

Abstract
Quaking (QKI) proteins belong to the signal transduction and activation of RNA (STAR) family of RNA‐binding proteins that have multiple functions in RNA biology. Here, we show that QKI‐5 is dramatically decreased in metastatic lung adenocarcinoma (LUAD). QKI‐5 overexpression inhibits TGF‐β‐induced epithelial–mesenchymal transition (EMT) and invasion, whereas QKI‐5 knockdown has the opposite effect. QKI‐5 overexpression and silencing suppresses and promotes TGF‐β‐stimulated metastasis in vivo, respectively. QKI‐5 inhibits TGF‐β‐induced EMT and invasion in a TGFβR1‐dependent manner. KLF6 knockdown increases TGFβR1 expression and promotes TGF‐β‐induced EMT, which is partly abrogated by QKI‐5 overexpression. Mechanistically, QKI‐5 directly interacts with the TGFβR1 3′ UTR and causes post‐transcriptional degradation of TGFβR1 mRNA, thereby inhibiting TGF‐β‐induced SMAD3 phosphorylation and TGF‐β/SMAD signaling. QKI‐5 is positively regulated by KLF6 at the transcriptional level. In LUAD tissues, KLF6 is lowly expressed and positively correlated with QKI‐5 expression, while TGFβR1 expression is up‐regulated and inversely correlated with QKI‐5 expression. We reveal a novel mechanism by which KLF6 transcriptionally regulates QKI‐5 and suggest that targeting the KLF6/QKI‐5/TGFβR1 axis is a promising targeting strategy for metastatic LUAD.
Keywords: KLF6, metastasis, QKI‐5, TGF‐β‐induced EMT, TGFβR1
Subject Categories: Cancer, RNA Biology, Signal Transduction
The zinc finger transcription factor KLF6 and the RNA‐binding protein Quaking 5 are dramatically reduced in metastatic lung adenocarcinoma. KLF6 transcriptionally activates QKI‐5 and QKI‐5 post‐transcriptionally represses TGFβR1, thereby inhibiting EMT and invasion.

Introduction
Improving clinical outcomes for lung cancer (LC) is still challenging due to its high metastasis and mortality (Jett et al, 1983; Siegel et al, 2019). Non‐small‐cell lung cancer (NSCLC) accounts for ~ 85% of all LC cases, which includes lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), and large cell carcinoma (Chen et al, 2014). LUAD represents the most common subtype of NSCLC and has the highest heterogeneity and aggressiveness (Kuhn et al, 2018). Thus, it is of great importance and urgency to elucidate the mechanisms underlying LUAD metastasis.
Epithelial–mesenchymal transition (EMT) has been demonstrated to associate with tumor initiation, metastasis, and drug resistance (Du & Shim, 2016; Pastushenko & Blanpain, 2019). When cells undergo EMT, epithelial markers (e.g., E‐cadherin) are down‐regulated while mesenchymal markers (e.g., N‐cadherin and Snail) are up‐regulated, accompanied by a cuboidal‐to‐spindle shape shift. These changes confer tumor cells with malignant characteristics, including increased metastatic capabilities (Dongre & Weinberg, 2019). Transforming growth factor‐β (TGF‐β)/SMAD signaling plays a predominant role in triggering EMT (Lamouille et al, 2014). Our previous studies indicated that activation of TGF‐β/SMAD signaling can drive TGF‐β‐induced EMT in LUAD cells (Wang et al, 2016; Wang et al, 2018; Tong et al, 2020). TGF‐β receptor type I (TGFβR1), an indispensable upstream receptor in TGF‐β/SMAD signal transduction, initiates a cascade reaction through phosphorylating R‐SMADs and thereby activating the pathway (Schmierer & Hill, 2007). There is convincing evidence showing that dysregulation and dysfunction of TGFβR1 can affect tumor metastasis by regulating TGF‐β/SMAD signaling in NSCLC and breast cancer (Fang et al, 2013; Wang et al, 2015; Kudinov et al, 2016).
Quaking (QKI) proteins, including three major isoforms QKI‐5, QKI‐6, and QKI‐7, belong to the signal transduction and activation of RNA (STAR) family of RNA‐binding proteins (RBPs). QKIs exert multiple functions in RNA biology, thereby affecting transcription, translation, shuttling between nucleus and cytoplasm, and non‐coding RNA processing (Larocque et al, 2002; Wang et al, 2013; Zong et al, 2014; Conn et al, 2015; Zhou et al, 2017). Unlike QKI‐6 and QKI‐7 found in the cytoplasm, QKI‐5 is mainly localized to the nucleus because it has a nuclear localization signal (Wu et al, 1999). It was reported that QKI‐5 was frequently reduced in lung cancer (Zong et al, 2014; Zhou et al, 2017), suggesting a tumor‐suppressive role of QKI‐5 in lung cancer. Most recently, Liang et al (2020) found that QKI‐5 exhibited anticancer potential by inhibiting miR‐196b‐5p in NSCLC. To date, QKI‐5 was identified to inhibit LUAD aggressiveness through preventing β‐catenin activation (Zhou et al, 2017). However, an involvement of QKI‐5 in TGF‐β/SMAD signaling and its underlying mechanisms remain unclear in LUAD. Moreover, the reason why QKI‐5 is down‐regulated in LUAD needs to be further elucidated.
Krüppel‐like factor 6 (KLF6) is a broadly expressed zinc finger transcription factor, which has been found to be frequently decreased or inactivated in several human cancers, such as prostate cancer, colorectal cancer, and NSCLC (Narla et al, 2001; Ito et al, 2004; Reeves et al, 2004). Given that we have previously demonstrated that the transcriptional regulation of oncogenes or tumor suppressors participates in TGF‐β‐induced EMT and NSCLC metastasis (Wang et al, 2016; Tong et al, 2020) and a series of computational algorithms predicted that two putative KLF6 binding sites (KBEs) are harbored in QKI‐5 gene promoter, it was proposed that KLF6 may transcriptionally regulate QKI‐5 during LUAD metastasis.
Here, we have screened for metastatic LUAD‐associated RBPs and investigated whether QKI‐5 contributes to TGF‐β‐induced EMT and metastasis by regulating TGF‐β/SMAD signaling in LUADs. We have explored the novel mechanism by which KLF6 transcriptionally regulates QKI‐5 in LUADs and find that QKI‐5 can prevent TGF‐β/SMAD signaling by post‐transcriptionally inhibiting TGFβR1 mRNA and thereby repressing TGF‐β‐induced EMT and metastasis of LUAD cells. Intriguingly, we also reveal that QKI‐5 is positively regulated by KLF6 at the transcriptional level. Our findings discover a KLF6/QKI‐5/TGFβR1 axis that blocks TGF‐β/SMAD signaling, providing new insight for a better understanding of TGF‐β‐induced EMT and metastasis in LUAD.
Results
Identification of metastatic LUAD‐associated QKI‐5
In order to identify LUAD‐associated RBPs, we firstly screened for the differentially expressed genes in 4 independent data sets, which meet the following criteria: (i) For TCGA_LUAD database (https://gdc.cancer.gov/), false discovery rate (FDR) < 0.01 and the absolute value of log2 fold change (abs.log2FC) > 1 were applied; (ii) for lung cancer data sets of GSE40791, GSE75037, and GSE7670 from Gene Expression Omnibus (GEO) database (Chen et al, 2009; Zhang et al, 2012; Girard et al, 2016), adjusted P‐value < 0.01 and abs.log2FC > 1 were applied. Subsequently, ATtRACT database (Giudice et al, 2016) containing all human RBPs was intersected with the aforementioned four data sets to yield the differentially expressed RBPs. In total, we identified four differentially expressed RBPs associated with LUAD: ADARB1, CELF2, QKI, and ZFP36 (Fig 1A). Next, we detected the expression of these 4 LUAD‐associated RBPs by qRT–PCR in 61 LUAD and matched paracarcinoma tissues, and LUAD tissues showed significant reduction in three RBPs (CELF2, QKI, and ZFP36) when compared with paracarcinoma tissues (Fig 1B upper panel and Appendix Table S1). However, we found that only QKI expression was significantly down‐regulated in metastatic LUAD tissues (n = 28) compared with that in non‐metastatic counterparts (n = 33), when 61 paired LUAD samples were divided into metastatic and non‐metastatic groups (Fig 1B lower panel and Appendix Table S1). Furthermore, clinical data analysis was performed to investigate the role of QKI in the progression and prognosis of LUAD. As a result, TCGA and CPTAC analysis (UALCAN, http://ualcan.path.uab.edu/) showed that QKI mRNA and protein levels were lower in LUAD tissues than those in normal lung tissues, and QKI expression was negatively correlated with the clinical stage of LUAD (Fig 1C and D). Kaplan–Meier survival analysis (http://kmplot.com/analysis/) showed that low expression of QKI was significantly associated with poor survival in LUAD patients, but not in lung squamous carcinoma (LUSC; Fig 1E–G). Collectively, these data suggest that QKI is linked to LUAD metastasis.
Figure 1. Identification of metastasis‐associated QKI in human LUAD.

- A
-
BThe upper panel showing qRT–PCR analysis of four identified LUAD‐associated RBPs (ADARB1, CELF2, QKI, and ZFP36) mRNA levels in 61 LUAD tissues and matched paracarcinoma tissues. The lower panel showing relative mRNA expression (T/N) of the four RBPs in metastatic (n = 28) and non‐metastatic (n = 33) LUAD tissues. LUAD tissues were classified into metastatic and non‐metastatic tissues as described in Materials and Methods. T, LUAD tissues; N, paracarcinoma tissues. Y axis represents the log10‐transformed fold change of mRNA expression (T/N) of four RBPs. Data are shown as the mean ± SD. ns, not significant; **P < 0.01 and ***P < 0.001 by unpaired Student's t‐test.
-
C, DComparison of QKI mRNA and protein expression between normal lung tissues and LUAD tissues, which were stratified by clinical stages (Stages 1–4) in TCGA database. Data are shown as the boxplot. Central band: median; upper and lower line of boxes: upper and lower quartile; upper and lower line of whiskers: maximum and minimum. P < 0.001 compared to normal lung tissues by unpaired Student's t‐test.
-
E–GKaplan–Meier survival curves (http://kmplot.com/analysis/) of LC, LUAD, and LUSC patients (n = 1,540, n = 865, and n = 675, respectively) with high or low expression levels of QKI. The log‐rank test was used to analyze the difference between two groups.
-
HqRT–PCR analysis of endogenous mRNA levels of QKI‐5, QKI‐6, and QKI‐7 in human bronchial epithelial cell line BEAS‐2B, LUAD cell lines A549 and H1299. Data are shown as the mean ± SD of n = 3 technical replicates. ***P < 0.001 compared to BEAS‐2B cells by unpaired Student's t‐test.
-
IWestern blot analysis of QKI‐5 protein levels in BEAS‐2B, A549, and H1299 cells. β‐actin was used as an internal control.
-
J, KqRT–PCR analysis of QKI‐5 mRNA in 61 LUAD tissues and matched paracarcinoma tissues (J) and T/N mRNA expression of QKI‐5 in 61 paired LUAD tissues (K). T, LUAD tissues; N, paracarcinoma tissues. Y axis represents the log10‐transformed fold change of mRNA expression of QKI‐5. In the violin plot, central dotted line represents median, and upper and lower dotted line represents quartile (J). ***P < 0.001 by unpaired Student's t‐test.
-
LViolin plots showing QKI‐5 mRNA expression (T/N) in metastatic (n = 28) and non‐metastatic (n = 33) LUAD tissues. Central dotted line of the violin plot represents median, and upper and lower dotted line represents quartile. *P < 0.05 by unpaired Student's t‐test.
Source data are available online for this figure.
