

Abstract
Severe cases of SARS‐CoV‐2 infection are characterized by hypercoagulopathies and systemic endotheliitis of the lung microvasculature. The dynamics of vascular damage, and whether it is a direct consequence of endothelial infection or an indirect consequence of an immune cell‐mediated cytokine storm remain unknown. Using a vascularized lung‐on‐chip model, we find that infection of alveolar epithelial cells leads to limited apical release of virions, consistent with reports of monoculture infection. However, viral RNA and proteins are rapidly detected in underlying endothelial cells, which are themselves refractory to apical infection in monocultures. Although endothelial infection is unproductive, it leads to the formation of cell clusters with low CD31 expression, a progressive loss of barrier integrity and a pro‐coagulatory microenvironment. Viral RNA persists in individual cells generating an inflammatory response, which is transient in epithelial cells but persistent in endothelial cells and typified by IL‐6 secretion even in the absence of immune cells. Inhibition of IL‐6 signalling with tocilizumab reduces but does not prevent loss of barrier integrity. SARS‐CoV‐2‐mediated endothelial cell damage thus occurs independently of cytokine storm.
Keywords: alveolar models, COVID‐19, interleukin‐6, organ‐on‐chip, vasculitis
Subject Categories: Immunology; Microbiology, Virology & Host Pathogen Interaction; Molecular Biology of Disease
In a human lung‐on‐chip model maintained at an air‐liquid interface exposed to SARS‐CoV‐2, endothelial lung microvascular cells are infected via alveolar epithelial cells, leading to persistent endothelial cell inflammation and loss of barrier integrity.
Introduction
Organ‐on‐chip technologies recreate key aspects of human physiology in a bottom‐up and modular manner (Bhatia & Ingber, 2014). In the context of infectious diseases, this allows for studies of cell dynamics (Villenave et al, 2017; Thacker et al, 2020), infection tropism (Ortega‐Prieto et al, 2018) and the role of physiological factors in disease pathogenesis in more native settings (Grassart et al, 2019). This is particularly relevant for the study of respiratory infectious diseases (Torrelles & Schlesinger, 2017), where the vast surface area of the alveoli poses a challenge to direct experimental observation.
COVID‐19, caused by the novel betacoronavirus SARS‐CoV‐2, first manifests as an infection of the upper airways. Severe cases are marked by progression into the lower airways and alveoli. Here, it manifests as an atypical form of acute respiratory distress syndrome (ARDS) characterized by good lung compliance measurements (Gattinoni et al, 2020; Marini & Gattinoni, 2020), elevated levels of coagulation markers such as D‐dimers (Yao et al, 2020), and pro‐inflammatory markers in the blood (Messner et al, 2020). Autopsy reports show numerous microvascular thrombi in the lungs of deceased patients together with evidence of the intracellular presence of the virus in vascular cells (Ackermann et al, 2020; Carsana et al, 2020). These reports suggest that infection of and alterations to lung microvasculature play a key role in COVID‐19 pathogenesis (Lang et al, 2020; Teuwen et al, 2020), which is further reinforced by the early observation of endothelial cell damage, platelet aggregation and the formation of microthrombi in an ante‐mortem study (preprint: Claudio et al, 2020). Yet, most in vitro studies have focused on monocultures of upper airway respiratory cells. In studies with alveolar epithelial cells, SARS‐CoV‐2 has been shown to replicate poorly both in the A549 lung adenocarcinoma cell line (Blanco‐Melo et al, 2020) and in the primary alveolar epithelial cells ex vivo (Hui et al, 2020) and has been reported to be unable to infect primary lung microvascular endothelial cells (Conde et al, 2020; Hou et al, 2020), which are at odds with the reported medical literature. There is therefore an urgent need for models that will help us develop a better understanding of the pathogenesis of SARS‐CoV‐2 in the human alveolar space and understand how this may be linked to the coagulatory phenotypes reported both in the clinic and in non‐human primate models. The lung‐on‐chip model is well suited to this purpose because it includes a vascular compartment maintained under flow and infection can occur at the air–liquid interface(Nawroth et al, 2020)—two key physiological features that are lacking in organoid models (Huang et al, 2020; preprint: Abo et al, 2020; Youk et al, 2020). We therefore establish a human lung‐on‐chip model for SARS‐CoV‐2 infections, and probe the viral growth kinetics, cellular localization, and responses to a low‐dose infection using qRT–PCR, ELISA, RNAscope, immunofluorescence, and confocal imaging (Appendix Fig S1A).
Results
SARS‐CoV‐2 infection of a human lung‐on‐chip alters expression of viral entry factors
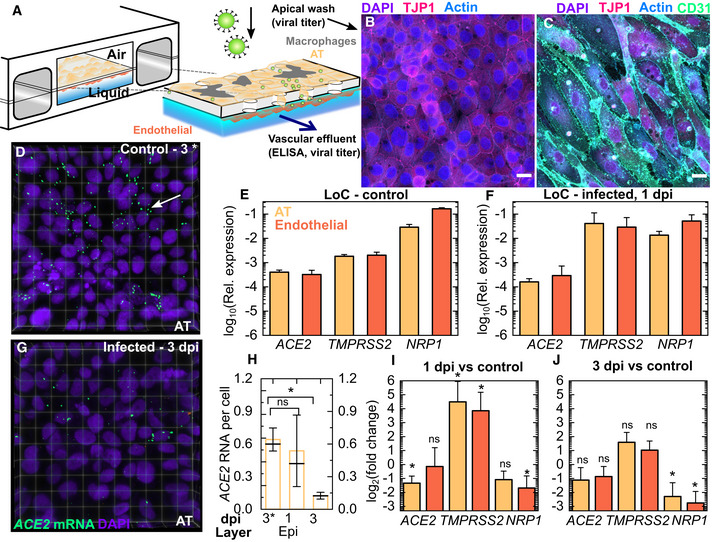
A human lung‐on‐chip (LoC) model for SARS‐CoV‐2 pathogenesis (Fig 1A) mimics the alveolar space using primary human alveolar epithelial cells (ATs) and lung microvascular endothelial cells (“endothelial”), which form confluent monolayers on the apical and vascular sides of the porous membrane in the chip (Fig 1B and C). The modular nature of the technology allowed us to recreate otherwise identical LoCs either without (“w/o”) (Fig 1B) or with the addition of CD14+ macrophages (representative image in Appendix Fig S1B) to the AT layer on the apical side of the chip. The latter configuration was used for a subset of experiments as indicated throughout the text. We first characterized the ATs and endothelial cells used, both in monoculture and on‐chip at the air–liquid interface to verify that the cells mimicked human alveolar physiology. In ATs, ACE2 expression was low both in monoculture (Fig EV1A) and on‐chip (Fig 1E) consistent with transcriptomic (Hou et al, 2020; Qi et al, 2020) and proteomic (Hikmet et al, 2020) analyses of human tissue. Expression on‐chip was not significantly different from monoculture (Fig EV1B). Low ACE2 expression was also observed from RNAscope assays on uninfected LoCs (Fig 1D). However, ACE2 expression was particularly low in endothelial cells in monoculture (Fig EV1A). Expression increased 10‐fold on‐chip (Fig EV1B) so that levels in ATs and endothelial cells were comparable (Fig 1E). This upregulation in ACE2 expression could possibly be due to the effects of shear stress and flow in the vascular channel (Song et al, 2020). Neuropilin‐1 (NRP1), an integrin‐binding protein(Valdembri et al, 2009), has recently been reported to be an alternative receptor for SARS‐CoV‐2 entry (Cantuti‐Castelvetri et al, 2020; Daly et al, 2020) that is highly expressed in pulmonary microvascular cells. Consistent with these reports, NRP1 expression was higher than ACE2 expression in both cell types in monoculture (Fig EV1A) and on‐chip (Fig 1E). On‐chip, NRP1 expression was 6‐fold higher in endothelial cells compared with ATs (Fig 1E). Lastly, the mature ATs retained differentiation in type I and type II cells, which we verified by immunostaining for type II (pro‐SPC) and type I (Podoplanin) markers in monocultures (Appendix Fig S2A–C), as well as through qRT–PCR measurements for type II (ABCA3, SFTPC) and type I (AQP5, PDPN, CAV1) markers in cells from the AT layer of uninfected LoCs (Appendix Fig S2D). Endothelial cells showed characteristic expression of platelet endothelial cell adhesion molecule (PECAM‐1 or CD31) at cell junctions (Fig 1C).
Figure 1. SARS‐CoV‐2 infection in a human lung‐on‐chip model.
-
A–C(A) Schematic of the LoC model for SARS‐CoV‐2. Maximum‐intensity projections show that confluent layers of alveolar epithelial cells (ATs) and endothelial cells with strong expression of tight junction markers populate the top (B) and bottom faces (C) of the porous membrane. Actin, CD31, TJP‐1 and nuclear labelling are shown in azure, spring green, bright pink and electric indigo LUTs, respectively.
-
DACE2 levels were characterized using RNAscope. A representative 3D view of a 232 × 232 µm2 field of view of the AT layer of an uninfected control at 3 days at air–liquid interface (3C) is shown. The nuclear labelling is indicated in electric indigo, and ACE2 mRNA is labelled in spring green LUT.
-
E, FqRT–PCR characterization of the AT and endothelial layers on‐chip for SARS‐CoV‐2 entry markers (ACE2, TMPRSS2 and NRP1) for cells extracted from the apical and vascular channels from uninfected LoCs (n = 3 biological replicates) (E) and infected LoCs (n = 4 biological replicates) at 1 day postinfection (F). In both cases, expression is normalized to GAPDH levels.
-
GSame as in (D) with infected LoC at 3 dpi (G) shown.
-
HACE2 expression decreases between 1 and 3 dpi in the LoC model, quantified on a per‐cell basis for n = 4 or 5 fields of view (technical replicates) across control and infected chips (n = 1 control and n = 1 infected chip) at each timepoint.
-
I, JA plot of the fold change in expression of host proteins required for viral entry in the AT and endothelial layer of LoCs at 1 day postinfection (n = 4 biological replicates) and 3 days postinfection (n = 3 biological replicates) versus uninfected control LoCs (n = 3 biological replicates).
Data information: The bars represent the mean value, the solid line represents the median value, and the error bars represent the standard deviation. P‐values are calculated using a Kruskal–Wallis one‐way ANOVA test, * represents P ≤ 0.05, ** represents P ≤ 0.01, and *** represents P ≤ 0.001. Scale bar = 20 µm.
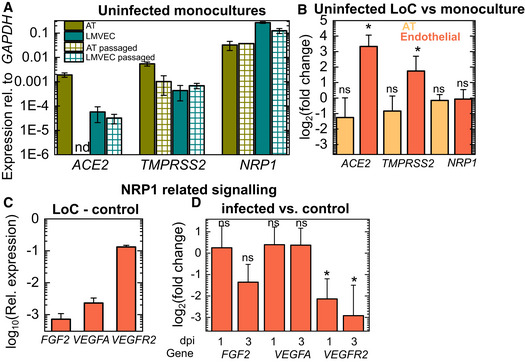
Figure EV1. Characterization of ACE2 and NRP1 expression on‐chip versus monocultures and alterations in NRP1‐related signalling upon infection.
-
APlots of expression of the cell receptors ACE2 and NRP1, and the protease TMPRSS2 relative to GAPDH expression for alveolar epithelial cells (ATs) obtained from a commercial supplier at passage 3, alveolar epithelial cells postpassage in the lab (“AT passaged”), freshly isolated lung microvascular endothelial cells from a commercial supplier (“LMVEC”) and lung microvascular endothelial cells postpassage (“LMVEC passaged”). “nd” refers to not detected. Data obtained from n = 2 technical replicates from n = 2 biological replicates.
-
BPlot of the fold change in expression of viral entry factors in cells from the epithelial and endothelial layers of uninfected control LoC (n = 3 biological replicates) versus monocultures (n = 2 biological replicates for ATs and n = 3 biological replicates for endothelial cells).
-
CPlot of the expression of basic fibroblast growth factor (FGF2), vascular endothelial growth factor A (VEGFA) and vascular endothelial growth factor receptor 2 (VEGFR2) relative to GAPDH from the endothelial layer of uninfected control LoCs (n = 3 biological replicates).
-
DPlot of the fold change in expression of FGF2, VEGFA and VEGFR2 in the endothelial layer of infected LoCs at 1 dpi (n = 3 biological replicates) and infected LoCs (n = 3 biological replicates) at 3 dpi versus uninfected controls (n = 3 biological replicates).
Data information: In all plots, the bars represent the mean values, and the error bars represent the standard deviation. P‐values are calculated using a one‐way Kruskal–Wallis ANOVA test, * represents P ≤ 0.05, ** represents P ≤ 0.01, and ** represents P ≤ 0.001.
A low‐dose infection (200‐300 plaque‐forming units (PFUs), multiplicity of infection (MOI)=0.003) of the apical surface of the LoC resulted in significant decreases in ACE2 expression in ATs and NRP1 expression and endothelial cells at 1 dpi (Fig 1F and I). At 3 dpi, expression of both cell surface receptors remained downregulated although only downregulation of NRP1 was statistically significant (Fig 1J). These results are consistent with observations using RNAscope assays, where ACE2 levels in the AT layer declined over subsequent days (Fig 1D, G and H, p = 0.014). On the other hand, infection resulted in increased expression of the TMPRSS2 protease (Hoffmann et al, 2020) required to cleave the spike protein for viral entry at 1 dpi in both cell types relative to uninfected controls (Fig 1F and I), with variable expression levels across chips. This trend was also maintained at 3 dpi (Fig 1J).
