

Abstract
Frontotemporal lobar degeneration with TPD-43-immunoreactive pathology (FTLD-TDP) is subclassified based on the type and cortical laminar distribution of neuronal inclusions. The relevance of these pathological subtypes is supported by the presence of relatively specific clinical and genetic correlations. Recent evidence suggests that the different patterns of pathology are a reflection of biochemical differences in the pathological TDP-43 species, each of which is influenced by differing genetic factors. As a result, patient FTLD-TDP subtype may be an important factor to consider when developing biomarkers and targeted therapies for frontotemporal dementia. In this review, we first describe the pathological features, clinical and genetic correlations of the currently recognized FTLD-TDP subtypes. We then discuss a number of novel patterns of TDP-43 pathology. Finally, we provide an overview of what is currently known about the biochemical basis of the different FTLD-TDP subtypes and how this may explain the observed phenotypic and pathological heterogeneity.
Keywords: frontotemporal dementia, frontotemporal lobar degeneration, TDP-43, FTLD-TDP
Introduction
Frontotemporal dementia (FTD) is a heterogeneous clinical syndrome characterized by progressive changes in behaviour, personality and/or language, with relative preservation of memory (1). Major clinical subtypes include the behavioural variant (bvFTD) and two forms of primary progressive aphasia (PPA); the non-fluent/agrammatic and semantic variants (nfvPPA, svPPA; respectively). In addition, FTD is often associated with motor features, either an extrapyramidal movement disorder (atypical parkinsonism or corticobasal syndrome, CBS) or motor neuron disease (MND, usually classical amyotrophic lateral sclerosis, ALS). A family history is present in 25–50% of cases, with autosomal dominant FTD caused by mutations in several different genes (2).
The neuropathology underlying clinical FTD is also heterogeneous. Relatively selective degeneration of the frontal and temporal lobes is a consistent feature and “frontotemporal lobar degeneration” (FTLD) is used as the generic term for those pathologies that commonly present as clinical FTD (3, 4). As with many other neurodegenerative conditions, the pathology of most cases of FTD includes the abnormal intracellular aggregation and accumulation of some pathological protein(s). Until quite recently, the vast majority of FTLD cases fell into two broad categories; those characterized by cellular inclusions composed of the microtubule associated protein tau (FTLD-tau) and those with tau-negative inclusions that could only be detected with immunohistochemistry (IHC) against the non-specific marker of pathological protein accumulation, ubiquitin (FTLD-U) (5). In 2006, two publications each described three distinct patterns of FTLD-U pathology, based on the anatomical distribution and morphology of ubiquitin immunoreactive (-ir) neuronal inclusions in the cerebral cortex (6, 7). Importantly, the pathological features that defined each of the subtypes in these two independent studies were almost identical, providing powerful validation of the results. The significance and legitimacy of the pathological subtypes was further supported by the finding of relatively specific correlations with different clinical phenotypes (6) and with the subsequent recognition that most of the newly identified genetic causes of FTD were each consistently associated with a specific type of FTLD-U pathology, including a novel (fourth) pattern that is only found in cases caused by mutations in the valosin containing protein gene (VCP) (8–10). A major breakthrough occurred, later in 2006, when the transactive response DNA binding protein with Mr 43 kD (TDP-43) was identified as the ubiquitinated pathological protein in most cases of FTLD-U (which now became FTLD-TDP) and in sporadic ALS, strengthening the concept that FTD and ALS are closely related conditions with overlapping pathogenesis (11, 12). Subsequent studies confirmed TDP-43 as the pathological protein in most clinical and genetic subtypes of FTLD-U and the same criteria were adopted for the pathological sub-classification of FTLD-TDP, with only minor modifications (13–15).
Over the past decade, the concept and utility of the current FTLD-TDP subtyping system has gained wide acceptance and has been repeatedly validated through its application in new case series and by the discovery of additional clinical, genetic and pathological correlations. Moreover, recent studies have demonstrated that cases with each of the different pathological subtypes are associated with different genetic risk factors and that the insoluble protein extracted from postmortem brain tissue has differing physical and biochemical properties (16–18). These findings suggest that accurate pathological subtyping of cases and a better understanding of their biochemical basis will likely be important to advance the development of biomarkers and targeted therapies for FTD.
Major FTLD-TDP pathological subtypes
Although the studies that originally described the FTLD-U subtypes evaluated ubiquitin-ir pathology in neocortex, hippocampus and (in one study) striatum (6, 7), the diagnostic criteria that are now commonly used to sub-classify FTLD-TDP cases are based exclusively on neocortical features (Table 1). Several studies have shown that these criteria are equally applicable and give comparable results regardless of whether the antibody used recognizes phosphorylated or phosphorylation-independent TDP-43 (15, 19). The two discordant numbering systems introduced in the original papers have since been replaced with the harmonized alphabetic classification that is used below (20).
Table 1.
FTLD-TDP subtypes: distinguishing pathological features*, associated phenotypes, causal mutations
| Type A | Type B | Type C | Type D | |
|---|---|---|---|---|
| TDP-ir pathology | ||||
| neocortex | II: cNCI, DN, NII | II-VI: dNCI | II-VI: long DN | II-VI:DN, NII |
| hippocampus | den: NII | den: dNCI | den: cNCI | |
| CA1: threads | ||||
| subcortical | WM: threads | WM: GCI | ||
| BG: DN, NII | BG: dNCI, GCI | BG: cNCI | BG: DN, NII | |
| SN: DN | SN: dNCI, GCI | SN: DN, NII | ||
| LMN: NCI | ||||
| phenotypes | bvFTD, nfvPPA | bvFTD, nfvPPA, ALS | svPPA | IBMPFD, ALS |
| mutations |
GRN C9orf72, TBK1 |
C9orf72, TBK1 | VCP |
see main text for full description of regional pathology. II, cortical lamina II; II-VI, cortical laminae II to VI; ALS, amyotrophic lateral sclerosis; BG, basal ganglia; bvFTD, behavioral variant frontotemporal dementia; C9orf72, chromosome 9 open reading frame 72 gene; CA1, cornu ammonis region 1; cNCI, compact neuronal cytoplasmic inclusions; den, dentate lamina of hippocampus; DN, dystrophic neurites (short unless otherwise specified); dNCI, diffuse NCI; GCI, glial cytoplasmic inclusions; GRN, granulin gene; IBMPFD, inclusion body myopathy with Paget disease of bone and frontotemporal dementia; LMN, lower motor neurons; NII, neuronal intranuclear inclusions; nfvPPA, non-fluent variant primary progressive aphasia; SN, substantia nigra; svPPA, semantic variant PPA; TBK1, TANK binding kinase 1 gene; TDP-ir, TDP-43 immunoreactive;VCP, valosin containing protein gene; WM, white matter.
Neocortical features
FTLD-TDP type A
Type A cases are characterized by abundant TDP-43-ir neuronal cytoplasmic inclusions (NCI) and short thick dystrophic neurites (DN) which are concentrated in the superficial cortical layers (Fig. 1). The NCI are mostly compact (cNCI) and have an oval or crescentic shape. Lentiform neuronal intranuclear inclusions (NII) are also usually present, but much less abundant.
Figure 1.
