

Abstract
Drug discovery and approval in oncology is mediated by the use of imaging to evaluate drug efficacy in clinical trials. Imaging is performed while patients receive therapy to evaluate their response to treatment. Response criteria, specifically Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST 1.1), are standardized and can be used at different time points to classify response into the categories of complete response, partial response, stable disease, or disease progression. At the trial level, categorical responses for all patients are summated into image-based trial endpoints. These outcome measures, including objective response rate (ORR) and progression-free survival (PFS), are characteristics that can be derived from imaging and can be used as surrogates for overall survival (OS). Similar to OS, ORR and PFS describe the efficacy of a drug. U.S. Food and Drug Administration (FDA) regulatory approval requires therapies to demonstrate direct evidence of clinical benefit, such as improved OS. However, multiple programs have been created to expedite drug approval for life-threatening illnesses, including advanced cancer. ORR and PFS have been accepted by the FDA as adequate predictors of OS on which to base drug approval decisions, thus substantially shortening the time and cost of drug development (1). Use of imaging surrogate markers for drug approval has become increasingly common, accounting for more than 90% of approvals through the Accelerated Approval Program and allowing for use of many therapies which have altered the course of cancer.
Keywords: Oncology, Tumor Response
RSNA, 2021
Keywords: Oncology, Tumor Response
Summary
Oncologic imaging in clinical drug trials provides acceptable markers of future response to treatment, which allows shortening the clinical trial length compared with use of standard measures of survival.
Essentials
■ The Response Evaluation Criteria in Solid Tumors version 1.1 is the most commonly used imaging response assessment criteria that is used in oncology clinical trials.
■ Clinical trial outcome measures that can be extracted from imaging include progression-free survival, time to progression, objective response rate, complete response, and best overall response.
■ Radiologic clinical trial outcome measures are surrogate endpoints that predict a clinical benefit before direct measures, such as survival, are assessed.
■ Expedited programs at the U.S. Food and Drug Administration commonly use radiologic clinical trial outcome measures to evaluate cancer drugs for regulatory approval in the setting of unmet needs.
Introduction
Measuring changes in tumor size with imaging can help determine if patients are responding to therapy, as well as if and when disease progression occurs. In the clinical trial setting, there is blending of patient care and scientific discovery. Quantification of radiologic changes in tumor size extends beyond contributing to individual patient treatment decisions. It can also provide a method to perform statistical analyses and attain regulatory drug approval of promising investigational therapeutic agents by objective clinical trial endpoints. Image-based treatment outcome measures are commonly used to assess if treatments are effective and, in the past few decades, have played an increasing role in the regulatory drug approval of a growing number of oncologic therapies. Therefore, it is imperative that the imaging community understands the application of these rule-based response criteria during image interpretation and how reported findings inform clinical trial results.
Oncology Innovation: Drug Development Process
The drug discovery and approval paradigm is a very time- and resource-intensive process. For U.S. Food and Drug Administration (FDA) approval of one therapy, typically anywhere between 5000 to 10 000 initial compounds are rigorously investigated in a structured process that usually takes more than $1 billion and 10 years to complete (2–4). Beyond the initial concept and basic drug development, promising agents must undergo extensive preclinical evaluation and endure multiple phases of clinical trials to ensure safety and efficacy. Preclinical testing is performed both in vitro and in vivo to evaluate a drug’s mechanisms of action, therapeutic index, and drug dosing (5).
The three main phases of clinical trials in drug development occur sequentially. Phase I is primarily designed to evaluate drug safety and dosing in a small number of patients but may also provide early insight into pharmacokinetics and antitumor effect (5). Promising therapies that emerge from phase I clinical trials will advance to phase II. Phase II clinical trials are generally small nonrandomized studies used to screen new agents for preliminary evidence of treatment efficacy. Objective measures of tumor response rate are commonly used to measure efficacy in phase II studies and determine if the potential treatment should move forward to the next phase of evaluation (5,6). Phase III clinical trials are commonly designed as large double-blinded and randomized studies with a main objective of rigorously evaluating a drug’s clinical benefit, such as improved survival (7,8). Imaging is often incorporated into all phases of drug development, with changes in tumor size measured over time while undergoing treatment to evaluate treatment efficacy or complement and support other clinical outcomes.
Imaging Treatment Response Assessment
Use of imaging has become integral to oncology clinical trial design and treatment response assessment. The World Health Organization criteria was the first consensus guideline established to standardize imaging interpretation in the oncology clinical trial setting, with a mission to ensure objective and uniform reporting of changes in tumor burden while receiving therapy. Since its inception in 1981, numerous adaptions have been made to these initial “rules” and have allowed for the creation of multiple additional response assessment criteria (9). Modifications have been made to customize interpretations to specific tumor types and/or classes of therapies.
Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST 1.1) is currently the most widely accepted and applied imaging response assessment criteria in oncology clinical trials for solid tumors (10). Adapted from the original RECIST criteria in 2009, this evidence- and consensus-based guideline provides guidance for image acquisition and systematic rules for the assessment of anatomic tumor burden change during study (10,11). Baseline imaging is performed just prior to initiating systemic therapy, and all anatomic sites of tumor involvement are identified and then categorized as either measurable or nonmeasurable disease (10,12). A maximum of five measurable target lesions (maximum of two lesions per organ) can be selected and measured. Measurable lesions must be 10 mm in the longest dimension for solid lesions and 15 mm in short axis for lymph nodes, with preference given to those lesions that are largest in size and most reproducible in measurement. Each target lesion’s relevant diameter will be added together as the “sum of diameters” and represent a surrogate for “overall tumor burden” (10) (Fig 1, A).
Figure 1:

Baseline and follow-up CT images in a 55-year-old man with metastatic melanoma. CT imaging of the chest, abdomen, and pelvis was from A (left column), clinical trial enrollment and, B (right column), evaluation for treatment response at 9 months. At each time, the following were assessed: left lower lobe mass (row 1, top), right lower lobe nodule (row 2, middle), and subcarinal lymph node (row 3, bottom). The sum of diameters at baseline was 89 mm and at follow-up was 45 mm (a decrease of 49%), which was categorized as partial response. Dotted line represents the measured longest diameter of a lymph node. Solid line represents short-axis measurement.
These selected target lesions are followed and remeasured at all imaging time points while a patient is receiving therapy. The change in the sum of diameters from baseline or nadir (smallest sum during study) primarily determines the imaging response category assigned for a given time point (Fig 1, B). All nontarget sites of disease will also be qualitatively assessed for unequivocal worsening or complete resolution at each time point (10). A single categorical response is reported as a summarization of changes in tumor burden by target and nontarget disease and presence of new lesions for each imaging time point (Figs 2 and 3). Radiologic findings related to target and/or nontarget, as well as new lesion responses and overall categorical response, are commonly documented and followed by tumor tracker and case report forms (Fig 4). The categorical responses and duration of the responses are what inform image-based outcome measures in oncology clinical trials.
Figure 2:
![Longitudinal response categorization. At each imaging time point, the patient will receive a single categorical response. When there is a partial treatment response (PR), the time point with the smallest tumor burden is the nadir (green arrow). Provided there are no changes in nontarget lesions and no new lesions, when the smallest tumor burden increases by more than 20% from nadir (or baseline [yellow arrow], if no nadir), this is the date of disease progression (PD) (red arrow). Lines on bottom images indicate tumor diameter.](https://www.ncbi.nlm.nih.gov/core/lw/2.0/html/tileshop_pmc/tileshop_pmc_inline.html?title=Click%20on%20image%20to%20zoom&p=PMC3&id=8183261_rycan.2021210008.fig2.jpg)
Longitudinal response categorization. At each imaging time point, the patient will receive a single categorical response. When there is a partial treatment response (PR), the time point with the smallest tumor burden is the nadir (green arrow). Provided there are no changes in nontarget lesions and no new lesions, when the smallest tumor burden increases by more than 20% from nadir (or baseline [yellow arrow], if no nadir), this is the date of disease progression (PD) (red arrow). Lines on bottom images indicate tumor diameter.
Figure 3:

Overall response is the sum of the categorical responses of the target lesions, nontarget lesions, and presence or absence of new lesions. CR = complete response, NE = inevaluable, PD = progressive disease, PR = partial response, RECIST 1.1 = Response Evaluation Criteria in Solid Tumors version 1.1, SD = stable disease.
Figure 4:

Tumor tracker. Target lesion measurements and nontarget responses are collected in tumor trackers longitudinally to allow comparison of imaging response assessment for each patient over time.
Defining Clinical Trial Outcome Measures
When designing a clinical trial, it is essential to understand the primary goal of therapy and the clinical benefit which is most desired. In the setting of advanced cancer, the primary desire of the patient is to live longer with improved quality of life. Therefore, clinical endpoints are used as surrogate endpoints to predict how a patient feels, functions, and/or survives (13,14). When assessing drug efficacy, the main purpose is to evaluate for a substantial improvement in overall survival (OS) and/or quality or life (6,13,14).
OS Measures
In oncology, the reference standard of clinical trial endpoints is OS, defined as the time from trial enrollment until death from any cause (15). This preferred outcome measure is considered the most precise, reliable, and unambiguous given its independence from investigator assessment and potential bias. However, OS analysis requires enrollment of a large number of participants and requires long-term follow-up (8,15). Additionally, trial-related confounders often limit extracting direct survival benefits from a particular treatment. Increasingly complex clinical trial designs, such as treatment crossover, prevent measuring direct effects on OS when a patient receives more than one therapy. Similarly, expanding treatment options and lines of therapy after progression dilute direct survival effects from the initial treatment (Fig 5) (8,13,15).