NRP1 has important roles in VEGF signalling in endothelial cells as a co‐receptor with vascular endothelial growth factor receptor 2 (VEGFR2) for vascular endothelial growth factor A (VEGFA) (Guo & Vander Kooi, 2015). We therefore examined the effect of downregulation of NRP1 expression on these pathways. At 1 dpi, expression of ligands (FGF2, VEGFA) for NRP1 was not significantly different from uninfected controls, with only a small decrease in VEGFA at 3 dpi (Fig EV1D, data for uninfected controls in Fig EV1C). Expression of VEGFR2, highly expressed in uninfected controls (Fig EV1C), was significantly lower at 1 dpi, and expression diminished further at 3 dpi (Fig EV1D). These observations are consistent with reports of the effect of NRP1 knockout on VEGFR2 levels (Gelfand et al, 2014), and overall show that both ACE2 and NRP1 likely play a role in SARS‐CoV‐2 pathogenesis in the alveolar space, aided by increased TMPRSS2 levels. Infection did not alter levels of type II or type I alveolar epithelial cell markers significantly (Appendix Fig S2E and F).
Infection of the alveolar space is characterized by a lack of productive infection, slow intracellular replication and transmission to the endothelial layer
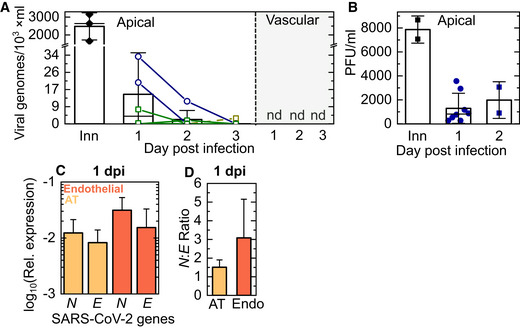
We first characterized the progression of infection by measuring the release of viral genomes via qRT–PCR and the number of infectious virions released via measurement of plaque‐forming units (PFUs). Infected LoCs were monitored daily for the release of infected viral progeny (i) apically—on the AT layer, and (ii) in the cell culture media flowed through the vascular channel (‘apical wash” and “vascular effluent” in Fig 1A). A low number of viral genomes were released apically from the AT layer, and the number of genomes detected decreased over 1–3 dpi (Fig 2A). Genome copy numbers were 100‐fold lower than the starting inoculum (between 200 and 300 PFUs). Low numbers of infectious virions were also detected via measurements of PFU from eight LoCs w/o macrophages at 1 dpi and two LoCs w/o macrophages at 2 dpi (Fig 2B). These results showed that, on the whole, infection of the AT cell layer did not lead to a large amplification of infectious virions, although instances of small numbers of AT cells with high levels of spike protein (Appendix Fig S4A and C–E) or viral genomic RNA (Fig EV4) were detected via microscopy and likely contribute to the low levels of infectious virions released.
Figure 2. Viral replication but lack of release of infectious virions characterizes SARS‐CoV‐2 infection in the LoC model.
-
A, BThe kinetics of release of SARS‐CoV‐2 progeny was assessed from samples obtained from a wash of the apical face of the chip (A, B), and the vascular effluent (A) collected at each day postinfection. (A) The number of viral genomes in each sample was quantified using a one‐step qRT–PCR assay for the SARS‐CoV‐2 N gene. The starting inoculum introduced to the apical side (labelled “Inn”) corresponded to 200‐300 PFU. Lines join datapoints from the same LoC, reconstituted with (green squares) and without (dark blue circles) macrophages. Viral genome numbers within the apical wash showed a declining trend over 3 days of infection. Viral genomes are not detected (“nd”) in the vascular effluent for all the chips shown. (B) Measurements of plaque‐forming units (PFUs) from the apical wash of LoCs reconstituted without macrophages (n = 8 biological replicates for 1 dpi, n = 2 biological replicates for 2 dpi) confirm the presence of infectious virions.
-
CQuantification of the intracellular levels of transcripts for the SARS‐CoV‐2 N and SARS‐CoV‐2 E genes in the AT and endothelial layers of infected LoCs at 1 dpi (n = 3 biological replicates) relative to expression of the eukaryotic housekeeping gene GAPDH.
-
DSARS‐CoV‐2N: SARS‐CoV‐2 E ratio in the AT and endothelial layers of the infected LoCs shown in (C), n = 3 biological replicates.
Data information: In all plots, the bars represent the mean value, and the error bars represent the standard deviation, the solid black line in (A) represents the median.
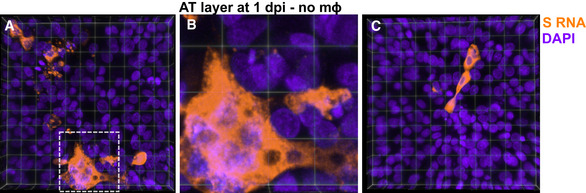
Figure EV4. RNAscope images of highly infected AT cells at 1 dpi.
-
A–C3D views of two representative 232 × 232 µm2 fields of view of the epithelial layer of an LoC reconstituted without macrophages at 1 dpi. S RNA is identified by RNAscope assay and false‐coloured in amber, and nuclear labelling is false‐coloured in indigo. Each field of view shows examples of heavily infected cells. (B) Zoom corresponding to the area marked by the white box in (A), a collection of heavily infected cells is visible together with syncytia formation.
No viral genomes were detected in the vascular effluent (Fig 2B), and no infectious virions were obtained from the effluent of two LoCs with and w/o macrophages by PFU assays. This ruled out the possibility of direct release of virions from the AT cell layer into the vascular channel and suggested that either dissemination of virions to the endothelial cell layer did not occur or that infection of the endothelial layer could occur, but no viral genomes or infectious virions were released apically into the vascular channel by endothelial cells.
To address this, we further quantified intracellular viral RNA loads from the total RNA extracted from cells of the apical and vascular channels of three infected LoCs w/o macrophages at 1 dpi. This revealed a significant number of viral transcripts in cells from both the AT and endothelial layers (Fig 2C, Appendix Fig S3C). Additional quantification of intracellular viral RNA from an infected LoC w/o macrophage using a SARS‐CoV‐2‐specific one‐step qRT–PCR kit revealed > 104 genomes in both ATs and endothelial cells (Appendix Fig S3A); genome copy numbers exceeded those for cellular housekeeping gene RNAseP (Appendix Fig S3B). Similar numbers of intracellular viral genome copy numbers have been reported for infections of ATs in monoculture (Hui et al, 2020), and these numbers are modest in comparison with cells such as airway epithelial cells and Vero E6 cells where SARS‐CoV‐2 replicates productively (Chu et al, 2020). The difference in replication kinetics can also be appreciated from the fact that whereas the N gene is most abundant during productive infection in Vero E6 cells (Kim et al, 2020), we obtained a ratio of SARS‐CoV‐2N to SARS‐CoV‐2 E < 5 (Fig 2D, Appendix Fig S3D), consistent with the observation of low numbers of extracellular infection virions (Fig 2B).
The detection of SARS‐CoV‐2 genomes in the endothelial layer within 1 dpi suggests rapid dissemination through basolateral transfer from the AT layer to the endothelial layer can occur. Although basolateral transmission has not been reported previously to be the significant mode of transmission for both SARS‐CoV‐2 (V’kovski et al, 2020) and SARS‐CoV‐1 (Tseng et al, 2005) infections of monocultures of upper airway cells at the air–liquid interface, this could reflect aspects of infection unique to ATs, or, more generally, of infection of cells where productive infection is rare.
SARS‐CoV‐2 infection of endothelial cells on‐chip but not in monoculture alters cell morphologies
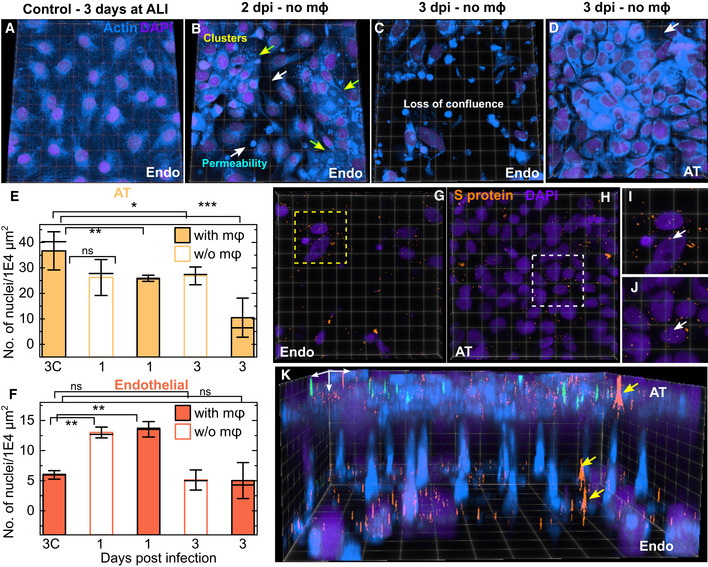
Recently published autopsy reports of COVID‐19 patients revealed features of exudative alveolar damage (Carsana et al, 2020), and a rapid loss of alveolar epithelial cell barrier integrity could account for the basolateral transfer to endothelial cells. The LoC model is compatible with microscopy‐based assays, which allowed us to assess changes in cellular physiology with high spatial resolution using confocal microscopy. Staining for F‐actin localization revealed striking changes to the morphology of cells in the vascular channel. In comparison with the confluent layer of cells in an uninfected control LoC (3D view of the epithelial layer in Fig 3A and of the endothelial layer in Fig EV2A), infected LoCs showed profound alterations in endothelial layer architecture (Fig 3B and C). At 2 dpi, cellular clusters characterized by an increased cell density and stronger nucleic acid staining (Fig 3B, yellow arrows) coexisted with areas with normal nucleic acid staining levels but reduced cell‐to‐cell contact (Fig 3B, white arrows). By 3 dpi, a significant loss of cell confluency was observed (Figs 3C and EV2B–D), with large cell clusters no longer visible. In contrast, even at 3 dpi, the AT layer maintained a relatively high degree of confluency (Figs 3D and EV2E), with only a few signs of cell death (Figs 3D and EV2E, white arrows). To quantify these changes across multiple areas of the chip, we enumerated the number of nuclei per unit area of the membrane surface for infected LoCs at 1 and 3 dpi and uninfected controls. In the AT layer, the density of nuclei declines progressively due to infection irrespective of the presence (P = 0.010, P = 9E‐4) or absence (P = 0.054, P = 0.029) of macrophages compared with uninfected controls (Fig 3E). In contrast, the density of cellular nuclei increases in the endothelial layer at 1 dpi before decreasing back to levels in the control samples at 3 dpi (Fig 3F), consistent with our observation of the transient formation of endothelial cell clusters. As a further measure of quantification, we measured the proportion of the surface area of the endothelial cell layer with low or no actin staining using a range of intensity thresholds (Fig EV2F). At 3 dpi, a much larger proportion of the surface area of the endothelial layer of infected LoCs had low or no actin staining compared with an uninfected control for all intensity thresholds (Fig EV2F).
Figure 3. SARS‐CoV‐2 infection rapidly disrupts confluency of the endothelial layer.
-
A–D3D views of representative 232 × 232 µm2 (A, B) and 155 × 155 µm2 (C, D) fields of view from confocal imaging of an uninfected control (A), and infected chips without macrophages at days 2 (B) and 3 (C, D) postinfection, respectively. Actin and nuclear labelling are shown in azure and indigo, respectively. (B) At 2 dpi, adjacent areas of endothelial cell clusters identified by a stronger nuclear stain and brighter actin staining (yellow arrows) and cells with increased permeability and loss of confluence (white arrows) are indicated. (C) At 3 dpi, a significant loss of barrier integrity is observed in the endothelial layer, whereas the AT layer directly above this field of view (D) is relatively intact. A small area of missing cells in the AT layer is indicated with a white arrow in (D).
-
E, FPlots of density of nuclei (number of nuclei per 1E4 µm2) on the AT (E) and endothelial (F) faces of infected LoCs both with and without macrophages for characterization of the cellular layer integrity. The bars represent the mean value, the solid line represents the median, and the error bars represent the standard deviation from at least 4 fields of view each from two independent LoCs for each timepoint. Data from uninfected controls corresponding to the 3 dpi timepoint are labelled “3C”.
-
G–JThe fields of view in (C, D) are shown also in (G, H), respectively, SARS‐CoV‐2 spike (S) proteins labelled via immunofluorescence are false‐coloured in amber. Zooms in (I) and (J) show clear evidence of intracellular spike protein within both endothelial and ATs, respectively.
-
KA 3D view of the fields of view in (C, D) and (G, H), respectively, highlights the relative damage to the endothelial layer versus the epithelium, as well as the basolateral transmission of S protein via junctions in the pores of the membrane (identified with yellow arrows). The white arrows define the XYZ axes.
Data information: P‐values are calculated using a one‐way Kruskal–Wallis ANOVA test, * represents P ≤ 0.05, ** represents P ≤ 0.01, and *** represents P ≤ 0.001.
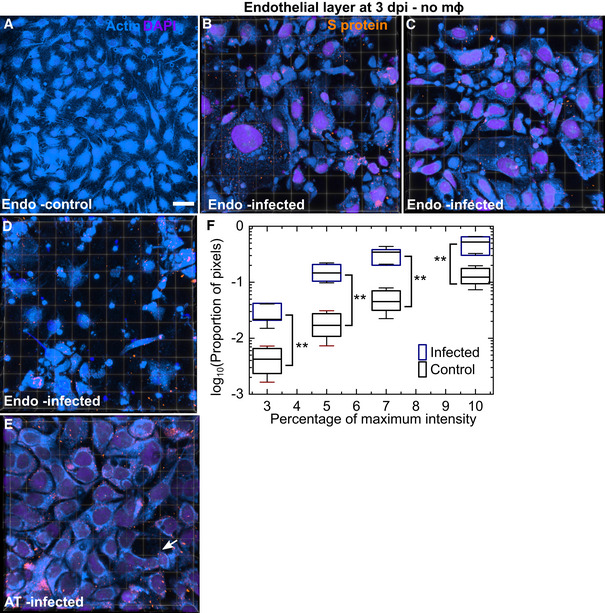
Figure EV2. Further characterization of the disruption of endothelial cell layer integrity.
