

Supplemental Digital Content is available in the text
Keywords: antiretroviral therapy, cytokine storm, ex-vivo tissues, extracellular vesicles, HIV, human herpesvirus, immune activation
Abstract
Objective:
HIV-1 infection triggers immune activation, as reflected by the upregulation of various cytokines. This immune activation remains elevated despite antiretroviral therapy (ART) and leads to early age-related diseases. Here, we addressed the mechanisms of sustained immune activation in HIV-1-infected human lymphoid tissues ex vivo.
Design/method:
We investigated several potential causes of immunoactivation, including: a proinflammatory effect of ART drugs themselves; an early HIV-1-triggered cytokine storm, which could in turn trigger a sustained cytokine dysregulation; herpesvirus reactivation; HIV-1 protein release; and production of defective virions and extracellular vesicles. Tissue immune activation was evaluated from measurements of cytokines in culture medium using multiplexed immunoassays.
Results:
Neither ART itself nor simulated cytokine storms nor exogenously added HIV-1 proteins triggered a sustained cytokine upregulation. In contrast, defective (replicative-incompetent) virions and extracellular vesicles induced sustained cytokine upregulation, as did infectious virus. Tissue immune activation was accompanied by reactivation of cytomegalovirus.
Conclusion:
The system of ex-vivo human lymphoid tissue allowed investigation, under laboratory-controlled conditions, of possible mechanisms involved in persistent immune activation in HIV-1 patients under ART. Mechanisms of this immunoactivation identified in ex-vivo tissues may indicate potential therapeutic targets for restoration of immune system homeostasis in HIV-1-infected patients.
Introduction
Persistent systemic immune activation in patients under antiretroviral therapy (ART) is one of the challenging problems in HIV-1 disease. Neither the causes nor the mechanisms of this phenomenon are well known [1–3].
Various hypotheses on the cause of this phenomenon have been proposed, including general immune dysregulation [4,5], bacterial gut translocation [6–8], persistent opportunistic infections [[9,10], nondetectable residual HIV-1 replication [11], and viral proteins, which continue to be present in different body fluids [12–17] in spite of ART. Also, it is possible that long-term ART itself may induce a proinflammatory environment.
Recently, we reported that the phenomenon of HIV-1-triggered persistent immune activation had been reproduced in ex-vivo human lymphoid tissues under controlled laboratory conditions [18]. As with what has been observed in patients, HIV-1 also triggered immune activation in these tissues, as evaluated by upregulation of cytokine release (’cytokine storm’), and these cytokines remained elevated despite suppression of HIV-1 replication by ART.
Here, we take advantage of this ex-vivo model to investigate possible mechanisms of sustained immune activation in HIV-1-infected individuals.
In this system, we found that extracellular vesicles as well as noninfectious HIV-1 particles released by ex-vivo HIV-1-infected lymphoid tissue are sufficient to prompt and sustain immune activation, as evaluated from cytokine release. This immune activation was associated with reactivation of cytomegalovirus (CMV) in tissues. In contrast, no sustained immune activation was triggered by antiretroviral drugs themselves or by the presence of viral proteins.
Materials and methods
Sample preparation
Human tonsillar tissues from routine tonsillectomies were obtained anonymously from the Children's National Medical Center in Washington DC, according to an Institutional Review Board-approved protocol. Tissues were dissected and cultured as previously described [19–21] and detailed in Supplemental Materials). Eighteen to 27 blocks were used for each experimental condition. Medium was collected and changed every 3 days.
HIV-1 infection and antiretroviral treatment
Matched lymphoid tissue blocks were uninfected or infected with HIV-1 X4LAI.04 [3.5 × 105 pg/ml (1×) multiplicity of infection of 0.05, or 3.5 × 104 pg/ml (0.1×)]. HIV-1 viral stock (5 μl) was deposited on top of each tissue block. For experiments including ART, tissues were treated with the protease inhibitor ritonavir (RTV), with two nucleotide retrotranscriptase inhibitors (NRTIs), zidovudine (ZDV) and lamivudine (3TC), or with nevirapine (NVP, nonNRTI) all at a final concentration of 5 μmol/l. Drugs were added at day 3 and subsequently at every medium change until the end of culture.
Evaluation of HIV-1 replication
HIV-1 replication was measured with an immunofluorescent cytometric bead assay for HIV-1 p24gag in tissue culture supernatants, as described previously [22] (and detailed in Supplemental Materials). Analysis was performed on a Luminex 200 (Luminex, Austin, Texas, USA) acquiring 100 beads/well.
Exogenous emulation of cytokine storm
Lymphoid tissues were treated at day 0 with cytokines known to be upregulated with HIV-1 infection: IFN-γ (1000 pg/ml), RANTES (1000 pg/ml), TNF-α (100 pg/ml), or with a mixture of these cytokines with IL-2 (25 pg/ml), IL-7 (100 pg/ml), MIP-1α (500 pg/ml), and MIP-1β (500 pg/ml).
Evaluation of endogenous herpesvirus replication
For each condition (uninfected control, HIV-1-infected, HIV-1-infected and treated with RTV) and each time point (days 0, 6, and 12), nine tissue blocks were collected and stored in RNAlater (Thermo Fisher) at −20 °C. We isolated DNA using QIAamp DNA Mini kits (Qiagen, Germantown, Maryland, USA) according to the manufacturer's guidelines.
We quantified DNA copies of several herpesviruses (HHVs) [Herpes Simplex Virus 2 (HSV-2), HHV-3, HHV-4 (or EBV), HHV-5 (or CMV), HHV-6, and HHV-7] using droplet digital PCR (BioRad, Hercules, California, USA). Ribonuclease P protein subunit p30 (RPP30) was used as a housekeeping gene to normalize HHV DNA numbers per cell. Primer-probe sequences are shown in Supplemental Table 1 and additional details are provided in Supplemental Materials).
Infectivity of CMV from cultures was evaluated by transfer of homogenized tissue blocks to MRC-5 cell line followed by microscopic evaluation (details in Supplemental Materials).
HIV-1 protein treatment
Lymphoid tissues were treated at day 0 and at every change of medium with recombinant HIV-1 protein gp120, Tat, or Nef (Abcam, Cambridge, Massachusetts, USA) at a final concentration of 5 ng/ml.
Extracellular vesicle isolation, characterization, and treatment
Extracellular vesicles were isolated by two methods: iodixanol gradients and membrane affinity spin columns (see details in Supplemental Materials). Extracellular vesicles and supernatants free of extracellular vesicles were collected for each experimental condition (uninfected, uninfected and treated with RTV or NVP, HIV-1-infected, and HIV-1-infected and treated with RTV or NVP), as well as from culture medium alone, as described in Supplemental Materials. Extracellular vesicle size and concentration were determined by NanoSight and evaluation of extracellular vesicle and non-extracellular vesicle markers, were assessed by flow cytometry (Supplemental Materials). Isolated extracellular vesicles were applied to fresh ex-vivo lymphoid tissue blocks on day 0 and day 6 at 50 extracellular vesicles/cell for highly purified iodixanol-isolated extracellular vesicles, and 500 extracellular vesicles/cell for spin column purified extracellular vesicles. RTV or NVP was added to tissues treated with spin column extracellular vesicles to prevent unwanted HIV-1 infection.
Inactivated HIV-1 preparation and treatment
Inactivated HIV-1 virions (X4LAI04) were prepared as described previously [23] with aldrithiol (AT-2), which covalently modifies the essential zinc fingers in the HIV-1 nucleocapsid, eliminating infectivity but maintaining the conformational and functional integrity of the viral surface proteins. Identical procedures were carried out in parallel on virus-free culture medium (AT-2 medium). These experimental control conditions showed that any residual AT-2 after concentration and washing did not mediate detectable effects on ex-vivo culture cell viability, HIV-1 replication, or total IgG production [23], and here we observed no change in cytokine secretion in tissues exposed to AT-2 medium.