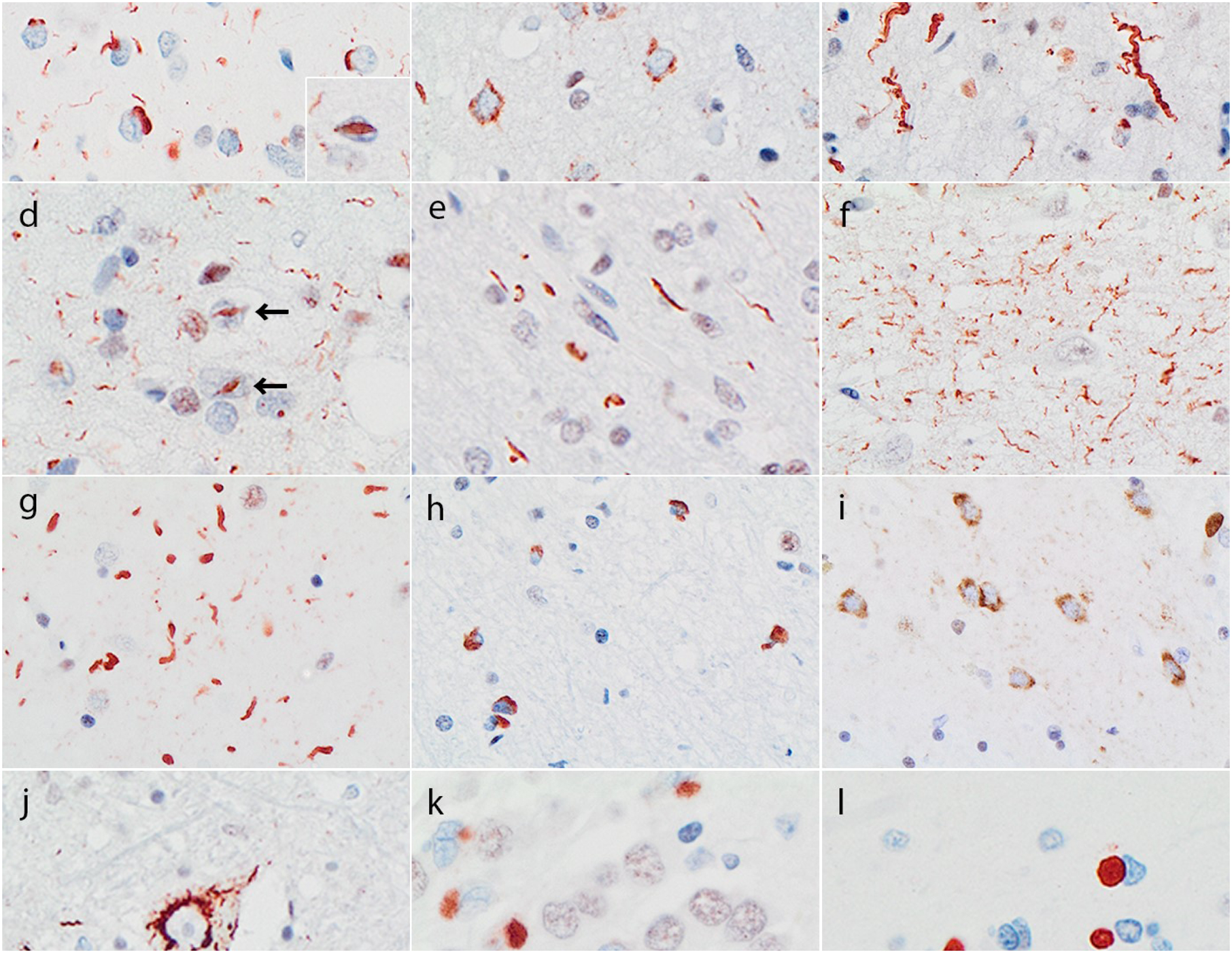
TDP-43 immunoreactive pathology in different FTLD-TDP subtypes. Subtypes are defined by the pattern in the neocortex: type A has compact neuronal cytoplasmic inclusions (cNCI), short dystrophic neurites (DN) and some lentiform neuronal intranuclear inclusions (NII, insert) concentrated in layer II (a); type B has diffuse granular NCI (dNCI) throughout the neocortex (b); type C has DN, many of which are long and tortuous DN (c), and type D has numerous NII (arrows) and delicate short DN (d). Each subtype also shows a characteristic pattern of pathology in the hippocampus and subcortical regions. Type A cases have thread pathology in the subcortic al white matter (e), delicate wispy threads in hippocampal CA1 (f) and a predominance of DN and occasional NII in striatum and other subcortical grey matter regions (g). Type B cases have glial cytoplasmic inclusions in the subcortical white matter (h), a predominance of dNCI in subcortical grey matter (i) and NCI in lower motor neurons of the medulla and spinal cord (j). Type C cases have compact “Pick body-like” NCI in dentate granule cells of the hippocampus (k) and striatum (l). Bar: 40 μm (a-c, f-j), 10 μm (a, insert), 30 μm (d, e, l), 25 μm (k). TDP-43 immunohistochemistry.
FTLD-TDP type B
Type B cases have at least moderate numbers of NCI in both superficial and deep cortical layers with relatively few DN and no NII. Most of the NCI have a diffuse granular morphology (dNCI), sometimes referred to as “pre-inclusions”. Importantly, some cases also have a background of delicate, small, TDP-43-ir threads and dots (ThD) which, when concentrated in layer II, may resemble the superficial laminar distribution that is typical of type A cases; however, this ThD pathology is neither consistent nor specific for type B cases.
FTLD-TDP type C
Type C cases have a predominance of DN with few if any NCI and no NII. DN are somewhat more abundant in superficial cortical layers and many have a unique long, tortuous morphology.
FTLD-TDP type D
The characteristic feature of FTLD-TDP type D pathology is an abundance of lentiform NII and delicate short DN which are somewhat concentrated in superficial laminae. cNCI are rare in this subtype.
Hippocampal and subcortical pathology
In addition to the characteristic neocortical features, most cases of FTLD-TDP are also found to have significant TDP-43-ir pathology in limbic and subcortical anatomical regions (Table 1) (8, 21, 22). Although not included in the diagnostic criteria, each of the neocortical subtypes shows a highly consistent pattern of subcortical involvement which may be helpful when classifying difficult cases, and which may help to explain the range of associated clinical features (22).
FTLD-TDP type A
A highly characteristic feature of type A cases is the presence of delicate TPD-43-ir threads in hippocampal CA1 region, which is often associated with significant pyramidal cell loss (hippocampal sclerosis) (Fig. 1). Type A cases also tend to have abundant white matter threads, a predominance of DN in subcortical grey matter regions and small numbers of NII in the hippocampus and striatum. Diffuse and compact NCI are also present in the hippocampal dentate and striatum, but tend to be less abundant than in type B or C cases.
FTLD-TDP type B
The most defining subcortical feature of type B cases is frequent NCI in LMN of the hypoglossal nucleus and spinal cord, which may have diffuse, compact or filamentous morphology. Moderate numbers of TDP-43-ir glial cytoplasmic inclusions (GCI) are present in the cerebral white matter. Many subcortical grey matter regions have abundant NCI which are predominantly diffuse, as well as more modest numbers of GCI.
FTLD-TDP type C
The hippocampal dentate gyrus and striatum consistently show numerous cNCI that have a unique “Pick body-like” morphology with uniform solid consistency and smooth round contour (in contrast to the cNCI found in some type A and B cases which usually appeared as a compact aggregate of coarse granules). The cerebral white matter is not involved and most other subcortical structures show only occasional DN.
FTLD-TDP type D
Modest numbers of DN and NII are present in the amygdala, basal ganglia, nucleus basalis, thalamus and midbrain. The pons, medulla and cerebellum are consistently spared. Notably, the dentate granule cells of the hippocampus are free of NCI.
Clinical correlations
There is significant overlap in the clinical features associated with each of the different major protein classes of FTLD (FTLD-tau, FTLD-TDP and FTLD-FET) and among the subtypes within each class (1, 23–25). Moreover, in cases within all the pathological groups, a patient’s phenotype often evolves as their disease progresses to include additional clinical features. In general, cases of svPPA and FTD combined with ALS are usually found to have underlying FTLD-TDP pathology, those with nfvPPA or prominent extrapyramidal features (particularly sporadic cases) more often have FTLD-tau; whereas, bvFTD can be associated with any of the FTLD pathologies. Within the FTLD-TDP group, each of the subtypes shows a number of important clinical correlations.
FTLD-TDP type A
Most cases present with features of bvFTD, often with prominent apathy and social withdrawl. An aphasic presentation is less common and may be nfvPPA or more difficult to classify. Executive dysfunction and some degree of memory impairment are not uncommon, particularly with older age at presentation. Neuropsychiatric manifestations (delusions, hallucinations or obsessive behaviors) are particularly common in those with an underlying GRN or C9orf72 mutation. Extrapyramidal features are reported in up to half of cases, but are rarely the presenting or predominant feature; whereas, ALS is highly unusual.
FTLD-TDP type B
This pathology underlies the vast majority of cases in which FTD occurs in combination with clinical features of ALS. The presenting dementia syndrome is most often bvFTD, while language problems usually develop later. Psychosis is particularly common in those caused by the C9orf72 repeat expansion where they may be the presenting feature in one third (26). Extrapyramidal features develop in at least half.
FTLD-TDP type C
There is a particularly strong correlation between this pathology and svPPA, with most cases of clinical svPPA having FTLD-TDP type C pathology. There are often some associated behavioral changes and cases with predominant right temporal involvement may present with loss of sympathy/empathy, hyposexuality, prosopagnosia and obsessive/compulsive behavior. Psychiatric features and extrapyramidal movement disorders are much less common than with the other subtypes. Although these cases do not develop ALS, they may have some upper motor neuron features. Patients with this pathology also tend to have a slower disease progression and older age at death compared to those with the other FTLD-TDP subtypes.
FTLD-TDP type D
This pathology is exclusively found in familial cases with VCP mutations in which there is variable penetrance of inclusion body myopathy (90%), Paget disease of bone (45%), FTD (30%) and ALS (10%) (27). The FTD syndrome is usually bvFTD with language dysfunction and extrapyramidal motor features being relatively uncommon.