-
AA maximum‐intensity projection of a 465 × 465 µm2 field of view of the endothelial layer in an uninfected chip at 3 days post‐air–liquid interface shows intact vasculature. Scale bar = 50 µm.
-
B–DAdditional examples of endothelial damage—3D views of 232 × 232 µm2 fields of view from LoCs infected without macrophages at 3 dpi. S protein identified via antibody labelling, actin and nuclear labelling is shown in amber, azure and electric indigo LUTs, respectively. Many areas with little or no actin staining are observed.
-
EAn additional example of a 3D view of a 155 × 155 µm2 field of view of the epithelial layer from the same LoC shown in (B–D). The integrity of the layer is better maintained, and cell loss is indicated by a white arrow.
-
FA plot of the proportion of pixels (shown on a logarithmic scale) with regions of low F‐actin intensity identified via cut‐off thresholds defined as a percentage of the maximum intensity in 4–5 fields of view (technical replicates) each for one control and infected LoC. The solid bar represents the median, the boxes represent the 25–75 percentile range, and the whiskers represent the standard deviation. P‐values are calculated using a one‐way Kruskal–Wallis ANOVA test, * represents P ≤ 0.05, ** represents P ≤ 0.01, and ** represents P ≤ 0.001.
Immunostaining for the viral S protein at 3 dpi (Fig 3G for endothelial layer and Fig 3H for AT layer, additional examples in Fig EV2B–D and E and controls with secondary antibody only in Fig EV3, EV4, EV5) showed additional evidence of individual infected ATs (Fig 3I) and endothelial cells (Fig 3J). In some cases, S proteins appear to co‐localize with the periphery of the nucleus (Fig 3I and J, white arrows). The transfer of viral proteins from the apical to vascular channel and difference in confluency between the AT and endothelial layers at 3 dpi can be better appreciated from a 3D view of the two channels together with the connecting pores in the PDMS membrane; S protein can be clearly localized to the interface of the two cell types in the pores of the membrane (yellow arrows, Fig 3K). Thus, ATs and endothelial cells exhibit very different physiological responses to low levels of infection, and vascular damage by 3 dpi was a consistent observation across all infected LoCs (n = 12 biological replicates) maintained until 3 dpi in this study.
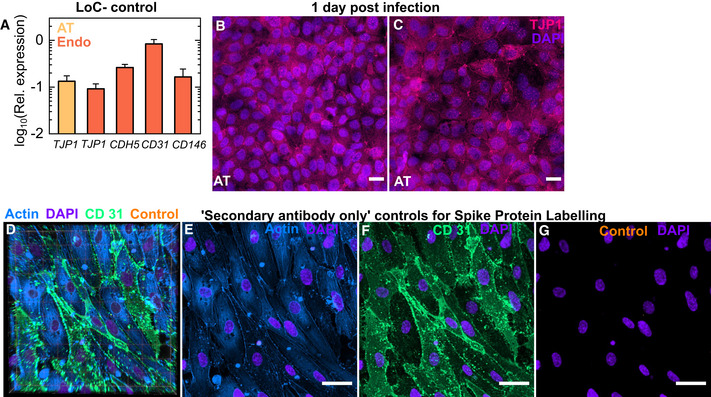
Figure EV3. Characterization of the endothelial cell monolayer on‐chip.
-
APlot of the expression of the tight junction markers PECAM‐1 (CD31), VE‐Cadherin (CDH5) and melanoma cell adhesion molecule MCAM (CD 146) in cells from the endothelial layer and of ZO‐1 (TJP1) in cells from both the endothelial and epithelial layers of uninfected controls (n = 3 biological replicates) relative to GAPDH expression. The bars represent the mean, and the error bars represent the standard deviation.
-
B, CMaximum‐intensity projection of two additional fields of view from the epithelial layer of an infected LoC reconstituted without macrophages at 1 dpi. TJP1 identified via antibody labelling and nuclear labelling is shown in bright pink and electric indigo LUTs, respectively.
-
D–GSecondary antibody only controls for spike protein labelling. (D) 3D view of a 116.36 × 116.36 µm2 field of view of the endothelial layer of an uninfected control LoC. Actin, CD31, secondary antibody only (“control”) and nuclear labelling are indicated in azure, spring green, amber and electric indigo LUTs, respectively. (E–G) Maximum‐intensity projections of actin (E), CD31 (F) and control staining (G) for the same field of view. Nuclear labelling is shown in all panels.
Data information: Scale bar = 20 µm.
Figure EV5. Quantification of levels of viral antisense RNA, genomic RNA and ACE2 mRNA via RNAscope per field of view.

-
A–DQuantification of viral antisense RNA (A, B), viral genomic RNA (C, D) and ACE2 mRNA from pairs of otherwise identical LoCs reconstituted without (A, C, E) and with macrophages (B, D) and analysed at 1 and 3 dpi. Plots show the cumulative number of spots imaged per field of view from 4 to 6 fields of view (technical replicates) detected using RNAscope and confocal imaging using identical imaging conditions for all chips.
-
ESARS‐CoV‐2 infection reduces ACE2 expression per field of view in both epithelial cells and endothelial cells by 3 dpi in LoCs reconstituted without macrophages. Data obtained from 4 to 6 fields of view (technical replicates) from one LoC at each timepoint.
Data information: Bars represent the mean value, the solid line represents the median, and error bars represent the standard deviation. P‐values are calculated using a Kruskal–Wallis one‐way ANOVA test, * represents P ≤ 0.05, ** represents P ≤ 0.01, and ** represents P ≤ 0.001.
We next sought to understand whether these responses were specific to the lung‐on‐chip physiology and co‐culture, and so examined infections of ATs and endothelial cells in monoculture. At 1 dpi, heavily infected ATs in monoculture with 1,000 PFU showed similar levels and intracellular distributions of spike protein (Appendix Fig S4B). In stark contrast, monoculture infections of endothelial cells with viral inocula up to 100‐fold greater than the apical inoculum used in the LoC experiments (Fig 2A) showed few foci of viral S proteins. Endothelial cells retained their morphology and confluency (Appendix Fig S5) up to 2 dpi, in agreement with recent reports of the inability of apical inoculations of SARS‐CoV‐2 to infect lung microvascular endothelial cells (Hou et al, 2020). This suggests that the site of contact of SARS‐CoV‐2 with endothelial cells (apical versus basolateral) may play an important role—a physiological factor that can be recapitulated in the LoC but not in simpler models.
SARS‐CoV‐2‐infected endothelial cells lose expression of tight junction markers and adopt a pro‐coagulatory phenotype
Actin staining definitively demonstrated the vascular damage caused as a result of loss of cell confluence, we therefore probed cells from both layers in infected and uninfected LoCs for changes in expression of tight junction proteins that might have caused these changes. Maximum‐intensity projections from the AT layer of an uninfected control showed strong expression of both actin (Fig 4A) and zonula occludens‐1 (ZO‐1 or TJP1) at the cell junctions (Fig 4B). Similarly, images from a confluent endothelial cell layer in an uninfected control LoC showed actin staining correlated with high expression of TJP1 (Fig 4C) and high expression of the endothelial cell‐specific marker CD31 at cell junctions (Fig 4D). These results were corroborated by qRT–PCR measurement of high levels of expression of TJP1 and endothelial cell‐specific markers CD31, VE‐cadherin (CDH5) and melanoma cell adhesion molecule (CD146) in uninfected controls (Fig EV3A).
Figure 4. SARS‐CoV‐2 infection leads to a loss of tight junctions, generation of CD31‐ endothelial cell clusters and a pro‐coagulatory environment.
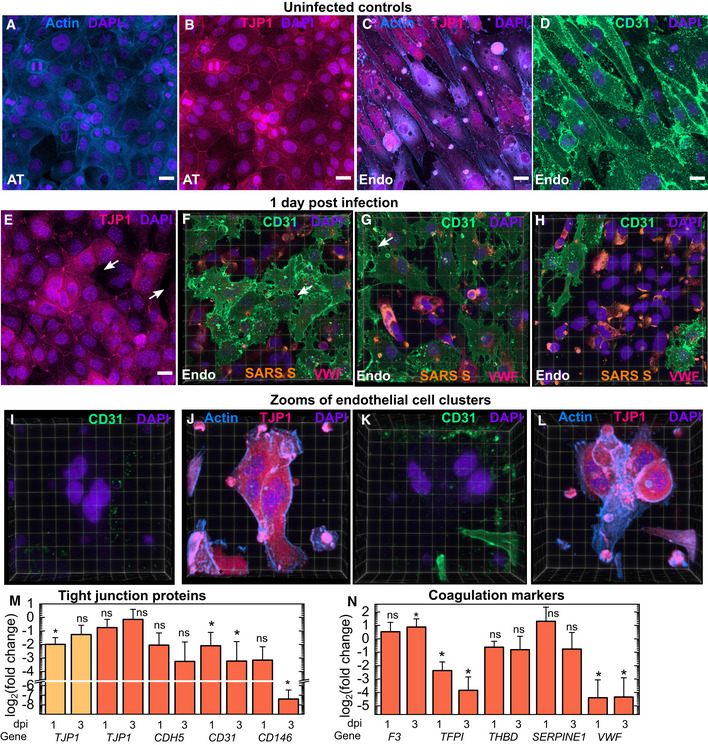
-
A–DMaximum‐intensity projects of 232 × 232 µm2 fields of view from the AT (A, B) and endothelial layers of an uninfected control LoC. ATs show strong actin (A) and TJP1 expression (B, identified via antibody staining) at the cell junction boundaries. Endothelial cells also show high levels of TJP‐1 expression (C) together with CD31 expression at cell junctions (D, identified via antibody labelling). Actin, TJP‐1, CD31 and nuclear labelling are indicated with the azure, bright pink, spring green and electric indigo LUTs, respectively.
-
E–H3D views of one 232 × 232 µm2 field of view of the AT layer (E) and (F‐H) three 232 × 232 µm2 fields of view of the endothelial layer of an infected LoC at 1 dpi. Areas with lower TJP1 expression in the AT layer in (E) are indicated by white arrows. (F‐H) Areas with loss of tight junctions between cells on the endothelial surface are indicated by white arrows. The proportion of the surface area covered by confluent cells decreases sequentially from (F) to (H). Endothelial cell clusters with low CD31 expression are clearly visible, and the proportion of the surface area covered by these clusters increases sequentially from (F) to (H). In (F‐H), spike protein is identified via an anti‐SARS‐spike antibody and shown in amber LUT, and von Willebrand Factor identified by antibody labelling is shown in bring pink LUT.
-
I–LZooms of two endothelial cell clusters (I, J and K, L) in the endothelial layer of another infected LoC at 1 dpi confirm that little or no CD31 expression is detected (I, K) despite high levels of actin and TJP‐1 protein expression in these cells. (J, L).
-
MPlot of the fold change in expression of TJP1 (AT and endothelial layers) and CDH5, CD31 and CD146 (endothelial layers only) in infected LoCs at 1 and 3 dpi (n = 3 biological replicates in each case) versus uninfected controls (n = 3 biological replicates).
-
NPlot of the fold change in gene expression of endothelial cell‐specific coagulation markers—tissue factor (F3), tissue factor pathway inhibitor (TFPI), thrombomodulin (THBD), plasminogen activator inhibitor (SERPINE1) and von Willebrand factor (VWF) in infected LoCs at 1 and 3 dpi (n = 3 biological replicates in each case) versus uninfected controls (n = 3 biological replicates).
Data information: In all plots, bars represent the mean value and error bars represent the standard deviation. Scale bar = 20 µm. P‐values are calculated using a one‐way Kruskal–Wallis ANOVA test, * represents P ≤ 0.05, ** represents P ≤ 0.01, and *** represents P ≤ 0.001.
Consistent with Fig 3, changes to the expression pattern of TJP1 in the AT layer at 1 dpi were subtle, TJP1 expression at cell junctions continued to be observed across much of the AT cell layer (Fig 4E, additional images in Fig EV3B and C). SARS‐CoV‐2 infection, on the other hand, profoundly altered CD31 expression and distribution in the endothelial layer. 3D views of regions of interest in the endothelial layer of an infected LoC showed this to be a dynamic process that resulted in two significant changes: first, an increase in vascular permeability from the loss of tight junctions between CD31+ cells (white arrows in Fig 4F and G), and second, the presence of cells that showed little or no CD31 expression (Fig 4F–H). Cells displaying the “CD31‐ phenotype” typically had higher levels of spike protein (shown in amber in Fig 4F and G), and an increased local cell density and nuclear labelling consistent with the endothelial cell clusters identified in Fig 3. This link is strengthened by examination of zooms of two endothelial cell clusters (Fig 4I–L) from another LoC at 1 dpi. This confirmed that cells in both clusters were CD31‐ (Fig 4I and K) but nevertheless express TJP1 and showed strong and altered actin distributions (Fig 4J and L), morphological features consistent with the clusters identified in independent experiments in Fig 3. These changes are further corroborated by qRT–PCR measurements, which show a progressive and statistically significant reduction in overall levels of CD31 in the endothelial layer at 1 dpi and 3 dpi, respectively, and CD146 at 3 dpi compared with uninfected controls (Fig 4M). CDH5 expression in the endothelial layer at 1 dpi and 3 dpi, respectively, was also lower compared with uninfected controls (Fig 4M). In contrast, the reduction in TJP1 expression at 1 dpi is significant only in ATs at 1 dpi and expression levels do not decline further at 3 dpi.
