

Abstract
Recent studies demonstrate that metabolic disturbance, such as augmented glycolysis, contributes to fibrosis. The molecular regulation of this metabolic perturbation in fibrosis, however, has been elusive. COUP‐TFII (also known as NR2F2) is an important regulator of glucose and lipid metabolism. Its contribution to organ fibrosis is undefined. Here, we found increased COUP‐TFII expression in myofibroblasts in human fibrotic kidneys, lungs, kidney organoids, and mouse kidneys after injury. Genetic ablation of COUP‐TFII in mice resulted in attenuation of injury‐induced kidney fibrosis. A non‐biased proteomic study revealed the suppression of fatty acid oxidation and the enhancement of glycolysis pathways in COUP‐TFII overexpressing fibroblasts. Overexpression of COUP‐TFII in fibroblasts also induced production of alpha‐smooth muscle actin (αSMA) and collagen 1. Knockout of COUP‐TFII decreased glycolysis and collagen 1 levels in fibroblasts. Chip‐qPCR revealed the binding of COUP‐TFII on the promoter of PGC1α. Overexpression of COUP‐TFII reduced the cellular level of PGC1α. Targeting COUP‐TFII serves as a novel treatment approach for mitigating fibrosis in chronic kidney disease and potentially fibrosis in other organs.
Keywords: acute kidney injury, COUP‐TFII, fibrosis, glycolysis, TGFβ
Subject Categories: Metabolism, Molecular Biology of Disease
This study reveals that upon injury, COUP‐TFII is upregulated in myofibroblasts and promotes fibrosis in human fibrotic diseases through augmented glycolysis of the myofibroblasts.

Introduction
Kidney fibrosis is a pathologic hallmark of chronic kidney disease (CKD), which affects ~ 10% of the world's adult population (Jha et al, 2013; Romagnani et al, 2017). The progressive deposition and expansion of the fibrotic matrix in kidney parenchyma ultimately lead to kidney failure (Bonventre & Yang, 2011; Rockey et al, 2015). Fibrosis also plays an important role in other chronic diseases, such as liver cirrhosis, idiopathic pulmonary fibrosis, and scleroderma (Wynn & Ramalingam, 2012). The global burden of fibrotic disease is high, with an estimated prevalence rate of one in four people (Zhao et al, 2019, 2020). Currently, there is no cure for fibrosis, highlighting the need for a new strategy and a better understanding of the molecular mechanisms underlying fibrosis. It is generally accepted that myofibroblasts are major cellular contributors to fibrotic disease (Tomasek et al, 2002; Bonventre & Yang, 2011; Duffield et al, 2013; Duffield, 2014). Myofibroblasts express alpha‐smooth muscle actin (αSMA) and contract, migrate, and produce excessive extracellular matrix (ECM) (Klingberg et al, 2013). Comprehensive genetic fate‐mapping studies in rodents indicate that kidney resident Foxd1 expressing pericytes/perivascular fibroblasts/mesenchymal stem cell‐like stromal cells are the main source of myofibroblasts after kidney injury (Humphreys et al, 2008; Lin et al, 2008; Picard et al, 2008; Humphreys et al, 2010; Kobayashi et al, 2014; Falke et al, 2015; Kramann et al, 2015).
Recently, an emerging body of evidence has demonstrated the link between metabolic dysregulation and fibrosis (Xie et al, 2015; Lan et al, 2016; Hou & Syn, 2018; Zank et al, 2018). Genome‐wide transcriptome profiling revealed inflammation and metabolism as the top dysregulated pathways in fibrotic human kidneys (Kang et al, 2015). A similar analysis of human skin fibrosis identified perturbations of fatty acid oxidation (FAO) and glycolysis pathways (Zhao et al, 2019). Inhibition of glycolysis or restoring FAO by genetic or pharmacological methods mitigated fibrosis in various animal models (Kang et al, 2015; Tran et al, 2016; Ding et al, 2017; Han et al, 2017; Zhao et al, 2019, 2020). Despite these preliminary findings, the exact mechanisms that regulate metabolic dysregulation, especially in myofibroblasts, remain largely unknown.
Chicken ovalbumin upstream promoter‐transcription factor II (COUP‐TFII, also known as NR2F2) is an orphan member of the nuclear receptor family with unknown endogenous ligands (Pereira et al, 1999). Since COUP‐TFII has been reported to regulate metabolic functions (Li et al, 2009; Planchais et al, 2015; Ashraf et al, 2019), we evaluated whether it played an important role in fibrosis. Downstream proteins under COUP‐TFII control tend to be involved in energy production, anabolic pathways, and cell cycle progression, all of which impact cell proliferation (Chen et al, 2012; Planchais et al, 2015; Wu et al, 2015). Up‐regulation of COUP‐TFII in numerous cancers, such as colon (Bao et al, 2014), pancreas (Polvani et al, 2014), prostate (Qin et al, 2013), and renal cell carcinoma (Fang et al, 2020), further supports the role of COUP‐TFII in promoting cell proliferation. We hypothesized that COUP‐TFII contributed to organ fibrosis via a regulatory role in the metabolism of the myofibroblast.
In this study, we demonstrate that COUP‐TFII facilitates the shift of myofibroblast metabolism toward enhanced glycolysis and generation of profibrotic mediators. Increased COUP‐TFII expression co‐localized with αSMA expression in stromal cells of fibrotic human kidneys, lungs, kidney organoids, and fibrotic mouse kidneys after injury. Ablation of COUP‐TFII in adult mice attenuated injury‐induced kidney fibrosis. Our findings demonstrate a previously unrecognized role of COUP‐TFII on regulating myofibroblast differentiation and highlight COUP‐TFII as a relevant therapeutic target to prevent organ fibrosis after injury.
Results
COUP‐TFII expression is increased in myofibroblasts in human fibrotic diseases and human kidney organoids
We interrogated previously published, human CKD microarray datasets and found a significant increase in COUP‐TFII mRNA levels (1.9‐fold) in renal biopsy tissues of 53 patients with CKD (GSE66494; Fig 1A) (Nakagawa et al, 2015). In another large cohort (n = 95) of microdissected human kidney samples from diabetic or hypertensive CKD subjects with pathology‐defined fibrosis, microarray transcription profiling also revealed a significant up‐regulation of COUP‐TFII by analysis of total kidney mRNA levels (1.33‐fold) (Kang et al, 2015). Results from these two independent cohorts of patients suggest an association between COUP‐TFII mRNA expression and CKD in humans. We confirmed COUP‐TFII protein expression and localization by immunofluorescent staining of normal and diseased human kidneys. In control “healthy” kidney tissues, which were obtained from the non‐tumor portions of total nephrectomy samples from patients with renal cell carcinoma, we found little scattered COUP‐TFII expression (Fig 1B). In the setting of kidney injury (patients with either acute thrombotic microangiopathy [TMA] or chronic diabetic nephropathy [DN]), however, the number of COUP‐TFII‐positive cells was significantly increased (Fig 1B). The majority of these cells were localized in the interstitial region and co‐localized with expanded αSMA‐positive areas of fibrosis (Fig 1B). Similar results were also identified in human fibrotic lungs from two patients with idiopathic pulmonary fibrosis (IPF; Fig 1C).
Figure 1. COUP‐TFII expression is increased in human fibrotic kidneys and lungs, and human kidney organoids.
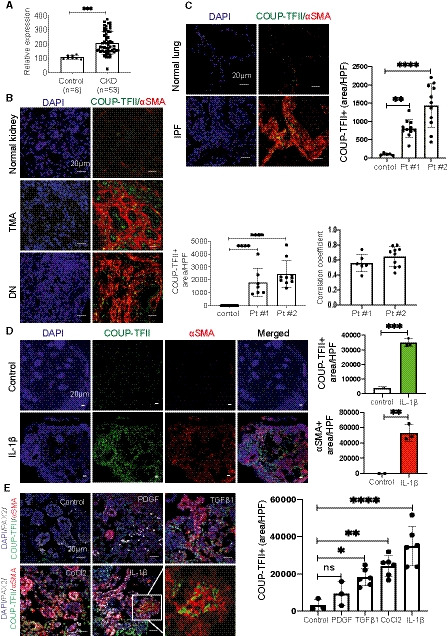
- COUP‐TFII mRNA levels in human kidney tissues of control (n = 8) and CKD (n = 53) subjects from GSE66494. ***P < 0.001 by t‐test.
- COUP‐TFII expression (green) is increased in human kidneys from patients with thrombotic microangiopathy (TMA) and diabetic nephropathy (DN; n = 2). Quantification of mean of 7–10 confocal images per patient at 400× hpf. ****P < 0.0001 by one‐way ANOVA; mean ± SD. The majority of these cells were localized in the interstitial region. More than 50% COUP‐TFII (+) cells (green) are co‐localized with expanded αSMA‐positive (red) areas of fibrosis. Pearson correlation coefficient (PCC) = 0.56 ± 0.1 for patient#1 and 0.65 ± 0.13 for patient#2.
- COUP‐TFII expression (green) is increased in lungs from patients with IPF (n = 2) by immunofluorescence. Quantification of mean of 7–10 confocal images per patient at 400× hpf. **P < 0.01; ****P < 0.0001 by one‐way ANOVA, mean ± SD.
- Human kidney organoids were treated with IL‐1β (10 ng/ml) for 96 h. Immunofluorescence reveals significantly increased COUP‐TFII (green) and αSMA (red) expression in IL‐1β‐treated organoids, compared with non‐treated organoids. Quantification by confocal micrographs in 200× hpf. **P < 0.01; ***P < 0.001 by t‐test; mean ± SD.
- COUP‐TFII expression increased significantly in organoids treated with TGFβ1 (10 ng/ml), cobalt chloride (100 μM), IL‐1β (10 ng/ml), but not PDGF (50 ng/ml). Quantification by confocal micrographs in 400× hpf. *P < 0.05; **P < 0.01; ****P < 0.0001 by one‐way ANOVA, mean ± SD. The higher magnification of the insert from IL‐1β showed co‐localization of COUP‐TFII (green) and αSMA (red) staining.
We also examined COUP‐TFII expression in human kidney organoids generated by directed differentiation of human pluripotent stem cells (Morizane et al, 2015). Human kidney organoids provide advantages of 3D nephron structures with multiple human kidney cell types and a rich stroma. Fibrosis of human kidney organoids was induced by incubation with IL‐1β for 96 h as previously reported (Lemos et al, 2018). As showed in Fig 1D, COUP‐TFII expression significantly increased in IL‐1β‐treated organoids compared with control organoids treated with vehicle. Most COUP‐TFII‐positive cells were located in the interstitial region and co‐localized with αSMA, as observed in the kidneys of human subjects with CKD. In addition to IL‐1β, we also treated organoids with PDGF, TGFβ1, or cobalt chloride (CoCl2, which induces hypoxia‐inducible factor [HIF] signaling, mimicking the response to hypoxia) in an attempt to screen for the upstream activators of COUP‐TFII. As shown in Fig 1E, COUP‐TFII expression increased significantly in organoids treated with TGFβ1 and cobalt chloride, but not PDGF. Together, these results indicate an association of increased COUP‐TFII expression with fibrosis in human kidney tissue. The potential link to fibrosis prompted us to investigate the spatial and temporal expression, and mechanistic implications of COUP‐TFII in non‐injured and injured kidneys in adult mice.