In fact, QKI has three major isoforms, including QKI‐5, QKI‐6, and QKI‐7. QKI‐5 is mainly localized in the nucleus due to its nuclear localization signal, while QKI‐6 and QKI‐7 are in the cytoplasm (Wu et al, 1999). In this study, QKI‐5 mRNA expression was far higher than QKI‐6 and QKI‐7 in LUAD cell lines A549 and H1299 (Fig 1H), and QKI‐5 mRNA and protein expression was significantly lower in A549 and H1299 cells than that in lung normal epithelial cell line BEAS‐2B (Fig 1H and I). Moreover, the data regarding QKI‐5 can be reflected in LUAD tissues of patients (n = 61), which showed a significant reduction in QKI‐5 mRNA expression (47/61, 77.1%) when compared with matched paracarcinoma tissues (Fig 1J and K). Importantly, QKI‐5 mRNA expression was lower in metastatic LUAD tissues than that in non‐metastatic counterparts (Fig 1L, Appendix Tables S2 and S3). Therefore, we will focus on QKI‐5 to explore the mechanisms behind its metastasis‐suppressing potential in LUADs.
QKI‐5 directly interacts with TGFβR1 mRNA by targeting TGFβR1 3′ UTR in LUAD cells
Next, as shown in Fig 2A, we identified 2,511 potential QKI‐targeted genes by integrating 3 RBP‐binding sites database (POSTAR, EuRBPDB, and doRiNA) (Blin et al, 2015; Zhu et al, 2019; Liao et al, 2020). Interestingly, Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of these 2511 QKI‐targeted genes revealed that 22 genes were part of the TGF‐β signaling pathway (Fig 2B). Among these 22 genes, TGFβR1, an upstream receptor that initiates TGF‐β/SMAD signaling, has attracted our attention because we are interested in studying the crucial role of TGF‐β signaling in NSCLC metastasis (Wang et al, 2016; Wang et al, 2018; Tong et al, 2020). Thus, we performed nuclear/cytosol fractionation experiments and found that QKI‐5 was predominantly distributed in the nucleus (Fig 2C), being consistent with the previous report (Wu et al, 1999). Subsequently, we performed RIP using anti‐QKI‐5 antibody and qRT–PCR assays in nuclear and cytoplasmic extracts from A549 and H1299 cells. The results indicated that QKI‐5 can specifically bind to TGFβR1 mRNA in the nucleus of A549 and H1299 cells (Fig 2D and E).
Figure 2. QKI‐5 directly interacts with TGFβR1 mRNA by targeting TGFβR1 3′ UTR in LUAD cells.

- The Venn diagram depicts the identification of 2,511 QKI‐targeted genes overlapping between three RBP‐binding sites databases (POSTAR, EuRBPDB, and doRiNA).
- KEGG enrichment analysis was performed by clusterProfiler in R language. Top 20 interested KEGG enrichment terms of 2,511 QKI‐targeted genes identified in (A) were shown in left panel. A total of 22 genes were enriched in TGF‐β signaling pathway, which were presented in right panel.
- Western blot analysis of QKI‐5 expression in nuclear and cytoplasmic fractions extracted from A549 and H1299 cells. Lamin B and β‐actin were used as internal controls in nuclear and cytoplasmic extracts, respectively.
- Anti‐QKI‐5 RIP was conducted in nuclear and cytoplasmic extracts from A549 cells and H1299 cells, and followed by RT–PCR and gel‐staining analyses to examine TGFβR1 mRNA enrichment. The input and anti‐IgG antibody were used as positive and negative controls, respectively. GAPDH served as an internal RNA control in RT–PCR analysis.
- After RIP assay, endogenous TGFβR1 mRNA was enriched from A549 cells and H1299 cells and determined by qRT–PCR. Results were shown as the percentage of the enrichment to input. Data are shown as the mean ± SD of n = 3 technical replicates. **P < 0.01; ***P < 0.001 by unpaired Student's t‐test.
- A conserved consensus QKI‐5 response element (QRE) was presented.
- Schematic drawing shows two predicted QREs in TGFβR1 mRNA 3′ UTR (upper panel). Boxed areas indicate several TGFβR1 3′ UTR segments (positions, 2,058–2,557) containing wild type (TGFβR1 3′ UTR‐WT) or mutants of QRE‐1/2 (TGFβR1 3′ UTR‐Mut1, TGFβR1 3′ UTR‐Mut2, and TGFβR1 3′ UTR‐Mut1*2; lower panel), which are prepared to subclone into pGL3‐Luc reporter vector. Numbers represent the nucleotide position of TGFβR1 mRNA.
- Western blot analyses of QKI‐5 expressed in A549 and H1299 cells stably overexpressing Flag‐tagged QKI‐5.
- Left panel: schematic drawing of the pGL3‐TGFβR1 3′ UTR luciferase reporter assay system. Middle and right panels: QKI‐5‐overexpressing and vector A549 and H1299 cells were transfected with pGL3‐Luc reporter plasmids that were made by fusing the above‐mentioned TGFβR1 3′ UTR segments. Relative luciferase activities were determined as described in Materials and Methods and normalized to those in vector cells. Data are shown as the mean ± SD of n = 3 technical replicates. *P < 0.05, **P < 0.01 and ***P < 0.001 by unpaired Student's t‐test.
- In vitro RNA pull‐down assays were performed using biotin‐labeled TGFβR1 3′ UTR segments in A549 and H1299 cells. After pull‐down, endogenous QKI‐5 enrichments were detected by Western blot analyses. β‐actin was used as the protein control.
Source data are available online for this figure.
It has been well documented that QKI regulates gene expression by binding to 3′ UTR of target mRNAs via a specific QKI response element [QRE, 5′‐A(C/U)UAAY] (Fig 2F) (Galarneau & Richard, 2005; Thangaraj et al, 2017). Sequence analysis revealed that there were two putative QREs at positions 2,108–2,113 and 2,486–2,491 in TGFβR1 3′ UTR, termed as QRE‐1 and QRE‐2 (Fig 2G upper panel). To verify whether the two predicted QREs affect the post‐transcriptional regulation of TGFβR1 mRNA through interacting with QKI‐5, we prepared a series of TGFβR1 3′ UTR fragments containing wild‐type or mutant QRE‐1/2 (Fig 2G lower panel and Appendix Table S4) and overexpressed QKI‐5 via lentiviral expression system in A549 and H1299 cells (Fig 2H). Then, we created various constructs encompassing above‐mentioned TGFβR1 3′ UTR fragments using Dual‐Luciferase reporter vector (Fig 2I left panel) and transfected them into A549 and H1299 cells overexpressing QKI‐5. Luciferase reporter assay showed that QKI‐5 overexpression resulted in a dramatic reduction in luciferase activity of TGFβR1 3′ UTR. Importantly, we found that either the QRE‐2 mutation or the combined mutations of QRE‐1 and QRE‐2, but not the QRE‐1 mutation, abolished the inhibitory effects of QKI‐5 overexpression on luciferase activity in A549 and H1299 cells (Fig 2I middle and right panels). To further confirm these luciferase data, we performed in vitro RNA pull‐down assays in A549 and H1299 cells and detected that QKI‐5 protein was obviously pulled down by biotin‐labeled TGFβR1 3′ UTR fragments carrying wild‐type QRE‐1/2 or mutant QRE‐1 (Fig 2J), suggesting that QRE‐2 of TGFβR1 mRNA is a target site of QKI‐5. Overall, these findings demonstrate that QKI‐5 directly interacts with TGFβR1 mRNA through binding to TGFβR1 3′ UTR in LUAD cells.
QKI‐5 negatively regulates TGFβR1 and TGF‐β/SMAD signaling in LUAD cells
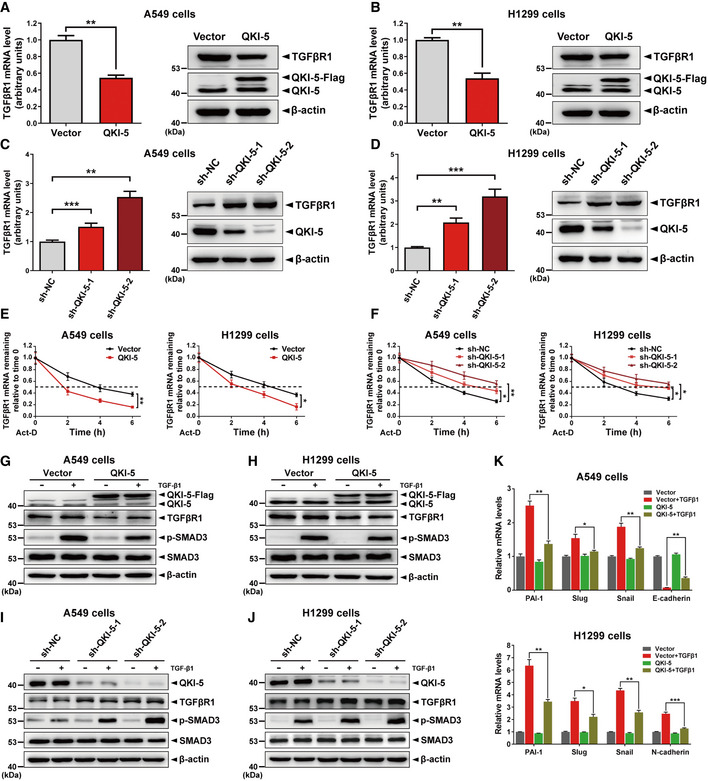
Based on the aforementioned findings, we speculated that QKI‐5 may post‐transcriptionally regulate the expression of TGFβR1 gene in LUAD cells. To test this, we performed qRT–PCR and Western blot assays to measure TGFβR1 mRNA and protein levels in A549 and H1299 cells stably overexpressing or silencing QKI‐5. We found that QKI‐5 overexpression dramatically reduced the expression of TGFβR1 at both mRNA and protein levels in A549 and H1299 cells (Fig 3A and B). On the contrary, knockdown of QKI‐5 significantly increased the TGFβR1 mRNA and protein expression in A549 and H1299 cells (Fig 3C and D). These data suggested that QKI‐5 suppressed TGFβR1 expression in LUAD cells. Accumulating evidence suggests that QKI plays critical and diverse biological roles in regulating RNA, including mRNA stability and translational efficiency (Saccomanno et al, 1999; Larocque et al, 2002; Wang et al, 2013; Zhou et al, 2017). In the present study, we conducted a standard mRNA stability assay (Rodrigues et al, 2016) to investigate whether QKI‐5 affected the stability of TGFβR1 mRNA. As expected, upon actinomycin D treatment, TGFβR1 mRNA expression was inhibited and decreased in time‐dependent manner (Fig 3E and F). Importantly, TGFβR1 mRNA reduction was significantly faster in QKI‐5‐overexpressing A549 and H1299 cells than that in control cells (Fig 3E), whereas the opposite tendency occurred in QKI‐5‐silenced A549 and H1299 cells (Fig 3F). In summary, these results indicate that QKI‐5 promotes TGFβR1 mRNA instability, supporting that QKI‐5 is capable of accelerating TGFβR1 mRNA degradation at post‐transcriptional level in LUAD cells.
Figure 3. QKI‐5 negatively regulates TGFβR1 and TGF‐β/SMAD signaling in LUAD cells.
-
A, BqRT–PCR and Western blot analyses of TGFβR1 mRNA and protein levels in QKI‐5‐overexpressing A549 and H1299 cells. Data are shown as the mean ± SD of n = 3 technical replicates. **P < 0.01 by unpaired Student's t‐test.
-
C, DExpression of TGFβR1 mRNA and protein in QKI‐5‐silenced A549 and H1299 cells. **P < 0.01 and ***P < 0.001 by unpaired Student's t‐test.
-
E, FThe mRNA stability of TGFβR1 was examined in QKI‐5‐overexpressing/‐silenced and control A549 and H1299 cells. Cells were treated with actinomycin D (Act‐D, 10 μg/ml) for 0, 2, 4, or 6 h, and mRNA levels of TGFβR1 were determined by qRT–PCR. The quantity value of qRT–PCR at 0 h was set 1. *P < 0.05 and **P < 0.01 by unpaired Student's t‐test.