These dramatic changes in expression of tight junction proteins also resulted in series of complex alterations to the expression of endothelial cell proteins with direct roles in coagulatory pathways (Fig 4N). For example, infection modestly increased expression of pro‐coagulatory tissue factor (F3) released by endothelial cells as part of the extrinsic coagulation pathway by 3 dpi (Zelaya et al, 2018), but induced a progressive and significant decrease in the expression of anti‐coagulatory tissue factor pathway inhibitor (TFPI) (Mast, 2016). A more modest decline was also observed for the expression of thrombomodulin (THBD) that also has an anti‐coagulatory function. Although infection also decreased the expression of von Willebrand factor (VWF), elevated levels of VWF protein were observed on the surface of the CD31‐ endothelial cells at 1 dpi via immunofluorescence (bright pink, Fig 4F and G). In contrast, expression of plasminogen inhibitor activation (PAI‐1) expressed by SERPINE1 was not significantly altered. Taken together with the loss of CD31 expression, these results point to the generation of a pro‐coagulatory environment in the vascular channel of infected chips.
SARS‐CoV‐2 persists in individual alveolar epithelial and endothelial cells in the alveolar space
Our results thus far showed that infection on‐chip itself leads to heterogeneous outcomes that were strongly cell‐type‐specific. We therefore performed a series of RNAscope assays with confocal microscopy to probe the spatial distribution and cell‐to‐cell variability in gene expression at the single‐cell level to complement observations from qRT–PCR measurements. We began by examining levels of viral RNA and antisense RNA generated during viral replication in situ using probes for S RNA and ORF1AB antisense RNA, respectively. Representative images for 232 × 232 µm2 fields of view from the apical and vascular channels of chips at 1 and 3 dpi are shown in Fig 5A–H; negative controls are shown in Appendix Fig S6. At 1 dpi, a few examples of heavily infected cells are visible on the AT layer (Fig EV4, EV5) and viral genomic RNA can be detected throughout the cytoplasm. Consistent with the observations in Appendix Fig S4, such cells are likely to be the source of infectious virions released in the apical wash in Fig 2A and B. However, in the vast majority of cells in the AT layer, the virus is detected only at low levels at 1 dpi (Fig 5A), and infection does not appear to be limited by (Fig 5C, white arrows) or even correlated to cellular ACE2 expression at the time of fixation (Fig 5D, yellow arrows). Both genomic and antisense RNA are detected in endothelial cells at 1 dpi (Fig 5B), indicating that intracellular viral replication can also occur in these cells. Heavily infected endothelial cells were not observed, in agreement with the lack of infectious virions in the vascular effluent (Fig 2C). Endothelial cell clusters are clearly identifiable by their morphological features, and zoomed‐in images (Fig 5E and F) show that viral infection of endothelial cells with normal and clustered morphology. As with the AT layer, instances of localization of viral RNA with the nucleus are also observed (Fig 5F, white arrowheads). At 3 dpi, infection in both layers (Fig 5G and H) and the loss of endothelial layer integrity are evident (compare Fig 5G versus Fig 5H). We did not detect heavily infected AT cells at 3 dpi, in agreement with the diminishing viral titre (Fig 2A).
Figure 5. RNAscope analysis reveals intracellular localization and slows accumulation of intracellular viral RNA in the LoC model.
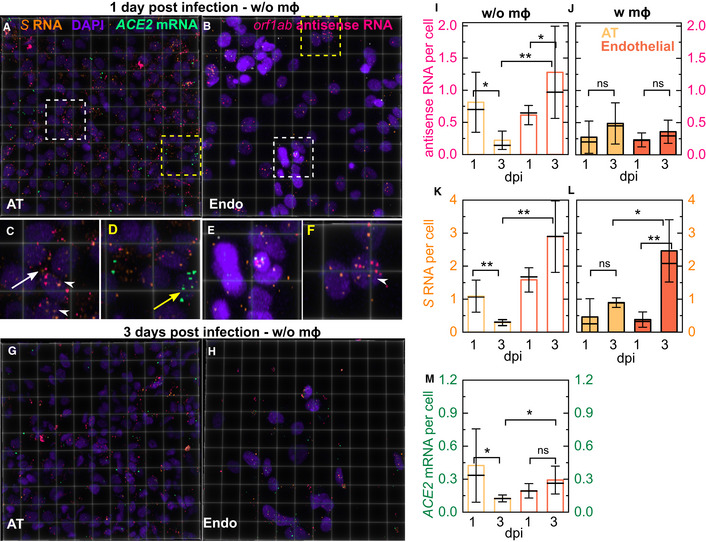
-
A–H3D views of 232 × 232 µm2 fields of view of the AT (A, G) and endothelial layer (B, H) from infected LoCs reconstituted without macrophages at 1 (A, B) and 3 dpi (G, H). Orf1ab antisense RNA, S RNA, ACE2 mRNA and nuclear staining with DAPI are false‐coloured in pink, amber, spring green and electric indigo, respectively. (C, D) Zooms corresponding to the region in (A) highlighted with white and yellow boxes, respectively. (C) An example of infection and intracellular replication in a cell with no detectable ACE2 expression (white arrow) and nuclear localization of viral RNA (white arrowhead). (D) An example of an uninfected cell with ACE2 mRNA expression (yellow arrow). (E, F) Zooms corresponding to the regions in (B) representing increased nucleic acid staining endothelial cell clusters and normal levels of nucleic acid staining highlighted with white and yellow boxes, respectively. (E) Examples of infection of clustered endothelial cells both with and without ACE2 expression. (F) Examples of endothelial cell infection with no ACE2 expression, nuclear localization of viral RNA in an infected endothelial cell is indicated (white arrowhead).
-
I–LQuantification of viral antisense RNA (I, J) and viral genomic RNA (K, L) from pairs (n = 2 biological replicates) of otherwise identical LoCs reconstituted without (I, K) and with macrophages (J, L) analysed at 1 and 3 dpi. Plots show the number of spots normalized by the total number of cells in 4–6 fields of view (technical replicates) detected using RNAscope and confocal imaging using identical imaging conditions for all chips.
-
MQuantification of ACE2 expression per cell in ATs and endothelial cells from 4 to 6 fields of view (technical replicates) in a pair of LoCs reconstituted without macrophages (n = 2 biological replicates) analysed at 1 and 3 dpi, respectively.
Data information: Bars represent the mean value, the solid line represents the median, and error bars represent the standard deviation. P‐values are calculated using a Kruskal–Wallis one‐way ANOVA test, * represents P ≤ 0.05, ** represents P ≤ 0.01, and *** represents P ≤ 0.001.
We normalized the number of RNA dots detected either on a per‐cell basis (Fig 5I–K) or per‐field‐of‐view basis (Fig EV5), to enable quantification of differences between the two layers while accounting for differences in cell numbers and cell death. The median values of intensities were similar across all conditions (Appendix Fig S7). In the AT layer, viral infection and replication per cell were either similar (Fig 5J and L) or lower by 3 dpi (Fig 5I, P = 0.018; Fig 5K, P= 0.006). In contrast, viral RNA accumulates in individual endothelial cells (Fig 5K and L, P = 0.004). The presence of macrophages generally leads to lower levels of viral RNA replication (Fig 5I and J) but does not prevent the accumulation of viral genomes intracellularly in endothelial cells (Fig 5L). At 3 dpi, ACE2 levels are higher on a per‐cell basis in endothelial versus AT cells (Fig 5M, P = 0.028), due to a reduction in ACE2 levels in the AT layer.
Normalization on a per‐field‐of‐view basis shows predominantly a declining trend for levels of replication in ATs (Fig EV5A and B) and intracellular viral RNA (Fig EV5C and D) in both cell types, as well as a decline in ACE2 expression (Fig EV5E), consistent with qRT–PCR measurements (Fig 1J). These results highlight the cumulative effect of differential cell numbers and cell death in these two layers (Fig 3E and F). Overall, virus levels are low and decline in both layers by 3 dpi, yet the virus persists and continues to replicate within individual infected cells, in agreement with reports of clinical infection.
Persistent NF‐KB inflammatory responses to SARS‐CoV‐2 infection in endothelial but not alveolar epithelial cells
Concurrently, we investigated host responses to the infection. Expression levels of NF‐KB‐related pro‐inflammatory genes (TNFA, IL6 and IL1B) at 1 dpi was an order of magnitude higher than that of interferon genes in both cell types, with little to no interferon stimulatory gene expression detected in both cell types (Fig 6A). Responses were also cell‐type‐specific; endothelial cells from infected LoCs showed significant upregulation of TNFA, IL6 and IFNB and more modest increases in IFNL1 and IFNL3 expression with greater variability between LoCs, whereas only expression of IFNB in AT cells was higher than uninfected controls (Fig 6B). In both cell types, expression of IP10 was significantly lower than uninfected controls, consistent with the action of a number of SARS‐CoV‐2 proteins in inhibiting type I interferon signalling (Miorin et al, 2020; Xia et al, 2020).
Figure 6. SARS‐CoV‐2 infection generates a persistent pro‐inflammatory response in endothelial cells.
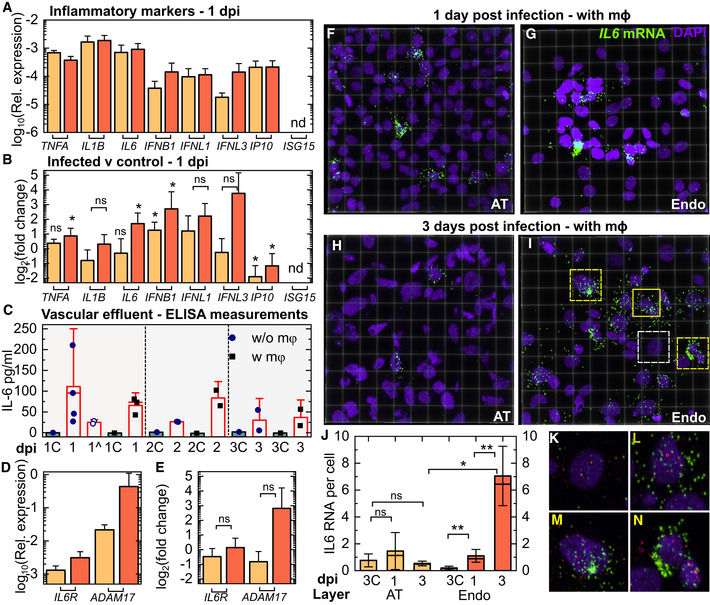
-
AExpression relative to GAPDH of pro‐inflammatory cytokines (TNFA, IL1B and IL6), interferon genes (IFNB, IFNL1 and IFNL3) and interferon‐stimulating genes (IP10 and ISG15) in the AT and endothelial layer of infected LoCs at 1 dpi reconstituted without macrophages (n = 3 biological replicates for IFNB, IFNL1 and IFNL3 in the AT layer, n = 4 biological replicates for all other data).
-
BFold change in the markers in (A) relative to uninfected controls (n = 3 biological replicates) (data in Appendix Fig S8) at the same timepoint. The bar represents the mean, and the error bars, the standard deviation.
-
CEnzyme‐linked immunosorbent assay (ELISA) measurements for IL‐6 in the vascular effluent for LoCs reconstituted with and without macrophages (at least n = 2 biological replicates at each timepoint). Uninfected controls for each condition are labelled with a “C” and grey bars, and LoCs with tocilizumab administration are indicated by ^. The height of the bars represents the mean, the median is represented by a solid line, and the error bars represent the standard deviation. Each dot represents the mean of two technical replicates.
-
DExpression of IL6R and ADAM17 relative to GAPDH in the AT and endothelial layer of infected LoCs at 1 dpi (n = 3 biological replicates for the AT layer and n = 4 biological replicates for endothelial layer).
-
EFold change in expression of IL6 and ADAM17 in the epithelial and endothelial layers of infected LoCs at 1 dpi relative to uninfected controls (n = 3 biological replicates for both layers).
-
F–I3D views of representative 232 × 232 µm2 fields of view of the AT (F, H) and endothelial layer (G, I) from infected LoCs reconstituted with macrophages at 1 (F, G) and 3 dpi (H, I). IL6 mRNA and nuclear staining are indicated with the chartreuse and electric indigo LUTs, respectively.
-
JQuantification of IL6 expression in ATs and endothelial cells from a pair of otherwise identical LoCs (n = 2 biological replicates) analysed at 1 and 3 dpi, respectively. Plots show the total number of spots normalized by the number of cells in 4–6 fields of view (technical replicates) detected using RNAscope assay using identical imaging conditions for all chips. Bars represent the mean value, the solid line represents the median, and error bars represent the standard deviation. Data from uninfected controls for 3 days at air–liquid interface are indicated by “C”.
-
K–NZooms corresponding to the regions in (I) highlighted with white (K) and yellow boxes with solid (L) and dashed lines (M, N), respectively. In these panels, orf1abantisense RNA (pink) and S RNA (amber) are also shown. The panels show examples of cells with similar levels of viral infection but with no (K), intermediate (L) and high levels (M, N) of IL6 expression.
Data information: The bar represents the mean, and the error bars, the standard deviation. P‐values are calculated using a Kruskal–Wallis one‐way ANOVA test, * represents P ≤ 0.05, ** represents P ≤ 0.01, and *** represents P ≤ 0.001.
The LoC platform also allows the simultaneous detection of secreted cytokines in the vascular effluent from the same chips. ELISAs for IL‐1B and IP10 did not detect these cytokines in the effluent, and the lack of IL‐1B secretion despite high expression (Fig 6B) suggests that it is not exported (Lopez‐Castejon & Brough, 2011). IL‐6, on the other hand, was consistently detected up to 3 dpi in the vascular effluent from LoCs both in the presence and in the absence of macrophages (Fig 6C). No IL‐6 was detected in the vascular effluent from uninfected controls. IL‐6 levels were not significantly different between these two categories of LoCs, which strongly suggested a non‐immune cell source for this cytokine (Fig 6C).
