

Abstract
Hepatitis B virus (HBV) persists by depositing a covalently closed circular DNA (cccDNA) in the nucleus of infected cells that cannot be targeted by available antivirals. Interferons can diminish HBV cccDNA via APOBEC3‐mediated deamination. Here, we show that overexpression of APOBEC3A alone is not sufficient to reduce HBV cccDNA that requires additional treatment of cells with interferon indicating involvement of an interferon‐stimulated gene (ISG) in cccDNA degradation. Transcriptome analyses identify ISG20 as the only type I and II interferon‐induced, nuclear protein with annotated nuclease activity. ISG20 localizes to nucleoli of interferon‐stimulated hepatocytes and is enriched on deoxyuridine‐containing single‐stranded DNA that mimics transcriptionally active, APOBEC3A‐deaminated HBV DNA. ISG20 expression is detected in human livers in acute, self‐limiting but not in chronic hepatitis B. ISG20 depletion mitigates the interferon‐induced loss of cccDNA, and co‐expression with APOBEC3A is sufficient to diminish cccDNA. In conclusion, non‐cytolytic HBV cccDNA decline requires the concerted action of a deaminase and a nuclease. Our findings highlight that ISGs may cooperate in their antiviral activity that may be explored for therapeutic targeting.
Keywords: APOBEC3A, chronic hepatitis B, HBV, interferon alpha, interferon gamma
Subject Categories: Immunology; Microbiology, Virology & Host Pathogen Interaction
Hepatitis B virus deposits a covalently closed circular (ccc)DNA in the nucleus of infected cells to persist. Interferons can purge cccDNA from the nucleus by inducing deaminases and the nuclease ISG20 that modify and subsequently degrade cccDNA.

Introduction
Despite the availability of an effective vaccine, more than 255 million humans are suffering from chronic HBV infection and are at risk of developing liver cirrhosis or hepatocellular carcinoma (Thomas, 2019; WHO, 2019) causing 880,000 deaths every year (Naghavi et al, 2015; WHO, 2019). Current antivirals control HBV replication but cannot target its persistence form, the HBV covalently closed circular (ccc) DNA, in the nucleus of infected cells (Lucifora & Protzer, 2016; Xia & Guo, 2020).
HBV is a small, enveloped virus that belongs to the Hepadnaviridae family, a group of pararetroviruses that replicate via reverse transcription (RT) and express viral proteins from their nuclear transcription template, the cccDNA (Hu & Liu, 2017). Its genomic DNA is a partially double‐stranded relaxed circular DNA (rcDNA) genome. Upon receptor‐mediated entry into hepatocytes, the viral capsid is released into the cytoplasm and rcDNA is transferred into the nucleus where it is completed into the fully double‐stranded and covalently closed form of HBV cccDNA that remains episomal (Nassal, 2015).
In livers of chronically infected ducks, a mean of 10 duck HBV cccDNA copies/cell is reported (Zhang et al, 2003), while in human cell cultures, copy numbers of human HBV cccDNA are estimated between 1.4 and 9.6 copies/cell (Tropberger et al, 2015; Ko et al, 2018). cccDNA serves as the transcription template for all viral RNAs: subgenomic mRNAs for expression of the small, medium, and large (S, M, and L) HBV envelope proteins (Seeger & Mason, 2015) and the HBV x protein (HBx) that is essential to initiate and maintain viral transcription (Lucifora et al, 2011; Decorsiere et al, 2016); the pregenomic RNA encoding the viral polymerase and the HBV core protein forming the viral capsid; and a precore RNA translated into a protein that is processed and secreted as hepatitis B e antigen (HBeAg) (Seeger & Mason, 2015). Within the newly forming capsid, pregenomic RNA is reverse‐transcribed by the viral polymerase into the genomic rcDNA (Summers & Mason, 1982). These DNA‐containing capsids can either be transported back to the nucleus to increase the cccDNA copy number or be enveloped and secreted. By this means, cccDNA provides the source for new virions and is thus a major therapeutic target in chronic hepatitis B (Nassal, 2015; Lucifora & Protzer, 2016).
Half‐life of HBV cccDNA has not been exactly defined. In cell culture, in the absence of cell division, cccDNA appears to be rather stable (Cai et al, 2012; Ko et al, 2018) independently of its transcriptional activity (Lucifora et al, 2011). In vivo in the liver, cccDNA half‐life is influenced by a number of factors. Cytotoxic activity of T cells, e.g., during acute hepatitis, depletes cccDNA by induction of hepatocyte death (Murray et al, 2005). Interferons and T cell‐derived cytokines destabilize and diminish cccDNA in a non‐cytolytic fashion (Lucifora et al, 2014; Xia et al, 2016; Koh et al, 2018). Since cccDNA does not have a nuclear re‐targeting signal, it is not self‐maintained and may be lost during cell division, e.g., during liver regeneration, as has been demonstrated in humanized mice (Allweiss et al, 2018). However, cccDNA may also survive through mitosis through a “nuclear refill” that occurs via reimport of rcDNA from newly formed viral capsids into the nucleus. A rebound of HBV replication upon withdrawal from nucleos(t)ide analogue treatment and traces of HBV DNA remaining detectable years after clinical recovery from acute hepatitis (Rehermann et al, 1996) demonstrate that HBV cccDNA may be very long‐lived in the human liver and can persist for months (Huang et al, 2021).
Cytokine signaling can control cccDNA transcription and its epigenetic state, and limits stability of HBV transcripts and pregenomic RNA‐containing capsids (summarized in Xia & Protzer, 2017). Recently, we demonstrated that cccDNA levels can be reduced without hepatocyte cell death by treatment with interferon alpha (IFNα) or lymphotoxin beta receptor (LTβR) stimulation (Lucifora et al, 2014), but also by the T‐cell cytokines interferon gamma (IFNγ) and tumor necrosis factor alpha (TNFα) (Xia et al, 2016). These cytokines or LTβR ligands on non‐lytic T cells induce the expression of apolipoprotein B editing complex 3 (APOBEC3) enzymes (Lucifora et al, 2014; Xia et al, 2016; Koh et al, 2018), which elicit antiviral activity against a broad range of DNA viruses and retroviruses (Stavrou & Ross, 2015). APOBEC3 proteins are a family of cytidine deaminases leading to conversion of cytosines to uracils in DNA. All members of the APOBEC3 family except APOBEC3DE can hypermutate HBV (Janahi & McGarvey, 2013), but only APOBEC3A (A3A) that is an ISG and APOBEC3B (A3B) that is additionally expressed in an NFκB‐mediated fashion can directly target nuclear HBV DNA (Lucifora et al, 2014). During cytokine‐mediated targeting of HBV cccDNA, deamination by A3A or A3B is an essential step (Lucifora et al, 2014; Xia et al, 2016).
While the essential role of deaminases has been demonstrated, downstream enzymatic processes that determine whether cccDNA is repaired or degraded after deamination have not yet been defined. Stenglein et al (2010) described that interferon‐induced A3A deaminating foreign double‐stranded DNA generates a substrate for uracil DNA glycosylase (UNG) 2, and speculated that APEX1 might function as an endonuclease finally degrading the DNA. After treatment with transforming growth factor beta, HBV cccDNA is deaminated by activation‐induced cytidine deaminase (AICDA) that triggers uracil excision by UNG (Qiao et al, 2016). In the duck model of HBV infection, AICDA and UNG also trigger reduction of cccDNA (Chowdhury et al, 2013). However, the nuclease responsible for the degradation of nuclear HBV DNA has not been identified so far.
Therefore, the aim of the present study was to identify the nuclease involved in interferon‐induced purging of HBV cccDNA. To this end, we searched for interferon‐induced nucleases in gene expression analyses and confirmed the expression of the strongest induced nuclease, ISG20 (interferon‐stimulated gene product of 20 kDa), in liver tissue samples from patients with acute but not chronic hepatitis B. Knockdown and overexpression experiments confirmed the requirement of ISG20 for interferon‐induced cccDNA reduction. ISG20 together with A3A was sufficient to induce cccDNA loss without any further treatment, thereby opening perspectives for therapeutic applications in chronic hepatitis B.
Results
A3A deaminates HBV cccDNA dependent on transcription but is not sufficient to induce its degradation
We and others have previously shown that deamination by A3A is an essential step in IFNα‐induced purging of DNA from the nucleus of cells (Lucifora et al, 2014; Li et al, 2017; Zhou et al, 2019). Here, we confirmed IFNα‐induced upregulation of A3A, A3F, and A3G, but not of other APOBEC3 proteins or AICDA (Fig EV1A). Among the upregulated APOBEC3 proteins, only A3A is expressed in the nucleus of cells (Muckenfuss et al, 2006), a pre‐requisite characteristic for nuclear HBV cccDNA regulation, and thus, we knocked down A3A by small interfering RNA (siRNA) (Fig EV1A). Accordingly, we observed IFNα‐induced reduction of cccDNA that is abolished by A3A knockdown (Fig 1A) without cytotoxicity (Fig EV1B). In contrast, knockdown of A3A did not rescue the reduction of total intracellular HBV DNA (Fig EV1C) or HBeAg (Fig EV1D) by IFNα, suggesting that additional antiviral mechanisms mediated by IFNα apply.
Figure EV1. IFNα induces APOBEC3 proteins, of which A3A is knocked down by siRNA.

-
AdHepaRG cells were infected with HBV at a multiplicity of infection (MOI) of 100 virions/cell, transfected with siRNA against A3A (siA3A) or control siRNA (−) on day 6 postinfection (p.i.), and treated with 300 U/ml IFNα 1 day later for 6 h. Expression of indicated genes was analyzed by qRT–PCR relative to TATA‐box binding protein (TBP) mRNA (n = 3 biological replicates). mRNA from PBMCs was used as positive control in qRT–PCR for AICDA.
-
B–DdHepaRG cells were infected with HBV at a multiplicity of infection (MOI) of 100 virions/cell and transfected with siRNA against A3A (siA3A) or control siRNA (−) on day 8 postinfection (p.i.). Cells were treated with 300 U/ml IFNα from days 9 to 15 p.i., and cell viability was determined thereafter by CellTiter‐Blue (CTB) assay (B), total intracellular HBV DNA by qPCR relative to the prion protein (Prnp) gene (C), and HBeAg by ELISA (D) (n = 4 biological replicates).
Data information: Data are represented as mean ± standard deviation (SD), nd: not detected. *P < 0.05, **P < 0.01, ***P < 0.001, and ns: not significant by Student’s unpaired t‐test with Welch’s correction.
Figure 1. A3A deaminates HBV cccDNA dependent on transcription but is not sufficient to induce its degradation.

-
AdHepaRG cells were infected with HBV at a multiplicity of infection (MOI) of 100 virions/cell and transfected with siRNA against A3A (siA3A) or control siRNA (−) on day 8 postinfection (p.i.). Cells were treated with 300 U/ml IFNα from days 9 to 15 p.i., and HBV cccDNA was determined thereafter by qPCR relative to the prion protein (Prnp) gene (n = 4 biological replicates).
-
B–DdHepaRG‐TR‐X cells were infected with an HBx‐deficient HBV (HBVΔx) at an MOI of 200 virions/cell. Cells were treated with tetracycline to induce expression of HBx (+HBx) and/or 1,000 U/ml IFNα from days 10 to 17 p.i. HBeAg (B) was measured by ELISA (OD: optical density) and HBV cccDNA (C) by qPCR relative to Prnp (n = 3 biological replicates). cccDNA amplicons were analyzed by differential DNA denaturation PCR (3D‐PCR) (D) to visualize deamination.
-
E–GdHepaRG‐TR‐A3A cells were infected with HBV at an MOI of 300 virions/cell. Cells were treated with tetracycline (Tet (A3A)) or/and 300 U/ml IFNα from days 9 to 18 p.i. cccDNA amplicons were analyzed by 3D‐PCR (E). Marked amplicons were sequenced and analyzed for nucleotide composition (F) in comparison with the wild‐type HBV sequence (n = 3–5 biological replicates). HBV infection level was determined by measuring cccDNA, intracellular HBV DNA by qPCR relative to Prnp, or HBeAg by ELISA (G) (n = 6 biological replicates from two independent experiments).
Data information: Data are represented as mean ± standard deviation (SD), nd: not detected. *P < 0.05, **P < 0.01, ***P < 0.001, and ns: not significant by Student’s unpaired t‐test with Welch’s correction.
Since A3A acts on single‐stranded DNA (Hoopes et al, 2016), we investigated whether HBV cccDNA must be transcriptionally active to be deaminated, which requires the presence of HBx (Lucifora et al, 2011). Therefore, we studied the effect of IFNα on an HBx‐deficient HBV (HBVΔx) in differentiated HepaRG (dHepaRG) cells, where HBx can be reconstituted by tetracycline‐induced expression (HepaRG‐TR‐X cells). As expected, HBVΔx infection alone led to HBeAg levels below the detection limit due to disabled transcription from the cccDNA template and reconstitution of HBx allowed HBeAg transcription that was reduced under IFNα treatment (Fig 1B). IFNα did not affect cccDNA levels of HBVΔx over a time period of 15 days (Appendix Fig S1A) but under HBx‐mediated transcription, the cccDNA amount was reduced (Fig 1C). Analogously, cccDNA deamination under IFNα treatment appeared only when HBVΔx infection was complemented with HBx expression as seen in lower denaturing temperatures in differential DNA denaturation PCR (3D‐PCR) (Fig 1D) indicating that active transcription is required for cccDNA purging and deamination by A3A.
To further investigate whether activation of A3A expression—in addition to proteins constitutively expressed within the host cell—is sufficient for cccDNA removal or whether additional cytokine‐induced host factors are required, we generated HepaRG cells for conditional, tetracycline‐induced overexpression of A3A (HepaRG‐TR‐A3A). We chose HepaRG cells as they, upon differentiation, most closely resemble interferon responses observed in primary human hepatocytes (Lucifora et al, 2014), while other hepatoma cell lines such as HuH7 or HepG2 show much less pronounced interferon responses (Appendix Fig S1B). HepaRG‐TR‐A3A cells maintained the ability to differentiate into biliary‐ and hepatocyte‐like cells after 4 weeks of cell culture (Appendix Fig S1C) with hepatocyte‐like cells being susceptible to HBV infection (Appendix Fig S1D). Tetracycline‐inducible expression of A3A was confirmed after 2 and 7 days on mRNA and protein levels (Appendix Fig S1E).