Figure 5:

Overall survival. A patient receives multiple additional lines of treatment after their first therapy. In this case, overall survival is not a direct measure of treatment efficacy by therapy 1, as it also includes effects from therapies 2 and 3. PFS = progression-free survival.
Biomarkers and Rationale for Surrogate Outcome Measures
While OS remains the clinical trial endpoint of choice for assessing therapeutic agents for advanced cancer, additional outcome measures can be used in place of or in addition to OS as a more efficient method for outcome assessment (8). These endpoints are collected in place of more direct measures of how a patient feels, functions, or survives and is instead used to predict an expected clinical benefit (16).
Surrogate endpoints are also commonly a subset of biomarkers (14). A biomarker is defined as an objective measure that can be used as a predictor of a normal or pathologic process or response. These are commonly molecular, histologic, radiologic, or physiologic characteristics (14,17). To be an effective marker, any effect on the surrogate endpoint should also result in a clinically meaningful effect on additional clinical endpoint(s) (13). In the noncancer setting, a common biomarker that has been validated and widely accepted in medicine is the reduction of elevated blood pressure, which is used as a substitute for incidence of stroke, congestive heart failure, and cardiovascular death (14). With cancer, this is commonly a predictor of clinical benefit, which is possible to measure earlier than irreversible morbidity or mortality (18).
Surrogate endpoints used to assess therapeutic efficacy in clinical trials for advanced cancer are often radiologic biomarkers. Imaging assessments such as tumor shrinkage in certain cancers have been shown to directly correlate with other direct measures of clinical benefit, including survival (6,8,10,19,20). These measurements are also already common practice in the standard clinical setting, where alterations in tumor size by imaging commonly impact treatment decisions (8). By indirectly measuring clinical benefit, use of image-based biomarkers as surrogate endpoints for survival can reduce the time- and resource-intensive process of a clinical trial (6). Commonly assessed radiologic image-based surrogate outcome measures include: progression-free survival (PFS), time to progression (TTP), objective response rate (ORR), and complete response (Fig 6).
Figure 6:
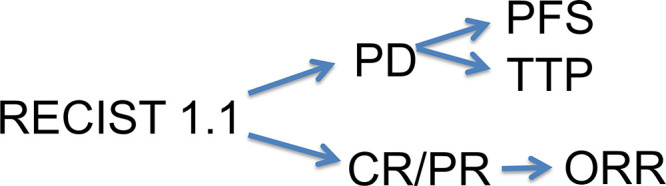
Image-based surrogate outcome measures. Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST 1.1) categorical treatment responses inform several image-based surrogate outcome measures. The time point of progressive disease (PD) is used to determine both progression-free survival (PFS) and time to progression (TTP). Categorical responses of complete response (CR) and partial response (PR) will determine objective response rate (ORR).
Image-based Surrogate Outcome Measures
PFS Measures
PFS is defined as the time from randomization until objective tumor progression or death (10). Tumor progression is determined by the response assessment criteria selected for the clinical trial, most often being RECIST 1.1 for solid tumors. As the most commonly used surrogate endpoint for OS, PFS is often used for drug approval (8,10,13). Given that OS is the summation of PFS and survival after progression, statistical models and prior analyses have shown that PFS demonstrates reasonable concordance with OS when median survival after progression is short (ie, 12 months or less) (13,21).
Evaluation of PFS may be a preferable measure when compared with OS, as it requires a smaller sample size to assess for a meaningful therapeutic response when compared with OS (8). Another advantage of PFS is that it is not affected by subsequent treatments, thus evaluating a direct effect of the treatment at hand (13). However, subsequent treatments after initial disease progression will also decrease the concordance between PFS and OS by lengthening survival after initial progression (13,22). Another limitation in using PFS is that radiologic changes in tumor size resulting in disease progression may not necessarily result in a similar effect in OS (13). Thus initial selection of the surrogate biomarker requires a priori knowledge of the natural disease course and molecular characteristics of the therapeutic agent of interest.
TTP Measures
Similar to PFS, TTP is defined as the time from randomization until the time of disease progression; however, it is different as it does not include deaths (Fig 7) (13). PFS still remains the preferred surrogate endpoint for drug approval, as by including deaths, it can be better correlated to OS (8). TTP is therefore a surrogate endpoint solely based on imaging findings and an imaging definition of disease progression. PFS and TTP are commonly used endpoints in randomized trials (15).
Figure 7:

Time to progression (TTP) is the total time from which the patient starts treatment until radiologic disease progression. In this example, the patient’s tumor grew 20% from nadir, reaching disease progression. TTP was 54 weeks. SOD = sum of diameters.