Given the pleiotropic nature of IL‐6 (Tanaka et al, 2014a), we also examined expression of the IL‐6R receptor and the metallopeptidase ADAM17, which sheds the TNF‐alpha receptor and the IL‐6R receptor (Zunke & Rose‐John, 2017) in both the AT and endothelial layer at 1 dpi (Fig 6D). Expression of ADAM17, which has been shown to enhance vascular permeability (Dreymueller et al, 2012), was on average 7‐fold higher in the endothelial layer at 1 dpi (Fig 6E), suggestive of the activation of the IL‐6 signalling pathway, although expression was highly variable between infected LoCs.
COVID‐19 patients with severe infection have elevated IL‐6 levels, and we therefore sought to examine the cell‐type‐specific IL6 expression at the single‐cell resolution. We therefore used RNAscope to probe the spatial distribution of IL6 expression in LoCs reconstituted with macrophages. Consistent with the qRT–PCR data (Appendix Fig S8), baseline IL6 expression in the control LoC is higher in the AT layer (Appendix Fig S9A) than in the endothelial layer (Appendix Fig S9B), and immunostaining with an anti‐CD45 antibody confirmed that the IL6 expression in the AT layer of the chip did not co‐localize with macrophages (Appendix Fig S9A and B). At 1 dpi, individual cells with higher levels of IL6 expression were observed in both AT and endothelial layers (Fig 6F and G), but only differences in expression in the endothelial layer were statistically significant (Fig 6J). Cells with high IL6 expression were also unlikely to be macrophages (Appendix Fig S9C–E).
By 3 dpi, IL6 expression in the AT layer had returned to the control levels (Fig 6H and J, Appendix Fig S10A), whereas expression increased dramatically and was more widespread in the endothelial layer (Fig 6I and J, Appendix Fig S10A and B). Expression did not appear to correlate with level of infection—zooms of cells with comparable levels of infection in Fig 6I with low (Fig 6K), medium (Fig 6L) and high (Fig 6M and N) IL6 expression are shown. Expression in the endothelial layer at 3 dpi was higher than that in the endothelial layer at 1 dpi, the AT layer at 3 dpi, and the uninfected controls, both on a per‐cell and on a per‐field‐of‐view basis (Fig 6J, Appendix Fig S10A, P = 0.004, P = 0.014, P = 0.008, respectively). Unlike viral RNA levels (Fig EV5B and D), IL6 expression in the endothelial layer does not diminish over 1‐3 dpi and would therefore appear to be the major contributor to IL‐6 secretion in the vascular effluent as infection progresses.
Inhibition of trans IL‐6 signalling reduces but does not eliminate endothelial cell inflammation
The activation of IL‐6 signalling observed suggested trans IL‐6 signalling as a potential target to ameliorate the vascular inflammation observed. A similar rationale has led to numerous clinical trials of the anti‐IL‐6R monoclonal antibody tocilizumab (Tanaka et al, 2014b) as a repurposed therapeutic for COVID‐19. Tocilizumab administration at 56 µg/ml via continuous perfusion over 2 days postinfection per se did not abrogate IL‐6 secretion, consistent with its mode of action (Fig 6C). We therefore undertook an investigation of cell morphologies in the endothelial layer of a treated and untreated LoC via actin staining. This revealed that whereas endothelial cell confluency was, on the whole, better retained in the treated chip (Fig 7A, zooms in Fig 7B, untreated control in Fig 7D, zoom in Fig 7E), tocilizumab administration did not prevent the occurrence of endothelial cell clusters (Fig 7A, zooms in Fig 7C, untreated control in Fig 7D, zoom in Fig 7F). Consistent with these results, immunostaining for CD31 in another treated LoC showed an improvement in the integrity of the endothelial cell layer with tight junctions observable at 2 dpi (3D views in Fig 7G and H), yet endothelial cell clusters with reduced CD31 expression and strong actin staining were nevertheless formed (3D views in Fig 7G and H, indicated by yellow arrows).
Figure 7. Tocilizumab treatment reduces vascular permeability but does not prevent formation of endothelial cell clusters with reduced CD31 expression.
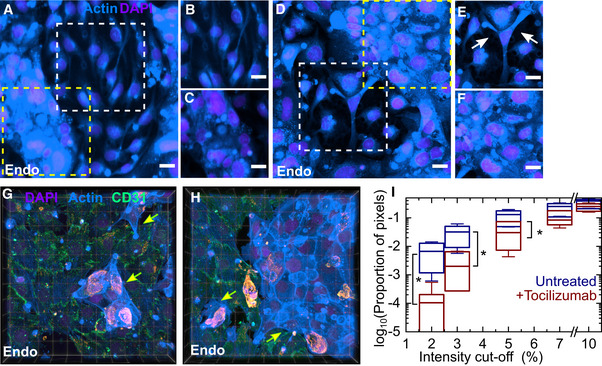
-
A–FMaximum‐intensity projections of representative 232 × 232 µm2 fields of view of the endothelial layer from an infected LoC reconstituted without macrophages at 2 dpi with (A) and without tocilizumab treatment (D). Actin and nuclear staining are indicated with the azure and electric indigo LUTs, respectively, the actin staining is saturated for the endothelial cell clusters to highlight cells with lower levels of actin. (B, C) Zooms corresponding to the region in (A) highlighted with white and yellow boxes, respectively. (B) A region with normal vascular profile, (C) A region of endothelial cell cluster formation with increased actin staining. (E, F) Zooms corresponding to the regions in (D) representing regions highlighted with white and yellow boxes, respectively. (E) An example of reduced tight junctions between endothelial cells; bare patches are indicated with arrows. (F) A region of endothelial cell clusters. Scale bar = 20 µm.
-
G, H3D views from two 232 × 232 µm2 fields of view of the endothelial layer of a tocilizumab‐treated infected LoC at 2 dpi, immunostaining for the tight junction protein CD31 is shown in the spring green LUT. Endothelial cell clusters with low CD31 expression and altered actin staining are also visible and indicated by yellow arrows.
-
IA plot of the proportion of pixels (shown on a logarithmic scale) with regions of low F‐actin intensity identified via cut‐off thresholds defined as a percentage of the maximum intensity in regions of interest from one LoC with and without tocilizumab administration. ROIs were chosen from between 6 and 7 fields of view (technical replicates), each field of view was 232 × 232 µm2. The ROIs were defined to exclude the occurrence of endothelial cell clusters. A significantly larger fraction of the surface area of the ROIs for the untreated chip has low F‐actin intensities.
Data information: The solid bar represents the median, the boxes represent the 25–75 percentile range, and the whiskers represent the standard deviation from 6 to 7 regions of interest (technical replicates) from each chip. P‐values are calculated using a Kruskal–Wallis one‐way ANOVA test, * represents P ≤ 0.05, ** represents P ≤ 0.01, and *** represents P ≤ 0.001.
To quantify the improvement in permeability, we compared regions of interest (ROIs) that excluded endothelial cell clusters across at least six fields of view from the endothelial layer of LoCs with and without tocilizumab perfusion and identified areas with low or no F‐actin staining as those with reduced confluence, as in Fig EV2F. A plot of the proportion of pixels with intensities below a defined cut‐off threshold (Fig 7I) showed that the untreated LoC did have a significantly higher proportion of pixels with intensities lower than 5% (P = 0.032) of the maximum intensity (P = 0.022 for 3% and P = 0.038 for 2%). Inhibition of IL‐6 signalling through tocilizumab is therefore able to ameliorate some but not all of the vascular damage observed.
Discussion
The alveolar space has a strikingly different physiology from that of the upper airway. ACE2 is the canonical entry receptor used by SARS‐CoV‐2 yet is expressed in only a small fraction of alveolar cells, predominantly type II ATs. It has been speculated that high levels of ACE2 expression may be necessary for export of mature virions (Klein et al, 2020). Consequently, because most virology assays measure viral titre, studies of infection of alveolar epithelial cells have either used cell lines such as A549 transfected with ACE2 or used type II AT alveolar organoids that do not capture the functional diversity of the lung (Hekman et al, 2020). As a consequence, the infection of type I ATs, the role of alternate receptors such as NRP1 that is more abundant in the lower airways and the effects of uptake via NRP1 versus ACE2 on the viral life cycle are relatively unknown.
The lung‐on‐chip architecture is well placed to mimic alveolar physiology. The endothelial layer is exposed to fluid flow and shear stress that is necessary for an accurate recreation of endothelial cell biology and is difficult to achieve in Transwell‐based systems. ATs can be maintained at an air–liquid interface, which is necessary for surfactant production. Although Zhang et al, (2021) recently reported on a lung‐on‐chip platform to study SARS‐CoV‐2 infection of the alveolar space, the platform was not maintained at an air–liquid interface and immortalized alveolar epithelial cell lines and a lung microvascular endothelial cell line were used. In contrast, we populate our system with the relevant primary human cells that includes a mix of type I and type II ATs. We make every effort to minimize in vitro passages of ATs to avoid the loss of type II ATs, although it is likely that the cell population does not completely reflect the type II: type I AT ratio found in the lung. However, we report low ACE2 expression and high NRP1 expression, consistent with human alveolar physiology. Similarly, most infections of alveolar epithelial cells are usually performed at high MOI (Blanco‐Melo et al, 2020). Instead, we used an infectious dose that better mimics either a direct inspiration of aerosols or the transfer of infectious virions from the upper respiratory tract.
Despite low ACE2 expression and low MOI, SARS‐CoV‐2 infection produced pronounced physiological changes on‐chip. This could be due to the high levels of NRP1 and aided by upregulation of TMPRSS2, which is known to be important in human SARS‐CoV‐2 infection. In contrast to models of productive replication, we observe slow intracellular viral replication without significant release of infectious virions, observations that are consistent with reports of recovery of viral RNA in patients after they cease to be infectious (Bussani et al, 2020; Cevik et al, 2021). Microscopy‐based analyses also revealed that responses to infection are highly heterogeneous; for example, the few foci of heavily infected cells might represent cells with constitutively higher levels of ACE2 expression at the time of infection. However, why alveolar epithelial cells show persistent but not productive infection is still unclear.
Infection of the AT layer rapidly led to an infection of the endothelial layer. Basolateral transmission cannot occur in typical monoculture experiments, but it has not been reported to occur to a large extent in monocultures of airway epithelial cells at the air–liquid interface (V’kovski et al, 2020). However, the basement membrane is much thinner in alveoli than in the upper airway(Weibel, 2015), with cell‐to‐cell contact between the ATs and endothelial cells that is recreated in the array of pores on‐chip. Infection of lung microvascular cells therefore might occur through uptake of vesicles as part of cell‐to‐cell communication between the layers (a receptor‐independent mechanism of uptake). Such mechanisms of cell–cell communication play an important role in maintaining homeostasis at the air–liquid interface, a physiological feature that our model incorporates. These observations are strengthened by the lack of observation of any signs of infection in lung microvascular endothelial cells when directly exposed to the virus on their apical surface, consistent with recent reports of the absence of infection in native primary endothelial cells from multiple tissues (Conde et al, 2020). Alternatively, infection of endothelial cells when exposed to the virus basolaterally might nevertheless occur due to receptor‐mediated uptake, with differences between apical and basolateral infection explained by differences in distribution of cell surface receptors. Notably, NRP1 is likely to be located along the basal surface of the endothelial cells as an integrin‐binding protein (Valdembri et al, 2009), and SARS‐CoV‐2 has been shown to infect vascularized organoids, albeit at high MOI (Monteil et al, 2020). The lack of endothelial cell infection and inflammation in Zhang et al (2021) may be explained by the absence of air–liquid interface, which would alter cell–cell communication between the endothelial and epithelial cell layers, as well as the maintenance of flow in the epithelial channel that would wash away virions or inflammatory stimuli secreted by the epithelial cells. Indeed, Wang et al (2020) reported that supernatants from infected alveolar epithelial cells were sufficient to induce changes in tight junction protein expression in endothelial cell monolayers, consistent with a role for cell–cell communication at the alveolar interface. Although the exact mechanisms remain to be elucidated, the consequences of endothelial infection may extend downstream to the persistent infection and endothelial cell‐specific cell damage observed in human patient samples.
A striking feature of SARS‐CoV‐2 infections in the LoC model was the cell‐type‐specific morphological changes. Infection altered endothelial cell morphology in two different but interconnected ways. First, the loss of tight junctions between cells is indicative of increased vascular permeability that could lead to exudative alveolar damage observed in autopsy reports. Exposed subendothelial collagen would likely trigger the contact activation or intrinsic pathway of coagulation. This was also accompanied by pro‐coagulatory changes in the extrinsic pathway through a modest increase in expression of tissue factor, a pro‐coagulatory factor and reduction in expression of anti‐coagulatory factors such as TFPI and thrombomodulin. These changes are in agreement both with reports of early vascular damage in the rhesus macaque model of infection(Aid et al, 2020) and in part consistent with reports of altered expression of coagulation markers in bronchoalveolar lavages from COVID‐19 patients (Mast et al, 2021). Interestingly, infection of human umbilical vein endothelial cells (HUVECs) with dengue virus has been shown to have the opposite effect (Jiang et al, 2007), with a strong increase in thrombomodulin expression consistent with the disease manifestation as a haemorrhagic fever. These results highlight the close link between coagulation and inflammation in this cell type. Second, a subpopulation of endothelial cells completely lost CD31 expression and adopted an altered morphology consisting of cell clusters. These clusters could form either through enhanced proliferation or from cell migration. The former is more likely, as a consistent increase in cell density was observed across multiple LoCs, but both possibilities are consistent with the role of NRP1 as a growth factor receptor and through alteration in VEGF signalling that we also report (Guo & Vander Kooi, 2015). The loss of CD31 expression could itself have a number of possible implications: CD31 itself plays important roles in leucocyte migration (Newman & Newman, 2003), in angiogenesis, in maintaining a anti‐coagulatory environment (Falati et al, 2006; Zhang et al, 2007) and in preventing endothelial cell death from immune cell‐mediated inflammation (Cheung et al, 2015). These CD31‐ cells could further contribute to a pro‐coagulatory environment in the microvasculature and lead to changes in vascular architecture reported in autopsies (Ackermann et al, 2020). A reduction in CD31 expression in endothelial cells will likely alter interactions with neutrophils and platelets, which also express CD31, potentially contributing to the formation of neutrophil extracellular traps and platelet hyperactivity reported in COVID‐19 (Manne et al, 2020; Zuo et al, 2020). These findings are also consistent with the observation of elevated serum levels of CD31 in patients with severe COVID‐19 (Li et al, 2020). The exact mechanisms for the loss of CD31 and other tight junction markers remain to be elucidated, but a possible mechanism could be through action of specific viral proteins themselves, either acting alone as is observed in the case of the K5 protein of the Kaposi sarcoma herpesvirus (Mansouri et al, 2006) that causes Kaposi sarcoma or in conjunction with a pro‐inflammatory host response.