-
G, HAfter being serum‐starved for 24 h, A549 and H1299 cells stably overexpressing QKI‐5 were treated with or without TGF‐β1 (5 or 10 ng/ml) for 24 h and subjected to Western blot assays to determine the protein levels of QKI‐5, TGFβR1, p‐SMAD3, and SMAD3. β‐actin was used as an internal control.
-
I, JQKI‐5‐silenced A549 and H1299 cells were treated as above. Then, Western blot analysis was conducted to examine the protein levels of QKI‐5, TGFβR1, p‐SMAD3, and SMAD3.
-
KQKI‐5‐overexpressing A549 and H1299 cells were treated as above and subjected to qRT–PCR assays to detect the mRNA expression of downstream genes of the TGF‐β/SMAD signaling, including PAI‐1, Slug, Snail, E‐cadherin, and/or N‐cadherin. Data are shown as the mean ± SD of n = 3 technical replicates. *P < 0.05, **P < 0.01 and ***P < 0.001 by unpaired Student's t‐test.
Source data are available online for this figure.
Given the fact that TGFβR1 acts as an indispensable receptor in TGF‐β/SMAD signal transduction (Schmierer & Hill, 2007), we are inspired to explore whether QKI‐5 regulates TGF‐β/SMAD signaling in LUAD cells. In addition, TGFβR1 directly phosphorylates SMAD3, which is a hallmark of TGF‐β/SMAD activation (ten Dijke & Hill, 2004). Therefore, we detected TGF‐β‐induced phosphorylation of SMAD3 (p‐SMAD3) in QKI‐5‐overexpressing or QKI‐5‐silenced LUAD cells. Upon TGF‐β1 stimulation, QKI‐5 overexpression significantly inhibited the expression levels of p‐SMAD3 under the precondition of not changing total SMAD3 protein levels in A549 and H1299 cells (Fig 3G and H). In contrast, shRNA‐mediated silencing of QKI‐5 enhanced the expression levels of p‐SMAD3 in the presence of TGF‐β1 (Fig 3I and J). Moreover, upon TGF‐β1 stimulation, QKI‐5‐overexpressing A549 and H1299 cells showed significant changes in the expression of downstream genes of TGF‐β/SMAD signaling, including PAI‐1, Slug, Snail, E‐cadherin, and/or N‐cadherin (Fig 3K). Taken together, our results reveal that QKI‐5 can negatively regulate TGFβR1, thereby undermining the TGF‐β/SMAD signaling pathway in LUAD cells.
QKI‐5 represses TGF‐β‐induced EMT and migration and invasion of LUAD cells
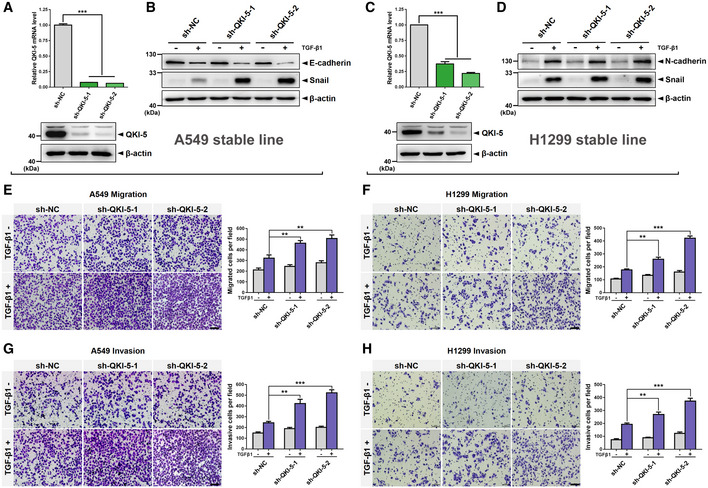
According to the above findings and our previous studies that TGF‐β/SMAD activation promotes EMT and invasion of NSCLC cells (Wang et al, 2016; Tong et al, 2020), we hypothesized that QKI‐5 may inhibit TGF‐β‐induced EMT and migration and invasion of LUAD cells. Upon TGF‐β1 stimulation, QKI‐5‐overexpressing A549 and H1299 cells exhibited significantly reduced protein levels of mesenchymal markers N‐cadherin and Snail (Fig 4A). Moreover, we carried out IF assays using F‐actin antibody to label the cytoskeleton of QKI‐5‐overexpressing LUAD cells with and without TGF‐β1 treatment and examined cell morphology. After treatment with TGF‐β1 for 24 h, A549 and H1299 cells expressing empty vector displayed the typical spindle‐shape mesenchymal morphology, whereas A549 and H1299 cells overexpressing QKI‐5 largely reversed the mesenchymal morphology (Fig 4B). Wound‐healing migration assay, as well as Transwell migration and invasion assays, showed that QKI‐5 overexpression significantly suppressed TGF‐β‐induced migratory and invasive capabilities of A549 and H1299 cells (Fig 4C–J). Furthermore, shRNA‐mediated silencing of QKI‐5 effectively promoted TGF‐β‐induced EMT by downregulating E‐cadherin or upregulating N‐cadherin and Snail in A549 and H1299 cells (Fig EV1A–D) and enhanced TGF‐β‐induced migratory and invasive abilities (Fig EV1E–H). Collectively, these results show that QKI‐5 inhibits TGF‐β‐induced EMT and migration and invasion of LUAD cells.
Figure 4. QKI‐5 overexpression inhibits TGF‐β‐induced EMT and migration and invasion of LUAD cells.

-
AAfter serum starvation for 24 h, A549 and H1299 cells overexpressing QKI‐5 were treated with or without TGF‐β1 (5 or 10 ng/ml) for 24 h. Then, EMT markers N‐cadherin and Snail expression were analyzed using Western blot. β‐actin served as an internal control.
-
BQKI‐5‐overexpressing A549 and H1299 cells and control cells were treated as above and incubated with a mouse anti‐F‐actin antibody and then stained with a FITC‐conjugated anti‐mouse IgG (green, for F‐actin). Cell nuclei were counterstained and visualized with DAPI (blue). Scale bar, 100 μm.
-
C–FWound‐healing migration assays were performed on QKI‐5‐overexpressing A549 and H1299 cells as well as control cells, which were treated as above. The wound healing was recorded (C, E) and quantitatively measured (D, F) at least six times. Data are shown as the mean ± SD of n = 6 technical replicates. **P < 0.01 and ***P < 0.001 by unpaired Student's t‐test. Scale bar, 100 μm.
-
G, HIn the presence or absence of TGF‐β1, QKI‐5‐overexpressing A549 and H1299 cells were allowed to migrate through a polycarbonate membrane in Transwells. After 24 h, migrated cells were stained, photographed, and counted in at least four random fields under a light microscope. Representative images (left) and the migrated cell numbers (right) were presented. Data are shown as the mean ± SD of n = 3 technical replicates. **P < 0.01 by unpaired Student's t‐test. Scale bar, 100 μm.
-
I, JQKI‐5‐overexpressing A549 and H1299 cells were treated as above and allowed to invade through Matrigel‐coated membrane in Transwells. Invasive cells were determined as described above. Scale bar, 100 μm. Data are shown as the mean ± SD of n = 3 technical replicates. **P < 0.01 by unpaired Student's t‐test.
Source data are available online for this figure.
Figure EV1. Silencing of QKI‐5 promotes TGF‐β‐induced EMT and LUAD cell invasion.
-
A–DAfter serum starvation for 24 h, QKI‐5‐silenced A549 cells and H1299 cells (A, C) were stimulated with or without TGF‐β1 (5 or 10 ng/ml) for 24 h. Data are shown as the mean ± SD of n = 3 technical replicates. ***P < 0.001 compared to sh‐NC by unpaired Student's t‐test. The expression of EMT markers (E‐cadherin or N‐cadherin and Snail) was determined using Western blot. β‐actin was used as an internal control (B, D).
-
E–HIn the presence or absence of TGF‐β1, the migratory and invasive abilities of QKI‐5‐silenced A549 and H1299 cells were evaluated by Transwell assays as described in Fig 4G–J. Migrated and invasive cells were stained and counted under a light microscope. Scale bar, 100 μm. Data are shown as the mean ± SD of n = 3 technical replicates. **P < 0.01 and ***P < 0.001 by unpaired Student's t‐test.
Source data are available online for this figure.
QKI‐5 attenuates TGF‐β‐stimulated LUAD cell metastasis in vivo
To further elucidate the function of QKI‐5 exerting in LUAD cell metastasis in vivo, we injected BALB/c nude mice intravenously (i.v.) with A549 cells stably overexpressing QKI‐5 and vector control A549 cells, followed by injecting intraperitoneally (i.p.) with TGF‐β1, established an in vivo model of LUAD metastasis (Fig 5A and B). At eight weeks post‐inoculation, the mice were euthanized and their lungs and livers were surgically resected for evaluation of metastases and histology (Fig 5A). As shown in Fig 5C and D, the mice injected with QKI‐5‐overexpressing A549 cells developed fewer metastatic lung nodules than those injected with control A549 cells. The metastatic liver nodules were macroscopically undetectable in mice. However, the results of hematoxylin‐eosin (H&E) staining showed that the injection of QKI‐5‐overexpressing A549 cells resulted in significantly fewer micrometastatic foci in lung and liver tissues as compared with vector control group (Fig 5E–H). Moreover, the mice injected with QKI‐5‐silenced A549 cells exhibited more metastatic lung nodules and micrometastatic foci than those injected with control cells (Fig 5E–K). Taken together, the in vivo metastasis assays show that QKI‐5 suppresses TGF‐β‐stimulated metastasis of LUAD cells, further supporting our in vitro findings.
Figure 5. QKI‐5 suppresses TGF‐β‐stimulated LUAD cell metastasis in vivo .

-
ASchematic flowchart of LUAD cell in vivo metastasis model. QKI‐5‐overexpression and control vector A549 cells (3 × 106 cells/mouse) were i.v. injected into BALB/c nude mice (10 mice per group). TGF‐β1 was injected i.p. at day 1, 6, 11, 16 after cell injection to achieve TGF‐β‐stimulated A549 cell metastasis.
-
BBefore cell injection, QKI‐5 was confirmed to overexpress in A549 cells using qRT–PCR and Western blot. Data are shown as the mean ± SD of n = 3 technical replicates. ***P < 0.001 by unpaired Student's t‐test.
-
CRepresentative images of lung metastatic nodules developed in mice 8 weeks after injection of QKI‐5‐overexpressing A549 cells or control A549 cells. The surgically resected lungs were stained as described in Materials and Methods. Red arrowheads indicate metastatic nodules established in lungs. Scale bar, 5 mm.
-
DComparison of the number of lung metastatic nodules between QKI‐5‐overexpressing group and vector group (n = 10 mice per group). Data are shown as the boxplot. Central band: median; upper and lower line of boxes: upper and lower quartile; upper and lower line of whiskers: maximum and minimum. Data are shown as the mean ± SEM. **P < 0.01 by unpaired Student's t‐test.
-
EH&E staining was performed for the evaluation of lung micrometastases. Representative images showing micrometastases of lung tissues from a pair of mice referred in (C). Blue and red arrowheads indicate lung micrometastases of vector group and QKI‐5‐overexpressing group, respectively. Scale bar: 100 μm.
-
FDot plots showing the difference of the micrometastases counts in lung tissues between QKI‐5‐overexpressing group and vector control group (n = 10 mice per group). Data are shown as the mean ± SEM. *P < 0.05 by unpaired Student's t‐test.
-
GRepresentative microscopic photographs of H&E staining for liver micrometastases in a pair of mice referred in (E). Blue and red arrowheads indicate liver micrometastases. Scale bar: 100 μm.
