

Abstract
Autism spectrum disorder (ASD) is a neurodevelopmental disorder characterized by deficits in social communication and the presence of restricted patterns of interest and repetitive behaviors. ASD is genetically heterogeneous and is believed to be caused by both inheritable and de novo gene variations. Studies have revealed an extremely complex genetic landscape of ASD, favoring the idea that mutations in different clusters of genes interfere with interconnected downstream signaling pathways and circuitry, resulting in aberrant behavior. In this review, we describe a select group of candidate genes that represent both syndromic and non‐syndromic forms of ASD and encode proteins that are important in transcriptional and translational regulation. We focus on the interplay between dysregulated translation and transcription in ASD with the hypothesis that dysregulation of each synthetic process triggers a feedback loop to act on the other, which ultimately exacerbates ASD pathophysiology. Finally, we summarize findings from interdisciplinary studies that pave the way for the investigation of the cooperative impact of different genes and pathways underlying the development of ASD.
Keywords: ASD, mRNA, transcription, translation
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Neuroscience; Protein Biosynthesis & Quality Control
Autism‐spectrum disorder (ASD) is a genetically complex and heterogenous disorder. Here, the authors review a select group of ASD‐associated genes and discuss the interplay between dysregulated translation and transcription in the pathophysiology of ASD.

Glossary
- 4E‐BP
eIF4E‐binding protein
- ADHD
attention deficit and hyperactivity disorder
- ADNP
activity‐dependent neuroprotective protein
- aNCSs
adult neural stem cells
- APOE
apolipoprotein E
- ASD
autism spectrum disorder
- BDNF
brain‐derived neurotrophic factor
- BNIP3
BCL2 and adenovirus E1B 19‐kDa‐interacting protein 3
- Brd4
bromodomain‐containing protein 4
- CACNA1E
calcium voltage‐gated channel subunit alpha1 E
- CACNB1
calcium voltage‐gated channel auxiliary subunit beta 1
- CaMKII
Ca2+/calmodulin‐dependent protein kinase II
- CNS
central nervous system
- CNVs
copy number variants
- CREB
cAMP response element‐binding protein
- CSF
cerebrospinal fluid
- CTGF
connective tissue growth factor
- CYFIP1
cytoplasmic FMRP‐interacting protein 1
- DA
dopamine
- DAT
dopamine transporter
- eIF4A
eukaryotic translation initiation factor 4 A
- eIF4E
eukaryotic initiation factor 4E
- eIF4G
eukaryotic translation initiation factor 4 G
- EN1
engrailed 1
- EN2
engrailed 2
- ERK
extracellular signal‐regulated kinase
- Fmr1
fragile X mental retardation 1
- FXS
fragile X syndrome
- GAP
GTPase‐activating protein
- GAPDH
glyceraldehyde 3‐phosphate dehydrogenase
- Grm5
glutamate metabotropic receptor 5
- HDVAS
Helsmoortel‐Van der Aa syndrome
- HIF1a
hypoxia‐inducible factor 1‐alpha
- HITS‐CLIP
high‐throughput sequencing of RNAs isolated by cross‐linking immunoprecipitation
- IGF‐1
insulin‐like growth factor 1
- IL‐6
interleukin 6
- KO
knockout
- LIF
leukemia inhibitory factor
- LTD
long‐term depression
- LTP
long‐term potentiation
- MeCP2
methyl‐CpG‐binding protein 2
- MEK
MAPK/ERK kinase
- mGluR
metabotropic glutamate receptor
- MNK
MAP kinase‐interacting kinase
- mTORC1
mammalian/mechanistic target of rapamycin complex 1
- Myl2
myosin regulatory light chain 2
- NMDAR
N‐methyl‐D‐aspartate receptor
- Nrxn3
neurexin 3
- PABP
poly(A)‐binding protein
- P‐CREB
phosphorylated CREB
- PDE4D
cAMP‐specific 3',5'‐cyclic phosphodiesterase 4D
- PDK1
3‐phosphoinositide‐dependent protein kinase 1
- PI3K
phosphoinositide 3‐kinase
- PIP3
phosphatidylinositol (3,4,5)‐trisphosphate
- PSD95
postsynaptic density protein 95
- PTEN
phosphatase and tensin homolog
- PV
parvalbumin
- Rack1
receptor for activated C kinase 1
- REDD1
regulated in development and DNA damage responses 1
- RHEB
ras homolog enriched in brain
- RYK
receptor tyrosine kinase
- S6K1
ribosomal protein S6 kinase beta‐1
- SCN1A
sodium voltage‐gated channel alpha subunit 1
- SIRT1
sirtuin 1
- SLC6A4
solute carrier family 6 member 4
- SWI/SNF
SWItch/Sucrose non‐fermentable
- TH
tyrosine hydroxylase
- TrKB
tropomyosin receptor kinase B
- TSC1
tuberous sclerosis complex 1
- TSC2
tuberous sclerosis complex 2
- ZFP161
zinc finger protein 161
Introduction
Autism spectrum disorder (ASD) comprises a class of heterogeneous neurodevelopmental diseases characterized by three salient features: early‐onset difficulties in communication, reduced social interaction, and repetitive, restricted behavior/interests (McPartland & Volkmar, 2012; Kulage et al, 2014). Individuals diagnosed with ASD often present with co‐morbid psychiatric and medical disorders, such as attention deficit and hyperactivity disorder (ADHD), intellectual disability, epilepsy, anxiety, eating problems, and alterations in mood and sleep and mood (Zoghbi & Bear, 2012; Mannion & Leader, 2013; Romero et al, 2016). In many cases, the co‐morbid conditions lead to more severe impairments as a result of the cumulative effects of these different disorders, as well as a synergistic action of the various molecular pathways involved.
ASD is highly heritable and yet is a genetically heterogeneous disorder (Hallmayer et al, 2011). Over the last few decades, great progress has been made in understanding the genetic etiology of ASD, spurred by a wide array of genome‐wide analyses and approaches (Iossifov et al, 2012; Neale et al, 2012; O’Roak et al, 2012; Sanders et al, 2012). These genetic studies have helped to reveal neurobiological mechanisms, as well as the contribution of environmental and epigenetic factors (Kim & Leventhal, 2015) involved in the pathophysiology of ASD. Advances in genetic and genomic methodologies have identified a large number of ASD‐associated de novo mutations, including single‐base pair mutations (single‐nucleotide variants; SNVs) and a number of copy number variants (CNVs) that disrupt protein‐coding genes (Sanders et al, 2012; Iossifov et al, 2014), as well as mutations located within intronic and intergenic regions (Zhou et al, 2019b). In this heterogeneous genetic architecture, syndromic forms of ASD (Box 1) caused by highly penetrant single‐gene mutations represent only a minority of the ASD cases (Sztainberg & Zoghbi, 2016). In contrast, most of the cases are idiopathic with common variants contributing to a significant fraction (40–60%) of genetic liability of ASD (Klei et al, 2012; Gaugler et al, 2014; Zhou et al, 2019b).
Box 1. Syndromic and non‐syndromic ASD.
ASD is a heterogeneous heritable neuropsychiatric disorder that can be classically categorized into two types: syndromic and non‐syndromic autism. Syndromic autisms are caused by mutations in a particular gene or a set of genes and are manifested within the context of neurological syndromes, such as Fragile X Syndrome, Tuberous Sclerosis, or Rett Syndrome. Non‐syndromic autism, which comprises a vast majority of autism cases, is not linked to other neurological diseases (or syndromes) but is also heritable with a number of genes that are linked to this class of autism.
To date, these large‐scale sequencing efforts certainly have contributed to unveiling the genetic etiology of ASD and have advanced our understanding of potential neurobiological mechanisms that give rise to this disorder. From this perspective, several studies have emphasized the categorization of ASD risk gene groups by their putative functions: ASD‐related mutations tend to be significantly enriched in genes involved in synaptic function and structure, and in genes encoding for transcripts that are important in transcriptional and translational regulation (De Rubeis et al, 2014; Krumm et al, 2014; de la Torre‐Ubieta et al, 2016).
Given the extremely complex genetic landscape of ASD, it has been proposed that mutations in these different clusters of genes interfere with interconnected downstream signaling pathways and circuitry in a cell type‐specific manner, resulting in ASD pathophysiology (Santini & Klann, 2014; de la Torre‐Ubieta et al, 2016; Mullins et al, 2016; Wang et al, 2018). The identification of a cluster of genes encoding for transcripts involved in different phases of mRNA translation (Darnell et al, 2011; Santoro et al, 2012) supports one of the unifying theories on ASD according to which dysregulation of translational control represents a common endpoint of familial and sporadic ASD‐associated signaling pathways (Kelleher & Bear, 2008; Darnell & Klann, 2013; Santini & Klann, 2014). Consistent with this notion, several signaling pathways upstream of translation, including PI3K (phosphoinositide 3‐kinase)/Akt/mTORC1 (mammalian/mechanistic target of rapamycin complex 1) and ERK (extracellular signal‐regulated kinase) are dysregulated in several types of ASD (Sonenberg & Hinnebusch, 2009; Darnell & Klann, 2013), resulting in increased net de novo protein synthesis, which in turn leads to altered synaptic plasticity and ASD‐like phenotypes (Auerbach et al, 2011; Gkogkas et al, 2013a; Santini et al, 2013). However, the physiological outcome of increased translation of specific mRNAs differs depending on the different regulatory processes and ASD‐linked genes tightly coalesce in modules that implicate distinct biological functions during development, including early transcriptional regulation and synaptic development (Parikshak et al, 2013). Interestingly, recent studies on monogenic forms of ASD suggest that not all excessively translating mRNAs contribute to pathological changes, raising the hypothesis that some of these mRNAs may be protective adaptations (Thomson et al, 2017). On the other hand, excessive translation of certain transcripts may result in widespread epigenetic misregulation (Korb et al, 2017) creating a positive feedback loop between translational and transcriptional processes that exacerbate neuronal dysfunction in ASD (Box 2).
Box 2. In need of answers.
ASD‐associated genes are often regulators of the expression of a large group of other genes with multiple molecular functions. No gene operates alone. The extremely complex genetic landscape of ASD favors the idea that mutations in different clusters of genes interfere with interconnected downstream signaling pathways and circuitry. Which synaptic and non‐synaptic regulatory processes are particularly susceptible to altered translation and transcription and are some downstream biological processes more relevant than others to drug treatments?
Does the epigenetic misregulation resulting from an excessive translation of certain transcripts act at a critical time in development, and when would treatment be most effective?
We have hypothesized a bidirectional regulation of translation and transcription in ASD and that mutations in genes that result in dysregulation of one of those processes triggers a positive feedback loop to act on the other, which exacerbates ASD pathophysiology. Is there a specific brain region where to identify a core set of mistranslated mRNAs that mediate the complex level of dynamic regulation between translation and transcription underlying ASD pathophysiology?
The direct effect of specific proteins encoded by genes implicated in ASD, regulating the translation of transcriptional regulators is a likely way to explain many of the changes in mRNA levels reported in ASD. The possibility that the observed regulatory interplay between translation and transcription may not be direct for some of those genes arise the question on whether changes observed in one process from disruptions in the other could likely represent a protective and compensatory mechanisms rather than a positive feedback loop exacerbating ASD pathophysiology, aiming further investigation.
Here, we examine the evidence for interplay between dysregulated translation and transcription in ASD. We review a select group of ASD‐associated genes representative of both syndromic and non‐syndromic forms of ASD that are involved in either translational or transcriptional regulation and describe the molecular function of their protein products, and the ASD‐associated clinical phenotypes caused by disruption of these genes. Then, we summarize findings from interdisciplinary studies that have queried the impact of dysregulated translational control on transcription and dysregulated transcriptional control on translation with respect to ASD pathophysiology.
Regulation of translation and ASD
Examination of the different genetic studies and transcriptomic analyses (Voineagu et al, 2011; De Rubeis et al, 2014; Gupta et al, 2014) that have been performed to date indicates that ASD‐associated genes are often regulators of the expression of a large group of other genes with multiple molecular functions. The phosphatidylinositol 3‐kinase (PI3K)‐mechanistic target of rapamycin (mTOR) signaling cascade is one of the most commonly studied pathways where mutations occur and give rise to ASD‐related phenotypes (Huber et al, 2015). Overactivation of the PI3K‐mTOR pathway has been described in fragile X syndrome (FXS) (Sharma et al, 2010; Hoeffer et al, 2012), and mutations in genes encoding for negative regulators of the mTOR pathway, such as tuberous sclerosis complex 1 (TSC1), tuberous sclerosis complex 2 (TSC2), and phosphatase and tensin homolog (PTEN) (Kwon et al, 2006) are consistent with increased mTOR activity in cells isolated from individuals with ASD (Onore et al, 2017). Because mTOR complex 1 (mTORC1) is a well‐known regulator of translation (Hay & Sonenberg, 2004; Ma & Blenis, 2009; Zoncu et al, 2011), deletion or loss‐of‐function mutations in genes encoding for proteins involved in regulating mTORC1 lead to alteration of the translational control machinery, ultimately resulting in increased de novo translation, altered protein synthesis‐dependent synaptic plasticity, and the appearance of behavioral defects consistent with ASD (Kelleher & Bear, 2008; Ebert & Greenberg, 2013; Santini & Klann, 2014). In the next section, we will briefly summarize the impact of dysregulated translational control on transcription focusing on three ASD‐associated genes involved in translation regulation: fragile X mental retardation 1 (FMR1), phosphatase and tensin homolog (PTEN), and tuberous sclerosis complex (TSC).
Fragile X mental retardation protein (FMR1)
Transcriptional silencing of the fragile X mental retardation 1 (FMR1) gene results in the loss of fragile X mental retardation protein (FMRP) and gives rise to fragile X syndrome (FXS) (Verkerk et al, 1991). This form of intellectual disability represents one of the most prominent single‐gene causes of ASD, accounting for an estimated 1% to 6% of all cases (Schaefer & Mendelsohn, 2013). Reduced expression or absence of FMRP leads to atypical brain development and dendritic spine anomalies (Irwin et al, 2000), resulting in cognitive and motor dysfunction in FXS individuals such as hyperactivity, anxiety, attention deficit disorder, speech perseveration, impulsive behavior, and stereotypical and repetitive movements (Martin & Bell, 1943; Dykens et al, 1989; Hodapp et al, 1990; Cordeiro et al, 2011).
