

SUMMARY
During the repeated cycles of damage and repair in many muscle disorders, including Duchenne muscular dystrophy (DMD), the muscle stem cell (MuSC) pool becomes less efficient at responding to and repairing damage. The underlying mechanism of such stem cell dysfunction is not fully known. Here, we demonstrate that the distinct early telomere shortening of diseased MuSCs in both mice and young DMD patients is associated with aberrant NF-κB activation. We find that prolonged NF-κB activation in MuSCs in chronic injuries leads to shortened telomeres and Ku80 dysregulation and results in severe skeletal muscle defects. Our studies provide evidence of a role for NF-κB in regulating stem-cell-specific telomere length, independently of cell replication, and could be a congruent mechanism that is applicable to additional tissues and/or diseases characterized by systemic chronic inflammation.
In brief
Tichy et al. reveal a role for NF-κB signaling in regulating telomere length in muscle stem cells (MuSCs) after chronic injuries. Persistent activation of NF-κB leads to shortened telomeres, Ku80 dysregulation, and muscle defects. The findings link stem cell dysfunction and NF-κB-dependent telomere shortening in Duchenne muscular dystrophy.
Graphical Abstract
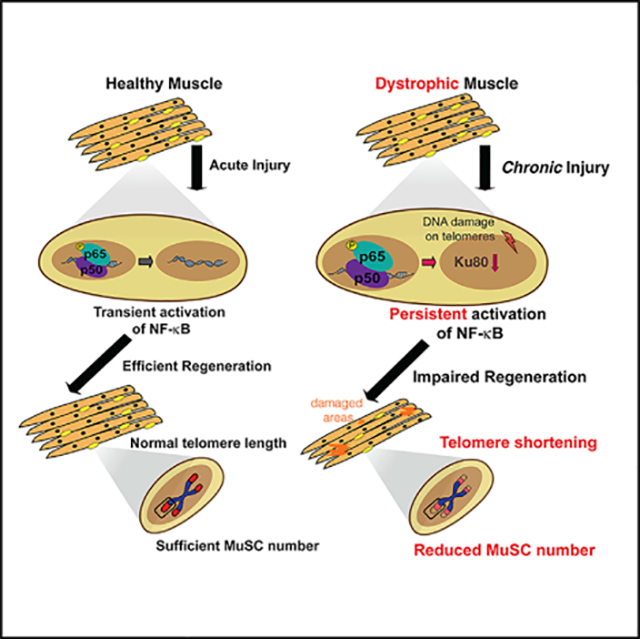
INTRODUCTION
The stages of skeletal muscle regeneration are well characterized following injury, starting with necrosis of damaged muscle tissue and the infiltration of immune cells (Frenette et al., 2000; Tidball, 2005). Inflammatory signals both directly and indirectly lead to activation of resident muscle stem cells (MuSCs; also called satellite cells), which respond to muscle damage by population expansion, followed by selective differentiation into myogenic progeny (Chazaud et al., 2003; Tidball and Wehling-Henricks, 2015).
Duchenne muscular dystrophy (DMD) is a fatal X-linked degenerative muscle disease, and it is among the most common lethally inherited disorders in children (Emery, 2002). DMD is caused by mutations in the dystrophin gene, which codes for a cytoskeletal protein that acts as a crucial bridging protein between the sarcomere and extracellular matrix (Hoffman et al., 1987). The complete or partial loss of dystrophin in muscular dystrophies impairs the ability of the muscle to contract and causes the muscle to break down. This acts as constant injury to the muscle that progressively affects the MuSC pool (Morgan and Zammit, 2010; Sacco et al., 2010), presumably due to an autonomous dysfunction (Dumont et al., 2015). Thus, MuSC function is severely affected in DMD, and although the disease is initiated by dystrophin deficiency, it is ultimately a stem cell disease (Dumont et al., 2015; Sacco et al., 2010). Previous work showed that accelerated telomere shortening occurs in muscular dystrophy in both skeletal muscle and cardiomyopathy (Mourkioti et al., 2013; Sacco et al., 2010). Recent studies have also revealed that MuSCs derived either from dystrophic mice or young DMD patients exhibit critically shortened telomeres early on during disease progression (Tichy et al., 2017). These observations suggest that modulation or maintenance of MuSC function can prevent or diminish destructive muscle processes in chronic injury conditions. However, the molecular mechanisms contributing to MuSC-specific telomere shortening are currently unknown.
Here, we show that persistent activation of nuclear factor kb (NF-κB) leads to telomere shortening in MuSCs under chronic injury conditions, and it is independent of proliferation. Instead, this mechanism involves Ku80 dysregulation and increased DNA damage, specifically on telomeres, that results in MuSC loss and, subsequently, skeletal muscle regenerative failure. Overall, our findings propose a previously unrecognized role of NF-κB in regulating MuSC dysfunction by affecting telomeres, a key event during the progression of DMD.
RESULTS
NF-κB is dysregulated in dystrophic MuSCs
To understand the mechanisms that affect telomere length and function in diseased MuSCs, we assayed their internal signaling pathways by flow cytometry (Table S1). We found that a higher proportion of the MuSCs from dystrophic mice exhibited higher NF-κB activation (Figure 1A) and accumulation of p-p65 (Figures 1B and 1C). We expanded upon these observations by assessing the timing of NF-κB activation following a single intramuscular injury. Muscle injury rapidly increases NF-κB activation in MuSCs (Figure 1D). But in dystrophic mice, p-p65+ MuSCs are more numerous at all time points, including baseline conditions (Figure 1D). Hence, the inherent endogenous injuries observed in dystrophic mice induce a constant elevated NF-κB signaling response in MuSCs.
Figure 1. NF-κB activation in dystrophic MuSCs.

(A) Higher NF-κB activation in FACS-isolated MuSCs from dystrophic mice, as shown by elevated phospho-p65 levels. n ≥ 3 mice (2–3 months old)/group. Percent of cells positive for p-p65 are displayed.
(B) Freshly isolated, cytospun dystrophic MuSCs exhibit accumulation of phospho-p65 compared to control MuSCs. Note the lack of staining in the no primary Ab (technical control), highlighting the specificity of the staining.
(C) Quantification shows higher levels of p-p65 in dystrophic MuSCs. n = 3 mice (2–3 months old) per genotype. n > 50 cells per condition.
(D) MuSCs from notexin-injured muscles were isolated at the indicated times. Phospho-p65+ MuSCs from dystrophic animals were elevated at all time points, including baseline conditions. n = 3 mice (3 months old)/condition. n > 1,000 cells per condition. Displayed is mean ± SEM for all graphs. Statistical analyses were performed using unpaired Student’s t test with Welch’s correction. *p ≤ 0.05.
MuSC-specific NF-κB activation does not alter muscles in non-injured conditions
To examine the in vivo impact of persistent NF-κB activation in regulating telomere length in MuSCs during chronic injury, we generated a mouse that allows expression of a constitutively active (CA) form of the IKK2 gene (Calado et al., 2010) specifically in MuSCs (Pax7ERT2Cre/IKK2CA or IKK2CAMuSC) (Figure S1A). To generate mice in which NF-κB signaling is abrogated in MuSCs, we crossed mice carrying a floxed allele of the NF-κB regulator NEMO (Schmidt-Supprian et al., 2000) with the same Pax7ERT2Cre mice (Murphy et al., 2011) to generate NEMO knockout (NEMOKOMuSC) mice. Characterization of these mice demonstrates that we obtained >90% Cre recombination efficiency after seven daily intraperitoneal tamoxifen injections, which resulted in significant upregulation of the IKK2 gene in IKK2CAMuSC mice and downregulation in NEMOKOMuSC mice (Figure S1B). The above genetic approaches allow for CA or inactivation of p65 in MuSCs, as shown by flow cytometry analysis (Figure 2A). Enforced expression of activated IKK2 alone or ablation of NEMO in MuSCs had no effect on muscle morphology (Figure S1C), muscle size (Figure S1D), function (Figure S1E), and/or telomere length (Figure S1F) under steady-state conditions. These data suggest that in the absence of injury, persistent activation of MuSC-specific NF-κB has minimal effects in skeletal muscles.
Figure 2. Persistent NF-κB activation in MuSCs leads to rapid telomere shortening.

(A) p65 activation in uninjured (green plots) or injured (red plots) control MuSCs (first column), MuSCs with kinase CA of the IKK2 protein (IKK2CAMuSC, second column), or MuSCs with inactivation of the NEMO protein (NEMOKOMuSC, third column). Blue plots are technical controls with no primary antibody. Percent of cells positive for p-p65 is displayed.
(B) Experimental scheme for weekly notexin injuries to mimic chronic injury.
(C) IKK2CA-derived MuSCs had much shorter telomere lengths after 6× injury, while inhibition of NF-κB (NEMOKOMuSC) maintains telomere length even after 20× injuries. Telomere length in MuSCs was normalized to telomere length of contralateral legs. n = 3–5 mice/genotype. Percent difference was calculated between normalized means.
(D–F) Distribution of telomere lengths of MuSCs following 3× (D), 6× (E), or 20× (F) injuries. Telomeres start to shorten after three injuries in IKK2CAMuSC cells (red bars), but this reduction becomes more severe with increasing number of injuries. n = 3 mice/condition. n > 100 cells. Mean ± SEM for each bin is displayed.
(G–I) Progressive MuSC-specific telomere shortening is associated with worsened histology in IKK2CAMuSC muscles at 3× (G), 6× (H), or 20× (I) injuries. n = 3–6 mice/condition. Mice were 2 months old at the time of the first injury.
Rapid telomere shortening in injured MuSCs with persistent NF-κB
To investigate the importance of injury in NF-κB-dependent telomere length regulation and to mimic the rounds of degeneration/regeneration occurring in the dystrophic condition (Morgan and Zammit, 2010), we implemented a multiple injury model in control, IKK2CAMuSC, and NEMOKOMuSC mice (Figure 2B). Assessment of telomere lengths of MuSCs isolated from these mice by MuQ-FISH (fluorescence in situ hybridization) (Tichy et al., 2017) uncovered a propensity of telomere shortening in IKK2CAMuSC-derived MuSCs (Figure 2C). However, NEMOKOMuSC MuSCs have unaltered telomere lengths even after many injuries (Figure 2C), demonstrating that NF-kB inhibition maintains telomere length. These findings further suggest that NF-κB activation is required for telomere shortening in injured muscles, providing strong support that prolonged NF-κB activation drives telomere shortening under chronic injuries. Interestingly, telomeres start to shorten after three injuries in IKK2CAMuSC cells, but this reduction becomes more severe with an increasing number of injuries (Figures 2D–2F and S2A). Moreover, the progressive telomere shortening observed after an increased number of injuries correlates well with the worsening histology (Figures 2G–2I) and fibrosis (Figures S2B and S2C) in IKK2CAMuSC muscles with an increased number of injuries. These studies support the notion that NF-κB-dependent telomere shortening contributes to the histopathology in skeletal muscles under repetitive injuries. Altogether, these data demonstrate an unconventional role for unrestrained NF-κB signaling in MuSCs to promote telomere attrition and impairment of regeneration under chronic injury conditions.