Genetic correlations
Patients with FTD due to mutations in GRN are consistently found to have FTLD-TDP type A pathology at autopsy (10, 13, 15), while those with VCP mutations always have type D (Table 1) (8, 28). In contrast, the C9orf72 repeat expansion has more variable TDP-43-ir pathology, with most studies reporting some cases with type A and others with type B FTLD-TDP (26, 29–32). Moreover, two recent studies, found that only half of C9orf72 mutation cases had either typical type A or type B pathology, while the largest group had the combined pathological features of both type A and type B (type A+B, see below) (15, 22). Although there are currently few reports describing the pathology in cases of FTD caused by mutations in the TANK binding kinase 1 gene (TBK1), these also seem to include both type A and type B cases (33–36). There are a number of other rare genetic causes of FTD that have been reported to have TDP-43 pathology, but for which there is currently insufficient information to define the specific pattern (e.g. TARDBP, CHCHD10, OPTN, SQSTM1)(2). Finally, in addition to causal mutations, genetic risk factors have been identified for FTLD-TDP, some of which are associated with a specific pathological subtype (e.g. a variant in UNC13A has was found to be associated with FTLD-TDP type B cases but not A or C)(17).
Other FTLD-TDP subtypes and patterns of pathology
Although the subtyping of FTLD-TDP cases has proven to be useful and the current criteria generally accepted, several reports have identified cases that are difficult to classify, either because the pattern of pathology does not to fit with any of the existing subtypes or because it shows overlapping features of more than one subtype (15, 22, 37–42). Although these cases represent a small minority in most series, they highlight some of the technical and interpretive differences that exist among neuropathologists in applying the current FTLD-TDP classification criteria.
FTLD-TDP cases with overlapping pathological features
In a series of 30 FTLD-TDP cases selected for a BrainNet Europe study, an initial panel of five neuropathologists designated three cases as “atypical” type B, four cases as having features of both type A and type B (A+B), and two cases that had insufficient TDP-43 pathology for typing (37). A follow-up analysis of this case series, involving a much larger group of investigators, found relatively poor agreement amongst the reviewers in assigning FTLD-TDP subtypes (~62%), with the worst agreement observed for FTLD-TDP type B cases. However, agreement was better (up to 85%) when raters were asked to simply dichotomize between type A or B versus type C, suggesting that the major difficulty was in differentiating between type A and type B. An earlier study by Armstrong et al., that used principal component analysis, and that included a combination of TDP-43-ir pathology and additional changes that are not part of the standard subtyping criteria (neuronal loss, neuronal enlargement, neuropil vacuolation, oligodendroglial inclusions) also found significant overlap among FTLD-TDP subtypes, particularly between type A and type B (43). Finally, a small series of four cases of FTD with delusions also reported two as having mixed type A+B pathology and two which were unclassifiable (39). Importantly, all four of these cases harbored the C9orf72 repeat expansion.
The issue of combined subtypes was addressed more specifically in a study design to compare the pathological features that define the subtypes based on the original ubiquitin-based criteria versus TDP-43 IHC (15). In this series of 78 FTLD-TDP cases, the majority (81%) were easily classified as types A, B, or C; however, 15 cases demonstrated mixed features of both FTLD-TDP type A and type B. These mixed cases were characterized by NII, NCI and short DN in layer II (type A features), as well as granular NCI in deeper neocortical layers that were at least as numerous as in layer II (type B features) (Fig. 2). Importantly, 12 of the 15 type A+B cases carried the C9orf72 repeat expansion, while the remaining three cases had clinical or pathologic evidence of MND. In fact, half of the C9orf72 mutation cases in this study had FTLD-TDP type A+B pathology with the other half were classified as pure type B.
Figure 2.
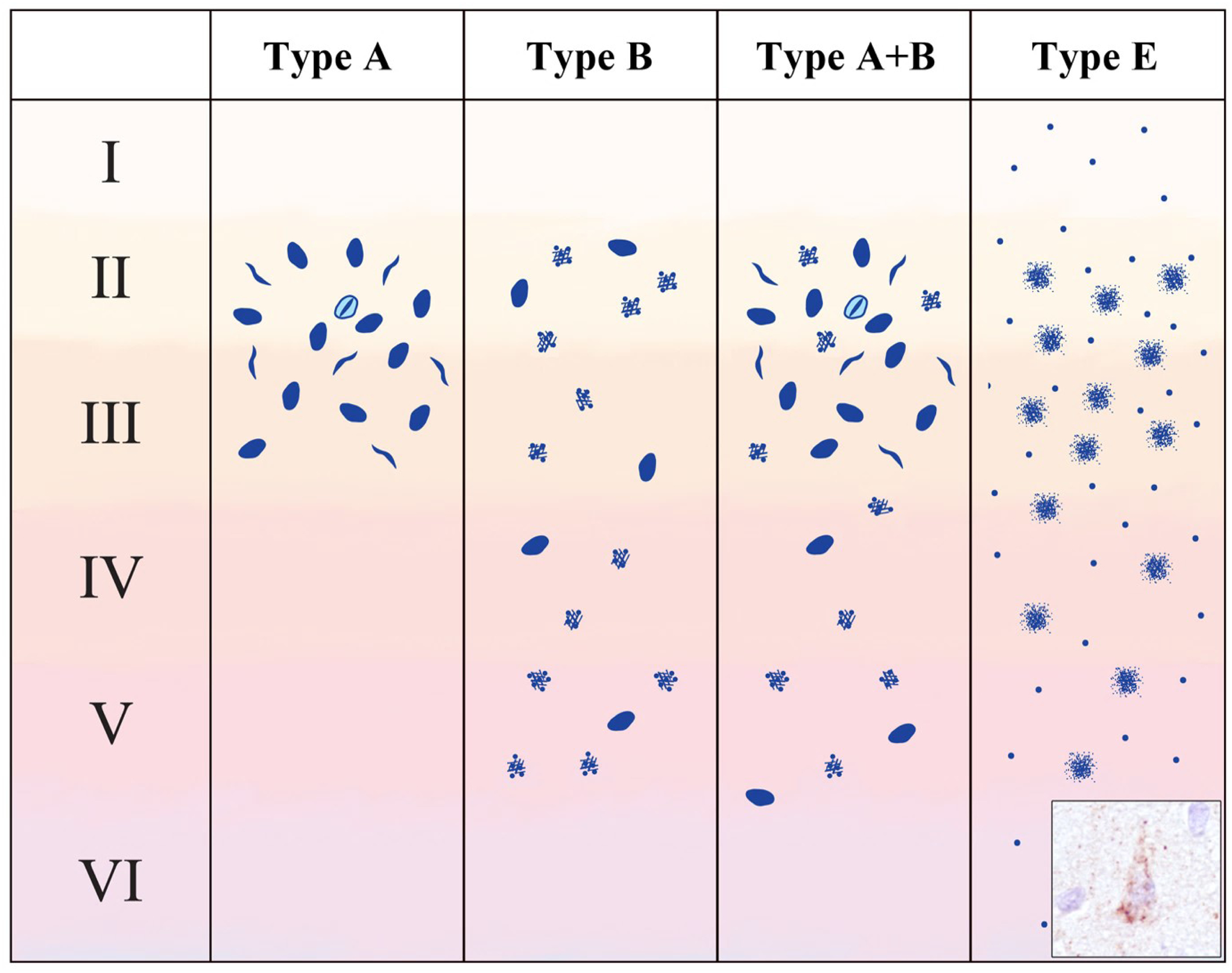
Schematic representation of TDP-43 inclusion morphologies and distribution in cases with mixed (A+B) and novel (E) subtypes. Type A+B cases show the characteristic features of type A (compact neuronal cytoplasmic inclusions (NCI), short dystrophic neurites and neuronal intranuclear inclusions, concentrated in layer II) as well as the characteristic features of type B (compact and diffuse granular NCI in deep and superficial layers). Type E cases exhibit granulofilamentous neuronal cytoplasmic inclusions and a background of and fine grains (inset photo) throughout the neocortex. Modified from Lee et al. 2017 (40).
A similar analysis of 89 cases by another group found that a higher proportion of cases (96%) could be readily subtyped as A, B or C; whereas, five cases were judged to have features that crossed FTLD-TDP subtypes, all of which also had concomitant MND pathology (44). One case with the C9orf72 mutation exhibited type B features with NII (type A+B) while another C9orf72 case exhibited a mixed type B+C pattern. The other three were non-C9orf72 cases and included one type C with NII (type A+C), and two type B with long DN (type B+C).