Lymphoid tissues were treated with different amounts and exposures of HIV-1 AT-2-inactivated virions: 5 μl of AT-2 virions (3.5 × 105 pg/ml, 1×) were applied as single inoculum at day 0, with repeated exposure of 1× virions every 3 days, a single inoculum of 5 μl of AT-2 X4LAI04–inactivated virions diluted 10 times (0.1×) at day 0, and repeated exposure with AT-2 medium every 3 days.
Cytokine measurement
An in-house multiplexed bead-based assay was used to measure the following 33 cytokines: IL (interleukin)-1α, IL-1β, IL-2, IL-4, IL-6, IL-7, IL-8, IL-10, IL-12p70, IL-13, IL-15, IL-16, IL-17, IL-18, IL-21, IL-22, IL-33, Calgranulin A (Calg A or S100A8), Eotaxin (CCL11), granulocyte--macrophage colony-stimulating factor (GM-CSF), growth-regulated alpha (GRO-α or CXCL1), interferon-γ (IFN-γ), interferon-γ-induced protein (IP-10 or CXCL10), interferon-inducible T-cell alpha chemoattractant (ITAC or CXCL11), macrophage colony-stimulating factor (M-CSF), monocyte chemoattractant protein-1 (MCP-1 or CCL2), monokine induced by IFN-γ (MIG or CXCL9), macrophage inflammatory protein-1α (MIP-1α or CCL3), MIP-1β (CCL4), MIP-3α (CCL20), regulated upon activation, normal T-cell expressed, and secreted (RANTES or CCL5), transforming growth factor-β (TGF-β), and tumor necrosis factor-α (TNF-α).
Assays were performed as described previously [24,25] (and detailed in Supplemental Materials). Plates were read on a Luminex 200 (Luminex) and analyzed with Bioplex Manager software (BioRad). Cytokine concentrations were determined using 5P regression algorithms.
Intracellular cytokine measurement
Flow cytometry was employed to assess cell types producing the increased cytokines IFN-γ, MIP-1α, MIP-1β, RANTES, and TNF-α (detailed in Supplemental Materials).
Cytokine production with toll-like receptor agonists and inhibitors
Tissues were treated at day 0 with 1 μg/ml toll-like receptor (TLR) 3 [polyinosinic-polytidylic acid (poly(I:C)], TLR7/8 (R848), or TLR9 (ODN2006 CpG-B) (all Invivogen, San Diego, California, USA) agonists and cytokines were measured over 12 days. Inhibition of cytokine production was investigated by pretreatment with 10 μmol/l TLR8 inhibitor (CU-CPT9a) or TLR9 antagonist (ODN TTAGGG) for 4 h followed by challenge with AT-2 HIV-1.
Statistical analysis
Cytokine measurements are represented as means ± SEM. Results were analyzed by pairwise comparison with the Wilcoxon signed-rank test between different pairs of treatments. Values of P less than 0.05 were considered statistically significant.
Results
Antiretroviral therapy alone did not lead to immune activation
To determine if ART alone might contribute to sustained immunoactivation, we compared cytokines released by ex-vivo human lymphoid tissues under ART (RTV, ZDV+3TC, or ZDV) with those released by donor-matched untreated controls. We previously reported that RTV did not lead to a significant increase in cytokines throughout the culture period [18]. Similar results were obtained here with ZDV+3TC and NVP: no increase in cytokine release was observed (Fig. 1a). The only two cytokines that were changed with ZDV+3TC, IL-1β, and IL-2, were actually decreased (by 22.95 and 36.79%, respectively, P < 0.05, n = 8; Fig. 1b).
Fig. 1.
Antiretroviral therapy does not upregulate cytokines in human lymphoid tissues.
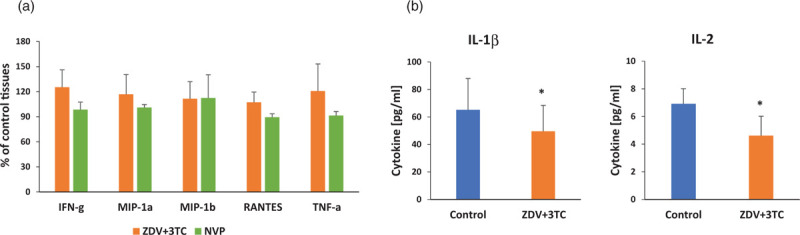
Blocks of lymphoid tissue were treated with 5 μmol/l of nucleotide retrotranscriptase inhibitors, ZDV (zidovudine) and 3TC (lamivudine) or NVP (nevirapine, NNRTI). Compounds were added every 3 days. Untreated donor-matched tissues were used as controls. (a) Cytokines upregulated by HIV-1 were not upregulated by ART alone. Presented are percentage of untreated control (±SEM) of cytokines measured at last day of culture (n = 8, ZDV+3TC; n = 8, NVP). (b) IL-1β and IL-2 were significantly downregulated by ZDV+3TC. Presented are mean concentrations (±SEM) of cytokines measured at the last day of culture (∗P < 0.05, n = 8). ART, antiretroviral therapy.
Initial cytokine storm did not trigger sustained cytokine upregulation
It is conceivable that an HIV-1-triggered upregulation of cytokines could stimulate immune cells to produce a long-term cascade of other cytokines even after HIV-1 is suppressed. To investigate this possibility, we simulated this situation by treating uninfected ex-vivo lymphoid tissues with a cocktail of cytokines that are upregulated in HIV-1 infection, namely, IL-2, IL-7, IFN-γ, MIP-1α, MIP-1β, RANTES, and TNF-α⋅
We found that tissue exposure to an exogenous cytokine combination did not result in a significant and sustained increase of cytokines, except for IL-13 (205.3 ± 27.6% of control, n = 6, P = 0.009) at day 12. Also, we exposed tissues to single cytokines (IFN-γ, RANTES, or TNF-α). None of these cytokines triggered sustained upregulation of endogenous cytokines.
HIV-1 co-pathogen reactivation is associated with immune activation
We tested whether sustained immune activation in HIV-1-infected human lymphoid tissues under ART is associated with herpesviruses (HHV) reactivation, HHVs 2–7 were quantified by droplet digital PCR in tissues infected with HIV-1 and treated with RTV. In these tissues, as we reported previously [18], despite viral suppression, IL-2, IL-7, IFN-γ, MIP-1α, MIP-1β, RANTES, and TNF-α remained elevated in HIV-1-infected RTV-treated ex-vivo lymphoid tissues compared with uninfected lymphoid tissues (1.5-fold to 5.3-fold increases, n = 8, P < 0.05, except for TNF-α, P = 0.07). HIV-1 replication in these tissues was inhibited by 99.1 ± 0.7% as measured from p24 released into the medium. In all HIV-1 infected tissues, the only increase was observed in CMV DNA: HIV-1 infection led to an almost 20-fold increase of CMV DNA copies in lymphoid tissues compared with uninfected control (19.1 ± 6.5-fold increase, n = 8, P = 0.023). Also, we observed an increase of CMV DNA in tissues with HIV-1 suppressed by RTV, but on a much smaller scale, and in these tissues, the increase did not reach statistical significance (3.8 ± 1.7-fold increase, n = 8, P = 0.191; Fig. 2).
Fig. 2.
Cytomegalovirus (CMV) is reactivated in HIV-1-infected ex-vivo human lymphoid tissues.