FMRP is an mRNA‐binding protein that operates as regulator of translation (Darnell & Klann, 2013), among other functions (Contractor et al, 2015). Studies of Fmr1 knockout (KO) mice indicate that FMRP has a crucial role in the activity‐dependent regulation of mRNA transport (Dictenberg et al, 2008), translation, and stability in neurons (Bassell & Warren, 2008; Darnell et al, 2011). Evidence from cells derived from FXS patients and FXS model mice suggests that FMRP represses the initiation and elongation steps of translation, which results in a net increase in protein synthesis when expression of FMRP is absent in FXS (Darnell & Klann, 2013). With respect to translation initiation, FMRP interacts with cytoplasmic FMRP‐interacting protein 1 (CYFIP1), which associates and sequesters the cap‐binding protein eukaryotic initiation factor 4E (eIF4E), thereby inhibiting the translation initiation of specific mRNAs (Napoli et al, 2008). FMRP also regulates translation elongation, where it has been shown that FMRP stalls ribosomal translocation on specific mRNAs during elongation (Darnell et al, 2011), although the molecular mechanisms responsible for this regulation remain to be determined.
Although the mechanisms of translational control by FMRP have been extensively described (Bassell & Warren, 2008; Costa‐Mattioli et al, 2009; Darnell et al, 2011) and several FMRP‐regulated transcripts and pathways have been identified (Darnell et al, 2001, 2011), the mRNAs with differential translation that contribute to neuronal function or that impact gene expression have not been evaluated comprehensively in FXS, although several recent studies provided significant progress in this direction (Ceolin et al, 2017; Korb et al, 2017; Thomson et al, 2017; Liu et al, 2018). New insights into genome‐wide gene expression changes resulting from FMRP deficiency come from a study on adult neural stem cells (aNCSs) derived from wild‐type and Fmr1‐deficient mice (Liu et al, 2018). Using simultaneous high‐resolution ribosome profiling and transcriptomic analysis, Liu and colleagues described diverse expression changes at both mRNA and translation levels, revealing a translational buffering mechanism by FMRP. A form of buffering whereby the loss of FMRP leads to increased translation of mRNAs, which then stimulates a feedback decrease in the levels of mRNA, in order to conserve the amount of protein produced. Accordingly, FMRP deficiency results in dysregulation of numerous mitosis and neurogenesis genes primarily at the RNA expression level, whereas the expression of many synaptic genes is mostly dysregulated at the translation level (Liu et al, 2018). Moreover, exaggerated translation of epigenetic or transcriptional transcripts, which are direct targets of FMRP, may indirectly result in the upregulated expression of transcription regulators such as Ndn, an intronless gene encoding for a growth suppressor protein in post‐mitotic neurons (Liu et al, 2018).
Several direct FMRP target mRNAs encoding transcription factors and chromatin modifiers (Darnell et al, 2011; Korb et al, 2017) have been identified by using high‐throughput sequencing of RNAs isolated by cross‐linking immunoprecipitation (HITS‐CLIP). Loss of FMRP in mice results in elevated expression of several chromatin‐associated proteins, including bromodomain‐containing protein 4 (Brd4). The upregulation of Brd4, as well as of other chromatin‐associated FMRP targets such as MLL1 and p300, results primarily in an increase in histone modifications (i.e. acetylation and methylation) and active chromatin (Korb et al, 2017). In addition, Brd4 acts as activator hub for several FMRP targets (Rahman et al, 2011; Shen et al, 2015) and its loss is correlated with the decreased expression of synaptic genes (Korb et al, 2015, 2017; Liu et al, 2018), which are increased in FXS (Darnell et al, 2011; Korb et al, 2017). Finally, inhibition of Brd4 using the small molecule JQ1 reversed several of the gene expression changes observed in FMRP‐lacking neurons and alleviated many phenotypes displayed by Fmr1 KO mice (Korb et al, 2017). Interestingly, many of the chromatin‐associated FMRP targets are also ASD‐linked genes; mutations in these genes result in either increased or decreased expression (Korb et al, 2017). Taken together, these observations provide insight into FMRP‐dependent translatome and transcriptome changes in FXS, suggesting that FMRP functions by regulating translation to modify transcription (Fig 1). Misregulation of gene expression at both the transcription and translation levels likely contribute to FXS, creating a positive feedback loop which exacerbates molecular and neuronal dysfunction. Given these observations, targeting the synergistic dysregulation of translational and transcriptional mechanisms, perhaps using transcription inhibitors (Korb et al, 2017), to rebalance global changes in gene expression may represent a successful therapeutic approach for treatment of FXS.
Figure 1. Dysregulation of translation and transcription in FXS.
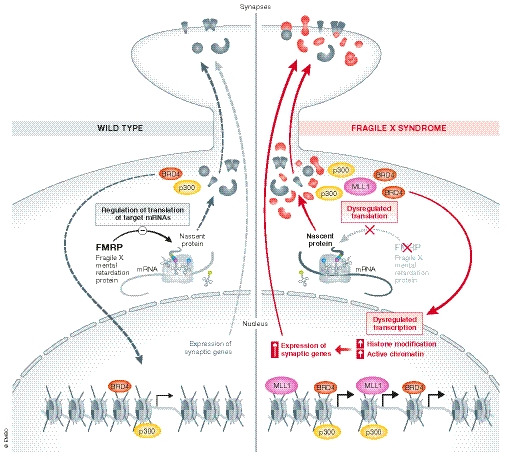
(Left) FMRP regulates the translation of certain mRNAs, including synaptic and chromatin‐associated proteins, such as Brd4 (red) and p300 (yellow), which facilitate the initiation and elongation phases of transcription by binding to activated chromatin at acetylated lysine residues. (Right) Dysregulation of translation in FXS impacts gene expression at the transcription level creating a feedback loop which exacerbates both molecular and neuronal dysfunction.
Phosphatase and tensin homolog (PTEN)
Originally identified as a cancer predisposition gene (Liaw et al, 1997), the phosphatase and tensin homolog (PTEN) gene located on chromosome 10q23 is an ASD risk gene (Butler et al, 2005; Buxbaum et al, 2007; Varga et al, 2009). Human genetic studies showed that ASD and macrocephaly are associated with germline heterozygous PTEN mutations, with PTEN‐associated ASD accounting for approximately 10% of macrocephalic ASD (Butler et al, 2005; Varga et al, 2009; Frazier et al, 2015). Nearly all individuals with PTEN‐associated ASD exhibit severe cognitive deficits, seizures, motor dysfunction, and verbal ability delay (Varga et al, 2009; Child & Cascino, 2013; Frazier et al, 2015), consistent with the macrocephaly white matter abnormalities that have been reported (Frazier et al, 2015).
PTEN encodes for a lipid phosphatase (PTEN) with specificity toward phosphatidylinositol (3,4,5)‐triphosphate (PIP3) (Parsons, 2004), which acts as a negative regulator of the PI3K/Akt/mTORC1 signaling pathway (Maehama & Dixon, 1998). PTEN inhibition of PIP3 results in decreased phosphorylation of Akt and germline PTEN mutations lead to reductions in PTEN protein levels, increased Akt phosphorylation, and subsequently a constitutively active PI3K/Akt/mTORC1 signaling pathway (Tan et al, 2011). Activated Akt also phosphorylates and inhibits TSC2 (also known as tuberin) (Inoki et al, 2002; Potter et al, 2002), thus removing the inhibition of mTORC1 by the TSC1/2 complex (Fig 2). Due to the important role of PI3K/Akt/mTORC1 pathway in the regulation of cell growth, survival, and proliferation, it is not surprising that PTEN inactivation has been observed in a broad spectrum of human cancers. However, ASD‐associated PTEN mutations affect Akt inhibition differently than those appearing in adults with predominantly cancer phenotypes and seem not to substantially abrogate PTEN activity in vivo (Rodríguez‐Escudero et al, 2011).
Figure 2. Mutations in PTEN and TSC1/2 are associated with dysregulation of translation and transcription.
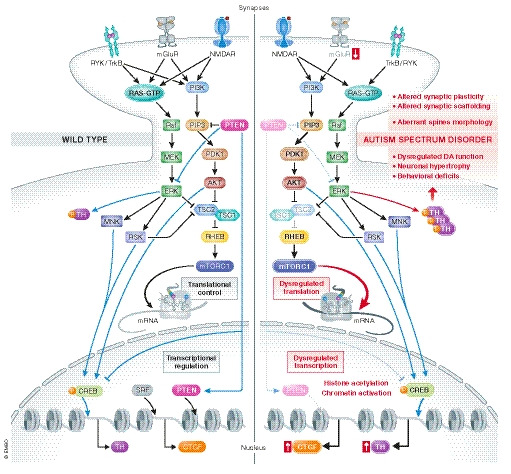
(Left) PTEN regulates translational/transcriptional systems, playing an important role in neuronal proliferation, migration, survival, morphology, and plasticity. As a lipid phosphatase, PTEN catalyzes the removal of the D3 phosphate from PIP3, which acts as a negative regulator of the PI3K/Akt/mTORC1 signaling pathway, whereas PTEN’s protein phosphatase activity targets a number of substrates, including MAPK signaling, which PTEN inhibits. The TSC1–TSC2 complex, through its GTPase‐activating protein activity towards the small G‐protein Rheb, is downstream effectors of the PI3K/Akt pathway and a critical negative regulator of mTORC1, regulating translation, transcription, and other cellular processes. (Right) Mutations in both PTEN and TSC1/2 trigger the activation of mTORC1, thereby regulating translation, transcription, and other cellular processes. Exaggerated mTORC1 signaling results in disinhibited protein synthesis. The loss of either PTEN or TSC1/2 activity directly impact gene transcription and the translation of specific mRNAs, such as TH and CTGF, and a number of synaptic scaffolding proteins such as PSD‐95 and SAPAP1.
Several studies have focused on the role of PTEN mutations in ASD by generating gene disruption models in mice to investigate PTEN neurobiology and unveil the cellular processes regulated by this phosphatase (reviewed in Tilot et al, 2015). PTEN heterozygous mice represent the most investigated model for PTEN loss in the CNS (Tilot et al, 2015). However, endophenotypes displayed by conditional PTEN knockout mice more closely resemble PTEN‐related ASDs and provide insight into the different brain regions and cell types involved in ASD. Nse‐Cre mice, where PTEN has been deleted selectively in a subset of neurons in cortex and hippocampus, exhibit social deficits and increased repetitive and perseverative behaviors (Kwon et al, 2006; Napoli et al, 2012) suggesting that PTEN simultaneously regulates proliferation and connectivity in the CNS. Pan‐neural PTEN loss results in hyperproliferation and increased cell size, which are due to the overactivation of Akt/mTORC1 signaling (Kwon et al, 2001; Amiri et al, 2012). Elevated mTORC1 signaling also is responsible for increased branching of the dendritic arbor, thickening of dendritic processes, and ectopic axonal projections exhibited by PTEN null neurons (Kwon et al, 2006; Fraser et al, 2008). Thus, morphological aberrations of neurons lacking PTEN may be responsible for the significant changes in firing properties and synaptic plasticity observed in different PTEN mouse models (Luikart et al, 2011). For example, PTEN heterozygous KO mice exhibit decreased long‐term potentiation (LTP) and impaired NMDA receptor‐dependent long‐term depression (LTD) at hippocampal CA1 synapses (Wang et al, 2006). GFAP‐Cre PTEN conditional KO mice also display decreased LTP at CA1 synapses (Fraser et al, 2008) as well as impaired metabotropic glutamate receptor‐dependent (mGluR)‐dependent LTD at dentate granule cell synapses(Takeuchi et al, 2013). However, it has been suggested that the loss of PTEN may not induce de novo spinogenesis but instead may increase synaptic activity by inducing morphological and functional maturation of spines (Haws et al, 2014). While all these studies imply that increased spine density or maturation underlie the changes in synaptic plasticity, mice with a CamKIIα‐Cre‐dependent postnatal deletion of PTEN exhibit LTP and LTD deficits at CA1 synapses in the hippocampal neurons, but no alterations in either neuronal or dendritic morphology, suggesting that these phenotypes are not necessarily dependent on each other (Sperow et al, 2012). Moreover, rapamycin, the pharmacological inhibitor mTORC1 signaling, rescues the neuronal hypertrophic phenotypes and behavioral abnormalities associated with loss of PTEN, but surprisingly the loss of S6K1, a downstream effector of mTORC1 and ERK does not result in the same outcome (Kwon et al, 2003; Chalhoub et al, 2006; Zhou et al, 2009).
Given the increasing evidence that common/shared biological pathways or brain circuits may account for the cellular and molecular mechanisms involved in ASD development, it has been proposed that although loss of PTEN in neurons is sufficient to cause social deficits in mice, one cannot exclude the possibility that other ASD‐related genes are necessary for the development of the complex autistic phenotype seen in patients carrying PTEN mutations (Zhou & Parada, 2012). Indeed, the deletion of PTEN alters the expression of the ASD‐related genes FMRP and MeCP2. The activation of the PI3K/Akt/mTORC1 pathway in the Nse‐Cre, PTEN mouse model, for example, results in a decreased number of synaptic scaffolding proteins such as PSD‐95 and SAPAP1, decreased mGluR, and increased FMRP expression (Backman et al, 2001; Kwon et al, 2001). Moreover, both PTEN and transcription factor MeCP2 reciprocally regulate the expression of each other via microRNAs (Lyu et al, 2016), 20–25‐nucleotide‐long noncoding RNAs that modulate gene expression and development by post‐transcriptionally targeting RNA‐induced silencing complexes (Bartel, 2004). Lyu and colleagues reported that deletion of PTEN leads to increased phosphorylation of Serine 133 of CREB (P‐CREB) and increased expression of its target microRNA‐132, which in turn inhibits the expression of MeCP2 by targeting the 3′UTR of MeCP2 mRNA. The loss of PTEN thus results in lower MeCP2 expression. Interestingly, it has been shown that microRNA‐132 inhibitors largely blocked the effects of CREB on dendrite maturation (Magill et al, 2010). On the other hand, knockdown of MeCP2 leads to upregulation of microRNA‐137, which in turn represses expression of PTENP (Lyu et al, 2016). Thus, PTEN is downregulated when MeCP2 is knocked down.