-
HDot plots showing the difference of the micrometastases counts in liver tissues between QKI‐5‐overexpressing group and control vector group (n = 10 mice per group). Data are shown as the mean ± SEM. *P < 0.05 by unpaired Student's t‐test.
-
ILeft part: Photographs of murine lungs with metastatic nodules after inoculation of sh‐QKI‐5‐2 or sh‐NC A549 cells. Scale bar, 5 mm. Right part: representative microscopic images of H&E staining for micrometastatic foci established in lungs from a pair of mice referred in left part. Scale bar, 1 mm. Red arrowheads indicate metastatic nodules and micrometastatic foci in lungs.
-
J, KDot plots showing the difference of the metastatic nodules (J) and micrometastases counts (K) in lung tissues between QKI‐5‐silenced group and control group (n = 5 mice per group). In the boxplot, central band represents median, and upper and lower line of boxes represents upper and lower quartile, upper and lower line of whiskers represents maximum and minimum (J). Data are shown as the mean ± SEM. ***P < 0.001 by unpaired Student's t‐test.
Source data are available online for this figure.
Knockdown of TGFβR1 suppresses TGF‐β‐induced EMT and invasion of LUAD cells
Since QKI‐5 overexpression effectively reduced TGFβR1 expression (Fig 3A and B) and repressed TGF‐β‐induced EMT and LUAD cell metastasis in vitro and in vivo (Figs 4A–J and 5C–H), we next explored whether TGFβR1 silencing could copy the phenotype caused by QKI‐5 overexpression. As illustrated in Fig EV2A–D, shRNA‐mediated silencing of TGFβR1 decreased the expression levels of p‐SMAD3 and mesenchymal markers N‐cadherin and Snail in A549 and H1299 treated with TGF‐β1. Furthermore, Transwell assays indicated that TGFβR1 silencing markedly inhibited TGF‐β‐induced migratory and invasive capabilities of A549 and H1299 cells (Fig EV2E–H). Taken together, our results show that TGFβR1 silencing can block TGF‐β/SMAD signaling and inhibit TGF‐β‐induced EMT and invasion of LUAD cells, which mimics the phenotypic changes resulted from QKI‐5 overexpression in LUADs.
Figure EV2. Knockdown of TGFβR1 suppresses TGF‐β‐induced EMT and invasion of LUAD cells.

-
A–DAfter serum starvation for 24 h, TGFβR1‐silenced A549 and H1299 cells (A, C) were stimulated with or without TGF‐β1 (5 or 10 ng/ml) for 24 h. Data are shown as the mean ± SD of n = 3 technical replicates. ***P < 0.001 compared to sh‐NC by unpaired Student's t‐test. The expression of p‐SMAD3, SMAD3, and EMT markers (N‐cadherin and Snail) was determined using Western blot. β‐actin was used as an internal control (B, D).
-
E–HIn the presence or absence of TGF‐β1, the migratory and invasive capabilities of TGFβR1‐silenced A549 and H1299 cells were evaluated by Transwell assays as described in Fig 4G–J. Migrated and invasive cells were stained and counted under a light microscope. Scale bar, 100 μm. Data are shown as the mean ± SD of n = 3 technical replicates. ***P < 0.001 by unpaired Student's t‐test.
Source data are available online for this figure.sou
QKI‐5 inhibits TGF‐β‐induced EMT and cell invasion via a TGFβR1‐dependent manner
Our aforementioned results indicate that QKI‐5 not only represses TGFβR1, but also inhibits TGF‐β‐induced EMT and invasion in LUAD cells. To address the question of whether QKI‐5 regulates these biological processes in a TGFβR1‐dependent manner, we first performed a direct rescue experiment in A549 and H1299 cells overexpressing QKI‐5 and TGFβR1 ORF‐only. TGFβR1 cDNA, lacks the 3′UTR containing QREs, not only restored the TGFβR1 expression repressed by QKI‐5 overexpression in A549 and H1299 cells (Fig 6A) but also abolished the inhibition of p‐SMAD3, N‐cadherin, and Snail caused by QKI‐5 overexpression in A549 and H1299 cells treated with TGF‐β1 (Fig 6B). Importantly, the TGFβR1 cDNA overexpression rescued TGF‐β‐induced migratory and invasive capabilities of A549 and H1299 cells overexpressing QKI‐5 in the presence of TGF‐β1 (Fig 6C–F). Moreover, TGFβR1 mRNA expression was significantly up‐regulated (51/61, 83.6%) in 61 LUAD tissues (Fig 6G and H) and negatively correlated with QKI‐5 mRNA expression (Figs 6I and 1K). Importantly, TGFβR1 mRNA expression was higher in metastatic LUAD tissues than that in non‐metastatic counterparts (Appendix Tables S2 and S3). Kaplan–Meier survival analysis (http://kmplot.com/analysis/) showed that LUAD patients with high expression of TGFβR1 had a significantly poor survival (Fig 6J).
Figure 6. QKI‐5 inhibits TGF‐β‐induced EMT and cell invasion via a TGFβR1‐dependent manner.

-
AA549 and H1299 cells were co‐transfected with QKI‐5 and ORF‐only of TGFβR1 and then subjected to Western blot assays for detection of QKI‐5 and TGFβR1 expression.
-
BAfter serum starvation for 24 h, the above‐treated A549 and H1299 cells were stimulated with or without TGF‐β1 (5 or 10 ng/ml) for 24 h. The expression of p‐SMAD3, SMAD3, and EMT markers (N‐cadherin and Snail) was determined using Western blot. β‐actin was used as an internal control.
-
C–FIn the presence or absence of TGF‐β1, the migratory and invasive capabilities of above‐treated A549 and H1299 cells were evaluated by Transwell assays as described in Fig 4G–J. Migrated and invasive cells were stained and counted under a light microscope. Data are shown as the mean ± SD of n = 3 technical replicates. **P < 0.01 and ***P < 0.001 by unpaired Student's t‐test. Scale bar, 100 μm.
-
G, HqRT–PCR analysis of TGFβR1 mRNA in 61 LUAD tissues and matched paracarcinoma tissues (G) and T/N mRNA expression of TGFβR1 in 61 paired LUAD tissues (H). T, LUAD tissues; N, paracarcinoma tissues. Y axis represents the log10‐transformed fold change of mRNA expression of TGFβR1. Central dotted line of the violin plot represents median, and upper and lower dotted line represents quartile (G). ****P < 0.0001 by unpaired Student's t‐test.
-
ICorrelation between QKI‐5 and TGFβR1 mRNA expression in 61 paired LUAD tissues. X and y axes represent the log10‐transformed fold change of T/N expression ratios of QKI‐5 and TGFβR1 mRNA, respectively. P < 0.0001 by Pearson's correlation test.
-
JKaplan–Meier survival curves (http://kmplot.com/analysis/) of LUAD patients (n = 865) with high or low expression levels of TGFβR1. The log‐rank test was used to compare the difference between two groups.
Source data are available online for this figure.
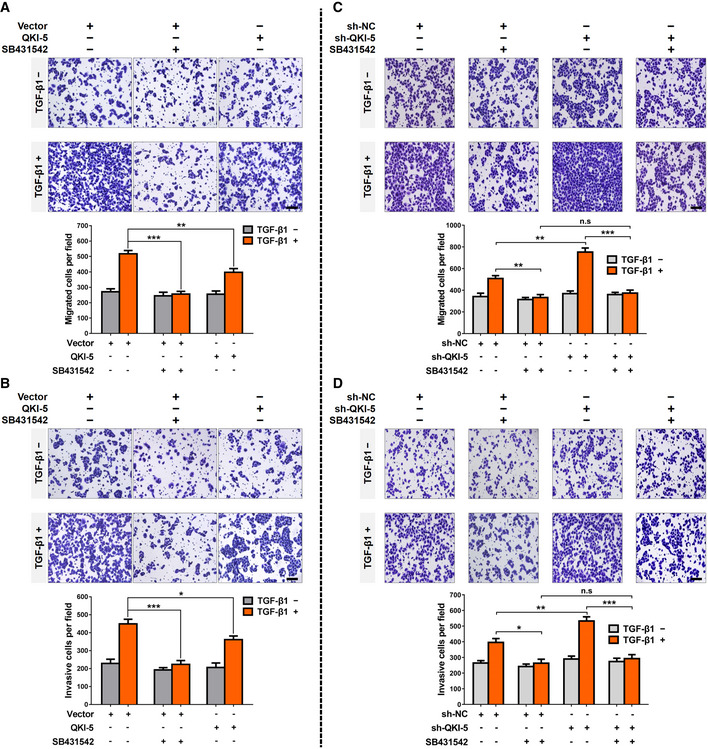
Furthermore, we treated A549 cells with SB431542, a specific TGFβR1 kinase inhibitor (Mikami et al, 2006; Muppala et al, 2017), blocked TGF‐β/SMAD signaling. Being consistent with the impact of QKI‐5 overexpression on cell invasion, SB431542 abolished TGF‐β‐mediated migratory and invasive abilities of A549 cells (Fig EV3A and B), indicating that the invasion‐suppressing action of QKI‐5 overexpression was recapitulated by TGFβR1 inhibitor. SB431542 almost completely offset the promotion effect of QKI‐5 silencing on TGF‐β‐induced migratory and invasive abilities of A549 cells (Fig EV3C and D), suggesting that TGFβR1 inhibitor abolished the effects of QKI‐5 silencing on TGF‐β‐induced cell invasion. Taken together, our results demonstrate that QKI‐5 inhibits TGF‐β‐induced EMT, LUAD cell migration, and invasion in a TGFβR1‐dependent manner.
Figure EV3. QKI‐5 inhibits TGF‐β‐induced EMT and LUAD cell invasion via a TGFβR1‐dependent manner.
-
A, BQKI‐5‐overexpressing and control vector A549 cells treated with or without SB431542 (a specific TGFβR1 inhibitor, 10 mM) were stimulated with TGF‐β1 (5 ng/ml) for 24 h and subjected to Transwell migration and invasion assays. Scale bar, 100 μm. Data are shown as the mean ± SD of n = 3 technical replicates. *P < 0.05, **P < 0.01 and ***P < 0.001 by unpaired Student's t‐test.
-
C, DQKI‐5‐silenced and control sh‐NC A549 cells treated with or without SB431542 were stimulated with TGF‐β1 (5 ng/ml) for 24 h and subjected to Transwell migration and invasion assays. Scale bar, 100 μm. Data are shown as the mean ± SD of n = 3 technical replicates. n.s, not significant; *P < 0.05, **P < 0.01 and ***P < 0.001 by unpaired Student's t‐test.
KLF6 transcriptionally regulates QKI‐5 gene in LUADs
Subsequently, we explored why QKI‐5 was frequently down‐regulated in LUADs. First, after intersecting the aforementioned four data sets (Fig 1A) with human TFs database JASPAR (Fornes et al, 2020), we obtained 25 differentially expressed TFs in LUADs, 10 of which were predicted to have potential binding sites in the QKI‐5 promoter (Fig 7A and B). Of note, knockdown of FLI1, KLF6, and TBX3 expression (Fig 7C) significantly affected QKI‐5 mRNA expression in A549 cells (Fig 7D). In this study, more attention was paid to KLF6 because of it was frequently reduced in various human cancers, including NSCLC (Narla et al, 2001; Ito et al, 2004; Reeves et al, 2004). Furthermore, we found that KLF6 knockdown also attenuated QKI‐5 protein levels in A549 cells (Fig 7E). These results suggested that KLF6 may positively regulate QKI‐5 at transcriptional level. By analyzing KLF6‐ChIP‐seq data (GSE96355), we found an obvious peak (position −100 to +100) flanking the QKI‐5 promoter (Fig 7F). Also, we predicted two putative KLF6‐binding elements (KBEs) at positions −346 to −336 (termed as KBE1) and +21 to +31 (termed as KBE2) in QKI‐5 promoter (QP) using PROMO and JASPAR (Fig 7G upper panel). Subsequently, ChIP assays in A549 cells showed that a DNA fragment containing KBE2 rather than KBE1 in ΔQP was enriched using anti‐KLF6 antibody (Fig 7G lower panel), indicating a direct interaction of KLF6 with QKI‐5 promoter via binding to KBE2.