COUP‐TFII protein is expressed in stromal cells in non‐injured and injured mouse kidneys
In healthy adult mouse kidneys, a few scattered COUP‐TFII‐positive cells were present within the interstitium. The majority of COUP‐TFII‐positive cells expressed PDGFRβ, a pericyte/fibroblast marker. COUP‐TFII was not observed in endothelial cells marked by CD31 (Fig 2A).
Figure 2. COUP‐TFII protein is expressed in pericytes/fibroblasts, both in non‐injured and injured mouse kidneys.
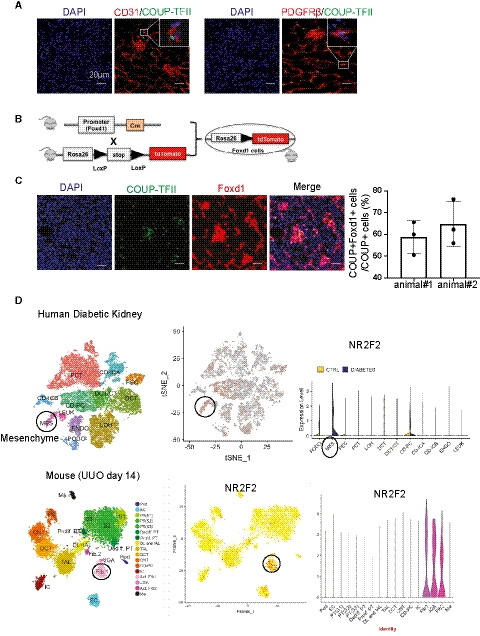
- COUP‐TFII+ cells (green) express pericyte/fibroblast marker (PDGFRβ) but not the endothelial cell marker (CD31).
- Strategy of genetically labeling Foxd1‐derived stromal cells in non‐injured mouse kidney.
- Most COUP‐TFII+ cells (green) were also positive for Foxd1 (tdTomato‐labeled as red) in the non‐injured mice kidney (n = 2). Quantification by confocal micrographs in 400× hpf.
- COUP‐TFII was enriched most in pericytes/fibroblasts in injured kidney, both in the human (Diabetic kidneys, (D) top) and mouse model (UUO day 14, (D) bottom) as analyzed from single‐cell sequencing database (http://humphreyslab.com/SingleCell/).
To further assess the developmental origin of COUP‐TFII+ cells, we crossed Foxd1‐Cre driver mice (Humphreys et al, 2010) to tdTomato reporter mice (Madisen et al, 2010) to genetically label the Foxd1‐derived stromal cells (Fig 2B). The majority of COUP‐TFII protein‐expressing cells (green) were also tdTomato‐labeled Foxd1‐derived stromal cells (red; Fig 2C), demonstrating that COUP‐TFII+ cells in non‐injured mice derive from the Foxd1 population. Analysis of an available single‐cell RNA sequencing database (http://humphreyslab.com/SingleCell/) (Wilson et al, 2019; Wu et al, 2019) confirmed that COUP‐TFII RNA was most enriched in pericyte/fibroblast cells in injured kidneys, both in human (diabetic kidney; Fig 2D top) and mouse (UUO day 14; Fig 2D bottom). Thus, the majority of COUP‐TFII+ cells are kidney stromal cells, both in non‐injured and injured kidneys.
COUP‐TFII expression is increased during the development of kidney fibrosis in various kidney injury models in mice
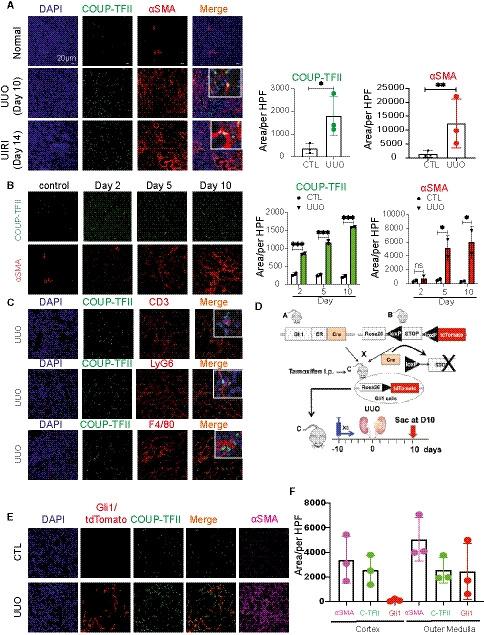
COUP‐TFII expression was significantly increased and co‐localized in αSMA+ cells within fibrotic regions in the injured kidney in two mouse kidney injury models: unilateral ureteral obstruction (UUO) and unilateral ischemia‐reperfusion injury (UIRI; Fig 3A). To further characterize the spatial and temporal expression kinetics of COUP‐TFII, we focused on the UUO model since it more reliably induces fibrosis in a short time frame. As shown in Fig 3B, COUP‐TFII expression was up‐regulated as early as day 2 after injury, which is well before up‐regulation of αSMA and any histologic evidence of fibrosis was evident. We further demonstrated that the increased COUP‐TFII expression is distinct from the inflammatory infiltrate that accompanies injury and fibrosis, as it does not overlap with markers of T lymphocytes (CD3), neutrophils (LyG6), or macrophages (F4/80; Fig 3C). Thus, we conclude that COUP‐TFII is up‐regulated specifically within the stromal compartment following kidney injury, and it precedes the expression of fibrotic markers, compatible with a potential causative role in the pathophysiology of fibrosis.
Figure 3. COUP‐TFII expression is increased in the injured mouse kidneys during the development of kidney fibrosis.
-
ACOUP‐TFII expression is significantly increased and co‐localized with αSMA+ cells at day 10 of UUO and day 14 of UIRI in mice (n = 3 animals per group). Quantification of mean of 8–10 confocal images per animal at 200× hpf. *P < 0.05 **P < 0.01 by paired t‐test; mean ± SD. CTL: contralateral kidney.
-
BIn a time course of UUO, COUP‐TFII expression increased as early as day 2 and preceded the increased expression of αSMA (n = 2 animals per time point). Quantification of mean of 8–10 confocal images per animal at 200× hpf. *P < 0.05; ***P < 0.001 by two‐way ANOVA test; mean ± SD.
-
CIn the UUO model, COUP‐TFII‐positive cells do not stain with markers of inflammatory cells (T cell marker CD3, neutrophil marker LyG6, or macrophage marker F4/80; n = 3 animals).
-
D–F(D) Protocol for fate tracing of Gli1+ cells in the UUO model, (E, F) Gli1‐tdTomato+ cells expanded primarily in the outer medullary region. In contrast, COUP‐TFII+ cells are distributed both in cortex and medulla. A subset of COUP‐TFII+ cells (green) overlap with genetically labeled Gli1+ pericyte/perivascular cells (red) in UUO injury model in the outer medullary region (n = 3 animals per group). CTL: contralateral kidney. Quantification of mean of 8–10 confocal images per animal at 200× hpf.
Genetic lineage tracing analysis demonstrated that Gli1 marks perivascular mesenchymal stem cells‐like cells, which are proposed to contribute to organ fibrosis (Kramann et al, 2015). We evaluated whether COUP‐TFII+ cells and Gli1+ cells represent the same or closely related populations. We used the Gli1‐CreERt2 line crossed with tdTomato reporter mice and induced genetic labeling 10 days prior to UUO injury (Fig 3D). Kidney tissues were collected at day 10 after UUO and stained for COUP‐TFII expression. As expected, Gli1‐derived cells expanded in number and acquired αSMA expressing by 10 days after UUO (Fig 3E). Co‐staining revealed that COUP‐TFII expression was indeed found in nearly all of Gli1‐tdTomato+ cells; however, there were also a large number of COUP‐TFII+ cells that were distinct from the Gli1 lineage (Fig 3E). Although some of these cells could result from incomplete labeling with the inducible Gli1‐Cre, there was also a noticeable difference in the spatial distribution between the COUP‐TFII+/tdTomato− and COUP‐TFII+/tdTomato+ populations. Interestingly, while COUP‐TFII+ cells were found throughout both the cortex and medulla, co‐localizing in αSMA+ cells in both regions, the Gli1‐tdTomato+ cells were largely restricted to the outer medullary region (Fig 3F). This is consistent with the previous observations (Kramann et al, 2015) that Gli1‐tdTomato+ cells represented only a small fraction of the total PDGFRβ+ population. Gli1 cells were enriched in the outer medulla with much less expression in pericytes and perivascular fibroblasts of the cortex (Kramann et al, 2015; Humphreys, 2018). As fibrosis is present both in cortex and outer medulla in the UUO model, our data suggest that COUP‐TFII functions more generally in regulating pericyte to myofibroblast differentiation, irrespective of the anatomic compartments.
Ablation of COUP‐TFII in adult mice attenuates injury‐induced kidney fibrosis
Since global knockout of COUP‐TFII in mice confers embryonic arrest at E10 due to defective angiogenesis and cardiac development (Pereira et al, 1999), we used a tamoxifen‐inducible Cre‐loxP system to knockout COUP‐TFII in adult mice. We generated COUP‐TFIIflox/+; Rosa26CreERT2/+ (F/+; Cre/+) and COUP‐TFIIflox/flox; Rosa26CreERT2/+ (F/F; Cre/+) mice. COUP‐TFII expression was maintained in these mice without tamoxifen (TAM). Three injections of TAM activated Cre recombinase and generated COUP‐TFII heterozygous (+/−) and homozygous (−/−) knockout mice. Ten days after TAM injection, mice were subjected to UUO and euthanized 7 days after UUO (Fig 4A). There was no identifiable phenotypic difference between WT and knockout mice without injury.
Figure 4. Genetic ablation of COUP‐TFII in adult mice attenuates injury‐induced kidney fibrosis.
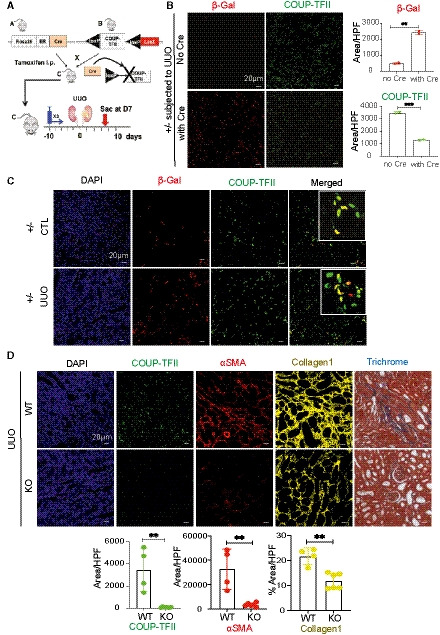
- Strategy of conditional knockdown of COUP‐TFII in adult mice. A LacZ knock‐in allele is inserted into the genomic COUP‐TFII locus after the second LoxP site.