Telomere shortening in MuSCs is not caused by enhanced proliferation
In proliferative cells, DNA polymerases fail to completely replicate telomeres following each cell division, which eventually leads to critical telomere shortening (Lee et al., 1998). To address whether the rapid MuSC-specific telomere shortening observed in injured IKK2CAMuSC mice was caused by MuSC proliferative changes, we injured control or IKK2CAMuSC mice repeatedly. At 3 days after the last injury, a timepoint considered the peak of MuSC proliferation (Morgan and Zammit, 2010), mice were pulsed in vivo with the nucleotide analog 5-ethynyl-2′-deoxyuridine (EdU) to label cells undergoing DNA replication (Figure 3A). In uninjured muscles, only a small portion of MuSCs was EdU+ (Figures 3B and 3C), consistent with the notion that adult MuSCs with higher NF-kB activation are largely quiescent under steady-state conditions. Interestingly, upon injury, both control and IKK2CAMuSC MuSCs were proliferating at comparable rates (Figures 3B and 3C). Similarly, an equivalent level of the proliferation marker Ki67, in combination with the stem cell marker Pax7 was found in injured control and IKK2CAMuSC muscles (Figures 3D and 3E). To ensure that the window of optimal in vivo proliferation was not missed by our previous assessments, as well as to confirm a large enough window of EdU incorporation was included, EdU was administered daily, for a total of three times, and MuSCs were harvested 3 or 5 days after the last injury (Figure 3F). Similar to the previous data, no significant increases in proliferation were evident in IKK2CAMuSC MuSCs (Figure 3G). We conclude that in contrast to other cell types (Sasaki et al., 2006), the constant activation of NF-κB in MuSCs does not result in higher rates of proliferation even under injury conditions. Altogether, these data unequivocally demonstrate a proliferation-independent mechanism of NF-κB to drive MuSC-specific telomere shortening.
Figure 3. Activation of NF-κB does not alter in vivo proliferation in MuSCs under injury conditions.

(A) Experimental scheme for in vivo 5-Ethynyl-2′deoxyuridine (EdU) incorporation assay. Mice were left uninjured or injured with notexin three times (once/week) and treated with EdU once intraperitoneally 2 days post-final injury. MuSCs were FACS-isolated 3 days post-final injury.
(B) Representative images of isolated MuSCs stained for EdU (red) and counterstained with 4’,6-diamidino-2-phenylindole (DAPI, blue).
(C) EdU quantification shows no changes in proliferation between genotypes. n = 3 mice/condition. n > 50 cells per mouse.
(D) Representative sections stained for Ki67 (red), Pax7 (green), and nuclei (DAPI, blue).
(E) Quantifications of Ki67+ MuSCs show similar proportions of proliferating MuSCs between control and IKK2CAMuSC muscles. n = 3 mice/condition. n > 80 cells per mouse.
(F) Experimental schemes of additional EdU injections. Top: control and IKK2CAMuSC mice injured with notexin 3× (once/week), followed by three daily EdU injections, and MuSC isolation on day 3 post-last injury (D3). Bottom: mice were notexin injured as above, followed by three daily EdU injections, and MuSC isolation on day 5 post-last injury (D5).
(G) Quantitation of EdU incorporation in MuSC-derived mice following either the D3 (left) or D5 (right) protocol. n = 3 mice (2–3 months old)/condition. n > 50 cells per mouse. No significant increases in proliferation were found in MuSCs from IKK2CAMuSC compared to control mice. Data from all graphs are depicted as mean ± SEM. All statistical analyses were performed using unpaired Student’s t test with Welch’s correction. n.s., non-significant.
NF-κB regulates Ku80 expression in MuSCs in chronic muscle injuries
Telomeres are not naked DNA but are instead associated with six proteins, called shelterins (TIN2, TPP1, POT1, RAP1, TRF1, and TRF2) (de Lange, 2005; Hsu et al., 1999), together with the DNA-binding proteins Kus (de Lange, 2005; Hsu et al., 1999) (Figure S4A). To delineate alternative mechanisms to rationalize the observed telomere shortening in IKK2CAMuSC MuSCs experiencing chronic injuries, we carried out in-silico analysis on promoters of genes, whose proteins are known to reside at telomeres. Given that Kus bind to telomeres, aid the localization of other shelterins, and regulate telomere length, we included them in our in-silico analysis (de Lange, 2005; Hsu et al., 1999). NF-κB functions by binding to promoter and enhancer regions containing kB sites to regulate gene expression (Dong et al., 2008; Hayden and Ghosh, 2004). We found that proteins bound to telomeric DNA contain kB sites in their promoters (Table S2), raising the intriguing possibility that NF-κB-dependent regulation of telomere binding proteins may have a significant impact on telomere length in MuSCs. We then performed qRT-PCR to validate the in-silico analysis and detected downregulation of telomeric XRCC5/Ku80 gene expression in MuSCs from injured muscles (Figure 4A), while all other telomeric genes had no significant differences (Figure 4A). Similarly, we found downregulation of Ku80 protein in MuSCs from IKK2CAMuSC muscles after 3× injury (Figures 4B and 4C), further demonstrating the NF-κB-dependent regulation of Ku80. In addition, we found undetectable tert levels in wild-type (WT) or IKK2CAMuSC MuSCs either in steady-state or upon injury conditions (Figure S3B). Since tert transcripts are detectable in embryonic stem cells (ESCs) within the same reaction (technical control), our data suggest that tert is either present at very low (undetectable) levels or expressed transiently in freshly isolated MuSCs, in contrast to what is known for cancer cell lines (Yin et al., 2000; Zuo et al., 2011). Importantly, Ku80 levels are unchanged in both IKK2CA uninjured MuSCs (Figure S3C; NF-κB activation alone without injury) and MuSCs from NEMOKOMuSC injured mice (Figure S3D; injury combined with NF-κB inactivation in MuSCs), demonstrating that NF-κB is required during injury to alter Ku80 levels (Figures 4B and 4C) and induce telomere shortening (Figures 2C–2F). Overall, these data indicate a previously unrecognized requirement of NF-κB in regulating Ku80 expression associated with telomere length in freshly isolated MuSCs from injured muscles.
Figure 4. Activation of NF-κB in MuSCs in the context of repetitive injuries leads to Ku80 dysregulation.

(A) Isolated MuSCs from 3× injured control and IKK2CAMuSC mice subjected to qRT-PCR for genes associated with telomeric function. Note that XRCC5, which encodes the DNA repair protein, Ku80, is downregulated in IKK2CAMuSC MuSCs. n = 4–7 mice (3–5 months old) per genotype.
(B) Representative Ku80 staining (red), VCAM (green), and nuclei (DAPI, blue) in muscle sections from control and IKK2CAMuSC mice after 3× injury.
(C) Quantification shows reduced levels of Ku80 in IKK2CAMuSC MuSCs compared to controls. n = 3 samples/group. Mean ± SEM. Statistical analysis was performed using unpaired Student’s t test with Welch’s correction. ***p ≤ 0.001.
(D) Representative image of TIFs (telomere-induced foci), where DNA damage (53BP1, green) co-localize with telomere (red) in VCAM-positive cells (white).
(E) Quantification shows higher TIFs in IKK2CA-MuSC compared to controls. n = 3 mice (4 months old) per genotype. n > 50 cells per mouse.
(F) Left: representative two-photon microscopy images. MuSCs are shown in green, while muscle fibers are visualized in red by second harmonic generation (SHG). Right: quantification of the number of Pax7 (EGFP+) MuSCs shows stem cell exhaustion over time under chronic injuries. n = 3 mice per genotype. All datasets displayed are mean ± SEM. Statistical analyses were performed using unpaired Student’s t test with Welch’s correction. *p ≤ 0.05; ***p ≤ 0.001.
MuSC telomere shortening leads to increased DNA damage in telomeres and stem cell exhaustion over time
In mammalian cells, DNA damage, in the form of double-stranded DNA breaks, is typically repaired by non-homologous end-joining (NHEJ) (Mao et al., 2008) and depends on the activity of Ku80 to recognize damaged DNA ends (Koike and Koike, 2008). Indeed, we observed a higher number of centralized nuclei in regenerating IKK2CAMuSC fibers that positively stained for the DNA damage marker 53BP1 (Figures S3E and S3F). Extensive telomere shortening can be recognized by DNA damage sensors as free DNA ends (Hsu et al., 2000). In fact, we did observe increased numbers of telomere-induced foci (TIFs) in chronically injured IKK2CAMuSC MuSCs (Figures 4D and 4E), as identified by the stem cell marker, vascular cell adhesion molecule (VCAM) (Figure S3G). These data are in agreement with the notion that telomere dysfunction is governed by proteins that also control DNA damage response (Takai et al., 2003).
To further investigate the in vivo consequence of the early telomere shortening in MuSCs in chronic injury, we crossed the MuSC-specific IKK2CAMuSC mice with Pax7EGFP reporter mice, which express EGFP under the control of the Pax7 promoter (Tichy et al., 2018). The Pax7EGFP/IKK2CAMuSC mouse allows for direct visualization and evaluation of MuSCs in their native environment on all fibers within the same animal in a spatiotemporal manner. Utilizing this method, we assessed the number of MuSCs by two-photon microscopy following chronic injury and recovery. We found that the number of MuSCs decreases over time in Pax7EGFP/IKK2CAMuSC skeletal muscles (Figure 4F), indicative of an exhaustion of the stem cell pool under chronic injury.
MuSC-specific NF-κB activation exacerbates the dystrophic muscle defects
To explore whether MuSC-specific activation of NF-κB could exacerbate the progression of dystrophy, we crossed IKK 2CAMuSC mice with a dystrophin mutant mouse (Im et al., 1996). The resulting double mutants are designated herein as mdx/IKK2CAMuSC. These mice have higher muscle damage, shown by the levels of creatine kinase (Figure 5A), an established damage diagnostic indicator in DMD (Zatz et al., 1991). The severity of muscle defects in mdx/IKK2CAMuSC mice is consistent with reduced muscle weight (Figures S4A and S4B), worse muscle morphology (Figure S4C), and reduced area of fibers with centralized nuclei (Figure S4D). Interestingly, we observed substantial kyphosis (Figure 5B), a classical clinical manifestation of DMD due to weakness of trunk muscles (Oda et al., 1993; Wilkins and Gibson, 1976) and impaired muscle performance in mdx/IKK2CAMuSC mice compared to mdx controls (Figures 5C and 5D). Importantly, MuSCs isolated from mdx/IKK2CAMuSC mice exhibited higher levels of p65 (Figure 5E), demonstrated shortened telomeres (Figures 5F and S4E), exhibited increased numbers of TIFs (Figure 5G), and had a reduced number of MuSCs (Figure S4F). We conclude that the persistent activation of NF-κB in MuSCs in the context of repetitive injuries as caused by the lack of dystrophin leads to the aggravation of the mdx phenotype, imitating the human disease.