Next, we studied the effect of A3A overexpression on cccDNA in HBV‐infected, dHepaRG‐TR‐A3A cells. A3A expression alone, IFNα treatment, or the combination of A3A expression and IFNα treatment was not toxic to the cells (Appendix Fig S1F) but resulted in lower denaturing temperatures of cccDNA amplicons in 3D‐PCR (Fig 1E), indicating cytosine deamination. Consistently, sequence analyses of amplicons at low denaturing temperatures revealed increased G‐to‐A conversions under treatments compared to controls (Appendix Fig S1G, Fig 1F). This indicated deamination of nuclear, minus‐strand HBV DNA during A3A expression, IFNα treatment, or the combination of both. To see whether HBV expression and replication levels were affected analogously, we measured HBV cccDNA, total intracellular HBV DNA, and HBeAg. As expected, IFNα treatment alone or combined with A3A expression reduced all viral markers. A3A expression on its own, however, did neither affect HBV DNA and cccDNA nor HBeAg levels (Fig 1G). Altogether, these results showed that A3A expression resulted in transcription‐dependent cccDNA deamination but was not sufficient to reduce cccDNA.
Differential gene expression analysis indicates ISG20 as candidate nuclease
As IFNα was required to reduce cccDNA, we hypothesized that IFNα induced at least one more factor enabling cccDNA degradation. To identify this factor, we performed gene expression analysis of HBV‐infected dHepaRG‐TR‐A3A cells comparing A3A induction alone to A3A induction in combination with IFNα treatment for six (Appendix Fig S2A) and 72 h (Appendix Fig S2B). Since non‐cytolytic purging of cccDNA by IFNα supposedly requires a nuclease (Lucifora et al, 2014), we focused on genes with known nuclease activity overexpressed in A3A‐expressing and IFNα‐treated cells compared to cells that were not treated with IFNα. After 6 h of treatment, expression levels of ISG20, PNPT1, XRN1, RAD9A, and PELO and after 72 h those of ISG20 and PNPT1 were increased (Fig 2A).
Figure 2. Differential gene expression analysis indicates ISG20 as candidate nuclease.

-
AA3A‐expressing dHepaRG‐TR‐A3A cells were infected with HBV at an MOI of 300 virions/cell and treated 7 days after infection with 300 U/ml IFNα (+IFNα) or without (−IFNα). Microarray data were analyzed for upregulated nucleases after 6 or 72 h of treatment, respectively.
-
B, CdHepaRG cells were infected with HBV at an MOI of 100 virions/cell and treated 9 days later with 300 U/ml IFNα (B) or 200 U/ml IFNγ (C) for indicated times (h: hours). Expression of indicated genes was analyzed by qRT–PCR relative to TATA‐box binding protein (TBP) mRNA (n = 6 biological replicates of two independent experiments).
-
DdHepaRG cells were infected with HBV at an MOI of 100 virions/cell or not infected and treated 7 days later with 300 U/ml IFNα for 6 h or 200 U/ml IFNγ for 24 h. ISG20 mRNA levels were measured by qRT–PCR relative to TBP mRNA (n = 6 biological replicates of two independent experiments).
-
EdHepaRG cells were infected with HBV at an MOI of 100 virions/cell and treated with 300 U/ml IFNα from days 7 to 9 p.i. or 200 U/ml IFNγ from days 7 to 10 p.i. Primary human hepatocytes were infected with HBV at an MOI of 100 virions/cell and treated with 500 U/ml IFNα or 200 U/ml IFNγ from days 4 to 6 p.i. Expression of ISG20 was detected by Western blot.
Data information: Data are represented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, and ns: not significant by Student’s unpaired t‐test with Welch’s correction.
Source data are available online for this figure.
Within the group of nucleases identified, we focused on ISG20, RAD9A, and PELO encoded enzymes that localize to the nucleus where cccDNA resides (Shamsadin et al, 2000; Yoshida et al, 2003; Chen et al, 2007; Lu et al, 2013; Nagarajan et al, 2013). Quantitative reverse transcription PCR (qRT–PCR) confirmed upregulation of ISG20 with a maximum 9 h after IFNα treatment (Fig 2B). Treatment with type II interferon IFNγ, that triggers similar signaling cascades as IFNα (Schneider et al, 2014) and shows the same effect on HBV cccDNA (Xia et al, 2016), also resulted in upregulation of ISG20 and to a minor extent RAD9A (Fig 2C) with slightly different kinetics. PELO, in contrast, was either unchanged or even downregulated by IFNα and IFNγ treatments (Fig 2B and C).
Since ISG20 was most prominently upregulated by both IFNα and IFNγ treatments, we first investigated whether HBV infection influenced the expression of this nuclease. HBV infection neither affected mRNA nor protein levels of ISG20 and also did not affect induction of ISG20 by IFNα or IFNγ treatment in dHepaRG cells and primary human hepatocytes (Fig 2D and E). This demonstrated that both type I and type II interferons induced expression of the sequence non‐specific, 3′ to 5′ exonuclease ISG20 in HBV‐infected cells, that targets RNA but to a lesser extent also DNA (Nguyen et al, 2001).
ISG20 is expressed in acute, self‐limiting HBV infection but not in chronic hepatitis B
Since ISG20 was the strongest upregulated nuclease in our analyses (84‐fold after 6 h and 4‐fold after 72 h by microarray, 6‐fold to 26‐fold by qRT–PCR) and was induced by both type I and type II interferons, we considered ISG20 as the prime candidate for DNA degradation in interferon‐induced purging of HBV cccDNA. To analyze whether ISG20 may be involved when immune responses restrict HBV infection from the majority of hepatocytes to a low percentage being infected (Guidotti et al, 1999), we analyzed liver tissue samples from HBV‐infected patients with different courses of infection by immunohistochemistry. We validated the antibody used herein by showing that it binds recombinant overexpressed ISG20‐mKate in HepG2‐NTCP cells (Appendix Fig S3A) and gives a specific signal in lymphoid and liver tissue at 1/100 dilution (Appendix Fig S3B). Indeed, we found strong ISG20 expression in livers of patients with acute, self‐limiting but not with chronic hepatitis B (Fig 3A). Quantitative analysis revealed a significantly higher percentage of ISG20‐positive areas per sample in acute hepatitis B compared to chronic hepatitis B or control samples (Fig 3B).
Figure 3. ISG20 is expressed in acute, self‐limiting HBV infection but not in chronic hepatitis B.
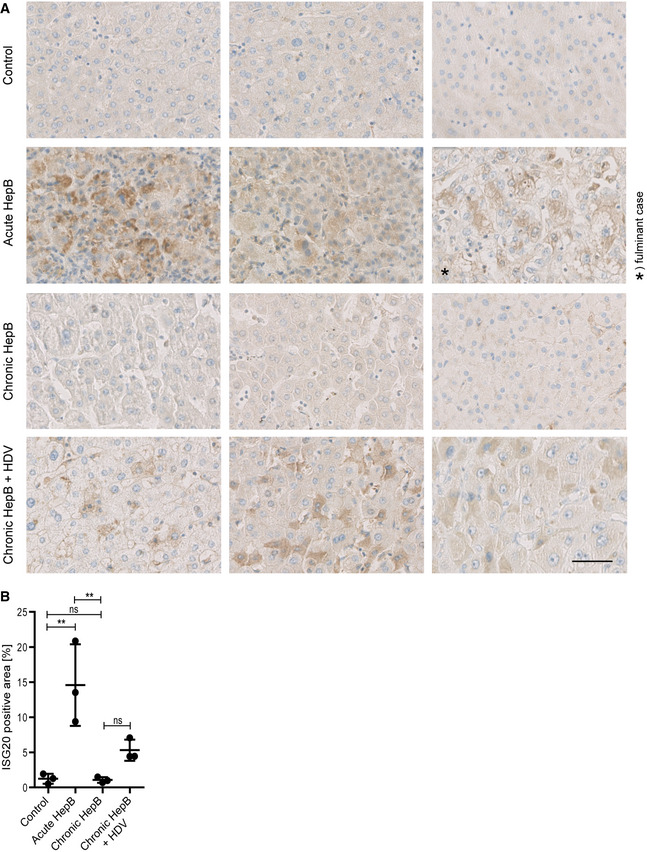
- Liver tissue samples obtained from HBV‐negative patients undergoing metastasis resection (control) or patients with acute, self‐limiting HBV infection or chronic hepatitis B, or chronic coinfection with HBV and hepatitis D virus (HDV) were stained for ISG20 by immunohistochemistry. For each clinical entity, tissue sections from three different patients are shown; scale bar: 50 µm.
- ISG20‐positive area of each sample (n = 3 biological replicates) was determined by Tissue IA image analysis software and is given in % of total tissue area scanned.
Data information: Data are represented as mean ± SD. **P < 0.01 and ns: not significant by one‐way ANOVA.
To determine whether triggering an immune response during chronic HBV infection can restore ISG20 expression, we additionally analyzed samples from chronic hepatitis B patients with hepatitis D virus (HDV) coinfection. HDV induces an innate immune response and expression of ISGs in livers of humanized mice (Giersch et al, 2015). HDV coinfection resulted in positive ISG20 staining (Fig 3A and B). In addition, liver tissue samples from patients mono‐ or coinfected with hepatitis C virus (HCV), which also activates an interferon response in a relevant proportion of patients (Heim & Thimme, 2014), stained positive for ISG20 (Fig EV2A and B). Thus, ISG20 was expressed in livers of patients with acute hepatitis B, acute hepatitis C or chronic hepatitis B upon coinfection with HDV or coinfection with HCV which both are known to suppress HBV infection (Cheng et al, 2020). This indicated that ISG20 is a relevant downstream molecule in immune control or interferon‐mediated control of HBV infection.
Figure EV2. ISG20 is expressed in HCV infection.

- Liver tissue samples obtained from HBV‐negative patients undergoing metastasis resection (control), with chronic hepatitis B (HepB) and HCV coinfection or acute hepatitis C (HepC) were stained for ISG20 by immunohistochemistry. For each clinical entity, tissue sections from three different patients are shown; scale bar: 50 µm.
- ISG20‐positive area of each sample was determined by Tissue IA image analysis software (n = 3 biological replicates) and is given in % of total tissue area scanned.
Data information: Data are represented as mean ± SD. ns: not significant by one‐way ANOVA.
ISG20 localizes to nucleoli after interferon treatment
To confirm that ISG20 localizes to the nucleus and to determine which compartment it localizes to, we investigated the localization of ISG20 within hepatocytes by immunofluorescence. We first validated specific detection of ISG20 when overexpressed as mKate fusion protein in HepG2‐NTCP cells (Appendix Fig S4A). As we detected additional punctuate, nuclear staining, we assumed that this was endogenous ISG20. To test this, we generated ISG20 knockout (HepaRG‐ISG20‐KO) cells. All three ISG20 knockouts and the non‐target control cells expressed MxA when treated with IFNα but only the control showed interferon‐induced ISG20 (Appendix Fig S4B and C).
Both IFNα and IFNγ induced ISG20 expression and localization into defined nuclear structures, in dHepaRG cells (Fig 4A), in primary human hepatocytes (Fig 4B), and in dHepG2NTCP cells (Appendix Fig S5A). We separated cytoplasm and nuclei of dHepaRG cells treated with IFNα or IFNγ to investigate the subcellular distribution of ISG20. Western blots for tubulin (cytoplasm fraction) and lamin A/C (nucleus fraction) confirmed efficient separation (Appendix Fig S5B), and ISG20 was present in both fractions after treatment with IFNα or IFNγ (Appendix Fig S5C). To evaluate whether nuclear ISG20 localizes to PML (promyelocytic leukemia) nuclear bodies, which are known to be induced by interferon and, e.g., control replication of negative‐strand RNA viruses (Chelbi‐Alix et al, 1998), we stained ISG20 and PML in HBV‐infected dHepaRG cells under interferon treatment. Both IFNα and IFNγ led to increased expression of ISG20 as well as PML in nuclei but ISG20 and PML showed distinct staining patterns and did not co‐localize (Fig 4C). Similarly, IFNα and IFNγ induced nuclear expressions of ISG20 and PML in dHepG2H1.3 cells but the two proteins did not co‐localize (Fig EV3A). To assure that ISG20 does not localize to PML nuclear bodies, we confirmed the identity of PML nuclear bodies by co‐staining PML and Daxx (death domain‐associated protein 6) (Fig EV3B).
Figure 4. ISG20 localizes to nucleoli after interferon treatment.

-
AdHepaRG cells were infected with HBV at an MOI of 100 virions/cell and treated with 600 U/ml IFNα or 400 U/ml IFNγ from days 10 to 14 p.i., and ISG20 was stained by immunofluorescence.
-
BPrimary human hepatocytes were infected with HBV at an MOI of 100 virions/cell and treated with 500 U/ml IFNα or 200 U/ml IFNγ from days 4 to 6 p.i., and ISG20 was stained by immunofluorescence.
-
C, DdHepaRG cells were infected with HBV at an MOI of 500 virions/cell, treated with 600 U/ml IFNα or 400 U/ml IFNγ from days 7 to 9 p.i., and stained by immunofluorescence for ISG20 and PML (C) or ISG20 and nucleophosmin (D).
-
EPrimary human hepatocytes were infected with HBV at an MOI of 100 virions/cell, treated with 500 U/ml IFNα or 200 U/ml IFNγ from days 4 to 6 p.i., and stained for ISG20 and nucleophosmin. White arrowheads indicate double‐positive nuclei.
Data information: Scale bars: 10 µm.
Figure EV3. ISG20 co‐localizes with nucleolar proteins and is expressed in HBV‐replicating cells.

-
A–DdHepG2H1.3 cells were treated with 1,500 U/ml IFNα or 1,000 U/ml IFNγ from days 3 to 4 after differentiation start and stained by immunofluorescence for ISG20 and PML (A), PML and Daxx (B), ISG20 and nucleophosmin (C), or ISG20 and a nucleolar marker protein (D). Scale bar: 10 µm.
