

Abstract
In eukaryotic cells, mitochondria are closely tethered to the endoplasmic reticulum (ER) at sites called mitochondria‐associated ER membranes (MAMs). Ca2+ ion and phospholipid transfer occurs at MAMs to support diverse cellular functions. Unlike those in yeast, the protein complexes involved in phospholipid transfer at MAMs in humans have not been identified. Here, we determine the crystal structure of the tetratricopeptide repeat domain of PTPIP51 (PTPIP51_TPR), a mitochondrial protein that interacts with the ER‐anchored VAPB protein at MAMs. The structure of PTPIP51_TPR shows an archetypal TPR fold, and an electron density map corresponding to an unidentified lipid‐like molecule probably derived from the protein expression host is found in the structure. We reveal functions of PTPIP51 in phospholipid binding/transfer, particularly of phosphatidic acid, in vitro. Depletion of PTPIP51 in cells reduces the mitochondrial cardiolipin level. Additionally, we confirm that the PTPIP51–VAPB interaction is mediated by the FFAT‐like motif of PTPIP51 and the MSP domain of VAPB. Our findings suggest that PTPIP51 is a phospholipid transfer protein with a MAM‐tethering function.
Keywords: endoplasmic reticulum, MAM, mitochondria, phospholipid, PTPIP51
Subject Categories: Membrane & Intracellular Transport, , Structural Biology
The crystal structure and biochemical analyses of PTPIP51, a mitochondrial protein localized at the mitochondria‐associated ER membrane (MAM), revealed its phospholipid binding and transfer activity.
Introduction
One of the remarkable features of eukaryotic cells is their partitioning into individual organelles with specific functions. Since some essential biochemical pathways function across multiple organelles, interorganelle communication through vesicular or non‐vesicular trafficking to translocate various cellular materials in cells is required (Gatta & Levine, 2017). In particular, translocation of many important cellular materials occurs at interorganelle contact sites and is closely related to cellular physiology and many human diseases (Bittremieux et al, 2016; Filadi et al, 2017; Gomez‐Suaga et al, 2018). Among the various organelle contact sites, the mitochondria–endoplasmic reticulum (ER) interaction site is one of the best characterized (Rowland & Voeltz, 2012; Phillips & Voeltz, 2016). Approximately 5–20% of the mitochondrial membrane is closely attached to the ER membrane with a gap of 10–30 nm; these membranes are named mitochondria‐associated ER membranes (MAMs) (Rizzuto et al, 1998; Rowland & Voeltz, 2012; Stoica et al, 2014; Paillusson et al, 2016; Hirabayashi et al, 2017). The formation of MAMs is mediated by several membrane‐anchored MAM‐tethering protein complexes. The vesicle‐associated membrane protein‐associated protein B (VAPB)–protein tyrosine phosphatase‐interacting protein 51 (PTPIP51), oxysterol‐binding protein‐related protein 5/8 (ORP5/8)–PTPIP51, B‐cell receptor‐associated protein 31 (BAP31)–mitochondrial fission 1 protein (FIS1), inositol 1,4,5‐trisphosphate receptor (IP3R)–glucose‐regulated protein 75 (GRP75)–voltage‐dependent anion channel (VDAC)–mitochondrial calcium uniporter (MCU), mitofusin (MFN2)–MFN1 (or MFN2), and ribosome‐binding protein (RRBP1)–synaptojanin 2‐binding protein (SYNJ2BP) complexes have been discovered in mammalian cells and have been studied (Gomez‐Suaga et al, 2018).
MAMs are intimately related to various cellular processes, including ER stress, mitochondrial dynamics, inflammation, autophagy, and apoptosis (Filadi et al, 2017). These functions mainly result from the exchange of cellular materials, such as Ca2+ ions and phospholipids (Rizzuto et al, 1998; Paillusson et al, 2016; Filadi et al, 2017). In detail, Ca2+ can be transferred from the ER, which functions as a Ca2+ reservoir in cells, to mitochondria through the IP3R–GRP75–VDAC Ca2+ uptake/release channel complex (Bittremieux et al, 2016; Phillips & Voeltz, 2016). These supplied Ca2+ ions can stimulate mitochondrial bioenergetic reactions, including NADH formation, respiratory chain activity, and the activity of some enzymes in the TCA cycle, which favors cell survival (Filadi et al, 2017). However, Ca2+ overload has opposite physiological effects, such as the induction of mitochondrial depolarization and release of cytochrome c, which induces apoptosis. Phospholipids are another major cellular material transferred at MAMs. While many phospholipids are usually synthesized in the ER membrane, some phospholipids, such as phosphatidylethanolamine (PE) and cardiolipin (CL), are synthesized in the inner mitochondrial membrane (IMM) after the transport of precursor phospholipids from other cellular organelles (Holthuis & Menon, 2014; Tamura et al, 2014; Tatsuta et al, 2014; Muallem et al, 2017). For example, the pathways by which PE and phosphatidylcholine (PC), major phospholipids in most cellular organelles, are synthesized function across the ER and mitochondria (Rowland & Voeltz, 2012; Tamura et al, 2014). Phosphatidic acid (PA) is converted to phosphatidylserine (PS) via cytidine diphosphate diacylglycerol (CDP‐DAG) as an intermediate at the ER membrane. The produced PS molecules are transferred to mitochondria because the enzyme PSD1, which catalyzes the conversion of PS to PE, localizes at the IMM (Tatsuta et al, 2014). The produced PE molecules move back to the ER membrane, and PC molecules are synthesized from PE. For synthesis of these phospholipids, each phospholipid must be properly transferred with the aid of lipid transfer proteins. In yeast cells, the ER–mitochondria encounter structure (ERMES) complex, which is composed of several synaptotagmin‐like mitochondrial lipid‐binding protein (SMP) domain‐containing proteins, interacts with various glycerophospholipids and governs phospholipid transportation at MAMs (Kornmann et al, 2009; Tatsuta et al, 2014; AhYoung et al, 2015). However, the protein complex in mammalian cells that corresponds to the ERMES complex has not yet been identified. Instead, phospholipid trafficking in mammalian cells may be mediated by different kinds of proteins. For example, MFN2 binds and transfers PS, and STARD7 transfers PC (Horibata & Sugimoto, 2010; Horibata et al, 2017; Hernandez‐Alvarez et al, 2019). Two oxysterol‐binding proteins, ORP5 and ORP8, are also localized to MAMs and related to the exchange of PS (Rochin et al, 2020).
Among the components of MAM‐tethering protein complexes, PTPIP51 (also called RMDN3) is a mitochondrial membrane‐anchored protein that interacts with ER membrane‐bound VAPB (De Vos et al, 2012; Stoica et al, 2014). PTPIP51 was originally identified as a cellular protein that regulates the differentiation, apoptosis, proliferation, and mitosis of cells by forming protein complexes with PTP1B or 14‐3‐3 proteins (Brobeil et al, 2017). Previous studies have suggested that the PTPIP51−VAPB association contributes to MAM formation and the regulation of Ca2+ homeostasis and autophagy (De Vos et al, 2012; Gomez‐Suaga et al, 2017), and disruption of this complex by TDP‐43 or GSK‐3β modulates cellular Ca2+ homeostasis and is related to neurodegenerative disease (Stoica et al, 2014; Stoica et al, 2016). Furthermore, PTPIP51 interacts with ORP5/8, suggesting a functional link to lipid transfer (Galmes et al, 2016). However, the exact biochemical functions of PTPIP51 beyond MAM tethering have not yet been elucidated. Here, we determined the crystal structure of the C‐terminal tetratricopeptide repeat (TPR) domain of PTPIP51 (PTPIP51_TPR), which is the largest globular domain in PTPIP51, to gain molecular and functional insights into MAM‐localized PTPIP51. Although the TPR domain has been suggested to be a protein–protein interaction module, structural analysis of PTPIP51_TPR provided evidence of a lipid binding function. Based on this finding, we revealed the phospholipid binding and transfer activity of PTPIP51. These results suggest that the PTPIP51−VAPB complex is a functional human counterpart of the yeast ERMES complex.
Results and Discussion
Structure of the PTPIP51 TPR domain
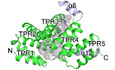
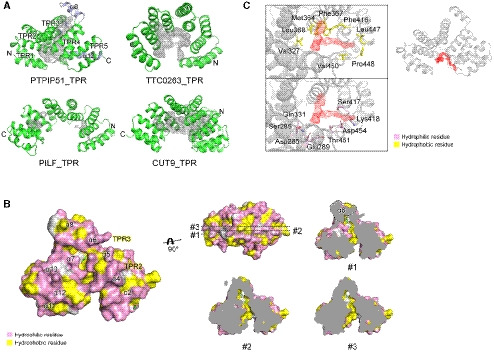
To understand the molecular functions of PTPIP51, we aimed to study the structure of PTPIP51 and successfully crystallized its C‐terminal domain (residues 236–470, Appendix Fig S1). The crystallized domain was predicted to be a TPR (PTPIP51_TPR) domain (Stenzinger et al, 2009). The crystal structure of PTPIP51_TPR was determined at 1.45 Å resolution by the single‐wavelength anomalous dispersion (SAD) method (Fig 1A; Appendix Table S1). PTPIP51_TPR adopts an all‐α‐helical structure and contains twelve α‐helices: α1 (239–250), α2 (252–270), α3 (272–290), α4 (294–314), α5 (317–335), α6 (338–358), α7 (362–379), α8 (381–390), α9 (398–412), α10 (418–431), α11 (434–446), and α12 (452–468). These α‐helices form several antiparallel TPR units, joined mainly by hydrophobic interactions within TPR1 (α1–α2), TPR2 (α3–α4), TPR3 (α5–α6), TPR4 (α7–α9), and TPR5 (α10–α11). The PTPIP51_TPR structure features two kinds of hydrophobic interactions: intra‐TPR and inter‐TPR interactions. In the former interaction, the residues that form the hydrophobic patches are directed toward the inside of each TPR structure: TPR1 (Leu241, Ala245, Leu248, Leu262, and Leu263), TPR2 (Leu277, Ala281, Ala310, and Ala311), TPR3 (Ala319, Ala326, Phe348, Ala355, and Leu358), TPR4 (Pro363, Phe367, Ala402, Ala409, and Leu412), and TPR5 (Val422 and Ala444). The latter inter‐TPR interactions involve four regions: TPR1–TPR2 (Phe260, Leu264, Leu280, and Met287), TPR3–TPR4 (Val352, Ile356, and Leu369), TPR4–TPR5 (Phe406 and Ile424), and TPR5–α12 (Val422, Ala437, Ala444, Leu461, Leu464, and Leu468) (Fig 1B). Ten of the twelve antiparallel α‐helices form double‐helical layers consisting of concave inner layers (α1, α3, α5, α7, and α10) and convex outer layers (α2, α4, α6, α9, and α11) (Fig 1A). Helix α8 generates a bulge, and helix α12 covers half of the concave region of PTPIP51_TPR. Analytical ultracentrifugation (AUC) experiments with PTPIP51_TPR or PTPIP51_ΔTM (residues 36–470) indicated that these proteins exist as monomers (PTPIP51_TPR) or tetramers (PTPIP51_ΔTM) in solution (Fig 1C). Interestingly, prediction of the coiled‐coil oligomeric state by LOGICOIL (http://coiledcoils.chm.bris.ac.uk/LOGICOIL/) (Vincent et al, 2013) also predicted tetramerization of PTPIP51. These results suggest that tetramerization of PTPIP51 may be mediated by the coiled‐coil domain of the protein. We also tried to measure the molecular weight of PTPIP51 without the TPR domain (PTPIP51_ΔTPR, residues 36–235 or 36–130). However, these constructs existed as high‐molecular‐weight aggregates in solution, and we could not estimate their oligomeric states. A DALI server (http://ekhidna2.biocenter.helsinki.fi/dali) search with PTPIP51_TPR identified structurally similar proteins (Holm & Laakso, 2016). The proteins with the greatest structural similarity are the hypothetical conserved protein TTC0263 (PDB ID 2PL2, Z score = 18.1) (Lim et al, 2007), the type 4 fimbrial biogenesis protein PILF (PDB ID 2FI7, Z score = 17.0) (Kim et al, 2006), and anaphase‐promoting complex subunit CUT9 (PDB ID 2XPI, Z score = 17.0) (Zhang et al, 2010). Although the amino acid sequence identities between PTPIP51_TPR and these three structures were very low (< 15%), their structural topologies were quite similar (Fig 1D).