Additional evidence for association between ASD‐linked PTEN mutations and dysregulated translational/transcriptional systems underlying ASD comes from a study on PTEN missense (PTENm3m4) knock‐in transgenic mice (He et al, 2015b). This study is based on the evidence that conditional Pten deletion in dopaminergic neurons of mice results in elevated expression of tyrosine hydroxylase (TH), the key enzyme in the dopamine (DA) biosynthesis pathway, associated with increased striatal DA content (Domanskyi et al, 2011). PTEN normally acts to suppress tyrosine hydroxylase (TH) transcription through the inhibitory effect on PI3K and P‐CREB but also inhibits TH phosphorylation by suppressing the ERK pathway. There is thus an increase in TH expression and phosphorylation in the cerebrum and in the striatum of PTENm3m4 mice (He et al, 2015b). Dopamine (DA) exerts a pivotal role in the modulation of locomotive function, learning and memory, and social interactions (Wise, 2004; Shohamy & Adcock, 2010; Rice et al, 2011; Matthews et al, 2016; Torquet et al, 2018). Dysregulation of the dopaminergic system has been linked to ASD pathophysiology, including exaggerated repetitive and perseverative behaviors (McDougle et al, 2005; DiCarlo et al, 2019). An augment in TH synthesis and function leads to the release of too much dopamine in the prefrontal cortex and midbrain, which can result in repetitive and perseverative behaviors (He et al, 2015a). Conditional deletion of PTEN in dopaminergic neurons results in neuronal hypertrophy, an increased number of dopaminergic neurons and social behavioral deficits (Clipperton‐Allen & Page, 2014). Consistent with studies of the PTENm3m4 mice, the ASD‐linked PTEN mutations H93R, F241S, and D252G were not able to suppress TH when overexpressed in PC12 cells. In addition, ectopic expression of two other PTEN missense mutations that either lack phosphatase function in general (C124S) or only lipid phosphatase function (G129E) failed to suppress TH in PC12 cells (He et al, 2015b). Together, these findings suggest that ASD‐associated PTEN mutations can enhance TH expression in two ways: the loss of the lipid phosphatase activity of PTEN will induce TH transcription and translation through the dysregulated PI3K/CREB pathway and the loss of the protein phosphatase activity of PTEN will enhance the phosphorylation of TH through the ERK pathway (He et al, 2015b). Therefore, the loss of both lipid and protein phosphatase activity of PTEN results in a synergic effect on TH regulation leading to ASD.
Finally, microarray studies have revealed changes in gene transcription profiles following PTEN depletion or overexpression (Matsushima‐Nishiu et al, 2001; Carver et al, 2011; Mulholland et al, 2012; Chen et al, 2014), and PTEN was proposed to repress transcription by maintaining a condensed chromatin structure through a physical interaction with histone H1. Loss of PTEN results in the displacement of histone H1, chromatin decondensation and the elevation of H4K16 acetylation (Chen et al, 2014), which opens chromatin structure for transcription (Chen et al, 2014).
Tuberous sclerosis complex (TSC)
Tuberous sclerosis complex (TSC) is an autosomal‐dominant inherited disorder caused by loss‐of‐function mutations in either TSC1 or TSC2 genes (Tsai & Sahin, 2011). TSC affects 1/6,000 newborns worldwide with ASD occurring in more than 50% of the individuals (Ehninger & Silva, 2011; Han & Sahin, 2011; Gipson et al, 2013) and accounting for 1–4% of all cases of ASD (Fombonne, 2003). Clinical manifestations associated with TSC are variable with respect to symptoms and disease severity, which is in part dependent on which TSC gene is affected. TSC patient populations are characterized by high prevalence of epilepsy (∼ 90%), intellectual disability, ASD, ADHD, sleep disruption, and additional psychiatric features in adults, such as anxiety and mood disorders (Ehninger & Silva, 2011; Han & Sahin, 2011). Neuropathologies associated with TSC include formation of cortical tubers, which are characterized by dysplastic neurons with immature electrophysiological properties (Cepeda et al, 2012), subependymal nodules, and subependymal giant cell astrocytomas (DiMario, 2004). Extensive studies of the TSC1 and TSC2 genes have revealed a wide spectrum of mutations in TSC individuals. These mutations comprise a mix of missense, nonsense, insertions, and deletions involving nearly all exons present in both the TSC1 and TSC2 genes (Rosset et al, 2017), and their impact on the clinical phenotypes is extremely variable (Ehninger & Silva, 2011; Han & Sahin, 2011). Individuals with heterozygous mutations in either of the TSC1 or TSC2 genes have a ~ 100‐fold increase in the probability of being diagnosed with ASD compared with the general population, which is phenotypically similar to ASD than other etiologies at the behavioral level (Hunt & Shepherd, 1993; Smalley, 1998; Ehninger & Silva, 2011).
TSC1 and TSC2 encode for hamartin (also referred to as TSC1) and tuberin (also referred to as TSC2), respectively, which are canonical components of the mTOR pathway (Tsai & Sahin, 2011; Lipton & Sahin, 2014). As mentioned above (see PTEN section), phosphorylation of TSC2 by Akt inhibits its function. This relieves the inhibition of TCS1/2 on mTORC1 and promotes its activity (Inoki et al, 2002; Takei et al, 2004). In neurons, TSC1 and TSC2 proteins form a heterodimer that regulates protein synthesis and cell size (Kwiatkowski & Manning, 2005). TSC1 functions as a regulator of TSC2 stability, preventing TSC2 degradation (Benvenuto et al, 2000), whereas TSC2, a GTPase‐activating protein (GAP), inactivates Rheb, a Ras family GTPase, and other small G proteins (Kwiatkowski & Manning, 2005). The phosphorylation of TSC2 by Akt results in Rheb activation, which in turn triggers the activation of mTORC1, thereby regulating translation, transcription, and other cellular processes (Kwiatkowski & Manning, 2005). Without a normally functioning TSC1/2 complex, mTORC1 is hyperactive, resulting in disinhibited protein synthesis and subsequent cell growth (Ruvinsky & Meyuhas, 2006; Wullschleger et al, 2006). Thus, mutations in TSC1 and TSC2 are consistent with the notion that dysregulated neuronal translation underlies several forms of ASD (Fig 2) (Ehninger & Silva, 2011).
Human neurons derived from TSC2‐deficient pluripotent stem cells exhibited developmental abnormalities that recapitulate pathological hallmarks of cortical malformations in patients such as altered synaptogenesis and synaptic transmission paralleled by molecular changes in pathways associated with ASD. TSC2 deletion leads to exaggerated mTORC1 signaling during differentiation of human neurons in a gene‐dosage‐dependent manner. Pharmacological inhibition of mTORC1 corrects developmental abnormalities and synaptic dysfunction, supporting the notion that the lack of TSC2 causes alterations that are dependent on hyperactive mTORC1 signaling (Costa et al, 2016). Synaptic abnormalities were also observed in astrocyte‐specific Tsc1 deletion mice and in the Eker rat model of TSC (Von Der Brelie et al, 2006). Several different heterozygous TSC1 or TSC2 rodent models exhibit behavioral abnormalities, including social interaction and learning deficits (Goorden et al, 2007, Waltereit et al, 2011, Von Der Brelie et al, 2006), which are reversed by acute rapamycin treatment. The behavioral deficits displayed by the various TSC model mice are often correlated with impaired synaptic plasticity. For example, mice with a heterozygous deletion of Tsc2 (TSC2 +/−) have deficient hippocampal mGluR‐LTD and decreased translation of Arc mRNA (Auerbach et al, 2011). Similarly, postnatal deletion of Tsc1 in hippocampal CA1 neurons in mice results in impaired mGluR‐LTD and increased excitatory synaptic function (Bateup et al, 2011). Notably, the hyperactivation of the mTORC1 pathway results in reduced mGluR‐LTD and reduced de novo protein synthesis in TSC model mice. To note, this is in contrast to Fmr1 KO mice, which exhibit increased net de novo translation, enhanced mTOR signaling and exaggerated mGluR‐LTD at CA1 synapses (Sharma et al, 2010). Moreover, inhibition of mTORC1 and augmentation of mGluR5 signaling in Tsc2 +/− mice restored mGluR‐LTD and normalized de novo protein synthesis. These findings suggest that TSC1/TSC2 and FMR1 mutations have opposite effects on the tight regulation of translation that underlies proper synaptic function. It has been suggested that TSC1/TSC2 and FMRP may differentially regulate the translation of different pools of mRNA and that their dysregulated transcripts may have a different outcome in terms of synaptic function. An alternative possibility is that enhanced mTORC1 activity in TSC may result in increased FMRP activation, resulting in suppression of specific mRNAs (Ebert & Greenberg, 2013). In line with this hypothesis, deleting Fmr1 in Tsc2 +/− mice prevented synaptic and behavioral deficits in the double mutant mice (Auerbach et al, 2011).
A delicate balance of mTORC1 activity also has been shown to be required for proper myelination in oligodendrocytes, where both inactivation and hyperactivation of mTORC1 results in a hypomyelination phenotype (Lebrun‐Julien et al, 2014; Carson et al, 2015). Interestingly, disruption of myelination during development has been implicated in a range of neurodevelopmental disorders including TSC. Moreover, TSC patients with ASD display impaired white matter integrity compared with TSC patients without ASD (Lewis et al, 2013; Peters et al, 2013). Similarly, mice lacking neuronal TSC1/2 have a hypomyelination phenotype (Meikle et al, 2007; Lebrun‐Julien et al, 2014; Carson et al, 2015). It has been shown that the loss of functional TSC1/2 in neurons results in blockade of oligodendrocyte development and myelination, which is mediated by neuronal connective tissue growth factor (CTGF) (Ercan et al, 2017). CTGF has been shown to bind to, antagonize insulin‐like growth factor, and stimulate oligodendrocyte development and, thus, can indirectly block the activation of mTORC1 signaling in oligodendrocytes (Stritt et al, 2009). mTORC1 in oligodendrocytes promotes initiation of myelination, myelin thickness by controlling lipogenesis, and translation of myelin proteins (Lebrun‐Julien et al, 2014). In addition, CTGF is highly expressed and secreted from neurons lacking TSC1/2 and blocks the development of oligodendrocytes. Thus, genetic deletion of CTGF in TSC1‐KO neurons improves myelination. Finally, TSC2‐deficient neurons exhibit a decrease of serum response factor (SRF) protein, which is the transcriptional repressor of the CTGF gene (Stritt et al, 2009) (Fig 2). Taken together these findings indicate that in addition to its well‐known role in the regulation of protein synthesis, TSC1/2 affects the transcription machinery in the context of myelination, adding an additional layer of regulatory control during development.
Regulation of transcription and ASD
Gene expression is regulated by orchestrated mechanisms that include the action of thousands of transcription factors, epigenetic factors (Day & Sweatt, 2011; Cholewa‐Waclaw et al, 2016; Marshall & Bredy, 2016) and three‐dimensional genomic factors such as chromatin structure or nuclear organization (Medrano‐Fernández & Barco, 2016; Rajarajan et al, 2016; Watson & Tsai, 2017) that act at the transcriptional and post‐transcriptional level. Perturbations of these transcriptional control programs likely causes a broad range of neurodevelopment diseases processes, including ASD. Several mutations associated with ASD have been shown to disrupt components and functions of signaling networks involved in the control of gene transcription, such as regulation of chromatin and expression of effector genes that act on synapses or that control calcium influx into neurons (Ebert & Greenberg, 2013). Herein, we summarize findings related to three ASD‐associated genes that act as key sensors of transcription regulation and highlight their impact on translational control underlying ASD pathophysiology: methyl‐CpG‐binding protein 2 (MECP2), activity‐dependent neuroprotective protein (ADNP), and engrailed 2 (EN2).
Methyl‐CpG‐binding protein 2 (MECP2)
Rett syndrome (RTT) is a neurodevelopmental disorder often associated with ASD and characterized by compromised brain function, severe intellectual disability, language and learning disabilities, repetitive stereotyped hand movements, and developmental regression (Liyanage & Rastegar, 2014). De novo mutations in the methyl‐CpG‐binding protein 2 (MECP2) gene, a transcriptional regulator, account for more than 95% of RTT cases (Amir et al, 1999; Chahrour & Zoghbi, 2007; Neul et al, 2010). Due to the X‐linked nature of the MECP2 gene, RTT is the leading cause of intellectual disability in females, affecting ~ 1:10,000 girls worldwide, with rare cases reported in males (Shahbazian & Zoghbi, 2002; Moog et al, 2003; Liyanage & Rastegar, 2014). Either loss or duplication of MECP2 results in neurodevelopmental disorders characterized by multiple autistic behaviors (Amir et al, 1999). Loss‐of‐function mutations in MECP2 can cause RTT (Chahrour & Zoghbi, 2007), whereas gain‐of‐function mutations result in MECP2 duplication syndrome (Van Esch et al, 2005), which is observed more frequently in males and is characterized by developmental delay, motor dysfunction, epilepsy, anxiety, frequent respiratory infections, and early death (Ramocki et al, 2010). Several studies suggest an association between mutations in MECP2 and ASD (Beyer et al, 2002; Shibayama et al, 2004; Swanberg et al, 2009), although whether MECP2 mutations identified in patients with ASD interfere with normal functions of MeCP2 remained unknown.
MeCP2 belongs to the DNA methyl‐binding protein (MBP) family (Lewis et al, 1992), and it is an epigenetic regulator that acts on chromatin structure (Nan et al, 1998) with crucial functions in controlling neuronal morphology, synaptic transmission, and synaptic plasticity (Chao et al, 2007; Monteggia & Kavalali, 2009). Although, initially proposed as a transcriptional repressor (Nan et al, 1998), evidence suggests that MeCP2 acts by facilitating global transcription (Chahrour et al, 2008; Ben‐Shachar et al, 2009). Indeed, MeCP2 has recently emerged as a multifunctional protein that bridges epigenetic marks in the DNA (cytosine methylation/hydroxymethylation) with chromatin structure and the regulation of gene expression (Fasolino & Zhou, 2017). The transcriptional regulatory role of MeCP2 appears to depend on its interacting protein partners, such as CREB, cohesin, and HP1, which contribute to MeCP2 multiple functions (de Paz & Ausió, 2017): transcription activation, chromatin loop formation, and chromatin architecture modulation, respectively (Fuks et al, 2003; Chahrour et al, 2008; Lyst & Bird, 2015). Moreover, de novo phosphorylation of MeCP2 at serine 421 (S421) by a CaMKII‐dependent mechanism controls the ability of MeCP2 to regulate dendritic patterning, spine morphogenesis, and the activity‐dependent induction of Bdnf transcription (Zhou et al, 2006) (Fig 3).