- Activation of Cre recombinase by tamoxifen (TAM) results in COUP‐TFII deletion and the expression of LacZ reporter (detected by immunostaining of β‐galactosidase (β‐Gal, red) in the UUO mice model (n = 2). Quantification of mean of 8–10 confocal images at 200× hpf. **P < 0.01; ***P < 0.001 by paired t‐test; mean ± SD.
- Using heterozygous mice (F/+; Cre/+), β‐Gal+ cells (red) increased after TAM injection in UUO kidneys, and co‐localized with COUP‐TFII+ cells (green; n = 3).
- COUP‐TFII+ cells decreased significantly in the UUO kidney in TAM‐treated homozygous (F/F;Cre/+) mice (KO group, n = 6) compared with wild‐type littermates (WT group, n = 4). Expression of αSMA (red) and collagen 1 (yellow) are also markedly reduced. Masson Trichrome staining shows less kidney fibrosis in KO compared with WT group. Quantification of mean of 8–10 confocal images per animal at 200× hpf. **P < 0.01 by unpaired t‐test, mean ± SD.
We first examined the efficacy of COUP‐TFII ablation. A LacZ knock‐in allele was inserted into the genomic COUP‐TFII locus after the second LoxP site (Takamoto et al, 2005). Treatment with Cre recombinase resulted in COUP‐TFII deletion and the expression of the LacZ reporter. Using this LacZ reporter, we were able to trace the COUP‐TFII lineage following deletion. As shown in Fig 4B, expression of β‐galactosidase (β‐Gal) increased after TAM injection in UUO kidneys of heterozygous mice with Cre allele, but not in the control mice with a flox allele but without Cre allele (WT), indicating the specificity of β‐Gal staining in the Cre‐LoxP system. Expression of COUP‐TFII decreased after TAM injection in heterozygous (+/−) mice. Ten days after TAM injection, β‐Gal expression co‐localized with COUP‐TFII expression in both contralateral and UUO kidneys, consistent with the deletion of one allele of COUP‐TFII (Fig 4C). Both β‐Gal expression and COUP‐TFII expression increased in UUO kidney compared with contralateral non‐injured kidney (Fig 4C). β‐Gal staining demonstrated that this population remained stable following TAM injection, indicating that COUP‐TFII deletion does not impact the survival or proliferation of these cells at baseline.
Next, we examined the effect of COUP‐TFII ablation on injury‐induced kidney fibrosis (UUO model) using knockout (KO, −/−) mice. As shown in Fig 4D, COUP‐TFII‐positive cells decreased significantly in a KO (−/−) mice after UUO compared with WT kidneys, indicating successful knockout of COUP‐TFII. Associated with much less COUP‐TFII expression, αSMA and collagen 1 expression was significantly decreased in KO compared with WT at 7 days after UUO. Histological evaluation demonstrated less kidney fibrosis in KO compared with WT by Masson Trichrome (Fig 4D). These data demonstrated that ablation of COUP‐TFII in adult mice attenuates injury‐induced kidney fibrosis.
COUP‐TFII regulates myofibroblast differentiation in vitro through augmented glycolysis
In order to dissect the mechanistic role of COUP‐TFII in myofibroblast differentiation, which leads to fibrosis, we generated COUP‐TFII loss‐ and gain‐of‐function cell lines using CRISPR‐Cas9 (clustered regularly interspaced short palindromic repeats [CRISPR]–CRISPR‐associated protein 9 [Cas9]) and an inducible lentiviral construct, respectively, in the pericyte‐like cell line C3H/10T1/2. C3H/10T1/2 is a mouse mesenchymal cell that has been used in studies related to pericyte biology and cell fate determination in vitro (Pinney & Emerson, 1989; Kale et al, 2005; Pecot et al, 2013). Naïve C3H/10T1/2 cells (WT) showed basal expression of COUP‐TFII, which we were able to successfully modulate using the knockout (KO) or overexpression (OE) systems (Fig 5A). Neither KO nor OE affected cell viability (Fig 5B), although a decreased proliferation rate in KO cells and increased proliferation rate in OE cells were observed compared with naïve (WT) cells (Fig 5C). Treatment of C3H/10T1/2 cells with TGFβ1 induces myofibroblast differentiation with up‐regulation of αSMA (Fig 5D). Interestingly, OE cells, in the absence of TGFβ1 stimulation, displayed an elongated fibroblast shape, similar to WT cells treated with TGFβ1 (Fig 5D). There was increased expression of αSMA and collagen 1 in COUP‐TFII OE cells even in the absence of TGFβ1 stimulation (Fig 5D and E), while their expression was significantly diminished in the KO cells. Collectively, these data further support a profibrotic function for COUP‐TFII in pericyte/fibroblast‐like cells.
Figure 5. Cellular knockout of COUP‐TFII in vitro in C3H/10T1/2 cells decreases cell proliferation and suppresses TGFβ1‐induced αSMA and collagen 1 expression. In contrast, overexpression of COUP‐TFII alone increases collagen 1 production.
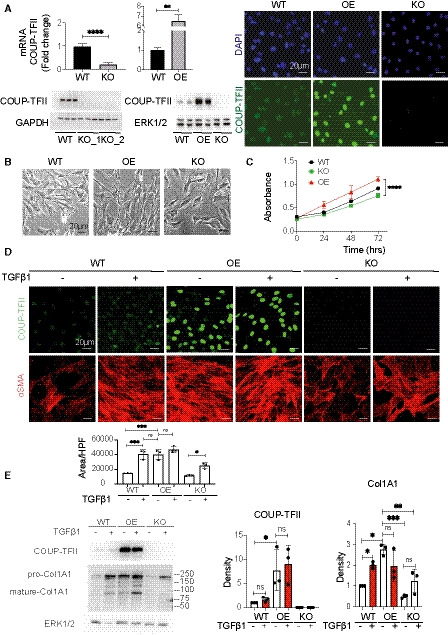
-
AVerification of COUP‐TFII loss‐ and gain‐of‐function cell lines generated by CRISPR‐Cas9 and an inducible lentiviral construct in pericyte‐like cell line C3H/10T1/2 in vitro (n = 6), ****P < 0.0001 by paired t‐test.
-
B, CCOUP‐TFII KO had no effect on cell viability, although there was decreased proliferation rate compared with naïve C3H/10T1/2 cells (WT; n = 6), ****P < 0.0001 by two‐way ANOVA.
-
DCOUP‐TFII‐OE cells, in the absence of TGFβ1 stimulation, displayed an elongated fibroblast shape with increased αSMA expression, similar to WT cells treated with TGFβ1. KO cells had morphology and aSMA expression similar to untreated WT cells. Quantification of mean of 8–10 confocal images at 400× hpf, *P < 0.05, **P < 0.01, ***P < 0.001 by one‐way ANOVA, mean ± SD.
-
EOverexpression of COUP‐TFII alone without TGFβ1 induces collagen 1 production (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 by one‐way ANOVA, mean ± SD.
To further interrogate the molecular mechanisms by which COUP‐TFII regulates myofibroblast differentiation, we evaluated the proteome of WT and OE cells that were differentiated with TGFβ1. This non‐biased approach unequivocally implicated cellular metabolic pathways as predominant direct or indirect targets of COUP‐TFII. Gene set enrichment analysis (GSEA) highlighted that the most significantly down‐regulated proteins in COUP‐TFII‐OE cells were enriched in mitochondrial electron transport chain and fatty acid oxidation (FAO) pathways (Fig 6A), which are involved in oxidative metabolism. Conversely, the top up‐regulated pathways in COUP‐TFII‐OE cells were associated with both extracellular matrix, including collagen fiber organization, cadherin binding and actin cytoskeleton, and glycolysis pathways (Fig 6A and B). Taken together, these data support a model in which COUP‐TFII enhances glycolysis and suppresses FAO, which may lead to increased extracellular matrix production and fibrosis.
Figure 6. COUP‐TFII enhances TGFβ1‐induced glycolysis during myofibroblast differentiation, which is essential for collagen production in vitro .
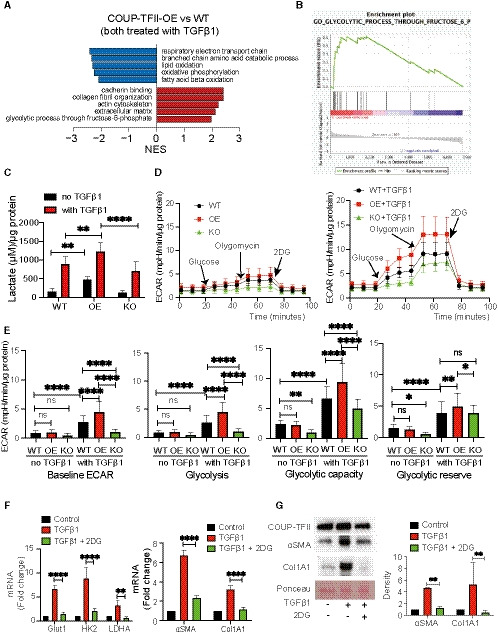
-
A, BNon‐biased proteomics of TGFβ1‐treated naïve (WT) and COUP‐TFII overexpressing (COUP‐TFII‐OE) cells revealed that COUP‐TFII promotes the expression of proteins enriched in metabolic process critical for myofibroblasts, in particular, suppression of FAO and enhancement of glycolysis. Top pathways ranked by NES (gene set enrichment with FDR < 0.05) altered in COUP‐TFII‐OE compared with naïve C3H/10T1/2 cells (n = 2).
-
CTGFβ1 treatment for 48 h significantly increased more lactate production by COUP‐TFII‐OE (OE) cells when compared to WT or COUP‐TFII‐KO (KO) cells treated with TGFβ1. Interestingly, overexpression of COUP‐TFII alone (without TGFβ1) significantly increased lactate production compared with WT (n = 3). **P < 0.01, ****P < 0.0001 by one‐way ANOVA, mean ± SD. Lactate was released by cells into 100 µl of media.
-
DWT, COUP‐TFII overexpression (OE), and COUP‐TFII knockout (KO) cells were seeded in Seahorse XF‐24 cell culture microplates. The cells were rendered quiescent in 0.5% FBS DMEM overnight and then treated with or without 10 ng/ml TGFβ1 for 24 h. All the cells were incubated in the glycolysis stress test medium without glucose and pyruvate, followed by sequential treatments with glucose (10 mM), oligomycin (5 μg/ml), and 2‐DG (50 mM). Real‐time extracellular acidification rate (ECAR) was recorded as the baseline (before glucose), the rate of glycolysis (after glucose), glycolytic capacity (after oligomycin), and glycolytic reserve (after 2‐DG; n = 10).