Figure 1. Crystal structure of PTPIP51_TPR.
- Domain architecture of PTPIP51 and overall structure of PTPIP51_TPR. The transmembrane (TM), coiled‐coil (CC), and tetratricopeptide repeat (TPR) domains are presented. Each TPR pair is marked with a different color: red, TPR1 (α1 and α2); orange, TPR2 (α3 and α4); yellow, TPR3 (α5 and α6); green, TPR4 (α7 and α9); and blue, TPR5 (α10 and α11). Two α‐helices that are not involved in a TPR (α8 and α12) are highlighted in light blue. The lower panel shows helical wheel projections for the PTPIP51_TPR structure showing one of the features typical of a multi‐TPR protein: TPR pairs forming a superhelical twist. The diagram was drawn using NetWheels (http://lbqp.unb.br/NetWheels).
- Detailed views of the hydrophobic interactions of TPRs. There are two kinds of hydrophobic interactions, intra‐TPR and inter‐TPR. The hydrophobic residues associated with inter‐TPR interactions are indicated with asterisks (*).
- Analytical ultracentrifugation (AUC) of PTPIP51_TPR and PTPIP51_ΔTM. The molecular weights of recombinant PTPIP51 calculated and estimated in AUC experiments are shown. All fusion tags were removed from the respective recombinant proteins for the AUC experiments.
- Topology diagrams of PTPIP51_TPR and other TPRs: TTC0263_TPR (PDB ID 2PL2), PILF_TPR (PDB ID 2FI7), and CUT9_TPR (PDB ID 2XPI). TPR pairs are presented in different color, which are identical to those in the topology diagrams.
- Electrostatic surface potentials of TTC0263_TPR, PILF_TPR, CUT9_TPR, and PTPIP51_TPR, showing two different views of the electrostatic surface potential of each TPR. The concave and convex regions of each TPR are shown. The electrostatic surface potentials were calculated using the APBS electrostatics plugin (https://www.poissonboltzmann.org/) at ±3 kT/e.
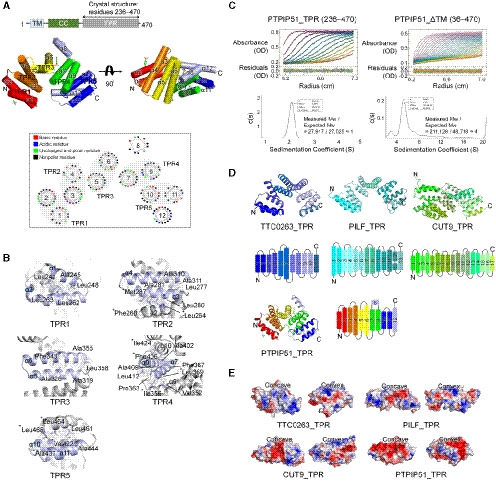
Among the three structures with the greatest similarity to PTPIP51_TPR identified by the DALI server search, the TPR domain of TTC0263 contains a weakly positively charged concave surface that may provide a docking site for its binding partners (Fig 1E) (Lim et al, 2007). However, the corresponding surfaces in the TPR domains of PILF, CUT9, and PTPIP51 are negatively charged (Fig 1E) (Kim et al, 2006; Zhang et al, 2010).
Identification of a serpentine electron density in the PTPIP51_TPR structure
Interestingly, an elongated, deep cavity generated by TPR2 (α3–α4) and TPR3 (α5–α6) and a bulge region by α8 was found in PTPIP51 (Fig 2A). The sizes of the cavities in PTPIP51_TPR and other structurally similar TPR proteins were analyzed with CASTp 3.0 (http://sts.bioe.uic.edu/castp/) (Tian et al, 2018), and the cavities in the TTC0263_TPR, CUT9_TPR, and PTPIP51_TPR structures were found to be large. The calculated sizes of the cavities in TTC0263_TPR, CUT9_TPR, and PTPIP51_TPR were 827.7, 3009.6, and 709.4 Å3, respectively. We could not find a large cavity in the PILF_TPR structure. Compared to the cavities in other TPR structures, the cavity in PTPIP51 forms a channel in the absence of α8, and this non‐TPR helix covers one end of the channel like a plug. Therefore, the resulting cavity resembles a cave with a wide entrance (Fig 2). The inside surface of the cavity is relatively hydrophobic and weakly positively charged (Fig 2B). In the cave‐like cavity, electron density maps for two nonprotein molecules were found, and based on the shapes of the electron density maps, we tentatively assigned these molecules as glycerol derived from the cryoprotectant solution and p‐toluenesulfonic acid (Fig 2B; Appendix Fig S2A and B). The p‐toluenesulfonic acid molecule in the cavity is mainly surrounded by hydrophobic residues (Trp372, Val376, and Leu395) and two hydrophilic residues (Arg341 and Gln375), which form water‐mediated interactions with this p‐toluenesulfonic acid molecule. An additional p‐toluenesulfonic acid molecule with few hydrophobic contacts (only Gln258) is located in another part of the protein structure. The glycerol molecule in the cavity participates in two direct hydrogen bonds, with Ser344 and Gln375, and two water‐mediated interactions, with Leu368 and Arg371.
Figure 2. Calculated cavities in the TPR structures and tube‐shaped electron density map in the PTPIP51_TPR structure.
- Cavities of TTC0263_TPR, PILF_TPR, CUT9_TPR, and PTPIP51_TPR. The cavities were calculated as solvent‐accessible regions using the CASTp program.
- The surface properties of the PTPIP51_TPR structure. Hydrophilic and hydrophobic residues are shown in pink and yellow, respectively. Sliced views of PTPIP51_TPR showing the cavity and its hydrophobic property. The α8 helix is highlighted with a dotted line in the sliced view.
- Detailed view of the electron density map indicative of a tube‐shaped molecule. Hydrophilic and hydrophobic amino acid residues adjacent to the electron density map are shown in pink and yellow, respectively. The mFo‐DFc omit maps were generated using the program “phenix.composite_omit_map” in the Phenix program suite. The contour level of the omit map is 0.8 σ.
Notably, a long tube‐shaped electron density map was found in the entrance of the cave‐like cavity (Fig 2C). The crystallization reservoir solution, which contained the Sokalan® CP42 polymer, suggests that the density map corresponds to part of this polymer. However, the electron density map was relatively weak, and we could not precisely determine its identity. Since many phospholipids can be exchanged at MAMs, and the electron density map looked like the hydrocarbon chain of a lipid, we presumed that this electron density could be assigned to part of an unknown phospholipid from E. coli, the expression host for the recombinant PTPIP51_TPR protein. We tried to identify lipids in the PTPIP51 structure by mass spectrometry with PTPIP51 crystals and identified various kinds of lipid species (Appendix Fig S3). However, we could not specify and assign this electron density map to a phospholipid because no electron density map for a phospholipid head group was seen. This tube‐shaped electron density map is surrounded by many hydrophobic and hydrophilic patches (Fig 2C). Therefore, our crystal structure suggests that the electron density map found in the PTPIP51 structure corresponds possibly to some lipid species and that the PTPIP51 may functionally associate with lipids, notwithstanding that the role of the cavity in the TPR domain is indefinitive.
PTPIP51 has phospholipid binding and phospholipid transfer activity
In addition to Ca2+ ion exchange, an important biochemical event at MAMs is the transfer of phospholipids for the synthesis of various kinds of phospholipids. Although the ERMES complex, a MAM‐tethering macromolecular complex involved in phospholipid exchange, has been relatively well studied in yeast (Phillips & Voeltz, 2016; Jeong et al, 2017; Muallem et al, 2017), MAM‐tethering complexes that also function in phospholipid exchange have not been fully identified in humans. Recently, the amino acid sequence of PDZD8, an SMP domain‐containing ER protein, was shown to partially align with that of yeast MMM1 (a component of the yeast ERMES complex), suggesting that PDZD8 is a human ortholog or paralog of the ERMES complex (Hirabayashi et al, 2017; Wideman et al, 2018). However, while the ERMES complex is known to regulate the transfer of phospholipids between the ER and mitochondria (Kornmann et al, 2009; AhYoung et al, 2015; Jeong et al, 2017; Cockcroft & Raghu, 2018), PDZD8 regulates Ca2+ dynamics in neurons (Hirabayashi et al, 2017).