Although the current subtyping criteria is based solely on pathological findings in neocortical sections, each of the different FTLD-TDP subtypes has also been reported to be associated with distinctive patterns of TDP-43 pathology in limbic and subcortical regions (see above) (21, 22). In a recent study, Mackenzie and Neumann investigated whether including pathological data from subcortical anatomical regions would allow for better classification of cases with a mixed pattern of neocortical TDP-43-ir pathology (22). Using standard observational assessment of neocortical sections, all of the non-C9orf72 mutation cases could be readily classified as type A, B or C, and these results were validated using non-biased hierarchical clustering analysis (HCA). Furthermore, HCA of the pathological data from subcortical regions found that these cases again formed three distinct clusters, which perfectly matched the neocortical type A, B and C groups. In contrast, using the neocortical data, only half of the C9orf72 mutation cases clustered with either the type A or type B cases, and the remaining 14 formed a distinct cluster exhibiting mixed features of type A and B. When the same group of C9orf72 mutations cases was analyzed using the limbic and subcortical TDP-43 pathology data, more of the cases segregated as type A or type B; however, five cases remained as a separate mixed A+B cluster.
The results of these studies indicate that, although the vast majority of FTLD-TDP cases can be readily sub-classified based on the current criteria, there exists a minority that are difficult to assign because they have a combination of pathological features that characterize more than one subtype. Interestingly, these mixed patterns of pathology seem to be particularly common in cases with the C9orf72 repeat expansion and sporadic cases that have features of both FTD and ALS (15, 22, 39, 41, 44), suggesting that there may be something unique about the mechanism of TDP-43 mis-metabolism in these clinical and genetic groups that result in greater pathological heterogeneity.
Novel FTLD-TDP subtypes
In 2017, Lee et al. described a series of seven cases that were difficult to categorize based on the 2011 harmonized FTLD-TDP classification, that they felt represented a unique subtype which they designated as type E (40). The neocortical TDP-43 pathology involved all cortical layers and consisted of weakly staining granulofilamentous neuronal cytoplasmic inclusions (GFNI) set in a background of very fine grain-like deposits (Fig. 2). In contrast to the NCI found in other FTLD-TDP subtypes, these GFNI were negative for ubiquitin and mostly negative for p62. GFNI and grain pathology, as well as TDP-43-ir oligodendroglial inclusions, were also present in a wide range of neocortical and subcortical regions, sparing only of the occipital neocortex and cerebellum. Motor neuron involvement was a consistent feature, although only one case was associated with clinical features of ALS. Interestingly, these FTLD-TDP type E cases were consistently associated with a rapid clinical course of one to three years duration. Some additional reports have described cases with pathology similarity to the type E of Lee et al. Takeuchi et al. reported a subset of sporadic ALS cases with NCI, granular or dot like DN, and a high density of GCI, involving motor cortex, other neocortical regions, basal ganglia and spinal cord (41). Ubiquitin and p62 IHC were not performed. The authors interpreted these findings as distinct from FTLD-TDP types A - D. More recently, two cases with one year duration of PPA and ALS were reported to exhibit FTLD-TDP type E pathology, consisting of TDP-43-ir, p62-negative GFNI and grains (42). Finally, a case of rapidly progressive Foix-Chavany-Marie syndrome (FCMS) has been reported to exhibit FTLD-TDP type E (45). FCMS, also known as bilateral opercular syndrome, is characterized by prominent motor dysfunction involving muscles of the face, tongue and pharynx. While the etiology of FCMS is diverse, sometimes being associated with bilateral opercular infarcts, progressive forms of FCMS share clinical similarities to FTD (46).
While FTLD-TDP type E may represent a distinct subtype, the association with ALS in some cases and the similarities with the pathological features of type B cases, raise the possibility that types E and B represent a continuum. Indeed, FTLD-TDP type B has been described as often having a predominance of granular rather than compact NCI, a synaptic pattern of neuropil inclusions, and abundant threads and dots (15, 21, 32). Given the relatively short disease duration of most cases with FTLD-TDP type E, one possibility is that these represent “early stage” disease when the TDP-43 inclusions are still immature and have not yet coalesced into a more typical FTLD-TDP type B morphology and become ubiquitinated. Alternatively, FTLD-TDP type E could represent a more virulent pathology which spreads through the brain and spinal cord quickly, resulting in rapid clinical disease progression and relatively immature inclusions.
Unique patterns of TDP-43 pathology in rare disorders
Unique patterns of TDP-43 proteinopathy have been described in a few rare neurodegenerative diseases, not typically classified as FTLD or ALS. A screen of non-neurodegenerative disease neuropathology specimens revealed that Rosenthal fibers and eosinophilic granular bodies, which may be present in reactive gliosis and in some low grade astrocytic brain tumors, label with TDP-43 IHC (47). Rosenthal fibers are protein aggregates within astrocytes that are composed primarily of glial fibrillary acidic protein (GFAP) and are also the defining pathological feature of Alexander disease, a leukodystrophy associated with GFAP mutations (48). A subsequent study demonstrated that Rosenthal fibers in Alexander disease are also TDP-43-ir (49). Thus, Alexander disease represents a unique TDP-43 proteinopathy in which neurodegeneration is associated exclusively with astrocytic inclusions. Another unique pattern of TDP-43 proteinopathy is found in Perry syndrome, a progressive neurodegenerative disease characterized by parkinsonism, psychiatric symptoms and hypoventilation, caused by mutations in the gene encoding dynactin-1 (DCTN1) (50). In addition to modest numbers of NCI composed of the dynactin subunit p50, cases of Perry syndrome exhibit TDP-43-ir NCI, DN, oligodendroglial GCI, axonal spheroids and perivascular astrocytic inclusions (51). Based on the very limited number of cases reported (n=3), the pattern of TDP-43 pathology in Perry syndrome seems to be distinct from FTLD-TDP, with a predisposition for the substantia nigra and other subcortical regions with only mild and inconsistent involvement of the cerebral cortex.
In both Alexander disease and Perry syndrome, TDP-43 protein aggregation is likely secondary to the accumulation and dysfunction of other proteins (GFAP and dynactin, respectively). None-the-less, these conditions are informative by demonstrating that TDP-43 proteinopathy may result from diverse mechanisms.
TDP-43 pathology in aging and common neurodegenerative disorders
Finally, it is important to recognize that some degree of TDP-43-ir pathology is a common finding in the limbic structures of the mesial temporal lobe in aging and in association with many common neurodegenerative disorders, including Alzheimer’s and Lewy body disease (52). The clinical relevance of this pathology and its relationship to FTLD-TDP is currently the topic of tremendous interest and controversy (53, 54), but is beyond the scope of the present review.
Biochemical basis of FTLD-TDP subtypes
Biochemical properties of TDP-43 aggregates and disease associated modifications
Aggregated TDP-43 isolated from human postmortem FTLD-TDP brain tissue is poorly detergent soluble and subject to a variety of disease associated posttranslational modifications (PTM). These result in a highly characteristic biochemical banding pattern by immunoblot analysis, with the presence of disease specific bands of ~25 kDa, ~45 kDa and a high molecular smear, in addition to the ~43 kDa band corresponding to normal TDP-43 (Fig. 3a) (11, 12). PTM of TDP-43 include N-terminal truncation, phosphorylation, ubiquitination, acetylation, cysteine oxidation and sumoylation (55). The characterization of the various PTM and their functional consequences are still poorly understood and not fully validated in human postmortem tissue; however, there is increasing evidence that TDP-43 PTM may play a crucial role in disease pathogenesis and that modulation of disease relevant PTM might be a promising avenue for future therapeutic approaches.
Figure 3.