Increased NF‐KB inflammatory responses could also account for the vascular changes observed, and the role of cytokine storms in COVID‐19 pathogenesis is still unclear. We observed that low levels of intracellular viral RNA generate an NF‐KB‐mediated pro‐inflammatory response, with suppression of antiviral interferon responses, consistent with reports of the effect of a number of viral proteins (Hadjadj et al, 2020; Thoms et al, 2020). Within each cell types, there were significant cell‐to‐cell differences in expression of inflammatory markers, clearly identifiable with single‐cell spatial resolution through the use of RNAscope and confocal imaging in the LoC model. These differences could be due to differences in the mode of uptake or differences in the intracellular localization of viral RNA and RNA replication, or differences in the levels of translation of viral proteins. We observe instances of co‐localization of viral RNA and S protein with the nucleus, which is consistent with bioinformatic predictions using algorithms for RNA localization (Wu et al, 2020), but has not been reported in studies using cell lines with productive infections. These atypical subcellular localizations may be characteristic of persistent infection and merit further investigation. Yet, once again the responses of ATs and endothelial cells contrasted sharply—inflammation in AT cells was transient, whereas it was persistent in endothelial cells.
Lastly, given the persistent IL6 expression in endothelial cells, we applied the model and our microscopy‐based approach to examine whether tocilizumab perfusion could reduce the occurrence of either increased vascular permeability or the formation of endothelial cell clusters or both. We found that tocilizumab was able to maintain cellular confluency but did not prevent the formation of endothelial cell clusters. These observations at the single‐cell level offer insights into reasons why outcomes with this drug in clinical trials have been modest and at times contradictory (Hermine et al, 2021; Rosas et al, 2021), and there is insufficient evidence of the efficacy of tocilizumab in reducing mortality (Parr, 2020). The lung‐on‐chip model is therefore able to make clinically testable predictions, highlighting the utility of this approach.
Many animal models reported for SARS‐CoV‐2 do not recreate all aspects of the human manifestation of COVID‐19 or lead to exaggerated outcomes (Zheng et al, 2021). Thus, an advanced cell culture platform such as the lung‐on‐chip complements these models and can serve as a useful intermediary between animal models and clinical studies (Adhikary et al, 2021). Yet, significant limitations of the model remain. These include the lack of a complete recapitulation of resident innate immunity and the absence of an adaptive immune response and other cell types necessary for the complete recapitulation of vascular function such as fibroblasts and pericytes. Nevertheless, it provides a platform in which complexity can be introduced in a tuneable manner, that is amenable to live‐cell imaging (Thacker et al, 2020), and where components of whole blood can be added to recreate aspects of coagulation (Barrile et al, 2018) or peripheral blood mononuclear cells or neutrophils via the vascular channel (preprint: Sharma et al, 2021).
In sum, SARS‐CoV‐2 infection of a model of the alveolar space shows a set of unique characteristics underlined by persistent infection, low levels of productive viral replication, inflammation and damage to endothelial barrier integrity and the formation of a pro‐coagulatory environment that are in good agreement with reports from animal models and of clinical disease. The physiological mimicry that the LoC model provides (air–liquid interface, co‐culture, perfusion through a vascular channel) and high‐resolution confocal imaging reveal insights into the dynamics and mechanisms that underlie these observations that are difficult to obtain in other experimental models and that will improve our understanding of the pathogenesis of this multi‐organ disease.
Materials and Methods
Cell culture
Primary human alveolar epithelial cells (ATs) and human lung microvascular endothelial cells (endothelial cells) were obtained from a commercial supplier (Cell Biologics, USA). Each vial of ATs comprised a mix of type I and type II cells at passage 3, and expression of cell‐type‐specific markers (AQP5, PDPN and CAV1 for type I and ABCA3 and SFTPC for type II) was verified via qRT–PCR on‐chip (Appendix Fig S2D). Both cell types were cultured in vitro in complete medium comprising base medium and supplements (Cell Biologics, USA) in 5% CO2 at 37 °C. Passage of ATs lowered ACE2 expression (Fig EV1A); therefore, all chips were reconstituted with ATs seeded directly on the lung‐on‐chip (see below), without any additional in vitro culture. In contrast, we observed no significant changes in viral entry factor expression in endothelial cells upon passage, and so therefore these cells were passaged between 3 and 5 times before seeding in the LoC devices.
PBMC isolation and macrophage differentiation
Peripheral blood mononuclear cells were obtained from buffy coat (Interregional Blood Transfusion SRC Ltd, Switzerland) obtained from anonymized donors. PBMCs were isolated using a Biocol Separation procedure as per the manufacturer’s instructions. Isolated PBMCs were subsequently cryopreserved in a solution of 70% heat‐inactivated foetal bovine serum (FBS, Gibco), 20% RPMI medium (Gibco) and 10% dimethyl sulfoxide (DMSO). One week prior to seeding the macrophages in the LoC devices, a cryopreserved aliquot was thawed and cultured in a T‐75 flask (TPP, Switzerland) in RPMI supplemented with 10% FBS. CD14+ monocytes were isolated using positive selection (CD14 Ultrapure Isolation Kit, Miltenyi Biosciences) and cultured in RPMI medium supplemented with 10% FBS and differentiated for 7 days with 20 ng/ml recombinant human macrophage‐colony stimulating factor protein (M‐CSF) (Thermo Fisher Scientific), and 100 U/l of penicillin‐streptomycin solution (Thermo Fisher Scientific) to avoid bacterial contamination. The cells were cultured in plastic Petri dishes without pre‐sterilization (Greiner Bio‐One) so that differentiated macrophages could be more easily detached.
Quantitative Real‐Time PCR (qRT–PCR) for cell characterization
AT cells obtained directly from the supplier were grown overnight in cell culture microdishes (Ibidi) or T‐25 cell culture flask (TPP, Switzerland). Passaged ATs were grown to confluency in a T‐75 cell culture flask (TPP). Growth media were removed from the flask, the cells were incubated with the appropriate volume of RNA lysis buffer (RNeasy Plus Micro Kit, Qiagen), and RNA was isolated as per the manufacturer’s instructions and resuspended in 25 μl of DEPC‐treated water. Approximately 1 μg of RNA was then used to generate cDNA using the SuperScript® IV First‐Strand Synthesis System with random hexamers (Invitrogen), which was stored at −20°C. Specific primers used for the qRT–PCR are listed in Appendix Table S1. qRT–PCRs were prepared with SYBR® Green PCR Master Mix (Applied Biosystems) with 500 nM primers and 1 μl cDNA. Reactions were run as absolute quantification on ABI PRISM® 7900HT Sequence Detection System (Applied Biosystems). Amplicon specificity was confirmed by melting‐curve analysis.
Quantitative Real‐Time PCR (qRT–PCR) for quantification of viral genomes in the supernatant
150 µl of cell‐free supernatant was processed using a kit to extract viral RNA as per the manufacturer’s instructions (NucleoSpin Dx Virus, Macherey‐Nagel). For the vascular effluent, this was taken from the approximately 1.5 ml of effluent generated daily by the flow through the vascular channel. In the case of the apical wash, the 30 µl collected from the apical channel was further diluted in cell culture media up to 150 µl and subsequently processed. Viral RNA was eluted in 50 ml of DEPC‐treated water. 5 ml of RNA was then used in a one‐step qRT–PCR kit for SARS‐CoV‐2 detection (2019‐nCoV TaqMan RT–PCR Kit, Norgen Biosciences) using primer and probe mixes corresponding to the nucleocapsid (N2) gene and the host RNAseP genes. Primer sequences are those listed by the US Centre for Disease Control (CDC).
Generation of SARS‐CoV‐2 stocks
Vero E6 cells and a clinical isolate of SARS‐CoV‐2 were a kind gift from the laboratory of Prof Carolyn Tapparel at the University of Geneva. SARS‐CoV‐2/Switzerland/GE9586/2020 was isolated from a clinical specimen in the University Hospital in Geneva in Vero‐E6. Cells were infected, and the supernatant was collected 3 days postinfection, clarified, aliquoted and frozen at −80°C and subsequently titrated by plaque assay in Vero‐E6. Virus used for all experiments in this manuscript was at passage 3 in Vero E6 cells.
Human lung‐on‐chip (LoC) model
LoC made of polydimethylsiloxane (PDMS) was obtained from Emulate (Boston, USA). Extracellular matrix (ECM) coating was performed as per the manufacturer’s instructions. Chips were activated using ER‐1 solution (Emulate) dissolved in ER‐2 solution at 0.5 mg/ml (Emulate) and exposed for 20 min under UV light. The chip was then rinsed with coating solution and exposed again to UV light for a further 20 min. Chips were then washed thoroughly with PBS before incubating with an ECM solution of 150 µg/ml bovine collagen type I (AteloCell, Japan) and 30 µg/ml fibronectin from human plasma (Sigma‐Aldrich) in PBS buffered with 15 mM HEPES solution (Gibco) for 1–2 h at 37°C. If not used directly, coated chips were stored at 4°C and pre‐activated before use by incubation for 30 min with the same ECM solution at 37°C. Endothelial cells were cultured overnight at 37°C and 5% CO2 in T‐75 cell culture flasks, detached with 0.05% trypsin, concentrated to 5–10 million cells/ml, and seeded on the bottom face of the PDMS membrane. The chip was then incubated for a short period at 37°C to allow the endothelial cells to spread and subsequently seeded with ATs. ATs were seeded directly from cryopreserved vials received from the supplier, each vial of 0.5 million cells could be used to seed between two and three chips. To increase reproducibility, vials were pooled and used to seed multiple chips. The chip was incubated overnight with complete epithelial and endothelial media in the epithelial and endothelial channels, respectively, under static conditions. The next day, the chip was washed and a reduced medium for the air–liquid interface (ALI) was flowed through the vascular channel using syringe pumps (Aladdin‐220, Word Precision Instruments) at 60 µl/h as described (Hassell et al, 2017). The composition of the ALI media used was as described in Hassell et al (2017) but with an FBS concentration of 2%. The epithelial face was incubated with epithelial base medium with 1 µM dexamethasone (Sigma‐Aldrich) without FBS supplementation to promote tight junction formation and surfactant expression as reported in previous lung‐on‐chip studies(Huh et al, 2010; Hassell et al, 2017). Flow was maintained over 2–3 days with daily replacement of the medium on the epithelial face (with dexamethasone supplementation). At the end of this period, in LoCs reconstituted with macrophages, CD14+ macrophages differentiated for 7 days in M‐CSF (described above) were detached from the Petri dish using 2 mM ethylenediaminetetraacetic acid (EDTA, Sigma‐Aldrich) in PBS at 4°C and mechanical scraping, centrifuged at 300 g for 5 min and resuspended in a small volume of epithelial cell media without dexamethasone. This solution containing macrophages was introduced onto the epithelial face and incubated for 30 min at 37°C and 5% CO2 to allow macrophages to attach to the AT cells. Medium on the epithelial face was then removed, and the chip was maintained overnight at an air–liquid interface (ALI). Chips were controlled to ensure that they successfully maintained the ALI overnight and were then transferred to the biosafety level 3 (BSL‐3) facility for SARS‐CoV‐2 infection. No antibiotics were used in any of the cell culture media for setting up the LoC model.
Tocilizumab perfusion in the LoC model
Tocilizumab/Rho‐Actemra (Roche, 20 mg/ml in saline) for intravenous administration were a kind gift from Prof Thomas Hugle at the University Hospital in Lausanne, Switzerland (CHUV). We simulated a single dose of 8 mg/kg given to a person of bodyweight 70 kg and a blood volume of 5 l to obtain a concentration of 56 µg/ml in the cell culture media. Tocilizumab perfusion was started immediately after infection and was flowed through the vascular channel of infected chips for a period of 2 days postinfection.
Characterization of infection in LoCs
Uninfected LoCs were maintained at an ALI either for 1 day (for total RNA extraction (two LoCs without macrophages) or for up to 3 days for RNAscope characterization (one LoC each was reconstituted with and without macrophages) after addition of macrophages, during which time ALI medium was flowed through the endothelial channel at 60 µl/h. In the case of the former, total RNA from the AT and endothelial layers was extracted separately by passing 350 µl of the lysis buffer reconstituted as per the manufacturer’s instructions (RNeasy Plus Micro Kit, Qiagen) through easy layer. Complete detachment of the cells in each layer was verified via optical microscopy. RNA was eluted in 14 µl of DEPC‐treated nuclease‐free water and used for subsequent qRT–PCRs.
In the case of LoCs processed for RNAscope, the chips were fixed for 1 h using a fresh solution of 4% paraformaldehyde in phosphate‐buffered saline (PBS), and subsequently dehydrated in a sequence of washes with 50% EtOH, 70% EtOH and 100% EtOH. The chips were then stored in 100% EtOH at −20°C until processed for RNAscope at the Histology Core Facility at EPFL.