Figure 7. KLF6 transcriptionally regulates QKI‐5 gene in LUADs.

-
AFour databases used in Fig 1A were intersected with human TFs database JASPAR to identify differentially expressed TFs in LUAD. The Venn diagram depicts the overlap between five independent data sets.
-
BListed are 25 LUAD‐related differentially expressed TFs. TFBSTools was used to predict potential binding sites of each TF in QKI‐5 promoter sequence (position −3,000 to +200). TFBSTools minscore > 95%; TFs, transcription factors.
-
C, DqRT–PCR analyses of mRNA expression of 10 TFs and QKI‐5 in A549 cells transfected with siRNAs of TFs, which have potential binding sites in QKI‐5 promoter. Data are shown as the mean ± SD of n = 3 technical replicates. ***P < 0.001 compared to si‐NC by unpaired Student's t‐test.
-
EWestern blot analysis of QKI‐5 protein levels in KLF6‐silenced A549 cells.
-
FAn overview of KLF6‐ChIP‐seq data (GSE96355) is illustrated using Integrative Genomics Viewer (IGV) software. An obvious peak was detected at position −100 to +100 in the QKI‐5 promoter.
-
GUpper panel: computational algorithms using PROMO and JASPAR predicted that two putative KBEs were harbored in ΔQP region (position −400 to +200). QP, QKI‐5 promoter; KBE, KLF6 binding element. Lower panel: anti‐KLF6 ChIP was conducted in A549 cells and followed by PCR and gel staining to confirm the presence of QKI‐5 promoter sequence containing KBE1 (position −346 to −336) or KBE2 (position +21 to +31). The input and anti‐IgG antibody were used as positive and negative controls, respectively.
-
HLeft panel: Schematic drawing shows the pGL3‐ΔQP luciferase reporter assay system. Boxed areas indicate four ΔQP segments containing wild type or mutants of KBE1/2 (Mutant 1, Mutant 2, and Mutant 1*2). Right panel: Four different pGL3‐ΔQP luciferase reporter constructs were co‐transfected with si‐NC or si‐KLF6 into A549 cells, and their luciferase activities were determined as described in Materials and Methods. Data are shown as the mean ± SD of n = 3 technical replicates. *P < 0.05 compared to si‐NC by unpaired Student's t‐test.
-
I, JqRT–PCR analysis of KLF6 mRNA in 61 LUAD tissues and matched paracarcinoma tissues (I) and T/N mRNA expression of KLF6 in 61 paired LUAD tissues (J). T, LUAD tissues; N, paracarcinoma tissues. Y axis represents the log10‐transformed fold change of mRNA expression of KLF6. Central dotted line of the violin plot represents median, and upper and lower dotted line represents quartile (I). ***P < 0.001 by unpaired Student's t‐test.
-
KCorrelation between KLF6 and QKI‐5 mRNA expression in 61 paired LUAD tissues. X and y axes represent the log10‐transformed fold change of T/N expression ratios of KLF6 and QKI‐5 mRNA, respectively. P = 0.002 by Pearson's correlation test.
-
LA proposed work model: KLF6/QKI‐5/TGFβR1 axis regulates TGF‐β/SMAD signaling during TGF‐β‐induced EMT.
Source data are available online for this figure.
Next, to clarify whether KLF6 contributes to QKI‐5 transcriptional activity through KBE2, we constructed four different pGL3‐ΔQP luciferase reporter plasmids containing wild type or mutants of KBE1 and 2 (Fig 7H left panel and Appendix Table S4) and co‐transfected them with KLF6 siRNAs into A549 cells. Results of luciferase reporter assay showed that pGL3‐ΔQP activity inhibition by KLF6 knockdown was abolished only when KBE2 was mutated (Fig 7H right panel), indicating that KBE2 of QKI‐5 promoter is a functional KLF6‐binding site. In support of these results, KLF6 mRNA expression was significantly down‐regulated (46/61, 75.4%) in 61 LUAD tissues (Fig 7I and J) and positively correlated with QKI‐5 mRNA expression (Figs 7K and 1K). Taken together, these results reveal a transcriptional regulation mechanism of QKI‐5 by KLF6, being responsible for the altered expression of QKI‐5 in LUADs.
KLF6 knockdown leads to an increase in TGFβR1 expression and promotes TGF‐β‐induced EMT and LUAD cell invasion, which is mediated by QKI‐5
Based on the aforementioned findings that QKI‐5 repressed TGFβR1 expression (Fig 3A and B) and inhibited TGF‐β‐induced EMT and invasion of LUAD cells (Fig 4A–J) while KLF6 positively regulated QKI‐5 expression at transcriptional level (Fig 7C–H), we were encouraged to investigate whether KLF6 reduces TGFβR1 expression and inhibits TGF‐β‐induced EMT in a QKI‐5‐dependent manner. Firstly, a rescue experiment combining KLF6 knockdown and QKI‐5 overexpression was performed to determine the expression of TGFβR1 and EMT markers. As a result, siRNA‐mediated knockdown of KLF6 caused a robust increase in mRNA and protein levels of TGFβR1, which was significantly abrogated by QKI‐5 overexpression in A549 cells (Fig EV4A and B). Moreover, knockdown of KLF6 increased the expression levels of p‐SMAD3 and mesenchymal markers N‐cadherin and Snail in A549 cells treated with TGF‐β1. However, QKI‐5 overexpression partly abolished the increase of p‐SMAD3, N‐cadherin, and Snail expression caused by KLF6 knockdown (Fig EV4C). Furthermore, wound‐healing and Transwell assays showed that knockdown of KLF6 markedly prompted TGF‐β‐induced migratory and invasive capabilities of A549 cells, whereas this promotion effect was significantly impaired by QKI‐5 overexpression (Fig EV4D–G). Collectively, these results indicate KLF6 regulates TGFβR1 expression and TGF‐β‐induced EMT as well as LUAD cell invasion, which were largely mediated by QKI‐5.
Figure EV4. KLF6 knockdown promotes TGFβR1 expression and TGF‐β‐induced EMT as well as LUAD cell invasion in a QKI‐5‐dependent manner.

-
A, BA549 control cells were transfected with control siRNA or siRNA against KLF6 (si‐KLF6‐2), and A549 cells overexpressing QKI‐5 were transfected with si‐KLF6‐2. Cells were then subjected to qRT–PCR and Western blot assays for detection of KLF6, QKI‐5, and TGFβR1 expression. Data are shown as the mean ± SD of n = 3 technical replicates. **P < 0.01 and ***P < 0.001 by unpaired Student's t‐test.
-
CAfter serum starvation for 24 h, the above‐mentioned A549 cells (A) were stimulated with or without TGF‐β1 (5 ng/ml) for 24 h. The expression of p‐SMAD3, SMAD3, and EMT markers (N‐cadherin and Snail) was determined using Western blot. β‐actin was used as an internal control. Densitometry values of each protein were normalized to β‐actin and shown below the corresponding bands.
-
D, EA549 cells were treated as above and subjected to wound‐healing migration assays. The wound healing was recorded (D) and quantitatively measured (E) at least six times. Scale bar, 100 μm. Data are shown as the mean ± SD of n = 6 technical replicates. ***P < 0.001 by unpaired Student's t‐test.
-
F, GThe migratory and invasive capabilities of A549 cells treated as above were determined by Transwell assays. Migrated (F) and invasive (G) cells were stained and counted in four microscopic fields. Scale bar, 100 μm. Data are shown as the mean ± SD of n = 3 technical replicates. *P < 0.05 and **P < 0.01 by unpaired Student's t‐test.
Source data are available online for this figure.
Discussion
So far, whether and how RBPs regulates TGF‐β/SMAD signaling and act on TGF‐β‐induced EMT and metastasis of LUAD cells remains poorly understood. In this study, we identified a metastatic LUAD‐associated RBP, QKI‐5, which exerts anti‐metastatic functions by destabilizing TGFβR1 mRNA and inactivating TGF‐β/SMAD signaling in LUADs. Of note, we established a novel mechanistic role of KLF6/QKI‐5/TGFβR1 axis controlling TGF‐β/SMAD signaling during TGF‐β‐induced EMT and LUAD metastasis (Fig 7L).
Using a series of computational algorithms and tissue‐based analyses, we verified that QKI is associated with tumor metastasis in LUAD because QKI expression was not only decreased in unclassified LUAD tissues but reduced in metastatic LUAD tissues (Fig 1A and B). QKI has three major isoforms, including nuclear QKI‐5 and cytoplasmic QKI‐6/‐7 (Wu et al, 1999). QKI‐6 and QKI‐7 were reported to be predominantly related to development and diseases of the nervous system (Ebersole et al, 1996; Larocque et al, 2009), while QKI‐5 was widely studied in various human cancers including NSCLC (Zhou et al, 2017; Li et al, 2018; Kim et al, 2019). In the present study, we found that QKI‐5 expression was far higher than QKI‐6 and QKI‐7 in LUAD cells (Fig 1H), suggesting that QKI‐5 is a predominant isoform of QKI expressed in LUAD cells. Moreover, low expression of QKI‐5 was observed in LUAD cells and tissues, especially in metastatic LUAD tissues. Therefore, these findings promoted us to focus on QKI‐5 and elucidate the mechanisms behind its metastasis‐suppressive properties in LUADs. We have recently reported that TGF‐β‐induced EMT plays an important role in LUAD cell metastasis (Wang et al, 2016; Wang et al, 2018; Tong et al, 2020). However, to the best of our knowledge, it has not been clear whether QKI‐5 can affect TGF‐β‐induced EMT during LUAD cell metastasis. Thus, we explored the involvement of QKI‐5 in TGF‐β‐induced EMT and its anti‐metastatic potential in LUAD cells. Gain‐ and loss‐of‐function experiments showed that QKI‐5 repressed TGF‐β‐induced EMT and invasion of LUAD cells (Figs 4, 5, and EV1). In support of this, demonstrated that QKI‐5 inhibited EMT and aggressiveness of LUAD cells; Kim and colleagues reported that knockdown of QKI induced EMT and enhanced cell invasion in oral squamous cell carcinoma (Kim et al, 2019).
QKIs perform diverse biological functions, such as regulation of RNA splicing and stability, or non‐coding RNA processing (Zong et al, 2014; Conn et al, 2015; Zhou et al, 2017). In our study, QKI‐5 can interact with TGFβR1 mRNA 3′ UTR and decrease RNA stability of TGFβR1, thereby deactivating TGF‐β/SMAD signaling (Figs 2 and 3). QKI isoforms including QKI‐5 may interact with argonaute 2 (Ago2) (Wang et al, 2010). Ago2 is the core of the RNA‐induced silencing complex (RISC) and may act as endonuclease activity in human cells, which mediates siRNA‐triggered degradation of target mRNAs (Meister & Tuschl, 2004; Okamura et al, 2004). Thus, there would be the possibility that QKI‐5 may destabilize TGFBR1 mRNA by interacting with and recruiting Ago2 to TGFBR1 3′‐UTR. Our investigation provided the first evidence for post‐transcriptional repression of TGFβR1 by QKI‐5. Recently, Zhou et al (2017) demonstrated that QKI‐5 inhibits aggressiveness of LUAD cells through promoting β‐catenin degradation. However, QKI‐5 was reported to increased migration and invasion of breast cancer cells by modulating RNA splicing during EMT (Pillman et al, 2018). This discrepancy is not surprising because QKI‐5 may play distinct roles in cancer progression depending on cellular contexts.