Figure 3. Regulation of transcriptional and translational machinery by MeCP2.

(Left) MeCP2 facilitates global transcription by interacting with its protein partners such as transcription activators, and proteins acting on chromatin loop formation and chromatin architecture modulation. De novo phosphorylation of MeCP2 at serine 421 (S421) by a CaMKII‐dependent mechanism controls the ability of MeCP2 to regulate dendritic patterning, spine morphogenesis, and the activity‐dependent induction of Bdnf transcription. MECP2 regulates translation initiation by interacting with components of the pre‐initiation complex, which is consistent with its ability to bind mRNA, regulates ribosome biogenesis by interacting with nucleolin, which in turn regulates rRNA synthesis/ribosome biogenesis by controlling RNA polymerase I activity, and promotes the posttranscriptional processing of particular miRNAs, such as miR‐199a which positively controls mTOR signaling by targeting inhibitors for mTOR signaling (e.g. PDE4D, HIF1a, SIRT1). (Right) Hypothetical connection between MeCP2‐mediated transcriptional regulation and mTORC1 activity, highlighting the cooperative network between translational control and transcriptional co‐regulation underlying ASD pathophysiology. Both loss of function mutations or duplication of MECP2 result in dysregulation of both processes, ultimately altering ribosome biogenesis (e.g. nucleolin), neuronal plasticity and synapses (e.g. reduced expression of CAMKIIα and PSD95), and spine formation. Impaired Akt/mTORC1 signaling is associated to reduced de novo translation and posttranscriptional processing of particular miRNAs in MeCP2‐mediated ASD.
Apart from mutations in the MECP2 coding region, mutations in the 3’UTR and increased DNA methylation of the MECP2 promoter have been found in ASD patients (Shibayama et al, 2004; Loat et al, 2008; Nagarajan et al, 2008; Xu et al, 2012). Misexpression of the MeCP2 protein is thought to contribute to neuropathology by causing dysregulation of plasticity. Findings from mice carrying mutations that abolish expression of MeCP2 strongly suggest that it regulates plasticity in development and adulthood (Krishnan et al, 2017; Karaca et al, 2018). Nevertheless, how MECP2 mutations affect the plasticity that underlies learning and memory is poorly understood. It has been suggested that MeCP2 is required for long‐term memory formation and that it controls the learning‐induced transcriptional response of hippocampal neurons required for memory consolidation (Karaca et al, 2018). Furthermore, Mecp2 mutations impair auditory cortical plasticity and maternal vocal perception in adult female mice by altering parvalbumin‐expressing cortical inhibitory interneurons (PV) (Krishnan et al, 2017), which appear to be especially vulnerable to loss of MeCP2 (Ito‐Ishida et al, 2015; Morello et al, 2018). It has been suggested that MeCP2 acts in cortical PV interneurons to coordinate stimulus‐specific, experience‐dependent cortical plasticity, thereby reshaping the output of deep layer pyramidal neurons and that mutations in MECP2 disrupt cortical activity by arresting modulation of cortical PV networks (Lau et al, 2020). Consistent with these findings, MeCP2 overexpression results in atypical response properties of the auditory cortex in mice (Zhou et al, 2019a).
As mentioned above, proper regulation of protein synthesis in neurons is crucial for the organization of synaptic structure and synaptic plasticity (Richter & Klann, 2009), and aberrant regulation of the mTOR pathways have been associated with molecular defects in different neurodevelopmental disorders. Evidence of the relationship between MECP2 mutations, translational control, and ASD comes from studies on Mecp2 mutant mice (Ricciardi et al, 2011) and human embryonic stem cell model of RTT (Li et al, 2013). Notably, pre‐symptomatic Mecp2 mutant mice exhibit downregulated Akt/mTORC1 signaling and reduced protein synthesis, consistent with deficits in translation (Fig 3). In addition, MECP2 may promote translation initiation by interacting with components of the pre‐initiation complex, which is consistent with its ability to bind mRNA (Young et al, 2005). The impaired translational control in Mecp2 mutant mice is not restricted to a specific subset of transcripts but affects molecules with pivotal roles in neuronal activity and plasticity (i.e. CAMKIIα and PSD95), as well as proteins with no clear role in neuronal function (i.e. Rack1 and GAPDH) (Ricciardi et al, 2011). Consistent with these findings, MECP2 deficiency in neurons is associated with compromised protein synthesis (Li et al, 2013), suggesting that reduced translation in MECP2‐deficient neurons contributes to the ASD phenotypes in RTT. As mentioned above, reduced de novo protein synthesis in MECP2 mutant neurons is accompanied with impaired Akt/mTORC1 signaling; conversely, its activation by exogenous growth factors such as BDNF and IGF‐1 (insulin‐like growth factor 1) or by depletion of PTEN (a negative regulator of PI3K, an upstream component of the AKT/mTOR pathway) promoted protein synthesis and ameliorated ASD phenotypes in RTT mutant neurons (Li et al, 2013). Furthermore, studies on RTT human cerebellum showed that common MECP2 mutations (T158M, R255X) affect pathways that impinge on ribosome biogenesis. Ribosomal RNA (rRNA) synthesis is a rate‐limiting step for ribosome biogenesis and it has been suggested that nucleolin, a regulator of rRNA transcription and processing might be a potential MeCP2 target (Olson et al, 2018). Nucleolin regulates rRNA synthesis/ribosome biogenesis by controlling RNA polymerase I activity, a process that is also controlled by mTORC1–S6K1 signaling. Although studies on murine models reported that the 28S and 18S rRNA transcripts are reduced in murine Mecp2‐deficient neurons (Gabel et al, 2015) and that the mTOR signaling is impaired in RTT models (Ricciardi et al, 2011), human RTT cerebellum neurons exhibited elevated rRNA transcripts and mTORC1–S61K signaling, pointing toward a potential over‐activation of these processes (Olson et al, 2018). Further evidence that links MeCP2 to mTORC1 signaling comes from a study investigating MeCP2 and its role in posttranscriptional processing of particular microRNAs (miRNAs) in RTT (Tsujimura et al, 2015). miR‐199a, a MeCP2‐regulated miRNA, positively controls mTOR signaling by targeting inhibitors of mTOR activity ameliorating RTT neuronal phenotypes, whereas miR‐199a inhibition blocks MeCP2 function and its genetic deletion led to a reduction of mTOR activity resulting in RTT phenotype in mice (Tsujimura et al, 2015).
All together, these studies suggest that both RTT and MECP2 duplication syndrome are among the growing number of neurodevelopmental disorders that are linked to dysregulated mTORC1 signaling, highlighting the cooperative network between translational control and transcriptional co‐regulation underlying ASD pathophysiology (Fig 3).
Activity‐dependent neuroprotective protein (ADNP)
Activity‐dependent neuroprotective protein (ADNP) represents a leading de novo mutated gene causing a constellation of features associated with a particular form of ASD termed Helsmoortel‐Van der Aa Syndrome (HVDAS). Heterozygous mutations in ADNP, which encodes a transcription factor, on the long arm of chromosome 20 (20q13.13), account for the etiology of 0.17% of patients with ASD (Helsmoortel et al, 2014) and ADNP levels in the plasma are significantly correlated with IQ (Malishkevich et al, 2016). Individuals with ADNP mutations share features such as global developmental delay, intellectual disability, facial anomalies, behavioral, and motor disturbances, as well as congenital cardiac defects, feeding difficulties, and visual problems (Helsmoortel et al, 2014; Pascolini et al, 2018). Moreover, the evidence that ADNP is required for brain formation (Pinhasov et al, 2003) coupled with the finding that a major phenotypic outcome of Adnp‐haploinsufficency in mice leads to cognitive impairments, placed ADNP as a key transcription factor required for normal brain function (Vulih‐Shultzman et al, 2007) (Fig 4).
Figure 4. Disruption of ADNP and EN2 intracellular pathways causes dysregulation of transcriptional and translational control in ASD.

(Left) ADNP and EN2, which act as key transcription regulators, also impact translation by interacting with factors such as eIF4E. ADNP binds directly to eIF4E, suggesting an active role of ADNP in cap‐dependent translational control differentially regulating the expression of several mRNAs in age‐dependent and sex‐dependent manner, such as SlLC6A4 (serotonin transporter), and CACNA1E (calcium channels) and BECN1 (autophagy regulator). ADNP binds and regulates ZFP161 and his mouse homolog zf5, which act as a transcriptional activator of DAT, IL‐6, LIF, and a transcriptional repressor of FMR1 by binding to promoters of these genes. EN2 contains a binding site for eIF4E and triggers the rapid phosphorylation of eIF4E and eIF4E‐binding protein (4E‐BP). EN2 modulates the pleiotropic effects of IGF‐1 by altering S6K1 activation and attenuates S6K1 phosphorylation and activation via mTORC1‐dependent and mTORC1‐independent pathways downstream of PI3K. (Right) Perturbations of transcriptional and translational control results in disruption of multiple signaling networks involved in the control of gene transcription and protein synthesis. Subsequent changes in the expression of synaptic proteins and proteins that control calcium influx into neurons results in altered E/I balance in ASD. Excessive EN2 signaling alters excitation/inhibition (E/I) balance by acting on glutamatergic and GABAergic dendritic branching, which is mediated by an increase in mTORC1‐dependent protein synthesis.
Originally associated with neuroprotection and neuroglia interactions (Gozes, 2007), ADNP is a member of the SWI/SNF (SWItch/Sucrose Non‐Fermentable) chromatin remodeling complex, with predominantly nuclear localization (Mandel & Gozes, 2007; Helsmoortel et al, 2014) and controls the expression of more than 400 genes during embryonic development (Mandel & Gozes, 2007). Besides the direct interaction with the SWI/SNF complex, ADNP is involved in multiple molecular mechanisms and its transcriptional control function is also associated with myosin regulatory light chain 2 (Myl2) (Mandel & Gozes, 2007), globin (Dresner et al, 2012), and the major Alzheimer’s disease risk gene apolipoprotein E (APOE) (Mandel & Gozes, 2007) in a sex‐dependent manner (Malishkevich et al, 2016). Therefore, ADNP shows a tight association with neurodevelopmental processes, exerting a key role in regulating cognitive functions which are not limited to ASD, but extend to schizophrenia (Merenlender‐Wagner et al, 2015) and Alzheimer's disease (Malishkevich et al, 2016).
ADNP can be localized to the cytoplasm and the axon and, thus, is not confined to the cell nucleus (Gennet et al, 2008), suggesting additional, non‐transcriptional activities of ADNP. Consistent with this idea, there is evidence that ADNP plays a role in the initiation step of protein synthesis (Malishkevich et al, 2015). ADNP has been reported to bind directly to eIF4E, suggesting an active role of ADNP in cap‐dependent translational control (Fig 4). Haploinsufficient ADNP (Adnp +/‐) mice mimic human HVDAS in terms of altered gene expression patterns and synapse density, as well as compromised developmental, motor, and cognitive ability (Vulih‐Shultzman et al, 2007; Amram et al, 2016). In contrast, complete deficiency in ADNP results in neural tube closure defects and death during gestation in Adnp −/− homozygous embryos (Mandel & Gozes, 2007). Altered eIF4E expression in the hippocampus of Adnp +/− male mice has been reported. The levels of eIF4E show a transient increase at a young age and are subsequently decreased, suggesting changes in translational control early in development (Malishkevich et al, 2015). Reduced Adnp levels thus correlate with increased eIF4E expression in mice. Interestingly, ADNP expression is reduced in female postmortem human hippocampal tissue (Malishkevich et al, 2015), but whether this changes eIF4E expression has not been investigated. In addition to the age‐dependent changes in eIF4E expression, ADNP seems to differentially regulate the expression of several mRNAs in a sex‐ dependent manner (Amram et al, 2016). It has been suggested that differences in ADNP expression may account for the sexual divergence associated with ASD (Malishkevich et al, 2015). Adnp‐deficient genotype shows sexual dichotomy and ADNP expression is modulated during the estrous cycle in the hypothalamus (Furman et al, 2004) in mice. Adnp +/− mice exhibit impaired hippocampal expression of key ASD‐linked genes including the serotonin transporter (SlLC6A4), the calcium channel (voltage‐dependent calcium channel, CACNB1), the calcium channel (CACNA1E), and the autophagy regulator, BECN1 (Beclin1), in a sex‐dependent manner. Finally, the ADNP‐interaction partner, zinc finger protein 161 homolog (ZFP161) contributes to the complex interplay between transcription and translation. ADNP binds and regulates ZFP161 and its mouse homolog zf5, which act as a transcriptional activator of dopamine transporter (DAT; SLC6A3), interleukin 6 (IL‐6), leukemia inhibitory factor (LIF), and a transcriptional repressor of FMR1 by binding to promoters of these genes (Malishkevich et al, 2015; Gozes, 2018) (Fig 4), arising questions on the impact of ADNP–ZFP161–FMR1 interaction on translational control. These observations suggest that ADNP is a multitasking regulatory protein at the interface between transcriptional and translational processes that underlie sexual divergence and pathophysiology associated with ASD.
Engrailed 2 (EN2)
Several studies suggest an association between intronic polymorphisms of human Engrailed 2 (EN2) gene and non‐syndromic forms of ASD (Benayed et al, 2005, 2009; Hnoonual et al, 2016). Human EN2 maps to distal chromosome 7 (7q36.3), a chromosomal region that has been linked to ASD (Allen et al, 1997; Liu et al, 2001; Alarcón et al, 2002). Notably, in ASD subjects, several SNPs in EN2 function as a transcriptional activator, resulting in increased levels of gene expression (Courchesne et al, 2001; Bauman & Kemper, 2005; Benayed et al, 2009). In addition, variations in the EN2 protein expression and abnormal cerebellar structure have been identified in ASD patients (Courchesne et al, 2001; Bauman & Kemper, 2005). The cerebellum is one of the first structures of the human brain to differentiate, and histopathological changes in its neuronal structure, such as the loss of Purkinje cells, have been observed in a number of post‐mortem brains of ASD individuals (Bailey et al, 1998; Palmen et al, 2004; Allen, 2005). Evidence suggests that very early cerebellar developmental defects may account for a number of neurological abnormalities reported in ASD individuals (see review by Bolduc and Limperopoulos (2009)). Indeed, cerebellar abnormalities have also been described in human and mouse studies of ASD‐associated phenotypes in tuberous sclerosis (Asano et al, 2001).