Since the presence of the unexplained serpentine electron density map in the crystal structure of PTPIP51_TPR suggests a possible phospholipid binding function, we sought to determine whether PTPIP51 carries out phospholipid‐related functions. First, we performed a series of in vitro phospholipid‐ and lipid‐binding assays with purified recombinant PTPIP51_ΔTM, PTPIP51_TPR, and PTPIP51_ΔTPR (residues 36–235) proteins using a dot blot overlay (PIP strips) assay (Fig 3A). Two of these PTPIP51 protein constructs—PTPIP51_ΔTM and PTPIP51_TPR—interacted with various phospholipids with different affinities. PTPIP51_ΔTM bound phosphatidylinositol (PtdIns) mono‐, di‐, and triphosphate, PA, PS, and CL. PTPIP51_TPR interacted with PtdIns(4)P, PtdIns(5)P, and PA. While PTPIP51_ΔTM bound CL and di‐PtdIns, PTPIP51_TPR barely bound these phospholipids. Two phospholipids (PA and PtdIns(4)P) bound both PTPIP51 constructs. PtdIns(5)P interacted with all three constructs of PTPIP51. Interestingly, PTPIP51 did not bind phospholipids with positively charged head groups, such as PE and PC. Other lipids, such as sphingomyelin and cholesterol, whose biosynthesis does not occur in mitochondria (Holthuis & Menon, 2014; Tatsuta et al, 2014), did not interact with PTPIP51.
Figure 3. Phospholipid binding and phospholipid transfer functions of PTPIP51.
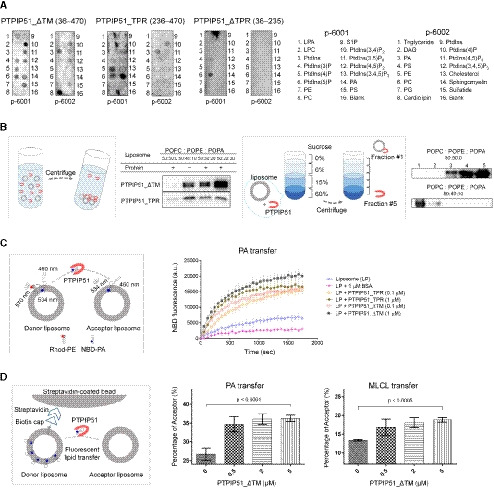
- PIP strip lipid‐binding assay with recombinant PTPIP51_ΔTM, PTPIP51_TPR, and PTPIP51_ΔTPR.
- PA binding of PTPIP51 (PTPIP51_ΔTM and PTPIP51_TPR) was monitored by a PA precipitation assay and a liposome flotation assay. The POPC:POPE:POPA ratios in each liposome were 50:50:0, 50:40:10, 50:30:20, and 50:20:30. Diagrams of the liposome precipitation and flotation assays are illustrated separately.
- The PA transfer activity of PTPIP51 (PTPIP51_ΔTM and PTPIP51_TPR) was monitored by a FRET‐based lipid transfer assay. A schematic cartoon of the FRET‐based in vitro lipid transfer assay is presented on the left. a.u.: arbitrary unit.
- The alternative bead pulldown‐based lipid transfer assay with PA and MLCL. A schematic cartoon representation of the fluorescent lipid transfer assay is shown on the left. The y‐axes in the graphs show the percentage of acceptor liposome fluorescence calculated using the following formula: 100×F acceptor/(F acceptor+F donor).
Data information: The data are presented as the mean ± SD of technical triplicate (C) or quadruplicate (D) from one representative experiment (n = 2), and P‐values calculated using one‐way ANOVA are shown.
Considering the cellular localization of PTPIP51 and the biological significance of PA transport between the ER and mitochondria, we further investigated the interaction between PTPIP51 and PA using a PA precipitation assay (Fig 3B). PTPIP51_ΔTM or PTPIP51_TPR was mixed with liposomes with different lipid contents (POPC:POPE:POPA = 50:50:0, 50:40:10, 50:30:20, and 50:20:30 (%)), and ultracentrifugation experiments were performed. PTPIP51_ΔTM showed increased binding to liposomes with increasing PA concentration. PTPIP51_TPR also interacted with liposomes containing PA, but this interaction did not depend on the PA concentration. These results suggest that the middle region of PTPIP51, which includes the coiled‐coil domain and FFAT (two phenylalanines (FF) in acidic tract)‐like motifs, partially contributes to its phospholipid binding and specificity (Appendix Fig S1). The interaction between PTPIP51 and PA was also confirmed by a sucrose gradient liposome flotation assay (Fig 3B). While PTPIP51_ΔTM floated in the less‐dense fractions of the sucrose gradient due to its binding to PA‐containing liposomes, it was distributed in the denser layers when mixed with liposomes not containing PA. These results showed that PTPIP51 selectively interacts with PA and not PC or PE, as also shown in the PIP strip assay. We also investigated the interaction between PTPIP51_ΔTM and other phospholipids that exhibited binding in the PIP strip assay. We selected PS, PtdIns(4)P, and CL for the liposome precipitation assay and found that PtdIns(4)P and CL interacted with PTPIP51_ΔTM (Fig EV1). In contrast to the PIP strip results, the interaction between PS and PTPIP51_ΔTM was not observed in the liposome precipitation assay. These data suggest that PTPIP51 has broad lipid binding specificity.
Figure EV1. Liposome precipitation‐based phospholipid‐binding assay.
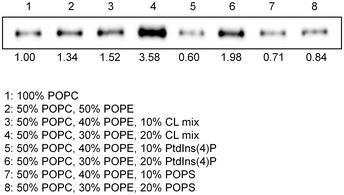
Results of the liposome precipitation assay of PTPIP51 with liposomes containing a CL mixture, PtdIns(4)P, and PS. The relative band intensities on Western blots were quantified using ImageJ (National Institutes of Health, USA).
Since phospholipid transfer is one of the important events at MAMs, it is quite likely that interactions between PTPIP51 and phospholipids relate to the transfer of phospholipids between the ER and mitochondrial membranes. Therefore, we also evaluated the PA transfer activity of PTPIP51 using a FRET‐based in vitro PA transfer assay (Fig 3C). Donor liposomes containing fluorescent nitrobenzoxadiazole‐labeled PA (NBD‐PA) and rhodamine‐labeled PE (Rhod‐PE) and acceptor liposomes not containing fluorescent lipids were used. When NBD‐PA and Rhod‐PE are contained in the same liposome, the NBD fluorescence signal is quenched by rhodamine; thus, if the PTPIP51 constructs transport NBD‐PA to the acceptor liposome, the fluorescence signal of NBD‐PA should not be quenched (Fig 3C). Compared to the controls (liposome only and liposome + bovine serum albumin (BSA)), both PTPIP51 constructs showed increased NBD fluorescence signals with increasing time, indicating PA transfer activity by PTPIP51. The TPR domain of PTPIP51 is responsible for its PA transfer function, as PTPIP51_TPR itself has PA transfer activity. The PA transfer activity increased with increasing protein concentration. The NBD fluorescence signal from NBD‐PA was higher with the PTPIP51_ΔTM construct than with the PTPIP51_TPR construct, suggesting that PTPIP51_ΔTM transfers PA more efficiently than PTPIP51_TPR, probably due to its oligomerization via the coiled‐coil domain. Although the PA transfer activity in this case was very slow to have physiological relevance, similar slow PA transfer rate has been reported in TRIAP1/PRELI complex (Miliara et al, 2015). These slow transfer rates are probably due to the fundamental limitations of in vitro fluorescence dequenching‐based transfer assay methods, which utilize liposomes with limited number of lipids, rendering it difficult to compare with natural ER and mitochondrial membranes containing numerous lipid species and membrane‐imbedded proteins. Nevertheless, it is noteworthy that our PA transfer assay showed a clear PTPIP51 dose‐dependent lipid transfer activity. Furthermore, an alternative bead pulldown‐based fluorescence in vitro lipid transfer assay was performed to reconfirm the PA transfer and to determine whether other phospholipids can be transferred by PTPIP51 (Fig 3D). Our results showed that PTPIP51_ΔTM can very efficiently transfer PA and monolyso‐CL (MLCL). Additionally, very weak but statistically significant transfer activity was observed for PS and PE, but PTPIP51 could not transfer PtdIns(4)P and CL (Fig EV2A and B). We also monitored the PA transfer activity of PTPIP51 with liposomes containing CL or PtdIns(4)P and found that the addition of these phospholipids increased the PA transfer activity of PTPIP51 (Fig EV3A and B). These results suggest that the presence of various PTPIP51‐binding phospholipids in the mitochondrial membrane can enhance the PA transfer activity of PTPIP51 at MAMs. A similar result was shown for PC transfer by Sec14 in the presence of PtdIns(4)P (Panagabko et al, 2019). At MAMs, the local concentration of PTPIP51 is high, and the mitochondrial membrane contains various kinds of PTPIP51‐interacting phospholipids. Therefore, we predict that the phospholipid transfer activity of PTPIP51 would be higher under physiological conditions than under in vitro conditions.
Figure EV2. Streptavidin–biotin bead‐based phospholipid transfer assay.
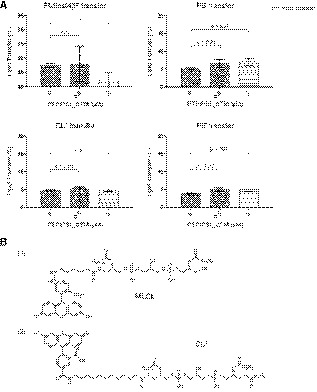
- Results of the in vitro phospholipid transfer assay with PTPIP51 and various phospholipids. The percentages of lipid transfer were calculated with the following formula: 100×Facceptor/(F acceptor+F donor). The data are presented as the mean ± SD of technical quadruplicate experiments from one representative experiment (n = 2), and P‐values calculated using Student’s t‐test are shown.
- Two kinds of fluorescent CLs were used in this assay. The fluorescent MLCL in Fig 3 has the chemical structure shown in (1), and fluorescent CL* has the chemical structure shown in (2).
Figure EV3. Lipid transfer assays in the presence of other PTPIP51‐interacting phospholipids.
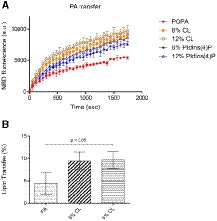
- FRET‐based phospholipid transfer assay: acceptor liposomes (POPC:POPE:POPA = 50:40:10 (%)) and donor liposomes (POPC:POPE:Rhod‐PE:NBD‐PA = 50:40:2:8, POPC:POPE:Rhod‐PE:NBD‐PA/CL = 50:32:2:8:8 and 50:28:2:8:12, and POPC:POPE:Rhod‐PE:NBD‐PA/PtdIns(4)P = 50:32:2:8:8 and 50:28:2:8:12 (%)).