In fact, TGF‐β/SMAD signaling acts as a metastasis promoter by inducing EMT in late‐stage tumors (Akhurst & Derynck, 2001; Xu & Pasche, 2007; Chen et al, 2015). In this study, TGFβR1, an essential receptor of TGF‐β/SMAD signaling, was found highly expressed in metastatic LUAD tissues. Moreover, TGFβR1 knockdown significantly suppressed TGF‐β‐induced EMT and metastasis of LUAD cells, which is supported by the finding that TGFβR1 knockdown attenuated metastatic ability and enhanced chemosensitivity to cisplatin in LUAD cells (Wang et al, 2015). Therefore, TGFβR1 knockdown showed similar effects as QKI‐5 overexpression in LUAD cells (Fig EV2), thus functionally testing the mechanistic role of QKI‐5 in TGFβR1 mRNA degradation. Furthermore, our rescue experiments demonstrated that QKI‐5 inhibited TGF‐β‐induced EMT and invasion of LUAD cells in a TGFβR1‐dependent manner (Figs 6 and EV3).
Why is QKI‐5 down‐regulated in LUADs? Zhou et al (2017) found that QKI‐5 promoter hypermethylation caused a reduction of QKI‐5 expression in LUAD cells. This may be one of the underlying mechanisms responsible for the downregulation of the QKI‐5 gene. In fact, the mechanisms behind it should be more complicated. Thus, we tried to find specific transcriptional factors regulating the QKI‐5 gene and saw that KLF6 positively regulated QKI‐5 at the transcriptional level in LUAD cells. The functional KLF6‐binding site we confirmed is localized at position +21 to +31 in the QKI‐5 gene (Fig 7G and H), which is different from the hypermethylation region (position −700 to +1) affecting QKI‐5 expression (Zhou et al, 2017). Besides, KLF6 was lowly expressed in LUAD tissues, especially in metastatic LUAD tissues (Appendix Tables S2 and S3), which is consistent with low expression of QKI‐5. These data further suggested that QKI‐5 is a target of KLF6. However, we cannot exclude the possibility that there are other factors activating the transcription of QKI‐5. For example, C/EBP can transcriptionally activate QKI‐5 during macrophage differentiation (Fu et al, 2012). Furthermore, our results showed that knockdown of KLF6 increased TGFβR1 expression and promoted TGF‐β‐induced EMT, which was partly abrogated by QKI‐5 overexpression in LUAD cells (Fig EV4). In contrast, in proximal tubule cells, KLF6 not only promoted EMT induced by TGF‐β1, but also KLF6 expression was up‐regulated by TGF‐β1 (Holian et al, 2008). Nevertheless, neither KLF6 nor QKI‐5 expression were significantly affected by TGF‐β1 in LUAD cells (Fig EV5), implying that the regulation of QKI‐5 by KLF6 is independent of TGF‐β1.
Figure EV5. KLF6 and QKI‐5 expressions are not affected by TGF‐β1 in LUAD cells.

-
A, BAfter being serum‐starved for 24 h, A549 and H1299 cells were treated with or without TGF‐β1 (5 or 10 ng/ml) for 24 h and subjected to qRT–PCR and Western blot assays for detecting the mRNA and protein expression of KLF6 and QKI‐5. β‐actin was used as an internal control. Data are shown as the mean ± SD of n = 3 technical replicates. ns, not significant; by unpaired Student's t‐test.
Source data are available online for this figure.
Although QKI‐5 blocks TGF‐β/SMAD signaling by degrading TGFβR1 mRNA, it is possible that QKI‐5 regulates other TGF‐β/SMAD signaling genes such as BMPR2, MAPK1, and RHOA (Fig 2B) via other mechanisms. In the present study, we have focused on TGFβR1 because it is an indispensable upstream receptor in TGF‐β/SMAD signaling. Further research is warranted to investigate other potential QKI‐targeted genes and possible mechanisms. Moreover, a future challenge will be the evaluation of the function of the isoforms QKI‐6/‐7 in LUAD development and progression.
Materials and Methods
Cell lines and cell culture
Human lung adenocarcinoma (LUAD) cell lines A549 and H1299, immortalized human bronchial epithelial cell line BEAS‐2B, and human embryonic kidney (HEK) 293T cells from the Cell Bank of Chinese Academy of Sciences were, respectively, cultured in RPMI 1640 medium (HyClone, South Logan, UT, USA) and Dulbecco's modified Eagle medium (DMEM; Thermo Fisher Scientific, Waltham, MA, USA) with 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA, USA) at 37°C in a 5% CO2 humidified incubator. A549 and H1299 cells were treated by TGF‐β1 to elicit EMT following serum starvation for 24 h.
Human LUAD tissue samples
Sixty‐one fresh‐frozen LUAD tissues and matched paracarcinoma tissues were obtained after informed consent from patients in the First Affiliated Hospital of Soochow University (Suzhou, China). LUAD patients were pathologically diagnosed and evaluated in accordance with the Revised International System for Staging Lung Cancer. The clinical characteristics of 61 LUAD patients are presented in Appendix Table S3, metastatic tissues (n = 28) were from LUAD patients with local lymph node metastasis (T1–4N1–2M0) or distant organ metastasis (T1–4NanyM1), and non‐metastatic tissues (n = 33) were from LUAD patients without any metastasis (T1–4N0M0). All of the LUAD patients enrolled in this study had not received either radiotherapy or chemotherapy before surgery. This work was authorized by the Ethics Committee of Soochow University.
RNA extraction, qRT–PCR, and mRNA half‐life assays
Total RNA isolation, cDNA synthesis, and qRT–PCR analysis were performed as previously described by us (Yang et al, 2015) with some modification. Expression of ADARB1, CELF2, QKI, ZFP36, QKI‐5/6/7, TGFβR1, E‐cadherin, N‐cadherin, PAI‐1, Slug, Snail, KLF6, etc., was normalized to β‐actin. Relative mRNA levels were calculated using the ΔΔCt method. For mRNA half‐life assay, 10 μg/ml actinomycin D (Sigma‐Aldrich, St. Louis, MO, USA) was added to A549 and H1299 cells. Total RNAs were extracted at the indicated time points for qRT–PCR assay. Primers used for qRT–PCR assay are listed in Appendix Table S5. Each RT–qPCR analysis was carried out three times.
Western blot assay
Total protein was isolated from cell lysate and subjected to Western blot analysis according to our previous methods (Yang et al, 2015). The primary antibodies employed in Western blot were as follows: goat anti‐TGFβR1 (R&D Systems, Minneapolis, MN, USA), mouse anti‐E‐cadherin and anti‐N‐cadherin (BD Biosciences, San Jose, CA, USA), mouse anti‐Snail, rabbit anti‐SMAD3 and anti‐p‐SMAD3 and anti‐Lamin B (Cell Signaling Technology, Danvers, MA, USA), rabbit anti‐QKI (Abcam, Cambridge, MA, USA), rabbit anti‐QKI‐5 (Millipore, Billerica, MA, USA), mouse anti‐KLF6 and anti‐β‐actin, and anti‐goat or anti‐rabbit or anti‐mouse secondary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA). The relative protein expression was normalized to β‐actin. Each Western blot analysis was performed in triplicate, and representative bands were presented.
Generation of LUAD cell lines stably overexpressing QKI‐5
To generate A549 and H1299 cell lines stably overexpressing QKI‐5, a ~ 1‐kb coding sequence (GenBank Accession number: NM_006775) of QKI‐5 was subcloned into a GV358 lentiviral expression vector (GeneChem Inc., Shanghai, China) containing a C‐terminal 3× Flag tag using endonucleases AgeI/BamhI (Fermentas, Glen Burnie, MD, USA). Primers used for amplifying the coding sequence of QKI‐5 were listed in Appendix Table S5. The empty lentivirus vector was served as a negative control. Subsequently, the QKI‐5 expression construct or empty vector was co‐transfected with packaging plasmids Helper 1.0 and Helper 2.0 (GeneChem Inc.) into HEK 293T cells using Lipofectamine 3000 (Invitrogen). At 48 h post‐incubation in DMEM with 10% FBS, the packaged lentiviruses were collected and used to infect A549 and H1299 cells for 72 h. Finally, Flag‐tagged QKI‐5‐overexpressing A549 and H1299 stable cells were selected with 2 μg/ml puromycin (Solarbio Lifesciences, Beijing, China).
Establishment of TGFβR1‐overexpressing LUAD cells
The coding sequence of TGFβR1 (GenBank Accession number: NM_004612.4) was amplified with the corresponding primers (Forward: ACCTCCATAGAAGATTCTAGAATGGAGGCGGCGGTCGCT; Reverse: AACATCGTATGGGTAGGATCCCATTTTGATGCCTTCCTGTTGA) and subcloned into a pcDNA3.1 vector using endonucleases XbaI/BamhI (New England Biolabs, Beverly, MA, USA) to generate pcDNA3.1‐TGFβR1 vectors. Then, A549 and H1299 cells were transiently transfected with above‐constructed vectors using Lipofectamine 3000 (Invitrogen). At 48 h post‐transfection, the cells were harvested or treated for further experiments.
Establishment of QKI‐5‐silenced and TGFβR1‐silenced stable LUAD cell lines
To establish QKI‐5‐silenced or TGFβR1‐silenced stable A549 and H1299 cell lines, the relative DNA fragments (sh‐QKI‐5‐1, sh‐QKI‐5‐2, sh‐TGFβR1‐1, and sh‐TGFβR1‐2; Appendix Table S6) were subcloned into a lentiviral vector pLKO.1‐puro (GENEWIZ Inc., Suzhou, China) with endonucleases AgeI/EcoRI (Fermentas). A scrambled sequence (sh‐NC; Appendix Table S6) was used as a negative control. Then, the construct containing sh‐QKI‐5 or sh‐TGFβR1 or sh‐NC was co‐transfected with packaging plasmids psPAX2 and pMD2.G into HEK 293T cells. The rest of the procedure is the same as that of generation of LUAD cell lines overexpressing QKI‐5 described above.
RNA interference
Two short interfering RNAs (siRNAs) for each gene were designed and synthesized (GenePharma, Shanghai, China). A scrambled sequence was served as a negative control (si‐NC). The sequences of synthesized siRNAs were listed in Appendix Table S6. A549 and H1299 cells were transiently transfected with 100 pmol of siRNAs using Lipofectamine 3000 (Invitrogen). At 72 h post‐transfection, the cells were harvested or treated for further experiments.
Luciferase reporter assay
To determine the effect of QKI‐5 on the luciferase activity of TGFβR1 3′ UTR, a series of constructs containing the wild‐type and mutated (Mut) 3′ UTR of TGFβR1 fused to the 3′‐end of the luciferase cDNA was generated using pGL3‐Luc reporter vector (Promega, Madison, WI, USA). Briefly, different fragments (Appendix Table S4) were directly synthesized (GENEWIZ Inc.) and subcloned into pGL3‐basic vector to create various constructs (pGL3‐Luc‐TGFβR1 3′ UTR wild type, pGL3‐Luc‐TGFβR1 3′ UTR‐Mut1, pGL3‐Luc‐TGFβR1 3′ UTR‐Mut2, and pGL3‐Luc‐TGFβR1 3′ UTR‐Mut1*2). Then, the above‐mentioned constructs and pRL‐TK plasmids were co‐transfected into A549 and H1299 stable cell lines with vector or QKI‐5 overexpression using Lipofectamine 3000 (Invitrogen).