EN2 belongs to the homeobox gene family and encodes a homeodomain‐containing transcription factor, which has a key role in the early development of the cerebellum. During neurodevelopment, EN2 regulates cerebellar growth and is important in the organization of the cerebellum, as well as the midbrain/hindbrain region (Sgaier et al, 2007). EN2 and its homolog EN1 have been shown to have different regulatory roles at different stages of development involving both transcriptional and translational activities (see review by Spatazza et al (2013)). Indeed, EN2 mRNA expression is downregulated during the first postnatal week in the hippocampus and in the cerebellum, a period associated with a sudden increase in synaptogenesis in rodents (Bury & Sabo, 2011). Several mouse models with alterations in EN2 expression have been generated to investigate its role in ASD pathophysiology. Interestingly, both homozygous En2 knockout (En2 KO) and En2 transgenic mice exhibit abnormal cerebellar development, such as cerebellar hypoplasia and decreased number of cerebellar neuronal cells (i.e. Purkinje cells), as well as cognitive impairments (Kuemerle et al, 2007; Tripathi et al, 2009), which are believed to be due to abnormal postnatal cerebellar development (Gharani et al, 2004). Finally, En2 KO mice exhibit ASD‐like behavioral traits such as decreased sociability, spatial learning deficits, and increased seizure susceptibility (Cheh et al, 2006; Tripathi et al, 2009; Brielmaier et al, 2012; Provenzano et al, 2014) associated with hippocampal dysregulation of neurofibromin‐dependent pathways (Provenzano et al, 2014).
Protein synthesis is crucial for spine morphology, as well as long‐term dendritic and synaptic plasticity (Klann & Dever, 2004; Sutton & Schuman, 2005; Lo & Lai, 2020), whose dysregulation has been associated with several forms of ASD (Hoeffer et al, 2012; Gkogkas et al, 2013b; Santini et al, 2013; Xu et al, 2020). Several studies reported that EN2 directly regulates translation. EN2 interacts with the translational machinery and activates cap‐dependent mRNA translation in axonal growth cones, a function that is crucial for axon turning (Brunet et al, 2005). Brunet et al, (2005) reported that secreted En2 generates an external gradient promoting growth cone turning in Xenopus. This depends on an interaction of internalized En2 with eIF4E and is blocked by protein synthesis but not transcription inhibitors. The interaction between En2 and eIF4E triggers the rapid phosphorylation of eIF4E and eIF4E‐binding protein (4E‐BP) (Brunet et al, 2005; Osborne & Borden, 2015) resulting in enhanced cap‐dependent translation (Fig 4). A mutant form of En2 lacking the putative eIF4E‐binding domain had no effect on growth cones. In addition, another study suggests that EN1 (the EN2 orthologue) increases mRNA translation in hippocampal cells, whereas EN2 is subjected to a tight regulation during development (Soltani et al, 2017).
Excessive EN2 signaling alters excitation/inhibition (E/I) balance by acting on glutamatergic and GABAergic dendritic branching, which is mediated by an increase in mTORC1‐dependent protein synthesis (Provenzano et al, 2014; Soltani et al, 2017) (Fig 4). Transcriptome analysis identified over 800 genes differentially expressed in the cerebellum and hippocampus of En2 KO mice and showed a significant convergence of neurobiological pathways previously linked to ASD pathology. Among the differentially expressed genes, Grm5, Nrxn3, and Scn1a, which encode for mGluR5, a neuronal adhesion protein of the Neurexin (NRXN) family, and the voltage‐gated sodium channel alpha subunit (SCN1A), respectively, are of particular interest for ASD (Sgadò et al, 2013). Finally, it was demonstrated that there is a functional interaction between EN2 expression and IGF1 signaling (Rossman et al, 2014), whose reduction in cerebrospinal fluid (CSF) of ASD patients has been reported (Riikonen et al, 2006), suggesting that EN2 modulates the pleiotropic effects of IGF‐1 by altering S6K1 activation in vitro (Rossman et al, 2014). It has been speculated that reduced EN2 expression increases S6K1 phosphorylation and activation via mTORC1‐dependent and mTORC1‐independent pathways downstream of PI3K, whereas in physiological conditions, EN2 attenuates mTORC1‐mediated proliferation (Rossman et al, 2014). Translational signaling pathways, including phosphorylation of eIF4E and the PI3K/Akt/mTORC1 and ERK (extracellular signal‐regulated kinase) signaling cascades, represent a common feature in various ASD‐associated gene alterations (Ebert & Greenberg, 2013), and it is intriguing that deletion of EN2, a transcription factor, shows dysregulated Akt‐mTORC1‐S6K1 signaling, which may account for increased mRNA translation and spine formation (Fig 4).
Conclusion and future outlook
The evidence highlighted in this review suggests that there is a complex level of dynamic regulation between translation and transcription that likely contribute to ASD pathophysiology. It should be noted that the interplay between translation and transcription is presented here as positive feedback loop that exacerbates ASD pathophysiology, but it is possible that this interplay could be compensatory and protective, which should be investigated in the future. We propose that there is bidirectional regulation of translation and transcription in ASD and that mutations in genes that result in dysregulation of one of those processes triggers a positive feedback loop to act on the other, which exacerbates ASD pathophysiology. This raises the possibility that rebalancing the system by targeting key players in this mutual regulatory loop could restore protein homeostasis, and ultimately, normal neuronal function. From this perspective, isolating and interpreting the changes in mRNA translation that affect neuronal physiology in terms of pathways, synaptic properties, and epigenetic outcomes represent a challenging but plausible route toward developing effective treatment in ASD. Moreover, it provides a mechanistic framework for the investigation of the synergistic contributions of different ASD‐associated genes and pathways underlying the development of ASD phenotypes.
Conflict of interest
The authors declare that they have no conflict of interest.
EMBO reports (2021) 22: e52110.
See the Glossary for abbreviations used in this article.
References
- Alarcón M, Cantor RM, Liu J, Gilliam TC, Geschwind DH, Consortium AGRE (2002) Evidence for a language quantitative trait locus on chromosome 7q in multiplex autism families. Am J Hum Genet 70: 60–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen G, Buxton RB, Wong EC, Courchesne E (1997) Attentional activation of the cerebellum independent of motor involvement. Science 275: 1940–1943 [DOI] [PubMed] [Google Scholar]
- Allen G (2005) The cerebellum in autism. Clin Neuropsychiatry 2: 321–337 [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY (1999) Rett syndrome is caused by mutations in X‐linked MECP2, encoding methyl‐CpG‐binding protein 2. Nat Genet 23: 185–188 [DOI] [PubMed] [Google Scholar]
- Amiri A, Cho W, Zhou J, Birnbaum SG, Sinton CM, McKay RM, Parada LF (2012) Pten deletion in adult hippocampal neural stem/progenitor cells causes cellular abnormalities and alters neurogenesis. J Neurosci 32: 5880–5890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amram N, Hacohen‐Kleiman G, Sragovich S, Malishkevich A, Katz J, Touloumi O, Lagoudaki R, Grigoriadis N, Giladi E, Yeheskel A (2016) Sexual divergence in microtubule function: the novel intranasal microtubule targeting SKIP normalizes axonal transport and enhances memory. Mol Psychiatry 21: 1467–1476 [DOI] [PubMed] [Google Scholar]
- Asano E, Chugani DC, Juhásza C, Muzik O, Chugani HT (2001) Surgical treatment of West syndrome. Brain Develop 23: 668–676 [DOI] [PubMed] [Google Scholar]
- Auerbach BD, Osterweil EK, Bear MF (2011) Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature 480: 63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backman SA, Stambolic V, Suzuki A, Haight J, Elia A, Pretorius J, Tsao M‐S, Shannon P, Bolon B, Ivy GO (2001) Deletion of Pten in mouse brain causes seizures, ataxia and defects in soma size resembling Lhermitte‐Duclos disease. Nat Genet 29: 396–403 [DOI] [PubMed] [Google Scholar]
- Bailey A, Luthert P, Dean A, Harding B, Janota I, Montgomery M, Rutter M, Lantos P (1998) A clinicopathological study of autism. Brain 121: 889–905 [DOI] [PubMed] [Google Scholar]
- Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116: 281–297 [DOI] [PubMed] [Google Scholar]
- Bassell GJ, Warren ST (2008) Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron 60: 201–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateup HS, Takasaki KT, Saulnier JL, Denefrio CL, Sabatini BL (2011) Loss of Tsc1 in vivo impairs hippocampal mGluR‐LTD and increases excitatory synaptic function. J Neurosci 31: 8862–8869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauman ML, Kemper TL (2005) Neuroanatomic observations of the brain in autism: a review and future directions. Int J Dev Neurosci 23: 183–187 [DOI] [PubMed] [Google Scholar]
- Benayed R, Gharani N, Rossman I, Mancuso V, Lazar G, Kamdar S, Bruse SE, Tischfield S, Smith BJ, Zimmerman RA et al (2005) Support for the homeobox transcription factor gene ENGRAILED 2 as an autism spectrum disorder susceptibility locus. Am J Hum Genet 77: 851–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benayed R, Choi J, Matteson PG, Gharani N, Kamdar S, Brzustowicz LM, Millonig JH (2009) Autism‐associated haplotype affects the regulation of the homeobox gene, ENGRAILED 2. Biol Psychiatry 66: 911–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben‐Shachar S, Chahrour M, Thaller C, Shaw CA, Zoghbi HY (2009) Mouse models of MeCP2 disorders share gene expression changes in the cerebellum and hypothalamus. Hum Mol Genet 18: 2431–2442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benvenuto G, Li S, Brown SJ, Braverman R, Vass WC, Cheadle JP, Halley DJ, Sampson JR, Wienecke R, DeClue JE (2000) The tuberous sclerosis‐1 (TSC1) gene product hamartin suppresses cell growth and augments the expression of the TSC2 product tuberin by inhibiting its ubiquitination. Oncogene 19: 6306–6316 [DOI] [PubMed] [Google Scholar]
- Beyer KS, Blasi F, Bacchelli E, Klauck SM, Maestrini E, Poustka A, Consortium IMGSoA (2002) Mutation analysis of the coding sequence of the MECP2 gene in infantile autism. Hum Genet 111: 305–309 [DOI] [PubMed] [Google Scholar]
- Bolduc ME, Limperopoulos C (2009) Neurodevelopmental outcomes in children with cerebellar malformations: a systematic review. Dev Med Child Neurol 51: 256–267 [DOI] [PubMed] [Google Scholar]
- Brielmaier J, Matteson PG, Silverman JL, Senerth JM, Kelly S, Genestine M, Millonig JH, DiCicco‐Bloom E, Crawley JN (2012) Autism‐relevant social abnormalities and cognitive deficits in engrailed‐2 knockout mice. PLoS One 7: e40914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet I, Weinl C, Piper M, Trembleau A, Volovitch M, Harris W, Prochiantz A, Holt C (2005) The transcription factor Engrailed‐2 guides retinal axons. Nature 438: 94–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bury LA, Sabo SL (2011) Coordinated trafficking of synaptic vesicle and active zone proteins prior to synapse formation. Neural development 6: 24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MG, Dasouki MJ, Zhou X‐P, Talebizadeh Z, Brown M, Takahashi TN, Miles JH, Wang C, Stratton R, Pilarski R (2005) Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet 42: 318–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum JD, Cai G, Chaste P, Nygren G, Goldsmith J, Reichert J, Anckarsäter H, Rastam M, Smith CJ, Silverman JM et al (2007) Mutation screening of the PTEN gene in patients with autism spectrum disorders and macrocephaly. Am J Med Genet B Neuropsychiatr Genet 144: 484–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson RP, Kelm ND, West KL, Does MD, Fu C, Weaver G, McBrier E, Parker B, Grier MD, Ess KC (2015) Hypomyelination following deletion of Tsc2 in oligodendrocyte precursors. Ann Clin Transl Neurol 2: 1041–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, Arora VK, Le C, Koutcher J, Scher H (2011) Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN‐deficient prostate cancer. Cancer Cell 19: 575–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceolin L, Bouquier N, Vitre‐Boubaker J, Rialle S, Severac D, Valjent E, Perroy J, Puighermanal E (2017) Cell Type‐Specific mRNA dysregulation in hippocampal CA1 pyramidal neurons of the fragile X syndrome mouse model. Front Mol Neurosci 10: 340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, André VM, Hauptman JS, Yamazaki I, Huynh MN, Chang JW, Chen JY, Fisher RS, Vinters HV, Levine MS (2012) Enhanced GABAergic network and receptor function in pediatric cortical dysplasia Type IIB compared with Tuberous Sclerosis Complex. Neurobiology of disease 45: 310–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahrour M, Zoghbi HY (2007) The story of Rett syndrome: from clinic to neurobiology. Neuron 56: 422–437 [DOI] [PubMed] [Google Scholar]
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY (2008) MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320: 1224–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalhoub N, Kozma SC, Baker SJ (2006) S6k1 is not required for Pten‐deficient neuronal hypertrophy. Brain Res 1100: 32–41 [DOI] [PubMed] [Google Scholar]
- Chao H‐T, Zoghbi HY, Rosenmund C (2007) MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron 56: 58–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheh MA, Millonig JH, Roselli LM, Ming X, Jacobsen E, Kamdar S, Wagner GC (2006) En2 knockout mice display neurobehavioral and neurochemical alterations relevant to autism spectrum disorder. Brain Res 1116: 166–176 [DOI] [PubMed] [Google Scholar]