- Bead pulldown‐based phospholipid transfer assay: acceptor liposomes (100% POPC) and donor liposomes (POPC/biotinyl cap‐PE:TopFluor TMA‐PA = 97:2:1, POPC:biotinyl cap‐PE:TopFluor TMA‐PA:CL = 94:2:1:3 and 92:2:1:5 (%)).
Data information: All data are presented as the mean ± SD of technical quadruplicate experiments from one representative experiment (n = 2). The P‐value was calculated using one‐way ANOVA.
PTPIP51 barely interacted with PC or PE (Fig 3A). This finding suggests that PTPIP51 may not be involved in the PE and PC biosynthetic pathways. PC is usually transferred from the ER to mitochondria by STARD7 (Horibata & Sugimoto, 2010; Horibata et al, 2017), and a recent study showed that PS, the major precursor of PE biosynthesis in mitochondria, is transferred at MAMs by ORP5/8 (Rochin et al, 2020). Similarly, previous reports on the yeast ERMES complex have shown different phospholipid binding preferences. While Zygosaccharomyces rouxii MMM1 broadly interacts with various glycerophospholipids (such as PG, PA, PC, and PS) (Jeong et al, 2017), Saccharomyces cerevisiae MDM12 selectively interacts with PC and PE (Jeong et al, 2016).
The TPR domain usually mediates protein–protein interactions and the assembly of many multiprotein complexes (D'Andrea & Regan, 2003). However, the TPR domain of PTPIP51 has both phospholipid binding and phospholipid transfer functions. Although in a similar example, the TPR domain of protein phosphatase 5 (PP5) was shown to interact with PtdIns and arachidonic acid for its activation (Chen & Cohen, 1997; Yang et al, 2005), the interaction between the TPR domain and lipids is quite unique.
The FFAT‐like motif of PTPIP51, not the TPR domain, mediates the interaction of PTPIP51 with VAPB at MAMs
Since PTPIP51 associates with VAPB from the ER and generates a tethering protein complex at MAMs, we investigated whether PTPIP51_TPR is responsible for the interaction of PTPIP51 with VAPB. In a GST pulldown experiment, PTPIP51_ΔTM interacted with VAPB_ΔTM (residues 1–220; a TM‐truncated construct), but PTPIP51_TPR did not (Fig 4A). This finding suggests that the middle region of PTPIP51 (residues 36–235) is responsible for the PTPIP51−VAPB interaction. PTPIP51 possesses tandem FFAT (FFAT #1, 157VYFTASS163; FFAT #2, 166TFTDAES172)‐like motifs between the coiled‐coil region and TPR domain (Fig 4B; Appendix Fig S1) (Mikitova & Levine, 2012). The N‐terminal globular domain of VAPB, the MSP domain, is a well‐known FFAT‐like motif‐interacting domain (Kaiser et al, 2005; Mikitova & Levine, 2012). Therefore, it is highly likely that VAPB interacts with the FFAT‐like motifs of PTPIP51 to contribute to MAM formation. Based on this rationale, we sought to determine whether VAPB interacts with the FFAT‐like motifs of PTPIP51. The quantitative binding affinity was measured by a fluorescence polarization (FP) binding assay with the FFAT‐like peptide of PTPIP51 (residues 151–174) and VAPB_ΔTM (Fig 4C). As expected, a direct interaction between the tandem FFAT‐like motif of PTPIP51 and VAPB_ΔTM was observed; the dissociation constant (Kd) for the interaction was approximately 52.87 (±2.11) µM, and the interaction was decreased or completely abolished when the tyrosine and phenylalanine residues in the tandem FFAT motif were substituted with alanine residues. The strictly conserved Tyr158 and Phe159 residues in FFAT#1 of PTPIP51 were more essential for the PTPIP51–VAPB interaction than was Phe167 in FFAT#2 of PTPIP51. These results show that PTPIP51 interacts with VAPB through its tandem FFAT‐like motifs for mitochondria–ER tethering. Considering the K d value determined in the FP experiment, the interaction between the PTPIP51 peptide and VAPB protein is likely to only weakly contribute to MAM tethering. However, it is possible that the tetrameric association of PTPIP51 and its expected high local concentration at MAMs may enhance the PTPIP51–VAPB interaction. In this context, the TPR domain of PTPIP51 functions in phospholipid transfer instead of participating in MAM tethering via the PTPIP51−VAPB interaction. Previous reports have shown that VAPB can exist as a homodimer or as a heterodimer with itself or VAPA (Kim et al, 2010; Di Mattia et al, 2018), and our size exclusion chromatography–multiangle light scattering (SEC‐MALS) results also suggested that VAPB_ΔTM exists primarily as a dimer (Appendix Fig S4). Therefore, PTPIP51−VAPB complex formation at MAMs is anticipated to occur through a tetramer−dimer association.
Figure 4. The interaction between the FFAT‐like motif of PTPIP51 and VAPB.
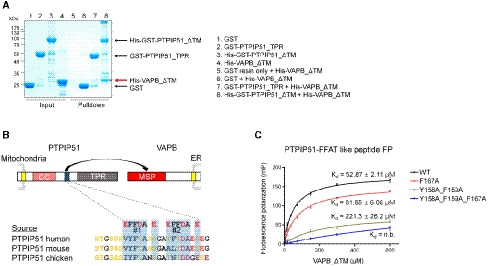
- A GST pulldown assay was performed to monitor the interaction between 6×His tag‐fused VAPB_ΔTM and GST‐fused PTPIP51_TPR (or GST‐fused PTPIP51_ΔTM). VAPB_ΔTM pulled down by PTPIP51_ΔTM is marked with an asterisk and highlighted with a red arrow.
- Schematic representation of the FFAT‐like motif of PTPIP51 and amino acid sequence alignment of the FFAT‐like motifs of human, mouse, and chicken proteins.
- Results of fluorescence polarization (FP) experiments to monitor the interaction between the FFAT‐like motif‐containing peptide of PTPIP51 and VAPB_ΔTM. The K d value of the interaction determined in the FP experiment was ~53 µM. Peptides with mutation of conserved phenylalanine and tyrosine residues in the FFAT motif were also tested. The data are presented as the mean ± SD of technical quadruplicate experiments.
Regulation of the mitochondrial CL concentration by PTPIP51
One of the interesting findings of the in vitro phospholipid‐binding assay was the interaction between PTPIP51 and CL (Figs 3A and EV1). Moreover, the MLCL transfer activity of PTPIP51 is worth noting (Fig 3D). CL is a unique mitochondria‐enriched dimeric phospholipid that is essential for the proper functioning of various mitochondrial proteins (Paradies et al, 2014). CL is synthesized and remodeled by various enzymes in the IMM using PA as a starting material (Schlattner et al, 2014; Tamura et al, 2014). Since PA is synthesized at the ER membrane due to the localization of the enzyme catalyzing its synthesis (AGPAT) (Holthuis & Menon, 2014), PA from the ER needs to be supplied to the IMM through the outer mitochondrial membrane (OMM) for CL biosynthesis (Holthuis & Menon, 2014). The PA‐shuttling proteins in the mitochondrial intermembrane space have been well characterized in yeast (UPS1/MDM35) and mammalian (TRIAP1/PRELI) cells (Connerth et al, 2012; Potting et al, 2013; Tatsuta et al, 2014; Watanabe et al, 2015). However, the proteins that mediate PA transfer between the ER and mitochondria have not yet been identified. Considering the PA binding and transfer functions of PTPIP51, we hypothesized that PTPIP51 functions in CL biosynthesis by supplying the starting material, PA, to mitochondria. Therefore, we sought to determine whether PTPIP51 influences the biosynthesis of CL. We measured the mitochondrial CL concentration in cells because CL is synthesized in and usually localized to mitochondria. For this analysis, we generated a doxycycline‐inducible PTPIP51‐knockdown HeLa cell line and restored the expression of full‐length PTPIP51 (Fig 5A). Then, mitochondrial CL levels were monitored by a probe‐based CL detection assay. The mitochondrial CL concentration was slightly reduced when PTPIP51 was depleted, and the CL level was restored by PTPIP51 supplementation (Fig 5A). Similar results were shown when PA transfer between the OMM and IMM was disrupted by depleting the TRIAP1/PRELI complex (Connerth et al, 2012; Potting et al, 2013; Tatsuta et al, 2014). We further analyzed the levels of various phospholipids in the mitochondrial membrane by mass spectrometry‐based lipidomics (Fig 5B; Appendix Fig S5). While the levels of most of the lipids were not greatly changed by PTPIP51 depletion and reconstitution, various CL levels were slightly reduced when PTPIP51 was depleted, although the changes in the relative intensities of the mass spectra were not statistically significant. The result was similar to that of the probe‐based CL detection assay. The CL levels, except for CL64:0, were significantly increased by PTPIP51 reconstitution. Interestingly, various mitochondrial CL levels were not decreased by reconstitution of PTPIP51_ΔFFAT (deletion of the tandem FFAT motifs (Ser151−Gly174)), suggesting that CL level changes mediated by changes in PTPIP51 were independent of its interaction with VAPB. Lipidomic analyses of some phospholipids whose biosynthesis occurs at MAMs (PS, PC, and PE) were also performed. Of note, the PS levels were inversely correlated with the CL levels except for the FFAT mutant (Appendix Fig S5A). However, we could not easily explain this result, although it is probably due to ORP5/8, which are likely to interact with PTPIP51 and are involved in PS transfer at MAMs (Galmes et al, 2016; Rochin et al, 2020). The PC and PE levels did not show notable patterns of change (Appendix Fig S5B and C).
Figure 5. Mitochondrial CL levels mediated by PTPIP51.
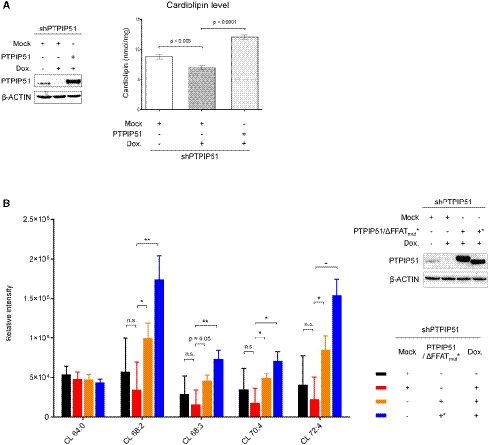
- CL levels after PTPIP51 depletion and reconstitution were monitored. A doxycycline‐induced shRNA expression system was used to deplete the PTPIP51 protein in HeLa cells. Left, Western blot analysis of HeLa cells in which PTPIP51 was depleted and restored. β‐ACTIN was used as the loading control. Right, the mitochondrial CL level (nmol/mg) according to PTPIP51 expression. To reconstitute PTPIP51, the full‐length PTPIP51 gene was transiently expressed. The data are presented as the mean ± SD of technical triplicate experiments from one representative experiment (n = 3), and P‐values calculated using Student’s t‐test are shown.