-
EPrimary human hepatocytes were infected with an MOI of 200 virions/cell, treated with 500 U/ml IFNα or 200 U/ml IFNγ from days 5 to 6 p.i., and stained for ISG20 and HBV core protein. Scale bar: 50 µm.
Subsequently, we tested whether ISG20 localizes to nucleoli. Thus, we stained HBV‐infected dHepaRG cells after interferon treatment for ISG20 and nucleophosmin. IFNα‐ and IFNγ‐induced ISG20 showed very similar staining patterns compared to nucleophosmin with co‐localization of the two proteins confirmed by z‐stack analysis (Fig 4D). Analogously, interferon‐induced ISG20 co‐localized with nucleophosmin in dHepG2H1.3 cells (Fig EV3C) and a further nucleolus‐marker protein (Fig EV3D). Finally, we confirmed the co‐localization of nucleophosmin as marker for nucleoli with ISG20 in HBV‐infected primary human hepatocytes upon induction of ISG20 by IFNα or IFNγ (Fig 4E).
To see whether HBV‐infected cells can still express ISG20 upon interferon treatment, we stained them for HBV core protein and ISG20. Under both IFNα and IFNγ treatments, we found double‐positive dHepaRG cells (Appendix Fig S5D) and primary human hepatocytes (Fig EV3E). Taken together, these results demonstrate that interferon‐induced ISG20 does not localize to PML nuclear bodies but to nucleoli within the nuclei of HBV‐replicating hepatocytes.
ISG20 knockdown mitigates interferon‐induced decline of cccDNA
To prove the functional relevance of ISG20, we established a knockdown via siRNA or short hairpin RNA (shRNA) targeting ISG20 in HBV‐infected dHepaRG cells and evaluated whether this influenced the antiviral effect of IFNα. Transfection of HBV‐infected dHepaRG cells with ISG20 siRNA decreased basal and IFNα‐induced ISG20 expression (Fig EV4A). As expected, IFNα reduced cccDNA but when ISG20‐specific siRNA was transfected, the effect was reverted (Fig EV4B). Due to short half‐life of siRNA, however, IFNα could only be applied for 72 h and thus only had a moderate effect on cccDNA levels, total intracellular HBV DNA (Fig EV4C), and HBeAg (Fig EV4D). Comparing the effect of ISG20 on HBV DNA and ssRNA, we found that ssRNA is degraded almost completely, while ssDNA and dU‐containing ssDNA are only partially degraded (Fig EV4E). The ISG20 knockdown only reverted the effect of IFNα on HBV cccDNA but neither that on HBV DNA nor that on HBeAg due to transcriptional and post‐transcriptional antiviral effects of interferon (Uprichard et al, 2003; Belloni et al, 2012).
Figure EV4. ISG20 is knocked down by siRNA and binds and degrades ssDNA.
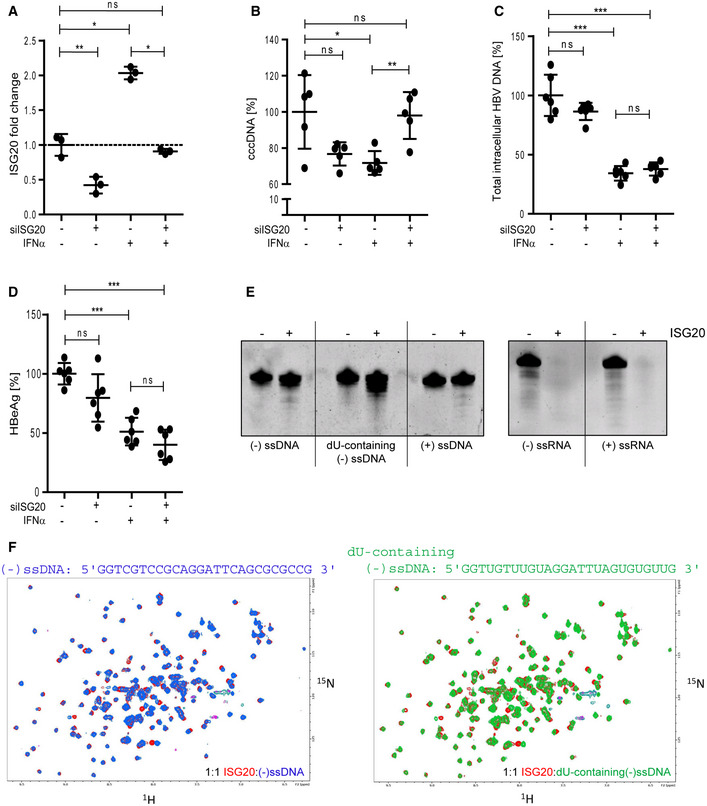
-
A–DdHepaRG cells were infected with HBV at an MOI of 100 virions/cell and transfected with siRNA targeting ISG20 (siISG20) or control siRNA (−) at day 7 p.i. and treated with 300 U/ml IFNα from days 8 to 11 p.i. ISG20 mRNA (A) was measured by qRT–PCR relative to TBP mRNA (n = 3 biological replicates). cccDNA (B) and total intracellular HBV DNA (C) were determined by qPCR relative to Prnp, and HBeAg (D) was determined by ELISA (n = 5–6 biological replicates from two independent experiments).
-
EssDNA oligomers with and without dU modifications (left panel) or ssRNA oligomers (right panel) were digested in vitro for 4 h using 7 µM oligomer and 2.5 µM recombinant ISG20 each and separated by polyacrylamide gel electrophoresis.
-
FNMR spectra of 15N‐labeled ISG20 bound to the (−) ssDNA and dU‐containing (−) ssDNA oligomers are shown. At a 1:1 ratio of oligomer to ISG20, both oligomers induce similar chemical shift perturbations in ISG20 amide resonances, as observed in the 1H,15N correlation NMR spectra.
Data information: Data are represented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, and ns: not significant by Student’s unpaired t‐test with Welch’s correction.
To reduce ISG20 expression levels in a more sustained fashion, we transduced HBV‐infected dHepaRG cells with adenoviral vectors expressing shRNA against ISG20. ISG20 protein expression was diminished by shRNA‐mediated knockdown after 7 days of IFNα or 12 days of IFNγ treatment (Fig 5A, Appendix Fig S6A). As expected, IFNα and IFNγ reduced cccDNA levels after 12 days of treatment but the shRNA‐mediated ISG20 knockdown partially (IFNα treatment) or even completely (IFNγ treatment) prevented cccDNA reduction (Fig 5B) proving that ISG20 is essential to purge cccDNA from HBV‐infected cells. Again, suppression of total intracellular HBV DNA as well as HBeAg by IFNα or IFNγ treatment was not rescued by ISG20 knockdown via shRNA (Fig 5C and D).
Figure 5. ISG20 knockdown mitigates interferon‐induced control of cccDNA.
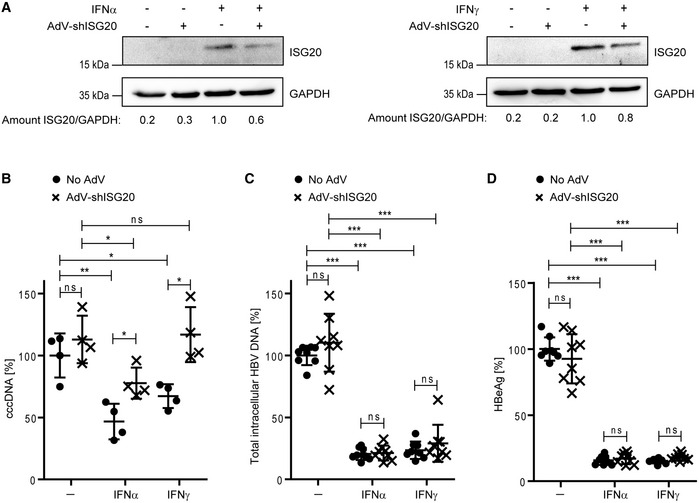
-
A–DdHepaRG cells were infected at an MOI of 100 virions/cell and transduced at day 7 p.i. with an adenoviral vector for expression of shRNA targeting ISG20 (AdV‐shISG20). After 2 days, cells were treated with 300 U/ml IFNα or 200 U/ml IFNγ. ISG20 expression (A) was analyzed by Western blot after 7 days of IFNα or 12 days of IFNγ treatment, respectively. The amount of ISG20/GAPDH was determined by signal density measurement. cccDNA (B) relative to Prnp (n = 4 biological replicates), total intracellular HBV DNA (C) relative to Prnp, and HBeAg (D) were measured 12 days after treatment (n = 8 biological replicates of two independent experiments). cccDNA was measured after T5 digestion of DNA.
Data information: Data are represented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, and ns: not significant by Student’s unpaired t‐test with Welch’s correction.
Source data are available online for this figure.
Southern blot analysis confirmed the elimination of capsid‐associated HBV DNA by interferon treatment (Appendix Fig S6B). No rescue of this effect was observed after ISG20 knockdown indicating that ISG20 directly targets cccDNA and the antiviral effects observed were not a consequence of targeting of pregenomic RNA which serves as template for HBV DNA reverse transcription within HBV capsids. In summary, ISG20 knockdown abolished the interferon‐triggered decline of cccDNA but had no effect on HBV DNA or HBeAg levels indicating that it specifically targets deaminated cccDNA.
ISG20 degrades double‐stranded (ds) and single‐stranded (ss) DNA and is enriched on deoxyuridine (dU)‐containing ssDNA
To test whether ISG20 can degrade HBV DNA sequences, we performed in vitro digestion experiments. First, we incubated recombinantly expressed and purified ISG20 (Appendix Fig S6C) with the 213 bp amplicon from 3D‐PCR, which is a dsDNA fragment of the HBV cccDNA. This double‐stranded 3D‐PCR amplicon (Fig 6A) and plus‐ and minus‐strand ssDNA and ssRNA oligonucleotides (Figs 6B and EV4E) were degraded in the presence of Mn2+, a cofactor necessary for ISG20 degradation activity (Nguyen et al, 2001). We selected the ssDNA oligonucleotides and the minus strand with deoxycytidines substituted by deoxyuridines (dU‐containing (−) ssDNA) as a 24‐nucleotide‐long fragment of the 3D‐PCR amplicon, because we observed substantial deamination by A3A and/or IFNα treatment in this region (Appendix Fig S1G). To confirm the direct interaction between the single‐stranded DNA oligomers and ISG20, we performed a single point binding mobility shift assay with the single‐stranded oligomers and ISG20 or recombinantly expressed and purified mA3A (mutant E72A, C171A) (Appendix Fig S6D). (−) ssDNA, dU‐containing (−) ssDNA, and (+) ssDNA all formed complexes with ISG20 (Fig 6C).
Figure 6. ISG20 degrades dsDNA and ssDNA and is enriched on deoxyuridine(dU)‐containing ssDNA.

- A 3D‐PCR amplicon was digested in vitro with recombinant ISG20 and increasing concentration of manganese chloride.
- A subsequence of the 3D‐PCR amplicon was used as (−) ssDNA oligomer, complementary (+) ssDNA, and a dU‐containing (−) ssDNA, where all cytosines were replaced by uracils. These oligomers were digested in vitro with recombinant ISG20 with or without EDTA as indicated.
- Recombinant ISG20 and mA3A (mutant E72A, C72A) were mixed with indicated oligomers and analyzed in a single point binding mobility shift assay.
- dHepG2H1.3 cells were treated for 1 day with 1,000 U/ml IFNγ, and harvested proteins were subjected to an affinity purification–mass spectrometry assay (n = 4 technical replicates). Oligomers used for pulldown were single‐stranded or double‐stranded DNA with and without dU as indicated. Significantly enriched protein groups in the volcano plot were identified via two‐sided Welch’s t‐tests (S0 = 1) corrected for multiple hypothesis testing applying a permutation‐based FDR (FDR < 0.01, 250 randomizations). Dashed line and blue colored points indicate a significant enrichment according to the Welch’s t‐test cut‐offs. Statistical significance of log2 iBAQ intensities of ISG20 enrichment was determined using a two‐sided Welch’s t‐test.
Data information: Data are represented as mean ± SD. ***P ≤ 0.001 and n.s.: not significant (P > 0.05) by two‐sided Welch’s t‐test.
To determine whether ISG20 recognizes the (−) ssDNA and dU‐containing (−) ssDNA oligomers differentially, we used NMR spectroscopy. In a single point titration, 1H,15N SOFAST‐HMQC experiments (Schanda & Brutscher, 2005) revealed that both oligomers induce similar chemical shift perturbations to amide resonances (Fig EV4F). These data corroborate previous findings by Nguyen et al (2001) that ISG20‐facilitated DNA and RNA degradation is not sequence‐specific.
Additionally, we performed affinity purification–mass spectrometry using the whole proteome of IFNγ‐treated dHepG2H1.3 cells and the oligomers (−) ssDNA and dU‐containing (−) ssDNA as baits. This resulted in ISG20 enrichment on the (−) ssDNA if it contained deoxyuridines as compared to the wild‐type (−) ssDNA sequence without deoxyuridines (Fig 6D) suggesting that other cellular proteins may support ISG20 binding to dU‐containing ssDNA. When we used the same oligomers and annealed them with the complementary (+) ssDNA oligomers to get dsDNA constructs as control, ISG20 was not enriched depending on deoxyuridine anymore (Fig 6D). Taken together, our in vitro analysis demonstrated that ISG20 can target both dsDNA and ssDNA for degradation and indicated that dU‐containing ssDNA is the primary target.