- Chen Z, Zhu M, Yang J, Liang H, He J, He S, Wang P, Kang XI, McNutt M, Yin Y et al (2014) PTEN interacts with histone H1 and controls chromatin condensation. Cell Rep 8: 2003–2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Child ND, Cascino GD (2013) Mystery case: Cowden syndrome presenting with partial epilepsy related to focal cortical dysplasia. Neurology 81: e98–e99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cholewa‐Waclaw J, Bird A, von Schimmelmann M, Schaefer A, Yu H, Song H, Madabhushi R, Tsai L‐H (2016) The role of epigenetic mechanisms in the regulation of gene expression in the nervous system. J Neurosci 36: 11427–11434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clipperton‐Allen AE, Page DT (2014) Pten haploinsufficient mice show broad brain overgrowth but selective impairments in autism‐relevant behavioral tests. Hum Mol Genet 23: 3490–3505 [DOI] [PubMed] [Google Scholar]
- Contractor A, Klyachko VA, Portera‐Cailliau C (2015) Altered neuronal and circuit excitability in fragile X syndrome. Neuron 87: 699–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordeiro L, Ballinger E, Hagerman R, Hessl D (2011) Clinical assessment of DSM‐IV anxiety disorders in fragile X syndrome: prevalence and characterization. J Neurodev Disord 3: 57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa V, Aigner S, Vukcevic M, Sauter E, Behr K, Ebeling M, Dunkley T, Friedlein A, Zoffmann S, Meyer CA et al (2016) mTORC1 inhibition corrects neurodevelopmental and synaptic alterations in a human stem cell model of tuberous sclerosis. Cell Rep 15: 86–95 [DOI] [PubMed] [Google Scholar]
- Costa‐Mattioli M, Sossin WS, Klann E, Sonenberg N (2009) Translational control of long‐lasting synaptic plasticity and memory. Neuron 61: 10–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courchesne E, Karns CM, Davis HR, Ziccardi R, Carper RA, Tigue ZD, Chisum HJ, Moses P, Pierce K, Lord C et al (2001) Unusual brain growth patterns in early life in patients with autistic disorder: an MRI study. Neurology 57: 245–254 [DOI] [PubMed] [Google Scholar]
- Darnell JC, Jensen KB, Jin P, Brown V, Warren ST, Darnell RB (2001) Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell 107: 489–499 [DOI] [PubMed] [Google Scholar]
- Darnell J, Van Driesche S, Zhang C, Hung K, Mele A, Fraser C, Stone E, Chen C, Fak J, Chi S et al (2011) FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146: 247–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, Klann E (2013) The translation of translational control by FMRP: therapeutic targets for FXS. Nat Neurosci 16: 1530–1536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day JJ, Sweatt JD (2011) Epigenetic mechanisms in cognition. Neuron 70: 813–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Ercument Cicek A, Kou Y, Liu Li, Fromer M, Walker S et al (2014) Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515: 209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiCarlo GE, Aguilar JI, Matthies HJG, Harrison FE, Bundschuh KE, West A, Hashemi P, Herborg F, Rickhag M, Chen H et al (2019) Autism‐linked dopamine transporter mutation alters striatal dopamine neurotransmission and dopamine‐dependent behaviors. J Clin Investig 129: 3407–3419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dictenberg JB, Swanger SA, Antar LN, Singer RH, Bassell GJ (2008) A direct role for FMRP in activity‐dependent dendritic mRNA transport links filopodial‐spine morphogenesis to fragile X syndrome. Dev Cell 14: 926–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMario FJ Jr (2004) Brain abnormalities in tuberous sclerosis complex. J Child Neurol 19: 650–657 [DOI] [PubMed] [Google Scholar]
- Domanskyi A, Geiβler C, Vinnikov IA, Alter H, Schober A, Vogt MA, Gass P, Parlato R, Schütz G (2011) Pten ablation in adult dopaminergic neurons is neuroprotective in Parkinson's disease models. FASEB J 25: 2898–2910 [DOI] [PubMed] [Google Scholar]
- Dresner E, Malishkevich A, Arviv C, Barak SL, Alon S, Ofir R, Gothilf Y, Gozes I (2012) Novel evolutionary‐conserved role for the ADNP protein family that is important for erythropoiesis. J Biol Chem 287: 40173–40185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dykens EM, Hodapp RM, Ort S, Finucane B, Shapiro LR, Leckman JF (1989) The trajectory of cognitive development in males with fragile X syndrome. J Am Acad Child Adolesc Psychiatry 28: 422–426 [DOI] [PubMed] [Google Scholar]
- Ebert DH, Greenberg ME (2013) Activity‐dependent neuronal signalling and autism spectrum disorder. Nature 493: 327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehninger D, Silva AJ (2011) Rapamycin for treating Tuberous sclerosis and Autism spectrum disorders. Trends Mol Med 17: 78–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ercan E, Han JM, Di Nardo A, Winden K, Han M‐J, Hoyo L, Saffari A, Leask A, Geschwind DH, Sahin M (2017) Neuronal CTGF/CCN2 negatively regulates myelination in a mouse model of tuberous sclerosis complexCTGF/CCN2 regulates myelination. J Exp Med 214: 681–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasolino M, Zhou Z (2017) The crucial role of DNA methylation and MeCP2 in neuronal function. Genes 8: 141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fombonne E (2003) Epidemiological surveys of autism and other pervasive developmental disorders: an update. J Autism Dev Disord 33: 365–382 [DOI] [PubMed] [Google Scholar]
- Fraser MM, Bayazitov IT, Zakharenko SS, Baker SJ (2008) Phosphatase and tensin homolog, deleted on chromosome 10 deficiency in brain causes defects in synaptic structure, transmission and plasticity, and myelination abnormalities. Neuroscience 151: 476–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazier TW, Embacher R, Tilot AK, Koenig K, Mester J, Eng C (2015) Molecular and phenotypic abnormalities in individuals with germline heterozygous PTEN mutations and autism. Mol Psychiatry 20: 1132–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuks F, Hurd PJ, Deplus R, Kouzarides T (2003) The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic Acids Res 31: 2305–2312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman S, Hill JM, Vulih I, Zaltzman R, Hauser JM, Brenneman DE, Gozes I (2004) Sexual dimorphism of activity‐dependent neuroprotective protein in the mouse arcuate nucleus. Neurosci Lett 373: 73–78 [DOI] [PubMed] [Google Scholar]
- Gabel HW, Kinde B, Stroud H, Gilbert CS, Harmin DA, Kastan NR, Hemberg M, Ebert DH, Greenberg ME (2015) Disruption of DNA‐methylation‐dependent long gene repression in Rett syndrome. Nature 522: 89–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaugler T, Klei L, Sanders SJ, Bodea CA, Goldberg AP, Lee AB, Mahajan M, Manaa D, Pawitan Y, Reichert J (2014) Most genetic risk for autism resides with common variation. Nat Genet 46: 881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gennet N, Herden C, Bubb V, Quinn J, Kipar A (2008) Expression of activity‐dependent neuroprotective protein in the brain of adult rats. Histol Histopathol 23: 309–317 [DOI] [PubMed] [Google Scholar]
- Gharani N, Benayed R, Mancuso V, Brzustowicz L, Millonig J (2004) Association of the homeobox transcription factor, ENGRAILED 2, 3, with autism spectrum disorder. Mol Psychiatry 9: 474–484 [DOI] [PubMed] [Google Scholar]
- Gipson TT, Gerner G, Wilson MA, Blue ME, Johnston MV (2013) Potential for treatment of severe autism in tuberous sclerosis complex. World J Clin Pediatr 2: 16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gkogkas CG, Khoutorsky A, Ran I, Rampakakis E, Nevarko T, Weatherill DB, Vasuta C, Yee S, Truitt M, Dallaire P et al (2013a) Autism‐related deficits via dysregulated eIF4E‐dependent translational control. Nature 493: 371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goorden SM, van Woerden GM, van der Weerd L, Cheadle JP, Elgersma Y (2007) Cognitive deficits in Tsc1+/− mice in the absence of cerebral lesions and seizures. Ann Neurol 62: 648–655 [DOI] [PubMed] [Google Scholar]
- Gozes I (2007) Activity‐dependent neuroprotective protein: from gene to drug candidate. Pharmacol Ther 114: 146–154 [DOI] [PubMed] [Google Scholar]
- Gozes I (2018) ADNP regulates cognition: a multitasking protein. Front Neurosci 12: 873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Ellis SE, Ashar FN, Moes A, Bader JS, Zhan J, West AB, Arking DE (2014) Transcriptome analysis reveals dysregulation of innate immune response genes and neuronal activity‐dependent genes in autism. Nat Commun 5: 5748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallmayer J, Cleveland S, Torres A, Phillips J, Cohen B, Torigoe T, Miller J, Fedele A, Collins J, Smith K (2011) Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry 68: 1095–1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JM, Sahin M (2011) TSC1/TSC2 signaling in the CNS. FEBS Lett 585: 973–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haws ME, Jaramillo TC, Espinosa F, J. Widman A, Stuber GD, Sparta DR, Tye KM, Russo SJ, Parada LF, Stavarache M et al (2014) PTEN knockdown alters dendritic spine/protrusion morphology, not density. J Comp Neurol 522: 1171–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay N, Sonenberg N (2004) Upstream and downstream of mTOR. Genes Dev 18: 1926–1945 [DOI] [PubMed] [Google Scholar]
- He X, Thacker S, Romigh T, Yu Q, Frazier TW, Eng C (2015a) Cytoplasm‐predominant Pten associates with increased region‐specific brain tyrosine hydroxylase and dopamine D2 receptors in mouse model with autistic traits. Mol Autism 6: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helsmoortel C, Vulto‐van Silfhout AT, Coe BP, Vandeweyer G, Rooms L, Van Den Ende J, Schuurs‐Hoeijmakers JH, Marcelis CL, Willemsen MH, Vissers LE (2014) A SWI/SNF‐related autism syndrome caused by de novo mutations in ADNP. Nat Genet 46: 380–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnoonual A, Sripo T, Limprasert P (2016) Whole‐exome sequencing identifies a novel heterozygous missense variant of the EN2 gene in two unrelated patients with autism spectrum disorder. Psychiatr Genet 26: 297–301 [DOI] [PubMed] [Google Scholar]
- Hodapp RM, Dykens EM, Hagerman RJ, Schreiner R, Lachiewicz AM, Leckman JF (1990) Developmental implications of changing trajectories of IQ in males with fragile X syndrome. J Am Acad Child Adolesc Psychiatry 29: 214–219 [DOI] [PubMed] [Google Scholar]
- Hoeffer CA, Sanchez E, Hagerman RJ, Mu Y, Nguyen DV, Wong H, Whelan AM, Zukin RS, Klann E, Tassone F (2012) Altered mTOR signaling and enhanced CYFIP2 expression levels in subjects with fragile X syndrome. Genes Brain Behav 11: 332–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Klann E, Costa‐Mattioli M, Zukin RS (2015) Dysregulation of mammalian target of rapamycin signaling in mouse models of autism. J Neurosci 35: 13836–13842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt A, Shepherd C (1993) A prevalence study of autism in tuberous sclerosis. J Autism Dev Disord 23: 323–339 [DOI] [PubMed] [Google Scholar]
- Inoki K, Li Y, Zhu T, Wu J, Guan K‐L (2002) TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4: 648–657 [DOI] [PubMed] [Google Scholar]
- Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee Y‐h, Narzisi G, Leotta A et al (2012) De novo gene disruptions in children on the autistic spectrum. Neuron 74: 285–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE et al (2014) The contribution of de novo coding mutations to autism spectrum disorder. Nature 515: 216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin SA, Galvez R, Greenough WT (2000) Dendritic spine structural anomalies in fragile‐X mental retardation syndrome. Cereb Cortex 10: 1038–1044 [DOI] [PubMed] [Google Scholar]
- Ito‐Ishida A, Ure K, Chen H, Swann JW, Zoghbi HY (2015) Loss of MeCP2 in parvalbumin‐and somatostatin‐expressing neurons in mice leads to distinct Rett syndrome‐like phenotypes. Neuron 88: 651–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neul JL, Kaufmann WE, Glaze DG, Christodoulou J, Clarke AJ, Bahi‐Buisson N, Leonard H, Bailey MES, Schanen NC, Zappella M et al (2010) Rett syndrome: revised diagnostic criteria and nomenclature. Ann Neurol 68: 944–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaca KG, Brito DV, Zeuch B, Oliveira AM (2018) Adult hippocampal MeCP2 preserves the genomic responsiveness to learning required for long‐term memory formation. Neurobiol Learn Mem 149: 84–97 [DOI] [PubMed] [Google Scholar]
- Kelleher RJ III, Bear MF (2008) The autistic neuron: troubled translation? Cell 135: 401–406 [DOI] [PubMed] [Google Scholar]
- Kim YS, Leventhal BL (2015) Genetic epidemiology and insights into interactive genetic and environmental effects in autism spectrum disorders. Biol Psychiatry 77: 66–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klann E, Dever TE (2004) Biochemical mechanisms for translational regulation in synaptic plasticity. Nat Rev Neurosci 5: 931–942 [DOI] [PubMed] [Google Scholar]
- Klei L, Sanders SJ, Murtha MT, Hus V, Lowe JK, Willsey AJ, Moreno‐De‐Luca D, Yu TW, Fombonne E, Geschwind D et al (2012) Common genetic variants, acting additively, are a major source of risk for autism. Mol Autism 3: 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korb E, Herre M, Zucker‐Scharff I, Darnell RB, Allis CD (2015) BET protein Brd4 activates transcription in neurons and BET inhibitor Jq1 blocks memory in mice. Nat Neurosci 18: 1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korb E, Herre M, Zucker‐Scharff I, Gresack J, Allis CD, Darnell RB (2017) Excess translation of epigenetic regulators contributes to fragile X syndrome and is alleviated by Brd4 inhibition. Cell 170: 1209–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan K, Lau BY, Ewall G, Huang ZJ, Shea SD (2017) MECP2 regulates cortical plasticity underlying a learned behaviour in adult female mice. Nat Commun 8: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumm N, O’Roak BJ, Shendure J, Eichler EE (2014) A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci 37: 95–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuemerle B, Gulden F, Cherosky N, Williams E, Herrup K (2007) The mouse Engrailed genes: a window into autism. Behav Brain Res 176: 121–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulage KM, Smaldone AM, Cohn EG (2014) How will DSM‐5 affect autism diagnosis? A systematic literature review and meta‐analysis. J Autism Dev Disord 44: 1918–1932 [DOI] [PubMed] [Google Scholar]