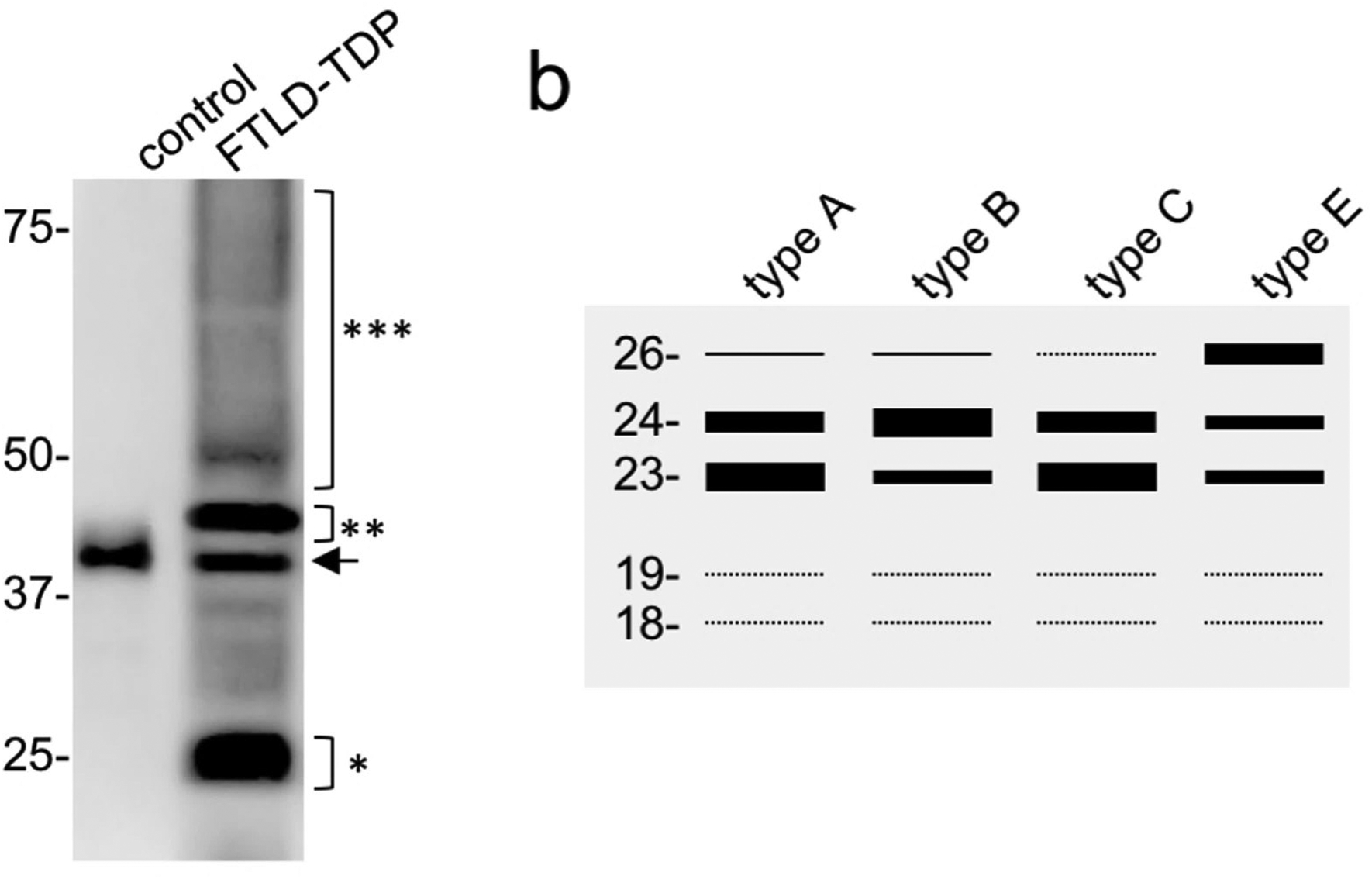
Immunoblot analysis of sarcosyl insoluble protein fractions from FTLD-TDP shows the disease-specific biochemical signature of TDP-43 with pathological bands −25 kDa (*), −45 kDa (**) and a high molecular smear (***), in addition to the physiological TDP-43 band (arrow) also present in control brains (a). Schematic representation of distinct banding patterns of C-terminal fragments among FTLD-TDP subtypes (type A, B and C based on Kawakami et al. 2018, type E based on Lee at al. 2017) (40, 84)(b).
N-terminal truncation and C-terminal fragments
The presence of short TDP-43 fragments of ~25 kDa are a hallmark feature of FTLD-TDP (11, 12). They are composed of N-terminally truncated TDP-43 species as demonstrated by absent labelling with antibodies raised against the N-terminus (amino acids 6–24) but detection with antibodies against the extreme C-terminus of TDP-43 (56). N-terminal sequencing of fragments isolated from human postmortem tissue has revealed arginine at position 208 (57), and mass spectrometry analysis of tryptic digests of isolated fragments has demonstrated aspartic acid residues at positions 219 and 247 (58) as potential cleavage sites. Although these experiments clearly demonstrate that these short species contain N-terminally truncated fragments that extend to the extreme C-terminus, it is still unclear whether they all include the entire C-terminal region. Moreover, the origins and pathomechanistic relevance of the C-terminal fragments (CTF) remain to be fully established. Most studies propose proteolytic cleavage/degradation by caspases (59, 60), asparaginyl endopeptidase (61) or calpains (62); although other explanations include alternative splicing events or usage of alternate translational start sites (63, 64). However, several proposed cleavage sites and generated fragments/isoforms in these studies do not match well with the fragments observed in human postmortem tissue, suggesting that additional enzymes and/or mechanisms might exist.
The potential role of TDP-43 CTF in disease pathogenesis is supported by findings of cellular toxicity upon overexpression of CTF in some cellular and animal models (57, 65); however, in several other model systems, the correlation is less clear (66). Moreover, while enrichment for CTF over full-length TDP-43 is a characteristic feature of most types of cellular inclusions in the cerebral cortex, CTF are less abundant or absent in inclusions in spinal cord LMN in FTLD-TDP/ALS (56) and in cortical pre-inclusions (67), thereby suggesting that the formation of CTF might not be mandatory for aggregation and toxicity.
Phosphorylation
Aberrant phosphorylation of TDP-43 has been recognized as one of the major PTM of pathological TDP-43 since its initial discovery as the disease protein in FTLD-TDP and ALS (11, 12). The fact that the majority of pathogenic TARDBP mutations either introduce or disrupt potential serine/threonine phosphorylation sites or introduce phosphomimic residues (glutamate/aspartate), suggest that alterations in the phosphorylation status of TDP-43 play a crucial role in the pathogenesis of TDP-43 proteinopathies (55). TDP-43 has 41 serine, 15 threonine and 8 tyrosine residues acting as potential phosphorylation sites. Mass spectrometry analysis of recombinant TDP-43 treated with casein kinase 1 and of aggregated TDP-43 isolated from human postmortem tissue has revealed several phosphorylated residues (68–70); however, so far only five sites at the C-terminus of TDP-43 (pS379, pS403, pS404, pS409, pS410) have been validated in pathological TDP-43 inclusions in human postmortem tissue with phosphorylation-site-specific antibodies (19, 68). Phosphorylation at these C-terminal serine residues (with pS409/410 as most studied sites) is a highly consistent and specific feature of aggregated TDP-43 in all types of pathological TDP-43 inclusions, in all sporadic and familial FTLD-TDP subtypes, and is considered an abnormal event due to the lack of phosphorylation of these sites under physiological conditions (15, 19, 68, 70, 71). The functional consequences of TDP-43 C-terminal phosphorylation are not fully resolved. While some experimental studies have described an association with decreased solubility of TDP-43 and greater toxicity (68, 72), others have reported the opposite effects with phospho-mimicking mutants showing increased solubility and reduced toxicity (73, 74). Further insights into the role of TDP-43 phosphorylation in regulating its physiological functions (e.g. RNA binding, dimerization) and the impact of abnormal phosphorylation events through the identification of the involved kinases and phosphatases, will be crucial steps to elucidate the pathological processes in TDP-proteinopathies.
Ubiquitination
Ubiquitination of TDP-43 aggregates is a key feature in FTLD-TDP; however, insights into the specific lysin residues that are ubiquitinated in human FTLD-TDP tissues and the functional consequences are still limited. The detection of TDP-43 Lys-48 and Lys-63 linked poly-ubiquitin chains in cellular models is suggestive of proteasomal and autophagosomal degradation of TDP-43 (75). Lysine residues 84, 95, 102, 114, 121, 140, 145, 160, 176, 181, 263 have been identified as ubiquitinated TDP-43 residues is cellular models; however, with some variability among studies, most likely reflecting the complexity of ubiquitin-proteasome regulation of TDP-43 in a highly context-dependent manner (76–79). Notably, ubiquitination of lysin 84 has been postulated as an important modifier of nuclear import of TDP-43 in mutagenesis experiments, and a complex interplay between TDP-43 ubiquitination at distinct sites and phosphorylation at pS409/410 has been observed (77). However, validation of any ubiquitination site in human postmortem FTLD-TDP tissue is lacking, and to date the only ubiquitinated residue identified by mass spectrometry of insoluble protein extracts from postmortem tissue (of an ALS patient) is lysine 79 (80).
Acetylation
Another modification of lysine residues is acetylation. So far, two acetylation sites have been identified in cellular models at lysine 145 (located in RRM1) and 192 (located in RRM2) (81). However, since mutation of TDP-43 at these two sites did not completely abrogated acetylation, additional acetylated lysine residues may be present. Acetylation at lysine 145 and 192 has been shown to impair the binding of TDP-43 to RNA and to promote TDP-43 phosphorylation at pS409/410 (81). The potential role of this modification in disease was demonstrated using an antibody specific for TDP-43 acetylated at lysine 145, which revealed acetylated TDP-43 as a biochemical component of the TDP-43 inclusions in ALS/FTD spinal cord, which are known to be composed of the full length protein, but not the inclusions in cerebral cortex, which are composed primarily of CTF that lack the epitope recognized by the antibody (81).