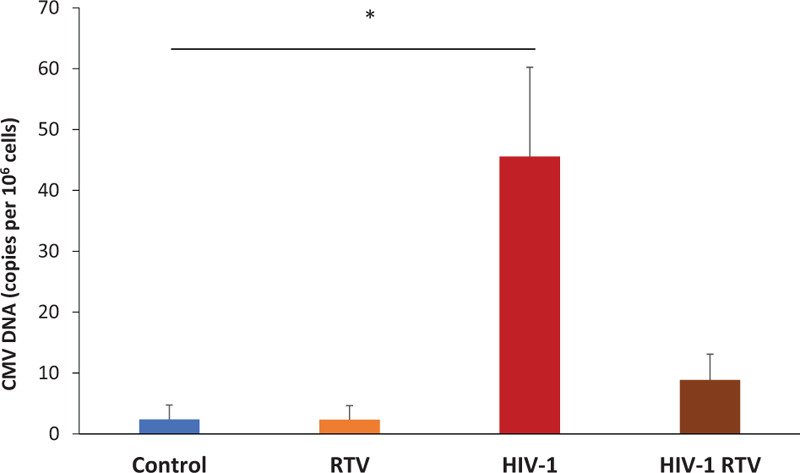
Donor-matched lymphoid tissue blocks were either treated with ritonavir (RTV), infected with HIV-1, or infected with HIV-1 and treated with RTV or not treated (control) and cultured for 12 days. Presented are the mean sums (± SEM) of the numbers of CMV DNA copies (per 106 cells contingent on RPP-30 DNA copies) at day 6 and day 12 (∗P = 0.023, n = 8).
We next investigated the infectivity of the reactivated CMV. Tissue blocks from control, HIV-1 infection, or HIV-1 infection with RTV were collected at day 12. Blocks were frozen and later thawed, homogenized, centrifuged, and supernatants applied for 4 h at 1 : 10 dilutions to MRC-5 cells, which are susceptible to CMV but not HIV-1 infection. Cells remained in culture for 13 days. Microscopy revealed that MRC-5 cells treated with HIV-1 or HIV-1 with RTV tissue homogenates showed signs of cytopathicity compared with cells treated with control cell homogenates (Supplemental Fig. 1).
HIV-1 proteins did not induce cytokine dysregulation
To test whether HIV-1 proteins might play a role in sustained immune activation when viral replication is suppressed, we treated ex-vivo tissues with gp120, Tat, or Nef at 5 ng/ml, a concentration that is comparable with that present in the viral inoculum used in our experiments. Cytokines were measured at each time-point and compared with untreated donor-matched tissues. Treatment with these HIV-1 proteins did not induce any significant sustained cytokine upregulation. In tissues treated with gp120, we observed a transient increase in Eotaxin at day 3 (271.9 ± 70.2% of control, n = 5, P = 0.020) and in IL-21 at day 9 (118.3 ± 5.6% of control, n = 5, P = 0.004). However, these increased cytokines returned to the control levels by day 12.
Extracellular vesicles from both HIV-1-infected lymphoid tissues and infected tissues treated with antiretroviral therapy induced sustained immune activation
Extracellular vesicles isolated from lymphoid tissue culture medium were characterized by NanoSight and flow cytometry. Extracellular vesicle size and concentrations were not significantly different between conditions as previously reported [18] and average extracellular vesicle size was 170.2 ± 4.34 nm (Supplemental Fig. 2). Extracellular vesicles contained various levels of tetraspanins (72.3% of CD9+, 45.4% of CD63+, and 88.9% CD81+), had lipid components as evidenced by staining with Cell Mask Deep Red, and did not possess non-extracellular vesicle markers including apolipoprotein A1 or B and albumin (Supplemental Fig. 3). Extracellular vesicle preparations from iodixonal gradients were verified to be free of HIV-1 virions (0 pg/ml p24, and no detectable infection in tissues treated with these extracellular vesicle preparations).
We tested whether extracellular vesicles from HIV-1-infected and ART (RTV or NVP)-treated tissues triggered immune activation. Extracellular vesicles isolated by membrane affinity spin columns were potentially contaminated with infectious virions, thus we added RTV or NVP to exclude possible infection. Indeed, a small amount of p24 was present in the extracellular vesicle preparations (0.017 ± 0.009 ng/ml for HIV-1 extracellular vesicles, 0.004 ± 0.001 ng/ml for HIV-1 with RTV extracellular vesicles, and 0.0 ± 0.0 ng/ml for HIV-1 with NVP), but ART prevented any HIV-1 infection during culture. In donor-matched tissues infected with HIV-1, the average cumulative p24 production was 22.6 ± 4.2 ng/ml (n = 9; Fig. 3a).
Fig. 3.
Extracellular vesicles from HIV-1-infected human tissues upregulate cytokines.
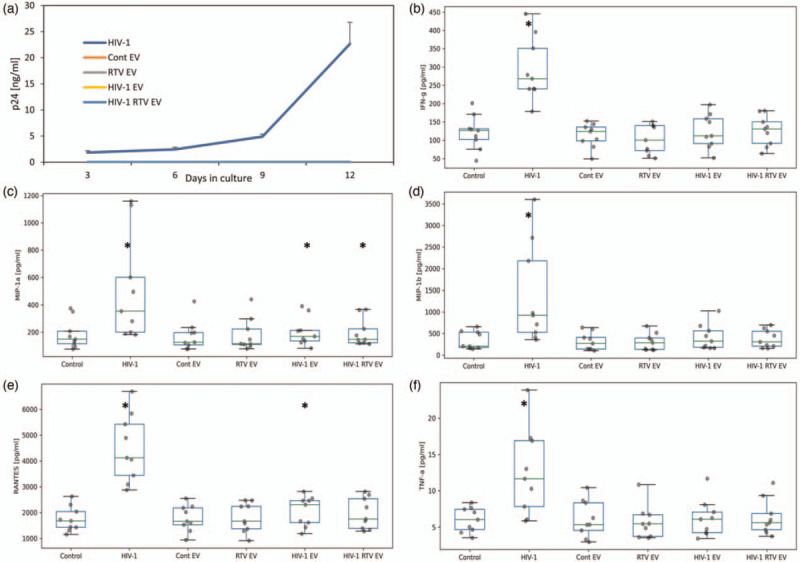
Extracellular vesicles (EVs) collected from donor-matched tissue cultures either infected with HIV-1, infected with HIV-1 and treated with ritonavir (RTV), treated only with RTV or uninfected (controls), and isolated using membrane affinity spin columns, were added to previously untreated (recipient) tissues and the release of cytokines was evaluated. EVs were collected from donor cultures on day 9 and added to the recipient cultures at day 0 and again on day 6 of culture. To exclude the possible effect of HIV-1 contamination in EVs collected from HIV-1-infected cultures, RTV was added to the tissue culture. Cytokine and p24 production were measured every 3 days and cumulative production was calculated. (a) HIV-1 replication in donor-matched tissues infected with HIV-1 or exposed to EVs. HIV-1 replication was assessed by p24gag released into the supernatant. Presented are mean cumulative releases (± SEM) over a 12 day period. (b--f) Cytokine release into the culture medium by tissues treated with EVs. Cytokine release is cumulative production over 12 days expressed in pg/ml. Presented are mean (±SEM) values (∗P < 0.05, n = 9).
In agreement with our earlier research results [18], there was a consistent elevation of cytokines in infected tissues. Here, we focused on upregulation of IFN-γ, MIP-1α, MIP-1β, RANTES, and TNF-α (P < 0.05, n = 9; Fig. 3b--f). We found that extracellular vesicles from tissues productively infected with HIV-1, and also from tissues in which HIV-1 replication was suppressed by RTV, induced significant sustained upregulation of MIP-1α, and there was a significant increase in RANTES for HIV-1 extracellular vesicle-treated tissues (P < 0.05; Fig. 3b--f). Extracellular vesicles isolated from control uninfected tissues or from uninfected tissues treated with RTV did not produce a significant sustained increase in these cytokines compared with control untreated tissues. See Supplemental Figure for kinetic graphs.