- Kwiatkowski DJ, Manning BD (2005) Tuberous sclerosis: a GAP at the crossroads of multiple signaling pathways. Hum Mol Genet 14: R251–R258 [DOI] [PubMed] [Google Scholar]
- Kwon C‐H, Zhu X, Zhang J, Knoop LL, Tharp R, Smeyne RJ, Eberhart CG, Burger PC, Baker SJ (2001) Pten regulates neuronal soma size: a mouse model of Lhermitte‐Duclos disease. Nat Genet 29: 404–411 [DOI] [PubMed] [Google Scholar]
- Kwon C‐H, Zhu X, Zhang J, Baker SJ (2003) mTor is required for hypertrophy of Pten‐deficient neuronal soma in vivo. Proc Natl Acad Sci 100: 12923–12928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon C‐H, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ, Parada LF (2006) Pten regulates neuronal arborization and social interaction in mice. Neuron 50: 377–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau BY, Krishnan K, Huang ZJ, Shea SD (2020) Maternal experience‐dependent cortical plasticity in mice is circuit‐and stimulus‐specific and requires MECP2. J Neurosci 40: 1514–1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebrun‐Julien F, Bachmann L, Norrmén C, Trötzmüller M, Köfeler H, Rüegg MA, Hall MN, Suter U (2014) Balanced mTORC1 activity in oligodendrocytes is required for accurate CNS myelination. J Neurosci 34: 8432–8448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JD, Meehan RR, Henzel WJ, Maurer‐Fogy I, Jeppesen P, Klein F, Bird A (1992) Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell 69: 905–914 [DOI] [PubMed] [Google Scholar]
- Lewis WW, Sahin M, Scherrer B, Peters JM, Suarez RO, Vogel‐Farley VK, Jeste SS, Gregas MC, Prabhu SP, Nelson CA et al (2013) Impaired language pathways in tuberous sclerosis complex patients with autism spectrum disorders. Cereb Cortex 23: 1526–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wang H, Muffat J, Cheng A, Orlando D, Lovén J, Kwok S‐M, Feldman D, Bateup H, Gao Q et al (2013) Global transcriptional and translational repression in human‐embryonic‐stem‐cell‐derived Rett syndrome neurons. Cell Stem Cell 13: 446–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liaw D, Marsh DJ, Li J, Dahia PLM, Wang SI, Zheng Z, Bose S, Call KM, Tsou HC, Peacoke M et al (1997) Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet 16: 64–67 [DOI] [PubMed] [Google Scholar]
- Lipton JO, Sahin M (2014) The neurology of mTOR. Neuron 84: 275–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Nyholt DR, Magnussen P, Parano E, Pavone P, Geschwind D, Lord C, Iversen P, Hoh J, Autism Genetic Resource Exchange Consortium T et al (2001) A genomewide screen for autism susceptibility loci. Am J Human Genet 69: 327–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Li Y, Stackpole EE, Novak A, Gao Y, Zhao Y, Zhao X, Richter JD (2018) Regulatory discrimination of mRNAs by FMRP controls mouse adult neural stem cell differentiation. Proc Natl Acad Sci USA 115: E11397–E11405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liyanage VR, Rastegar M (2014) Rett syndrome and MeCP2. NeuroMol Med 16: 231–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo LH‐Y, Lai K‐O (2020) Dysregulation of protein synthesis and dendritic spine morphogenesis in ASD: studies in human pluripotent stem cells. Mol Autism 11: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loat C, Curran S, Lewis C, Duvall J, Geschwind D, Bolton P, Craig I (2008) Methyl‐CpG‐binding protein 2 polymorphisms and vulnerability to autism. Genes Brain Behav 7: 754–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luikart BW, Schnell E, Washburn EK, Bensen AL, Tovar KR, Westbrook GL (2011) Pten knockdown in vivo increases excitatory drive onto dentate granule cells. J Neurosci 31: 4345–4354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyst MJ, Bird A (2015) Rett syndrome: a complex disorder with simple roots. Nat Rev Genet 16: 261–275 [DOI] [PubMed] [Google Scholar]
- Lyu J‐W, Yuan B, Cheng T‐L, Qiu Z‐L, Zhou W‐H (2016) Reciprocal regulation of autism‐related genes MeCP2 and PTEN via microRNAs. Sci Rep 6: 20392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XM, Blenis J (2009) Molecular mechanisms of mTOR‐mediated translational control. Nat Rev Mol Cell Biol 10: 307–318 [DOI] [PubMed] [Google Scholar]
- Maehama T, Dixon JE (1998) The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3, 4, 5‐trisphosphate. J Biol Chem 273: 13375–13378 [DOI] [PubMed] [Google Scholar]
- Magill ST, Cambronne XA, Luikart BW, Lioy DT, Leighton BH, Westbrook GL, Mandel G, Goodman RH (2010) microRNA‐132 regulates dendritic growth and arborization of newborn neurons in the adult hippocampus. Proc Natl Acad Sci USA 107: 20382–20387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malishkevich A, Amram N, Hacohen‐Kleiman G, Magen I, Giladi E, Gozes I (2015) Activity‐dependent neuroprotective protein (ADNP) exhibits striking sexual dichotomy impacting on autistic and Alzheimer’s pathologies. Transl Psychiatry 5: e501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malishkevich A, Marshall GA, Schultz AP, Sperling RA, Aharon‐Peretz J, Gozes I (2016) Blood‐borne activity‐dependent neuroprotective protein (ADNP) is correlated with premorbid intelligence, clinical stage, and Alzheimer’s disease biomarkers. J Alzheimers Dis 50: 249–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandel S, Gozes I (2007) Activity‐dependent neuroprotective protein constitutes a novel element in the SWI/SNF chromatin remodeling complex. J Biol Chem 282: 34448–34456 [DOI] [PubMed] [Google Scholar]
- Mannion A, Leader G (2013) Comorbidity in autism spectrum disorder: a literature review. Res Autism Spectr Disord 7: 1595–1616 [Google Scholar]
- Marshall P, Bredy TW (2016) Cognitive neuroepigenetics: the next evolution in our understanding of the molecular mechanisms underlying learning and memory? NPJ Sci Learn 1: 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JP, Bell J (1943) A pedigree of mental defect showing sex‐linkage. J Neurol Psychiatry 6: 154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushima‐Nishiu M, Unoki M, Ono K, Tsunoda T, Minaguchi T, Kuramoto H, Nishida M, Satoh T, Tanaka T, Nakamura Y (2001) Growth and gene expression profile analyses of endometrial cancer cells expressing exogenous PTEN. Can Res 61: 3741–3749 [PubMed] [Google Scholar]
- Matthews G, Nieh E, Vander Weele C, Halbert S, Pradhan R, Yosafat A, Glober G, Izadmehr E, Thomas R, Lacy G et al (2016) Dorsal raphe dopamine neurons represent the experience of social isolation. Cell 164: 617–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDougle CJ, Scahill L, Aman MG, McCracken JT, Tierney E, Davies M, Arnold LE, Posey DJ, Martin A, Ghuman JK et al (2005) Risperidone for the core symptom domains of autism: results from the study by the autism network of the research units on pediatric psychopharmacology. Am J Psychiatry 162: 1142–1148 [DOI] [PubMed] [Google Scholar]
- McPartland J, Volkmar FR (2012) Autism and related disorders. In Handbook of Clinical Neurology, Aminoff MJ, Boller F, Swaab DF (eds), Vol. 106, pp 407–418. Elsevier; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medrano‐Fernández A, Barco A (2016) Nuclear organization and 3D chromatin architecture in cognition and neuropsychiatric disorders. Mol Brain 9: 83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meikle L, Talos DM, Onda H, Pollizzi K, Rotenberg A, Sahin M, Jensen FE, Kwiatkowski DJ (2007) A mouse model of tuberous sclerosis: neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced myelination, seizure activity, and limited survival. J Neurosci 27: 5546–5558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merenlender‐Wagner A, Malishkevich A, Shemer Z, Udawela M, Gibbons A, Scarr E, Dean B, Levine J, Agam G, Gozes I (2015) Autophagy has a key role in the pathophysiology of schizophrenia. Mol Psychiatry 20: 126–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteggia LM, Kavalali ET (2009) Rett syndrome and the impact of MeCP2 associated transcriptional mechanisms on neurotransmission. Biol Psychiatry 65: 204–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moog U, Smeets EE, van Roozendaal KE, Schoenmakers S, Herbergs J, Schoonbrood‐Lenssen AM, Schrander‐Stumpel CT (2003) Neurodevelopmental disorders in males related to the gene causing Rett syndrome in females (MECP2). Eur J Paediatr Neurol 7: 5–12 [DOI] [PubMed] [Google Scholar]
- Morello N, Schina R, Pilotto F, Phillips M, Melani R, Plicato O, Pizzorusso T, Pozzo‐Miller L, Giustetto M (2018) Loss of Mecp2 causes atypical synaptic and molecular plasticity of parvalbumin‐expressing interneurons reflecting Rett syndrome–like sensorimotor defects. Eneuro 5: ENEURO.0086‐18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulholland DJ, Kobayashi N, Ruscetti M, Zhi A, Tran LM, Huang J, Gleave M, Wu H (2012) Pten loss and RAS/MAPK activation cooperate to promote EMT and metastasis initiated from prostate cancer stem/progenitor cells. Can Res 72: 1878–1889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins C, Fishell G, Tsien RW (2016) Unifying views of autism spectrum disorders: a consideration of autoregulatory feedback loops. Neuron 89: 1131–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan RP, Patzel KA, Martin M, Yasui DH, Swanberg SE, Hertz‐Picciotto I, Hansen RL, Van de Water J, Pessah IN, Jiang R (2008) MECP2 promoter methylation and X chromosome inactivation in autism. Autism Res 1: 169–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan X, Ng H‐H, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A (1998) Transcriptional repression by the methyl‐CpG‐binding protein MeCP2 involves a histone deacetylase complex. Nature 393: 386–389 [DOI] [PubMed] [Google Scholar]
- Napoli E, Ross‐Inta C, Wong S, Hung C, Fujisawa Y, Sakaguchi D, Angelastro J, Omanska‐Klusek A, Schoenfeld R, Giulivi C (2012) Mitochondrial dysfunction in Pten haplo‐insufficient mice with social deficits and repetitive behavior: interplay between Pten and p53. PLoS One 7: e42504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napoli I, Mercaldo V, Boyl PP, Eleuteri B, Zalfa F, De Rubeis S, Di Marino D, Mohr E, Massimi M, Falconi M et al (2008) The fragile X syndrome protein represses activity‐dependent translation through CYFIP1, a new 4E‐BP. Cell 134: 1042–1054 [DOI] [PubMed] [Google Scholar]
- Neale BM, Kou Y, Liu Li, Ma’ayan A, Samocha KE, Sabo A, Lin C‐F, Stevens C, Wang L‐S, Makarov V et al (2012) Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485: 242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD et al (2012) Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485: 246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson CO, Pejhan S, Kroft D, Sheikholeslami K, Fuss D, Buist M, Ali Sher A, Del Bigio MR, Sztainberg Y, Siu VM et al (2018) MECP2 mutation interrupts nucleolin–mTOR–P70S6K signaling in rett syndrome patients. Front Genet 9: 635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onore C, Yang H, Van de Water J, Ashwood P (2017) Dynamic Akt/mTOR signaling in children with autism spectrum disorder. Front Pediatr 5: 43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne MJ, Borden KL (2015) The eukaryotic translation initiation factor eIF 4E in the nucleus: taking the road less traveled. Immunol Rev 263: 210–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmen SJ, van Engeland H, Hof PR, Schmitz C (2004) Neuropathological findings in autism. Brain 127: 2572–2583 [DOI] [PubMed] [Google Scholar]
- Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V, Horvath S, Geschwind DH (2013) Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 155: 1008–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons R (2004) Human cancer, PTEN and the PI‐3 kinase pathway. Semin Cell Dev Biol 15: 171–176 [DOI] [PubMed] [Google Scholar]
- Pascolini G, Agolini E, Majore S, Novelli A, Grammatico P, Digilio MC (2018) Helsmoortel‐Van der Aa Syndrome as emerging clinical diagnosis in intellectually disabled children with autistic traits and ocular involvement. Eur J Paediatr Neurol 22: 552–557 [DOI] [PubMed] [Google Scholar]
- de Paz AM, Ausió J (2017) MeCP2, a modulator of neuronal chromatin organization involved in Rett syndrome. In Neuroepigenomics in Aging and Disease, Delgado‐Morales R (ed), pp 3–21. Springer; [DOI] [PubMed] [Google Scholar]
- Peters JM, Taquet M, Vega C, Jeste SS, Fernández IS, Tan J, Nelson CA, Sahin M, Warfield SK (2013) Brain functional networks in syndromic and non‐syndromic autism: a graph theoretical study of EEG connectivity. BMC Med 11: 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinhasov A, Mandel S, Torchinsky A, Giladi E, Pittel Z, Goldsweig AM, Servoss SJ, Brenneman DE, Gozes I (2003) Activity‐dependent neuroprotective protein: a novel gene essential for brain formation. Dev Brain Res 144: 83–90 [DOI] [PubMed] [Google Scholar]
- Potter CJ, Pedraza LG, Xu T (2002) Akt regulates growth by directly phosphorylating Tsc2. Nat Cell Biol 4: 658–665 [DOI] [PubMed] [Google Scholar]
- Provenzano G, Pangrazzi L, Poli A, Pernigo M, Sgado P, Genovesi S, Zunino G, Berardi N, Casarosa S, Bozzi Y (2014) Hippocampal dysregulation of neurofibromin‐dependent pathways is associated with impaired spatial learning in engrailed 2 knock‐out mice. J Neurosci 34: 13281–13288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman S, Sowa ME, Ottinger M, Smith JA, Shi Y, Harper JW, Howley PM (2011) The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol Cell Biol 31: 2641–2652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajarajan P, Gil SE, Brennand KJ, Akbarian S (2016) Spatial genome organization and cognition. Nat Rev Neurosci 17: 681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramocki MB, Tavyev YJ, Peters SU (2010) The MECP2 duplication syndrome. Am J Med Genet A 152: 1079–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricciardi S, Boggio EM, Grosso S, Lonetti G, Forlani G, Stefanelli G, Calcagno E, Morello N, Landsberger N, Biffo S et al (2011) Reduced AKT/mTOR signaling and protein synthesis dysregulation in a Rett syndrome animal model. Hum Mol Genet 20: 1182–1196 [DOI] [PubMed] [Google Scholar]