Sumoylation
Evidence for sumoylation of TDP-43 comes mainly from a proteomics approach that revealed SUMO-2/3 in complex with insoluble TDP-43 in a cellular model system overexpressing a CTF (75); however, sumoylation of TDP-43 has not yet been directly demonstrated in human disease tissue.
Cysteine oxidation
Upon exposure to oxidative stressors, TDP-43 has been reported to undergo cysteine oxidation and disulfide crosslinking in vitro and in cellular models, resulting in enhanced TDP-43 aggregation and alterations in subcellular distribution (82). TDP-43 has six cysteine residues and there is experimental evidence that all sites contribute to proper folding, self-assembly and oligomerization of TDP-43 (55). While increased levels of cross-linked TDP-43 species are present in FTLD-TDP brains (82), the pathomechanistic role of cysteine oxidation and cross-linking remains to be fully determined.
Biochemical diversity of TDP-43 aggregates in FTLD-TDP subtypes and evidence for TDP-43 strains
A crucial open question in the FTLD-TDP research field is the molecular basis behind the huge clinical and neuropathological phenotypic variability, as well as the selective vulnerability in FTLD-TDP subtypes and ALS. The concept that distinct self-propagating conformers of an aggregated protein (‘strains’) represent the basis for phenotypic diversity in a neurodegenerative disease was first established in prion diseases (83). By analogy, a popular hypothesis to explain the heterogeneity in FTLD-TDP is the presence of different conformational types of misfolded TDP-43 (‘TDP-43 strains’) that can propagate in a prion-like manner (84). In fact, there is a growing body of evidence supporting this idea.
Biochemical heterogeneity of aggregated TDP-43 has already been recognized in the initial report on the discovery of TDP-43 as the disease protein (11). Briefly, monoclonal antibodies (clones 182 and 406) generated against insoluble protein fractions from FTLD-TDP brains each labelled distinct bands of the N-terminally truncated TDP-43 species by immunoblot, specific for either type A or type B FTLD-TDP cases (then referred to as FTLD-U type 3 or 1, respectively). This suggested that each antibody was recognizing either a specific conformation or a specific pattern of PTM of aggregated TDP-43 species, each being specific for a different FTLD-TDP subtype. Several studies have been performed since then to further characterize and correlate an immunoblot banding pattern of TDP-43 CTF with distinct FTLD-TDP subtypes, with most employing antibodies against pS409/410 (19, 40, 68, 85). Using high percentage polyacrylamide gel electrophoresis, distinct CTF with up to three major bands (23, 24, and 26 kDa) and two minor bands (18 and 19 kDa) can be present in sarkosyl-insoluble lysates of FTLD-TDP brains, with some studies demonstrating subtle differences in the banding pattern among FTLD-TDP subtypes (Fig. 3b) (40, 68, 85). Briefly, in type A the most intense major band is at 23 kDa, in type B it is at 24 kDa, type C lacks the 26 kDa band and has a more prominent 23 kDa band, and type E shows three major bands with the most intense at 26 kDa. However, significant variability within and overlap between subtypes exists (19); and so, the biochemical classification of subtypes remains challenging and more sensitive methods of detection, quantification and analysis of various CTF and their PTM are required.
Nevertheless, it is tempting to speculate that the different banding patterns in FTLD-TDP may correspond to different conformational species of abnormal TDP-43. In strong support of this idea, protease treatment of insoluble TDP-43 aggregates has revealed different patterns of protease-resistant cores among FTLD-TDP subtypes, highly suggestive of different conformers (18). More recently, a new extraction method, termed ‘SarkoSpin’, has been developed that allows extraction of pathological TDP-43 species from postmortem tissue with improved separation from physiological TDP-43, compared to the previous sequential extraction protocols (16). This approach has revealed additional insights into distinct biophysical properties of aggregated TDP-43 among the TDP-43 proteinopathies, with TDP-43 from FTLD-TDP type C found to exhibit a higher intrinsic density and protease resistant CTF core compared to that from cases of type A or ALS (type B not examined).
In addition to the observed biochemical/structural differences, crucial support for the idea that distinct pathological TDP-43 species may (at least partially) explain the clinical and pathological variability in FTLD-TDP comes from the observations that TDP-43 extracted from different FTLD-TDP subtypes exhibits different levels of seeding activity and toxicity in vitro and in vivo. The first such evidence was provided by Nokanko et al. who reported that seeding activity of TDP-43 extracted from human postmortem tissue in a cell culture model was more efficient when using extracts from type A and B cases compared to type C (86). Interestingly, the banding pattern of insoluble CTF extracted from the seeded cell lysates resembled that from the corresponding FTLD-TDP subject used as the seed, suggestive of a prion-like self-templating process of TDP-43 aggregation. These results were validated and expanded in a report where TDP-43 aggregates extracted using the SarkoSpin protocol from FTLD-TDP type A cases demonstrated templated seeding and toxicity in cultured primary neurons, while those from subtype C seemed inert (16). While in these studies no differences between sporadic and genetic cases were mentioned, Porta et al. reported that lysates from GRN mutation carriers had the highest seeding activity in their cellular screening assay, followed by C9orf72 mutation carriers and sporadic FTLD-TDP type A and B cases (87). Biochemical analyses of the lysates revealed a correlation between the presence of two minor CTF bands of 18 and 19 kDa and seeding activity, suggesting that distinct fragments and/or conformational TDP-43 species seem to be more potent (87). Most importantly, this study provided the first in vivo evidence for propagation of TDP-43 pathology in a prion-like manner by demonstrating the induction and spreading of de-novo TDP-43 pathology following the intracerebral injection of FTLD-TDP aggregates isolated from human FTLD-TDP type A tissue into transgenic mice expressing cytoplasmic human TDP-43 and non-transgenic mice (87).
Therefore, current insights are consistent with the idea that the progression of FTLD-TDP pathology involves self-templating seeded aggregation and cell-to-cell spreading of pathological TDP-43 that exists in different conformations. However, more extensive biochemical, biophysical and seeding studies are needed to strengthen the hypothesis that different TDP-43 conformers/species indeed contribute to the phenotypic heterogeneity in FTLD-TDP patients (e.g. by demonstrating whether distinct FTLD-TDP subtype derived TDP-43 aggregates can reproduce their distinct clinical and neuropathological characteristics in animal models).
Finally, in addition to biochemical differences of TDP-43 itself, co-aggregation of other proteins into TDP-43 inclusions might contribute to the diversity among FTLD-TDP subtypes. This hypothesis is supported by double-label immunohistochemical findings with co-localization of hnRNP E2 and TDP-43 in FTLD-TDP subtype C and subsets of FTLD-TDP type A inclusions but not in type B cases (88, 89). However, in-depths biochemical characterization of the protein composition of TDP-43 inclusions are required to further address this.
Summary
The current criteria for the pathological sub-classification of FTLD-TDP are widely accepted and show a number of highly relevant clinical and genetic associations. However, the presence of a small proportion of cases with novel patterns of TDP-43-ir pathology indicate the need for additional correlative studies. Investigations to date suggest that the basis for the different subtypes is, at least partially, biochemical and/or conformational variation in the aggregating protein. Further studies to more fully elucidate the nature of the subtype-specific pathological species of TDP-43 will be crucial to the development of useful biomarkers and targeted therapies.
Acknowledgements
This work was supported by the NOMIS foundation (MN) and the Canadian Institutes of Health Research (IRM).