Immunofluorescence characterization
For immunofluorescence labelling, the fixed LoCs were incubated with a blocking solution of 2% bovine serum albumin (BSA) in PBS (“blocking buffer”), followed by overnight incubation with the primary antibody in the blocking buffer at 4°C. A list of primary antibodies and concentrations used is included in Appendix Table S2. In addition, the infected LoC shown in Fig 4E–G was immunostained with rabbit anti‐SARS‐CoV spike antisera (McBride et al, 2007), which was a kind gift of Prof. Carolyn Machamer at John Hopkins University. The following day, the chip was washed extensively with fresh blocking buffer before inoculation with the secondary antibody at a dilution of 1:300 for 1 h at room temperature. The secondary antibodies used were as follows: donkey anti‐mouse Alexa Fluor 488 (A21206, Thermo Fisher), or donkey anti‐mouse Alexa Fluor 568 (A10037, Thermo Fisher), or donkey anti‐mouse Alexa Fluor 647 (A31573, Thermo Fisher), and were chosen to complement the fluorophores already assigned to RNAscope labelling. F‐actin on both the AT and endothelial face was stained using Sir‐Actin dye in the far‐red (Spherochrome) at 1 µM for 30 min concurrently with Hoechst staining, as described above. Confocal images were obtained on a Leica SP8 microscope in the inverted optical configuration at the EPFL BIOP core facility.
Infection of the LoC with SARS‐CoV‐2
LoCs for infection were assembled into a sealed Tupperware box modified to contain inlets and outlets for the flow of medium through the vascular channel and were maintained throughout the course of the experiment by use of a syringe pump. On the day of the experiment, an aliquot of virus‐containing supernatant was thawed from the stock at −80°C and diluted approximately 35‐fold in epithelial cell media without FBS to generate the inoculum that corresponded to an infectious dose of 200 – 300 plaque‐forming units (PFUs) in a volume of 30 µl. 30 µl of this solution was then added to the apical channel of each LoC, and the LoC was incubated for an hour at 37°C and 5% CO2 to enable infection. Thereafter, the medium in the apical channel was removed. The LoC was returned to air–liquid interface, and the inlets of the infected chip were sealed with solid pins as a safety precaution. The media flowing through the vascular channel of each LoC were collected in a separate sealed 15‐ml Falcon tube, and each tube was emptied daily, and the effluent was stored at −80°C; for further processing. At this time, the metal pins sealing the apical channel of each chip were removed, and 30 µl of epithelial cell media without FBS was briefly added to the channel before being withdrawn and collected as the “apical wash”. The metal pins were reinserted, and the LoCs returned to the cell culture incubator. Both the vascular effluent and the apical wash from each chip were collected for each day postinfection.
Infection was terminated at specified time points either by treatment with the lysis buffer for RNA extraction (one LoC reconstituted without macrophages) or by fixing with freshly prepared 4% paraformaldehyde for a period of 1 h (all other infected LoCs) for RNAscope assay preparation. The chips were then dehydrated as described above and immersed in 100% EtOH before removal from the BSL‐3 facility.
RNAscope assay
RNAscope Multiplex Fluorescent V2 Assay (Bio‐techne, Cat. No. 323110) was performed according to the manufacturer's protocol directly on chips. They were hybridized with the Hs 3plex‐positive control (Bio‐techne, Cat. No. 320861), or the DapB‐negative control (Bio‐techne, Cat. No. 310043) or a combination of the following probes V‐nCoV2019‐orf1ab‐sense (Bio‐techne, Cat. 859151), Hs‐ACE2‐C2 (Bio‐techne, Cat. 848151‐C2), V‐nCoV2019‐S‐C3 (Bio‐techne, Cat. 848561‐C3), HS‐Il1b‐C1 (Bio‐techne, Cat. 310361) and Hs‐Il6‐C2 (Bio‐techne, Cat. No. 310371‐C2) simultaneously at 40°C for 2 h. The different channels were revealed with TSA Opal570 (Akoya Biosciences, Cat. No. FP1488001KT), TSA Opal650 (Akoya Biosciences, Cat. No. FP1488001KT) and TSA Opal690 (Akoya Biosciences, Cat. No. FP1497001KT). Cells were counterstained with DAPI and mounted with ProLong Gold Antifade Mountant (Thermo Fisher, P36930).
ELISAs
Frozen vascular effluent from infected chips was thawed, incubated at 58°C for 30 min to ensure sterilization and then removed from the BSL‐3 for further processing. Levels of human IL‐6 were determined by enzyme‐linked immunosorbent assays (BD Biosciences) as per the manufacturers’ instructions.
Confocal imaging and image analysis
Infected and control LoCs were imaged using a Leica SP8 confocal microscope with a white light laser. For chips processed via RNAscope and labelled with Opal 650 and Opal 690, the excitation and emission windows were carefully chosen to minimize overlap of signal. LoCs were imaged with a 25× water immersion objective (NA = 0.95, Leica), with standard settings across chips labelled the same way. Z‐stacks were subsequently deconvolved using the Huygens Deconvolution Software (Scientific Volume Imaging), and 3D views were rendered using the FIJI ClearVolume plugin (Royer et al, 2015). Custom‐written software in MATLAB was used to segment and identify the 3D volume, mean intensity and number of RNA dots in each field of view using the nestedSortStruct algorithm for MATLAB written by the Hughey Lab (https://www.github.com/hugheylab/nestedSortStruct, GitHub.
Statistical analysis
Statistical analysis was performed using Origin 9.2 (OriginLabs). P‐values were calculated using a Kruskal–Wallis one‐way ANOVA test, with the null hypothesis that the medians of each population were equal.
Author contributions
VVT conceptualized the project, contributed to software, performed formal analysis and data curation, contributed to visualization and supervision, and wrote the original draft of the manuscript. VVT, KS, ND and JS‐D contributed to methodology and performed validation. VVT, KS, ND, G‐FM and JS‐D contributed to investigation. VVT, KS, ND, JS‐D and JDM reviewed and edited the manuscript. VVT and JDM contributed to project administration and funding acquisition. None of the authors contributed to resources.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
We gratefully acknowledge assistance from Dr Muhammet Fatih Gulen for the ELISA assays. We are also grateful to the members of the BioImaging Core Facility (BIOP) for assistance with confocal microscopy and to Prof Dr Thomas Hugle at the Centre Hospitalier Universitaire Vaudois (CHUV) for donation of tocilizumab. This research was supported by funding to V.V.T from the EPFL COVID‐19 Research Committee, from a grant to V.V.T from the Novartis Foundation for Medical‐Biological Research (#20C240) and a grant to J.D.M. from the Swiss National Science Foundation (grant number 310030B_176397).
EMBO reports (2021) 22: e52744.
Data availability
No primary datasets have been generated and deposited as part of this study. The source data generated during and/or analysed during the current study are available from the corresponding authors on reasonable request and will be made available at the EPFL Community Page on Zenodo upon publication; the data will be made available to all users as requested publication.
References
- Abo KM, Ma L, Matte T, Huang J, Alysandratos KD, Werder RB, Mithal A, Lou BM, Lindstrom‐Vautrin J, Mostoslavsky G et al (2020) Human iPSC‐derived alveolar and airway epithelial cells can be cultured at air‐liquid interface and express SARS‐CoV‐2 host factors. bioRxiv 10.1101/2020.06.03.132639 [PREPRINT] [DOI]
- Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, Vanstapel A, Werlein C, Stark H, Tzankov A et al (2020) Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid‐19. N Engl J Med 383: 120–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikary PP, Ul Ain Q, Hocke AC, Hedtrich S (2021) COVID‐19 highlights the model dilemma in biomedical research. Nat Rev Mater 6: 374–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aid M, Busman‐Sahay K, Vidal SJ, Maliga Z, Bondoc S, Starke C, Terry M, Jacobson CA, Wrijil L, Ducat S et al (2020) Vascular disease and thrombosis in SARS‐CoV‐2‐infected rhesus macaques. Cell 183: 1354–1366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrile R, van der Meer AD, Park H, Fraser JP, Simic D, Teng F, Conegliano D, Nguyen J, Jain A, Zhou M et al (2018) Organ‐on‐chip recapitulates thrombosis induced by an anti‐CD154 monoclonal antibody: translational potential of advanced microengineered systems. Clin Pharmacol Ther 104: 1240–1248 [DOI] [PubMed] [Google Scholar]
- Bhatia SN, Ingber DE (2014) Microfluidic organs‐on‐chips. Nat Biotechnol 32: 760–772 [DOI] [PubMed] [Google Scholar]
- Blanco‐Melo D, Nilsson‐Payant BE, Liu W‐C, Uhl S, Hoagland D, Møller R, Jordan TX, Oishi K, Panis M, Sachs D et al (2020) Imbalanced host response to SARS‐CoV‐2 drives development of COVID‐19. Cell 181: 1036–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussani R, Schneider E, Zentilin L, Collesi C, Ali H, Braga L, Volpe MC, Colliva A, Zanconati F, Berlot G et al (2020) Persistence of viral RNA, pneumocyte syncytia and thrombosis are hallmarks of advanced COVID‐19 pathology. EBioMedicine 61: 103104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantuti‐Castelvetri L, Ojha R, Pedro LD, Djannatian M, Franz J, Kuivanen S, van der Meer F, Kallio K, Kaya T, Anastasina M et al (2020) Neuropilin‐1 facilitates SARS‐CoV‐2 cell entry and infectivity. Science 370: 856–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carsana L, Sonzogni A, Nasr A, Rossi RS, Pellegrinelli A, Zerbi P, Rech R, Colombo R, Antinori S, Corbellino M et al (2020) Pulmonary post‐mortem findings in a series of COVID‐19 cases from northern Italy: a two‐centre descriptive study. Lancet Infect Dis 20: P1135–P1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cevik M, Tate M, Lloyd O, Maraolo AE, Schafers J, Ho A (2021) SARS‐CoV‐2, SARS‐CoV, and MERS‐CoV viral load dynamics, duration of viral shedding, and infectiousness: a systematic review and meta‐analysis. Lancet Microbe 2: e13–e22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung K, Ma L, Wang G, Coe D, Ferro R, Falasca M, Buckley CD, Mauro C, Marelli‐Berg FM (2015) CD31 signals confer immune privilege to the vascular endothelium. Proc Natl Acad Sci USA 112: E5815–E5824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu H, Chan J‐W, Yuen T‐T, Shuai H, Yuan S, Wang Y, Hu B, Yip C‐Y, Tsang J‐L, Huang X et al (2020) Comparative tropism, replication kinetics, and cell damage profiling of SARS‐CoV‐2 and SARS‐CoV with implications for clinical manifestations, transmissibility, and laboratory studies of COVID‐19: an observational study. Lancet Microbe 1: e14–e23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claudio D, Claudia R, Giulio R, Alessandra D, Federica P, Sara P, Antonio V, Lorenza P, Franco S, Stefano M et al (2020) Acute lung injury evolution in Covid‐19. medRxiv 10.1101/2020.08.09.20170910 [PREPRINT] [DOI]
- Conde JN, Schutt WR, Gorbunova EE, Mackow ER (2020) Recombinant ACE2 expression is required for SARS‐CoV‐2 to infect primary human endothelial cells and induce inflammatory and procoagulative responses. MBio 11: e03185‐20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly JL, Simonetti B, Klein K, Chen K‐E, Williamson MK, Antón‐Plágaro C, Shoemark DK, Simón‐Gracia L, Bauer M, Hollandi R et al (2020) Neuropilin‐1 is a host factor for SARS‐CoV‐2 infection. Science 370: 861–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreymueller D, Martin C, Kogel T, Pruessmeyer J, Hess FM, Horiuchi K, Uhlig S, Ludwig A (2012) Lung endothelial ADAM17 regulates the acute inflammatory response to lipopolysaccharide. EMBO Mol Med 4: 412–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falati S, Patil S, Gross PL, Stapleton M, Merrill‐Skoloff G, Barrett NE, Pixton KL, Weiler H, Cooley B, Newman DK et al (2006) Platelet PECAM‐1 inhibits thrombus formation in vivo. Blood 107: 535–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattinoni L, Coppola S, Cressoni M, Busana M, Rossi S, Chiumello D (2020) COVID‐19 does not lead to a “typical” acute respiratory distress syndrome. Am J Respir Crit Care Med 201: 1299–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelfand MV, Hagan N, Tata A, Oh WJ, Lacoste B, Kang KT, Kopycinska J, Bischoff J, Wang JH, Gu C (2014) Neuropilin‐1 functions as a VEGFR2 co‐receptor to guide developmental angiogenesis independent of ligand binding. Elife 3: e03720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassart A, Malardé V, Gobba S, Sartori‐Rupp A, Kerns J, Karalis K, Marteyn B, Sansonetti P, Sauvonnet N (2019) Bioengineered human organ‐on‐chip reveals intestinal microenvironment and mechanical forces impacting shigella infection. Cell Host Microbe 26: 435–444 [DOI] [PubMed] [Google Scholar]
- Guo HF, Vander Kooi CW (2015) Neuropilin functions as an essential cell surface receptor. J Biol Chem 290: 29120–29126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, Péré H, Charbit B, Bondet V, Chenevier‐Gobeaux C et al (2020) Impaired type I interferon activity and inflammatory responses in severe COVID‐19 patients. Science 369: 718–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassell BA, Goyal G, Lee E, Sontheimer‐Phelps A, Levy O, Chen CS, Ingber DE (2017) Human organ chip models recapitulate orthotopic lung cancer growth, therapeutic responses, and tumor dormancy in vitro. Cell Rep 21: 508–516 [DOI] [PubMed] [Google Scholar]