Figure 5. Activation of NF-κB in MuSCs exacerbates the mdx phenotype.

To explore whether MuSC-specific activation of NF-κB could exacerbate the progression of dystrophy, mdx mice were bred to IKK2CAMuSC mice, and different metrics were assessed.
(A) mdx/IKK2CAMuSC exhibited more muscle damage as shown by higher serum creatine kinase activity, a strong indicator of skeletal muscle damage. n > 10 mice (8–12 months old) per genotype.
(B) Increased skeletal deformity (kyphosis), as shown by whole body CT images. n = 3–4 mice (12 months old) per genotype.
(C) mdx/IKK2CAMuSC mice were substantially impaired in the grip test. n = 4–5 mice (8–12 months old) per genotype.
(D) Reduced strength of mdx/IKK2CAMuSC mice compared to mdx mice. n = 3–5 mice (8–12 months old) per genotype.
(E) Higher levels of phospho-p65 in isolated MuSCs (upper) and quantification of p-p65 (lower). n = 3–4 mice (4–5 months old) per genotype. n > 30 cells per condition.
(F) Telomere length assessment by MuQ-FISH in isolated MuSCs (insert: representative image of telomere staining) shows reduced telomere length in mdx/IKK2CAMuSC mice. n = 3–4 mice (5 months old) per genotype. n > 70 cells per condition.
(G) Increased TIFs specifically in MuSCs. n = 3 mice (5 months old) per genotype. n > 70 cells per condition. All datasets are displayed as mean ± SEM. All statistical analyses were performed using unpaired Student’s t test with Welch’s correction. *p ≤ 0.05; **p ≤ 0.01.
Downregulation of Ku80 alone or in the context of chronic injuries reduces telomere length in MuSCs
To gain more insight into the involvement of Ku80 in telomere shortening in myogenic cells, we first investigated telomere length in the C2C12 mouse myoblast cell line (Figure S5A). Using short hairpin RNA (shRNA) vectors targeting XRCC5, which encodes the Ku80 protein, we observed significant knockdown at the protein level when compared to cells transfected with empty vector (Figure S5B). Upon performing MuQ-FISH, we uncovered that Ku80 downregulation is sufficient to induce telomere shortening in myogenic cells with two different shRNAs (Figures S5C and S5D). These data demonstrate that downregulation of Ku80 alone is sufficient to reduce telomere length in myogenic cells. Next, we extended our findings by investigating the effect of Ku80 manipulation on telomere length in vivo, utilizing a Ku80 heterozygous mouse line (Zhu et al., 1996). We found that MuSCs derived from this mouse exhibited only ~20% reduction of Ku80 protein expression compared to Ku80 WT littermates (Figure S5E). Nevertheless, we still observed a reduction in telomere lengths of these Ku80 heterozygous mice after 3× injury (Figure S5F), suggesting that even a mild reduction of Ku80 protein is sufficient to shorten telomeres in MuSCs. To further demonstrate the involvement of Ku80 in telomere length modulation in vivo in more severe chronic injuries, we bred mdx mice with Ku80+/− mice. Interestingly, MuSCs in the mdx/Ku80+/− muscles have a pronounced shift toward shorter telomeres (Figure S5G), increased muscle damage (Figure S5H), and impaired muscle performance (Figure S5I). These data suggest that the mdx/Ku80+/− mice exhibit similar muscle defects as the ones seen in mdx/IKK2CAMuSC mice (Figure 5). Altogether, these results demonstrate that Ku80 is a major regulator of telomere length in myogenic cells, both in vitro and in vivo, either alone or in the context of dystrophy.
Ku80 dysregulation and telomere damage in human dystrophic MuSCs
We next investigated whether our mouse findings recapitulate what happens in the human dystrophic condition. We found that human DMD MuSCs express significantly higher levels of p-p65 (Figures 6A and 6B), and a larger portion is localized in the nucleus (Figure 6C), further supporting the notion of persistent elevated NF-κB signaling in dystrophic human MuSCs. Interestingly, human DMD MuSCs have significantly decreased Ku80 levels (Figures 6D and 6E), as shown by immunohistochemistry analysis on human muscles from DMD patients. Furthermore, in corroboration with the mouse data, we found shortened telomeres (Figure 6F), higher levels of DNA damage (Figures S6A and S6B), and increased numbers of TIFs (Figures 6G and 6H) in human diseased MuSCs compared to aged-matched healthy controls. Altogether, these striking human findings, which are consistent with the mouse results, provide further evidence that persistent NF-κB activation specifically in MuSCs leads to Ku80 dysregulation and predisposes these cells to premature telomere shortening and an increased number of TIFs that ultimately contribute to the severe dystrophic skeletal muscle phenotype.
Figure 6. Persistent NF-κB activation in human DMD MuSCs leads to Ku80 dysregulation and predisposes these cells to premature telomere shortening and increased number of TIFs.

(A) In-cell western representative image of phospho-p65 staining of human MuSCs. MuSCs from three healthy and three DMD patients 10–15 years old were analyzed with five technical replicates each. Green, phospho-p65 staining; red, Draq5 (nuclear) staining.
(B) Quantification shows nuclear accumulation of p-p65 in hMuSCs. The total signal intensity in the IR 800 channel (p-p65) was normalized to the total intensity of the Draq5 (nuclear channel) per sample. n = 3 samples/group. n = 6 wells per sample.
(C) Higher NF-κB activity in NCAM+ human MuSCs from young DMD patients, as shown by the p-p65 nuclear accumulation compared to age-matched control MuSCs.
(D) n = 3 samples/group. Representative Ku80 staining (green), Pax7 (red), and nuclei (DAPI, blue) in human healthy and DMD muscle sections (21–26 years old).
(E) Quantification shows reduced levels of Ku80 in dystrophic MuSCs compared to controls. n = 3 samples/group. n > 30 cells per sample.
(F) Reduced telomere length in human MuSCs, assessed by MuQ-FISH of telomere staining (insert, representative image). n = 3 samples/group. n > 80 cells per sample.
(G) Representative images of TIF assay in human MuSCs.
(H) Quantification analysis reveals increased number of TIFs in human MuSCs.
n = 3 samples/group. n > 60 cells per sample. Graphed data are displayed as mean ± SEM. Statistical analyses in (B), (E), and (H) were performed using unpaired Student’s t test with Welch’s correction. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
DISCUSSION
Previous work had revealed that MuSCs progressively become dysfunctional in dystrophy, and telomere shortening is a major hallmark of the disease (Dumont et al., 2015; Sacco et al., 2010; Tichy et al., 2017). Here, we provide evidence of the in vivo molecular basis for telomere shortening of stem cells under chronic injury conditions. These findings provide insights into the fundamental regulatory mechanisms of MuSC-specific telomere shortening under chronic injuries.
Indications of perturbation of NF-κB signaling exist in various muscle diseases, such as muscle atrophy conditions (Cai et al., 2004; Mourkioti et al., 2006; Mourkioti and Rosenthal, 2008), cancer cachexia (He et al., 2013), dystrophinopathies such as Limb-girdle muscular dystrophy type 2A (LGMD2A) (Baghdiguian et al., 1999), and DMD (Acharyya et al., 2007; Hammers et al., 2016; Messina et al., 2011). In addition to its role in regulating myogenesis (Mourkioti and Rosenthal, 2008), NF-κB is likely to modulate immune responses, muscle damage, and repair in DMD as well as in other muscle disorders. However, the survival effect of NF-κB in LGMD2A muscles, compared with its rather negative contribution in DMD, reflects the complexity of this pathway in chronic muscle diseases. Previous genetically modified animal models generated to interfere with NF-κB signaling only assessed NF-κB function regarding cancer cachexia (He et al., 2013) and aging (Oh et al., 2016; Proto et al., 2017).
By targeting the MuSC compartment in adult mice, we demonstrate that the inherent endogenous injuries observed in dystrophic mice induce a constant elevated NF-κB signaling response in MuSCs in both mdx and, most importantly, in human DMD MuSCs. Furthermore, we showed that persistent MuSC-specific NF-κB activation in injured muscles leads to telomere shortening and impairment of regeneration, as shown by the worsened histopathology of skeletal muscles during the progression of the chronic injury.
Additionally, we demonstrate that persistent MuSC-specific NF-κB activation in injured muscles drives telomere shortening via a mechanism that does not raise proliferation levels. Instead, we describe an NF-κB-dependent Ku80 dysregulation and increased DNA damage on telomeres in MuSCs from both dystrophic mice and human patients. While the effects of Ku80 in NHEJ are well documented (Fell and Schild-Poulter, 2015), its potential mechanistic involvement in telomere length alteration is less clear. Indeed, the absence of Ku80 has been associated with both telomere lengthening and shortening, depending on the cell line used; the organism (i.e., yeast, Drosophila, mouse, plant); or the context (embryonic development, cancer, or aging) (Boulton and Jackson, 1998; Chai et al., 2002; d’Adda di Fagagna et al., 2001; Didier et al., 2012; Espejel et al., 2004; Gallego et al., 2003; Indiviglio and Bertuch, 2009; Li et al., 2007; Melnikova et al., 2005). Furthermore, it is still unclear when Ku80 could function alone or as a heterodimer of both Ku80 and Ku70 subunits (Fell and Schild-Poulter, 2015). It was previously shown that loss of a single allele of Ku80 leads to dysfunction of muscle-derived cells and accelerated aging (Didier et al., 2012). In this study, we demonstrate that NF-κB regulates Ku80 expression in MuSCs in chronic muscle injuries and dystrophic muscles in both mouse and human. This NF-kB-dependent in vivo telomere regulatory mechanism is different from the modulation of telomeric proteins Tert or Rap1 that have been shown in cancer cells (Teo et al., 2010; Yin et al., 2000; Zuo et al., 2011). Additional work, to generate more targeted molecular tools for Kus and to investigate the exact role of Ku80 in the regulation of telomere integrity (Indiviglio and Bertuch, 2009), will be instrumental in order to identify other contributing molecular mechanisms and to develop concepts that could maintain MuSC telomere length under injured conditions.
