotoxicity via Gadd45β‐dependent regulation of JNK signaling in mice. J Hepatol 2015;63:75‐82. [DOI] [PubMed] [Google Scholar]
- 22. Goldring CEP, Kitteringham NR, Elsby R, Randle LE, Clement YN, Williams DP, et al. Activation of hepatic Nrf2 in vivo by acetaminophen in CD‐1 mice. Hepatology 2004;39:1267‐1276. [DOI] [PubMed] [Google Scholar]
- 23. Chan K, Han X‐D, Kan YW. An important function of Nrf2 in combating oxidative stress: detoxification of acetaminophen. Proc Natl Acad Sci U S A 2001;98:4611‐4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ni H‐M, Boggess N, McGill MR, Lebofsky M, Borude P, Apte U, et al. Liver‐specific loss of Atg5 causes persistent activation of Nrf2 and protects against acetaminophen‐induced liver injury. Toxicol Sci 2012;127:438‐450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pang C, Zheng Z, Shi L, Sheng Y, Wei H, Wang Z, et al. Caffeic acid prevents acetaminophen‐induced liver injury by activating the Keap1‐Nrf2 antioxidative defense system. Free Radic Biol Med 2016;91:236‐246. [DOI] [PubMed] [Google Scholar]
- 26. Reisman SA, Aleksunes LM, Klaassen CD. Oleanolic acid activates Nrf2 and protects from acetaminophen hepatotoxicity via Nrf2‐dependent and Nrf2‐independent processes. Biochem Pharmacol 2009;77:1273‐1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stanhope KL. Role of fructose‐containing sugars in the epidemics of obesity and metabolic syndrome. Annu Rev Med 2012;63:329‐343. [DOI] [PubMed] [Google Scholar]
- 28. Kim M‐S, Krawczyk SA, Doridot L, Fowler AJ, Wang JX, Trauger SA, et al. ChREBP regulates fructose‐induced glucose production independently of insulin signaling. J Clin Invest 2016;126:4372‐4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang YI, Pan Y, Xiong R, Zheng J, Li Q, Zhang S, et al. FGF21 mediates the protective effect of fenofibrate against acetaminophen‐induced hepatotoxicity via activating autophagy in mice. Biochem Biophys Res Comm 2018;503:474‐481. [DOI] [PubMed] [Google Scholar]
- 30. Vispute SG, Bu P, Le Y, Cheng X. Activation of GR but not PXR by dexamethasone attenuated acetaminophen hepatotoxicities via Fgf21 induction. Toxicology 2017;378:95‐106. [DOI] [PubMed] [Google Scholar]
- 31. Dushay JR, Toschi E, Mitten EK, Fisher FM, Herman MA, Maratos‐Flier E. Fructose ingestion acutely stimulates circulating FGF21 levels in humans. Mol Metab 2014;4:51‐57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pashkov V, Huang J, Parameswara VK, Kedzierski W, Kurrasch DM, Tall GG, et al. Regulator of G protein signaling (RGS16) inhibits hepatic fatty acid oxidation in a carbohydrate response element‐binding protein (ChREBP)‐dependent manner. J Biol Chem 2011;286:15116‐15125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Minn AH, Hafele C, Shalev A. Thioredoxin‐interacting protein is stimulated by glucose through a carbohydrate response element and induces β‐cell apoptosis. Endocrinology 2005;146:2397‐2405. [DOI] [PubMed] [Google Scholar]
- 34. Ma L, Robinson LN, Towle HC. ChREBP• Mlx is the principal mediator of glucose‐induced gene expression in the liver. J Biol Chem 2006;281:28721‐28730. [DOI] [PubMed] [Google Scholar]
- 35. Sae‐Lee C, Moolsuwan K, Chan L, Poungvarin N. ChREBP regulates itself and metabolic genes implicated in lipid accumulation in β‐cell line. PLoS One 2016;11:e0147411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gimeno RE, Moller DE. FGF21‐based pharmacotherapy–potential utility for metabolic disorders. Trends Endocrinol Metab 2014;25:303‐311. [DOI] [PubMed] [Google Scholar]
- 37. Minard A, Tan S‐X, Yang P, Fazakerley D, Domanova W, Parker B, et al. mTORC1 is a major regulatory node in the FGF21 signaling network in adipocytes. Cell Rep 2016;17:29‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kwon D, Kim S‐M, Correia MA. Cytochrome P450 endoplasmic reticulum‐associated degradation (ERAD): therapeutic and pathophysiological implications. Acta Pharm Sin B 2020;10:42‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pabarcus MK, Hoe N, Sadeghi S, Patterson C, Wiertz E, Correia MA. CYP3A4 ubiquitination by gp78 (the tumor autocrine motility factor receptor, AMFR) and CHIP E3 ligases. Arch Biochem Biophys 2009;483:66‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kwon D, Kim S‐M, Jacob P, Liu Y 3rd, Correia MA. Induction via functional protein stabilization of hepatic cytochromes P450 upon gp78/autocrine motility factor receptor (AMFR) ubiquitin E3‐ligase genetic ablation in mice: therapeutic and toxicological relevance. Mol Pharmacol 2019;96:641‐654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Baraille F, Planchais J, Dentin R, Guilmeau S, Postic C. Integration of ChREBP‐mediated glucose sensing into whole body metabolism. Physiology (Bethesda) 2015;30:428‐437. [DOI] [PubMed] [Google Scholar]
- 42. Tong X, Zhang D, Shabandri O, Oh J, Jin E, Stamper K, et al. DDB1 E3 ligase controls dietary fructose‐induced ChREBPα stabilization and liver steatosis via CRY1. Metabolism 2020;107:154222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ma X, Chang Y, Zhang Y, Muhammad I, Shi C, Li R, et al. Effects of C2‐ceramide and oltipraz on hepatocyte nuclear factor‐1 and glutathione S‐transferase A1 in acetaminophen‐mediated acute mice liver injury. Front Pharmacol 2018;9:1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lee FY, de Aguiar Vallim TQ, Chong HK, Zhang Y, Liu Y, Jones SA, et al. Activation of the farnesoid X receptor provides protection against acetaminophen‐induced hepatic toxicity. Mol Endocrinol 2010;24:1626‐1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Blazka ME, Germolec DR, Simeonova P, Bruccoleri A, Pennypacker KR, Luster MI. Acetaminophen‐induced hepatotoxicity is associated with early changes in NF‐kB and NF‐IL6 DNA binding activity. J Inflamm 1995. ‐1996;47:138‐150. [PubMed] [Google Scholar]
- 46. Bricambert J, Miranda J, Benhamed F, Girard J, Postic C, Dentin R. Salt‐inducible kinase 2 links transcriptional coactivator p300 phosphorylation to the prevention of ChREBP‐dependent hepatic steatosis in mice. J Clin Investi 2010;120:4316‐4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li Y, Yang D, Tian NA, Zhang P, Zhu Y, Meng J, et al. The ubiquitination ligase SMURF2 reduces aerobic glycolysis and colorectal cancer cell proliferation by promoting ChREBP ubiquitination and degradation. J Biol Chem 2019;294:14745‐14756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Postic C. Conversion of a dietary fructose: new clues from the gut microbiome. Nat Metab 2020;2:217‐218. [DOI] [PubMed] [Google Scholar]
- 49. Kim M, Astapova II, Flier SN, Hannou SA, Doridot L, Sargsyan A, et al. Intestinal, but not hepatic, ChREBP is required for fructose tolerance. JCI insight 2017;2.e96703. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S12
Supplementary Material