Similar experiments were conducted with extracellular vesicles from tissues treated with NVP. HIV-1 replicated in tissues with average cumulative p24 production of 28.3 ± 5.4 ng/ml (n = 12; Supplemental Fig. 4a) whereas no HIV-1 replication was observed in any extracellular vesicle-treated cultures. Cytokines were significantly upregulated with HIV-1 infection, and tissues treated with HIV-1 extracellular vesicles induced significant upregulation of MIP-1α, MIP-1β, and RANTES (P < 0.05; Supplemental Fig. 4b--f). Tissues treated with extracellular vesicles from HIV-1-infected, NVP-treated tissues slightly increased a few cytokines but not significantly. Extracellular vesicles isolated from control uninfected tissues or from uninfected tissues treated with NVP did not increase these cytokines compared with control untreated tissues, and a small but significant decrease in TNF-α was observed in control extracellular vesicle-treated tissues (Supplemental Fig. 4f).
We further verified that the effect observed was because of extracellular vesicles, rather than residual HIV-1 virions, by performing similar experiments with extracellular vesicles purified on iodixanol gradients, which were completely free of HIV-1 virions. No infection was observed in tissues treated with extracellular vesicles (without addition of ART); whereas infection was observed with HIV-1 (Fig. 4a). Isolation of these extracellular vesicles required large volumes of starting material and are technically difficult, so only a limited number of experiments could be performed, which did not provide significance with two-tailed testing. However, one-tailed tests for increases only showed that, as in above experiments, tissues treated with HIV-1 significantly increased all five cytokines (Fig. 4b--f). Tissue cultures treated with extracellular vesicles from tissues productively infected with HIV-1, and also from tissues in which HIV-1 replication was suppressed by RTV, induced significant sustained upregulation of MIP-1α, MIP-1β, and RANTES, and HIV-1 with RTV extracellular vesicle-treated tissues additionally increased TNF-α (Fig. 4b--f) (P < 0.05, one-tailed test; n = 5). See Supplemental Fig. 5 for kinetic graphs. Extracellular vesicles isolated from control uninfected tissues or from uninfected tissues treated with RTV did not produce a significant sustained increase in these cytokines compared with control untreated tissues.
Fig. 4.
Cytokine upregulation is not due to residual HIV-1 in extracellular vesicle fractions.
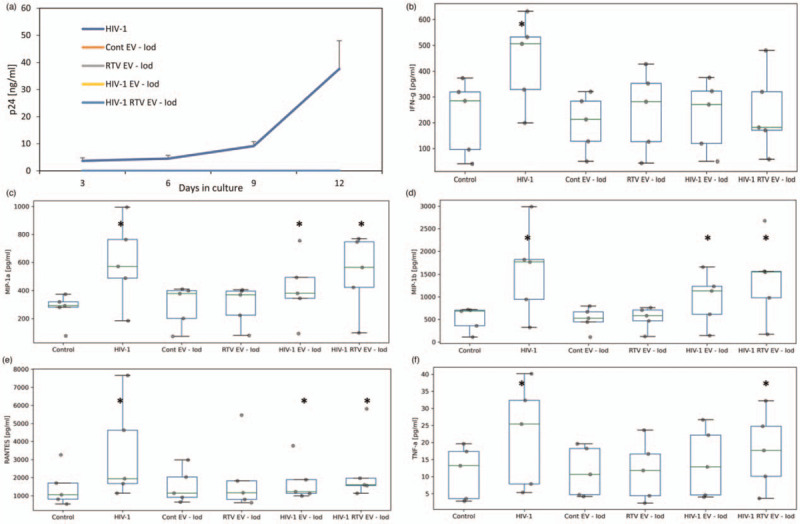
Extracellular vesicles (EVs) collected from tissue cultures either infected with HIV-1, infected with HIV-1 and treated with RTV, treated with RTV only, or uninfected (controls) were purified on iodixanol (Iod) gradients and verified to be free of HIV-1. EVs were added to recipient tissues on day 0 and day 6 and the release of cytokines was evaluated. Cytokine and p24 production were measured every 3 days and cumulative production was calculated. (a) HIV-1 replication in donor-matched tissues infected with HIV-1 or exposed to EVs. HIV-1 replication was assessed by p24gag released into the supernatant. Presented are mean cumulative releases (±SEM) over a 12 day period. (b--f) Cytokine release into the culture medium by tissues treated with EVs. Cytokine release is cumulative production over 12 days expressed in pg/ml. Presented are mean (±SEM) values (∗P < 0.05, one-sided t-test, n = 5).
In control experiments, tissues were treated with extracellular vesicle and supernatant fractions of fresh culture medium, as well as extracellular vesicle-free supernatant fractions from control, RTV, HIV-1-infected, and HIV-1-infected and RTV-treated tissue culture supernatants. No significant increase in cytokine production was observed in tissues treated with these fractions (Supplemental Fig. 6).
Noninfectious virions led to sustained immune activation
Next, we investigated whether inactivated replication-deficient virions can trigger immune activation. We inactivated HIV-1 with AT-2 according to published protocols [23] and treated tissues with these virions.
Control donor-matched tissues were inoculated with HIV-1 and became productively infected with an average cumulative p24 production of 18.0 ± 3.4 ng/ml (n = 7). Tissues infected with 0.1× HIV-1 were also productively infected but at lower levels, with average cumulative p24 production of 2.8 ± 0.3 ng/ml. Inoculation with AT-2-inactivated virions in the same amount as with infectious virus did not result in productive infection: p24 remained the same as it was in the inoculum (0.4–4.5 ng/ml cumulative p24), thus confirming that the inactivated virions were noninfectious (Fig. 5a).
Fig. 5.
Inactivated HIV-1 virions upregulate cytokines in human lymphoid tissue ex vivo.
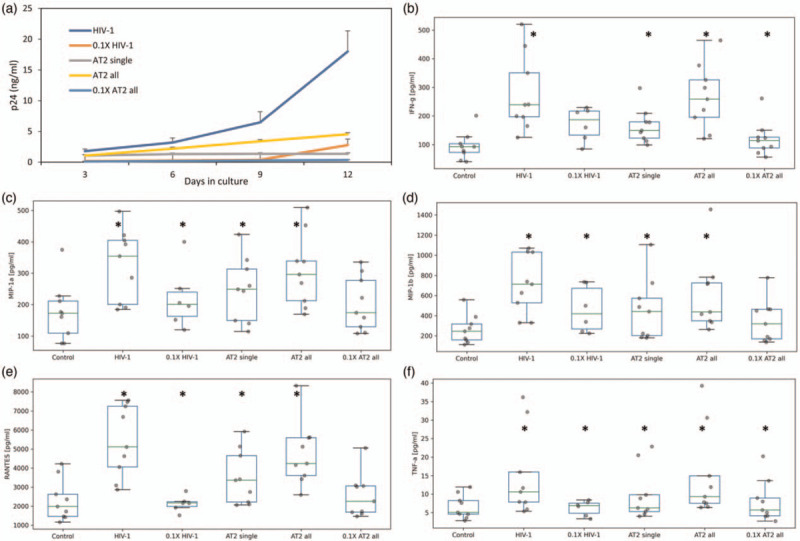
HIV-1 virions inactivated with AT-2 were added at two concentrations to previously untreated human lymphoid tissues every 3 days with a medium change starting from day 0 of culture or exposed once at the beginning of the culture. Cytokine and p24 release by these tissues were evaluated. (a) Cumulative p24 production of donor-matched tissues inoculated with HIV-1 or with AT-2-inactivated HIV-1. Tissues were inoculated with infectious HIV-1 (1× or 0.1×) or AT-2-inactivated HIV-1 (1× or 0.1×) either added every 3 days with medium change (all) or only once at the beginning of the culture (single). (b--f) Cytokine release into the culture medium by tissues treated with virions. Cytokine release is cumulative production over 12 days expressed in pg/ml. Presented are mean (± SEM) values (∗P < 0.05, n = 9).