- Quantification of CL in mitochondria using lipidomic analysis after PTPIP51 depletion and reconstitution. Color codes for each sample were presented in the lower right subpanel. Experiments were technically repeated three times, and the results are represented as the means ± SD of technical replicates (n = 3). *P < 0.05; **P < 0.01 (Student’s t‐test). Quality control (QC) samples, which were pooled identical aliquots of the mitochondria samples, were measured four times throughout the run for data reproducibility. Upper right, Western blot analysis of HeLa cells in which PTPIP51 WT or ΔFFAT mutant was depleted and restored. β‐actin was used as a loading control.
CL and MLCL molecules contain one and two PA moieties, respectively (Appendix Fig S6). In our study, whereas PA interacted with both tetrameric PTPIP51_ΔTM and monomeric PTPIP51_TPR, CL interacted with only tetrameric PTPIP51_ΔTM (Fig 3A). Therefore, it is likely that the binding of CL to PTPIP51 requires multimeric association of PTPIP51 and that this interaction is mediated by the PA moiety of the CL molecule. Furthermore, the in vitro MLCL transfer activity of PTPIP51 and the changes in the CL level mediated by changes in PTPIP51 also suggest that PTPIP51 may function in the biosynthesis and remodeling of CL. If PTPIP51 plays critical roles in CL production, the CL production rate would be expected to decrease substantially when PTPIP51 is depleted. However, considering that the amount of reduction of CL levels was not large in our result, we cannot rule out the presence of other PA transport systems which compensate for the PTPIP51 depletion. Therefore, further extensive cellular investigations regarding the function of PTPIP51 in CL production are required.
In this study, we determined the crystal structure of the C‐terminal TPR domain of PTPIP51, a mitochondrial protein that associates with VAPB from the ER at MAMs. Although previous studies have suggested that the PTPIP51−VAPB complex functions in Ca2+ homeostasis via MAM formation, we unexpectedly found the phospholipid binding and phospholipid transfer functions of PTPIP51 on the basis of insight from the crystal structure. Our findings regarding the PA and MLCL phospholipid transfer functions of PTPIP51 provide new insight into understanding the mitochondria–ER association and its roles in CL biosynthesis/maturation. Based on our present study, we suggest a model for the function of PTPIP51 at MAMs (Fig EV4). Although neither PTPIP51 nor VAPB contains a SMP domain, and these proteins are structurally distinct from any components of the yeast ERMES complex, the human PTPIP51−VAPB complex is a functional counterpart of the yeast ERMES complex with functions in both membrane tethering and phospholipid transfer at MAMs.
Figure EV4. Model of the PTPIP51−VAPB complex at MAMs and its phospholipid transfer function.
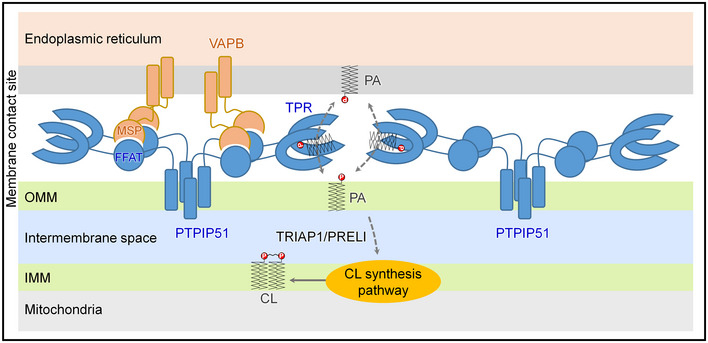
The interaction between tetrameric PTPIP51 and dimeric VAPB mediated by the FFAT‐like motif and MSP domain of each protein is shown. The PA transfer function of the TPR domain of PTPIP51 is also presented.
Materials and Methods
Protein expression and purification
The PTPIP51_TPR‐encoding gene was amplified by PCR (template DNA was distributed from DNASU, USA) and cloned into the BamHI/EcoRI sites in the pQlinkH vector (#13667, Addgene, USA) (Scheich et al, 2007). The PTPIP51_TPR protein was expressed in E. coli Rosetta2(DE3) cells (Novagen, USA) cultured in Terrific broth. After the cells had grown to an OD600 of 0.6 at 37°C, 0.1 mM isopropyl‐D‐thiogalactoside was added to the cells. The cells were further cultured at 16°C for approximately 14 h. The cells were resuspended in lysis buffer (20 mM Tris–HCl [pH 7.5], 150 mM NaCl, 5% glycerol, 5 mM β‐mercaptoethanol, and 1 mM phenylmethylsulfonyl fluoride) and lysed in a high‐pressure homogenizer (Micronox, South Korea). After centrifugation at 13,000 g for 2 h, the supernatant was added to Ni‐NTA resin (GE Healthcare, USA). Proteins were eluted from the Ni‐NTA resin using elution buffer (20 mM Tris–HCl [pH 7.5], 150 mM NaCl, 5% glycerol, 5 mM β‐mercaptoethanol, and 300 mM imidazole). The hexahistidine (6×His) tag at the N‐terminus was cleaved by tobacco etch virus (TEV) protease, and the cleaved PTPIP51_TPR proteins were collected after passing through the Ni‐NTA column. The proteins were further purified on a HiLoad 16/600 Superdex 200 prep grade column (GE Healthcare, USA) with buffer consisting of 20 mM Tris–HCl [pH 7.5], 150 mM NaCl, 5% glycerol, and 2 mM dithiothreitol. PTPIP51_TPR proteins were concentrated to 50 mg/ml for crystallization or concentrated to 5–10 mg/ml for biochemical assays. Selenomethionine‐substituted PTPIP51_TPR was prepared with the same method used to prepare native proteins, except the recombinant protein was overexpressed in selenomethionine‐containing M9 minimal medium. The PTPIP51_ΔTM gene was cloned into pFASTBacHisGST (a modified pFASTBacHTb vector containing a tandem 6×His‐GST tag at the N‐terminus of the protein). After baculovirus production, PTPIP51_ΔTM was expressed in Sf9 cells. PTPIP51_ΔTM expressed in Sf9 cells was purified using Ni‐NTA, glutathione‐Sepharose, and Superdex 200 prep grade columns (GE Healthcare, USA). The gene encoding GST‐PTPIP51_TPR was cloned into the pQLinkG2 vector, and genes for two PTPIP51‐ΔTPR constructs and VAPB_ΔTM were cloned into pQlinkH (Scheich et al, 2007). The recombinant proteins of these constructs were overexpressed in E. coli BL21(DE3) cells at 18°C under 0.1 mM IPTG induction. These proteins were purified using affinity chromatography resin (GST resin or Ni‐NTA resin) and a HiLoad 16/600 Superdex 200 (or 75) prep grade column (GE Healthcare, USA). To estimate the oligomeric state of the proteins, AUC or SEC‐MALS experiments were carried out. The ProteomeLab XL‐A system with an AN60‐Ti rotor (Beckman Coulter, USA) was used. The rotor speed was 35,000 rpm, and the PTPIP51 protein sample concentration was approximately 0.5 mg/ml. AUC data sets were analyzed with SEDFIT software (Ver. 16.1c) (Schuck, 2000). Oligomeric states of VAPB_ΔTM were estimated using a DAWN HELEOS II system (Wyatt, USA). All fusion tags on recombinant proteins were removed for all experiments except the PIP strip lipid‐binding assays and GST pulldown experiments. The purities of the purified recombinant proteins are shown in Appendix Fig S7.
Crystallization and structural determination
For crystallization, 0.5 μl of 50 mg/ml PTPIP51_TPR was mixed with the same volume of crystallization screening solution. Crystals of PTPIP51_TPR were grown in reservoir solution consisting of 100 mM Tris–HCl [pH 8.0] and 30% (w/v) SOKALAN® CP‐42 at 4°C. Prior to X‐ray data collection, the crystals were soaked in cryoprotectant solution (reservoir solution supplemented with 30% glycerol). Diffraction data were collected with an ADSC Quantum Q315r detector (Area Detector Systems Corporation, USA) at the Pohang Light Source (Beamline 5C) and processed with HKL2000 software (Otwinowski & Minor, 1997). The structure of PTPIP51_TPR was determined by the SAD method with diffraction data from a selenomethionine‐labeled PTPIP51_TPR crystal collected at 0.979028 Å (anomalous peak data set). After three selenium sites had been identified, automated model building was performed with the program Phenix (Adams et al, 2010). The initial structural model was transferred to the native data sets, and further manual model building was performed with the program COOT (Emsley et al, 2010). Structural refinement was conducted using the program REFMAC5 (Murshudov et al, 1997). All data collection, phasing, and refinement statistics are listed in Appendix Table S1. Structural similarities were calculated using the DALI server, and the oligomeric state of the coiled‐coil domain of PTPIP51 was predicted with the LOGICOIL server (Vincent et al, 2013; Holm & Laakso, 2016). Analysis and visualization of the cavity of each protein were performed using CASTp (Tian et al, 2018).
PIP strip experiment
To test the lipid‐binding property of PTPIP51, a PIP strip lipid‐binding assay was performed. Purified 6×His tag‐fused PTPIP51_ΔTM, PTPIP51_TPR, and PTPIP51_ΔTPR proteins were incubated with membrane lipid strips (cat. nos. P‐6001 and P‐6002, Echelon, USA) according to the manufacturer’s protocol. Briefly, membranes were blocked with 3% BSA in 1× TBST (25 mM Tris–HCl [pH 7.5], 150 mM NaCl, and 0.05% Tween 20) for 1 h. Then, each protein (0.5 μg/ml) was incubated with the membrane in TBST containing 3% BSA for 1 h with gentle agitation. The membrane was subsequently washed three times with TBST. Immunoblotting was performed to visualize the bound proteins using an anti‐His tag antibody (cat. no. G020, Abm, Canada) and an HRP‐conjugated secondary antibody (cat. no. 172‐1011, Bio‐Rad, USA). All steps were carried out at room temperature.