ORR Measures
ORR is defined as the proportion of patients who experience a prespecified degree of tumor shrinkage for a predefined or longer duration of time (8). These predetermined measures are commonly elicited from prior historical results. For solid tumors evaluated with RECIST 1.1, ORR is the summation of those patients with complete response or partial response (Fig 8) (8). Evaluation of ORR in phase II clinical trials has been shown to successfully select those therapies worth continuing forward for investigation in larger phase III clinical trials, deemed the “go, no-go” decision (6,23). In addition, ORR has been shown to be a robust endpoint for regulatory approval of post-first-line therapies (13,24). Duration of response is commonly reported with ORR, which is the time from initial response (complete or partial response) to the time of disease progression (Fig 9) (8).
Figure 8:

Objective response rate reflects the degree of tumor shrinkage and is defined as the proportion of patients who experience a complete response (CR) or partial response (PR) while receiving treatment. A, A patient who underwent chest CT that revealed a single site of disease at baseline experienced, B, marked decreased size of the right paratracheal lesion, and thus is an objective responder. C, Objective responders are the percentage of patients with CR and PR. Arrow indicates the single site of disease, mediastinal lymphadenopathy. PD = progressive disease, RECIST 1.1 = Response Evaluation Criteria in Solid Tumors version 1.1, SD = stable disease.
Figure 9:

Duration of response (DOR) is defined as the time from initial response (partial or complete response) until the time of disease progression. In this example, the sum of diameters was less than 30% of baseline at week 18 (defined as partial response), and disease progression occurred at week 54. Therefore DOR was 36 weeks.
An advantage in using ORR as an outcome measure is that it can be assessed by a single-arm study, as any reduction in tumor size is a direct measure of the antitumor effect of treatment (8,25). However, given that this is a direct measure of change in tumor burden while receiving treatment, only those patients with measurable disease at baseline should be included in clinical trials in which ORR is a primary endpoint of interest (10). This results in exclusion of some patients from trial enrollment if disease at baseline is not measurable.
ORR may not be the ideal surrogate outcome measure of OS in some cases. For example, biologic mechanisms of cytostatic agents may dampen the degree of tumor shrinkage, thus eliciting only stable disease in a larger proportion of patients. While certain targeted treatments, such as sorafenib for metastatic renal cell carcinoma, result in clinically meaningful improvements in OS, they may have a low ORR (13). Without inclusion of other outcome measures in the phase II trial setting, these therapies could inadvertently be deemed ineffective and thus not move forward for further evaluation of efficacy. This concept of minor response, in which tumor shrinkage does not meet the 30% threshold to be captured by ORR but still results in a meaningful clinical benefit and/or long-term disease control, has also been previously described in immunotherapy (26). For lymphoma, the International Working Group consensus response criteria, Response Evaluation Criteria in Lymphoma (RECIL), acknowledges that stable disease represents a wide range of tumor change (+20 to –30% change in size), and these thresholds may be too broad, as prior studies had found meaningful treatment response before the partial response threshold. Therefore RECIL has introduced minor response as a categorical treatment response, defined as a 10% or greater decrease in the sum of the longest diameters of target lesions but not a partial response (< 30%) (27).
Accurate and reproducible tumor measurements must be ascertained while receiving treatment to determine objective response. ORR may not extrapolate to clinical outcomes in certain primary tumors with indistinct margins and reduced reproducibility in measurement, such as pleural or peritoneal mesothelioma, pancreatic cancer, and certain brain tumors (8).
Complete Response
Complete response, as assessed by imaging or histopathologic assessment, is defined as resolution of all detectable tumor (8). In certain circumstances, complete response rates can be used for drug approval (8,15). For example, therapies for acute leukemia that result in durable complete response are associated with longer OS and would also likely improve quality of life through both a reduction in infection rates and decreased need for blood product support (15).
Of note, complete response by RECIST 1.1 may still include a measurable sum of diameters greater than 0 when lymph nodes are included as target lesions. Complete response of lymph nodes (targeted and nontargeted) is defined as a decrease in size of pathologic lymph nodes to a short axis of less than 10 mm (10).
Best Overall Response
Best overall response is the best categorical treatment response obtained for each patient while they are participating in the study. Given such, this outcome measure can only be calculated once the study has concluded. This takes into account changes in target and nontarget disease over time as well as development of new lesions. Best overall response as an outcome measure is a conclusion made at the patient level, not an analysis at the study level. If objective response is the primary endpoint in a nonrandomized clinical trial, best overall response must be confirmed at the subsequent imaging time point (10).
Regulatory History of Surrogate Outcome Measures
To facilitate approval and availability of new therapies, the FDA has created multiple programs to expedite drug approval. These include the Accelerated Approval, Priority Review, Fast Track, and Breakthrough therapy designations and programs (18,28). The mission of these programs is to expedite treatments that can address currently unmet medical needs of serious or life-threatening diseases (18,29). Through these programs, imaging-based outcome measures ORR and PFS are common clinical trial primary endpoints that support the drug approval process of cancer treatments.