- Rice ME, Patel JC, Cragg SJ (2011) Dopamine release in the basal ganglia. Neuroscience 198: 112–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter JD, Klann E (2009) Making synaptic plasticity and memory last: mechanisms of translational regulation. Genes Dev 23: 1–11 [DOI] [PubMed] [Google Scholar]
- Riikonen R, Makkonen I, Vanhala R, Turpeinen U, Kuikka J, Kokki H (2006) Cerebrospinal fluid insulin‐like growth factors IGF‐1 and IGF‐2 in infantile autism. Dev Med Child Neurol 48: 751–755 [DOI] [PubMed] [Google Scholar]
- Rodríguez‐Escudero I, Oliver MD, Andrés‐Pons A, Molina M, Cid VJ, Pulido R (2011) A comprehensive functional analysis of PTEN mutations: implications in tumor‐and autism‐related syndromes. Hum Mol Genet 20: 4132–4142 [DOI] [PubMed] [Google Scholar]
- Romero M, Aguilar JM, Del‐Rey‐Mejías Á, Mayoral F, Rapado M, Pecina M, Barbancho MÁ, Ruiz‐Veguilla M, Lara JP (2016) Psychiatric comorbidities in autism spectrum disorder: a comparative study between DSM‐IV‐TR and DSM‐5 diagnosis. Int J Clin Health Psychol 16: 266–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosset C, Netto CBO, Ashton‐Prolla P (2017) TSC1 and TSC2 gene mutations and their implications for treatment in Tuberous Sclerosis Complex: a review. Genet Mol Biol 40: 69–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossman IT, Lin L, Morgan KM, DiGiovine M, Van Buskirk EK, Kamdar S, Millonig JH, DiCicco‐Bloom E (2014) Engrailed2 modulates cerebellar granule neuron precursor proliferation, differentiation and insulin‐like growth factor 1 signaling during postnatal development. Mol Autism 5: 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruvinsky I, Meyuhas O (2006) Ribosomal protein S6 phosphorylation: from protein synthesis to cell size. Trends Biochem Sci 31: 342–348 [DOI] [PubMed] [Google Scholar]
- Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan‐Sencicek AG, DiLullo NM, Parikshak NN, Stein JL (2012) De novo mutations revealed by whole‐exome sequencing are strongly associated with autism. Nature 485: 237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Huynh TN, MacAskill AF, Carter AG, Pierre P, Ruggero D, Kaphzan H, Klann E (2013) Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature 493: 411–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Klann E (2014) Reciprocal signaling between translational control pathways and synaptic proteins in autism spectrum disorders. Sci Signal 7: re10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro MR, Bray SM, Warren ST (2012) Molecular mechanisms of fragile X syndrome: a twenty‐year perspective. Annu Rev Pathol 7: 219–245 [DOI] [PubMed] [Google Scholar]
- Schaefer GB, Mendelsohn NJ (2013) Clinical genetics evaluation in identifying the etiology of autism spectrum disorders: 2013 guideline revisions. Genet Med 15: 399 [DOI] [PubMed] [Google Scholar]
- Sgadò P, Provenzano G, Dassi E, Adami V, Zunino G, Genovesi S, Casarosa S, Bozzi Y (2013) Transcriptome profiling in engrailed‐2 mutant mice reveals common molecular pathways associated with autism spectrum disorders. Mol Autism 4: 51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgaier SK, Lao Z, Villanueva MP, Berenshteyn F, Stephen D, Turnbull RK, Joyner AL (2007) Genetic subdivision of the tectum and cerebellum into functionally related regions based on differential sensitivity to engrailed proteins. Development 134: 2325–2335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbazian MD, Zoghbi HY (2002) Rett syndrome and MeCP2: linking epigenetics and neuronal function. Am J Hum Genet 71: 1259–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Hoeffer CA, Takayasu Y, Miyawaki T, McBride SM, Klann E, Zukin RS (2010) Dysregulation of mTOR signaling in fragile X syndrome. J Neurosci 30: 694–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen C, Ipsaro JJ, Shi J, Milazzo JP, Wang E, Roe J‐S, Suzuki Y, Pappin DJ, Joshua‐Tor L, Vakoc CR (2015) NSD3‐short is an adaptor protein that couples BRD4 to the CHD8 chromatin remodeler. Mol Cell 60: 847–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibayama A, Cook EH Jr, Feng J, Glanzmann C, Yan J, Craddock N, Jones IR, Goldman D, Heston LL, Sommer SS (2004) MECP2 structural and 3′‐UTR variants in schizophrenia, autism and other psychiatric diseases: a possible association with autism. Am J Med Genet B Neuropsychiatr Genet 128: 50–53 [DOI] [PubMed] [Google Scholar]
- Shohamy D, Adcock RA (2010) Dopamine and adaptive memory. Trends Cogn Sci 14: 464–472 [DOI] [PubMed] [Google Scholar]
- Smalley SL (1998) Autism and tuberous sclerosis. J Autism Dev Disord 28: 407–414 [DOI] [PubMed] [Google Scholar]
- Soltani A, Lebrun S, Carpentier G, Zunino G, Chantepie S, Maïza A, Bozzi Y, Desnos C, Darchen F, Stettler O (2017) Increased signaling by the autism‐related Engrailed‐2 protein enhances dendritic branching and spine density, alters synaptic structural matching, and exaggerates protein synthesis. PLoS One 12: e0181350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonenberg N, Hinnebusch AG (2009) Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136: 731–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spatazza J, Di Lullo E, Joliot A, Dupont E, Moya KL, Prochiantz A (2013) Homeoprotein signaling in development, health, and disease: a shaking of dogmas offers challenges and promises from bench to bed. Pharmacol Rev 65: 90–104 [DOI] [PubMed] [Google Scholar]
- Sperow M, Berry RB, Bayazitov IT, Zhu G, Baker SJ, Zakharenko SS (2012) Phosphatase and tensin homologue (PTEN) regulates synaptic plasticity independently of its effect on neuronal morphology and migration. J Physiol 590: 777–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stritt C, Stern S, Harting K, Manke T, Sinske D, Schwarz H, Vingron M, Nordheim A, Knöll B (2009) Paracrine control of oligodendrocyte differentiation by SRF‐directed neuronal gene expression. Nat Neurosci 12: 418 [DOI] [PubMed] [Google Scholar]
- Sutton MA, Schuman EM (2005) Local translational control in dendrites and its role in long‐term synaptic plasticity. J Neurobiol 64: 116–131 [DOI] [PubMed] [Google Scholar]
- Swanberg SE, Nagarajan RP, Peddada S, Yasui DH, LaSalle JM (2009) Reciprocal co‐regulation of EGR2 and MECP2 is disrupted in Rett syndrome and autism. Hum Mol Genet 18: 525–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sztainberg Y, Zoghbi HY (2016) Lessons learned from studying syndromic autism spectrum disorders. Nat Neurosci 19: 1408 [DOI] [PubMed] [Google Scholar]
- Takei N, Inamura N, Kawamura M, Namba H, Hara K, Yonezawa K, Nawa H (2004) Brain‐derived neurotrophic factor induces mammalian target of rapamycin‐dependent local activation of translation machinery and protein synthesis in neuronal dendrites. J Neurosci 24: 9760–9769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi K, Gertner MJ, Zhou J, Parada LF, Bennett MV, Zukin RS (2013) Dysregulation of synaptic plasticity precedes appearance of morphological defects in a Pten conditional knockout mouse model of autism. Proc Natl Acad Sci USA 110: 4738–4743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M‐H, Mester J, Peterson C, Yang Y, Chen J‐L, Rybicki LA, Milas K, Pederson H, Remzi B, Orloff MS et al (2011) A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands. Am J Hum Genet 88: 42–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson SR, Seo SS, Barnes SA, Louros SR, Muscas M, Dando O, Kirby C, Wyllie DJA, Hardingham GE, Kind PC et al (2017) Cell‐type‐specific translation profiling reveals a novel strategy for treating fragile X syndrome. Neuron 95: 550–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilot AK, Frazier TW, Eng C (2015) Balancing proliferation and connectivity in PTEN‐associated autism spectrum disorder. Neurotherapeutics 12: 609–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torquet N, Marti F, Campart C, Tolu S, Nguyen C, Oberto V, Benallaoua M, Naudé J, Didienne S, Debray N et al (2018) Social interactions impact on the dopaminergic system and drive individuality. Nat Commun 9: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre‐Ubieta L, Won H, Stein JL, Geschwind DH (2016) Advancing the understanding of autism disease mechanisms through genetics. Nat Med 22: 345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi P, Sgadò P, Scali M, Viaggi C, Casarosa S, Simon H, Vaglini F, Corsini GU, Bozzi Y (2009) Increased susceptibility to kainic acid–induced seizures in Engrailed‐2 knockout mice. Neuroscience 159: 842–849 [DOI] [PubMed] [Google Scholar]
- Tsai P, Sahin M (2011) Mechanisms of neurocognitive dysfunction and therapeutic considerations in tuberous sclerosis complex. Curr Opin Neurol 24: 106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimura K, Irie K, Nakashima H, Egashira Y, Fukao Y, Fujiwara M, Itoh M, Uesaka M, Imamura T, Nakahata Y et al (2015) miR‐199a links MeCP2 with mTOR signaling and its dysregulation leads to Rett syndrome phenotypes. Cell Rep 12: 1887–1901 [DOI] [PubMed] [Google Scholar]
- Van Esch H, Bauters M, Ignatius J, Jansen M, Raynaud M, Hollanders K, Lugtenberg D, Bienvenu T, Jensen LR, Gécz J et al (2005) Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am J Hum Genet 77: 442–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga EA, Pastore M, Prior T, Herman GE, McBride KL (2009) The prevalence of PTEN mutations in a clinical pediatric cohort with autism spectrum disorders, developmental delay, and macrocephaly. Genet Med 11: 111–117 [DOI] [PubMed] [Google Scholar]
- Verkerk AJ, Pieretti M, Sutcliffe JS, Fu Y‐H, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang F (1991) Identification of a gene (FMR‐1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65: 905–914 [DOI] [PubMed] [Google Scholar]
- Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, Geschwind DH (2011) Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474: 380–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Der Brelie C, Waltereit R, Zhang L, Beck H, Kirschstein T (2006) Impaired synaptic plasticity in a rat model of tuberous sclerosis. Eur J Neurosci 23: 686–692 [DOI] [PubMed] [Google Scholar]
- Vulih‐Shultzman I, Pinhasov A, Mandel S, Grigoriadis N, Touloumi O, Pittel Z, Gozes I (2007) Activity‐dependent neuroprotective protein snippet NAP reduces tau hyperphosphorylation and enhances learning in a novel transgenic mouse model. J Pharmacol Exp Ther 323: 438–449 [DOI] [PubMed] [Google Scholar]
- Waltereit R, Japs B, Schneider M, de Vries PJ, Bartsch D (2011) Epilepsy and Tsc2 haploinsufficiency lead to autistic‐like social deficit behaviors in rats. Behav Genet 41: 364–372 [DOI] [PubMed] [Google Scholar]
- Wang Y, Cheng A, Mattson MP (2006) The PTEN phosphatase is essential for long‐term depression of hippocampal synapses. NeuroMol Med 8: 329–335 [DOI] [PubMed] [Google Scholar]
- Wang P, Zhao D, Lachman HM, Zheng D (2018) Enriched expression of genes associated with autism spectrum disorders in human inhibitory neurons. Transl Psychiatry 8: 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson LA, Tsai L‐H (2017) In the loop: how chromatin topology links genome structure to function in mechanisms underlying learning and memory. Curr Opin Neurobiol 43: 48–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise RA (2004) Dopamine, learning and motivation. Nat Rev Neurosci 5: 483–494 [DOI] [PubMed] [Google Scholar]
- Wullschleger S, Loewith R, Hall MN (2006) TOR signaling in growth and metabolism. Cell 124: 471–484 [DOI] [PubMed] [Google Scholar]
- Xu X, Xu Q, Zhang Y, Zhang X, Cheng T, Wu B, Ding Y, Lu P, Zheng J, Zhang M (2012) A case report of Chinese brothers with inherited MECP2‐containing duplication: autism and intellectual disability, but not seizures or respiratory infections. BMC Med Genet 13: 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z‐X, Kim GH, Tan J‐W, Riso AE, Sun Y, Xu EY, Liao G‐Y, Xu H, Lee S‐H, Do N‐Y (2020) Elevated protein synthesis in microglia causes autism‐like synaptic and behavioral aberrations. Nat Commun 11: 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JI, Hong EP, Castle JC, Crespo‐Barreto J, Bowman AB, Rose MF, Kang D, Richman R, Johnson JM, Berget S (2005) Regulation of RNA splicing by the methylation‐dependent transcriptional repressor methyl‐CpG binding protein 2. Proc Natl Acad Sci USA 102: 17551–17558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Hong EJ, Cohen S, Zhao W‐N, Ho H‐Y, Schmidt L, Chen WG, Lin Y, Savner E, Griffith EC et al (2006) Brain‐specific phosphorylation of MeCP2 regulates activity‐dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron 52: 255–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Blundell J, Ogawa S, Kwon C‐H, Zhang W, Sinton C, Powell CM, Parada LF (2009) Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural‐specific Pten knock‐out mice. J Neurosci 29: 1773–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Parada LF (2012) PTEN signaling in autism spectrum disorders. Curr Opin Neurobiol 22: 873–879 [DOI] [PubMed] [Google Scholar]
- Zhou C, Yan S, Qian S, Wang Z, Shi Z, Xiong Y, Zhou Y (2019a) Atypical response properties of the auditory cortex of awake MECP2‐overexpressing mice. Front Neurosci 13: 439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Park CY, Theesfeld CL, Wong AK, Yuan Y, Scheckel C, Fak JJ, Funk J, Yao K, Tajima Y (2019b) Whole‐genome deep‐learning analysis identifies contribution of noncoding mutations to autism risk. Nat Genet 51: 973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi H, Bear M (2012) Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb Perspect Biol 4: a009886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R, Efeyan A, Sabatini DM (2011) mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 12: 21–35 [DOI] [PMC free article] [PubMed] [Google Scholar]