ISG20 overexpression together with A3A is sufficient for reduction of transcriptionally active cccDNA
To determine whether expression of ISG20 in addition to A3A is sufficient to purge HBV cccDNA, we overexpressed ISG20 by adenoviral transduction. Western blotting confirmed the expression of ISG20 after adenoviral transduction and tetracycline‐induced expression of A3A in dHepaRG‐TR‐A3A cells (Fig 7A). This allowed us to obtain either individual expression of A3A or ISG20 or the simultaneous expression of both proteins. Neither A3A nor ISG20 expression alone significantly affected cccDNA levels, but the combination of both enzymes led to a significant reduction of cccDNA to < 50% of the control (Fig 7B) without cytotoxicity (Appendix Fig S7A). Total intracellular HBV DNA was also reduced by concerted A3A and ISG20 expression, whereby already ISG20 alone led to a slight decrease of HBV DNA (Fig 7C). In addition, all HBV RNAs were moderately reduced by ISG20 as described before (Leong et al, 2016; Liu et al, 2017) or by combined ISG20 and A3A expression in infected dHepaRG‐TR‐A3A cells (Appendix Fig S7B).
Figure 7. ISG20 overexpression together with A3A is sufficient for reduction of transcriptionally active cccDNA.
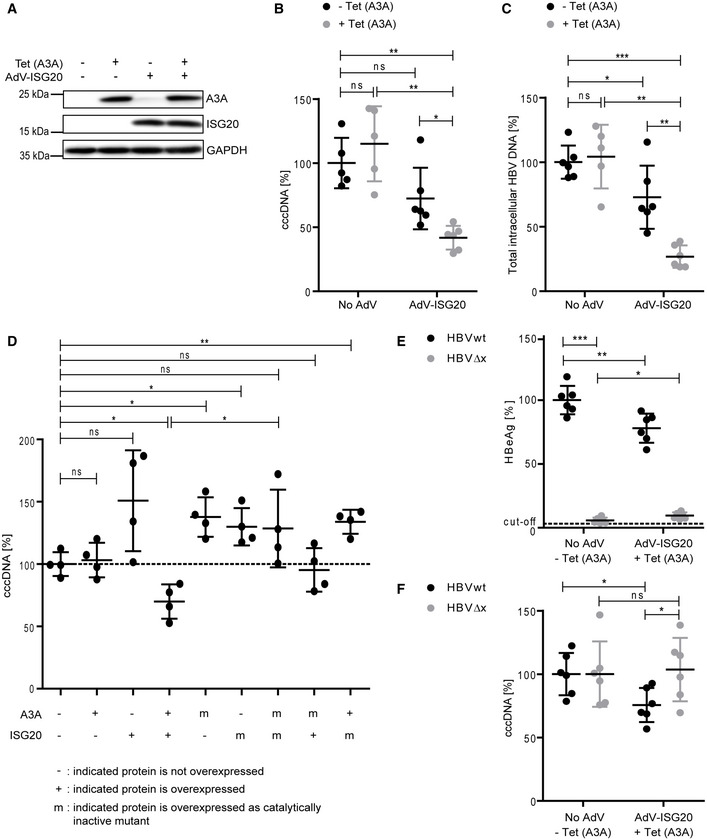
-
AdHepaRG‐TR‐A3A cells were treated with tetracycline to induce A3A expression (Tet(A3A)) or transduced with an adenoviral vector for expression of ISG20 (AdV‐ISG20). After 4 days, expression of A3A and ISG20 was detected by Western blot.
-
B, CdHepaRG‐TR‐A3A cells were infected with HBV at an MOI of 300 virions/cell and at day 7 p.i. transduced with AdV‐ISG20 and treated with tetracycline to induce A3A expression as indicated (± Tet (A3A)) for 7 days. Cell lysates were analyzed for cccDNA after T5 digest (B) and total intracellular HBV DNA (C) by qPCR relative to Prnp (n = 5–6 biological replicates from two independent experiments).
-
DPrimary human hepatocytes were infected with HBV at an MOI of 100 virions/cell and transfected on day 3 p.i. with mRNAs of A3A, ISG20, or catalytically inactive mutants thereof as indicated. Four days after transfection, cccDNA after T5 digest was measured by qPCR relative to Prnp (n = 4 biological replicates).
-
E, FdHepaRG‐TR‐A3A cells were infected with wild‐type HBV (HBVwt) or HBVΔx at an MOI of 300 virions/cell and transduced on day 7 p.i. with AdV‐ISG20 and treated with tetracycline to induce A3A expression as indicated (± Tet (A3A)) for 7 days. HBeAg (E) was measured by ELISA and cccDNA after T5 digest (F) by qPCR relative to Prnp (n = 6 biological replicates from two independent experiments).
Data information: Data are represented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, and ns: not significant by Student’s unpaired t‐test with Welch’s correction.
Source data are available online for this figure.
To confirm the effect, we overexpressed ISG20 in dHepG2H1.3‐A3A cells. These cells constantly express A3A and replicate HBV from a 1.3‐fold overlength genome supporting cccDNA formation via reimport of HBV‐genome‐containing capsids into the nucleus (Lucifora et al, 2014). After expression of ISG20 in dHepG2H1.3‐A3A cells using the adenoviral vector (Fig EV5A), cccDNA levels were reduced (Fig EV5B) without cytotoxicity (Appendix Fig S7C), confirming that the combined expression of A3A and ISG20 is sufficient to reduce HBV cccDNA levels.
Figure EV5. ISG20 overexpression together with A3A reduces cccDNA similar to interferon treatments.

-
A, BdHepG2H1.3‐A3A cells constantly expressing A3A (A3A Ctrl) were transduced 10 days after differentiation with an empty adenoviral vector (AdV) or AdV‐ISG20. ISG20 levels (A) were determined by qRT–PCR relative to TBP mRNA 1 day after transduction (n = 4 biological replicates). cccDNA (B) was measured after T5 digest 7 days after transduction by qPCR relative to Prnp (n = 4 biological replicates).
-
CdHepaRG‐TR‐A3A cells were infected with HBV at an MOI of 300 virions/cell and transduced on day 7 p.i. with AdV‐ISG20 and treated with tetracycline to induce A3A expression, 300 U/ml IFNα or 200 U/ml IFNγ. Seven days after transduction, cccDNA was measured after T5 digest by qPCR relative to Prnp (n = 5 biological replicates from two independent experiments).
-
D, EdHepaRG‐TR‐A3A cells were infected with wild‐type HBV (HBVwt) or HBVΔx at an MOI of 300 virions/cell and transduced on day 7 p.i. with AdV‐ISG20 and treated with tetracycline to induce A3A expression as indicated (± Tet (A3A)) for 7 days. Cell viability (D) was measured by CTB assay and total intracellular HBV DNA (E) by qPCR relative to Prnp (n = 6 biological replicates from two independent experiments).
Data information: Data are represented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, and ns: not significant by Student’s unpaired t‐test with Welch’s correction.
We tested further catalytically inactive mutants of A3A (mA3A) and ISG20 (mISG20) by transfection of in vitro‐transcribed (IVT) mRNAs for protein expression in primary human hepatocytes. Importantly, cccDNA levels were only reduced when both A3A and ISG20 were expressed in their catalytically active form but not if one or both of the expressed enzymes were mutated (Fig 7D). This result indicates that the enzymatic function of both the deaminase (A3A) and the nuclease (ISG20) are required to purge cccDNA. By measuring comparable amounts of mitochondrial DNA between the combined expression of A3A and ISG20 and the controls, we excluded a cytotoxic effect (Appendix Fig S7D).
To see whether the effect of combined A3A and ISG20 expression can be enhanced by addition of an interferon, we tested the different combinations in dHepaRG cells. IFNα or IFNγ treatment alone or combined with A3A and ISG20 expression led to cccDNA reduction without cytotoxicity (Appendix Fig S7E) but there was no increased effect when A3A and ISG20 overexpression was combined with interferon treatments (Fig EV5C). This might be a hint that A3A and ISG20 comprise the main axis of the interferon effect on cccDNA.
Finally, we tested whether cccDNA has to be transcriptionally active to be accessible for A3A and ISG20, as our initial experiments revealed that IFNα is only effective in reducing cccDNA when it is transcriptionally active (Fig 1C). Similarly, we infected dHepaRG‐TR‐A3A cells with HBVwt and HBVΔx, that lacks the HBx protein necessary for transcription from cccDNA (Lucifora et al, 2011), and expressed A3A and ISG20 in these cells without cytotoxicity (Fig EV5D). As expected, modest amounts of HBeAg (Fig 7E) and intracellular HBV DNA (mainly rcDNA) (Fig EV5E) were expressed from HBVΔx in comparison with HBVwt and combined A3A and ISG20 reduced both viral markers in the HBVwt infection. Importantly, only cccDNA from HBVwt was reduced by concerted A3A and ISG20 expression but not cccDNA from HBVΔx (Fig 7F) indicating that cccDNA has to be transcriptionally active to be vulnerable to A3A and ISG20 action.
In summary, these results show that catalytically active A3A and ISG20 together are sufficient to reduce transcriptionally active cccDNA without further treatment.
Discussion
IFNα (Lucifora et al, 2014) and IFNγ (Xia et al, 2016) can trigger a non‐cytolytic removal of HBV cccDNA from the nucleus of infected hepatocytes in an A3A deaminase‐dependent fashion. The nuclease which served to degrade the deaminated DNA, however, was unknown. In the present study, we identify ISG20 as the key nuclease involved in interferon‐triggered cccDNA purging. We show that ISG20 is expressed upon IFNα and IFNγ treatments in differentiated hepatocytes and in liver tissue samples of patients with acute, self‐limiting hepatitis B. However, it was not detected in chronic infection unless either HDV or HCV, RNA viruses that trigger IFN responses, coinfects the liver. Upon interferon induction, ISG20 localized to nucleoli rather than to PML nuclear bodies, where it may target HBV cccDNA. The knockdown of ISG20 blocked the interferon‐induced loss of cccDNA, and ISG20 in concert with A3A expression was sufficient to reduce transcriptionally active cccDNA when both enzymes were catalytically active. ISG20 was enriched on deoxyuridine‐containing single‐stranded DNA that mimicked A3A‐deaminated HBV DNA. Thus, we provide strong evidence that ISG20 is the key downstream nuclease, which finally cleaves “damaged” cccDNA.
ISG20 is long known to be induced by cellular responses to interferon signaling (Gongora et al, 1997; Espert et al, 2006; Liu et al, 2017) and is found in nuclei and cytoplasm of hepatocytes in response to IFNα treatment (Lu et al, 2013). ISG20 is a 3′ to 5′ exonuclease that—as the deaminase A3A—primarily acts on single‐stranded RNA and DNA substrates (Nguyen et al, 2001). Our study shows that similarly to A3A it can bind to single‐stranded DNA and efficiently degrades ssRNA and to a certain extent also single‐ and double‐stranded DNA in vitro. When the whole cellular proteome was present, ISG20 was enriched on deoxyuridine‐containing single‐stranded DNA, which mimicked A3A‐deaminated cccDNA in a transcriptionally active status. During active transcription, cccDNA needs to form a transcription bubble, which renders cccDNA partially single‐stranded (Liu et al, 2010; Nassal, 2015). This is in line with the observation that IFNα treatment and concerted A3A and ISG20 expression are only effective in reducing cccDNA levels when the HBx protein is expressed that enables transcription from cccDNA (Lucifora et al, 2011). The fact that ISG20 is an exonuclease (Nguyen et al, 2001) implies that its HBV DNA substrate has been damaged previously, i.e., the uracils introduced by APOBEC3 deaminases were removed and DNA strand breaks were formed. Accordingly, ISG20 expression alone was not sufficient to affect cccDNA levels in our experiments. However, a concerted expression of ISG20 and A3A reduced cccDNA. This effect was only achieved by expressing native A3A and ISG20 but not using catalytically inactive variants (Nguyen et al, 2001; Stenglein et al, 2010), demonstrating that deaminase and nuclease activities are required for cccDNA purging. Since A3A only deaminates cytosines to uracils, it seems reasonable that cellular factors constitutively expressed remove the uracils from the DNA leading to the formation of apurinic/apyrimidinic (AP) sites and introduce strand breaks. The resulting damaged cccDNA then becomes a substrate for ISG20. In line with this, AP sites have been shown to be formed during cccDNA purging induced by IFNα treatment or LTβR activation (Lucifora et al, 2014). AP sites have been reported to be generated by the glycosylase UNG in plasmid DNA that is degraded in an interferon‐dependent fashion (Stenglein et al, 2010) and to be involved in cccDNA loss after deamination by AICDA (Chowdhury et al, 2013; Qiao et al, 2016). Thus, it seems reasonable that deamination by APOBEC3 proteins is followed by further enzymatic steps generating damaged cccDNA, which is finally degraded by ISG20.
Recent studies have described additional mechanisms of viral inhibition by ISG20. Weiss et al, (2018) describe that ISG20 exerts indirect antiviral activity acting as a master regulator of type I interferon response proteins that prevent translation of infecting alphavirus RNAs in the cytoplasm. Wu et al, (2019) demonstrate that ISG20 inhibits translation of vesicular stomatitis virus RNA without degrading the RNA. They found that ISG20 exerts control over a large panel of non‐self RNAs while sparing endogenous transcripts and apparently is able to discriminate self from non‐self origin of cellular RNA through translation modulation.
Beside these indirect effects, degradation of viral RNAs has been described as ISG20’s most prominent antiviral effect. Expression of ISG20 inhibits HBV replication by diminishing HBV RNAs, whereby the exonuclease activity of ISG20 is required (Leong et al, 2016; Ma et al, 2016; Liu et al, 2017). Recently, it has been shown that degradation of HBV RNA depends on N6‐methyladenosine modification of nucleotide A1907 present twice in the terminally redundant genomic HBV RNA (Imam et al, 2020). Hereby, binding of ISG20 to the HBV RNA is mediated by a N6‐methyladenosine reader protein. Consistent with these findings, HBV RNAs as well as total intracellular HBV DNA were reduced in our experiments, when ISG20 was expressed in HBV‐infected cells. Intracellular HBV DNA consists mainly of cytoplasmic rcDNA, which is exclusively located inside capsids and therefore most likely protected from nuclease attack. Thus, the reduction of cytoplasmic HBV DNA may also be explained by direct degradation of pregenomic RNA by ISG20 (Leong et al, 2016; Liu et al, 2017; Imam et al, 2020) or steric inhibition of pregenomic RNA encapsidation by ISG20 (Liu et al, 2017). This, however, is not sufficient to explain the reduction of nuclear HBV cccDNA.