References
- 1.Woollacott IO, Rohrer JD. The clinical spectrum of sporadic and familial forms of frontotemporal dementia. J Neurochem. 2016;138 Suppl 1:6–31. [DOI] [PubMed] [Google Scholar]
- 2.Pottier C, Ravenscroft TA, Sanchez-Contreras M, Rademakers R. Genetics of FTLD: overview and what else we can expect from genetic studies. J Neurochem. 2016;138 Suppl 1:32–53. [DOI] [PubMed] [Google Scholar]
- 3.Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, et al. Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol. 2009;117(1):15–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol. 2010;119(1):1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mackenzie IR, Shi J, Shaw CL, Duplessis D, Neary D, Snowden JS, et al. Dementia lacking distinctive histology (DLDH) revisited. Acta Neuropathol. 2006;112(5):551–9. [DOI] [PubMed] [Google Scholar]
- 6.Mackenzie IR, Baborie A, Pickering-Brown S, Du Plessis D, Jaros E, Perry RH, et al. Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: classification and relation to clinical phenotype. Acta Neuropathol. 2006;112(5):539–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sampathu DM, Neumann M, Kwong LK, Chou TT, Micsenyi M, Truax A, et al. Pathological heterogeneity of frontotemporal lobar degeneration with ubiquitin-positive inclusions delineated by ubiquitin immunohistochemistry and novel monoclonal antibodies. Am J Pathol. 2006;169(4):1343–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Forman MS, Mackenzie IR, Cairns NJ, Swanson E, Boyer PJ, Drachman DA, et al. Novel ubiquitin neuropathology in frontotemporal dementia with valosin-containing protein gene mutations. J Neuropathol Exp Neurol. 2006;65(6):571–81. [DOI] [PubMed] [Google Scholar]
- 9.Holm IE, Englund E, Mackenzie IR, Johannsen P, Isaacs AM. A reassessment of the neuropathology of frontotemporal dementia linked to chromosome 3. J Neuropathol Exp Neurol. 2007;66(10):884–91. [DOI] [PubMed] [Google Scholar]
- 10.Mackenzie IR, Baker M, Pickering-Brown S, Hsiung GY, Lindholm C, Dwosh E, et al. The neuropathology of frontotemporal lobar degeneration caused by mutations in the progranulin gene. Brain. 2006;129(Pt 11):3081–90. [DOI] [PubMed] [Google Scholar]
- 11.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–3. [DOI] [PubMed] [Google Scholar]
- 12.Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–11. [DOI] [PubMed] [Google Scholar]
- 13.Cairns NJ, Neumann M, Bigio EH, Holm IE, Troost D, Hatanpaa KJ, et al. TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol. 2007;171(1):227–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davidson Y, Kelley T, Mackenzie IR, Pickering-Brown S, Du Plessis D, Neary D, et al. Ubiquitinated pathological lesions in frontotemporal lobar degeneration contain the TAR DNA-binding protein, TDP-43. Acta Neuropathol. 2007;113(5):521–33. [DOI] [PubMed] [Google Scholar]
- 15.Mackenzie IR, Neumann M. Reappraisal of TDP-43 pathology in FTLD-U subtypes. Acta Neuropathol. 2017;134(1):79–96. [DOI] [PubMed] [Google Scholar]
- 16.Laferriere F, Maniecka Z, Perez-Berlanga M, Hruska-Plochan M, Gilhespy L, Hock EM, et al. TDP-43 extracted from frontotemporal lobar degeneration subject brains displays distinct aggregate assemblies and neurotoxic effects reflecting disease progression rates. Nat Neurosci. 2019;22(1):65–77. [DOI] [PubMed] [Google Scholar]
- 17.Pottier C, Ren Y, Perkerson RB 3rd, Baker M, Jenkins GD, van Blitterswijk M, et al. Genome-wide analyses as part of the international FTLD-TDP whole-genome sequencing consortium reveals novel disease risk factors and increases support for immune dysfunction in FTLD. Acta Neuropathol. 2019;137(6):879–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsuji H, Arai T, Kametani F, Nonaka T, Yamashita M, Suzukake M, et al. Molecular analysis and biochemical classification of TDP-43 proteinopathy. Brain. 2012;135(Pt 11):3380–91. [DOI] [PubMed] [Google Scholar]
- 19.Neumann M, Kwong LK, Lee EB, Kremmer E, Flatley A, Xu Y, et al. Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol. 2009;117(2):137–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mackenzie IR, Neumann M, Baborie A, Sampathu DM, Du Plessis D, Jaros E, et al. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 2011;122(1):111–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Josephs KA, Stroh A, Dugger B, Dickson DW. Evaluation of subcortical pathology and clinical correlations in FTLD-U subtypes. Acta Neuropathol. 2009;118(3):349–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mackenzie IR, Neumann M. Subcortical TDP-43 pathology patterns validate cortical FTLD-TDP subtypes and demonstrate unique aspects of C9orf72 mutation cases. Acta Neuropathol. 2020;139(1):83–98. [DOI] [PubMed] [Google Scholar]
- 23.Josephs KA, Hodges JR, Snowden JS, Mackenzie IR, Neumann M, Mann DM, et al. Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol. 2011;122(2):137–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perry DC, Brown JA, Possin KL, Datta S, Trujillo A, Radke A, et al. Clinicopathological correlations in behavioural variant frontotemporal dementia. Brain. 2017;140(12):3329–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rohrer JD, Lashley T, Holton J, Revesz T, Urwin H, Isaacs AM, et al. The clinical and neuroanatomical phenotype of FUS associated frontotemporal lobar degeneration. J Neurol Neurosurg Psychiatry. 2011;82(12):1405–7. [DOI] [PubMed] [Google Scholar]
- 26.Snowden JS, Rollinson S, Thompson JC, Harris JM, Stopford CL, Richardson AM, et al. Distinct clinical and pathological characteristics of frontotemporal dementia associated with C9ORF72 mutations. Brain. 2012;135(Pt 3):693–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Al-Obeidi E, Al-Tahan S, Surampalli A, Goyal N, Wang AK, Hermann A, et al. Genotype-phenotype study in patients with valosin-containing protein mutations associated with multisystem proteinopathy. Clin Genet. 2018;93(1):119–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neumann M, Mackenzie IR, Cairns NJ, Boyer PJ, Markesbery WR, Smith CD, et al. TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. J Neuropathol Exp Neurol. 2007;66(2):152–7. [DOI] [PubMed] [Google Scholar]
- 29.Bigio EH, Weintraub S, Rademakers R, Baker M, Ahmadian SS, Rademaker A, et al. Frontotemporal lobar degeneration with TDP-43 proteinopathy and chromosome 9p repeat expansion in C9ORF72: clinicopathologic correlation. Neuropathology. 2013;33(2):122–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boeve BF, Boylan KB, Graff-Radford NR, DeJesus-Hernandez M, Knopman DS, Pedraza O, et al. Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain. 2012;135(Pt 3):765–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mahoney CJ, Beck J, Rohrer JD, Lashley T, Mok K, Shakespeare T, et al. Frontotemporal dementia with the C9ORF72 hexanucleotide repeat expansion: clinical, neuroanatomical and neuropathological features. Brain. 2012;135(Pt 3):736–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murray ME, Dejesus-Hernandez M, Rutherford NJ, Baker M, Duara R, Graff-Radford NR, et al. Clinical and neuropathologic heterogeneity of c9FTD/ALS associated with hexanucleotide repeat expansion in C9ORF72. Acta Neuropathol. 2011;122(6):673–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gijselinck I, Van Mossevelde S, van der Zee J, Sieben A, Philtjens S, Heeman B, et al. Loss of TBK1 is a frequent cause of frontotemporal dementia in a Belgian cohort. Neurology. 2015;85(24):2116–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hirsch-Reinshagen V, Alfaify OA, Hsiung GR, Pottier C, Baker M, Perkerson RB 3rd, et al. Clinicopathologic correlations in a family with a TBK1 mutation presenting as primary progressive aphasia and primary lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2019:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koriath CA, Bocchetta M, Brotherhood E, Woollacott IO, Norsworthy P, Simon-Sanchez J, et al. The clinical, neuroanatomical, and neuropathologic phenotype of TBK1-associated frontotemporal dementia: A longitudinal case report. Alzheimers Dement (Amst). 2017;6:75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lamb R, Rohrer JD, Real R, Lubbe SJ, Waite AJ, Blake DJ, et al. A novel TBK1 mutation in a family with diverse frontotemporal dementia spectrum disorders. Cold Spring Harb Mol Case Stud. 2019;5(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alafuzoff I, Pikkarainen M, Neumann M, Arzberger T, Al-Sarraj S, Bodi I, et al. Neuropathological assessments of the pathology in frontotemporal lobar degeneration with TDP43-positive inclusions: an inter-laboratory study by the BrainNet Europe consortium. J Neural Transm. 2015;122(7):957–72. [DOI] [PubMed] [Google Scholar]