In tissues productively infected with HIV-1, there was upregulation of IFN-γ MIP-1α, MIP-1β, RANTES, and TNF-α as above. Cumulative concentrations of these cytokines were significantly higher than in matched untreated control tissues (P < 0.05, n = 9; Fig. 5b--f). Tissues treated with 10-fold diluted (0.1×) HIV-1 also increased these same cytokines; however, there was a delay in the kinetics with increases in cytokines beginning only at day 9; all cytokines except IFN-γ were significantly increased. As with infectious virus, repeated AT-2 HIV-1 exposure of tissues significantly upregulated all five of the above-listed cytokines. Even a single exposure of tissue to AT-2-inactivated HIV-1 triggered sustained significant upregulation of these cytokines, although at lower levels. Also, exposure of tissues to 10-fold-diluted AT-2-treated HIV-1 resulted in cytokine upregulation, although only IFN-γ and TNF-α were significant (P < 0.05, n = 9). See Supplemental Figure 7 for kinetic graphs.
Cytokines are produced predominantly by CD4+ and CD8+ T cells
Next, we investigated the cellular source of the upregulated cytokines IFN-γ, MIP-1α, MIP-1β, RANTES, and TNF-α by flow cytometry. Control and HIV-1-infected tissues were dissociated at day 3 and cells were stained for phenotypic markers and for intracellular cytokines. We found that a variety of cells increased production of these cytokines: IFN-γ secretion by CD4+, CD8+, and natural killer cells (NK) increased, MIP-1α by CD4+ and CD8+, MIP-1β by macrophages, CD4+ and CD8+, RANTES by CD4+ and CD8+, and TNF-α by macrophages (Supplemental Fig. 8).
TLR7/8 and TLR9 agonists trigger cytokine increases
The increase of cytokines in response to HIV-1, and particularly to AT-2-inactivated HIV-1, may be the result of TLR stimulation. To test this, we stimulated tissues with TLR3, TLR7/8, and TLR9 agonists and followed their cytokine production. None of the agonists significantly increased IFN-γ and RANTES; however, TLR7/8 and TLR9 did generate significant increases over control tissues in MIP-1α and MIP-1β, and TLR7/8 also increased TNF-α (P < 0.05, n = 9). Although these increases were not to the same extent as triggered by infectious virus, these increases were not significantly different from those triggered by AT-2-inactivated HIV-1 (Supplemental Fig. 9a).
We further investigated the role of TLR signaling by pretreating tissues with TLR8 and 9 inhibitors before challenge with AT-2-inactivated virus. TLR8 inhibitor significantly decreased IFN-γ, MIP-1α, and TNF-α responses to AT-2-inactivated HIV-1 by 24.2, 28.7 and 33%, respectively (P < 0.05, n = 7), whereas TLR9 antagonist also lowered some cytokine production, but no decreases were significant (Supplemental Fig. 9b).
Discussion
Improper immune activation accompanies many human diseases and is associated with the release of high concentrations of cytokines (’cytokine storm’) [26]. Such a storm is characteristic of various viral infections, in particular SARS-CoV-2, in which it is considered to be one of the major causes of lethality [27]. Although pathogenesis of HIV-1 is different from SARS-CoV-2, HIV-1 infection also triggers immune activation as reflected in upregulation of various cytokines [18]. Immune activation is considered to be the driving force of HIV-1 disease [28], resulting in premature age-related diseases, such as cardiovascular disease or dementia [26]. HIV-1-triggered immune activation continues for years even after HIV-1 replication is successfully suppressed by ART. Specifically, several studies have shown that the cytokine networks in plasma and semen of HIV-1-infected patients do not normalize after ART [28,29]. Understanding the mechanisms prompting immune activation in HIV-1-suppressed patients is paramount to the development of efficient therapies able to fully restore the immune system homeostasis and possibly averting various diseases that are prematurely developed in such individuals.
Various factors are hypothesized to trigger persistent immune activation in HIV-1-infected individuals. Here, we used human ex-vivo lymphoid tissue [19] to test several of these hypotheses. In particular that immune activation may be the result of proinflammatory effects of certain antiretroviral drugs themselves; an HIV-1-triggered initial cytokine storm could modify the normal cytokine homeostasis, which remains dysregulated even after HIV-1 replication is suppressed; HIV-1 infection reactivates endogenous viruses, in particular, HHVs that may continue to replicate and induce immune activation after HIV is suppressed; HIV-1 proteins that continue to be released, in spite of ART, activate the immune system [17]; immune activation is supported by defective virions or extracellular vesicles that carry viral molecules rather than residual infectious HIV-1.
As in our earlier research [18], in testing these hypotheses, we evaluated tissue immune activation as measured by upregulation of cytokine release.
Before approval for clinical use, ART was tested for various side-effects. It has been shown that RTV may facilitate the generation of reactive oxygen species (ROS) [30,31], which in turn could establish the production of pro-inflammatory cytokines [32]. Also, ZDV and 3TC at high concentrations have been shown to interact with mitochondrial DNA polymerase [33], resulting in cellular damage. NVP has been associated with skin and hepatic hypersensitivity reactions [34]. The ability of ART to suppress HIV-1 replication far overweighs these and other side-effects, and thus it is successfully used in various clinical protocols. However, as HIV-1-infected patients have to remain on ART for the rest of their lives, it is conceivable that in the long-run, these drugs may have other effects that contribute to persistent immune activation [35].
Here, we tested the ability of ART to upregulate cytokines on the scale of the lifespan of ex-vivo human lymphoid tissues. We treated tissues either with RTV, a protease inhibitor, a combination of two NRTIs (ZDV and 3TC), or NVP, an NNRTI. The drugs in our tissue explants are added at somewhat higher concentrations than those used in vivo; however, there are differences in drug bioavailability, clearance, and bioaccumulation that make comparisons difficult. Also, we add drugs every 3 days, while patients take them daily. We chose the concentration of the drug so that, similar to in vivo, virus replication is completely suppressed. In our experiments, none of these drugs increased cytokine release by human lymphoid tissues ex vivo. ZDV+3TC did decrease IL-1β and IL-2, which may result from the disruption of mitochondrial bioenergetics, which can alter T-cell and macrophage activation. Thus, in ART regimens, the effect of drugs themselves did not explain persistent immune activation in HIV-1-infected ex-vivo human lymphoid tissues under ART [18].
Acute HIV-1 infection in vivo triggers an intense cytokine storm that may itself cause a permanent and irreversible disruption of the normal cytokine set points [9]. The upregulation of cytokines observed in HIV-1-infected human lymphoid tissues ex vivo[18] provides a model to test this possibility. In our experiments, an HIV-1-induced cytokine storm was simulated by treatment of ex-vivo lymphoid tissues with exogenous cytokines. Treating ex-vivo tissues with single cytokines or with combinations of the exogenous cytokines that were the most upregulated after HIV-1 infection [18] resulted in temporary increases of several endogenous cytokines but these increases were not sustained. Thus, under our experimental conditions, an early cytokine storm, distinctive of HIV-1 infection, did not support persistent release of cytokines.