In our experiments, siRNA‐ or shRNA‐mediated knockdown of ISG20 could rescue cccDNA levels but did not influence interferon‐mediated reduction of HBV DNA or HBeAg. This strongly indicated that ISG20 activity targets cccDNA directly and not indirectly by reducing pregenomic RNA or the reimport of rcDNA‐containing HBV capsids into the nucleus. In addition, dHepaRG cells lack cccDNA amplification by nuclear capsid reimport or reinfection (Hantz et al, 2009). The effect of interferon on HBeAg and HBV rcDNA is explained by its known additional antiviral effects, such as IFNα‐mediated epigenetic silencing of cccDNA resulting in transcriptional inhibition (Belloni et al, 2012; Tropberger et al, 2015), the cytokine‐induced destabilization of HBV RNA (Heise et al, 1999), or the inhibition of the formation or accelerated decay of HBV nucleic acid‐containing capsids by type I or II interferons (Wieland et al, 2005; Xu et al, 2010). Taken together, this indicates that interferon‐induced ISG20 can directly deplete the nuclear cccDNA pool, further strengthening the notion that ISG20‐mediated cccDNA loss under interferon treatment is not an artifact of reduced cytoplasmic rcDNA levels but a consequence of interferon‐induced ISG20 targeting nuclear HBV cccDNA.
In our experiments, shRNA‐mediated knockdown of ISG20 abolished IFNγ‐induced cccDNA loss completely, but IFNα‐mediated reduction was only partially rescued. The weaker ISG20‐knockdown effect under IFNα treatment might reflect an incomplete knockdown. Another possible explanation is a compensation by other nucleases, which may assist the function of ISG20. Stenglein et al (2010) hypothesized that the nuclease APEX1 degrades foreign DNA after deamination by A3A (Stenglein et al, 2010), but a knockdown of APEX1 did not affect IFNγ‐mediated reduction of cccDNA in our experiments (Xia et al, 2016). Gene expression analyses by microarrays displayed the induction of additional IFNα‐induced nucleases. However, addition of interferons to cells that expressed both A3A and ISG20 could not enhance the effect on cccDNA decline caused already by A3A and ISG20.
By genome‐wide gene expression analysis and gene ontology analysis, we identified a number of interferon‐induced nucleases that could potentially affect cccDNA stability. However, their involvement is unlikely since they localize to the wrong cellular compartment or were not or only minimally induced by interferon in further experiments. RAD9A is a DNase with preference for single over double strands (Bessho & Sancar, 2000) localizing to the nucleus (Yoshida et al, 2003), but it was only weakly induced in dHepaRG cells by interferons. PNPT1 encodes for the RNase human polynucleotide phosphorylase (hPNPase) that localizes to the mitochondrial intermembrane space (Chen et al, 2007), and the exoribonuclease XRN1 is a cytoplasmic RNase (Nagarajan et al, 2013). PELO (protein pelota homolog) has a nuclear localization signal (Shamsadin et al, 2000), also localizes to the cytoskeleton, and might degrade aberrant mRNAs (Burnicka‐Turek et al, 2010), but we could not confirm its upregulation by interferons in dHepaRG cells.
In studies similar to ours, overexpression of the deaminase AICDA was reported to be sufficient to reduce cccDNA levels (Chowdhury et al, 2013; Qiao et al, 2016). We could not confirm an induction of AICDA and detect no decrease of cccDNA levels when only A3A was overexpressed, indicating that A3A alone is not sufficient to enable cccDNA degradation. When ISG20 was co‐expressed, cccDNA was diminished in our cell culture experiments in dHepG2H1.3‐A3A cells that were differentiated to stabilize cccDNA pools (Protzer et al, 2007), as well as in dHepaRG‐TR‐A3A cells, in which cccDNA is only derived from incoming virus (Hantz et al, 2009). We found the combined A3A and ISG20 expression to be sufficient to reduce cccDNA levels to less than 50% of control without further treatment. This finding may open the opportunity for therapeutic intervention by specifically triggering A3A and ISG20 expression or direct delivery of A3A and ISG20 to hepatocytes and bears the chance to avoid the side effects of systemic IFNα therapy (Janssen et al, 2005).
Studies with chimpanzees showed that ISG20 was expressed in the liver of acutely infected animals during viral clearance (Wieland et al, 2004), suggesting that ISG20 would be available for the elimination of HBV. We confirmed regulation of ISG20 through in vivo histological examination for ISG20 expression in liver tissue samples from patients with acute, self‐limiting hepatitis B. Notably, during chronic hepatitis B the expression level of ISG20 was as low as in control samples. During acute hepatitis B a strong, polyfunctional T‐cell response is observed (Bertoletti & Ferrari, 2012; Said & Abdelwahab, 2015) that has cytolytic, but also non‐cytolytic effects on HBV (Murray et al, 2005). Mathematical simulation of HBV clearance in chimpanzees supported the notion that during the early phase of HBV clearance, non‐cytopathic T‐cell effector mechanisms inhibit viral replication and greatly shorten the half‐life of the long‐lived cccDNA, thereby limiting the extent to which cytopathic T‐cell effector functions and tissue destruction are required to terminate acute HBV infection (Murray et al, 2005). In line with this observation, in situ hybridization showed increased mRNA amounts of A3A and A3B in human liver tissue in acute but not in chronic hepatitis B (Xia et al, 2016) and T‐cell cytokines IFNγ and tumor necrosis factor alpha (TNFα) (Xia et al, 2016) or T cell‐mediated LTβR activation (Koh et al, 2018) lead to non‐cytolytic cccDNA reduction, most likely via induction of A3 deaminases and ISG20.
During chronic HBV infection, in contrast, the T‐cell response is weak. HBV‐specific T cells are scarce and partially exhausted, and thus, there is little cytokine secretion (Gehring & Protzer, 2019; Rehermann & Thimme, 2019). Coinfection with HDV or HCV during chronic hepatitis B restored ISG20 expression in liver tissue samples. HDV (Giersch et al, 2015) and HCV (Su et al, 2002; Heim & Thimme, 2014) trigger innate immune responses and induce interferons. Accordingly, chronic hepatitis B patients responding to IFNα therapy showed increased ISG20 expression as compared to non‐responders (Xiao et al, 2012; Lu et al, 2013). Taken together, clinical data strongly support the notion that ISG20 is critically involved in HBV elimination in vivo and acts as the DNase targeting the HBV persistence form cccDNA during viral clearance.
In summary, this study shows that ISG20 is the nuclease responsible for an interferon‐induced decline of HBV cccDNA. Co‐expression of catalytically active ISG20 and A3A is sufficient to reduce HBV cccDNA, opening new avenues for therapeutic targeting of the HBV persistence form, which remains a difficult target for antiviral therapies that aim at curing chronic hepatitis B.
Materials and Methods
Materials availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the corresponding author, Ulrike Protzer (protzer@tum.de). Viral vectors and cell lines generated in this study will be available for research purposes on demand. This study did not generate additional new unique reagents.
Cell culture
HepaRG cells and derivates were cultured similar to described procedures (Lucifora et al, 2010) and grown for 2 weeks in supplemented William’s E medium (10% FBS Fetalclone II, 100 U/ml penicillin/streptomycin, 2 mM glutamine, 0.023 U/ml human insulin, 0.0047 mg/ml hydrocortisone, 0.08 mg/ml gentamicin) and differentiation for 2 weeks by adding 1.8% DMSO (Sigma‐Aldrich, Steinheim, Germany) to the growth medium. Generation and characterization of HepaRG‐TR‐X cells were described previously (Lucifora et al, 2011). To generate tetracycline‐inducible HepaRG‐TR‐A3A cells for overexpression of A3A, A3A cDNA was amplified from IFNγ‐treated, dHepaRG cells and cloned into pLenti4‐TO plasmid between a tetracycline‐regulated minimal CMV promoter and a V5 tag. Using this plasmid, lentiviral vectors were produced and HepaRG‐TR cells, which express the tetracycline repressor (TR), were transduced as described (Lucifora et al, 2011). Analogously, HepaRG cells with tetracycline‐inducible expression of NTCP (HepaRG‐TR‐NTCP) were generated. HepaRG‐ISG20‐KO cells were generated using LentiCRISPRv2‐plasmid and lentiviral transduction as published (Sanjana et al, 2014; Shalem et al, 2014) and gRNA duplexes as indicated in Appendix Table S1. The empty plasmid backbone was used as non‐target control. The dHepaRG cells were treated with 5 µg/ml tetracycline (Applichem, Darmstadt, Germany), 300–1,000 U/ml IFNα (interferon alpha‐2a/Roferon‐A, Roche, Vienna, Austria), or 200–400 U/ml IFNγ (interferon gamma‐1b/Imukin, Boehringer Ingelheim, Vienna, Austria) as indicated in figure legends.
HuH7 and HepG2‐based cells were cultured in supplemented DMEM (10% FCS, 100 U/ml penicillin/streptomycin, 2 mM glutamine, 1× non‐essential amino acids, 1 mM sodium pyruvate), and HepG2‐based cells were grown on collagen‐coated vessels. Undifferentiated HuH7, HepG2, and HepG2H1.3 cells, a hepatoma cell line replicating HBV from a single integrate of a 1.3‐fold overlength genome (Jost et al, 2007; Protzer et al, 2007), were treated with 1,000 U/ml IFNα. HepG2H1.3 and HepG2H1.3‐A3A cells, additionally expressing constantly A3A under a CMV promoter (Lucifora et al, 2014), were differentiated in DMEM by adding 1.74% DMSO and reducing FCS to 1%. HepG2NTCP cells (clone K7) were differentiated with 2.5% DMSO as described (Ko et al, 2018). dHepG2NTCP or dHepG2H1.3 cells were treated with 1,500 U/ml IFNα or 1,000 U/ml IFNγ.
Primary human hepatocytes were obtained from the Department of General, Visceral, and Transplant Surgery at Hannover Medical School and were isolated as described before (Kleine et al, 2014). Liver tissue was derived from donors undergoing partial hepatectomy upon written informed consent (approved by the ethic commission of Hannover Medical School/Ethik‐Kommission der MHH, #252‐2008). Primary human hepatocytes were cultured in HepaRG differentiation medium and treated with 500 U/ml IFNα or 200 U/ml IFNγ.
HBV infection
HBV stocks were prepared as described (Xia et al, 2016), and DNA‐containing enveloped particles (virions) were determined for calculation of the MOI. dHepaRG cells or primary human hepatocytes were infected overnight at an MOI of 100–1,000 virions/cell—as indicated in figure legends—in William’s E differentiation medium with 5% PEG6000 (Merck, Hohenbrunn, Germany). dHepG2NTCP cells were infected overnight at an MOI of 400 virions/cell in DMEM supplemented with 2.5% DMSO and 4% PEG6000 as described (Ko et al, 2018). The HBx‐deficient HBVΔx stock was produced as described (Lucifora et al, 2011; Xia et al, 2016), and dHepaRG cells were infected overnight at an MOI of 200 virions/cell in William’s E differentiation medium with 5% PEG8000 (Merck) or at an MOI of 300 virions/cell with 5% PEG6000 (Merck) as indicated.
Measurement of HBV markers
Total cellular DNA was extracted (NucleoSpin Tissue kit, Macherey‐Nagel, Düren, Germany), digested with T5 exonuclease (New England Biolabs, Ipswich, MA, USA) where indicated and used for measuring cccDNA by quantitative real‐time PCR (qPCR) on a LightCycler 480 system (Roche, Mannheim, Germany) as described (Xia et al, 2017). Total intracellular HBV DNA and Prnp as a reference gene were quantified using primer sets listed in Appendix Table S1 and cycling programs as described (Xia et al, 2017). Secreted HBeAg was measured with a commercial immunoassay (BEP III, Siemens Molecular Diagnostics, Marburg, Germany).
Differential DNA denaturation PCR (3D‐PCR)
To investigate deamination of nuclear HBV DNA, 3D‐PCR was applied as described (Xia et al, 2017). Briefly, 1/50‐diluted products from cccDNA qPCR were used as template in nested PCR with the primers 5′‐HBxin and 3′‐HBxin (Appendix Table S1) using a denaturing temperature gradient. Cytosine deamination results in GC‐to‐AT conversions and therefore lower melting temperatures of double‐stranded DNA allowing differential amplification of AT‐rich sequences (Suspene et al, 2005). Amplicons from 3D‐PCR were visualized on a 2% agarose gel, extracted, cloned in sequencing vectors, and analyzed by multiple sequence alignment and nucleotide composition.
Knockdown of A3A and ISG20
dHepaRG cells were transfected overnight with 0.75 µl DharmaFECT 1 siRNA Transfection Reagent (GE Healthcare Dharmacon, Lafayette, CO, USA) per well of a 12‐well plate according to the manufacturer’s instructions. 20 nM Accell human APOBEC3A (200315) siRNA SMARTpool (#E‐017432‐00‐0005, GE Healthcare Dharmacon), 20 nM Accell human ISG20 siRNA SMARTpool (#E‐015994‐00‐0005, GE Healthcare Dharmacon), or 20 nM Accell non‐targeting pool siRNA (#D‐001910‐10‐05, GE Healthcare Dharmacon) was applied. Alternatively, synthetic oligonucleotides (sequences adapted from The RNAi Consortium Collection, Mission shRNA, Sigma‐Aldrich; listed in Appendix Table S1) were annealed and ligated into a HindIII‐ and SalI‐digested pEntry backbone with H1 promoter to generate adenoviral vectors following the Gateway approach (pAd/PL‐DEST Gateway Vectors and Virapower Adenoviral Expression Systems, Invitrogen, Carlsbad, CA, USA). Resulting constructs were transfected in HEK293 cells to amplify the adenoviral vector AdV‐shISG20, and infectious units (i.u.) were determined in HEK293 cells. dHepaRG cells were transduced using an MOI of 3 i.u./cell in DMEM without supplements for 2 h.