It has been suggested that MuSCs could not keep up with the high regeneration demand under chronic injury conditions (Sacco et al., 2010). However, other studies have shown that MuSC numbers are elevated in mdx muscles in mice or DMD patients (Boldrin et al., 2015; Dumont et al., 2015; Kottlors and Kirschner, 2010). All these studies were dependent on prospective cell isolation methods, which rely on enzymatic digestion efficiency, using fluorescence-activated cell sorting (FACS) based on cell-surface markers (Tichy et al., 2017) or ex vivo methods to isolate single muscle fibers, in combination with Pax7 antibody staining (Sacco et al., 2010). Both methods result in cell loss due to either incomplete tissue digestion and/or cell death during these long procedures, and the cell loss is likely exacerbated in diseased muscles that are inherently more fibrotic and fragile. Additionally, only the intact/non-broken fibers are used for quantification and, therefore, single-fiber isolation selects for MuSC measurements in somewhat “healthier” fibers. To overcome these limitations and to evaluate the consequences of telomere shortening in MuSCs, we used the Pax7EGFP reporter mouse (Tichy et al., 2018) to study changes in the number of MuSCs during disease conditions. Our analysis revealed that there is a loss of MuSC over time in both the IKK2CAMuSC after repetitive injuries and the mdx/IKK2CAMuSC mice, which contributes to the muscle regeneration impairment seen in the context of chronic injuries as caused by the lack of dystrophin.
In summary, this study establishes a link between stem cell functional exhaustion and NF-κB-dependent telomere shortening. Our data suggest that maintaining telomere length of MuSCs could boost the regenerative ability of MuSCs in disease-related chronic injuries such as DMD. It was recently proposed that chronic inflammation can accelerate aging via exacerbation of telomere dysfunction in multiple tissues (Jurk et al., 2014). Therefore, the molecular mechanism presented here for DMD could also be a more generalized mechanism that lowers regenerative potential in other tissues with systemic chronic inflammation and/or accelerates normal and pathological aging.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Foteini Mourkioti (fmour@pennmedicine.upenn.edu)
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability statement
This study did not generate any unique datasets or code.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Animals
All animals were housed and bred in accordance with the University of Pennsylvania IACUC guidelines. Mice used in this study include C57BL/6 (Jackson Labs-JAX Stock #000664), IKK2 constitutively active mice (IKK2CA); breeding of JAX stock 008242 for Lox-stop-Lox IKK2CA and PaxERT2CRE (JAX Stock #017763) (Murphy et al., 2011), mdx4cv mice (JAX stock #002378), or mdx4cv bred to IKK2CA. NEMO floxed mice and genotyping protocols were a gift from Dr. Pasparakis. Ku80+/− mice were a gift from Dr. Hasty. For several experiments, Pax7EGFP mice were bred to the aforementioned mouse lines. Mice were genotyped according to established protocols using ear punches as starting material. To induce Cre expression, mice were injected with 250uL tamoxifen (Sigma; 20mg/mL in ethanol/corn oil in a 1/10 ratio) IP twice weekly for a total of 7 injections, starting at 6 to 8 weeks of age. For all experiments, mice of the same sex and similar age were compared. The age of mice for each experiment is reported in Figure Legends.
Chronic induced injury
Muscles were injured with 10 μg/mL notexin (Latoxan) weekly. The tibialis anterior (TA), gastrocnemius, and quadriceps muscles of one hind limb were injured with 10 μL (TA) or 20 μL (gastrocnemius and quadriceps) notexin. Muscles were collected post-injury at three days (for MuSC isolation) or at seven days (for staining with Hematoxylin and Eosin).
Human samples
Human samples were a gift from Dr. Sacco (Tichy et al., 2017). Human muscle biopsies from healthy and DMD patients were obtained from the lower extremity muscles during surgical procedures as part of the patient’s clinical care plan at Rady’s Children’s Hospital, San Diego. Written informed consent from patients, parent or guardian was obtained for all subjects. The protocol was approved by the University of California, San Diego Human Research Protectants Program and Institutional Review Board in accordance with the requirements of the Code of Federal Regulations on the Protection of Human Subjects. All materials were collected from de-identified males.
METHOD DETAILS
Muscle stem cell isolation
MuSCs were FACS-isolated as previously described (Tichy et al., 2017). Briefly mice were sacrificed, and the tibialis anterior, quadriceps and gastrocnemius muscles were dissected from both hind limbs. Muscle was finely minced and placed in a C tube (Miltenyi) containing 0.15% collagenase in 10 mL DMEM. Tubes were loaded into a MACS Dissociator (Miltenyi) and the manufacturer’s muscle-01 program was run. Tubes were incubated in at 37°C for 30 min, subjected to the muscle-01 program again, and incubated at 37°C for an additional 30min. Seventy-five microliters of 2% collagenase (Sigma-Aldrich) and 75 μL of 4.8 U/mL dispase (Roche) was added, and tubes were vortexed at maximum speed prior to a 30min incubation at 37°C. Cells were passed through a 21-gauge needle until all muscle was broken apart. The remaining cell slurry was filtered through a 40 μM cell strainer that was prewet with 10 mL of cold myoblast media (DMEM:F12; 15% FBS, 1X anti-anti) the strainer was rinsed with an additional 10 mL of cold myoblast media, and cells were pelleted at 300xg/4°C. Cells were incubated with 1mL 1X red cell lysis buffer (ThermoFisher) for 5 min at room temperature and 9 mL cold FACS buffer (2.5% goat serum, 2 mM EDTA, pH 8.0 in 1X PBS) was added. Cells were spun and resuspended in 1mL of FACS buffer containing biotin-conjugated antibodies raised against antigens CD45, CD31, CD11b, and Sca1 prior to an incubation on ice for 45 min. Cells were centrifuged and resuspended in 100 μL of FACS buffer containing antibodies CD34, and α7-integrin, as well as streptavidin-PE-Cy7. Cells were incubated in the dark for 90 min on ice, with agitation occurring every 30 min. FACS buffer was added up to 1 mL final volume, and cells pelleted and resuspended in FACS buffer, and the viability dye 7-aminoactinomycin D (7-AAD) was added (final concentration 4 mg/mL). Cells were placed in falcon tubes with cell strainers before collection by FACS. Murine MuSCs were identified as CD11b−/CD31−/CD45−/Sca1−/α7-Integrin+/CD34+. Primary human MuSCs were provided as described (Tichy et al., 2017). Briefly, human calf muscle biopsies were enzymatically dissociated in 0.2% collagenase (Sigma-Aldrich) and 0.02 units/mL dispase (Roche) for 45 min at 37°C, minced under a dissection microscope and incubated for an additional 45 min at 37°C. The resulting cell suspension was filtered through a 70 μm cell strainer and incubated with biotinylated anti-human CD45, CD11b and CD31. Samples were washed with FACS buffer and incubated with anti-NCAM/CD56 and Streptavidin APC-Cy7. Human MuSCs were CD11b−/CD31−/CD45−/NCAM+.
Flow cytometry
FACS-isolated MuSCs from age and sex-matched animals were fixed in 1.6% PFA/PBS for 10’ on ice, spun at 350xg at 4°C, and stored in cold 100% methanol (final concentration 90%) at −80°C until the staining commenced. MuSCs were further permeabilized in 1% Igepal CA-630/PBS, washed with PBS, and blocked with 1% BSA/PBS/0.1% Triton X-100, and stained with phospho-p65 antibody (Abcam, 1/500), or phospho-JNK (1/100, Cell Signaling), phospho-p38 (1/100, BD Biosciences), phospho-S6K (1/100, Cell Signaling), phospho-AKT (1/100, Cell Signaling), or Bcl2 (1/100, BD Biosciences) in 1%BSA/PBS/0.1% Triton X-100 overnight at 4°C in V-bottom 96-well plates. MuSCs were washed in BSA buffer, stained with goat anti-mouse or goat anti-rabbit IgG Alexa Fluor 488 antibody (Thermo, 1/500), and analyzed on an LSRII flow cytometer using the 488nm laser line 530/30 bandpass filter set. Data were analyzed with FlowJo. Unbiased analysis was performed with the investigators blinded to genotypes and/or conditions. Results were consistent and reproducible with different batches of Abs, using MuSC isolation from different experimental mice performed on different days. Antibody specificity was determined by several methods, including using unstained controls, no primary antibody-stained controls, and FMO controls.
Muscle isolation and sectioning
Tibialis Anterior muscles were dissected and fixed in 4% PFA/PBS and incubated overnight in 30% sucrose prior to being embedded in OCT medium (Richard Allen Scientific) and frozen in chilled 2-methylbutane. Cryosections were cut at a thickness of 10 μm and placed on Superfrost plus slides. Sections were stained with Hematoxylin and Eosin. More specifically, slides were stained with Hematoxylin for 1min, rinsed in water, and incubated with acidic alcohol (1% hydrochloric acid in 100% ethanol). After rinsing in water, slides were immersed in bluing, water, and 70% ethanol for 30sec each. Next, slides were exposed to eosin for 1min and gradually dehydrated with a graduated ethanol series. After a final incubation in xylene, coverslips were mounted with Cytoseal-xyl. Sections were imaged on a Nikon Ni widefield epifluorescence microscope. Muscle fiber areas were manually determined using ImageJ/Fiji.
Immunofluorescence staining
Isolated cells or tissue cryosections were permeabilized in 0.5% Triton X-100/PBS for 10min and washed with PBS. If Pax7 staining was required, sections were processed as in Tichy et al. (2018). Primary antibodies for phospho-p65 (1/100; Abcam), Ki67 (1/250; Abcam) and/or 53BP1 (1/1000; Novus Biologicals) were incubated overnight at 4°C in antibody dilution buffer (1% BSA/PBS/0.1% Triton X-100). Secondary antibodies used were: Alexa Fluor 488 and Alexa Fluor 647-conjugated goat anti-mouse or anti-rabbit staining (1/300–500 in antibody dilution buffer) for 1hr in the dark at room temperature. For quantifications in Figure 1, p-p65 expression represent nuclear signal intensities normalized to DAPI intensities, to account for any possible differences in cell cycle position or nuclear size. A negative staining control (with no primary antibody) was included to further demonstrate the specificity of the p-p65 staining. For human sections, 10um formalin-fixed paraffin-embedded skeletal muscle from young normal donors or DMD patients were deparaffinized and rehydrated by standard procedures. Samples were permeabilized with 0.5% Triton X100/PBS, underwent heat-mediated antigen retrieval with acidic citrate, and were blocked first with the avidin/biotin blocking kit (Vector Labs), then with 3% BSA/PBS/0.1% Triton X-100. Sections were stained with Ku80 antibody (Proteintech) and Pax7 (Santa Cruz) overnight at 4°C in 1% BSA/PBS/0.1% Triton X-100. Slides were washed with PBS and the Pax7 signal was amplified with a mouse-on mouse kit (Vector labs). Sections were stained with Alexa Fluor 488 Goat anti-Rabbit IgG and Alexa Fluor 647-conjugated streptavidin for 1hr. All slides washed with PBS and coverslips were mounted with prolong gold with DAPI. Sections were imaged with a Nikon Ni widefield epifluorescence microscope under the same conditions and intensities of Ku80 in Pax7 cells were normalized to controls.