For analyzing transcriptional regulation of KLF6 on QKI‐5, various QKI‐5 promoter fragments (Appendix Table S4) were synthesized and subcloned into pGL3‐basic vector to generate the constructs (pGL3‐QKI‐5 promoter‐wild‐type‐Luc, pGL3‐QKI‐5 promoter‐mutant 1‐Luc, pGL3‐QKI‐5 promoter‐mutant 2‐Luc, and pGL3‐QKI‐5 promoter‐mutant 1*2‐Luc). Subsequently, the above‐mentioned constructs and pRL‐TK plasmids were co‐transfected into A549 and H1299 cells with si‐NC or si‐KLF6. At 48 h post‐transfection, cells were harvested and luciferase activity was assessed using a Dual‐Luciferase® Reporter Assay System (Promega). Results are presented as relative luciferase activities (firefly/Renilla), and each luciferase reporter analysis was performed in triplicate.
In vitro RNA pull‐down assay
The TGFβR1 3′ UTRs (Appendix Table S4) and an antisense sequence of TGFβR1 3′ UTR wild type were synthesized and labeled using biotin (RiboBio Biotech, Guangzhou, China). In vitro RNA pull‐down assay was performed using Pierce™ Magnetic RNA‐Protein Pull‐down Kit (Thermo Fisher Scientific) according to the manufacturer's protocol. Briefly, cell lysate was incubated with the beads containing biotin‐labeled RNAs for 1 h at 4°C. After washing the beads, the bound proteins were eluted and boiled in 1 × SDS lysis buffer and subjected to Western blot analysis for determining the presence of QKI‐5. Each experiment was done in triplicate.
RNA‐binding protein immunoprecipitation
RNA‐binding protein immunoprecipitation (RIP) assay was conducted using EZ‐Magna RIP Kit (Millipore) according to the manufacturer's protocol. The QKI‐5‐RIP experiments were performed in A549 and H1299 cells. Briefly, cells were lysed using RIP lysis buffer with protease inhibitor cocktail and RNase inhibitor (Millipore), and then, the RIP lysates were mixed with protein A/G magnetic beads incubated with rabbit anti‐QKI‐5 antibody or anti‐IgG antibody (Millipore) at 4°C overnight for immunoprecipitation. The rabbit IgG antibody was used as a negative control. The immunoprecipitated RNAs were extracted and purified, and then subjected to RT–PCR and gel‐staining analyses to determine the relative enrichment of interested mRNA immunoprecipitated by QKI‐5 protein. Specific RIP primers used for PCR were summarized in Appendix Table S5.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP)‐IT® Express Kit (Active Motif, Rixensart, Belgium) was used for ChIP analysis. The detailed protocol was performed as described earlier by us with modification (Tong et al, 2020). In this ChIP analysis, the chromatin fraction from cell nuclei was incubated with Protein G Agarose and anti‐KLF6 antibody (Santa Cruz Biotechnology) overnight at 4°C. The ChIP‐DNA was purified and used for PCR amplification with QKI‐5‐specific primers, which were provided in Appendix Table S5.
Immunofluorescence (IF) analysis
Immunofluorescent analysis was performed as described by us (Wang et al, 2016) with somewhat modification. Briefly, cell samples were fixed with 4% paraformaldehyde and permeabilized with 0.5% Triton X‐100 for 15 min and then blocked with 5% BSA for 1.5 h. Subsequently, cell samples were incubated with mouse anti‐F‐actin (Millipore) primary antibodies at 4°C overnight, followed by incubating with FITC‐conjugated anti‐mouse secondary antibody for 2.5 h. Cell nuclei were counterstained with 4′,6‐diamidino‐2‐phenylindole (DAPI). Finally, the cell samples were photographed using a LSM 700 confocal laser scanning microscope (Carl Zeiss, Jena, Germany), and representative images were presented.
Wound‐healing migration assay
A549 and H1299 cells were seeded and cultivated in six‐well plates until they grew into a 90% confluent monolayer. Cells were then scratched by a sterilized pipette tip and cultured in serum‐free medium with or without TGF‐β1 (5 or 10 ng/ml) for 24 h. Cells were photographed in 6 random fields under a microscope. Cell migration distances into the scratched area were determined and analyzed using ImageJ Launcher software (National Institutes of Health, Bethesda, MA, USA). Each wound‐healing analysis was repeated in triplicate.
Transwell migration and invasion assays
Transwell migration and invasion assays were performed as described previously (Wang et al, 2016) with somewhat modification. Briefly, 5 × 104 cells in 100 μl RPMI 1640 medium with 1% FBS were added to the upper chamber of Transwell plates (BD Biosciences), and 700 μl RPMI 1640 medium with 10% FBS was placed as a chemoattractant in the lower chamber. After 6 h, TGF‐β1 (5 or 10 ng/ml) was added to the lower chambers. After incubation for 24 h (migration) or 30 h (invasion) in 37°C, the inserts were removed and the non‐migrating or non‐invading cells were removed with cotton swabs. Cells that migrated or invaded to the lower chamber were fixed and stained with 1% crystal violet. Cells were photographed and counted in at least four random fields. Each Transwell migration and invasion analysis was performed in triplicate.
In vivo metastasis assays
Four‐week‐old female BALB/c nude mice were purchased from Laboratory Animal Center of Soochow University and housed under specific pathogen‐free conditions. The mice were divided into four groups, including control vector groups and QKI‐5 overexpression group, control sh‐NC group and sh‐QKI‐5 group (10 or 5 mice per group). QKI‐5‐overexpressing, QKI‐5‐silenced, and control A549 cells (3 × 106 cells/mouse) in 200‐μl of PBS were intravenously (i.v.) injected into the tail vein of mice. At every 5th day post‐inoculation, TGF‐β1 (4 μg/kg bodyweight) was intraperitoneally (i.p.) injected as previously described (Wang et al, 2018) to promote tumor cell metastasis. Eight weeks later, the mice were euthanized; then their lungs and livers were taken out and fixed in Bouin's solution for macroscopically metastatic nodule analysis. Lung and liver tissues were histologically examined by hematoxylin‐eosin (H&E) staining for tumor cell micrometastases analysis. Animal studies were approved and supervised by the Animal Ethics Committee of Soochow University.
Statistical analysis
Unpaired t‐test (two‐tailed) was used to assess comparisons between two groups in vitro data, in vivo data, and LUAD sample data. Differences between multiple groups in the clinicopathological character of LUAD were assessed by non‐parametric tests (Unpaired t‐test for two groups, and Kruskal–Wallis test for ≥ 3 groups). The association between two groups of LUAD sample data was analyzed by Pearson's correlation test. Data were expressed as mean ± SD/SEM. P values of < 0.05 were considered to be statistically significant. GraphPad Prism 7.01 (GraphPad, San Diego, CA, USA) was used for all statistical analyses.
Bioinformatic analysis of public data sets
TCGA_LUAD data set from Genomic Data Commons (GDC) database (https://gdc.cancer.gov/) and data sets including GSE40791, GSE75037, and GSE7670 from Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) are downloaded to screen for LUAD‐associated RBPs and TFs. Differentially expressed genes (DEGs) are statistically considered as significance with FDR < 0.01 and abs.log2FC > 1 in TCGA database, and adjusted P‐value < 0.01 and abs.log2FC > 1 in GEO database. The DEGs of the above‐mentioned four data sets are intersected with human RBPs database ATtRACT (Giudice et al, 2016) or with TFs database JASPAR (Fornes et al, 2020) to yield LUAD‐associated RBPs or TFs. TFBSTools is conducted to predict potential binding sites of each LUAD‐associated TFs in QKI‐5 promoter sequence (position −3,000 to +200). TFBSTools minscore > 95% is applied as selection criteria.
Kaplan–Meier Plotter (http://www.kmplot.com) database and log‐rank tests are performed to evaluate the prognostic value of QKI (data from 236154_at) and TGFβR1 (data from TCGA) in lung cancer patients.
Three QKI‐RIP‐seq data from POSTAR, EuRBPDB, and doRiNA databases (Blin et al, 2015; Zhu et al, 2019; Liao et al, 2020) are overlapped to identify potential QKI‐targeted genes. Subsequently, KEGG enrichment analysis of these potential QKI‐targeted genes is performed by clusterProfiler in R language. A conserved consensus QKI‐5 response element (QRE) is computationally generated using RBPmap database (http://rbpmap.technion.ac.il/index.html).
For ChIP‐seq data analysis, KLF6‐ChIP‐seq reads from GEO database (GSE96355) are aligned to human genome reference (hg38) using Bowtie2 with default parameters. The mapping reads from ChIP‐seq are preprocessed by Samtools and then submitted to MACS2 for peaks calling. The characterized peaks are annotated by the R package ChIPseeker and visualized with Integrative Genomics Viewer (IGV) software.
Author contributions
Conception and design: SW and H‐TZ; Development of methodology: SW, XT, and CL; Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): SW, XT, CL, EJ, ZhS, ZeS, WZ, and ZL; Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): SW, XT, and CL; Writing, review, and/or revision of the manuscript: SW and H‐TZ. Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): SW, XT, and CL. Study supervision: H‐TZ.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PD
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We are grateful for participation and cooperation from LUAD patients. Funding was provided by grants from National Natural Science Foundation of China (81872343, 81672277), and Suzhou Key Laboratory for Molecular Cancer Genetics (SZS201209), and A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
EMBO reports (2021) 22: e52079.
Data availability
No primary data sets have been generated and deposited.