In vivo, HIV-1 infection is known to reactivate endogenous viruses, including normally silent HHVs [36–39]. In particular, CMV, a virus that is not overly pathogenic in healthy individuals, can be reactivated [40], possibly supporting sustained immune activation. Earlier, we showed that in ex-vivo tissue, infection with exogenous CMV is augmented by HIV-1 co-infection [41]. Here, we evaluated the reactivation of endogenous HHVs upon HIV-1 infection and/or treatment with RTV. We found that out of all six measured HHVs, only endogenous CMV is reactivated with HIV-1 infection. After HIV-1 replication was inhibited by RTV in ex-vivo tissues, CMV reactivation continued, albeit at a lower level. Reactivation of CMV was also evidenced by cytopathic effects observed when tissue homogenates from HIV-1-infected or HIV-1-infected and RTV-treated tissues were applied to CMV-susceptible MRC-5 lung fibroblasts, whereas control tissue homogenates were not cytopathic.
CMV is a common HIV-1 co-pathogen in vivo and has been implicated as a driving force of continuing immune activation [42]. It was reported that myeloid cells, fibroblasts, and other cell types are the sources of latent CMV [43,44], and is has been hypothesized that the loss of CD4+ T cells by HIV-1 results in failure to provide help to CD8+ T cells that may normally keep CMV replication under control [45,46]. In-vivo HIV-1 replication is not fully suppressed by ART, and spikes of HIV-1 in lymphoid tissue have been reported [37,47]. Extrapolating the results of our experiments with HIV-1-infected lymphoid tissue ex vivo to the situation in vivo, it is possible that these spikes allow CMV to remain reactivated and to support immune activation.
Although ART prevents HIV-1 spreading, it is known that in vivo a reservoir of infected cells remains for many years [11,48,49]. These infected cells release HIV-1 proteins [50,51], which may induce tissue immune activation. In particular, significant concentrations of Nef found in vivo may make T cells hyperresponsive to stimulation and promote induction of inflammatory cytokines [52]. Here, we evaluated whether Nef, as well as two other major HIV-1 proteins, Tat and gp120, were capable of eliciting a sustained release of proinflammatory cytokines in ex-vivo lymphoid tissues. In our experiments with these ex-vivo tissues, no sustained increases in cytokine release occurred when tissues were treated with any of these three proteins. We chose to use concentrations of these proteins that were lower than what has been reported in vivo as our goal was not only to evaluate the effect of the HIV-1 proteins at a level that exists during HIV infection but to check whether, after suppression of HIV-1 replication, these proteins at their residual concentrations may affect immune activation.
Infected cells release various extracellular vesicles that carry HIV-1 components. Such vesicles may be classified as defective HIV-1 virions [53–55] or as extracellular vesicles carrying HIV-1 proteins [56–58]. It has been shown that HIV-1-infected patients under ART continue to release extracellular vesicles that carry HIV-1 components and that such extracellular vesicles may be responsible for neurocognitive and immunological dysfunction [57]. Also, we previously showed that some extracellular vesicles released from HIV-1-infected cells carry gp120 and facilitate HIV-1 infection [53]. It is conceivable that extracellular vesicles released by HIV-1-infected tissues, including those that carry HIV proteins, may contribute to immune activation.
Therefore, we investigated here whether defective virions or extracellular vesicles isolated from ex-vivo lymphoid tissues, treated or not with RTV or NVP, upregulated the production of cytokines when transferred to an untreated tissue culture. In control experiments with extracellular vesicles from uninfected tissues or from uninfected tissues treated with RTV or NVP, we did not observe any significant cytokine increases. In contrast, in tissues treated with extracellular vesicles from HIV-1-infected tissues, there was a significant and sustained increase of MIP-1α, MIP-1β, and RANTES. Tissues treated with extracellular vesicles from HIV-1-infected and RTV-treated tissues increased MIP-1α compared with controls. This was confirmed by experiments with more highly purified extracellular vesicles that demonstrated increases of the same cytokines in tissues treated with extracellular vesicles from HIV-1-infected tissues. Tissues treated with highly purified HIV-1 RTV extracellular vesicles increased MIP-1α, MIP-1β, RANTES, and TNF-α. Tissues treated with extracellular vesicles from HIV-1-infected and NVP-treated tissues demonstrated small increases in cytokines but these increases did not reach significance. Mechanisms by which extracellular vesicles deliver their cargo including cytokines, miRNAs and RNA, and trigger cell signals mediated by these and other extracellular vesicle molecules, such as MHC class I and II and HIV-1 proteins, are not fully understood but are the subject of extensive investigations [59–62].
To further simulate various types of viral-related particles that are released by HIV-1-infected tissues, we treated ex-vivo human lymphoid tissues with inactivated HIV-1 X4LAI.04 virions. We inactivated HIV-1 with AT-2, which abolishes infectivity by modifying HIV-1 nucleocapsid zinc fingers, while maintaining its structure, functionality, and like extracellular vesicles, ability to fuse with cells [23]. As with extracellular vesicles isolated from infected tissues, exposure of uninfected tissues to AT-2 HIV-1 upregulated several key cytokines, in particular, IFN-γ, MIP-1α, MIP-1β, TNF-α, and RANTES. Exposure to lower doses of both infectious and inactivated virions also resulted in cytokine upregulation albeit at lower levels.
Massive production of various defective virus particles and of extracellular vesicles carrying viral components released during infection is a hallmark of HIV-1 infection in vivo[55]. As such defective particles are produced in HIV-1-infected patients under ART [17], our results suggest that this would be sufficient to sustain immune activation. Extracellular vesicles and defective virions are part of a continuum of cell-released particles and are difficult to separate as they can both contain host cell proteins, viral components, and nucleic acids [55].
These upregulated cytokines are produced by a variety of cells in our system. We observed increases in the number of macrophages, NK cells, and CD4+ and CD8+ T cells producing these cytokines; however, it is possible that other cell types are responsible for the increased cytokine production.
The cells producing these upregulated cytokines are likely stimulated by dendritic cells following their interaction with HIV-1, and TLR7/8-signaling is known to be involved in response to HIV-1 [63,64]. TLR7/8 and TLR9 seemed also to be partially responsible for HIV-1-induced immunoactivation in our tissue system as TLR7/8 and TLR9 agonists triggered some of the same cytokine increases as HIV-1, though not to the same extent. In contrast, TLR3 was likely not involved. Experiments with AT-2 HIV-1 suggest that TLR signaling would be through TLR7/8, which senses ssRNA, and not through TLR9, which senses DNA, as AT-2 inactivated virus produces no viral DNA, as it does not initiate reverse transcription. This was confirmed by blocking signaling of TLR8 and TLR9 with inhibitors, followed by challenge with AT-2-inactivated HIV-1. Tissues treated with a TLR8 inhibitor demonstrated lower cytokine levels compared with tissues treated with a TLR9 antagonist. Thus, signaling through TLR, in particular TLR8, seemed to be one of the mechanisms mediating sustained immune activation by defective HIV-1 virions, although it, alone, does not seem to be sufficient.
In conclusion, the ex-vivo lymphoid tissue system allowed the investigation of possible mechanisms involved in persistent immune activation in HIV-1 patients under ART, under laboratory-controlled conditions. Our results with isolated lymphoid tissue indicate that the mechanisms of sustained immune activation in these patients may include the presence of defective (replication-incompetent) virions and of extracellular vesicles, probably together with reactivated HHVs, in particular, CMV.
These elements constitute potential therapeutic targets to combat the progression of various disorders in HIV-1-infected individuals after HIV-1 itself has been successfully suppressed.
Acknowledgements
V.M., W,F, and C.V. designed and performed experiments, analyzed data, and contributed to the manuscript. I.M. performed statistical analysis. L.M. supervised the project, designed experiments, analyzed data, and contributed to writing the manuscript. All authors participated in review and critique of the manuscript.