- Hekman RM, Hume AJ, Goel RK, Abo KM, Huang J, Blum BC, Werder RB, Suder EL, Paul I, Phanse S et al (2020) Actionable Cytopathogenic host responses of human alveolar type 2 cells to SARS‐CoV‐2. Mol Cell 80: 1104–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermine O, Mariette X, Tharaux P‐L, Resche‐Rigon M, Porcher R, Ravaud P, Bureau S, Dougados M, Tibi A, Azoulay E et al (2021) Effect of tocilizumab vs usual care in adults hospitalized with COVID‐19 and moderate or severe pneumonia: a randomized clinical trial. JAMA Intern Med 181: 32–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hikmet F, Méar L, Edvinsson Å, Micke P, Uhlén M, Lindskog C (2020) The protein expression profile of ACE2 in human tissues. Mol Syst Biol 16: e9610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M, Kleine‐Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu N‐H, Nitsche A et al (2020) SARS‐CoV‐2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181: 271–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou YJ, Okuda K, Edwards CE, Martinez DR, Asakura T, Dinnon KH, Kato T, Lee RE, Yount BL, Mascenik TM et al (2020) SARS‐CoV‐2 reverse genetics reveals a variable infection gradient in the respiratory tract. Cell 182: 429–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Hume AJ, Abo KM, Werder RB, Villacorta‐Martin C, Alysandratos KD, Lou BM, Simone‐Roach C, Olejnik J, Suder E et al (2020) SARS‐CoV‐2 infection of pluripotent stem cell‐derived human lung alveolar type 2 cells elicits a rapid epithelial‐intrinsic inflammatory response. Cell Stem Cell 27: 962–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh D, Matthews BD, Mammoto A, Montoya‐Zavala M, Hsin HY, Ingber DE (2010) Reconstituting organ‐level lung functions on a chip. Science 328: 1662–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui KPY, Cheung M‐C, Perera RAPM, Ng K‐C, Bui CHT, Ho JCW, Ng MMT, Kuok DIT, Shih KC, Tsao S‐W et al (2020) Tropism, replication competence, and innate immune responses of the coronavirus SARS‐CoV‐2 in human respiratory tract and conjunctiva: an analysis in ex‐vivo and in‐vitro cultures. Lancet Respir Med 8: 687–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z, Tang X, Xiao R, Jiang L, Chen X (2007) Dengue virus regulates the expression of hemostasis‐related molecules in human vein endothelial cells. J Infect 55: e23–e28 [DOI] [PubMed] [Google Scholar]
- Kim D, Lee JY, Yang JS, Kim JW, Kim VN, Chang H (2020) The architecture of SARS‐CoV‐2 transcriptome. Cell 181: 914–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein S, Cortese M, Winter SL, Wachsmuth‐Melm M, Neufeldt CJ, Cerikan B, Stanifer ML, Boulant S, Bartenschlager R, Chlanda P (2020) SARS‐CoV‐2 structure and replication characterized by in situ cryo‐electron tomography. Nat Commun 11: 5885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang M, Som A, Carey D, Reid N, Mendoza DP, Flores EJ, Li MD, Shepard J‐AO, Little BP (2020) Pulmonary vascular manifestations of COVID‐19 pneumonia. Radiol Cardiothorac Imaging 2: e200277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Huang M, Shen J, Wang Y, Wang R, Yuan C, Jiang L, Huang M (2020) Serum levels of soluble PECAM‐1 in COVID‐19 patients are associated with disease severity. J Infect Dis 223: 178–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Castejon G, Brough D (2011) Understanding the mechanism of IL‐1β secretion. Cytokine Growth Factor Rev 22: 189–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manne BK, Denorme F, Middleton EA, Portier I, Rowley JW, Stubben C, Petrey AC, Tolley ND, Guo Li, Cody M et al (2020) Platelet gene expression and function in patients with COVID‐19. Blood 136: 1317–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansouri M, Douglas J, Rose PP, Gouveia K, Thomas G, Means RE, Moses AV, Früh K (2006) Kaposi sarcoma herpesvirus K5 removes CD31/PECAM from endothelial cells. Blood 108: 1932–1940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marini JJ, Gattinoni L (2020) Management of COVID‐19 respiratory distress. J Am Med Assoc 323: 2329–2330 [DOI] [PubMed] [Google Scholar]
- Mast AE (2016) Tissue factor pathway inhibitor: multiple anticoagulant activities for a single protein. Arterioscler Thromb Vasc Biol 36: 9–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast AE, Wolberg AS, Gailani D, Garvin MR, Alvarez C, Miller JI, Aronow B, Jacobson D (2021) SARS‐CoV‐2 suppresses anticoagulant and fibrinolytic gene expression in the lung. Elife 10: e64330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride CE, Li J, Machamer CE (2007) The Cytoplasmic tail of the severe acute respiratory syndrome coronavirus spike protein contains a novel endoplasmic reticulum retrieval signal that binds COPI and promotes interaction with membrane protein. J Virol 81: 2418–2428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messner CB, Demichev V, Wendisch D, Michalick L, White M, Freiwald A, Textoris‐Taube K, Vernardis SI, Egger A‐S, Kreidl M et al (2020) Ultra‐high‐throughput clinical proteomics reveals classifiers of COVID‐19 infection. Cell Syst 11: 11–24.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miorin L, Kehrer T, Sanchez‐Aparicio MT, Zhang Ke, Cohen P, Patel RS, Cupic A, Makio T, Mei M, Moreno E et al (2020) SARS‐CoV‐2 Orf6 hijacks Nup98 to block STAT nuclear import and antagonize interferon signaling. Proc Natl Acad Sci USA 117: 28344–28354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteil V, Kwon H, Prado P, Hagelkrüys A, Wimmer RA, Stahl M, Leopoldi A, Garreta E, Hurtado del Pozo C, Prosper F et al (2020) Inhibition of SARS‐CoV‐2 infections in engineered human tissues using clinical‐grade soluble human ACE2 in brief clinical‐grade recombinant human ACE2 can reduce SARS‐CoV‐2 infection in cells and in multiple human organoid models. Inhibition of SARS‐CoV‐2 infections in engineered human tissues using clinical‐grade soluble human ACE2. Cell 181: 905–913.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawroth JC, Lucchesi C, Cheng D, Shukla A, Ngyuen J, Shroff T, Varone A, Karalis K, Lee H‐H, Alves S et al (2020) A microengineered airway lung chip models key features of viral‐induced exacerbation of asthma. Am J Respir Cell Mol Biol 63: 591–600 [DOI] [PubMed] [Google Scholar]
- Newman PJ, Newman DK (2003) Signal transduction pathways mediated by PECAM‐1: new roles for an old molecule in platelet and vascular cell biology. Arterioscler Thromb Vasc Biol 23: 953–964 [DOI] [PubMed] [Google Scholar]
- Ortega‐Prieto AM, Skelton JK, Wai SN, Large E, Lussignol M, Vizcay‐Barrena G, Hughes D, Fleck RA, Thursz M, Catanese MT et al (2018) 3D microfluidic liver cultures as a physiological preclinical tool for hepatitis B virus infection. Nat Commun 9: 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parr JB (2020) Time to reassess tocilizumab’s role in COVID‐19 pneumonia. JAMA Int Med 181: 12–15 [DOI] [PubMed] [Google Scholar]
- Qi F, Qian S, Zhang S, Zhang Z (2020) Single cell RNA sequencing of 13 human tissues identify cell types and receptors of human coronaviruses. Biochem Biophys Res Commun 526: 135–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas IO, Bräu N, Waters M, Go RC, Hunter BD, Bhagani S, Skiest D, Aziz MS, Cooper N, Douglas IS et al (2021) Tocilizumab in hospitalized patients with severe covid‐19 pneumonia. N Engl J Med 384: 1503–1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer LA, Weigert M, Günther U, Maghelli N, Jug F, Sbalzarini IF, Myers EW (2015) ClearVolume: open‐source live 3D visualization for light‐sheet microscopy. Nat Methods 12: 480–481 [DOI] [PubMed] [Google Scholar]
- Sharma K, Dhar N, Thacker VV, Simonet TM, Knott G, McKinney JD (2021) Dynamic persistence of intracellular bacterial communities of uropathogenic Escherichia 1 coli in a human bladder‐chip model of urinary tract infections 2. bioRxiv 10.1101/2021.01.03.424836 [PREPRINT] [DOI] [PMC free article] [PubMed]
- Song J, Hu B, Qu H, Wang L, Huang X, Li M, Zhang M (2020) Upregulation of angiotensin converting enzyme 2 by shear stress reduced inflammation and proliferation in vascular endothelial cells. Biochem Biophys Res Commun 525: 812–818 [DOI] [PubMed] [Google Scholar]
- Tanaka T, Narazaki M, Kishimoto T (2014a) Il‐6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol 6: 16295–16296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Narazaki M, Ogata A, Kishimoto T (2014b) A new era for the treatment of inflammatory autoimmune diseases by interleukin‐6 blockade strategy. Semin Immunol 26: 88–96 [DOI] [PubMed] [Google Scholar]
- Teuwen LA, Geldhof V, Pasut A, Carmeliet P (2020) COVID‐19: the vasculature unleashed. Nat Rev Immunol 20: 389–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thacker VV, Dhar N, Sharma K, Barrile R, Karalis K, McKinney JD (2020) A lung‐on‐chip model of early M. tuberculosis infection reveals an essential role for alveolar epithelial cells in controlling bacterial growth. Elife 9: e59961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoms M, Buschauer R, Ameismeier M, Koepke L, Denk T, Hirschenberger M, Kratzat H, Hayn M, Mackens‐Kiani T, Cheng J et al (2020) Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS‐CoV‐2. Science 369: 1249–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrelles JB, Schlesinger LS (2017) Integrating Lung physiology, immunology, and tuberculosis. Trends Microbiol 25: 688–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng C‐TK, Tseng J, Perrone L, Worthy M, Popov V, Peters CJ (2005) Apical entry and release of severe acute respiratory syndrome‐associated coronavirus in polarized calu‐3 lung epithelial cells. J Virol 79: 9470–9479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- V'kovski P, Gultom M, Steiner S, Kelly J, Russeil J, Mangeat B, Cora E, Pezoldt J, Holwerda M, Kratzel A et al (2020) Disparate temperature‐dependent virus – host dynamics for SARS‐CoV‐2 and SARS‐CoV in the human respiratory epithelium. PLOS Biol 19: e3001158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdembri D, Caswell PT, Anderson KI, Schwarz JP, König I, Astanina E, Caccavari F, Norman JC, Humphries MJ, Bussolino F et al (2009) Neuropilin‐1/GIPC1 signaling regulates α5β1 integrin traffic and function in endothelial cells. PLoS Biol 7: e1000025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villenave R, Wales SQ, Hamkins‐Indik T, Papafragkou E, Weaver JC, Ferrante TC, Bahinski A, Elkins CA, Kulka M, Ingber DE (2017) Human Gut‐On‐A‐Chip supports polarized infection of coxsackie B1 virus in vitro. PLoS One 12: e0169412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Luo R, Zhang M, Wang Y, Song T, Tao T, Li Z, Jin L, Zheng H, Chen W et al (2020) A cross‐talk between epithelium and endothelium mediates human alveolar–capillary injury during SARS‐CoV‐2 infection. Cell Death Dis 11: 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weibel ER (2015) On the tricks alveolar epithelial cells play to make a good lung. Am J Respir Crit Care Med 191: 504–513 [DOI] [PubMed] [Google Scholar]
- Wu K, Zou J, Chang HY (2020) RNA‐GPS predicts SARS‐CoV‐2 RNA localization to host mitochondria and nucleolus. Cell Syst 11: 102–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia H, Cao Z, Xie X, Zhang X, Chen JYC, Wang H, Menachery VD, Rajsbaum R, Shi PY (2020) Evasion of type I interferon by SARS‐CoV‐2. Cell Rep 33: 108234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Cao J, Wang Q, Shi Q, Liu K, Luo Z, Chen X, Chen S, Yu K, Huang Z et al (2020) D‐dimer as a biomarker for disease severity and mortality in COVID‐19 patients: a case control study. J Intensive Care 8: 49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youk J, Kim T, Evans KV, Jeong Y‐I, Hur Y, Hong SP, Kim JH, Yi K, Kim SY, Na KJ et al (2020) Robust three‐dimensional expansion of human adult alveolar stem cells and SARS‐CoV‐2 infection. Cell Stem Cell 27: 905–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelaya H, Rothmeier AS, Ruf W (2018) Tissue factor at the crossroad of coagulation and cell signaling. J Thromb Haemost 16: 1941–1952 [DOI] [PubMed] [Google Scholar]
- Zhang JJ, Kelm RJ, Biswas P, Kashgarian M, Madri JA (2007) PECAM‐1 modulates thrombin‐induced tissue factor expression on endothelial cells. J Cell Physiol 210: 527–537 [DOI] [PubMed] [Google Scholar]
- Zhang M, Wang P, Luo R, Wang Y, Li Z, Guo Y, Yao Y, Li M, Tao T, Chen W et al (2021) Biomimetic human disease model of SARS‐CoV‐2‐induced lung injury and immune responses on organ chip system. Adv Sci 8: 2002928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J, Wong L‐Y, Li K, Verma AK, Ortiz ME, Wohlford‐Lenane C, Leidinger MR, Knudson CM, Meyerholz DK, McCray PB et al (2021) COVID‐19 treatments and pathogenesis including anosmia in K18‐hACE2 mice. Nature 589: 603–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zunke F, Rose‐John S (2017) The shedding protease ADAM17: Physiology and pathophysiology. Biochim Biophys Acta ‐ Mol Cell Res 1864: 2059–2070 [DOI] [PubMed] [Google Scholar]
- Zuo Yu, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, Blair CN, Weber A, Barnes BJ, Egeblad M et al (2020) Neutrophil extracellular traps in COVID‐19. JCI . Insight 5: e138999 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Data Availability Statement
No primary datasets have been generated and deposited as part of this study. The source data generated during and/or analysed during the current study are available from the corresponding authors on reasonable request and will be made available at the EPFL Community Page on Zenodo upon publication; the data will be made available to all users as requested publication.