Surrogate imaging endpoints are most commonly mentioned in conjunction with the Accelerated Approval Program (13). Prior to its inception, the only option for drug approval was through the traditional drug approval pathway, which requires a therapy to demonstrate evidence of a direct clinical benefit, such as improved OS, quality of life, or symptoms (8). In certain cancers, a surrogate endpoint could still be applied when it is a well-established predictor of clinical benefit. For example, a large improvement in PFS or appreciably durable ORR could also be used for regular approval when evaluated in conjunction with a positive benefit-risk profile of improvements in clinical symptoms and/or low drug toxicity (8,25,30). The Accelerated Approval Program was created in 1992 in response to the AIDS epidemic and need to bring promising novel therapies to those in need in a more timely fashion (16). Unlike regular approval, for those serious conditions with an unmet need, treatments could be approved on the basis of less well-established surrogate or intermediate endpoints which are “reasonably likely” to correlate with a clinical outcome (8). Given that many advanced cancers are serious and life-threatening illnesses, oncology drugs may also be eligible for accelerated approval (31). Once accelerated approval has been granted, additional clinical trials must then be performed to confirm the original findings and clinical benefit (31).
Since the implementation of the FDA’s Accelerated Approval Program, ORR has been the most commonly used surrogate outcome measure for accelerated approval (8,31). PFS has also been increasingly used as a surrogate endpoint in the evaluation of drug efficacy in phase III trials in advanced cancer (10,31,32). A review of the Accelerated Approval Program in 2018 revealed that in its 25 years of existence, 64 cancer therapies were approved for 93 new indications (31). The majority of approval endpoints were image-based surrogates, including ORR (87%), PFS and/or TTP (9%), and disease-free survival (4%). For drugs approved through the Accelerated Approval Program, confirmation of clinical benefit and transition to regular approval commonly also used image-based surrogate markers the majority of the time, most commonly PFS and/or TTP (39%) (31). In comparison, during this same time, OS was the most commonly used approval endpoint for traditional approval (35%). However, image-based surrogate endpoints were still frequently used for traditional approval, including PFS (34%) and ORR and/or duration of response (25%) (31).
Use of image-based surrogate endpoints have led to regulatory approval of numerous cancer therapies through the other FDA expedited programs and designations. The Breakthrough Therapy Designation is devoted to expediting approval of promising treatments on the basis of early evidence of promising therapies that have shown a substantially improved clinical benefit above those already available treatment options (16,30). For metastatic non–small cell lung cancer (mNSCLC), PFS and ORR were successfully used as approval endpoints for the approval of crizotinib for anaplastic lymphoma kinase–positive mNSCLC, as well as erlotinib and osimertinib in epidermal growth factor receptor L858R and T790 mutation–positive mNSCLC. ORR and disease-free survival were also approval endpoints in the release of imatinib for gastrointestinal stromal tumor (30).
Similar early access programs are also available through the European Medicines Agency, including priority medicines (PRIME), conditional approval, authorization under exceptional circumstances, and accelerated assessment (33–35).
Limitations and Future
Oncologic imaging has proven to play a major role in the primary analysis of oncology clinical trial results as well as primary endpoints for drug approval. RECIST 1.1 is most commonly applied as a method to capture both treatment response, as well as disease progression, and should correlate with the clinical status of the patient (10,13). Thresholds of a greater than 30% decrease for partial response and a greater than 20% increase for disease progression have been retrospectively validated by the RECIST working group and well correlated with OS (36–38). These thresholds provide outcomes captured at the clinical trial level. At the same time, these trials provide clinical care to the patients who volunteer to participate in the studies. Imaging interpretation for clinical trials contributes directly to their clinical care, and when imaging categorizes response as disease progression, many protocols require cessation of that therapy. On an individual level, changes in tumor size of 19% or 21% likely do not correlate with differences in clinical symptoms but result in a change of categorical response from stable disease to disease progression (22). Sole reliance on imaging reduces the opportunity to consider other meaningful clinical outcomes. For prostate cancer, a more holistic approach to treatment response has been proposed by the Prostate Cancer Clinical Trials Working Group 3, which places greater emphasis on determining when patients are no longer clinically benefiting from treatment as opposed to the first evidence of progression. This consensus guideline proposes future integration of serum markers and patient-reported outcomes with imaging-based markers to better assess disease heterogeneity and tumor resistance (27).