The authors would like to thank James Erickson at the Laboratory of Molecular Virology at George Mason University for his generous assistance with the technique of EV isolation on iodixanol gradients.
The work of V.M., W.F., C.V, and L.M. supported by the Intramural Program of the National Institute of Child Health and Human Development.
Conflicts of interest
Conflicts of interest and source of funding: The authors declare no conflicts of interest.
Supplementary Material
Vincenzo Mercurio and Wendy Fitzgerald contributed equally.
Written work prepared by employees of the Federal Government as part of their official duties is, under the U.S. Copyright Act, a “work of the United States Government” for which copyright protection under Title 17 of the United States Code is not available. As such, copyright does not extend to the contributions of employees of the Federal Government.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Website (http://www.AIDSonline.com).
References
- 1.Ambrosioni J, Nicolas D, Sued O, Aguero F, Manzardo C, Miro JM. Update on antiretroviral treatment during primary HIV infection. Expert Rev Anti Infect Ther 2014; 12:793–807. [DOI] [PubMed] [Google Scholar]
- 2.Archin NM, Sung JM, Garrido C, Soriano-Sarabia N, Margolis DM. Eradicating HIV-1 infection: seeking to clear a persistent pathogen. Nat Rev Microbiol 2014; 12:750–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garbelli A, Riva V, Crespan E, Maga G. How to win the HIV-1 drug resistance hurdle race: running faster or jumping higher?. Biochem J 2017; 474:1559–1577. [DOI] [PubMed] [Google Scholar]
- 4.Brites-Alves C, Luz E, Netto EM, Ferreira T, Diaz RS, Pedroso C, et al. Immune activation, proinflammatory cytokines, and conventional risks for cardiovascular disease in HIV patients: a case-control study in Bahia, Brazil. Front Immunol 2018; 9:1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wada NI, Jacobson LP, Margolick JB, Breen EC, Macatangay B, Penugonda S, et al. The effect of HAART-induced HIV suppression on circulating markers of inflammation and immune activation. AIDS 2015; 29:463–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ortiz AM, Flynn JK, DiNapoli SR, Sortino O, Vujkovic-Cvijin I, Belkaid Y, et al. Antiretroviral therapy administration in healthy rhesus macaques is associated with transient shifts in intestinal bacterial diversity and modest immunological perturbations. J Virol 2019; 93:e00472–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marchetti G, Tincati C, Silvestri G. Microbial translocation in the pathogenesis of HIV infection and AIDS. Clin Microbiol Rev 2013; 26:2–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Voeght A, Martens H, Renard C, Vaira D, Debruche M, Simonet J, et al. Exploring the link between innate immune activation and thymic function by measuring sCD14 and TRECs in HIV patients living in Belgium. Plos One 2017; 12:e0185761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beltran LM, Rubio-Navarro A, Amaro-Villalobos JM, Egido J, Garcia-Puig J, Moreno JA. Influence of immune activation and inflammatory response on cardiovascular risk associated with the human immunodeficiency virus. Vasc Health Risk Manag 2015; 11:35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Achappa B, Unnikrishnan DM, Venugopal B. Immune reconstitution inflammatory syndrome (IRIS) in HIV positive patients initiated on antiretroviral therapy (ART). Retrovirology 2012; 9: Suppl 1: P21. [Google Scholar]
- 11.Paiardini M, Muller-Trutwin M. HIV-associated chronic immune activation. Immunol Rev 2013; 254:78–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferdin J, Goricar K, Dolzan V, Plemenitas A, Martin JN, Peterlin BM, et al. Viral protein Nef is detected in plasma of half of HIV-infected adults with undetectable plasma HIV RNA. PLoS One 2018; 13:e0191613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Collini PJ, Bewley MA, Mohasin M, Marriott HM, Miller RF, Geretti AM, et al. HIV gp120 in the lungs of antiretroviral therapy-treated individuals impairs alveolar macrophage responses to Pneumococci. Am J Respir Crit Care Med 2018; 197:1604–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eden A, Fuchs D, Hagberg L, Nilsson S, Spudich S, Svennerholm B, et al. HIV-1 viral escape in cerebrospinal fluid of subjects on suppressive antiretroviral treatment. J Infect Dis 2010; 202:1819–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Henderson LJ, Johnson TP, Smith BR, Reoma LB, Santamaria UA, Bachani M, et al. Presence of Tat and transactivation response element in spinal fluid despite antiretroviral therapy. AIDS 2019; 33: Suppl 2: S145–S157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hladnik A, Ferdin J, Goricar K, Deeks GS, Peterlin MB, Plemenitas A, et al. Trans-activation response element RNA is detectable in the plasma of a subset of aviremic HIV-1-infected patients. Acta Chim Slov 2017; 64:530–536. [DOI] [PubMed] [Google Scholar]
- 17.Imamichi H, Smith M, Adelsberger JW, Izumi T, Scrimieri F, Sherman BT, et al. Defective HIV-1 proviruses produce viral proteins. Proc Natl Acad Sci U S A 2020; 117:3704–3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mercurio V, Fitzgerald W, Molodtsov I, Margolis L. Persistent immune activation in HIV-1-infected ex vivo model tissues subjected to antiretroviral therapy: soluble and extracellular vesicle-associated cytokines. J Acquir Immune Defic Syndr 2020; 84:45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Introini A, Fitzgerald W, Vanpouille C, Margolis L. Histoculture and infection with hiv of functional human lymphoid tissue on Gelfoam ((R)). Methods Mol Biol 2018; 1760:187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grivel JC, Margolis L. Use of human tissue explants to study human infectious agents. Nat Protoc 2009; 4:256–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Introini A, Vanpouille C, Fitzgerald W, Broliden K, Margolis L. Ex vivo infection of human lymphoid tissue and female genital mucosa with human immunodeficiency virus 1 and histoculture. J Vis Exp 2018; 2018:57013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Biancotto A, Brichacek B, Chen SS, Fitzgerald W, Lisco A, Vanpouille C, et al. A highly sensitive and dynamic immunofluorescent cytometric bead assay for the detection of HIV-1 p24. J Virol Methods 2009; 157:98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fitzgerald W, Sylwester AW, Grivel JC, Lifson JD, Margolis LB. Noninfectious X4 but not R5 human immunodeficiency virus type 1 virions inhibit humoral immune responses in human lymphoid tissue ex vivo. J Virol 2004; 78:7061–7068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fitzgerald W, Freeman ML, Lederman MM, Vasilieva E, Romero R, Margolis L. A system of cytokines encapsulated in extracellular vesicles. Sci Rep 2018; 8:8973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Biancotto A, Grivel JC, Iglehart SJ, Vanpouille C, Lisco A, Sieg SF, et al. Abnormal activation and cytokine spectra in lymph nodes of people chronically infected with HIV-1. Blood 2007; 109:4272–4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Margolis L. Immunoactivation at the crossroads of human disease. Am J Med 2015; 128:562–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ, et al. HLH Across Speciality Collaboration, UK. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet 2020; 395:1033–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lederman MM, Funderburg NT, Sekaly RP, Klatt NR, Hunt PW. Residual immune dysregulation syndrome in treated HIV infection. Adv Immunol 2013; 119:51–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vanpouille C, Introini A, Morris SR, Margolis L, Daar ES, Dube MP, et al. Distinct cytokine/chemokine network in semen and blood characterize different stages of HIV infection. AIDS 2016; 30:193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Conklin BS, Fu W, Lin PH, Lumsden AB, Yao Q, Chen C. HIV protease inhibitor ritonavir decreases endothelium-dependent vasorelaxation and increases superoxide in porcine arteries. Cardiovasc Res 2004; 63:168–175. [DOI] [PubMed] [Google Scholar]