-
EWith TGFβ treatment COUP‐TFII OE significantly increased, while COUP‐TFII KO decreased baseline and glucose‐stimulated rate of glycolysis, glycolytic capacity, and glycolytic reserve when compared to WT cells. Without TGFβ1 treatment, there is no difference on the baseline and glucose‐stimulated glycolysis among WT, OE, and KO cells; however, COUP‐TFII KO significantly decreased glycolytic capacity and reserve compared with WT (n = 10), *P < 0.05; **P < 0.01; ****P < 0.0001 by one‐way ANOVA, mean ± SD.
-
F2‐deoxyglucose (2‐DG) treatment inhibited TGFβ1‐induced expression of genes involved in glycolysis (left) and fibrosis (right) in COUP‐TFII‐OE cells quantitated by qRT–PCR (n = 6). **P < 0.01; ****P < 0.0001 by one‐way ANOVA, mean ± SD. 2‐DG suppressed TGFβ1‐induced proteins of αSMA and ColA1 in COUP‐TFII OE cells without change the protein of COUP‐TFII by western blot (n = 3). **P < 0.01 by one‐way ANOVA.
Indeed, we found that COUP‐TFII overexpression significantly increased lactate production with or without TGFβ1 stimulation compared with WT cells (Fig 6C), consistent with augmented glycolysis in these cells. Furthermore, we performed a glycolysis stress test using the Seahorse X24 extracellular flux analyzer. All three types of cells (WT, OE, and KO), treated with or without TGFβ1, were incubated in the glycolysis stress test medium (no glucose and pyruvate). Then, the cells received serial exposures to glucose, oligomycin, and 2‐deoxyglucose (2‐DG; a glucose analog that inhibits glycolysis through competitive binding to hexokinase2 (HK2), the first enzyme in the glycolytic pathway; Fig 6D). The extracellular acidification rate (ECAR) at baseline, the rate of glycolysis, glycolytic capacity, and glycolytic reserve were determined. Without TGFβ1 treatment, COUP‐TFII overexpression did not change the baseline ECAR, the rate of glycolysis, glycolytic capacity, or glycolytic reserve. Interestingly, COUP‐TFII knockout significantly decreased glycolytic capacity and reserve compared with WT (Fig 6E). TGFβ1 significantly increased baseline ECAR, glycolysis, glycolytic capacity, and reserve in WT cells (Fig 6E). This effect of TGFβ1 on ECAR was significantly enhanced by COUP‐TFII overexpression and reduced by COUP‐TFII knockout (Fig 6E). Of note, unlike the lactate assay, COUP‐TFII OE cells did not increase glycolysis without TGFβ1 stimulation (Fig 6E). This was likely due to the fact that the base medium used in Seahorse experiments did not have glucose and pyruvate. Collectively, these data demonstrated increased glycolysis in COUP‐TF OE cells in response to TGFβ1 stimulation.
As shown in Fig 6F, TGFβ1 up‐regulated genes involved in glycolysis, such as glucose transporter 1 (Glut1), HK2, and lactate dehydrogenase A (LDHA), as well as genes activated in fibrosis (αSMA and ColA1). To directly test the link between glycolytic metabolism and myofibroblast differentiation, we treated COUP‐TFII OE cells with 2‐DG. 2‐DG attenuated the TGFβ1‐induced expression of genes involved in glycolysis, as well as αSMA and collagen 1. We confirmed that 2‐DG suppressed TGFβ1‐induced proteins of αSMA and ColA1 in COUP‐TFII OE cells without changes in the level of COUP‐TFII protein (Fig 6G). These data suggest that COUP‐TFII enhances TGFβ1‐induced glycolysis during myofibroblast differentiation and that the enhancement of glycolysis is essential for this process.
Our proteomic data suggested that COUP‐TFII overexpression significantly down‐regulated proteins involved in fatty acid oxidation (FAO) pathways (Fig 6A). Among those proteins, pyruvate dehydrogenase kinase 4 (PDK4) was significantly decreased in COUP‐TFII OE cells in response to TGFβ1 (Fig 7A). PDK4 plays a central role in cellular energy metabolism through inhibition of pyruvate dehydrogenase complex (PDC), a mitochondrial multi‐enzyme complex that converts pyruvate to acetylcoenzyme A (AcetylCoA). Glucose and fatty acid compete with each other for oxidation through PDC. Lower PDK4 leads to greater PDC activity and, therefore, increases in glucose oxidation and inhibition of fatty acid oxidation (Zhang et al, 2014). Quantitative real‐time PCR data revealed that overexpressing COUP‐TFII resulted in decreased transcription of PGC1α and PDK4, and knockout of COUP‐TFII increased PGC1α and PDK4 in vitro (Fig 7B).
Figure 7. Overexpressing COUP‐TFII resulted in decreased transcription of PGC1α and PDK4, and knockout of COUP‐TFII increased PGC1α and PDK4 in vitro. Overexpression of PGC1α, however, is not sufficient to suppress TGFβ1‐induced glycolysis and COUP‐TFII induced myofibroblast differentiation.
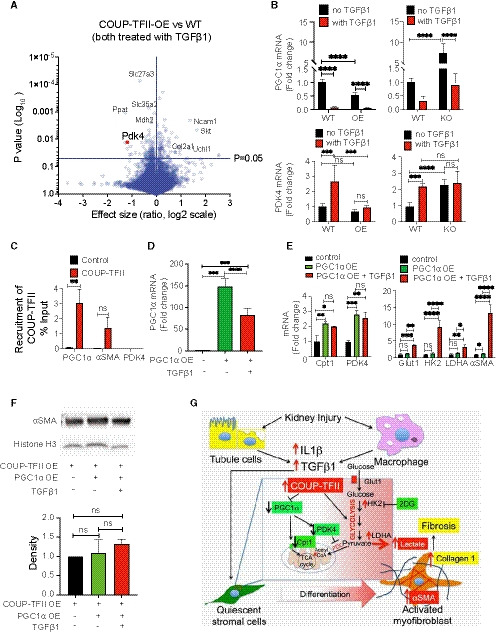
- Volcano plot of all detected proteins from proteomics of TGFβ1‐treated naïve (WT) and COUP‐TFII overexpressing (COUP‐TFII‐OE) cells. PDK4 expression is significantly decreased in OE cells (n = 2).
- Without TGFβ treatment, overexpression of COUP‐TFII inhibited, and knockout of COUP‐TFII increased, mRNA of PGC1α. Although overexpression of COUP‐TFII did not change the mRNA of PDK4, knockout of COUP‐TFII significantly increased the expression of PDK4 (n = 6). ***P < 0.001, ****P < 0.0001 by one‐way ANOVA, mean ± SD.
- Chip‐qPCR analysis on C3H/10T1/2 cells in vitro revealed binding of COUP‐TFII on the promoter of PGC1α, but not αSMA or PDK4 (n = 2). **P < 0.01, mean ± SD.
- Overexpression of PGC1α was achieved through adenovirus transduction in C3H/10T1/2 cells (WT) and confirmed by qRT–PCR (n = 6). ***P < 0.001, ****P < 0.0001 by one‐way ANOVA, mean ± SD.
- PGC1α OE significantly increased expression of Cpt1 and PDK4. Both are involved in key steps of FAO. However, overexpression of PGC1α did not abrogate the TGFβ1 up‐regulated glycolytic genes and αSMA (n = 6). *P < 0.05; **P < 0.01; ****P < 0.0001 by one‐way ANOVA, mean ± SD.
- PGC1α OE did not decrease αSMA protein neither with nor without TGFβ1 in COUP‐TFII OE cells by Western Blot analysis (n = 3).
- Schematic model of the role of COUP‐TFII in metabolic reprogramming during myofibroblast differentiation and fibrosis formation after injury. PDC: pyruvate dehydrogenase complex.
We then asked whether COUP‐TFII regulates gene expression of PGC1α, PDK4, or αSMA through direct binding to their promoters. Our Chip‐qPCR results showed that PGC1α is a target of COUP‐TFII, but αSMA or PDK4 are not (Fig 7C). Therefore, decreased PDK4 expression observed with COUP‐TFII overexpression might be indirectly due to suppression of PGC1α expression. To determine whether activation of FAO pathways is sufficient to mitigate the effect of COUP‐TFII on glycolysis and myofibroblast differentiation, we overexpressed PGC1α through adenovirus transduction in C3H/10T1/2 cells (WT; Fig 7D). As shown in Fig 7E, PGC1α OE significantly increased expression of Cpt1 and PDK4, two key contributors to FAO. However, overexpression of PGC1α did not abrogate the TGFβ1 up‐regulated genes in glycolysis and αSMA. Furthermore, we infected COUP‐TFII OE cells with the PGC1α adenovirus (Fig 7F). PGC1α OE did not decrease αSMA protein neither in the absence or presence of TGFβ1 in COUP‐TFII OE cells. These data suggest that COUP‐TFII OE suppressed FAO pathway, although activation of the FAO pathway is not sufficient to reverse TGFβ1‐induced glycolysis during myofibroblast differentiation.
Taken together, these data demonstrated that COUP‐TFII regulates cell metabolism through suppression of FAO and enhancement of glycolysis in myofibroblasts, thereby promoting myofibroblast differentiation, and collagen 1 production after injury (Fig 7G).
Discussion
Pericyte/perivascular cells are important contributors to kidney fibrosis after injury (Lin et al, 2008; Humphreys et al, 2010). These cells are the main source of myofibroblasts, effector cells for fibrosis (Duffield, 2014; Humphreys, 2018). Blocking the differentiation of pericyte/perivascular cells to myofibroblasts is an attractive strategy to reduce fibrosis. TGFβ, a master regulator of myofibroblast differentiation, has been extensively studied as a therapeutic target (Akhurst & Hata, 2012; Meng et al, 2012; Rangarajan et al, 2016). Direct inhibition of TGFβ, however, has led to more toxicity than benefit (Li et al, 2006; Principe et al, 2014). Therefore, alternative molecular approaches to the regulation of myofibroblast differentiation during fibrosis development are desirable for anti‐fibrotic drug development.
We demonstrate that COUP‐TFII is markedly increased in human kidneys with fibrosis or cells in human kidney organoids activated with IL‐1β, TGFβ, or cobalt chloride to enhance stromal fibrosis. COUP‐TFII is expressed in pericytes/perivascular cells in the adult non‐injured kidney and co‐localizes with αSMA expression during fibrosis formation after kidney injury. Our data suggest that COUP‐TFII can be a marker of myofibroblasts after injury and be a regulator of myofibroblast differentiation. Another protein proposed to be important in converting pericytes/perivascular cells to myofibroblasts is Gli1 (Kramann et al, 2015). Compared with Gli1, COUP‐TFII is seen in more αSMA+ cells, including many that are not Gli1‐tdTomato+, especially in the kidney cortex. Therefore, COUP‐TFII serves a different functional role than Gli1. Genetic depletion of COUP‐TFII reduced αSMA‐positive cells and attenuated kidney fibrosis after injury. Our results demonstrate that COUP‐TFII plays a pivotal role in myofibroblast differentiation and kidney fibrosis. We also show increased COUP‐TFII expression in myofibroblasts in fibrotic lungs of patients with idiopathic lung fibrosis (IPF). These findings suggest that the fibrogenic response may share a common pathway in kidney and lung injury and failure.