RECIST 1.1 is applied in oncology to provide objective measures of change in tumor burden. However, differences in image interpretation and interreader variation can result from a multitude of factors, including tumor measurement, target lesion selection, and qualitative nontarget lesion assessment, as well as from differences in identifying and interpreting lesions as new tumor. Tumors that are ill-defined in appearance inherently will have higher variations in inter- and intrareader measurements. The selection of which target lesions are assessed may itself lead to substantial discrepancies in interreader agreement. A prospective reader study found that discordance in target lesion selection among readers resulted in a disagreement in the classification of progressive versus nonprogressive disease in 45% of patients (39). Similarly, a retrospective review of combined trials in which dual reader with adjudication paradigm was used resulted in, on average, 40% reader disagreement (40,41). These discrepancies were most commonly due to disagreement in presence of new lesion, tumor measurements, and perception of progression of nontarget lesions (41). While RECIST disease progression as a result of qualitative nontarget disease alone was thought to be rare, an analysis of a large clinical trial by Coy et al of a cohort of patients with metastatic renal cell cancer demonstrated that almost 20% of disease progression was due to nontarget disease alone. This unexpected result demonstrates a not insubstantial percentage of cases that rely solely on qualitative assessments to inform outcome measures such as PFS and TTP (42).
Multiple commercial products are currently available to assist in radiologic interpretation, standardized response assessment, and data management of imaging performed as part of an oncology clinical trial. Implementation of electronic case report forms during RECIST 1.1 interpretation was previously found to reduce nonconformity errors by 10-fold (43). Artificial intelligence is increasingly embedded within many of these platforms, including automation for tumor measurement, target and nontarget location labeling, tumor localization at follow-up, and response assessment categorization. A recent study by Smith et al demonstrated that automation of these processes resulted in a 99% reduction in major errors, a 50% reduction in imaging interpretation time, and a 45% increase in total interobserver agreement (44). Given the above-described discrepancies in response assessment as a result of different target lesion selection, future implementation of artificial intelligence for tumor localization and measurement could also facilitate measurement of total tumor burden, a task that would otherwise be too time-intensive. In doing so, intertumor or interpatient disease heterogeneity found in mixed response could be better represented and considered when determining treatment response.
RECIST 1.1 relies upon unidimensional measurements for tumor assessment. However, the RECIST Working Group acknowledges that other imaging biomarkers, including functional imaging and tumor volume, could also provide valuable prognostic information in the future but currently lack clinical validation and sufficient standardization (10). The Quantitative Imaging Biomarkers Alliance recently released a profile for FDG PET/CT to characterize and reduce the variability of standardized uptake values and provide thresholds of meaningful change. Future multisite validation of these findings could result in functional imaging becoming even more embedded in oncology trials and playing a larger role in response assessment beyond lymphoproliferative disease (45).
Conclusions
Imaging is deeply woven into the routine practice of oncology and treatment response assessment in advanced cancers. In the clinical trial setting, uniform response criteria are used at every imaging time point to provide a standardized response for each patient. These categorical responses can then be used to compare responses at different time points within the same patient as well as compare among groups of patients. On a trial level, these imaging responses can be summated to inform image-based surrogate outcome measures of survival, including PFS and ORR. Imaging surrogate outcome measures have increasingly been applied not only to clinical trials to evaluate for clinical benefits but also to support FDA drug approval through expedited pathways.
K.R. supported by an AUR-GE Radiology Research Academic Fellowship.
Disclosures of Conflicts of Interest: K.R. disclosed no relevant relationships. M.B.A. disclosed no relevant relationships. M.D. disclosed no relevant relationships. V.S. disclosed no relevant relationships. A.G. disclosed no relevant relationships. R.D. disclosed no relevant relationships. J.G. Activities related to the present article: disclosed no relevant relationships. Activities not related to the present article: author is founder and board member of MEDQIA. Other relationships: disclosed no relevant relationships.
Abbreviations:
- FDA
- Food and Drug Administration
- mNSCLC
- metastatic non–small cell lung carcinoma
- ORR
- objective response rate
- OS
- overall survival
- PFS
- progression-free survival
- RECIL
- Response Evaluation Criteria in Lymphoma
- RECIST
- Response Evaluation Criteria in Solid Tumors
- TTP
- time to progression
References
- 1.Hsiue EHC, Moore TJ, Alexander GC. Estimated costs of pivotal trials for U.S. Food and Drug Administration-approved cancer drugs, 2015-2017. Clin Trials 2020;17(2):119–125. [DOI] [PubMed] [Google Scholar]
- 2.Adams CP, Brantner VV. Spending on new drug development1. Health Econ 2010;19(2):130–141. [DOI] [PubMed] [Google Scholar]
- 3.National Academies of Sciences, Engineering, and Medicine . The Drug Development Paradigm in Oncology: Proceedings of a Workshop. Washington, DC: The National Academies Press, 2018. [PubMed] [Google Scholar]
- 4.DiMasi JA. The value of improving the productivity of the drug development process: faster times and better decisions. Pharmacoeconomics 2002;20(Suppl 3):1–10. [DOI] [PubMed] [Google Scholar]
- 5.Eisenhauer E, Vermorken J. Principles and process of cancer drug development. CME J Gynecol Oncol 2000;18(1):344–350. https://www.researchgate.net/publication/265355221_Principles_and_process_of_cancer_drug_development. [Google Scholar]
- 6.El-Maraghi RH, Eisenhauer EA. Review of phase II trial designs used in studies of molecular targeted agents: outcomes and predictors of success in phase III. J Clin Oncol 2008;26(8):1346–1354. [DOI] [PubMed] [Google Scholar]
- 7.Ananthakrishnan R, Menon S. Design of oncology clinical trials: a review. Crit Rev Oncol Hematol 2013;88(1):144–153. [DOI] [PubMed] [Google Scholar]
- 8.U.S. Department of Health and Human Services; Food and Drug Administration; Oncology Center of Excellence; Center for Biologics Evaluation and Research; Center for Drug Evaluation and Research . Clinical Trial Endpoints for the Approval of Cancer Drugs and Biologics: Guidance for Industry. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-trial-endpoints-approval-cancer-drugs-and-biologics. Published December 2018. Accessed July 15, 2020.