Quantitative reverse transcription PCR (qRT–PCR)
RNA was extracted according to the manufacturer’s instructions (NucleoSpin RNA isolation kit, Macherey‐Nagel) and reverse‐transcribed into cDNA (SuperScript III First‐Strand Synthesis SuperMix for qRT–PCR, Invitrogen). 1/10‐diluted cDNA was used with LightCycler 480 SYBR Green I Master Mix (Roche) in qPCR (95°C, 300 s; (95°C, 15 s; 60°C, 10 s; 72°C, 25 s, single acquisition) *45 cycles; melting curve with continuous acquisition). Primer sequences are listed in Appendix Table S1. qPCRs ran on a LightCycler 480 system (Roche), and data were analyzed by advanced relative quantification considering primer efficiency and normalization to a reference gene.
Cell viability assays
In dHepaRG cells, viability was measured by CellTiter‐Blue Cell Viability Assay (Promega, Madison, WI, USA) according to the manufacturer’s instructions. Alternatively, CytoTox‐ONE Homogeneous Membrane Integrity Assay (Promega) was used for dHepG2H1.3‐A3A cells to determine the percentage of cell death relative to a complete lysis control according to the manufacturer’s instructions.
Microarray‐based gene expression analysis
Total RNA was extracted from cell culture samples using the AllPrep DNA/RNA Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions. RNA integrity was assessed using the 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) in combination with the RNA 6000 Nano Kit (Agilent Technologies): All samples showed a RIN of > 8.5. Global gene expression profiling was performed using SurePrint G3 Human Gene Expression 8 × 60k microarrays (v2, AMADID 039494, Agilent Technologies) according to the manufacturer’s protocol with an input of 25 ng of total RNA (one‐color Low Input Quick Amp Labeling Kit, Agilent Technologies). Hybridized microarrays were scanned with a G2505C Sure Scan Microarray Scanner (Agilent Technologies), and raw data were extracted using the Feature Extraction 10.7 software (Agilent Technologies). Data quality assessment, preprocessing, normalization, and differential expression analyses were conducted with the R Bioconductor packages limma (Ritchie et al, 2015) and Agi4x44PreProcess, whereas Benjamini–Hochberg‐adjusted P‐values (FDR (Benjamini & Hochberg, 1995)) smaller than 0.1 were considered statistically significant. Gene ontology (GO) terms of the resulting gene lists were extracted deploying the Ensembl BioMart database. The data have been deposited in NCBI's Gene Expression Omnibus (Edgar et al, 2002) and are accessible through GEO Series accession number GSE134929 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE134929).
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and Western blot analyses
For A3A and MxA Western blots, cells were lysed in M‐PER Mammalian Protein Extraction Reagent (Thermo Scientific, Rockford, IL, USA) with protease inhibitors (Complete, Roche) and mixed with a loading dye (LDS Sample Buffer, Non‐Reducing (4X), Thermo Scientific) before protein separation in a 12.5% SDS–PAGE or 7.5% SDS–PAGE, respectively. Proteins were blotted onto a methanol‐activated PVDF membrane (Amersham Hybond PVDF Membrane, GE Healthcare Life Sciences, Freiburg, Germany) in a semi‐dry manner. For ISG20 and MxA (if shown together with ISG20) Western blot, cells were lysed in Pierce RIPA buffer (Thermo Scientific) with protease inhibitors. Proteins were separated in a 12% SDS–PAGE and blotted onto a methanol‐activated PVDF membrane (Bio‐Rad, Hercules, CA, USA) in a wet blot procedure. Following antibodies were used: MxA (#13750‐1‐AP, ProteinTech Group, IL, USA) in 1/1,000‐dilution, β‐actin (#A5441, Sigma‐Aldrich) in 1/10,000‐dilution, V5 tag (#ab27671, Abcam, Cambridge, United Kingdom) in 1/5,000‐dilution for detection of V5‐tagged A3A, GAPDH (#ACR001PT, Acris, Herford, Germany) in 1/5,000‐dilution, ISG20 (#ab154393, Abcam) in 1/500‐dilution, α‐tubulin (#T5168, Sigma‐Aldrich) in 1/5,000‐dilution, lamin A/C (#sc‐376248, Santa Cruz Biotechnology) in 1/200‐dilution, a peroxidase‐labeled, secondary antibody against mouse (#A0168, Sigma‐Aldrich) in 1/10,000‐dilution, and a peroxidase‐labeled, secondary antibody against rabbit (#A0545, Sigma‐Aldrich) in 1/10,000‐dilution. All blots were developed using Amersham ECL Prime Western Blotting Detection Reagent (GE Healthcare Life Sciences). Where applicable, cells were separated in a cytoplasm and a nucleus fraction by CelLytic NuCLEAR Extraction Kit (NXTRACT, Sigma‐Aldrich) and subjected to Western blot in a wet blot procedure as described above.
Immunofluorescence staining
For immunofluorescence, dHepaRG cells, dHepG2NTCP cells or dHepG2H1.3 cells were fixed for 10 min using 4% paraformaldehyde (Roth, Karlsruhe, Germany) and permeabilized with PBS/0.5% saponin (Roth) for 10 min at room temperature. Blocking was performed with PBS/0.1% saponin/10% goat serum (Abcam). Fixed cells were incubated with following primary antibodies at 4°C overnight: rabbit‐anti‐ISG20 (#ab154393, Abcam) diluted 1/100, mouse‐anti‐PML (#sc‐966, Santa Cruz Biotechnology, Santa Cruz, CA, USA) diluted 1/300, rabbit‐anti‐Daxx (#07‐471, Upstate/Merck Millipore, Billerica, MA, USA) diluted 1/200, mouse‐anti‐nucleophosmin (#325200, Life Technologies/Invitrogen) diluted 1/200, mouse‐anti‐nucleolus (#ab190710, Abcam) diluted 1/200, and mouse‐anti‐core protein (#sc‐23945, Santa Cruz Biotechnology) diluted 1/50. After washing (PBS/0.1% saponin), an appropriate secondary antibody (goat‐anti‐rabbit 568 in 1/500‐dilution, goat‐anti‐rabbit 488 in 1/1,000‐dilution, goat‐anti‐rabbit 594 in 1/1,000‐dilution, goat‐anti‐mouse 647 in 1/1,000‐dilution) was added for 2 h at room temperature before slides were covered with DAPI mounting medium (Roche). Fluorescence microscopy was carried out with a confocal laser scanning microscope (Olympus FluoView 1000; Olympus, Hamburg, Germany) or a spinning disk microscope (Nikon Ti Eclipse microscope equipped with a PerkinElmer UltraView Vox System (Yokogawa CSU‐X1)). Spinning disk microscopy images were analyzed with the Volocity 6.2 software package (PerkinElmer, Waltham, USA).
Immunohistochemistry
Liver and lymphoid tissues were provided by DZIF Gewebebank Heidelberg and handled according to the guidelines of the tissue bank of the University of Heidelberg after a positive vote of the local ethics commission. Paraffin‐embedded liver tissue sections came from patients with acute or chronic hepatitis B, chronic hepatitis B with HDV coinfection, chronic hepatitis B with HCV coinfection or acute hepatitis C. As control, liver tissues obtained from metastasis resection but distant to the metastasis and from otherwise healthy patients were used. For each clinical entity, liver tissue samples from three different patients were stained with an antibody against ISG20 (H‐50, #sc‐66937, Santa Cruz Biotechnology) in 1/200‐dilution as described (Lu et al, 2013). Lymphoid and liver tissue for antibody dilution testing were stained accordingly. For quantification, image analysis software Tissue IA (Slidepath, Leica, Wetzlar, Germany) was used with optimized color detection and quantification algorithms at 40× magnification. Data are presented as DAB positive area in % of total recognizable tissue area (each sample 3–20 mm2 tissue area analyzed).
ISG20 in vitro digestion and binding assays
The 3D‐PCR amplicon was generated as described above and purified from an agarose gel. The 24‐nucleotide oligomers used herein are listed in Appendix Table S1 as 033, 034, 035, 036, 039, and 040 and were purified on 20% urea‐denaturing polyacrylamide gels. The oligomers were extracted from the gel using electroelution, followed by equilibration against water (for degradation and binding assays) or buffer containing 150 mM NaCl and 25 mM sodium phosphate, pH 6.5 (for NMR evaluation).
ISG20 was expressed and purified as previously described (Nguyen et al, 2001) and equilibrated in buffer containing 150 mM NaCl, 25 mM sodium phosphate, pH 6.5, and 5 mM DTT. mA3A contained two point mutations for increased solubility (E72A, C171A) (Bohn et al, 2015).
Degradation assays were carried out as described previously by Nguyen et al, (2001). Briefly, oligomers were incubated with ISG20 at a 3:1 molar ratio at 37°C for up to 6 h in buffer containing 13.5 mM NaCl, 6.5 mM HEPES pH 8, 2 mM MgCl2, and 2 mM MnCl2. Inhibition assays included 5 mM EDTA (as indicated in the figures). Reactions were quenched with 5 mM EDTA, supplemented with denaturing loading dye (8 M urea, 100 mM Tris pH 8, 0.1% xylene cyanol, and 0.1% bromophenol blue), and loaded on an 8% (3D‐PCR amplicon) or 20% (24‐nucleotide oligomers) urea‐denaturing polyacrylamide gel. Gels were run for 1.5 h at 12 W, followed by staining with SYBR Gold for 5 min prior to imaging.
Binding shift assays were performed by incubating 0.2 µM of each oligomer with 2.5 µM ISG20 or mA3A (in the same buffer used for degradation assays but supplemented with 5% glycerol), on ice for 10 min prior to loading on a 20% native polyacrylamide gel.
For nuclear magnetic resonance (NMR) spectroscopy, single point titration 1H,15N SOFAST‐HMQC experiments (Schanda & Brutscher, 2005) with 30 µM 15N‐labeled ISG20 (90% H2O/10% D2O) and 24‐nucleotide oligomers 033 and 035 were carried out on an 800 MHz Bruker NMR spectrometer equipped with a cryogenic probe at 298 K.
Affinity purification–mass spectrometry
HepG2H1.3 cells were differentiated for 10 days and treated for 1 day with 1,000 U/ml IFNγ. PBS‐washed cells were harvested with a cell scraper, and pellets were snap‐frozen in liquid nitrogen. DNA oligomers used herein are listed in Appendix Table S1 as 033, 034, and 036. Oligomers (033 and 034) were denatured (99°C; 10 min) and chilled on ice (single‐stranded versions) or mixed in a 1:1 ratio (033:036 and 034:036), denatured, and slowly cooled down at room temperature (double‐stranded versions). StrepTactin sepharose slurry (IBA, Göttingen, Germany) was washed three times and reconstituted in TAP buffer (50 mM Tris/HCl pH 7.5, 5% glycerol, 0.2% NP‐40, 1.5 mM MgCl2, 100 mM NaCl) in 10x volume. Per sample, 1 µg ssDNA or 2 µg dsDNA was incubated with 100 µl reconstituted StrepTactin beads at 4°C for 1 h. Cell pellets were lysed in TAP buffer including protease inhibitors on ice for 20 min and centrifuged thereafter (10,000 g, 15 min, 4°C). Supernatants were then cleared by incubation with washed and reconstituted StrepTactin beads (3.3× volume TAP buffer) in a 10:1 ratio (15 min, 4°C, rotary wheel) and centrifugation. Protein concentration was determined by absorbance at 280 nm (NanoDrop, Thermo Scientific) and Pierce 660 nm Protein Assay (Thermo Scientific). ssDNA‐ or dsDNA‐bound beads were washed three times with TAP buffer, resuspended in TAP buffer including protease inhibitors, and incubated with 2 mg protein lysate per sample (1 h, 4°C). After washing in TAP buffer and TAP buffer without NP‐40, beads were harvested (2,000 g, 1 min, 4°C). Enriched proteins on agarose beads were denatured, reduced, and alkylated by sequential addition of 40 µl U/T buffer (6 M urea, 2 M thiourea, 10 mM HEPES, pH 8.0), 10 mM DTT (30 min), and 55 mM IAA (20 min, in the dark). For protein digestion, 160 µl digestion buffer (50 mM ammonium bicarbonate, pH 8.0) containing 0.5 µg LysC (FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan) and 0.5 µg trypsin (Progema) was added to the beads and incubated at 25°C overnight. Peptides were desalted and concentrated using three layers of C18 StageTips (3 M) as described previously (Rappsilber et al, 2007). Purified peptides were loaded onto a 50 cm reverse‐phase analytical column (75 µm diameter; ReproSil‐Pur C18‐AQ 1.9 µm resin; Dr. Maisch GmbH, Ammerbuch, Germany) and separated using an EASY‐nLC 1200 system (Thermo Fisher Scientific) with a binary buffer system of buffer a (0.1% formic acid), buffer b (80% ACN, 0.1% formic acid), and a 120‐min gradient (5–30% buffer b for 90 min, 30% to 95% for 20 min, wash‐out at 95% for 5 min, readjustment to 5% in 5 min) at a flow rate of 300 nl per min. Eluting peptides were directly analyzed on a Q‐Exactive HF mass spectrometer (Thermo Fisher Scientific). Data‐dependent acquisition included repeating cycles of one MS1 full scan (300–1,600 m/z, resolution of 60,000) at an ion target of 3 × 106, followed by 15 MS2 scans of the highest abundant isolated and higher‐energy collisional dissociation (HCD) fragmented peptide precursors (resolution of 15,000). For MS2 scans, collection of isolated peptide precursors was limited by an ion target of 1 × 105 and a maximum injection time of 120 ms. Isolation and fragmentation of the same peptide precursor was eliminated by dynamic exclusion for 20 s. Raw files were processed with MaxQuant (version 1.6.10.43; Cox Lab, Max‐Planck‐Institute of Biochemistry, Martinsried, Germany) using the standard settings, label‐free quantification (iBAQ; LFQ, LFQ min ratio count 1, normalization type none, stabilize large LFQ ratios disabled) and match between run options enabled. Spectra were searched against forward and reverse sequences of the reviewed human proteome including isoforms (UniProtKB, release 10.2019) and HBV proteins by the built‐in Andromeda search engine (Tyanova et al, 2016a).