Trichrome staining
For Masson’s trichrome staining, tissue cryosections were fixed with Bouin’s solution overnight, rinsed with water, and incubated with Weigert’s Hematoxylin staining reagent (Electron Microscopy Sciences) for 5 min. Slides were washed with water for 10 min and stained with a commercially available kit (Sigma Aldrich). Briefly, slides were first stained with Biebrich Scarlet-Acid Fuchsin Solution (Sigma Aldrich) for 5 min, rinsed in three changes of water, followed by staining with phosphotungstic/phosphomolybdic acid solution (Sigma Aldrich) for 10 min. Slides were then stained with aniline blue solution (Sigma Aldrich) for 5min to stain collagen in blue, differentiated with 1% glacial acetic acid for 2min, and then dehydrated with an ascending series of ethyl alcohol followed by xylene before they were mounted. Sections were imaged under the same conditions and intensities using a Nikon Ni widefield epifluorescence microscope with an objective lens magnification at 20×. Analysis of fibrotic area was conducted in Fiji software, using color deconvolution and thresholding function to define the areas of collagen and the muscle fiber. The fibrotic area percentage is calculated as (collagen area/muscle fiber area) × 100.
Telomere length analysis of FACS isolated MuSCs (MuQ-FISH)
Telomere length was measured using quantitative fluorescent in situ hybridization of Cy3-conjugated PNA telomere probe (Tel-C, PNA probe). MuSCs were isolated and processed for telomere length measurements as previously described (Tichy et al., 2017). MuSCs were imaged using a Nikon eclipse 90i wide-field epifluorescence microscope equipped with a Prior Proscan III motorized stage, a Photometrics Coolsnap HQ2 14-bit digital camera and a Nikon 100x/1.40 Plan Apo VC objective. Initially, control samples are imaged to determine each channel’s exposure time in a way that situates the intensities for all samples in the mid-intensity range. This control prevents overexposure or loss of signal detection with the selected exposure times during sample imaging. Once an optimal exposure time is defined, image acquisition is utilized at the same exposure settings for the telomere and DAPI or centromeric signals for imaging of individual experiments. For each experiment, the intensity settings are not changed between sample groups. Images are taken without binning. Imaging must take place in the same time window for each experiment, and combination of multiple experiments performed at different days is not recommended, due to the inherent caveat of fluorescent intensity measurements differing between experiments. Telomere length was assessed as the total sum or mean telomeric probe intensity divided by the sum intensity of either the DAPI or the centromeric signal. Unbiased analysis with the investigators blinded to genotypes and/or conditions was conducted using the ImageJ plug-in Telometer.
Telomere length analysis of tissues sections
PFA-fixed cryosections were permeabilized for 10min with 0.5% Triton X-100, washed with PBS, and underwent heat-mediated antigen retrieval with TET buffer (10mM Tris, pH 7.5, 1mM EDTA, pH 8.0, 0.05% Tween-20). Sections were incubated with Tel C-Alexa Fluor 647 and CENPB-Cy3 (PNA bio, 1/600) in QFISH cryo-buffer (15% ethylene carbonate, 20% dextran sulfate, 600mM NaCl, 0.1% citric acid-based antigen retrieval buffer). Probe in buffer was added to slides, covered with a coverslip, and QFISH was carried out using a Thermobrite programmable slide warmer (Leica): 10min prehybridization step at 67°C, followed by 90min hybridization at 42C. Afterward, slides were washed twice, 5min each in buffers with decreasing salt concentrations at 55°C (2X SSC/0.1% Tween 20, 1X SSC/0.1% Tween 20, 0.5X SSC/0.1% Tween 20, 0.25X SSC/0.1% Tween 20). Slides were rinsed with PBS, blocked with 1% BSA/0.1% Triton X-100/1X PBS for 1hr at room temperature. Antibody to VCAM (ThermoFisher, 1/100) was added to the blocking buffer and slides were incubated overnight at 4°C. The following day, slides were washed with PBS and stained with Alexa Fluor 488-conjugated Donkey anti-goat IgG antibody (ThermoFisher) in blocking buffer for 1hr at room temperature. Slides were washed with PBS and coverslips were mounted with Prolong gold plus DAPI. Cryosections were imaged with a Nikon Ni widefield epifluorescence microscope equipped with a 100x Plan Apo objective and Nikon elements software. Analysis of telomere length was assessed with the investigators blinded to genotypes and/or conditions in VCAM+ cells using Telometer. At least 40 cells per mouse were analyzed.
MuSC proliferation assays
Mice were injected with EdU (100mg/kg) IP, following the experimental design shown in Figure 3. MuSCs were plated overnight following sorting on laminin-coated 8-well chamber slides (~2500 cells) in DMEM/F12 supplemented with 15% FBS, 1X antibiotic/antimycotic, 1X Glutamax, 1X NEAA. Cells were fixed the following day in 3.7% formaldehyde/PBS for 15min and processed for EdU incorporation using the Click-iT EdU Alexa Fluor 594 Imaging kit (ThermoFisher), according to the manufacturer’s instructions.
Grip test
Mice were inverted on a cross-hatched metal platform and the time required for mice to release themselves from the platform was quantified. Three repetition measurements were recorded per mouse, giving the mouse 1hr rest in between measurements.
Creatine kinase assay
Blood was collected from mice by cardiac puncture and allowed to clot. After centrifugation for 10min at 600 ×g, serum was diluted 1/10 with PBS and assayed and analyzed using a commercially available kit (Sekisui Diagnostics) according to the manufacturer’s instructions.
Quantitative real time-PCR (qRT-PCR)
RNA was isolated from FACS-sorted MuSCs using a RNeasy Plus micro kit (QIAGEN), according to the manufacturer’s instructions. cDNA was generated with the Protoscript II First Strand cDNA synthesis kit (New England Biolabs). qRT-PCR was carried out on a Quantstudio 6 instrument (Applied Biosystems) using Taqman primers and Taqman Universal PCR Master Mix. Reactions were carried out in triplicate with at least three mice analyzed per experiment. Gene expression was determined using the ΔΔCt method. Primers purchased from Applied Biosystems and included IKBKB-FAM (Gene assay ID: Mm01222247_m1), CHUK-FAM (Mm00432529_m1), IKBKG-FAM (Mm00494927_m1), XRCC5-FAM (Mm00550142_m1), XRCC6-FAM (Mm00487458_m1), TERF2-FAM (Mm01253555_m1), TERF1-FAM (Mm00436928_m1), TERF2IP-FAM (Mm01243676_m1), TPP1-FAM (Mm00487016_m1), TINF2-FAM (Mm00461166_g1), Pot1B-FAM (Mm01278790_m1), Tert-FAM (Mm00436931_m1), and GAPDH-VIC (4352339e).
In silico analysis of putative p65 binding sites in promoters
The potential of the p65 transcription factor to bind to selected murine gene promoters was analyzed using the Lasagna tool (https://biogrid.engr.uconn.edu/lasagna_search/) with the following conditions: using the transfac model, search range of −2000 to 0, p less than or equal to 0.001. Displayed are the
Telomere-induced foci (TIF) assay
For chronic injury assessment, mice were injured in the Tibialis Anterior muscle with 10uL of 10ug/mL notexin weekly, for three times and harvested 3 days after the last injury. For dystrophic models, mice were injured once and harvested 3 days post-injury. 10 μm cryosections were stained as described in “telomere length analysis in tissue sections” above using a Tel-C Alexa Fluor 647-conjugated probe (PNA bio). After staining and processing, slides were blocked for 2 hours at room temperature with 3% BSA in PBS, followed by overnight incubation at 4°C with antibody to VCAM (1/100 of 1 μg/μL stock ThermoFisher) and 53BP1 (1/2000, Novus Biologicals). Slides were rinsed in PBS and incubated with donkey anti-goat Alexa fluor-conjugated secondary antibodies (1/300) for 1 hour at room temperature. Slides were washed with PBS three times before mounting coverslips with prolong gold with DAPI. For imaging, 1 μm slices were captured with a Zeiss Confocal 710 microscope with Zen software. TIFs were defined as 53BP1 foci overlapping or contacting telomere foci. For TIF assessment of human-derived MuSCs, cells were plated on collagen-coated chamber slides and stained with TelC-Alexa 647 PNA probe (according to Telomere Length Analysis of FACS Isolated MuSCs (MuQ-FISH) above). Slides were blocked with 3% BSA in PBS for 1 hour and stained with 53BP1 antibody (1/2000) overnight at 4°C, followed by staining with Alexa Fluor 488-conjugated secondary antibody for 1 hour at room temperature. Coverslips were mounted with fluoromount G with DAPI (Southern Biotech). Cells were imaged on a Nikon widefield epifluorescence microscope and quantitated for TIFs.
In-cell western
Human-derived MuSCs were plated in collagen-coated 96-well optical bottom plates in equal numbers, fixed in 4% PFA/PBS, permeabilized with 1% IGEPAL CA-630, blocked in 10% goat serum/1% BSA/PBS and stained with antibody against phospho-p65 serine 536 (1/100; Abcam) overnight at 4°C. Cells were washed with PBS and stained with Dylight 800 goat anti-rabbit (1/1,000; Rockland) and DNA was stained with DRAQ5 (1/5,000; Biolegend). Signal intensities were measured on an Odyssey Infrared Imaging System, with Image Studio software. Antibody intensity to nuclear intensity ratios were analyzed to normalize for cell content/growth differences.
Imaging using two-photon microscopy
To define MuSC number in their endogenous environment, Control (Pax7EGFP) and genetically modified mice bred to Pax7EGFP mice were injured 3X with notexin, starting around 3 months of age, and allowed to recover for ~10 weeks. TA muscles were collected, fixed in 4% PFA/PBS and rinsed in PBS. Muscles were mounted on a custom-made chamber and high-resolution serial sections were collected from the middle of the muscle, using a Leica SP* Confocal/Multiphoton Microscope system equipped with a Chameleon Vision II Sapphire laser. A 910 nm laser was focused through a 20x HCX APO L Lens. Emission light was collected with a FITC (BP 525/50) filter, and muscle fibers were marked via second-generation harmonics. Serial optical sections were collected every 1.5 μm, flattened, and normalized to volume scanned.
uCT imaging
Each mouse was scanned in vivo using a preclinical μCT system (vivaCT 40, Scanco Medical AG, Brüttisellen, Switzerland) at 38μm nominal voxel size. The scanner was used at 55kVp energy and 145μA intensity. Images were generated with an integration time of 200ms and 1 signal average. During the scans, mice were anesthetized using 1%–1.5% isoflurane in 1–1.5mL/min of oxygen. The mouse was immobilized using a customized holder to ensure minimal motion. Before each scan, a 2D scout view was used to select the scan region. The average scan time was approximately 50min.