- 9.Tirkes T, Hollar MA, Tann M, Kohli MD, Akisik F, Sandrasegaran K. Response criteria in oncologic imaging: review of traditional and new criteria. RadioGraphics 2013;33(5):1323–1341. [DOI] [PubMed] [Google Scholar]
- 10.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009;45(2):228–247. [DOI] [PubMed] [Google Scholar]
- 11.Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 2000;92(3):205–216. [DOI] [PubMed] [Google Scholar]
- 12.Nishino M, Jagannathan JP, Ramaiya NH, Van den Abbeele AD. Revised RECIST guideline version 1.1: What oncologists want to know and what radiologists need to know. AJR Am J Roentgenol 2010;195(2):281–289. [DOI] [PubMed] [Google Scholar]
- 13.Wilson MK, Karakasis K, Oza AM. Outcomes and endpoints in trials of cancer treatment: the past, present, and future. Lancet Oncol 2015;16(1):e32–e42. [DOI] [PubMed] [Google Scholar]
- 14.Biomarkers Definitions Working Group . Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther 2001;69(3):89–95. [DOI] [PubMed] [Google Scholar]
- 15.Pazdur R. Endpoints for assessing drug activity in clinical trials. Oncologist 2008;13(Suppl 2):19–21. [DOI] [PubMed] [Google Scholar]
- 16.U.S. Food and Drug Administration . Surrogate Endpoint Resources for Drug and Biologic Development. https://www.fda.gov/drugs/development-resources/surrogate-endpoint-resources-drug-and-biologic-development. Published 2018. Updated July 24, 2018. Accessed August 31, 2020.
- 17.FDA-NIH Biomarker Working Group . BEST (Biomarkers, EndpointS, and other Tools) Resource. Silver Spring, Md: Food and Drug Administration, Bethesda, Md: National Institutes of Health, 2016. https://www.ncbi.nlm.nih.gov/books/NBK326791/. [PubMed] [Google Scholar]
- 18.U.S. Department of Health and Human Services; Food and Drug Administration; Center for Biologics Evaluation and Research (CDER); Center for Drug Evaluation and Research (CBER) . Guidance for Industry: Expedited Programs for Serious Conditions – Drugs and Biologics. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/expedited-programs-serious-conditions-drugs-and-biologics. Published May 2014. Accessed October 27, 2020.
- 19.Paesmans M, Sculier JP, Libert P, et al. Response to chemotherapy has predictive value for further survival of patients with advanced non-small cell lung cancer: 10 years experience of the European Lung Cancer Working Party. Eur J Cancer 1997;33(14):2326–2332. [DOI] [PubMed] [Google Scholar]
- 20.Buyse M, Thirion P, Carlson RW, Burzykowski T, Molenberghs G, Piedbois P. Relation between tumour response to first-line chemotherapy and survival in advanced colorectal cancer: a meta-analysis. Meta-Analysis Group in Cancer. Lancet 2000;356(9227):373–378. [DOI] [PubMed] [Google Scholar]
- 21.Broglio KR, Berry DA. Detecting an overall survival benefit that is derived from progression-free survival. J Natl Cancer Inst 2009;101(23):1642–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Booth CM, Eisenhauer EA. Progression-free survival: meaningful or simply measurable? J Clin Oncol 2012;30(10):1030–1033. [DOI] [PubMed] [Google Scholar]
- 23.Roberts TG Jr, Lynch TJ Jr, Chabner BA. The phase III trial in the era of targeted therapy: unraveling the “go or no go” decision. J Clin Oncol 2003;21(19):3683–3695. [DOI] [PubMed] [Google Scholar]
- 24.Tsimberidou AM, Braiteh F, Stewart DJ, Kurzrock R. Ultimate fate of oncology drugs approved by the us food and drug administration without a randomized Trial. J Clin Oncol 2009;27(36):6243–6250. [DOI] [PubMed] [Google Scholar]
- 25.Blumenthal GM, Pazdur R. Response Rate as an Approval End Point in Oncology: Back to the Future. JAMA Oncol 2016;2(6):780–781. [DOI] [PubMed] [Google Scholar]
- 26.Ribas A, Chmielowski B, Glaspy JA. Do we need a different set of response assessment criteria for tumor immunotherapy? Clin Cancer Res 2009;15(23):7116–7118. [DOI] [PubMed] [Google Scholar]