Liposomes
The following phospholipids were purchased from Avanti Polar Lipids (USA): 1‐palmitoyl‐2‐oleoyl‐sn‐glycero‐3‐phosphocholine (POPC, cat. no. 850457C), 1‐palmitoyl‐2‐oleoyl‐sn‐glycero‐3‐phosphoethanolamine (POPE, cat. no. 850757C), 1‐palmitoyl‐2‐oleoyl‐sn‐glycero‐3‐phosphate (sodium salt) (POPA, cat. no. 840857C), 1‐palmitoyl‐2‐oleoyl‐sn‐glycero‐3‐phospho‐L‐serine (sodium salt) (POPS, cat. no. 840034P), cardiolipin (sodium salt) (CL, cat. no. C0563), 1‐palmitoyl‐2‐oleoyl‐sn‐glycero‐3‐phospho‐(1'‐myo‐inositol‐4'‐phosphate) (ammonium salt) (PtdIns(4)P, cat. no. 850157P), 1‐oleoyl‐2‐(12‐[(7‐nitro‐2‐1,3‐benzoxadiazole‐4‐yl)amino]dodecanoyl)‐sn‐glycero‐3‐phosphate (NBD‐PA, cat. no. 810176C), and L‐a‐phosphatidylethanolamine‐N‐(lissamine rhodamine B sulfonyl) (Rhod‐PE, cat. no. 810146C), 1,2‐dioleoyl‐sn‐glycero‐3‐phosphoethanolamine‐N‐(cap biotinyl) (sodium salt) (biotinyl cap‐PE, cat. nos. 870273P and 870277P), 1‐oleoyl‐2‐{6‐[4‐(dipyrrometheneboron difluoride)butanoyl]amino}hexanoyl‐snglycero‐3‐phosphoinositol‐4‐phosphate (ammonium salt) (TopFluor®‐PtdIns(4)P, cat. no. 810185P), 1‐oleoyl‐2‐(6‐((4,4‐difluoro‐1,3‐dimethyl‐5‐(4‐methoxyphenyl)‐4‐bora‐3a,4a‐diaza‐s‐indacene‐2‐propionyl)amino)hexanoyl)‐sn‐glycero‐3‐phosphate (ammonium salt) (TopFluor® TMR‐PA, cat. no. 810240C), 1‐palmitoyl‐2‐(dipyrrometheneboron difluoride)undecanoyl‐sn‐glycero‐3‐phospho‐L‐serine (ammonium salt) (TopFluor®‐PS, cat. no. 810283P), and 1‐palmitoyl‐2‐(dipyrrometheneboron difluoride)undecanoyl‐sn‐glycero‐3‐phosphoethanolamine (TopFluor®‐PE, cat. no. 810282C). The following fluorescence‐labeled CLs were purchased from Echelon Biosciences (USA): fluorescein monolysocardiolipin (cat. no. L‐M16F) and sn‐1‐fluorescein‐labeled cardiolipin (cat. no. L‐C16F). Lipid solutions in chloroform were mixed to the desired composition, and the solvent was dried using a vacuum concentrator (Labconco, USA). The liposome film was dissolved in buffer (20 mM Tris–HCl [pH 8.0], 150 mM NaCl, and 2 mM EDTA). The liposomes were extruded 25 times through a 0.1‐μm polycarbonate filter using a mini‐extruder set (cat. no. 61000, Avanti Polar Lipids, USA).
Liposome‐binding assay
A liposome precipitation assay was performed in a manner similar to that described in a previous report (Julkowska et al, 2013). Briefly, 20 μM liposomes (POPC:POPE:POPA=50:50:0, 50:40:10, 50:30:20, and 50:20:30; POPC:POPE:POPS = 50:40:10 and 50:30:20; POPC:POPE:PtdIns(4)P = 50:40:10 and 50:30:20; and POPC:POPE:CL = 50:40:10 and 50:30:20) were incubated with 10 μM PTPIP51_ΔTM and PTPIP51_TPR in 100 μl of buffer (20 mM Tris–HCl [pH 8.0], 150 mM NaCl, and 2 mM EDTA) at 4°C for 30 min and centrifuged at 16,600 g for 1 h. Pellets were analyzed by Western blotting using an anti‐PTPIP51 (anti‐RMDN3) antibody (cat. no. HPA009975, Atlas Antibody, Sweden).
A liposome flotation assay with PA was performed as described in a previous report with slight modifications (Connerth et al, 2012; Watanabe et al, 2015). Liposomes (200 µM, POPC:POPE:POPA=50:50:0, 50:40:10, and 50:30:20) were mixed with 40 μM PTPIP51_ΔTM in 400 μl of flotation buffer (20 mM Tris–HCl [pH 8.0], 150 mM NaCl, and 2 mM EDTA) at 4°C for 30 min. Then, 1.6 ml of 60% sucrose dissolved in flotation buffer was added to the sample after incubation. The liposome/protein mixture was put into a 13‐ml ultracentrifuge tube and overlaid with 4 ml of 15% sucrose, 4 ml of 6% sucrose, and finally 2 ml of flotation buffer without sucrose. After centrifugation at 200,000 g for 3 h, fractions were collected from the top to the bottom, and trichloroacetic acid precipitation was performed. The resulting fractions were analyzed by Western blotting using an anti‐PTPIP51 antibody.
Lipid transfer assays
A PA transfer assay was also performed as described in previous reports (Miliara et al, 2015; Watanabe et al, 2015). The PA transfer activity of the PTPIP51 constructs was monitored by a FRET‐based fluorescence dequenching assay. Donor liposomes (6.25 μM; POPC:POPE:Rhod‐PE:NBD‐PA = 50:40:2:8 (%)) were incubated with acceptor liposomes (25 μM; POPC:POPE:POPA = 50:40:10 (%)) with or without 1 μM and 0.1 μM purified PTPIP51 constructs in 50 μl of assay buffer (20 mM Tris–HCl [pH 8.5], 150 mM NaCl, and 2 mM EDTA) at 25°C. The reactions were initiated by the addition of proteins. NBD fluorescence was measured every 1 min for 30 min at 460 nm (excitation) and 534 nm (emission) using an Infinite 200 Pro fluorometer (Tecan, Switzerland).
Bead pulldown‐based fluorescent lipid transfer assays were performed according to a previous report (Yu et al, 2016; Rochin et al, 2020). Briefly, after binding of the donor liposome (2% biotinyl cap‐PE; 1% fluorescent phospholipids, such as TopFluor‐lipid; and 97% POPC) and streptavidin‐conjugated beads (Dynabeads Streptavidin T1, cat. no. 65601), 25 μM donor liposomes and 25 μM acceptor liposomes (100% POPC) were incubated with several concentrations of PTPIP51_ΔTM (0, 2, and 5 μM) for 16 h at 4°C with gentle agitation. Supernatants containing acceptor liposomes were separated from the bead mixtures using magnetic racks. Donor liposomes were solubilized from the beads after treatment with 0.4% (w/v) n‐dodecyl‐β‐D‐maltoside. The fluorescence of acceptor and donor liposomes was also measured in an Infinite 200 PRO microplate reader (Tecan, Switzerland). Fluorescence was measured at excitation/emission wavelengths of 520/580 nm, 450/510 nm, and 470/530 nm for TopFluor®‐TMA‐PA, TopFluor®‐phospholipids (PtdIns(4)P, PS, and PE), and fluorescein‐CL, respectively. Lipid transfer was determined using the formula 100×F acceptor/(F acceptor+F donor).
Cell culture, chemicals, and antibodies
HeLa cells were purchased from ATCC (USA) and cultured in MEM (HyClone, USA) supplemented with 10% heat‐inactivated fetal bovine serum (HyClone, USA) and 5 μg/ml Cellmaxin (GenDEPOT, USA) at 37°C in a humidified 5% CO2 atmosphere. Doxycycline (Dox.) was purchased from Sigma‐Aldrich (USA). Conditional knockdown and reconstitution of PTPIP51 in HeLa cells were performed as previously reported (Bong et al, 2020). Huma PTPIP51‐targeting small hairpin RNA (shRNA) sequences (5’‐ATGACTTGATGCCACTATTTA‐3’) were inserted into the Tet‐pLKO‐blasticidin vector. Lentiviruses were produced according to a method described previously (Kim et al, 2018). HeLa cells infected with lentiviruses were selected with blasticidin (10 μg/ml, Invitrogen, USA) for at least 7 days and named HeLa Tet‐on‐shPTPIP51 cells. The genes encoding human full‐length PTPIP51 and PTPIP51_ΔFFAT were cloned into the pCAG‐Flag‐IRES‐puro vector. HeLa Tet‐on‐shPTPIP51 cells were transfected with pCAG‐Flag‐IRES‐puro empty vector (Mock), PTPIP51, and PTPIP51_ΔFFAT using Lipofectamine 3000 (Life Technologies, USA). Transfected cells were selected with puromycin (2 μg/ml, Amresco, USA) for at least 4 days. For endogenous PTPIP51 knockdown, doxycycline (1 μg/ml, Sigma‐Aldrich, USA) was added every 2 days. Because the shRNAs were designed to target the 3′ UTR of PTPIP51, exogenously added constructs were not targeted. All experiments were carried out in accordance with protocols approved by the National Cancer Center Institutional Biosafety Committee (approval number 20‐NCCIBC‐02). Anti‐β‐ACTIN (#A2228) antibody was purchased from Sigma‐Aldrich.
Monitoring of mitochondrial CL levels and lipidomic analyses
Mitochondria were isolated from HeLa cells using a mitochondria isolation kit for tissues (cat. no. 89874, Thermo Fisher Scientific, USA) according to the manufacturer’s instructions. Mitochondrial pellets were washed and stored in 1× TBS buffer supplemented with 0.1% CHAPS on ice until further use. Isolated mitochondrial fractions were used for the CL detection assay. Mitochondrial CL levels were measured by a spectrophotometric assay kit (cat. no. K944, BioVision, USA) using an Infinite 200 Pro fluorometer (Tecan, Switzerland). Briefly, isolated mitochondria (40 μg) were incubated with the CL probe for 5 min at room temperature. CL probe fluorescence was measured at 340 nm (excitation) and 480 nm (emission). All assays were performed in three independent experiments.