Our proteomic data revealed that COUP‐TFII promotes the expression of proteins enriched in metabolic processes critical for myofibroblasts, in particular, suppression of FAO and enhancement of glycolysis. Our data link COUP‐TFII to the metabolism deregulation leading to a proliferative and profibrotic function of myofibroblast. Overexpression of COUP‐TFII alone (without TGFβ1 treatment) is sufficient to increase glycolysis and collagen 1 expression. Knockout of COUP‐TFII dampened TGFβ1‐induced glycolysis and decreased αSMA and collagen 1 expression. The phenotypic resemblance between COUP‐TFII overexpressed cells and WT cells treated with TGFβ1 suggests that COUP‐TFII is essential to establish the myofibroblast phenotype through augmented glycolysis. High glycolytic flux is important for the self‐renewal of progenitor cells (Liu et al, 2017). Interestingly, COUP‐TFII expression is abundant in the mesenchymal compartment of developing organs during embryonic organogenesis but declines significantly right after birth. This suggests that COUP‐TFII might be important to maintain the de‐differentiation status of cells through augmented glycolysis during development and that injury may recapitulate these developmental programs.
We demonstrated that COUP‐TFII increases glycolysis in myofibroblast differentiation. Enhanced glycolysis drives collagen production and fibrosis formation. Besides supporting energy needs in a hypoxic environment, glycolysis also provides macromolecules required for cell proliferation, migration, and amino acid and nucleotide synthesis for DNA replication and RNA transcription (Xie et al, 2015; Zhao et al, 2019; Henderson et al, 2020). In this way, the reprogramming of cell metabolism ensures sufficient building blocks for biosynthesis and facilitates survival and proliferation of myofibroblasts in a harsh hypoxic and nutrient‐deprived microenvironment. Furthermore, glycolysis is clearly linked to ECM production (Ding et al, 2017; Zhao et al, 2019, 2020). Collagen 1, the predominant structural protein found in kidney fibrosis, is synthesized through multiple steps, including hydroxylation of amino acids, disulfide bonding, and glycosylation (Basak et al, 2016). The major amino acid components of collagen are glycine, proline, and lysine. Glycine is mainly produced from glycolysis (de Paz‐Lugo et al, 2018). In addition, collagen hydroxylation and glycosylation are dependent on glycolysis (Im et al, 1976). Our data, together with studies from others (Xie et al, 2015; Ding et al, 2017), demonstrate that 2‐DG (a hexokinase inhibitor) drastically decreases collagen 1 production in myofibroblasts in vitro. There is emerging evidence showing that augmented glycolysis in cancer stromal cells (also call cancer‐associated fibroblasts) supports cancer progression by secreting lactate and other glycolytic intermediates (Avagliano et al, 2018). In addition, a decrease in microenviromental pH from lactic acid accumulation has been associated with increased TGFβ activity (Kottmann et al, 2012). Therefore, targeting glycolysis in myofibroblasts would be expected to not only inhibit the activation of stromal cells, but also to modify the microenvironment of fibrosis foci.
Defective fatty acid oxidation (FAO) plays an important role in mechanisms of renal fibrosis (Kang et al, 2015) and DN (Proctor et al, 2006). Recent studies revealed that kidney injury down‐regulated expression of PGC1α, a master regulator of mitochondrial biogenesis and oxidative metabolism (Tran et al, 2016; Dumesic et al, 2019). PGC1α knockout mice exhibited more tubular injury and worse renal function after kidney ischemia‐reperfusion injury (Tran et al, 2016). Overexpression of PGC1α in renal tubular cells provided renal protection (Tran et al, 2016; Han et al, 2017). We found that COUP‐TFII OE significantly decreased, while COUP‐TFII KO increased, transcription of PGC1α in C3H/10T1/2 cells. Our Chip‐qPCR results revealed that COUP‐TFII directly binds to the PGC1α promoter, consistent with other reports (Li et al, 2009; Wu et al, 2015). Therefore, we speculate that COUP‐TFII‐mediated suppression of FAO may relate to decreased PGC1α expression. In our experiments, overexpression of PGC1α increased the expression of PDK4 and Cpt1, key components of FAO (Zhang et al, 2014; Schlaepfer & Joshi, 2020). PGC1α overexpression, however, did not suppress TGFβ‐induced glycolysis and fibrosis in myofibroblasts. Furthermore, overexpression of PGC1α did not reverse the effect of COUP‐TFII overexpression on αSMA production. Therefore, enhanced glycolysis by COUP‐TFII is not dependent on decreases in expression of PGC1α. COUP‐TFII can drive glycolysis and myofibroblast differentiation even in the absence of reductions in PGC1α expression. Renal proximal tubular cells are well known to depend mainly on oxidative metabolism. PGC1α overexpression is protective but this may be due to protection during the injury phase (Tran et al, 2016; Han et al, 2017). In contrast, podocytes and fibroblasts rely on glycolysis (Rabelink & Carmeliet, 2018; Brinkkoetter et al, 2019). Therefore, overexpression PGC1α in fibroblast might not be sufficient to suppress TGFβ1‐induced glycolysis and COUP‐TFII induced myofibroblast differentiation.
Since COUP‐TFII is expressed primarily in myofibroblasts, targeting it may be selectively effective to suppress myofibroblast differentiation and function without affecting epithelial or endothelial cells. There are no overt phenotypes in mice where COUP‐TFII has been knocked out at the adult stage (Xie et al, 2016; Ceni et al, 2017) further supporting the potential safety profile of COUP‐TFII inhibition. We demonstrate that COUP‐TFII is markedly increased in stroma cells in human kidney organoids treated with IL‐1β, TGFβ, or CoCl2, which mimic inflammation or ischemia during injury. These ex vivo human cell systems can be used to test potential inhibitors of COUP‐TFII.
In conclusion, our study provides compelling evidence that COUP‐TFII regulates myofibroblast differentiation and fibrosis in models of kidney injury. The profibrotic function is mediated through augmented glycolysis and resultant differentiation of pericytes/fibroblasts to myofibroblast after injury. Reducing COUP‐TFII is effective in diminishing myofibroblast differentiation and limiting fibrosis in vivo. The fibrogenic response may share a common pathway in different organ systems. Targeting COUP‐TFII may serve as a novel treatment approach for mitigating fibrosis in chronic kidney disease and fibrosis of other organs.
Materials and Methods
Human kidney biopsy sample preparation and immunostaining
Normal kidney samples used for immunostaining studies were obtained from surgical sections from patients undergoing nephrectomy due to renal cell carcinoma (RCC) under institutional review board‐approved protocols. Injured kidney samples were obtained from kidney biopsy samples from patients with TMA and DN.
Human kidney organoids generation and IL‐1β stimulation
Kidney organoids were derived from H9 human embryonic stem cells as previously described (Morizane et al, 2015). Briefly, H9 cells were cultured in StemFit (Ajinomoto) supplemented with 10 ng/ml FGF2 (PeproTech). Differentiation was started with 8 μM CHIR (TOCRIS) for 4 days, followed sequentially by Activin for 3 days and FGF9 for 1–2 days. Subsequently, cells were dissociated with Accutase (Stem Cell Technologies) and plated into U‐shaped bottoms 96‐well plates (Corning) at 100,000 cells per well in a medium supplemented with 3 μM CHIR and 10 ng/ml FGF9. Two days later, the medium was changed to one supplemented with only 10 ng/ml FGF9. After 3–4 days, the medium was changed to basal medium without additional growth factors. At day 51, matured organoids were treated with IL‐1β 10 ng/ml (Sigma) for 96 h. Organoids were collected and fixed in 4% PFA for 30 min followed by 20% sucrose overnight. Cryosections (7 μm) were incubated with antibodies for immunofluorescence. Images were captured by confocal microscopy using a Nikon C1 microscope running EZ‐C1 software.
Mouse strain and animal experiments
All mouse experiments were performed under the animal use protocol approved by the Institutional Animal Care and Use Committee of the Brigham and Women's hospital. Gli1‐CreERt2 (JAX# 007913), Rosa26tdTomato (JAX# 007909), Foxd1‐GFP‐Cre (Jax# 012463), and Rosa26‐CreERt2 (JAX# 008463) were purchased from Jackson Laboratories (Bar harbor, ME). COUP‐TFII flox/+ mice were purchased from Mutant Mouse Resource & Research Centers (MMRRC; B6;129S7‐Nr2f2tm2Tsa/Mmmh, Cat# 032805‐MU). The COUP‐TFII flox/+ mouse strain was maintained in a mixed genetic background (129/Sv × C57BL/6) and received standard rodent chow. To induce COUP‐TFII deletion in the adult, 8‐ to 12‐week‐old mice were intraperitoneally injected with three doses 0.1 mg/g body weight tamoxifen in corn oil/3% ethanol (Sigma) every other day starting 14 days before surgery.
Murine kidney fibrosis models were performed as previously described (Yang et al, 2010). Briefly, mice were anesthetized with pentobarbital sodium (60 mg/kg body weight, intraperitoneally). For the unilateral ureteral obstruction (UUO) surgery, a flank incision was made and the left ureter was tied off at the level of the lower pole with two 4.0 silk ties. For the unilateral ischemia‐reperfusion injury (IRI), the left kidney was exposed through a flank incision and subjected to ischemia by clamping the renal pedicle with non‐traumatic microaneurysm clamps (Roboz, Rockville, MD) for 30 min. Reperfusion was confirmed by visual color change. Body temperatures were controlled at 36.5–37.5°C throughout the procedure. One milliliter of warm (body temperature) saline was instilled in the retroperitoneum after surgery for volume supplementation. Buprenorphine was used for pain control (0.1 mg/kg body weight, intraperitoneally). Mice were sacrificed at day 2, 5, or 10 after UUO and day 14 after unilateral IRI.
Histology and immunofluorescence staining
Mice were anesthetized with isofluorane (Baxter) and subsequently perfused via the left ventricle with 4°C PBS for 1 min. Kidneys from adult mice were fixed with 4% paraformaldehyde (PFA), dehydrated, and embedded in paraffin. Hematoxylin/eosin, PAS, and Masson's trichrome staining were performed using standard protocols (Yang et al, 2010).
Immunofluorescence staining of mouse kidneys was performed on paraffin sections as previously described (Yang et al, 2010). Briefly, the tissue sections were deparaffinized, followed by antigen retrieval and rehydration. Then tissue antigens were labeled with primary antibodies to COUP‐TFII (Abcam, diluted 1: 200), αSMA (Sigma, 1:400), collagen 1 (EMD Millipore, 1:400), CD31 (Abcam 1: 200), PDGF receptor beta (PDGFRβ, Abcam 1:400), or β‐galactosidase (Abcam 1:100), followed by FITC or Cy3‐labeled secondary antibodies (Jackson ImmunoResearch). Some immunostaining was performed on frozen sections. Cryosections (7 μm) were fixed in 4% PFA for 2 h and then washed in 30% sucrose solution overnight. Primary antibodies used in cryosections recognized the following proteins: CD3 (eBioscience 1:100), LyG6, and F4/80 (Thermo Fisher). Images were captured by a confocal (Nikon C1) or standard fluorescent microscope (Nikon TE 1000).