- 27.Younes A, Hilden P, Coiffier B, et al. International Working Group consensus response evaluation criteria in lymphoma (RECIL 2017). Ann Oncol 2017;28(7):1436–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huber M, Huber B. Innovation in Oncology Drug Development. J Oncol 2019;20199683016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnson JR, Ning YM, Farrell A, Justice R, Keegan P, Pazdur R. Accelerated approval of oncology products: the food and drug administration experience. J Natl Cancer Inst 2011;103(8):636–644. [DOI] [PubMed] [Google Scholar]
- 30.Blumenthal GM, Kluetz PG, Schneider J, Goldberg KB, McKee AE, Pazdur R. Oncology Drug Approvals: Evaluating Endpoints and Evidence in an Era of Breakthrough Therapies. Oncologist 2017;22(7):762–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beaver JA, Howie LJ, Pelosof L, et al. A 25-year experience of us food and drug administration accelerated approval of malignant hematology and oncology drugs and biologics: A review. JAMA Oncol 2018;4(6):849–856. [DOI] [PubMed] [Google Scholar]
- 32.Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 2000;92(3):205–216. [DOI] [PubMed] [Google Scholar]
- 33.Senderowicz AM, Pfaff O. Similarities and differences in the oncology drug approval process between FDA and European Union with emphasis on in vitro companion diagnostics. Clin Cancer Res 2014;20(6):1445–1452. [DOI] [PubMed] [Google Scholar]
- 34.Martinalbo J, Bowen D, Camarero J, et al. Early market access of cancer drugs in the EU. Ann Oncol 2016;27(1):96–105. [DOI] [PubMed] [Google Scholar]
- 35.Cox EM, Edmund AV, Kratz E, Lockwood SH, Shankar A. Regulatory Affairs 101: Introduction to Expedited Regulatory Pathways. Clin Transl Sci 2020;13(3):451–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Litière S, Isaac G, Vries ED, et al. Validation of RECIST 1.1 for use with cytotoxic agents and targeted cancer agents (TCA): Results of a RECIST Working Group analysis of a 50 clinical trials pooled individual patient database. J Clin Oncol 2017;35(15_suppl):2534. [Google Scholar]
- 37.An MW, Dong X, Meyers J, et al. Evaluating Continuous Tumor Measurement-Based Metrics as Phase II Endpoints for Predicting Overall Survival. J Natl Cancer Inst 2015;107(11):djv239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mandrekar SJ, An MW, Meyers J, Grothey A, Bogaerts J, Sargent DJ. Evaluation of alternate categorical tumor metrics and cut points for response categorization using the RECIST 1.1 data warehouse. J Clin Oncol 2014;32(8):841–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuhl CK, Alparslan Y, Schmoee J, et al. Validity of RECIST Version 1.1 for Response Assessment in Metastatic Cancer: A Prospective, Multireader Study. Radiology 2019;290(2):349–356. [DOI] [PubMed] [Google Scholar]
- 40.Ford RR, O’Neal M, Moskowitz SC, Fraunberger J. Adjudication Rates between Readers in Blinded Independent Central Review of Oncology Studies. J Clin Trials 2016;6(5):289. [Google Scholar]
- 41.Beaumont H, Evans TL, Klifa C, et al. Discrepancies of assessments in a RECIST 1.1 phase II clinical trial - association between adjudication rate and variability in images and tumors selection. Cancer Imaging 2018;18(1):50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coy H, Michael D, Ruchalski K et al. Components of Radiologic Progressive Disease Defined by RECIST 1.1 in Patients with Metastatic Clear Cell Renal Cell Carcinoma. Radiology 2019;292(1):103–109. [DOI] [PubMed] [Google Scholar]
- 43.Beaumont H, Bertrand AS, Klifa C, et al. Radiology workflow for RECIST assessment in clinical trials: Can we reconcile time-efficiency and quality? Eur J Radiol 2019;118(257):263. [DOI] [PubMed] [Google Scholar]
- 44.Smith AD, Allen BC, Abou Elkassem A, et al. Multi-institutional comparative effectiveness of advanced cancer longitudinal imaging response evaluation methods: Current practice versus artificial intelligence-assisted. J Clin Oncol 2020;38(15_suppl):2010.32352857 [Google Scholar]
- 45.Kinahan PE, Perlman ES, Sunderland JJ, et al. The QIBA Profile for FDG PET/CT as an Imaging Biomarker Measuring Response to Cancer Therapy. Radiology 2020;294(3):647–657. [DOI] [PMC free article] [PubMed] [Google Scholar]