Myoblast culture and shRNA knockdown
C2C12 myoblast cells were grown in DMEM high glucose supplemented with 10% fetal bovine serum, 1X antibiotic/antimycotic, 1X Glutamax, and 1X nonessential amino acids. Equal numbers of cells were plated at low density (3×104 cells/well) into 24 well plates and transfected with shRNA vectors using lipofectamine 3000, according to the manufacturer’s instructions. Vectors used were shEMPTY (pLKO1.puro empty; Addgene plasmid 8453), mouse XRCC5–1 (Sigma SHCLNG-NM_009533; TRCN000031295), mouse XRCC5–2 (Sigma SHCLNG-NM_009533; TRCN0000071044). Transfected cells were re-plated in 8 well chamber slides 24 hours post-transfection, cultured a further 24 hours, and either subjected to MuQ-FISH or stained with antibody to Ku80 (Santa Cruz, B4, Alexa Fluor 647-conjugated) and imaged. Data were generated from 3 independently transfected wells which were stained, imaged and analyzed at the same end points. Analysis of nuclear Ku80 expression was determined using a semi-automated protocol in ImageJ.
Functional measurements
Muscle contractility was tested at the Penn Muscle Institute Muscle Physiology Assessment Core of the University of Pennsylvania. Eccentrics (ECCs) were assessed on freshly isolated EDL muscles using an Aurora Mouse 1200A System equipped with Dynamic Muscle Control v.5.5 software. EDL muscles were maintained in constantly oxygenated Ringer’s solution (100mM NaCl, 4.7mM KCl, 3.4mM CaCl2, 1.2mM KH2PO4, 1.2mM MgSO4, 25mM HEPES and 5.5mM D-glucose) at 24 C. Briefly, a series of two ECCs was applied using a tetanic stimulation of 80Hz and stretching the muscle by 10% of L0 over 200ms. The force drop was calculated as the percent of the force at the initial ECC.
QUANTIFICATION AND STATISTICAL ANALYSIS
Dataset values are described in the figure legends and are presented as mean ± SEM (standard error of the mean). n = number of mice per genotype. n = numbers of cells analyzed per mouse. Specific details on mouse numbers, age and statistics can be found in the figure legends. Analyses were performed using GraphPad Prism 6 software. *p ≤ 0.05; **p ≤ 0.01, *** p < 0.001, **** p < 0.0001. All investigators were blinded when analyzing data.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-Alpha 7 Integrin Alexa Fluor 647, clone R2F2 | ablab.ca | Cat# 67-0010-05 |
| Alexa Fluor 488-conjugated mouse anti-phospho-p38 MAPK threonine 180/tyrosine 182 | BD Biosciences | Cat# 612594; RRID: AB_399877 |
| Alexa Fluor 488 Donkey anti-goat IgG | Thermo Fisher Scientific | Cat# A11055; RRID:AB_2534102 |
| Alexa Fluor 488 Goat anti-rabbit IgG | Thermo Fisher Scientific | Cat# A-11034; RRID: AB_2576217 |
| Alexa Fluor 488 Goat anti-mouse IgG | Thermo Fisher Scientific | Cat# A-11001; RRID: AB_2534069 |
| Alexa Fluor 555 Donkey anti-goat IgG | Thermo Fisher Scientific | Cat# A21432; RRID:AB_2535853 |
| Alexa Fluor 555 Donkey anti-rabbit IgG | Thermo Fisher Scientific | Cat# A31572; RRID:AB_162543 |
| Alexa Fluor 555 Goat anti-rabbit IgG | Thermo Fisher Scientific | Cat# A21428; RRID:AB_2535849 |
| Alexa Fluor 647-conjugated mouse anti-Ku86 | Santa Cruz | Cat# sc-515736 AF645 |
| Alexa Fluor 647 Goat-anti-mouse IgG | Thermo Fisher Scientific | Cat# A-21235; RRID: AB_2535804 |
| Dylight 800-conjugated Goat-anti-rabbit IgG | Rockland | Cat# 611-145-122; RRID:AB_1057618 |
| Goat anti-mouse Vcam1 | Fisher Scientific | Cat# PIPA547029 |
| Hamster anti-mouse Bcl2 | BD Biosciences | Cat# 556537 RRID:AB_396457 |
| Ki67 antibody | abcam | Cat# ab15580; RRID:AB_443209 |
| Mouse anti-human CD11b | Thermo Fisher Scientific | Cat# 14-0118-82; RRID:AB_467120 |
| Mouse anti-human CD31 | Thermo Fisher Scientific | Cat# 14-0319-82; RRID:AB_467204 |
| Mouse anti-human CD45 | Thermo Fisher Scientific | Cat# 14-0459-82; RRID:AB_467274 |
| Mouse anti-human NCAM | Thermo Fisher Scientific | Cat# 17-0567-42; RRID:AB_10597454 |
| Mouse anti-Pax7 | Santa Cruz | Cat# sc-81648; RRID:AB_2159836 |
| Mouse anti-phospho-SAPK/JNK threonine 183/tyrosine 185 | Cell Signaling | Cat# 9255; RRID: AB_2307321 |
| Rabbit anti-53bp1 | Novus Biologicals | Cat# NB100-304; RRID:AB_10003037 |
| Rabbit anti-Ku80 | Proteintech | Cat# 16389-1-AP; RRID:AB_2257509 |
| Rabbit anti-phospho-Akt serine 473 | Cell Signaling | Cat# 4058; RRID:AB_331168 |
| Rabbit anti-phospho-p65 serine 529 | abcam | Cat# ab97726; RRID:AB_10681170 |
| Rabbit anti-phospho-p65 serine 536 | abcam | Cat# ab86299; RRID:AB_1925243 |
| Rabbit anti-phospho-p65 serine 536 | abcam | Cat# ab131109; RRID:AB_11160495 |
| Rabbit anti-phospho-p70 s6 kinase threonine 421/serine 424 | Cell Signaling | Cat# 9204; RRID: AB_2265913 |
| Rat anti-CD11b-biotin, clone M1/70 | BD Biosciences | Cat# 553309; RRID:AB_394773 |
| Rat anti-CD31-biotin | Fisher Scientific | Cat# 13-0311-85; RRID:AB_466421 |
| Rat anti-mouse CD34-BV421, Clone RAM34 | BD Biosciences | Cat# 562608; RRID:AB_11154576 |
| Rat anti-CD45-biotin | BD Biosciences | Cat# 553078; RRID:AB_394608 |
| Rat anti-Ly-6A/E-biotin (Sca1), clone E13-161.7 | BD Biosciences | Cat# 553334; RRID:AB_394790 |
| Biological samples | ||
| Primary human MuSCs | From collaborator Tichy et al., 2017 | N/A |
| Human skeletal muscle paraffin sections | Obtained in-house and from US Biomax | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| 16% paraformaldehyde | Electron Microscopy Sciences | Cat# 15710 |
| 2-Mercaptoethanol | Bio-Rad | Cat#1610710 |
| 2-Methylbutane | Honeywell | Cat# M32631 |
| 4% paraformaldehyde | Thermo Scientific | Cat# J19943-K2 |
| 5-Ethynyl-2′-deoxyuridine (EdU) | abcam | Cat# ab146186 |
| 7-aminoactinomycin D (7-AAD) | Fisher Scientific | Cat# A1310 |
| Accumax | Fisher Scientific | Cat# SCR006 |
| Acetic Acid, glacial | Fisher Scientific | Cat# A38S |
| Alcoholic Eosin | American MasterTech Scientific | Cat# STE0157 |
| Antibiotic-antimycotic | GIBCO | Cat# 15240-062 |
| Antigen unmasking solution | Vector Labs | Cat# H-3300 |
| Avidin/Biotin blocking solution | Vector Labs | Cat# SP2001 |
| Bluing | American MasterTech Scientific | Cat# HXB00242E |
| Bouin’s solution | Sigma-Aldrich | Cat# HT10132 |
| Bovine Serum Albumin (Fraction V) | Gemini Bio-Products | Cat# 700-100P |
| Cenpb-Cy3 | PNA Bio | Cat# F3002 |
| Collagen | Sigma-Aldrich | Cat# C8919 |
| Collagenase type 1A | Sigma-Aldrich | Cat# C9891 |
| Corn Oil | Millipore-Sigma | Cat# C8267 |
| Cytoseal-Xyl | Thermo Scientific | Cat# 8312-4 |
| DAPI | Sigma-Aldrich | Cat# D9542 |
| DRAQ5 | Biolegend | Cat# 424101 |
| Dextran sulfate sodium salt | Sigma-Aldrich | Cat# D8906 |
| Dispase II | Roche | Cat# 04942078001 |
| DMEM high glucose | Corning | Cat# 10-017-CV |
| DMEM/F-12 | ThermoFisher Scientific | Cat#11330-032 |
| EDTA | Invitrogen | Cat#15575-020 |
| Ethanol, 200 proof | Decon Labs | Cat# 2701 |
| ESGRO-2i | EMD Millipore | Cat# ESG1121 |
| Ethylene Carbonate | Sigma-Aldrich | Cat# E26258 |