The output of MaxQuant was analyzed with Perseus (version 1.6.10.43 (Tyanova et al, 2016b)), R (version 3.6.0), and RStudio (version 1.2.1335). Detected protein groups identified as known contaminants, reverse sequence matches or only identified by site as well as replicate samples with poor quality were excluded from the analysis. Following log2 transformation, iBAQ intensities were normalized to correct for technical variation by using the intensity of unspecifically bound proteins that were quantified in all samples. Following normalization, proteins without quantification in at least three replicates of one condition were removed and missing values were imputed for each replicate individually by sampling values from a normal distribution calculated from the original data distribution (width = 0.3×SD, downshift = −1.8×SD). Differentially expressed protein groups in the volcano plot were identified via two‐sided Welch’s t‐tests (S0 = 1) corrected for multiple hypothesis testing applying a permutation‐based FDR (FDR < 0.01, 250 randomizations). Statistical significance of log2 iBAQ intensities of ISG20 enrichment was determined using a two‐sided Welch’s t‐test: ***P ≤ 0.001 and n.s.: not significant (P > 0.05). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez‐Riverol et al, 2019) partner repository with the dataset identifier PXD019285.
Overexpression of ISG20 and A3A
For overexpression of ISG20, ISG20 cDNA was PCR‐amplified from IFNγ‐treated dHepaRG cells and ligated into a HindIII‐ and BamHI‐digested pEntry backbone with CMV promoter to generate adenoviral vectors following the Gateway approach (pAd/PL‐DEST Gateway Vectors and Virapower Adenoviral Expression Systems, Invitrogen). Resulting constructs were transfected in HEK293 cells to amplify the adenoviral vector AdV‐ISG20 and an empty pEntry‐CMV vector‐derived control vector (AdV). dHepaRG or dHepG2H1.3‐A3A cells were transduced using an MOI of 3 i.u./cell in DMEM without supplements for 2 h.
For overexpression of ISG20‐mKate fusion protein, the backbone was gained from pNTCP‐mKate2 (Van de Wiel et al, 2016) by EcoRI and BamHI digestion. The ISG20 insert was PCR‐amplified from above‐mentioned pEntry‐ISG20, digested with EcoRI, BamHI, and DpnI, and ligated into the backbone to get pISG20‐mKate. pISG20‐mKate was transfected into HepG2NCTP cells using 3 µl lipofectamine 2000 (Thermo Scientific) and 5 µg plasmid DNA per well of a 12‐well plate and fixed 3 days later for staining and/or fluorescence detection.
For overexpression of proteins via IVT mRNA transfection, following templates were used: For A3A and ISG20, cDNA from IFNγ ‐treated dHepaRG cells in plasmid backbones was used as original template. For mISG20, three point mutations were inserted (D11G, D94G, D154G) to get a catalytically inactive mutant (Nguyen et al, 2001) into the plasmid template. For mA3A, an inactivating point mutation was inserted (E72A) (Stenglein et al, 2010) into the plasmid template. These plasmid templates were subjected to PCRs to introduce the T7 promoter and a NLS sequence with the primers listed in Appendix Table S1 and used thereafter as templates for IVT mRNA synthesis. 300 ng of each purified template (High Pure PCR Product Purification Kit, Roche) was used for IVT mRNA synthesis with the HiScribe T7 ARCA mRNA Kit (with tailing) (New England Biolabs) according to the manufacturer’s instructions but with addition of 1.25 mM pseudo‐UTP and 1.25 mM 5‐methyl‐CTP (Jena Bioscience, Jena, Germany) per reaction. Cleaned IVT mRNAs (RNA Clean and Concentrator‐25, Zymo Research, Freiburg, Germany) were then transfected overnight to primary human hepatocytes using 22.5 µl Lipofectamine MessengerMAX (Thermo Fisher Scientific) and 75 ng per single IVT mRNA per well of a 12‐well plate. IVT mRNA of red fluorescent protein was used as control for transfection.
Southern blot analysis for intracellular capsid‐associated HBV DNA and northern blot analysis for HBV RNAs
Southern blot to detect intracellular capsid‐associated HBV DNA was performed as described (Ko et al, 2018). Briefly, after cell lysis (50 mM Tris–HCl (pH 8.0), 100 mM NaCl, 1 mM EDTA, 1% NP‐40), samples were incubated for 15 min on ice and centrifuged at 15,322 g for 15 min at 4 °C. Supernatants were adjusted to 11 mM MgCl2, 200 µg/ml RNaseA, and 0.02 U/ml DNase I, incubated at 37°C for 3 h, and centrifuged at 15,322 g for 15 min at 4°C. HBV capsids were precipitated from supernatants with PEG8000 overnight on ice. After centrifugation (15,322 g, 15 min, 4°C), viral DNA was extracted from the pellet using the QIAamp DNA Mini Kit (Qiagen) according to the manufacturer’s instructions. Viral DNA forms were separated on a 1.3% agarose gel, transferred to a nylon membrane (Hybond‐XL, Amersham Biosciences), hybridized with a digoxigenin‐labeled HBV probe, and developed with the DIG Luminescent Detection Kit (Roche). Northern blot was performed as described before (Ko et al, 2018). Briefly, total RNA was extracted from cell culture using TRIzol Reagent (Ambion/Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s instructions, separated through a 1% formaldehyde agarose gel, blotted onto a nylon membrane (Hybond‐XL, Amersham Biosciences), and hybridized as described above.
Statistical analysis
Numeric values are represented as means with standard deviation (SD); P‐values were calculated with Student’s unpaired two‐tailed t‐test with Welch’s correction or one‐way ANOVA using Prism 5.01 software (Graph Pad, La Jolla, CA, USA). Statistical analyses of microarray and mass spectrometry data are given in the respective sections.
Author contributions
Conceptualization: DS, MH, SS, AP, RBa, MS, KU, UP; Methodology: DS, MK, ANJ, JH, CU, YX, AO, CK, WMC, AG, JMW, SS; Investigation: DS, MK, ANJ, JH, CU, JS, YX, AO, FN, RBe, FL, MR, CK, W‐MC, MAvdK, JMW. Formal analysis: DS, JH, CU, MR; Resources: PS, AP, KU; Data Curation: JH, CU, AP, KU; Writing Original Draft: DS, UP; Writing – Reviewing & Editing: MK, ANJ, JH, CU, YX, AO, MR, CK, WMC, MAvdK, PS, MH, SS, AP, RBa, MS, KU; Visualization: DS, MK, ANJ, JH, CU, YX, MR; Project administration: DS, UP; Supervision: AP, RBa, MS, KU, UP; Funding Acquisition: UP.
Conflict of interest
The position of Daniela Stadler was in part financed by a research grant from ALIOS BioPharma. Ulrike Protzer is shareholder and board member of SCG Cell Therapy. The remaining authors declare no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Appendix
Review Process File
Source Data for Figure 2E
Source Data for Figure 5A
Source Data for Figure 7A
Acknowledgements
We thank Nehal Omar, Aaron Selmeier, and Veronika Geißler for excellent technical support and Lotte Glück for coordination of liver tissue stainings. We thank Julie Lucifora (INSERM, Lyon, France) for generating HepaRG‐TR‐NTCP cells and Florian Vondran (Medical School Hannover, Germany) for providing primary human hepatocytes. This work was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—Projektnummer 272983813—TRR 179 (to UP, RBa, AP, SS, and MH) and TRR 237 (to AP) and by a research grant of ALIOS BioPharma to Technical University of Munich. Open access funding enabled and organized by Projekt DEAL.
EMBO reports (2021) 22: e49568.
Data availability
The datasets produced in this study are available in the following databases:
Gene expression data (microarray): Gene Expression Omnibus GSE134929 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE134929).
Mass spectrometry proteomics data: PRIDE PXD019285 (http://www.ebi.ac.uk/pride/archive/projects/PXD019285).
References
- Allweiss L, Volz T, Giersch K, Kah J, Raffa G, Petersen J, Lohse AW, Beninati C, Pollicino T, Urban S et al (2018) Proliferation of primary human hepatocytes and prevention of hepatitis B virus reinfection efficiently deplete nuclear cccDNA in vivo. Gut 67: 542–552 [DOI] [PubMed] [Google Scholar]
- Belloni L, Allweiss L, Guerrieri F, Pediconi N, Volz T, Pollicino T, Petersen J, Raimondo G, Dandri M, Levrero M (2012) IFN‐α inhibits HBV transcription and replication in cell culture and in humanized mice by targeting the epigenetic regulation of the nuclear cccDNA minichromosome. J Clin Invest 122: 529–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B 57: 289–300 [Google Scholar]
- Bertoletti A, Ferrari C (2012) Innate and adaptive immune responses in chronic hepatitis B virus infections: towards restoration of immune control of viral infection. Gut 61: 1754–1764 [DOI] [PubMed] [Google Scholar]
- Bessho T, Sancar A (2000) Human DNA damage checkpoint protein hRAD9 Is a 3′ to 5′ exonuclease. J Biol Chem 275: 7451–7454 [DOI] [PubMed] [Google Scholar]
- Bohn MF, Shandilya SM, Silvas TV, Nalivaika EA, Kouno T, Kelch BA, Ryder SP, Kurt‐Yilmaz N, Somasundaran M, Schiffer CA (2015) The ssDNA mutator APOBEC3A is regulated by cooperative dimerization. Structure 23: 903–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnicka‐Turek O, Kata A, Buyandelger B, Ebermann L, Kramann N, Burfeind P, Hoyer‐Fender S, Engel W, Adham IM (2010) Pelota interacts with HAX1, EIF3G and SRPX and the resulting protein complexes are associated with the actin cytoskeleton. BMC Cell Biol 11: 28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Mills C, Yu W, Yan R, Aldrich CE, Saputelli JR, Mason WS, Xu X, Guo J‐T, Block TM et al (2012) Identification of disubstituted sulfonamide compounds as specific inhibitors of hepatitis B virus covalently closed circular DNA formation. Antimicrob Agents Chemother 56: 4277–4288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelbi‐Alix MK, Quignon F, Pelicano L, Koken MH, de The H (1998) Resistance to virus infection conferred by the interferon‐induced promyelocytic leukemia protein. J Virol 72: 1043–1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H‐W, Koehler CM, Teitell MA (2007) Human polynucleotide phosphorylase: location matters. Trends Cell Biol 17: 600–608 [DOI] [PubMed] [Google Scholar]
- Cheng X, Uchida T, Xia Y, Umarova R, Liu C‐J, Chen P‐J, Gaggar A, Suri V, Mücke MM, Vermehren J et al (2020) Diminished hepatic IFN response following HCV clearance triggers HBV reactivation in coinfection. J Clin Invest 130: 3205–3220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury S, Kitamura K, Simadu M, Koura M, Muramatsu M (2013) Concerted action of activation‐induced cytidine deaminase and uracil‐DNA glycosylase reduces covalently closed circular DNA of duck hepatitis B virus. FEBS Lett 587: 3148–3152 [DOI] [PubMed] [Google Scholar]
- Decorsière A, Mueller H, van Breugel PC, Abdul F, Gerossier L, Beran RK, Livingston CM, Niu C, Fletcher SP, Hantz O et al (2016) Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature 531: 386–389 [DOI] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE (2002) Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30: 207–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espert L, Eldin P, Gongora C, Bayard B, Harper F, Chelbi‐Alix MK, Bertrand E, Degols G, Mechti N (2006) The exonuclease ISG20 mainly localizes in the nucleolus and the Cajal (Coiled) bodies and is associated with nuclear SMN protein‐containing complexes. J Cell Biochem 98: 1320–1333 [DOI] [PubMed] [Google Scholar]
- Gehring AJ, Protzer U (2019) Targeting innate and adaptive immune responses to cure chronic HBV infection. Gastroenterology 156: 325–337 [DOI] [PubMed] [Google Scholar]
- Giersch K, Allweiss L, Volz T, Helbig M, Bierwolf J, Lohse AW, Pollok JM, Petersen J, Dandri M, Lutgehetmann M (2015) Hepatitis delta co‐infection in humanized mice leads to pronounced induction of innate immune responses in comparison to HBV mono‐infection. J Hepatol 63: 346–353 [DOI] [PubMed] [Google Scholar]
- Gongora C, David G, Pintard L, Tissot C, Hua TD, Dejean A, Mechti N (1997) Molecular cloning of a new interferon‐induced PML nuclear body‐associated protein. J Biol Chem 272: 19457–19463 [DOI] [PubMed] [Google Scholar]
- Guidotti LG, Rochford R, Chung J, Shapiro M, Purcell R, Chisari FV (1999) Viral clearance without destruction of infected cells during acute HBV infection. Science 284: 825–829 [DOI] [PubMed] [Google Scholar]
- Hantz O, Parent R, Durantel D, Gripon P, Guguen‐Guillouzo C, Zoulim F (2009) Persistence of the hepatitis B virus covalently closed circular DNA in HepaRG human hepatocyte‐like cells. J Gen Virol 90: 127–135 [DOI] [PubMed] [Google Scholar]
- Heim MH, Thimme R (2014) Innate and adaptive immune responses in HCV infections. J Hepatol 61: S14–25 [DOI] [PubMed] [Google Scholar]
- Heise T, Guidotti LG, Cavanaugh VJ, Chisari FV (1999) Hepatitis B virus RNA‐binding proteins associated with cytokine‐induced clearance of viral RNA from the liver of transgenic mice. J Virol 73: 474–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoopes JI, Cortez LM, Mertz TM, Malc EP, Mieczkowski PA, Roberts SA (2016) APOBEC3A and APOBEC3B preferentially deaminate the lagging strand template during DNA replication. Cell Rep 14: 1273–1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Liu K (2017) Complete and incomplete hepatitis B virus particles: formation, function, and application. Viruses 9: 56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q, Zhou B, Cai D, Zong Y, Wu Y, Liu S, Mercier A, Guo H, Hou J, Colonno R et al (2021) Rapid turnover of HBV cccDNA indicated by monitoring emergence and reversion of signature‐mutation in treated chronic hepatitis B patients. Hepatology 73: 41–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imam H, Kim GW, Mir SA, Khan M, Siddiqui A (2020) Interferon‐stimulated gene 20 (ISG20) selectively degrades N6‐methyladenosine modified Hepatitis B Virus transcripts. PLoS Pathog 16: e1008338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janahi EM, McGarvey MJ (2013) The inhibition of hepatitis B virus by APOBEC cytidine deaminases. J Viral Hepatitis 20: 821–828 [DOI] [PubMed] [Google Scholar]