References
- Akhurst RJ, Derynck R (2001) TGF‐beta signaling in cancer–a double‐edged sword. Trends Cell Biol 11: S44–S51 [DOI] [PubMed] [Google Scholar]
- Blin K, Dieterich C, Wurmus R, Rajewsky N, Landthaler M, Akalin A (2015) DoRiNA 2.0–upgrading the doRiNA database of RNA interactions in post‐transcriptional regulation. Nucleic Acids Res 43: D160–D167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CH, Lai JM, Chou TY, Chen CY, Su LJ, Lee YC, Cheng TS, Hong YR, Chou CK, Whang‐Peng J et al (2009) VEGFA upregulates FLJ10540 and modulates migration and invasion of lung cancer via PI3K/AKT pathway. PLoS One 4: e5052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Fillmore CM, Hammerman PS, Kim CF, Wong KK (2014) Non‐small‐cell lung cancers: a heterogeneous set of diseases. Nat Rev Cancer 14: 535–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Yang T, Lei Z, Wang L, Yang H, Tong X, Yang WT, Zhao J, Gu Y, Chen Y et al (2015) RNF111/Arkadia is regulated by DNA methylation and affects TGF‐beta/Smad signaling associated invasion in NSCLC cells. Lung Cancer 90: 32–40 [DOI] [PubMed] [Google Scholar]
- Conn SJ, Pillman KA, Toubia J, Conn VM, Salmanidis M, Phillips CA, Roslan S, Schreiber AW, Gregory PA, Goodall GJ (2015) The RNA binding protein quaking regulates formation of circRNAs. Cell 160: 1125–1134 [DOI] [PubMed] [Google Scholar]
- ten Dijke P, Hill CS (2004) New insights into TGF‐beta‐Smad signalling. Trends Biochem Sci 29: 265–273 [DOI] [PubMed] [Google Scholar]
- Dongre A, Weinberg RA (2019) New insights into the mechanisms of epithelial‐mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol 20: 69–84 [DOI] [PubMed] [Google Scholar]
- Du B, Shim JS (2016) Targeting epithelial‐mesenchymal transition (EMT) to overcome drug resistance in cancer. Molecules 21: 965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebersole TA, Chen Q, Justice MJ, Artzt K (1996) The quaking gene product necessary in embryogenesis and myelination combines features of RNA binding and signal transduction proteins. Nat Genet 12: 260–265 [DOI] [PubMed] [Google Scholar]
- Fang Y, Chen Y, Yu L, Zheng C, Qi Y, Li Z, Yang Z, Zhang Y, Shi T, Luo J et al (2013) Inhibition of breast cancer metastases by a novel inhibitor of TGFbeta receptor 1. J Natl Cancer Inst 105: 47–58 [DOI] [PubMed] [Google Scholar]
- Fornes O, Castro‐Mondragon JA, Khan A, van der Lee R, Zhang X, Richmond PA, Modi BP, Correard S, Gheorghe M, Baranasic D et al (2020) JASPAR 2020: update of the open‐access database of transcription factor binding profiles. Nucleic Acids Res 48: D87–D92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H, Yang G, Wei M, Liu L, Jin L, Lu X, Wang L, Shen L, Zhang J, Lu H et al (2012) The RNA‐binding protein QKI5 is a direct target of C/EBPalpha and delays macrophage differentiation. Mol Biol Cell 23: 1628–1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galarneau A, Richard S (2005) Target RNA motif and target mRNAs of the Quaking STAR protein. Nat Struct Mol Biol 12: 691–698 [DOI] [PubMed] [Google Scholar]
- Girard L, Rodriguez‐Canales J, Behrens C, Thompson DM, Botros IW, Tang H, Xie Y, Rekhtman N, Travis WD, Wistuba II et al (2016) An expression signature as an aid to the histologic classification of non‐small cell lung cancer. Clin Cancer Res 22: 4880–4889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giudice G, Sanchez‐Cabo F, Torroja C, Lara‐Pezzi E (2016) ATtRACT—a database of RNA‐binding proteins and associated motifs. Database (Oxford) 2016: baw035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holian J, Qi W, Kelly DJ, Zhang Y, Mreich E, Pollock CA, Chen XM (2008) Role of Kruppel‐like factor 6 in transforming growth factor‐beta1‐induced epithelial‐mesenchymal transition of proximal tubule cells. Am J Physiol Renal Physiol 295: F1388–F1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito G, Uchiyama M, Kondo M, Mori S, Usami N, Maeda O, Kawabe T, Hasegawa Y, Shimokata K, Sekido Y (2004) Kruppel‐like factor 6 is frequently down‐regulated and induces apoptosis in non‐small cell lung cancer cells. Cancer Res 64: 3838–3843 [DOI] [PubMed] [Google Scholar]
- Jett JR, Cortese DA, Fontana RS (1983) Lung cancer: current concepts and prospects. CA Cancer J Clin 33: 74–86 [DOI] [PubMed] [Google Scholar]
- Kim EJ, Kim JS, Lee S, Lee H, Yoon JS, Hong JH, Chun SH, Sun S, Won HS, Hong SA et al (2019) QKI, a miR‐200 target gene, suppresses epithelial‐to‐mesenchymal transition and tumor growth. Int J Cancer 145: 1585–1595 [DOI] [PubMed] [Google Scholar]
- Kudinov AE, Deneka A, Nikonova AS, Beck TN, Ahn YH, Liu X, Martinez CF, Schultz FA, Reynolds S, Yang DH et al (2016) Musashi‐2 (MSI2) supports TGF‐beta signaling and inhibits claudins to promote non‐small cell lung cancer (NSCLC) metastasis. Proc Natl Acad Sci USA 113: 6955–6960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn E, Morbini P, Cancellieri A, Damiani S, Cavazza A, Comin CE (2018) Adenocarcinoma classification: patterns and prognosis. Pathologica 110: 5–11 [PubMed] [Google Scholar]
- Lamouille S, Xu J, Derynck R (2014) Molecular mechanisms of epithelial‐mesenchymal transition. Nat Rev Mol Cell Biol 15: 178–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larocque D, Pilotte J, Chen T, Cloutier F, Massie B, Pedraza L, Couture R, Lasko P, Almazan G, Richard S (2002) Nuclear retention of MBP mRNAs in the quaking viable mice. Neuron 36: 815–829 [DOI] [PubMed] [Google Scholar]
- Larocque D, Fragoso G, Huang J, Mushynski WE, Loignon M, Richard S, Almazan G (2009) The QKI‐6 and QKI‐7 RNA binding proteins block proliferation and promote Schwann cell myelination. PLoS One 4: e5867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Choi PS, Chaffer CL, Labella K, Hwang JH, Giacomelli AO, Kim JW, Ilic N, Doench JG, Ly SH et al (2018) An alternative splicing switch in FLNB promotes the mesenchymal cell state in human breast cancer. Elife 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang G, Meng W, Huang X, Zhu W, Yin C, Wang C, Fassan M, Yu Y, Kudo M, Xiao S et al (2020) miR‐196b‐5p‐mediated downregulation of TSPAN12 and GATA6 promotes tumor progression in non‐small cell lung cancer. Proc Natl Acad Sci USA 117: 4347–4357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao JY, Yang B, Zhang YC, Wang XJ, Ye Y, Peng JW, Yang ZZ, He JH, Zhang Y, Hu K et al (2020) EuRBPDB: a comprehensive resource for annotation, functional and oncological investigation of eukaryotic RNA binding proteins (RBPs). Nucleic Acids Res 48: D307–D313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister G, Tuschl T (2004) Mechanisms of gene silencing by double‐stranded RNA. Nature 431: 343–349 [DOI] [PubMed] [Google Scholar]
- Mikami F, Lim JH, Ishinaga H, Ha UH, Gu H, Koga T, Jono H, Kai H, Li JD (2006) The transforming growth factor‐beta‐Smad3/4 signaling pathway acts as a positive regulator for TLR2 induction by bacteria via a dual mechanism involving functional cooperation with NF‐kappaB and MAPK phosphatase 1‐dependent negative cross‐talk with p38 MAPK. J Biol Chem 281: 22397–22408 [DOI] [PubMed] [Google Scholar]
- Muppala S, Xiao R, Krukovets I, Verbovetsky D, Yendamuri R, Habib N, Raman P, Plow E, Stenina‐Adognravi O (2017) Thrombospondin‐4 mediates TGF‐beta‐induced angiogenesis. Oncogene 36: 5189–5198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narla G, Heath KE, Reeves HL, Li D, Giono LE, Kimmelman AC, Glucksman MJ, Narla J, Eng FJ, Chan AM et al (2001) KLF6, a candidate tumor suppressor gene mutated in prostate cancer. Science 294: 2563–2566 [DOI] [PubMed] [Google Scholar]
- Okamura K, Ishizuka A, Siomi H, Siomi MC (2004) Distinct roles for Argonaute proteins in small RNA‐directed RNA cleavage pathways. Genes Dev 18: 1655–1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastushenko I, Blanpain C (2019) EMT transition states during tumor progression and metastasis. Trends Cell Biol 29: 212–226 [DOI] [PubMed] [Google Scholar]
- Pillman KA, Phillips CA, Roslan S, Toubia J, Dredge BK, Bert AG, Lumb R, Neumann DP, Li X, Conn SJ et al (2018) miR‐200/375 control epithelial plasticity‐associated alternative splicing by repressing the RNA‐binding protein Quaking. EMBO J 37: e99016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves HL, Narla G, Ogunbiyi O, Haq AI, Katz A, Benzeno S, Hod E, Harpaz N, Goldberg S, Tal‐Kremer S et al (2004) Kruppel‐like factor 6 (KLF6) is a tumor‐suppressor gene frequently inactivated in colorectal cancer. Gastroenterology 126: 1090–1103 [DOI] [PubMed] [Google Scholar]
- Rodrigues DC, Kim DS, Yang G, Zaslavsky K, Ha KC, Mok RS, Ross PJ, Zhao M, Piekna A, Wei W et al (2016) MECP2 is post‐transcriptionally regulated during human neurodevelopment by combinatorial action of RNA‐binding proteins and miRNAs. Cell Rep 17: 720–734 [DOI] [PubMed] [Google Scholar]
- Saccomanno L, Loushin C, Jan E, Punkay E, Artzt K, Goodwin EB (1999) The STAR protein QKI‐6 is a translational repressor. Proc Natl Acad Sci USA 96: 12605–12610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmierer B, Hill CS (2007) TGFbeta‐SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol 8: 970–982 [DOI] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A (2019) Cancer statistics, 2019. CA Cancer J Clin 69: 7–34 [DOI] [PubMed] [Google Scholar]
- Thangaraj MP, Furber KL, Gan JK, Ji S, Sobchishin L, Doucette JR, Nazarali AJ (2017) RNA‐binding protein quaking stabilizes Sirt2 mRNA during oligodendroglial differentiation. J Biol Chem 292: 5166–5182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong X, Wang S, Lei Z, Li C, Zhang C, Su Z, Liu X, Zhao J, Zhang HT (2020) MYOCD and SMAD3/SMAD4 form a positive feedback loop and drive TGF‐beta‐induced epithelial‐mesenchymal transition in non‐small cell lung cancer. Oncogene 39: 2890–2904 [DOI] [PubMed] [Google Scholar]
- Wang Y, Lacroix G, Haines J, Doukhanine E, Almazan G, Richard S (2010) The QKI‐6 RNA binding protein localizes with the MBP mRNAs in stress granules of glial cells. PLoS One 5: e12824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Vogel G, Yu Z, Richard S (2013) The QKI‐5 and QKI‐6 RNA binding proteins regulate the expression of microRNA 7 in glial cells. Mol Cell Biol 33: 1233–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Chen X, Meng Q, Jing H, Lu H, Yang Y, Cai L, Zhao Y (2015) MiR‐181b regulates cisplatin chemosensitivity and metastasis by targeting TGFbetaR1/Smad signaling pathway in NSCLC. Sci Rep 5: 17618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Yang H, Lei Z, Zhao J, Chen Y, Chen P, Li C, Zeng Y, Liu Z, Liu X et al (2016) Repression of TIF1gamma by SOX2 promotes TGF‐beta‐induced epithelial‐mesenchymal transition in non‐small‐cell lung cancer. Oncogene 35: 867–877 [DOI] [PubMed] [Google Scholar]
- Wang L, Tong X, Zhou Z, Wang S, Lei Z, Zhang T, Liu Z, Zeng Y, Li C, Zhao J et al (2018) Circular RNA hsa_circ_0008305 (circPTK2) inhibits TGF‐beta‐induced epithelial‐mesenchymal transition and metastasis by controlling TIF1gamma in non‐small cell lung cancer. Mol Cancer 17: 140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Zhou L, Tonissen K, Tee R, Artzt K (1999) The quaking I‐5 protein (QKI‐5) has a novel nuclear localization signal and shuttles between the nucleus and the cytoplasm. J Biol Chem 274: 29202–29210 [DOI] [PubMed] [Google Scholar]
- Xu Y, Pasche B (2007) TGF‐beta signaling alterations and susceptibility to colorectal cancer. Hum Mol Genet 16: R14–R20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang L, Zhao J, Chen Y, Lei Z, Liu X, Xia W, Guo L, Zhang HT (2015) TGF‐beta‐activated SMAD3/4 complex transcriptionally upregulates N‐cadherin expression in non‐small cell lung cancer. Lung Cancer 87: 249–257 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Foreman O, Wigle DA, Kosari F, Vasmatzis G, Salisbury JL, van Deursen J, Galardy PJ (2012) USP44 regulates centrosome positioning to prevent aneuploidy and suppress tumorigenesis. J Clin Invest 122: 4362–4374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Li X, Sun C, Shi C, Hua D, Yu L, Wen Y, Hua F, Wang Q, Zhou Q et al (2017) Quaking‐5 suppresses aggressiveness of lung cancer cells through inhibiting beta‐catenin signaling pathway. Oncotarget 8: 82174–82184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Xu G, Yang YT, Xu Z, Chen X, Shi B, Xie D, Lu ZJ, Wang P (2019) POSTAR2: deciphering the post‐transcriptional regulatory logics. Nucleic Acids Res 47: D203–D211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong FY, Fu X, Wei WJ, Luo YG, Heiner M, Cao LJ, Fang Z, Fang R, Lu D, Ji H et al (2014) The RNA‐binding protein QKI suppresses cancer‐associated aberrant splicing. PLoS Genet 10: e1004289 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PD
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Data Availability Statement
No primary data sets have been generated and deposited.