| Fetal Bovine Serum | Corning | Cat# MT35-010-CV |
| Fluoromount G plus DAPI | Southern Biotech | Cat# 0100-20 |
| Formaldehyde solution, 37% | Sigma-Aldrich | Cat# F1635 |
| Formamide | Sigma-Aldrich | Cat# 47670 |
| Gill’s Hematoxylin | Cancer Diagnostics | Cat# CM5951 |
| Glutamax | GIBCO | Cat# 35050-061 |
| Igepal CA-630 | Sigma-Aldrich | Cat# I8896 |
| Laminin | Sigma-Aldrich | Cat#11243217001 |
| Luria Broth, Miller | Sigma-Aldrich | Cat# L3152 |
| Maleic acid | Sigma-Aldrich | Cat# M0375 |
| MEM Nonessential amino acids | GIBCO | Cat# 11140-050 |
| Methanol | Fisher Scientific | Cat# A412-4 |
| Magnesium chloride | Sigma-Aldrich | Cat# M8266 |
| Mouse on mouse kit | Vector Labs | Cat# BMK-2202 |
| Normal Goat Serum | ThermoFisher Scientific | Cat# 16210-072 |
| Notexin | Accurate Chemical/Latoxan | Cat# TXL8104-100 |
| OCT | Thermo Scientific | Cat# 6502G |
| Phosphate buffered saline | Made in house | N/A |
| Prolong gold plus DAPI | ThermoFisher Scientific | Cat# P36935 |
| Red cell lysis buffer | Invitrogen | Cat# 50-112-9751 |
| Ringers solution | Made in house | N/A |
| RNase A | Fisher Scientific | Cat# AM9780 |
| SSC buffer, 20X | Corning | Cat# 46-020-CM |
| Streptavidin Alexa Fluor 488 | Biolegend | Cat# 405235 |
| Streptavidin APC-Cy7 | BD Biosciences | Cat# 554063 |
| Streptavidin PE-Cy7 | Biolegend | Cat# 405206 |
| Tamoxifen | Sigma-Aldrich | Cat# T5648 |
| Taqman universal 2x master mix | Applied Biosystems | Cat# 4304437 |
| TelC-Alexa Fluor 647 | PNA Bio | Cat# F1013 |
| TelC-Cy3 | PNA Bio | Cat# F1002 |
| Telomere blocking buffer | Millipore-Sigma | Cat# 11096176001 |
| Triton X-100 | Fisher Scientific | Cat# BP151 |
| Tris-Cl | Quality Biological | Cat#351-006-131 |
| Trypsin-EDTA | GIBCO | Cat# 25200-056 |
| Tween 20 | Fisher Scientific | Cat# BP337 |
| Weigert’s Iron Hematoxylin A | Electron Microscopy Sciences | Cat# 26386-02 |
| Weigert’s Iron Hematoxylin B | Electron Microscopy Sciences | Cat# 26386-03 |
| Xylene | Fisher Scientific | Cat# X5-4 |
| Critical commercial assays | ||
| Click-iT EdU Alexa Fluor 594 Imaging Kit | Life Technologies | Cat# C10339 |
| Creatine Kinase Assay | Sekisui Diagnostics | Cat# 326-10 |
| Protoscript II cDNA First Strand Synthesis Kit | New England Biolabs | Cat# E6560S |
| Qiafilter Plasmid Midi Kit | QIAGEN | Cat# 12243 |
| Rneasy Plus Micro Kit | QIAGEN | Cat# 74034 |
| Trichrome Staining Kit | Sigma-Aldrich | Cat# HT15-1KT |
| Experimental models: Cell lines | ||
| Human MuSCs, primary (NCAM+/CD31−/CD45−/CD11b−) | Collaborator: (Tichy et al., 2017) | N/A |
| Mouse C2C12 Cells | ATCC | Cat# CRL-1772; RRID:CVCL_0188 |
| Mouse Embryonic Stem Cells (W4) | University of Pennsylvania iPS cell core | RRID:CVCL_Y634 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | Jackson Labs | Stock# 000664; RRID:IMSR_JAX:000664 |
| Mouse: IKK2CA: B6.Cg-Gt(ROSA)26Sortm4(Ikbkb)Rsky/J | Jackson Labs | Stock # 008242; RRID:IMSR_JAX:008242 |
| Mouse: Ku80+/− | Collaborator: Zhu et al., 1996 | N/A |
| Mouse: mdx4cv: B6Ros.Cg-Dmdmdx-4Cv/J | Jackson Labs | Stock# 002378; RRID:IMSR_JAX:002378 |
| Mouse: NEMOKOFlox | Described in Schmidt-Supprian et al., 2000 | N/A |
| Mouse: Pax7EGFP | Generated In-house Tichy et al., 2018 | N/A |
| Mouse: Pax7ERT2Cre: B6.Cg-Pax7tm1(cre/ERT2)Gaka/J | Jackson Labs | Stock # 017763; RRID:IMSR_JAX:017763 |
| Oligonucleotides | ||
| CHUK- FAM | Applied Biosystems | Assay ID# Mm00432529_m1 |
| Gapdh-VIC-MGB | ThermoFisher Scientific | Cat# 4352339e |
| IKBKB -FAM | Applied Biosystems | Assay ID# Mm01222247_m1 |
| IKBKG-FAM | Applied Biosystems | Assay ID# Mm00494927_m1 |
| POT1B-FAM | Applied Biosystems | Assay ID# Mm01278790_m1 |
| TERF1-FAM | Applied Biosystems | Assay ID# Mm00436928_m1 |
| TERF2-FAM | Applied Biosystems | Assay ID# Mm01253555_m1 |
| TERF2IP-FAM | Applied Biosystems | Assay ID# Mm01243676_m1 |
| TERT-FAM | Applied Biosystems | Assay ID# Mm00436931_m1 |
| TINF2-FAM | Applied Biosystems | Assay ID# Mm00461166_g1 |
| TPP1-FAM | Applied Biosystems | Assay ID# Mm00487016_m1 |
| XRCC5-FAM | Applied Biosystems | Assay ID# Mm00550142_m1 |
| XRCC6-FAM | Applied Biosystems | Assay ID# Mm00487458_m1 |
| Recombinant DNA | ||
| shEmpty (pLKO.1 puro) | Addgene | RRID: Addgene_8453 |
| shXRCC5-1 | Sigma-Aldrich | Cat#:SHCLNG-NM_00953; ID#: TRCN0000312925 |
| shXRCC5-2 | Sigma-Aldrich | Cat#:SHCLNG-NM_00953; ID#: TRCN0000071044 |
| Software and algorithms | ||
| Fiji | Open Source | https://imagej.net/Fiji |
| Flowjo v. 10.7.1 | Licensed Software | https://www.flowjo.com |
| Graphpad Prism v. 6.0h | Licensed Software | https://www.graphpad.com |
| ImageJ | Open Source | https://imagej.net/Welcome |
| Image Studio | Licensed Software | https://www.licor.com/bio/image-studio/ |
| Lasagna | Open Source | https://biogrid-lasagna.engr.uconn.edu/lasagna_search/index.php |
| Leica Application Suite X | Licensed Software | https://www.leica-microsystems.com/products/microscope-software/p/leica-las-x-ls/ |
| NIS-Elements | Licensed Software | https://www.microscope.healthcare.nikon.com/products/software/nis-elements |
| Odyssey Infared Imaging Software, v. 3.0.30 | Licensed Software | https://www.licor.com |
| Telometer ImageJ Plugin | Open Source | https://demarzolab.pathology.jhmi.edu/telometer/ |
| Zeiss Zen Imaging Software | Licensed Software | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html |
| Other | ||
| 40 μm cell strainers | VWR | Cat# 10199-654 |
| 70 μm cell strainers | Thermo Fisher Scientific | Cat# 08-771-2 |
| 96 well plate, black, optical flat bottom | Thermo Fisher Scientific | Cat# 4311971 |
| 96 well plate, v bottom | Greiner Bio-One | Cat# 651160 |
| C-tubes | Miltenyi | Cat# 130-096-334 |
| Falcon tube with cell strainer cap | Fisher Scientific | Cat# 0877123 |
| Insulin Syringe | Exel International | Cat# 26028 |
| Lipofectamine 3000 | Thermo Fisher Scientific | Cat# L3000008 |
| Nunc Lab-Tek II 8-well chamber slides | Thermo Fisher Scientific | Cat# 125658 |
| Superfrost plus microscope slides | Thermo Fisher Scientific | Cat# 1255015 |
Highlights.
NF-κB is dysregulated in MuSCs of dystrophic mice and DMD patients
Persistent NF-κB activation in MuSCs leads to telomere shortening
Unrestrained NF-κB signaling in MuSCs does not alter proliferation
NF-κB activation in MuSCs in chronic injuries leads to Ku80 dysregulation
ACKNOWLEDGMENTS
We thank A. Sacco for providing the human samples, M. Pasparakis for providing the NEMOfl/fl mice, and W.J. Tseng and S. Liu (PCMD Imaging Core) for assistance with the μCT experiments. We would like to thank H. Papaioannou, A. Scaramella, I. Paez, Y. Lee, and G. Wang for unbiased validation and analysis assistance. The authors were supported by startup funds from the Perelman School of Medicine, the McCabe Award, the NIH Pilot Grant (P30 AR069619), NASA (18-FG_ind_2-0022), and the NIH (R01HL146662) to F.M.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109098.