For lipidomic analysis, ultra‐performance liquid chromatography (UPLC) coupled with quadrupole time‐of‐flight mass spectrometry (QTOF‐MS) was used to analyze the lipid composition of mitochondria‐isolated HeLa cells after PTPIP51 depletion and reconstitution with WT PTPIP51 or the ΔFFAT mutant. For cell analysis, 5 × 106 cells were quickly washed two times with phosphate‐buffered saline (PBS) buffer and scraped into 1 ml of 100 mM hydrochloric acid solution in methanol/water (8:2, v:v), and then, 1 ml of cell extract was transferred into a 5 ml tube. These washing steps were repeated twice. The cell extract was mixed with 3 ml of water/chloroform (1:2 (v/v)) for 1 min and incubated at −20°C for 20 min to precipitate proteins. After centrifugation at 21,191×g and 4°C for 20 min, 1.8 ml of the lower phase was collected. For mitochondrial analysis, isolated mitochondria from 2 × 107 cells were mixed with 500 µl of 100 mM hydrochloric acid solution in methanol/water (8:2 (v/v)), 700 µl of chloroform, and 500 µl of 100 mM hydrochloric acid solution in water. The mitochondrial sample was homogenized with 2.8 mm zirconium oxide beads for 5 min. After centrifugation at 30,130×g and 4°C for 15 min, 600 µl of the lower phase was collected. Both the cell and mitochondrial extracts were dried under a gentle nitrogen stream at room temperature and reconstituted in 300 µl of isopropanol/acetonitrile/water (2:1:1 (v/v/v)). Eighty microliters of each sample was mixed with 20 µl of SPLASH LIPIDOMIX internal standard mix (Avanti Polar Lipids, USA). Finally, 5 µl of each sample was injected into the UPLC‐QTOF‐MS system.
Chromatographic separation of lipids in cells and mitochondria was performed with an Acquity UPLC system (Waters, USA) using an Acquity UPLC CSH C18 column (2.1 × 100 mm, 1.7 µm; Waters) at 55°C and a flow rate of 0.4 ml/min. The mobile phase for positive ion mode comprised 10 mM ammonium formate in water/acetonitrile (40:60 (v/v)) containing 0.1% formic acid (solvent A) and isopropanol/acetonitrile (90:10 (v/v)) containing 0.1% formic acid (solvent B). The mobile phase for negative ion mode comprised 10 mM ammonium acetate in water/acetonitrile (60:40 (v/v)) (solvent A) and isopropanol/acetonitrile (90:10 (v/v)) (solvent B). The UPLC gradient was programmed as follows: 40–43% B from 0 min to 2 min, 43–50% B from 2 min to 2.1 min, 50–54% B from 2.1 min to 12 min, 54–70% B from 12 min to 12.1 min, 70–99% B from 12.1 min to 18 min, 99–40% B from 18 min to 18.1 min, and 40% B for 2 min to equilibrate for the next run.
A Triple TOF 5600 MS (Sciex, Canada) equipped with a DuoSpray ion source operating in positive and negative ESI modes was used for lipid detection with an m/z range of 80 – 1,500. The following parameter settings were used: ion spray voltage, 5,500 V (positive mode) and 4,500 V (negative mode); source temperature, 500°C; nebulizer gas pressure, 50 psi; drying gas pressure, 60 psi; and curtain gas pressure, 30 psi. An atmospheric pressure chemical ionization calibration solvent was used to maintain mass accuracy with an automated calibrant delivery system (Sciex). Information‐dependent acquisition (IDA) was used to acquire MS/MS spectra for ions. All samples were pooled in equal amounts to generate quality control (QC) samples, which were injected after every four samples to calculate the coefficient of variation (CV) and assess analytical reproducibility.
MarkerViewTM software (Sciex) was used to find peaks, perform alignment, normalize peaks (total area normalization), and generate peak tables of m/z and retention times. Then, lipids were identified according to accurate mass, fragment ions, and/or retention time using MS‐DIAL software version 4.38, an in‐house library, and LIPID MAPS. The 315 identified lipids included carnitine (CAR), cholesteryl ester (CE), ceramide (Cer), CL, diacylglycerol (DG), free fatty acid (FA), lysophosphatidylcholine (LPC), lysophosphatidylethanolamine (LPE), lysophosphatidylglycerol (LPG), lysophosphatidylinositol (LPI), PC, PE, phosphatidylglycerol (PG), PtdIns, PS, sphingomyelin (SM), and triacylglycerol (TG). The list of identified lipids is shown in Dataset EV1.
GST pulldown and FP assays
To test the direct interactions between the proteins, GST pulldown experiments with purified proteins were conducted. Equal amounts of His‐GST‐PTPIP51_ΔTM, GST‐PTPIP51_TPR, and GST proteins (approximately 9.4 nmol each) were used for pulldown assays. Purified His‐VAPB_ΔTM was also prepared. Glutathione‐agarose resin (GE Healthcare, USA) was used for the GST pulldown assays. To confirm that the FFAT motif of PTPIP51 is essential for the PTPIP51–VAPB interaction, an FP binding assay was performed according to our previous report (Kim et al, 2014). Briefly, the fluorescein isothiocyanate‐labeled tandem FFAT peptide of PTPIP51 (FITC‐151STGSSSVYFTASSGATFTDAESEG174) and three mutant peptides with mutation of conserved phenylalanine and tyrosine residues in the FFAT motif were mixed with VAPB_ΔTM at various concentrations and incubated in FP reaction buffer (10 mM HEPES [pH 7.5], 50 mM NaCl, 1 mM EDTA, 1 mM DTT, and 0.0025% (v/v) Tween 20). The final peptide concentration was 20 nM. Thirty minutes after the peptides and proteins were mixed in a 96‐well black bottom plate, FP was analyzed using an Infinite F200 Pro instrument (Tecan, Switzerland) at wavelengths of 485 nm (excitation) and 535 nm (emission). Binding curves were fitted using GraphPad Prism 6 software (GraphPad Software).
Author contributions
BIL, HKY, and THP designed the experiments. BIL, HKY, THP, HJ, GSH, SER, HSB, and HKS analyzed and reviewed the data. HKY, THP, HYK, JL, HSB, SHP, and HJY performed the experiments. BIL, HKY, and THP wrote the manuscript with input from all coauthors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Review Process File
Acknowledgements
We thank the beamline staff of Pohang Light Source (Beamline 5C) for their assistance during X‐ray data collection. This work was supported by National Research Foundation of Korea (NRF) grants funded by the Ministry of Science and ICT (NRF‐2019R1A2C1002545 and NRF‐2018R1A5A2023127 to BIL), a Korea Basic Science Institute grant (C170200 to GSH), and a National Cancer Center research grant (1910031 to BIL).
EMBO reports (2021) 22: e51323.
Data availability
Atomic coordinates and structural factors have been deposited in the Protein Data Bank (PDB) under accession number 7CC7 (https://www.rcsb.org/structure/7CC7). The mass spectrometry data from this publication have been deposited to the Metabolomics Workbench database and assigned Project ID PR001101 (https://www.metabolomicsworkbench.org/data/DRCCMetadata.php?Mode=Project&ProjectID=PR001101).
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse‐Kunstleve RW et al (2010) PHENIX: a comprehensive Python‐based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66: 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- AhYoung AP, Jiang J, Zhang J, Khoi Dang X, Loo JA, Zhou ZH, Egea PF (2015) Conserved SMP domains of the ERMES complex bind phospholipids and mediate tether assembly. Proc Natl Acad Sci USA 112: E3179–3188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittremieux M, Parys JB, Pinton P, Bultynck G (2016) ER functions of oncogenes and tumor suppressors: Modulators of intracellular Ca(2+) signaling. Biochim Biophys Acta 1863: 1364–1378 [DOI] [PubMed] [Google Scholar]
- Bong SM, Bae S‐H, Song B, Gwak HyeRan, Yang S‐W, Kim S, Nam S, Rajalingam K, Oh SJ, Kim TW et al (2020) Regulation of mRNA export through API5 and nuclear FGF2 interaction. Nucleic Acids Res 48: 6340–6352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brobeil A, Dietel E, Gattenlohner S, Wimmer M (2017) Orchestrating cellular signaling pathways‐the cellular "conductor" protein tyrosine phosphatase interacting protein 51 (PTPIP51). Cell Tissue Res 368: 411–423 [DOI] [PubMed] [Google Scholar]
- Chen MX, Cohen PT (1997) Activation of protein phosphatase 5 by limited proteolysis or the binding of polyunsaturated fatty acids to the TPR domain. FEBS Lett 400: 136–140 [DOI] [PubMed] [Google Scholar]
- Cockcroft S, Raghu P (2018) Phospholipid transport protein function at organelle contact sites. Curr Opin Cell Biol 53: 52–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connerth M, Tatsuta T, Haag M, Klecker T, Westermann B, Langer T (2012) Intramitochondrial transport of phosphatidic acid in yeast by a lipid transfer protein. Science 338: 815–818 [DOI] [PubMed] [Google Scholar]
- D'Andrea LD, Regan L (2003) TPR proteins: the versatile helix. Trends Biochem Sci 28: 655–662 [DOI] [PubMed] [Google Scholar]
- De Vos KJ, Morotz GM, Stoica R, Tudor EL, Lau KF, Ackerley S, Warley A, Shaw CE, Miller CC (2012) VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum Mol Genet 21: 1299–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Mattia T, Wilhelm LP, Ikhlef S, Wendling C, Spehner D, Nominé Y, Giordano F, Mathelin C, Drin G, Tomasetto C et al (2018) Identification of MOSPD2, a novel scaffold for endoplasmic reticulum membrane contact sites. EMBO Rep 19: e45453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66: 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filadi R, Theurey P, Pizzo P (2017) The endoplasmic reticulum‐mitochondria coupling in health and disease: molecules, functions and significance. Cell Calcium 62: 1–15 [DOI] [PubMed] [Google Scholar]
- Galmes R, Houcine A, van Vliet AR, Agostinis P, Jackson CL, Giordano F (2016) ORP5/ORP8 localize to endoplasmic reticulum‐mitochondria contacts and are involved in mitochondrial function. EMBO Rep 17: 800–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatta AT, Levine TP (2017) Piecing together the patchwork of contact sites. Trends Cell Biol 27: 214–229 [DOI] [PubMed] [Google Scholar]