- 31.Ganta KK, Chaubey B. Endoplasmic reticulum stress leads to mitochondria-mediated apoptosis in cells treated with anti-HIV protease inhibitor ritonavir. Cell Biol Toxicol 2019; 35:189–204. [DOI] [PubMed] [Google Scholar]
- 32.Naik E, Dixit VM. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J Exp Med 2011; 208:417–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bienstock RJ, Copeland WC. Molecular insights into NRTI inhibition and mitochondrial toxicity revealed from a structural model of the human mitochondrial DNA polymerase. Mitochondrion 2004; 4:203–213. [DOI] [PubMed] [Google Scholar]
- 34.McKoy JM, Bennett CL, Scheetz MH, Differding V, Chandler KL, Scarsi KK, et al. Hepatotoxicity associated with long- versus short-course HIV-prophylactic nevirapine use: a systematic review and meta-analysis from the Research on Adverse Drug events And Reports (RADAR) project. Drug Saf 2009; 32:147–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hileman CO, Funderburg NT. Inflammation, immune activation, and antiretroviral therapy in HIV. Curr HIV/AIDS Rep 2017; 14:93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang CC, Crane M, Zhou J, Mina M, Post JJ, Cameron BA, et al. HIV and co-infections. Immunol Rev 2013; 254:114–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Christensen-Quick A, Vanpouille C, Lisco A, Gianella S. Cytomegalovirus and HIV persistence: pouring gas on the fire. Aids Res Hum Retrovirus 2017; 33:S23–S30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Biancotto A, Grivel JC, Lisco A, Vanpouille C, Markham PD, Gallo RC, et al. Evolution of SIV toward RANTES resistance in macaques rapidly progressing to AIDS upon coinfection with HHV-6A. Retrovirology 2009; 6:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shmagel KV, Korolevskaya LB, Saidakova EV, Shmagel NG, Chereshnev VA, Margolis L, et al. HCV coinfection of the HIV-infected patients with discordant CD4(+) T-cell response to antiretroviral therapy leads to intense systemic inflammation. Dokl Biol Sci 2017; 477:244–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maidji E, Somsouk M, Rivera JM, Hunt PW, Stoddart CA. Replication of CMV in the gut of HIV-infected individuals and epithelial barrier dysfunction. PLoS Pathog 2017; 13:e1006202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Biancotto A, Iglehart SJ, Lisco A, Vanpouille C, Grivel JC, Lurain NS, et al. Upregulation of human cytomegalovirus by HIV type 1 in human lymphoid tissue ex vivo. AIDS Res Hum Retroviruses 2008; 24:453–462. [DOI] [PubMed] [Google Scholar]
- 42.Hunt PW, Martin JN, Sinclair E, Epling L, Teague J, Jacobson MA, et al. Valganciclovir reduces T cell activation in HIV-infected individuals with incomplete CD4+ T cell recovery on antiretroviral therapy. J Infect Dis 2011; 203:1474–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Forte E, Zhang Z, Thorp EB, Hummel M. Cytomegalovirus latency and reactivation: an intricate interplay with the host immune response. Front Cell Infect Microbiol 2020; 10:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sinclair J. Human cytomegalovirus: latency and reactivation in the myeloid lineage. J Clin Virol 2008; 41:180–185. [DOI] [PubMed] [Google Scholar]
- 45.Freeman ML, Lederman MM, Gianella S. Partners in crime: the role of CMV in immune dysregulation and clinical outcome during HIV infection. Curr HIV/AIDS Rep 2016; 13:10–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gianella S, Letendre S. Cytomegalovirus and HIV: a dangerous Pas de Deux. J Infect Dis 2016; 214: Suppl 2: S67–S74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Freeman ML, Mudd JC, Shive CL, Younes SA, Panigrahi S, Sieg SF, et al. CD8 T-cell expansion and inflammation linked to CMV coinfection in ART-treated HIV infection. Clin Infect Dis 2016; 62:392–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cohn LB, Chomont N, Deeks SG. The biology of the HIV-1 latent reservoir and implications for cure strategies. Cell Host Microbe 2020; 27:519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martinez-Picado J, Deeks SG. Persistent HIV-1 replication during antiretroviral therapy. Curr Opin HIV AIDS 2016; 11:417–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Makhathini KB, Abboussi O, Mabandla MV, Daniels WMU. The effects of repetitive stress on tat protein-induced pro-inflammatory cytokine release and steroid receptor expression in the hippocampus of rats. Metab Brain Dis 2018; 33:1743–1753. [DOI] [PubMed] [Google Scholar]
- 51.Planes R, Serrero M, Leghmari K, BenMohamed L, Bahraoui E. HIV-1 envelope glycoproteins induce the production of TNF-alpha and IL-10 in human monocytes by activating calcium pathway. Sci Rep 2018; 8:17215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Glushakova S, Grivel JC, Suryanarayana K, Meylan P, Lifson JD, Desrosiers R, Margolis L. Nef enhances human immunodeficiency virus replication and responsiveness to interleukin-2 in human lymphoid tissue ex vivo. J Virol 1999; 73:3968–3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Arakelyan A, Fitzgerald W, Zicari S, Vanpouille C, Margolis L. Extracellular vesicles carry HIV Env and facilitate HIV infection of human lymphoid tissue. Sci Rep 2017; 7:1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Arakelyan A, Fitzgerald W, King DF, Rogers P, Cheeseman HM, Grivel JC, et al. Flow virometry analysis of envelope glycoprotein conformations on individual HIV virions. Sci Rep 2017; 7:948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nolte-’t Hoen E, Cremer T, Gallo RC, Margolis LB. Extracellular vesicles and viruses: are they close relatives?. Proc Natl Acad Sci U S A 2016; 113:9155–9161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Puzar Dominkus P, Ferdin J, Plemenitas A, Peterlin BM, Lenassi M. Nef is secreted in exosomes from Nef.GFP-expressing and HIV-1-infected human astrocytes. J Neurovirol 2017; 23:713–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.DeMarino C, Pleet ML, Cowen M, Barclay RA, Akpamagbo Y, Erickson J, et al. Antiretroviral drugs alter the content of extracellular vesicles from HIV-1-infected cells (vol 8, 7653, 2018). Sci Rep 2018; 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chettimada S, Lorenz DR, Misra V, Dillon ST, Reeves RK, Manickam C, et al. Exosome markers associated with immune activation and oxidative stress in HIV patients on antiretroviral therapy. Sci Rep 2018; 8:7227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bernard MA, Zhao H, Yue SC, Anandaiah A, Koziel H, Tachado SD. Novel HIV-1 miRNAs stimulate TNFalpha release in human macrophages via TLR8 signaling pathway. PLoS One 2014; 9:e106006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Konadu KA, Chu J, Huang MB, Amancha PK, Armstrong W, Powell MD, et al. Association of cytokines with exosomes in the plasma of HIV-1-seropositive individuals. J Infect Dis 2015; 211:1712–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Raymond AD, Campbell-Sims TC, Khan M, Lang M, Huang MB, Bond VC, Powell MD. HIV type 1 Nef is released from infected cells in CD45(+) microvesicles and is present in the plasma of HIV-infected individuals. AIDS Res Hum Retroviruses 2011; 27:167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Robbins PD, Morelli AE. Regulation of immune responses by extracellular vesicles. Nat Rev Immunol 2014; 14:195–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Meier A, Alter G, Frahm N, Sidhu H, Li B, Bagchi A, et al. MyD88-dependent immune activation mediated by human immunodeficiency virus type 1-encoded Toll-like receptor ligands. J Virol 2007; 81:8180–8191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Silvin A, Manel N. Innate immune sensing of HIV infection. Curr Opin Immunol 2015; 32:54–60. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.