REFERENCES
- Acharyya S, Villalta SA, Bakkar N, Bupha-Intr T, Janssen PM, Carathers M, Li ZW, Beg AA, Ghosh S, Sahenk Z, et al. (2007). Interplay of IKK/NF-kappaB signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophy. J. Clin. Invest. 117, 889–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baghdiguian S, Martin M, Richard I, Pons F, Astier C, Bourg N, Hay RT, Chemaly R, Halaby G, Loiselet J, et al. (1999). Calpain 3 deficiency is associated with myonuclear apoptosis and profound perturbation of the IkappaB alpha/NF-kappaB pathway in limb-girdle muscular dystrophy type 2A. Nat. Med. 5, 503–511. [DOI] [PubMed] [Google Scholar]
- Boldrin L, Zammit PS, and Morgan JE (2015). Satellite cells from dystrophic muscle retain regenerative capacity. Stem Cell Res. (Amst.) 14, 20–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulton SJ, and Jackson SP (1998). Components of the Ku-dependent non-homologous end-joining pathway are involved in telomeric length maintenance and telomeric silencing. EMBO J. 17, 1819–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Frantz JD, Tawa NE Jr., Melendez PA, Oh BC, Lidov HG, Hasselgren PO, Frontera WR, Lee J, Glass DJ, and Shoelson SE (2004). IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell 119, 285–298. [DOI] [PubMed] [Google Scholar]
- Calado DP, Zhang B, Srinivasan L, Sasaki Y, Seagal J, Unitt C, Rodig S, Kutok J, Tarakhovsky A, Schmidt-Supprian M, and Rajewsky K (2010). Constitutive canonical NF-κB activation cooperates with disruption of BLIMP1 in the pathogenesis of activated B cell-like diffuse large cell lymphoma. Cancer Cell 18, 580–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai W, Ford LP, Lenertz L, Wright WE, and Shay JW (2002). Human Ku70/80 associates physically with telomerase through interaction with hTERT. J. Biol. Chem. 277, 47242–47247. [DOI] [PubMed] [Google Scholar]
- Chazaud B, Sonnet C, Lafuste P, Bassez G, Rimaniol AC, Poron F, Authier FJ, Dreyfus PA, and Gherardi RK (2003). Satellite cells attract monocytes and use macrophages as a support to escape apoptosis and enhance muscle growth. J. Cell Biol. 163, 1133–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d’Adda di Fagagna F, Hande MP, Tong WM, Roth D, Lansdorp PM, Wang ZQ, and Jackson SP (2001). Effects of DNA nonhomologous end-joining factors on telomere length and chromosomal stability in mammalian cells. Curr. Biol. 11, 1192–1196. [DOI] [PubMed] [Google Scholar]
- de Lange T (2005). Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 19, 2100–2110. [DOI] [PubMed] [Google Scholar]
- Didier N, Hourdé C, Amthor H, Marazzi G, and Sassoon D (2012). Loss of a single allele for Ku80 leads to progenitor dysfunction and accelerated aging in skeletal muscle. EMBO Mol. Med. 4, 910–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Jimi E, Zhong H, Hayden MS, and Ghosh S (2008). Repression of gene expression by unphosphorylated NF-kappaB p65 through epigenetic mechanisms. Genes Dev. 22, 1159–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont NA, Wang YX, von Maltzahn J, Pasut A, Bentzinger CF, Brun CE, and Rudnicki MA (2015). Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat. Med. 21, 1455–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery AE (2002). Muscular dystrophy into the new millennium. Neuromuscul. Disord. 12, 343–349. [DOI] [PubMed] [Google Scholar]
- Espejel S, Klatt P, Ménissier-de Murcia J, Martín-Caballero J, Flores JM, Taccioli G, de Murcia G, and Blasco MA (2004). Impact of telomerase ablation on organismal viability, aging, and tumorigenesis in mice lacking the DNA repair proteins PARP-1, Ku86, or DNA-PKcs. J. Cell Biol. 167, 627–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fell VL, and Schild-Poulter C (2015). The Ku heterodimer: function in DNA repair and beyond. Mutat. Res. Rev. Mutat. Res. 763, 15–29. [DOI] [PubMed] [Google Scholar]
- Frenette J, Cai B, and Tidball JG (2000). Complement activation promotes muscle inflammation during modified muscle use. Am. J. Pathol. 156, 2103–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallego ME, Jalut N, and White CI (2003). Telomerase dependence of telomere lengthening in Ku80 mutant Arabidopsis. Plant Cell 15, 782–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammers DW, Sleeper MM, Forbes SC, Coker CC, Jirousek MR, Zimmer M, Walter GA, and Sweeney HL (2016). Disease-modifying effects of orally bioavailable NF-κB inhibitors in dystrophin-deficient muscle. JCI Insight 1, e90341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden MS, and Ghosh S (2004). Signaling to NF-kappaB. Genes Dev. 18, 2195–2224. [DOI] [PubMed] [Google Scholar]
- He WA, Berardi E, Cardillo VM, Acharyya S, Aulino P, Thomas-Ahner J, Wang J, Bloomston M, Muscarella P, Nau P, et al. (2013). NF-κB-mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J. Clin. Invest. 123, 4821–4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman EP, Brown RH Jr., and Kunkel LM (1987). Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51, 919–928. [DOI] [PubMed] [Google Scholar]
- Hsu HL, Gilley D, Blackburn EH, and Chen DJ (1999). Ku is associated with the telomere in mammals. Proc. Natl. Acad. Sci. USA 96, 12454–12458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu HL, Gilley D, Galande SA, Hande MP, Allen B, Kim SH, Li GC, Campisi J, Kohwi-Shigematsu T, and Chen DJ (2000). Ku acts in a unique way at the mammalian telomere to prevent end joining. Genes Dev. 14, 2807–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im WB, Phelps SF, Copen EH, Adams EG, Slightom JL, and Chamberlain JS (1996). Differential expression of dystrophin isoforms in strains of mdx mice with different mutations. Hum. Mol. Genet. 5, 1149–1153. [DOI] [PubMed] [Google Scholar]
- Indiviglio SM, and Bertuch AA (2009). Ku’s essential role in keeping telomeres intact. Proc. Natl. Acad. Sci. USA 106, 12217–12218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurk D, Wilson C, Passos JF, Oakley F, Correia-Melo C, Greaves L, Saretzki G, Fox C, Lawless C, Anderson R, et al. (2014). Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat. Commun. 2, 4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike M, and Koike A (2008). Accumulation of Ku80 proteins at DNA double-strand breaks in living cells. Exp. Cell Res. 314, 1061–1070. [DOI] [PubMed] [Google Scholar]
- Kottlors M, and Kirschner J (2010). Elevated satellite cell number in Duchenne muscular dystrophy. Cell Tissue Res. 340, 541–548. [DOI] [PubMed] [Google Scholar]
- Lee HW, Blasco MA, Gottlieb GJ, Horner JW 2nd, Greider CW, and DePinho RA (1998). Essential role of mouse telomerase in highly proliferative organs. Nature 392, 569–574. [DOI] [PubMed] [Google Scholar]
- Li H, Vogel H, Holcomb VB, Gu Y, and Hasty P (2007). Deletion of Ku70, Ku80, or both causes early aging without substantially increased cancer. Mol. Cell. Biol. 27, 8205–8214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Z, Bozzella M, Seluanov A, and Gorbunova V (2008). Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair (Amst.) 7, 1765–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnikova L, Biessmann H, and Georgiev P (2005). The Ku protein complex is involved in length regulation of Drosophila telomeres. Genetics 170, 221–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messina S, Vita GL, Aguennouz M, Sframeli M, Romeo S, Rodolico C, and Vita G (2011). Activation of NF-kappaB pathway in Duchenne muscular dystrophy: relation to age. Acta Myol. 30, 16–23. [PMC free article] [PubMed] [Google Scholar]
- Morgan JE, and Zammit PS (2010). Direct effects of the pathogenic mutation on satellite cell function in muscular dystrophy. Exp. Cell Res. 316, 3100–3108. [DOI] [PubMed] [Google Scholar]
- Mourkioti F, and Rosenthal N (2008). NF-kappaB signaling in skeletal muscle: prospects for intervention in muscle diseases. J. Mol. Med. (Berl.) 86, 747–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourkioti F, Kratsios P, Luedde T, Song YH, Delafontaine P, Adami R, Parente V, Bottinelli R, Pasparakis M, and Rosenthal N (2006). Targeted ablation of IKK2 improves skeletal muscle strength, maintains mass, and promotes regeneration. J. Clin. Invest. 116, 2945–2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourkioti F, Kustan J, Kraft P, Day JW, Zhao MM, Kost-Alimova M, Protopopov A, DePinho RA, Bernstein D, Meeker AK, and Blau HM (2013). Role of telomere dysfunction in cardiac failure in Duchenne muscular dystrophy. Nat. Cell Biol. 15, 895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MM, Lawson JA, Mathew SJ, Hutcheson DA, and Kardon G (2011). Satellite cells, connective tissue fibroblasts and their interactions are crucial for muscle regeneration. Development 138, 3625–3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda T, Shimizu N, Yonenobu K, Ono K, Nabeshima T, and Kyoh S (1993). Longitudinal study of spinal deformity in Duchenne muscular dystrophy. J. Pediatr. Orthop. 13, 478–488. [DOI] [PubMed] [Google Scholar]
- Oh J, Sinha I, Tan KY, Rosner B, Dreyfuss JM, Gjata O, Tran P, Shoelson SE, and Wagers AJ (2016). Age-associated NF-κB signaling in myofibers alters the satellite cell niche and re-strains muscle stem cell function. Aging (Albany NY) 8, 2871–2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proto JD, Lu A, Dorronsoro A, Scibetta A, Robbins PD, Niedernhofer LJ, and Huard J (2017). Inhibition of NF-κB improves the stress resistance and myogenic differentiation of MDSPCs isolated from naturally aged mice. PLoS ONE 12, e0179270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacco A, Mourkioti F, Tran R, Choi J, Llewellyn M, Kraft P, Shkreli M, Delp S, Pomerantz JH, Artandi SE, and Blau HM (2010). Short telomeres and stem cell exhaustion model Duchenne muscular dystrophy in mdx/mTR mice. Cell 143, 1059–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Derudder E, Hobeika E, Pelanda R, Reth M, Rajewsky K, and Schmidt-Supprian M (2006). Canonical NF-kappaB activity, dispensable for B cell development, replaces BAFF-receptor signals and promotes B cell proliferation upon activation. Immunity 24, 729–739. [DOI] [PubMed] [Google Scholar]
- Schmidt-Supprian M, Bloch W, Courtois G, Addicks K, Israë l A, Rajewsky K, and Pasparakis M (2000). NEMO/IKK gamma-deficient mice model incontinentia pigmenti. Mol. Cell 5, 981–992. [DOI] [PubMed] [Google Scholar]
- Takai H, Smogorzewska A, and de Lange T (2003). DNA damage foci at dysfunctional telomeres. Curr. Biol. 13, 1549–1556. [DOI] [PubMed] [Google Scholar]
- Teo H, Ghosh S, Luesch H, Ghosh A, Wong ET, Malik N, Orth A, de Jesus P, Perry AS, Oliver JD, et al. (2010). Telomere-independent Rap1 is an IKK adaptor and regulates NF-kappaB-dependent gene expression. Nat. Cell Biol. 12, 758–767. [DOI] [PubMed] [Google Scholar]
- Tichy ED, Sidibe DK, Tierney MT, Stec MJ, Sharifi-Sanjani M, Hosalkar H, Mubarak S, Johnson FB, Sacco A, and Mourkioti F (2017). Single Stem Cell Imaging and Analysis Reveals Telomere Length Differences in Diseased Human and Mouse Skeletal Muscles. Stem Cell Reports 9, 1328–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tichy ED, Sidibe DK, Greer CD, Oyster NM, Rompolas P, Rosenthal NA, Blau HM, and Mourkioti F (2018). A robust Pax7EGFP mouse that enables the visualization of dynamic behaviors of muscle stem cells. Skelet. Muscle 8, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidball JG (2005). Inflammatory processes in muscle injury and repair. Am. J. Physiol. Regul. Integr. Comp. Physiol 288, R345–R353. [DOI] [PubMed] [Google Scholar]
- Tidball JG, and Wehling-Henricks M (2015). Shifts in macrophage cytokine production drive muscle fibrosis. Nat. Med. 21, 665–666. [DOI] [PubMed] [Google Scholar]
- Wilkins KE, and Gibson DA (1976). The patterns of spinal deformity in Duchenne muscular dystrophy. J. Bone Joint Surg. Am. 58, 24–32. [PubMed] [Google Scholar]
- Yin L, Hubbard AK, and Giardina C (2000). NF-kappa B regulates transcription of the mouse telomerase catalytic subunit. J. Biol. Chem. 275, 36671–36675. [DOI] [PubMed] [Google Scholar]
- Zatz M, Rapaport D, Vainzof M, Passos-Bueno MR, Bortolini ER, Pavanello, Rde C, and Peres CA (1991). Serum creatine-kinase (CK) and pyruvate-kinase (PK) activities in Duchenne (DMD) as compared with Becker (BMD) muscular dystrophy. J. Neurol. Sci. 102, 190–196. [DOI] [PubMed] [Google Scholar]
- Zhu C, Bogue MA, Lim DS, Hasty P, and Roth DB (1996). Ku86-deficient mice exhibit severe combined immunodeficiency and defective processing of V(D)J recombination intermediates. Cell 86, 379–389. [DOI] [PubMed] [Google Scholar]
- Zuo QP, Liu SK, Li ZJ, Li B, Zhou YL, Guo R, and Huang LH (2011). NF-kappaB p65 modulates the telomerase reverse transcriptase in the HepG2 hepatoma cell line. Eur. J. Pharmacol. 672, 113–120. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.