Cell culture and treatment
The C3H/10T1/2 (American Type Culture Collection) were cultured in DMEM medium supplemented with 10% FBS until the cells were 80% confluent. Cell morphology was examined and captured using live cell imaging (Nikon). For induction of myofibroblast differentiation, subconfluent cells were incubated in DMEM medium containing 0.5% FBS overnight, then treated with TGFβ1 (10ng/ml; R&D).
CRISPR/Cas9 knockout
COUP‐TFII guide RNAs were created using a guide design web tool (http://crispr.mit.edu): COUP‐TFII guide 1, TATATCCGGACAGGTACGAG; COUP‐TFII guide 2, GAGGGGGTCCCCGTTGGTCA. sgRNA oligos were cloned into pSpCas9(BB)‐2A‐GFP (Addgene, 48138). The protocol was performed according to published methods (Ran et al, 2013). C3H/10T1/2 cells (5 × 104) were transfected with lipofectamine 3000. Cells were grown for 48 h and then sorted for GFP and positive cells seeded as single cells in 96‐well plates. Cells were then returned to the incubator, and cultures were allowed to expand for 2–3 weeks.
Tet‐inducible COUP‐TFII expression
To generate a lentiviral transfer plasmid for inducible gain‐of‐function experiments, we used high‐fidelity PCR (iProof, Bio‐Rad) to amplify full‐length mouse COUP‐TFII cDNA, flanked with attB1 and attB2 sites on 5′ and 3′ ends, respectively, from a cDNA library from adult mouse kidney. The resulting PCR band was purified from a 0.8% agarose gel using the QIAquick Gel Extraction kit (Qiagen). The purified PCR product was then cloned into pDONR221 (Thermo Fisher Scientific) using BP Clonase II (Invitrogen) according to manufacturer's instructions. We then shuttled the COUP‐TFII cDNA into the destination vector pInducer20 (a gift from Stephen Elledge, Addgene #44010) using LR Clonase II (Invitrogen). All cloning steps were verified using Sanger sequencing (Genewiz, Inc.). C3H/10T1/2 cells were infected with lentivirus in the presence of 10 μg/ml polybrene (Sigma). Infected cells were selected with puromycin (Sigma).
Adenovirus‐mediated PGC1α expression
Adenovirus encoding GFP or tagless mouse PGC1α (PGC1α1 isoform, (Ruas et al, 2012)) was made using the virapower adenoviral expression system (invitrogen) and titered using the adeno‐x rapidtiter kit (clontech). C3H/10T1/2 cells were transduced with an adenovirus at an MOI of 300 overnight.
Cell proliferation assay
Cells were seeded on a 96‐well plate at a concentration of 1 × 104 per well. Three parallel wells of cells were studied for each group. After incubation for 1, 2, or 3 days, 20 μl MTT ((3‐(4,5‐Dimethyl‐2‐thiazolyl)‐2,5‐diphenyltetrazolium Bromide), promega) was added. After 2 h of MTT exposure, cells were washed and subjected for colorimetric measurement. The OD values were obtained by a microplate reader (SpectraMax M5, Molecular Devices) at 570 nm.
Quantitative real‐time PCR (qRT–PCR)
At indicated times, total RNA was extracted using TRIzol (Sigma) as described (Yang et al, 2010). Subsequently, 2 μg of total RNAs was reverse‐transcribed to cDNA with random primers using reverse transcriptase (Invitrogen). A 1:5 dilution of cDNA was then amplified by real‐time qPCR in a CFX96 real‐time system (Bio‐Rad) using SYBR green. Relative gene expression was calculated by the ΔΔCt method, and final results were expressed as the fold difference relative to control conditions in gene expression normalized to Ribosomal Protein L32 (RPL32). Primers for individual gene expression were listed in Table 1.
Table 1.
Primers for qRT–PCR and ChIP‐qPCR.
| Target | Primer | Sequence |
|---|---|---|
| qRT–PCR | ||
| mRPL32 | Forward | GCTGCCATCTGTTTTACGG |
| mRPL32 | Reverse | TGACTGGTGCCTGATGAACT |
| mHexokinase 2 | Forward | CAACTCCGGATGGGACAG |
| mHexokinase 2 | Reverse | CACACGGAAGTTGGTTCCTC |
| mGlut1 | Forward | GCT TCT CCA ACT GGA CCT CAA AC |
| mGlut1 | Reverse | ACG AGG AGC ACC GTG AAG ATG A |
| mLDHA | Forward | ACGCAGACAAGGAGCAGTGGAA |
| mLDHA | Reverse | ATGCTCTCAGCCAAGTCTGCCA |
| mPGC1α | Forward | AGTCCCATACACAACCGCAG |
| mPGC1α_ | Reverse | CCCTTGGGGTCATTTGGTGA |
| mαSMA | Forward | CTGACAGAGGCACCACTGAA |
| mαSMA | Reverse | CATCTCCAGAGTCCAGCACA |
| mCollagen1 | Forward | TGACTGGAAGAGCGGAGAGT |
| mCollagen1 | Reverse | GTTCGGGCTGATGTACCAGT |
| ChIP‐qPCR | ||
| mPGC1α | Forward | TTGCCTCCCCTCCTACCTAC |
| mPGC1α | Reverse | GCATGTTTGCTGGTTGCGTA |
| mPDK4 | Forward | GATCCCGACACGGTTTCCAT |
| mPDK4 | Reverse | AAGTCCTAGCGACCTGGGAT |
| mαSMA | Forward | TGTAGCCGTAGCATCTTGGC |
| mαSMA | Reverse | TAACCAACACACCAGGAGCC |
ChIP assay
Chromatin immunoprecipitation and real‐time PCR quantification were performed as described (Mukhopadhyay et al, 2008). The rabbit polyclonal COUP‐TFII antibody (Millipore, Cat# ABE 1426) and rabbit IgG1 antibody (Zymed) were used for immunoprecipitation. After purification of DNA from C3H/10T1/2 cells, bound sequences were determined by quantitative real‐time PCR (Table 1 for the primer sequences for the ChIP assay).
Western Blot analysis
48 h after transfection, C3H/10T1/2 cells were lysed with 1xRIPA buffer containing a protease inhibitor cocktail (Roche Applied Science) for 30 min on ice. After centrifugation for 15 min, the supernatant was collected, and protein content of the samples was analyzed according to the Bradford method. Proteins were loaded onto SDS–polyacrylamide gels and blotted onto PVDF membrane (Bio‐Rad Laboratories). Western blots were performed using antibodies directed against COUP‐TFII (abcam, 1: 2,000), αSMA (Sigma, 1:4,000), and ERK2 (Cell signaling). HRP‐conjugated secondary antibodies were purchased from DAKO. Enhanced chemiluminescence was performed according to the manufacturer's instructions. ERK1/2 protein was used to ensure equivalent loading of protein samples.
Proteomics
Naïve (WT) and COUP‐TFII overexpression (OE) cells were both treated with TGFβ1 (10 ng/ml) for 48 h to induce myofibroblast differentiation. After trypsinization, cells were washed twice in PBS. Cell pellets were frozen in −80°C for proteomics experiments.
Protein extraction and digest
PBS was removed, followed by the addition of SDS lysis buffer (2% SDS, 150 mM NaCl, 50 mM Tris, pH 8.7) containing protease inhibitors (Complete, Roche). Lysates were homogenized over Qiashredder columns (Qiagen, ref. 79656) and centrifuged at 16,000 g for 1 min at room temperature. Reductive methylation of cysteine residues was performed by adding dithiothreitol (DTT) to a final concentration of 5 mM and heating to 37°C for 1 h, followed by alkylation with iodoacetamide at a final concentration of 15 mM and incubation at room temperature in the dark for 30 min and quenching with DTT. Protein concentration was determined using a Micro BCATM Protein Assay Kit (Thermo Fisher, Catalog# 23235). Detergent was removed by methanol/chloroform protein precipitation as described previously (Wessel & Flugge, 1984). Lys‐C protease digests (Wako, Catalog# 129‐02541) in 2 M urea 20 mM EPPS, pH 8.5 at 37°C for 3 h were followed by further digestion at 37°C for 6 h with trypsin (Promega, Catalog# V5113). Missed cleavage rate was determined by LC‐MS/MS.
Tandem mass tag labeling, ratio check and HPLC fractionation
Equal amounts of protein were removed from each sample and labeled using a TMT11plex Mass Tag Labeling Kit (Thermo Fisher, Catalog# A34808). Tandem mass tag (TMT) labeling efficiency and ratio checks were determined by LC‐MS3 analysis. Quenched TMT labeling reactions were combined and de‐salted using a SepPak tC18 Vac RC Cartridge (50 mg, Waters, Catalog# WAT054960). HPLC fractionation was performed using an Agilent 1200 Series instrument with a flow rate of 600 µl/min over a period of 75 min. Peptides were collected in a 96‐well plate over a 65 min‐gradient of 13–44 %B with Buffer A comprising 5% acetonitrile, 10 mM ammonium bicarbonate, pH 8 and Buffer B comprising 90% acetonitrile, 10 mM ammonium bicarbonate, pH 8. Fractions were then pooled into 24 samples, followed by sample cleanup using the Stage Tip protocol. This protocol uses C18 EmporeTM Extraction Disks (Fisher Scientific, Catalog# 14‐386‐2). Samples were dried before re‐suspension in MS Loading Buffer (3% acetonitrile, 5% FA).
LC‐MS
Peptides were separated over a 30 cm, 100 µm (internal diameter) column using an EASY‐nLC 1200 HPLC system. Samples from the HPLC were injected into an Orbitrap Fusion Lumos Tribrid MS (Thermo Fisher, Catalog# FSN02‐10000) and measured using a multi‐notch MS3 method (Ting et al, 2011; McAlister et al, 2014). MS scans were performed in the Orbitrap over a scan range of 400–1,400 m/z. The top 10 ions with charge states from 2 to 6 were selected for MS/MS. Turbo rate scans were performed in the Ion Trap with a collision energy of 35% and a maximum injection time of 250 ms. TMT quantification was performed using SPS‐MS3 in the Orbitrap with a scan range of 100–1,000 m/z, and an HCD collision energy of 55%. Orbitrap resolution was 50,000 (dimensionless units) with a maximum injection time of 300 ms.
Proteomic data analysis
Raw data were converted to mzXML format and peptide ID used Sequest (Eng et al, 1994) (version 28 (http://fields.scripps.edu/yates/wp/?page_id=17)) with searches against the mouse proteome UniProt database (July 2014). The database search included reversed protein sequences and known contaminants such as keratins that were excluded for subsequent analyses. Linear discriminant analysis was performed (Elias & Gygi, 2007) and peptide false discovery rate (FDR) was < 1% after applying a target‐decoy database search strategy. Filtering was performed as described previously (McAlister et al, 2014). Variable modification for oxidized methionine (+15.99 Da) was used during searches. For protein identification and quantification, shared peptides were collapsed into the minimally sufficient number of proteins using rules of parsimony. Peptides with a total TMT value of > 200 and an isolation specificity of > 0.7 were included for quantification.