- Janssen HLA, van Zonneveld M, Senturk H, Zeuzem S, Akarca US, Cakaloglu Y, Simon C, So TMK, Gerken G, de Man RA et al (2005) Pegylated interferon alfa‐2b alone or in combination with lamivudine for HBeAg‐positive chronic hepatitis B: a randomised trial. Lancet 365: 123–129 [DOI] [PubMed] [Google Scholar]
- Jost S, Turelli P, Mangeat B, Protzer U, Trono D (2007) Induction of antiviral cytidine deaminases does not explain the inhibition of hepatitis B virus replication by interferons. J Virol 81: 10588–10596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleine M, Riemer M, Krech T, DeTemple D, Jäger MD, Lehner F, Manns MP, Klempnauer J, Borlak J, Bektas H et al (2014) Explanted diseased livers – A possible source of metabolic competent primary human hepatocytes. PLoS One 9: e101386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko C, Chakraborty A, Chou W‐M, Hasreiter J, Wettengel JM, Stadler D, Bester R, Asen T, Zhang Ke, Wisskirchen K et al (2018) Hepatitis B virus genome recycling and de novo secondary infection events maintain stable cccDNA levels. J Hepatol 69: 1231–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh S, Kah J, Tham CYL, Yang N, Ceccarello E, Chia A, Chen M, Khakpoor A, Pavesi A, Tan AT et al (2018) Non‐lytic lymphocytes engineered to express virus‐specific T‐cell receptors limit HBV infection by activating APOBEC3. Gastroenterology 155: 188–193 [DOI] [PubMed] [Google Scholar]
- Leong CR, Funami K, Oshiumi H, Mengao D, Takaki H, Matsumoto M, Aly HH, Watashi K, Chayama K, Seya T (2016) Interferon‐stimulated gene of 20 kDa protein (ISG20) degrades RNA of Hepatitis B virus to impede the replication of HBV in vitro and in vivo. Oncotarget 7: 68179–68193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Xia Y, Han M, Chen G, Zhang D, Thasler WE, Protzer U, Ning Q (2017) IFN‐alpha‐mediated base excision repair pathway correlates with antiviral response against hepatitis B virus infection. Sci Rep 7: 12715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Bushnell DA, Wang D, Calero G, Kornberg RD (2010) Structure of an RNA polymerase II–TFIIB complex and the transcription initiation mechanism. Science 327: 206–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Nie H, Mao R, Mitra B, Cai D, Yan R, Guo JT, Block TM, Mechti N, Guo H (2017) Interferon‐inducible ribonuclease ISG20 inhibits hepatitis B virus replication through directly binding to the epsilon stem‐loop structure of viral RNA. PLoS Pathog 13: e1006296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Qin B, Ma Q, Yang C, Gong XY, Chen LM (2013) Differential expression of ISG20 in chronic hepatitis B patients and relation to interferon‐alpha therapy response. J Med Virol 85: 1506–1512 [DOI] [PubMed] [Google Scholar]
- Lucifora J, Arzberger S, Durantel D, Belloni L, Strubin M, Levrero M, Zoulim F, Hantz O, Protzer U (2011) Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J Hepatol 55: 996–1003 [DOI] [PubMed] [Google Scholar]
- Lucifora J, Durantel D, Testoni B, Hantz O, Levrero M, Zoulim F (2010) Control of hepatitis B virus replication by innate response of HepaRG cells. Hepatology 51: 63–72 [DOI] [PubMed] [Google Scholar]
- Lucifora J, Protzer U (2016) Attacking hepatitis B virus cccDNA – The holy grail to hepatitis B cure. J Hepatol 64: S41–S48 [DOI] [PubMed] [Google Scholar]
- Lucifora J, Xia Y, Reisinger F, Zhang K, Stadler D, Cheng X, Sprinzl MF, Koppensteiner H, Makowska Z, Volz T et al (2014) Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 343: 1221–1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q, Qin B, Lu X, Kong L (2016) Interferon stimulated exonuclease gene 20 kDa promotes the activity of interferon‐alpha on the inhibition of hepatitis B virus replication via its exonuclease activity. Int J Clin Exp Med 9: 17940–17945 [Google Scholar]
- Muckenfuss H, Hamdorf M, Held U, Perkovic M, Löwer J, Cichutek K, Flory E, Schumann GG, Münk C (2006) APOBEC3 proteins inhibit human LINE‐1 retrotransposition. J Biol Chem 281: 22161–22172 [DOI] [PubMed] [Google Scholar]
- Murray JM, Wieland SF, Purcell RH, Chisari FV (2005) Dynamics of hepatitis B virus clearance in chimpanzees. Proc Natl Acad Sci USA 102: 17780–17785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan VK, Jones CI, Newbury SF, Green PJ (2013) XRN 5′ → 3′ exoribonucleases: Structure, mechanisms and functions. Biochim Biophys Acta 1829: 590–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naghavi M, Wang H, Lozano R, Davis A, Liang X, Zhou M, Vollset SE, Ozgoren AA, Abdalla S, Abd‐Allah F et al (2015) Global, regional, and national age‐sex specific all‐cause and cause‐specific mortality for 240 causes of death, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 385: 117–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassal M (2015) HBV cccDNA: viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 64: 1972–1984 [DOI] [PubMed] [Google Scholar]
- Nguyen LH, Espert L, Mechti N, Wilson DM 3rd (2001) The human interferon‐ and estrogen‐regulated ISG20/HEM45 gene product degrades single‐stranded RNA and DNA in vitro. Biochemistry 40: 7174–7179 [DOI] [PubMed] [Google Scholar]
- Perez‐Riverol Y, Csordas A, Bai J, Bernal‐Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M et al (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47: D442–d450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protzer U, Seyfried S, Quasdorff M, Sass G, Svorcova M, Webb D, Bohne F, Hosel M, Schirmacher P, Tiegs G (2007) Antiviral activity and hepatoprotection by heme oxygenase‐1 in hepatitis B virus infection. Gastroenterology 133: 1156–1165 [DOI] [PubMed] [Google Scholar]
- Qiao Y, Han X, Guan G, Wu N, Sun J, Pak V, Liang G (2016) TGF‐beta triggers HBV cccDNA degradation through AID‐dependent deamination. FEBS Lett 590: 419–427 [DOI] [PubMed] [Google Scholar]
- Rappsilber J, Mann M, Ishihama Y (2007) Protocol for micro‐purification, enrichment, pre‐fractionation and storage of peptides for proteomics using StageTips. Nat Protoc 2: 1896–1906 [DOI] [PubMed] [Google Scholar]
- Rehermann B, Ferrari C, Pasquinelli C, Chisari FV (1996) The hepatitis B virus persists for decades after patients' recovery from acute viral hepatitis despite active maintenance of a cytotoxic T–lymphocyte response. Nat Med 2: 1104–1108 [DOI] [PubMed] [Google Scholar]
- Rehermann B, Thimme R (2019) Insights from antiviral therapy into immune responses to hepatitis B and C virus infection. Gastroenterology 156: 369–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK (2015) limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res 43: e47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Said ZNA, Abdelwahab KS (2015) Induced immunity against hepatitis B virus. World J Hepatol 7: 1660–1670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, Zhang F (2014) Improved vectors and genome‐wide libraries for CRISPR screening. Nat Meth 11: 783–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schanda P, Brutscher B (2005) Very fast two‐dimensional NMR spectroscopy for real‐time investigation of dynamic events in proteins on the time scale of seconds. J Am Chem Soc 127: 8014–8015 [DOI] [PubMed] [Google Scholar]
- Schneider WM, Chevillotte MD, Rice CM (2014) Interferon‐stimulated genes: a complex web of host defenses. Annu Rev Immunol 32: 513–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger C, Mason WS (2015) Molecular biology of hepatitis B virus infection. Virology 480: 672–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi Xi, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG et al (2014) Genome‐scale CRISPR‐Cas9 knockout screening in human cells. Science 343: 84–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamsadin R, Adham IM, von Beust G, Engel W (2000) Molecular cloning, expression and chromosome location of the human pelota gene PELO. Cytogenet Genome Res 90: 75–78 [DOI] [PubMed] [Google Scholar]
- Stavrou S, Ross SR (2015) APOBEC3 proteins in viral immunity. J Immunol 195: 4565–4570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenglein MD, Burns MB, Li M, Lengyel J, Harris RS (2010) APOBEC3 proteins mediate the clearance of foreign DNA from human cells. Nat Struct Mol Biol 17: 222–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su AI, Pezacki JP, Wodicka L, Brideau AD, Supekova L, Thimme R, Wieland S, Bukh J, Purcell RH, Schultz PG et al (2002) Genomic analysis of the host response to hepatitis C virus infection. Proc Natl Acad Sci USA 99: 15669–15674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers J, Mason WS (1982) Replication of the genome of a hepatitis B–like virus by reverse transcription of an RNA intermediate. Cell 29: 403–415 [DOI] [PubMed] [Google Scholar]
- Suspene R, Henry M, Guillot S, Wain‐Hobson S, Vartanian JP (2005) Recovery of APOBEC3‐edited human immunodeficiency virus G‐>A hypermutants by differential DNA denaturation PCR. J Gen Virol 86: 125–129 [DOI] [PubMed] [Google Scholar]
- Thomas DL (2019) Global elimination of chronic hepatitis. N Engl J Med 380: 2041–2050 [DOI] [PubMed] [Google Scholar]
- Tropberger P, Mercier A, Robinson M, Zhong W, Ganem DE, Holdorf M (2015) Mapping of histone modifications in episomal HBV cccDNA uncovers an unusual chromatin organization amenable to epigenetic manipulation. Proc Natl Acad Sci USA 112: E5715–5724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Cox J (2016a) The MaxQuant computational platform for mass spectrometry‐based shotgun proteomics. Nat Protoc 11: 2301–2319 [DOI] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J (2016b) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13: 731–740 [DOI] [PubMed] [Google Scholar]
- Uprichard SL, Wieland SF, Althage A, Chisari FV (2003) Transcriptional and posttranscriptional control of hepatitis B virus gene expression. Proc Natl Acad Sci USA 100: 1310–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Wiel S, Merkx M, Van de Graaf S (2016) Real time monitoring of intracellular bile acid dynamics using a genetically encoded FRET‐based bile acid sensor. J Vis Exp e53659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss CM, Trobaugh DW, Sun C, Lucas TM, Diamond MS, Ryman KD, Klimstra WB (2018) The interferon‐induced exonuclease ISG20 exerts antiviral activity through upregulation of type I interferon response proteins. mSphere 3: e00209‐18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO (2019) Hepatitis B, Fact sheet, Updated 18 July 2019.
- Wieland SF, Eustaquio A, Whitten‐Bauer C, Boyd B, Chisari FV (2005) Interferon prevents formation of replication‐competent hepatitis B virus RNA‐containing nucleocapsids. Proc Natl Acad Sci USA 102: 9913–9917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieland S, Thimme R, Purcell RH, Chisari FV (2004) Genomic analysis of the host response to hepatitis B virus infection. Proc Natl Acad Sci USA 101: 6669–6674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N, Nguyen X‐N, Wang Li, Appourchaux R, Zhang C, Panthu B, Gruffat H, Journo C, Alais S, Qin J et al (2019) The interferon stimulated gene 20 protein (ISG20) is an innate defense antiviral factor that discriminates self versus non‐self translation. PLoS Pathog 15: e1008093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y, Guo H (2020) Hepatitis B virus cccDNA: Formation, regulation and therapeutic potential. Antiviral Res 180: 104824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y, Protzer U (2017) Control of hepatitis B virus by cytokines. Viruses 9: 18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y, Stadler D, Lucifora J, Reisinger F, Webb D, Hosel M, Michler T, Wisskirchen K, Cheng X, Zhang K et al (2016) Interferon‐gamma and tumor necrosis factor‐alpha produced by T cells reduce the HBV persistence form, cccDNA, without cytolysis. Gastroenterology 150: 194–205 [DOI] [PubMed] [Google Scholar]
- Xia Y, Stadler D, Ko C, Protzer U (2017) Analyses of HBV cccDNA quantification and modification. In Hepatitis B virus: methods and protocols, Guo H, Cuconati A (eds.), pp 59–72. New York, NY: Springer; [DOI] [PubMed] [Google Scholar]
- Xiao C, Qin B, Chen L, Liu H, Zhu Y, Lu X (2012) Preactivation of the interferon signalling in liver is correlated with nonresponse to interferon alpha therapy in patients chronically infected with hepatitis B virus. J Viral Hepatitis 19: e1–10 [DOI] [PubMed] [Google Scholar]
- Xu C, Guo H, Pan X‐B, Mao R, Yu W, Xu X, Wei L, Chang J, Block TM, Guo J‐T (2010) Interferons accelerate decay of replication‐competent nucleocapsids of hepatitis B virus. J Virol 84: 9332–9340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Wang HG, Miki Y, Kufe D (2003) Protein kinase Cdelta is responsible for constitutive and DNA damage‐induced phosphorylation of Rad9. EMBO J 22: 1431–1441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YY, Zhang BH, Theele D, Litwin S, Toll E, Summers J (2003) Single‐cell analysis of covalently closed circular DNA copy numbers in a hepadnavirus‐infected liver. Proc Natl Acad Sci USA 100: 12372–12377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Ren JH, Cheng ST, Xu HM, Chen WX, Chen DP, Wong VKW, Law BYK, Liu Y, Cai XF et al (2019) A functional variant in UBE2L3 contributes to HBV infection and maintains cccDNA stability by inducing degradation of APOBEC3A protein. Hepatology 69: 1885–1902 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Appendix
Review Process File
Source Data for Figure 2E
Source Data for Figure 5A
Source Data for Figure 7A
Data Availability Statement
The datasets produced in this study are available in the following databases:
Gene expression data (microarray): Gene Expression Omnibus GSE134929 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE134929).
Mass spectrometry proteomics data: PRIDE PXD019285 (http://www.ebi.ac.uk/pride/archive/projects/PXD019285).