- Gomez‐Suaga P, Bravo‐San Pedro JM, Gonzalez‐Polo RA, Fuentes JM, Niso‐Santano M (2018) ER‐mitochondria signaling in Parkinson's disease. Cell Death Dis 9: 337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez‐Suaga P, Paillusson S, Stoica R, Noble W, Hanger DP, Miller CCJ (2017) The ER‐mitochondria tethering complex VAPB‐PTPIP51 regulates autophagy. Curr Biol 27: 371–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández‐Alvarez MI, Sebastián D, Vives S, Ivanova S, Bartoccioni P, Kakimoto P, Plana N, Veiga SR, Hernández V, Vasconcelos N et al (2019) Deficient endoplasmic reticulum‐mitochondrial phosphatidylserine transfer causes liver disease. Cell 177: 881–895.e17 [DOI] [PubMed] [Google Scholar]
- Hirabayashi Y, Kwon SK, Paek H, Pernice WM, Paul MA, Lee J, Erfani P, Raczkowski A, Petrey DS, Pon LA et al (2017) ER‐mitochondria tethering by PDZD8 regulates Ca(2+) dynamics in mammalian neurons. Science 358: 623–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, Laakso LM (2016) Dali server update. Nucleic Acids Res 44: W351–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holthuis JC, Menon AK (2014) Lipid landscapes and pipelines in membrane homeostasis. Nature 510: 48–57 [DOI] [PubMed] [Google Scholar]
- Horibata Y, Ando H, Satou M, Shimizu H, Mitsuhashi S, Shimizu Y, Itoh M, Sugimoto H (2017) Identification of the N‐terminal transmembrane domain of StarD7 and its importance for mitochondrial outer membrane localization and phosphatidylcholine transfer. Sci Rep 7: 8793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horibata Y, Sugimoto H (2010) StarD7 mediates the intracellular trafficking of phosphatidylcholine to mitochondria. J Biol Chem 285: 7358–7365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong H, Park J, Lee C (2016) Crystal structure of Mdm12 reveals the architecture and dynamic organization of the ERMES complex. EMBO Rep 17: 1857–1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong H, Park J, Jun Y, Lee C (2017) Crystal structures of Mmm1 and Mdm12‐Mmm1 reveal mechanistic insight into phospholipid trafficking at ER‐mitochondria contact sites. Proc Natl Acad Sci USA 114: E9502–E9511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julkowska MM, Rankenberg JM, Testerink C (2013) Liposome‐binding assays to assess specificity and affinity of phospholipid‐protein interactions. Methods Mol Biol 1009: 261–271 [DOI] [PubMed] [Google Scholar]
- Kaiser SE, Brickner JH, Reilein AR, Fenn TD, Walter P, Brunger AT (2005) Structural basis of FFAT motif‐mediated ER targeting. Structure 13: 1035–1045 [DOI] [PubMed] [Google Scholar]
- Kim HY, Kim DK, Bae S‐H, Gwak HyeRan, Jeon JH, Kim JK, Lee BI, You HJ, Shin DH, Kim Y‐H et al (2018) Farnesyl diphosphate synthase is important for the maintenance of glioblastoma stemness. Exp Mol Med 50: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Shim J, Ji M‐J, Jung Y, Bong SM, Jang Y‐J, Yoon E‐K, Lee S‐J, Kim KG, Kim YH et al (2014) The condensin component NCAPG2 regulates microtubule‐kinetochore attachment through recruitment of Polo‐like kinase 1 to kinetochores. Nat Commun 5: 4588 [DOI] [PubMed] [Google Scholar]
- Kim K, Oh J, Han D, Kim EE, Lee B, Kim Y (2006) Crystal structure of PilF: functional implication in the type 4 pilus biogenesis in Pseudomonas aeruginosa . Biochem Biophys Res Commun 340: 1028–1038 [DOI] [PubMed] [Google Scholar]
- Kim S, Leal SS, Ben Halevy D, Gomes CM, Lev S (2010) Structural requirements for VAP‐B oligomerization and their implication in amyotrophic lateral sclerosis‐associated VAP‐B(P56S) neurotoxicity. J Biol Chem 285: 13839–13849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornmann B, Currie E, Collins SR, Schuldiner M, Nunnari J, Weissman JS, Walter P (2009) An ER‐mitochondria tethering complex revealed by a synthetic biology screen. Science 325: 477–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim H, Kim K, Han D, Oh J, Kim Y (2007) Crystal structure of TTC0263, a thermophilic TPR protein from Thermus thermophilus HB27. Mol Cells 24: 27–36 [PubMed] [Google Scholar]
- Mikitova V, Levine TP (2012) Analysis of the key elements of FFAT‐like motifs identifies new proteins that potentially bind VAP on the ER, including two AKAPs and FAPP2. PLoS One 7: e30455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miliara X, Garnett JA, Tatsuta T, Abid Ali F, Baldie H, Pérez‐Dorado I, Simpson P, Yague E, Langer T, Matthews S (2015) Structural insight into the TRIAP1/PRELI‐like domain family of mitochondrial phospholipid transfer complexes. EMBO Rep 16: 824–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muallem S, Chung WY, Jha A, Ahuja M (2017) Lipids at membrane contact sites: cell signaling and ion transport. EMBO Rep 18: 1893–1904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum‐likelihood method. Acta Crystallogr D Biol Crystallogr 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X‐ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326 [DOI] [PubMed] [Google Scholar]
- Paillusson S, Stoica R, Gomez‐Suaga P, Lau DHW, Mueller S, Miller T, Miller CCJ (2016) There's something wrong with my MAM; the ER‐mitochondria axis and neurodegenerative diseases. Trends Neurosci 39: 146–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panagabko C, Baptist M, Atkinson J (2019) In vitro lipid transfer assays of phosphatidylinositol transfer proteins provide insight into the in vivo mechanism of ligand transfer. Biochim Biophys Acta 1861: 619–630 [DOI] [PubMed] [Google Scholar]
- Paradies G, Paradies V, De Benedictis V, Ruggiero FM, Petrosillo G (2014) Functional role of cardiolipin in mitochondrial bioenergetics. Biochim Biophys Acta 1837: 408–417 [DOI] [PubMed] [Google Scholar]
- Phillips MJ, Voeltz GK (2016) Structure and function of ER membrane contact sites with other organelles. Nat Rev Mol Cell Biol 17: 69–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potting C, Tatsuta T, Konig T, Haag M, Wai T, Aaltonen MJ, Langer T (2013) TRIAP1/PRELI complexes prevent apoptosis by mediating intramitochondrial transport of phosphatidic acid. Cell Metab 18: 287–295 [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T (1998) Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280: 1763–1766 [DOI] [PubMed] [Google Scholar]
- Rochin L, Sauvanet C, Jääskeläinen E, Houcine A, Arora A, Kivelä AM, Xingjie M, Marien E, Dehairs J, Bars RL et al (2020) ORP5 transfers phosphatidylserine to mitochondria and regulates mitochondrial calcium uptake at endoplasmic reticulum ‐ mitochondria contact sites. bioRxiv 10.1101/695577 [PREPRINT] [DOI]
- Rowland AA, Voeltz GK (2012) Endoplasmic reticulum‐mitochondria contacts: function of the junction. Nat Rev Mol Cell Biol 13: 607–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheich C, Kummel D, Soumailakakis D, Heinemann U, Bussow K (2007) Vectors for co‐expression of an unrestricted number of proteins. Nucleic Acids Res 35: e43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlattner U, Tokarska‐Schlattner M, Rousseau D, Boissan M, Mannella C, Epand R, Lacombe ML (2014) Mitochondrial cardiolipin/phospholipid trafficking: the role of membrane contact site complexes and lipid transfer proteins. Chem Phys Lipids 179: 32–41 [DOI] [PubMed] [Google Scholar]
- Schuck P (2000) Size‐distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J 78: 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenzinger A, Schreiner D, Koch P, Hofer HW, Wimmer M (2009) Cell and molecular biology of the novel protein tyrosine‐phosphatase‐interacting protein 51. Int Rev Cell Mol Biol 275: 183–246 [DOI] [PubMed] [Google Scholar]
- Stoica R, De Vos KJ, Paillusson S, Mueller S, Sancho RM, Lau K‐F, Vizcay‐Barrena G, Lin W‐L, Xu Y‐F, Lewis J et al (2014) ER‐mitochondria associations are regulated by the VAPB‐PTPIP51 interaction and are disrupted by ALS/FTD‐associated TDP‐43. Nat Commun 5: 3996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoica R, Paillusson S, Gomez‐Suaga P, Mitchell JC, Lau DH, Gray EH, Sancho RM, Vizcay‐Barrena G, De Vos KJ, Shaw CE et al (2016) ALS/FTD‐associated FUS activates GSK‐3beta to disrupt the VAPB‐PTPIP51 interaction and ER‐mitochondria associations. EMBO Rep 17: 1326–1342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura Y, Sesaki H, Endo T (2014) Phospholipid transport via mitochondria. Traffic 15: 933–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsuta T, Scharwey M, Langer T (2014) Mitochondrial lipid trafficking. Trends Cell Biol 24: 44–52 [DOI] [PubMed] [Google Scholar]
- Tian W, Chen C, Lei X, Zhao J, Liang J (2018) CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Res 46: W363–W367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent TL, Green PJ, Woolfson DN (2013) LOGICOIL–multi‐state prediction of coiled‐coil oligomeric state. Bioinformatics 29: 69–76 [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Tamura Y, Kawano S, Endo T (2015) Structural and mechanistic insights into phospholipid transfer by Ups1‐Mdm35 in mitochondria. Nat Commun 6: 7922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wideman JG, Balacco DL, Fieblinger T, Richards TA (2018) PDZD8 is not the ‘functional ortholog’ of Mmm1, it is a paralog. F1000Res 7: 1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Roe SM, Cliff MJ, Williams MA, Ladbury JE, Cohen PT, Barford D (2005) Molecular basis for TPR domain‐mediated regulation of protein phosphatase 5. EMBO J 24: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Liu Y, Gulbranson DR, Paine A, Rathore SS, Shen J (2016) Extended synaptotagmins are Ca<sup>2+</sup>‐dependent lipid transfer proteins at membrane contact sites. Proc Natl Acad Sci USA 113: 4362–4367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Kulkarni K, Hanrahan SJ, Thompson AJ, Barford D (2010) The APC/C subunit Cdc16/Cut9 is a contiguous tetratricopeptide repeat superhelix with a homo‐dimer interface similar to Cdc27. EMBO J 29: 3733–3744 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Review Process File
Data Availability Statement
Atomic coordinates and structural factors have been deposited in the Protein Data Bank (PDB) under accession number 7CC7 (https://www.rcsb.org/structure/7CC7). The mass spectrometry data from this publication have been deposited to the Metabolomics Workbench database and assigned Project ID PR001101 (https://www.metabolomicsworkbench.org/data/DRCCMetadata.php?Mode=Project&ProjectID=PR001101).