Gene set enrichment analysis
We analyzed our proteome datasets from four independent experiments (WT and COUP‐TFII‐OE, all in duplicates) using the GSEA software developed by Broad Institute (Cambridge, MA) (Subramanian et al, 2005). All the analyses were performed in default setting based on all GO terms (c5.all.v7.0.symbols.gmt).
Real‐time cell metabolism assay
Naïve (WT), COUP‐TFII‐OE and COUP‐TFII‐KO C3H/10T1/2 cells were plated in XF‐24 Cell Culture Microplates (Seahorse Bioscience) at a cellular density of 20,000 cells per well, then serum starved for 24 h, and stimulated with or without TGFβ1 (10 ng/ml) for 24 h. Real‐time analysis of extracellular acidification rate (ECAR) was analyzed using XF Extracellular Flux Analyzer (Seahorse Bioscience). The cells were incubated in basal media followed by sequential injections with glucose (10 mM), oligomycin (5 μg/ml), and 2‐DG (50 mM).
Extracellular lactate assays
Naive (WT), COUP‐TFII‐OE, and COUP‐TFII‐KO C3H/10T1/2 cells were grown in a 96‐well plate untill 80% confluent, then serum‐starved for 24 h, and stimulated with or without TGFβ1 (10 ng/ml) for 48 h in 100 μl DMEM media. Extracellular levels of lactate were determined using the lactate assay kit (BioVision, Milpitas, CA) according to the manufacturer's instructions.
Statistical analysis
Results are expressed as mean ± SD. Group means were compared by one‐way analysis of variance (ANOVA), followed by the Tukey post‐test using GraphPad Prism (GraphPad software) for multiple comparisons or by Student's t‐test. A P value < 0.05 was considered statistically significant.
Author contributions
Experiment design: LL and JVB; Experiments: LL, PG, XX, ACF‐R, DT, MK, DG‐S, MSC, NNL, TI, YM, MTV, JW, and DRL; Proteomic data analysis: MK and JJ‐KL; Manuscript writing: LL; Manuscript revision: LL, PG, JVB, ERE, and KWM; Preparation and reading the manuscript: All authors.
Conflict of interest
Dr. Bonventre is cofounder and holds equity in Goldfinch Bio. He is co‐inventor on KIM‐1 and kidney organoid patents assigned to Mass General Brigham. Dr. Bonventre's interests were reviewed and are managed by BWH and Partners HealthCare in accordance with their conflict of interest policies. The other authors declare that they have no conflict of interest.
Supporting information
Review Process File
Acknowledgements
This work was supported by the National Institutes of Health, National Institutes of Diabetes, Digestive and Kidney Diseases (NIDDK) Grants R37DK039773, R01DK072381 and UH3TR002155 (to J.V.B.), National Institute of Biomedical Imaging and Bioengineering (NIBIB), Organ Design and Engineering Training Grant 1T32EB016652, Dialysis Clinics (2019‐06) (to L.L). P.G received support from Monahan Foundation, Fondation pour la Recherche Médicale, Groupe Pasteur Mutualité, Société Francophone de Transplantation, Arthur Sachs fellowship, Philippe Foundation, Fulbright Scholarship, ATIP Avenir program. X.X was supported by China Scholarship Council fellowship. We thank Dr. Edy Kim from Brigham and Women's hospital who provided human idiopathic pulmonary fibrosis samples for immunostaining. We thank Xiaoming Sun for providing technique support on mice kidney injury models. N.N.L. received support from a NIH training grant (T32HL007609).
EMBO reports (2021) 22: e51169.
Contributor Information
Li Li, Email: lli29@bwh.harvard.edu.
Joseph V Bonventre, Email: jbonventre@bwh.harvard.edu.
Data availability
The proteomics data from this publication have been deposited to the PRIDE archive and can be accessed under ProteomeXchange accession number: PXD024035 (http://www.ebi.ac.uk/pride/archive/projects/PXD024035).
References
- Akhurst RJ, Hata A (2012) Targeting the TGFβ signalling pathway in disease. Nat Rev Drug Discov 11: 790–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashraf UM, Sanchez ER, Kumarasamy S (2019) COUP‐TFII revisited: Its role in metabolic gene regulation. Steroids 141: 63–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avagliano A, Granato G, Ruocco MR, Romano V, Belviso I, Carfora A, Montagnani S, Arcucci A (2018) Metabolic reprogramming of cancer associated fibroblasts: the slavery of stromal fibroblasts. Biomed Res Int 2018: 6075403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Y, Gu D, Feng W, Sun X, Wang X, Zhang X, Shi Q, Cui G, Yu H, Tang C et al (2014) COUP‐TFII regulates metastasis of colorectal adenocarcinoma cells by modulating Snail1. Br J Cancer 111: 933–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basak T, Vega‐Montoto L, Zimmerman LJ, Tabb DL, Hudson BG, Vanacore RM (2016) Comprehensive characterization of glycosylation and hydroxylation of basement membrane collagen IV by high‐resolution mass spectrometry. J Proteome Res 15: 245–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonventre JV, Yang L (2011) Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest 121: 4210–4221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkkoetter PT, Bork T, Salou S, Liang W, Mizi A, Özel C, Koehler S, Hagmann HH, Ising C, Kuczkowski A et al (2019) Anaerobic glycolysis maintains the glomerular filtration barrier independent of mitochondrial metabolism and dynamics. Cell Rep 27: 1551–1566.e1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceni E, Mello T, Polvani S, Vasseur‐Cognet M, Tarocchi M, Tempesti S, Cavalieri D, Beltrame L, Marroncini G, Pinzani M et al (2017) The orphan nuclear receptor COUP‐TFII coordinates hypoxia‐independent proangiogenic responses in hepatic stellate cells. J Hepatol 66: 754–764 [DOI] [PubMed] [Google Scholar]
- Chen X, Qin J, Cheng CM, Tsai MJ, Tsai SY (2012) COUP‐TFII is a major regulator of cell cycle and Notch signaling pathways. Mol Endocrinol 26: 1268–1277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H, Jiang L, Xu J, Bai F, Zhou Y, Yuan Q, Luo J, Zen K, Yang J (2017) Inhibiting aerobic glycolysis suppresses renal interstitial fibroblast activation and renal fibrosis. Am J Physiol Renal Physiol 313: F561–F575 [DOI] [PubMed] [Google Scholar]
- Duffield JS, Lupher M, Thannickal VJ, Wynn TA (2013) Host responses in tissue repair and fibrosis. Annu Rev Pathol 8: 241–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffield JS (2014) Cellular and molecular mechanisms in kidney fibrosis. J Clin Invest 124: 2299–2306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumesic PA, Egan DF, Gut P, Tran MT, Parisi A, Chatterjee N, Jedrychowski M, Paschini M, Kazak L, Wilensky SE et al (2019) An evolutionarily conserved uORF regulates pgc1α and oxidative metabolism in mice, flies, and Bluefin tuna. Cell Metab 30: 190–200.e196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias JE, Gygi SP (2007) Target‐decoy search strategy for increased confidence in large‐scale protein identifications by mass spectrometry. Nat Methods 4: 207–214 [DOI] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, Yates JR (1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom 5: 976–989 [DOI] [PubMed] [Google Scholar]
- Falke LL, Gholizadeh S, Goldschmeding R, Kok RJ, Nguyen TQ (2015) Diverse origins of the myofibroblast‐implications for kidney fibrosis. Nat Rev Nephrol 11: 233–244 [DOI] [PubMed] [Google Scholar]
- Fang X, Liu CX, Zeng XR, Huang XM, Chen WL, Wang Y, Ai F (2020) Orphan nuclear receptor COUP‐TFII is an oncogenic gene in renal cell carcinoma. Clin Transl Oncol 22: 772–781 [DOI] [PubMed] [Google Scholar]
- Han SH, Wu MY, Nam BY, Park JT, Yoo TH, Kang SW, Park J, Chinga F, Li SY, Susztak K (2017) PGC‐1alpha protects from notch‐induced kidney fibrosis development. J Am Soc Nephrol 28: 3312–3322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson J, Duffy L, Stratton R, Ford D, O'Reilly S (2020) Metabolic reprogramming of glycolysis and glutamine metabolism are key events in myofibroblast transition in systemic sclerosis pathogenesis. J Cell Mol Med 24: 14026–14038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou W, Syn WK (2018) Role of metabolism in hepatic stellate cell activation and fibrogenesis. Front Cell Dev Biol 6: 150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphreys BD, Valerius MT, Kobayashi A, Mugford JW, Soeung S, Duffield JS, McMahon AP, Bonventre JV (2008) Intrinsic epithelial cells repair the kidney after injury. Cell Stem Cell 2: 284–291 [DOI] [PubMed] [Google Scholar]
- Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, Valerius MT, McMahon AP, Duffield JS (2010) Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol 176: 85–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphreys BD (2018) Mechanisms of renal fibrosis. Annu Rev Physiol 80: 309–326 [DOI] [PubMed] [Google Scholar]
- Im MJ, Freshwater MF, Hoopes JE (1976) Enzyme activities in granulation tissue: energy for collagen synthesis. J Surg Res 20: 121–125 [DOI] [PubMed] [Google Scholar]
- Jha V, Garcia‐Garcia G, Iseki K, Li Z, Naicker S, Plattner B, Saran R, Wang AY, Yang CW (2013) Chronic kidney disease: global dimension and perspectives. Lancet 382: 260–272 [DOI] [PubMed] [Google Scholar]
- Kale S, Hanai J, Chan B, Karihaloo A, Grotendorst G, Cantley L, Sukhatme VP (2005) Microarray analysis of in vitro pericyte differentiation reveals an angiogenic program of gene expression. FASEB J 19: 270–271 [DOI] [PubMed] [Google Scholar]
- Kang HM, Ahn SH, Choi P, Ko Y‐A, Han SH, Chinga F, Park ASD, Tao J, Sharma K, Pullman J et al (2015) Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med 21: 37–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingberg F, Hinz B, White ES (2013) The myofibroblast matrix: implications for tissue repair and fibrosis. J Pathol 229: 298–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi A, Mugford JW, Krautzberger AM, Naiman N, Liao J, McMahon AP (2014) Identification of a multipotent self‐renewing stromal progenitor population during mammalian kidney organogenesis. Stem Cell Rep 3: 650–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottmann RM, Kulkarni AA, Smolnycki KA, Lyda E, Dahanayake T, Salibi R, Honnons S, Jones C, Isern NG, Hu JZ et al (2012) Lactic acid is elevated in idiopathic pulmonary fibrosis and induces myofibroblast differentiation via pH‐dependent activation of transforming growth factor‐beta. Am J Respir Crit Care Med 186: 740–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramann R, Schneider RK, DiRocco DP, Machado F, Fleig S, Bondzie PA, Henderson JM, Ebert BL, Humphreys BD (2015) Perivascular Gli1+ progenitors are key contributors to injury‐induced organ fibrosis. Cell Stem Cell 16: 51–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan R, Geng H, Singha PK, Saikumar P, Bottinger EP, Weinberg JM, Venkatachalam MA (2016) Mitochondrial pathology and glycolytic shift during proximal tubule atrophy after ischemic AKI. J Am Soc Nephrol 27: 3356–3367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos DR, McMurdo M, Karaca G, Wilflingseder J, Leaf IA, Gupta N, Miyoshi T, Susa K, Johnson BG, Soliman K et al (2018) Interleukin‐1beta activates a MYC‐dependent metabolic switch in kidney stromal cells necessary for progressive tubulointerstitial fibrosis. J Am Soc Nephrol 29: 1690–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA (2006) Transforming growth factor‐beta regulation of immune responses. Annu Rev Immunol 24: 99–146 [DOI] [PubMed] [Google Scholar]
- Li L, Xie X, Qin J, Jeha GS, Saha PK, Yan J, Haueter CM, Chan L, Tsai SY, Tsai MJ (2009) The nuclear orphan receptor COUP‐TFII plays an essential role in adipogenesis, glucose homeostasis, and energy metabolism. Cell Metab 9: 77–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SL, Kisseleva T, Brenner DA, Duffield JS (2008) Pericytes and perivascular fibroblasts are the primary source of collagen‐producing cells in obstructive fibrosis of the kidney. Am J Pathol 173: 1617–1627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Edgington‐Giordano F, Dugas C, Abrams A, Katakam P, Satou R, Saifudeen Z (2017) Regulation of nephron progenitor cell self‐renewal by intermediary metabolism. J Am Soc Nephrol 28: 3323–3335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR et al (2010) A robust and high‐throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13: 133–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister GC, Nusinow DP, Jedrychowski MP, Wuhr M, Huttlin EL, Erickson BK, Rad R, Haas W, Gygi SP (2014) MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal Chem 86: 7150–7158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng XM, Huang XR, Xiao J, Chung AC, Qin W, Chen HY, Lan HY (2012) Disruption of Smad4 impairs TGF‐beta/Smad3 and Smad7 transcriptional regulation during renal inflammation and fibrosis in vivo and in vitro . Kidney Int 81: 266–279 [DOI] [PubMed] [Google Scholar]
- Morizane R, Lam AQ, Freedman BS, Kishi S, Valerius MT, Bonventre JV (2015) Nephron organoids derived from human pluripotent stem cells model kidney development and injury. Nat Biotechnol 33: 1193–1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay A, Deplancke B, Walhout AJ, Tissenbaum HA (2008) Chromatin immunoprecipitation (ChIP) coupled to detection by quantitative real‐time PCR to study transcription factor binding to DNA in Caenorhabditis elegans . Nat Protoc 3: 698–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa S, Nishihara K, Miyata H, Shinke H, Tomita E, Kajiwara M, Matsubara T, Iehara N, Igarashi Y, Yamada H et al (2015) Molecular markers of tubulointerstitial fibrosis and tubular cell damage in patients with chronic kidney disease. PLoS One 10: e0136994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Paz‐Lugo P, Lupianez JA, Melendez‐Hevia E (2018) High glycine concentration increases collagen synthesis by articular chondrocytes in vitro: acute glycine deficiency could be an important cause of osteoarthritis. Amino Acids 50: 1357–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecot CV, Rupaimoole R, Yang Da, Akbani R, Ivan C, Lu C, Wu S, Han H‐D, Shah MY, Rodriguez‐Aguayo C et al (2013) Tumour angiogenesis regulation by the miR‐200 family. Nat Commun 4: 2427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira FA, Qiu Y, Zhou G, Tsai MJ, Tsai SY (1999) The orphan nuclear receptor COUP‐TFII is required for angiogenesis and heart development. Genes Dev 13: 1037–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard N, Baum O, Vogetseder A, Kaissling B, Le Hir M (2008) Origin of renal myofibroblasts in the model of unilateral ureter obstruction in the rat. Histochem Cell Biol 130: 141–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinney DF, Emerson CP Jr (1989) 10T1/2 cells: an in vitro model for molecular genetic analysis of mesodermal determination and differentiation. Environ Health Perspect 80: 221–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planchais J, Boutant M, Fauveau V, Qing LD, Sabra‐Makke L, Bossard P, Vasseur‐Cognet M, Pegorier JP (2015) The role of chicken ovalbumin upstream promoter transcription factor II in the regulation of hepatic fatty acid oxidation and gluconeogenesis in newborn mice. Am J Physiol Endocrinol Metab 308: E868–E878 [DOI] [PubMed] [Google Scholar]
- Polvani S, Tarocchi M, Tempesti S, Mello T, Ceni E, Buccoliero F, D'Amico M, Boddi V, Farsi M, Nesi S et al (2014) COUP‐TFII in pancreatic adenocarcinoma: clinical implication for patient survival and tumor progression. Int J Cancer 134: 1648–1658 [DOI] [PubMed] [Google Scholar]
- Principe DR, Doll JA, Bauer J, Jung B, Munshi HG, Bartholin L, Pasche B, Lee C, Grippo PJ (2014) TGF‐beta: duality of function between tumor prevention and carcinogenesis. J Natl Cancer Inst 106: djt369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proctor G, Jiang T, Iwahashi M, Wang Z, Li J, Levi M (2006) Regulation of renal fatty acid and cholesterol metabolism, inflammation, and fibrosis in Akita and OVE26 mice with type 1 diabetes. Diabetes 55: 2502–2509 [DOI] [PubMed] [Google Scholar]
- Qin J, Wu SP, Creighton CJ, Dai F, Xie X, Cheng CM, Frolov A, Ayala G, Lin X, Feng XH et al (2013) COUP‐TFII inhibits TGF‐beta‐induced growth barrier to promote prostate tumorigenesis. Nature 493: 236–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabelink TJ, Carmeliet P (2018) Renal metabolism in 2017: Glycolytic adaptation and progression of kidney disease. Nat Rev Nephrol 14: 75–76 [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR‐Cas9 system. Nat Protoc 8: 2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangarajan S, Kurundkar A, Kurundkar D, Bernard K, Sanders YY, Ding Q, Antony VB, Zhang J, Zmijewski J, Thannickal VJ (2016) Novel mechanisms for the antifibrotic action of nintedanib. Am J Respir Cell Mol Biol 54: 51–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockey DC, Bell PD, Hill JA (2015) Fibrosis–a common pathway to organ injury and failure. N Engl J Med 372: 1138–1149 [DOI] [PubMed] [Google Scholar]
- Romagnani P, Remuzzi G, Glassock R, Levin A, Jager KJ, Tonelli M, Massy Z, Wanner C, Anders HJ (2017) Chronic kidney disease. Nat Rev Dis Primers 3: 17088 [DOI] [PubMed] [Google Scholar]
- Ruas J, White J, Rao R, Kleiner S, Brannan K, Harrison B, Greene N, Wu J, Estall J, Irving B et al (2012) A PGC‐1α isoform induced by resistance training regulates skeletal muscle hypertrophy. Cell 151: 1319–1331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer IR, Joshi M (2020) CPT1A‐mediated fat oxidation, mechanisms, and therapeutic potential. Endocrinology 161: 1–14 [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha Vk, Mukherjee S, Ebert Bl, Gillette Ma, Paulovich A, Pomeroy Sl, Golub Tr, Lander Es et al (2005) Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamoto N, You LR, Moses K, Chiang C, Zimmer WE, Schwartz RJ, DeMayo FJ, Tsai MJ, Tsai SY (2005) COUP‐TFII is essential for radial and anteroposterior patterning of the stomach. Development 132: 2179–2189 [DOI] [PubMed] [Google Scholar]
- Ting L, Rad R, Gygi SP, Haas W (2011) MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat Methods 8: 937–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA (2002) Myofibroblasts and mechano‐regulation of connective tissue remodelling. Nat Rev Mol Cell Biol 3: 349–363 [DOI] [PubMed] [Google Scholar]
- Tran MT, Zsengeller ZK, Berg AH, Khankin EV, Bhasin MK, Kim W, Clish CB, Stillman IE, Karumanchi SA, Rhee EP et al (2016) PGC1alpha drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 531: 528–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessel D, Flugge UI (1984) A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal Biochem 138: 141–143 [DOI] [PubMed] [Google Scholar]
- Wilson PC, Wu H, Kirita Y, Uchimura K, Ledru N, Rennke HG, Welling PA, Waikar SS, Humphreys BD (2019) The single‐cell transcriptomic landscape of early human diabetic nephropathy. Proc Natl Acad Sci USA 116: 19619–19625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S‐P, Kao C‐Y, Wang L, Creighton CJ, Yang J, Donti TR, Harmancey R, Vasquez HG, Graham BH, Bellen HJ et al (2015) Increased COUP‐TFII expression in adult hearts induces mitochondrial dysfunction resulting in heart failure. Nat Commun 6: 8245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Kirita Y, Donnelly EL, Humphreys BD (2019) Advantages of single‐nucleus over single‐cell RNA sequencing of adult kidney: rare cell types and novel cell states revealed in fibrosis. J Am Soc Nephrol 30: 23–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA, Ramalingam TR (2012) Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med 18: 1028–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie N, Tan Z, Banerjee S, Cui H, Ge J, Liu RM, Bernard K, Thannickal VJ, Liu G (2015) Glycolytic reprogramming in myofibroblast differentiation and lung fibrosis. Am J Respir Crit Care Med 192: 1462–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Tsai SY, Tsai MJ (2016) COUP‐TFII regulates satellite cell function and muscular dystrophy. J Clin Invest 126: 3929–3941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV (2010) Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med 16: 535–543. 531p following 143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zank DC, Bueno M, Mora AL, Rojas M (2018) Idiopathic pulmonary fibrosis: aging, mitochondrial dysfunction, and cellular bioenergetics. Front Med (Lausanne) 5: 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Hulver MW, McMillan RP, Cline MA, Gilbert ER (2014) The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility. Nutr Metab 11: 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Psarianos P, Ghoraie LS, Yip K, Goldstein D, Gilbert R, Witterick I, Pang H, Hussain A, Lee JH et al (2019) Metabolic regulation of dermal fibroblasts contributes to skin extracellular matrix homeostasis and fibrosis. Nat Metab 1: 147–157 [DOI] [PubMed] [Google Scholar]
- Zhao X, Kwan JYY, Yip K, Liu PP, Liu FF (2020) Targeting metabolic dysregulation for fibrosis therapy. Nat Rev Drug Discov 19: 57–75 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Review Process File
Data Availability Statement
The proteomics data from this publication have been deposited to the PRIDE archive and can be accessed under ProteomeXchange accession number: PXD024035 (http://www.ebi.ac.uk/pride/archive/projects/PXD024035).