- 38.Pikkarainen M, Hartikainen P, Alafuzoff I. Neuropathologic Features of Frontotemporal Lobar Degeneration With Ubiquitin-Positive Inclusions Visualized With Ubiquitin-Binding Protein p62 Immunohistochemistry. J Neuropathol Exp Neurol. 2008;67(4):280–98. [DOI] [PubMed] [Google Scholar]
- 39.Shinagawa S, Naasan G, Karydas AM, Coppola G, Pribadi M, Seeley WW, et al. Clinicopathological Study of Patients With C9ORF72-Associated Frontotemporal Dementia Presenting With Delusions. J Geriatr Psychiatry Neurol. 2015;28(2):99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee EB, Porta S, Michael Baer G, Xu Y, Suh E, Kwong LK, et al. Expansion of the classification of FTLD-TDP: distinct pathology associated with rapidly progressive frontotemporal degeneration. Acta Neuropathol. 2017;134(1):65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takeuchi R, Tada M, Shiga A, Toyoshima Y, Konno T, Sato T, et al. Heterogeneity of cerebral TDP-43 pathology in sporadic amyotrophic lateral sclerosis: Evidence for clinicopathologic subtypes. Acta Neuropathol Commun. 2016;4(1):61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tan RH, Guennewig B, Dobson-Stone C, Kwok JBJ, Kril JJ, Kiernan MC, et al. The underacknowledged PPA-ALS: A unique clinicopathologic subtype with strong heritability. Neurology. 2019;92(12):e1354–e66. [DOI] [PubMed] [Google Scholar]
- 43.Armstrong RA, Ellis W, Hamilton RL, Mackenzie IR, Hedreen J, Gearing M, et al. Neuropathological heterogeneity in frontotemporal lobar degeneration with TDP-43 proteinopathy: a quantitative study of 94 cases using principal components analysis. J Neural Transm (Vienna). 2010;117(2):227–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nishihira Y, Gefen T, Mao Q, Appin C, Kohler M, Walker J, et al. Revisiting the utility of TDP-43 immunoreactive (TDP-43-ir) pathology to classify FTLD-TDP subtypes. Acta Neuropathol. 2019;138(1):167–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Clark CN, Quaegebeur A, Nirmalananthan N, MacKinnon AD, Revesz T, Holton JL, et al. Foix-Chavany-Marie syndrome due to type E TDP43 pathology. Neuropathol Appl Neurobiol. 2019. [DOI] [PubMed] [Google Scholar]
- 46.Ihori N, Araki S, Ishihara K, Kawamura M. A case of frontotemporal lobar degeneration with progressive dysarthria. Behav Neurol. 2006;17(2):97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee EB, Lee VM, Trojanowski JQ, Neumann M. TDP-43 immunoreactivity in anoxic, ischemic and neoplastic lesions of the central nervous system. Acta Neuropathol. 2008;115(3):305–11. [DOI] [PubMed] [Google Scholar]
- 48.Brenner M, Johnson AB, Boespflug-Tanguy O, Rodriguez D, Goldman JE, Messing A. Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nat Genet. 2001;27(1):117–20. [DOI] [PubMed] [Google Scholar]
- 49.Walker AK, Daniels CM, Goldman JE, Trojanowski JQ, Lee VM, Messing A. Astrocytic TDP-43 pathology in Alexander disease. J Neurosci. 2014;34(19):6448–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Farrer MJ, Hulihan MM, Kachergus JM, Dachsel JC, Stoessl AJ, Grantier LL, et al. DCTN1 mutations in Perry syndrome. Nat Genet. 2009;41(2):163–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mishima T, Koga S, Lin WL, Kasanuki K, Castanedes-Casey M, Wszolek ZK, et al. Perry Syndrome: A Distinctive Type of TDP-43 Proteinopathy. J Neuropathol Exp Neurol. 2017;76(8):676–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Josephs KA, Murray ME, Whitwell JL, Parisi JE, Petrucelli L, Jack CR, et al. Staging TDP-43 pathology in Alzheimer’s disease. Acta Neuropathol. 2014;127(3):441–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Josephs KA, Mackenzie I, Frosch MP, Bigio EH, Neumann M, Arai T, et al. LATE to the PART-y. Brain. 2019;142(9):e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K, et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain. 2019;142(6):1503–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Buratti E TDP-43 post-translational modifications in health and disease. Expert Opin Ther Targets. 2018;22(3):279–93. [DOI] [PubMed] [Google Scholar]
- 56.Igaz LM, Kwong LK, Xu Y, Truax AC, Uryu K, Neumann M, et al. Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am J Pathol. 2008;173(1):182–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Igaz LM, Kwong LK, Chen-Plotkin A, Winton MJ, Unger TL, Xu Y, et al. Expression of TDP-43 C-terminal Fragments in Vitro Recapitulates Pathological Features of TDP-43 Proteinopathies. J Biol Chem. 2009;284(13):8516–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nonaka T, Kametani F, Arai T, Akiyama H, Hasegawa M. Truncation and pathogenic mutations facilitate the formation of intracellular aggregates of TDP-43. Hum Mol Genet. 2009;18(18):3353–64. [DOI] [PubMed] [Google Scholar]
- 59.Zhang YJ, Xu YF, Dickey CA, Buratti E, Baralle F, Bailey R, et al. Progranulin mediates caspase-dependent cleavage of TAR DNA binding protein-43. J Neurosci. 2007;27(39):10530–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dormann D, Capell A, Carlson AM, Shankaran SS, Rodde R, Neumann M, et al. Proteolytic processing of TAR DNA binding protein-43 by caspases produces C-terminal fragments with disease defining properties independent of progranulin. J Neurochem. 2009;110(3):1082–94. [DOI] [PubMed] [Google Scholar]
- 61.Herskowitz JH, Gozal YM, Duong DM, Dammer EB, Gearing M, Ye K, et al. Asparaginyl endopeptidase cleaves TDP-43 in brain. Proteomics. 2012;12(15–16):2455–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yamashita T, Hideyama T, Hachiga K, Teramoto S, Takano J, Iwata N, et al. A role for calpain-dependent cleavage of TDP-43 in amyotrophic lateral sclerosis pathology. Nat Commun. 2012;3:1307. [DOI] [PubMed] [Google Scholar]
- 63.Xiao S, Sanelli T, Chiang H, Sun Y, Chakrabartty A, Keith J, et al. Low molecular weight species of TDP-43 generated by abnormal splicing form inclusions in amyotrophic lateral sclerosis and result in motor neuron death. Acta Neuropathol. 2015;130(1):49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nishimoto Y, Ito D, Yagi T, Nihei Y, Tsunoda Y, Suzuki N. Characterization of alternative isoforms and inclusion body of the TAR DNA-binding protein-43. J Biol Chem. 2010;285(1):608–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang YJ, Xu YF, Cook C, Gendron TF, Roettges P, Link CD, et al. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc Natl Acad Sci U S A. 2009;106(18):7607–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Berning BA, Walker AK. The Pathobiology of TDP-43 C-Terminal Fragments in ALS and FTLD. Front Neurosci. 2019;13:335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Josephs KA, Zhang YJ, Baker M, Rademakers R, Petrucelli L, Dickson DW. C-terminal and full length TDP-43 specie differ according to FTLD-TDP lesion type but not genetic mutation. Acta Neuropathol Commun. 2019;7(1):100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hasegawa M, Arai T, Nonaka T, Kametani F, Yoshida M, Hashizume Y, et al. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann Neurol. 2008;64(1):60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kametani F, Nonaka T, Suzuki T, Arai T, Dohmae N, Akiyama H, et al. Identification of casein kinase-1 phosphorylation sites on TDP-43. Biochem Biophys Res Commun. 2009;382(2):405–9. [DOI] [PubMed] [Google Scholar]
- 70.Inukai Y, Nonaka T, Arai T, Yoshida M, Hashizume Y, Beach TG, et al. Abnormal phosphorylation of Ser409/410 of TDP-43 in FTLD-U and ALS. FEBS Lett. 2008;582(19):2899–904. [DOI] [PubMed] [Google Scholar]
- 71.Tan RH, Shepherd CE, Kril JJ, McCann H, McGeachie A, McGinley C, et al. Classification of FTLD-TDP cases into pathological subtypes using antibodies against phosphorylated and non-phosphorylated TDP43. Acta Neuropathol Commun. 2013;1:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim KY, Lee HW, Shim YM, Mook-Jung I, Jeon GS, Sung JJ. A phosphomimetic mutant TDP-43 (S409/410E) induces Drosha instability and cytotoxicity in Neuro 2A cells. Biochem Biophys Res Commun. 2015;464(1):236–43. [DOI] [PubMed] [Google Scholar]
- 73.Brady OA, Meng P, Zheng Y, Mao Y, Hu F. Regulation of TDP-43 aggregation by phosphorylation and p62/SQSTM1. J Neurochem. 2011;116(2):248–59. [DOI] [PubMed] [Google Scholar]
- 74.Li HY, Yeh PA, Chiu HC, Tang CY, Tu BP. Hyperphosphorylation as a defense mechanism to reduce TDP-43 aggregation. PLoS One. 2011;6(8):e23075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Seyfried NT, Gozal YM, Dammer EB, Xia Q, Duong DM, Cheng D, et al. Multiplex SILAC analysis of a cellular TDP-43 proteinopathy model reveals protein inclusions associated with SUMOylation and diverse polyubiquitin chains. Mol Cell Proteomics. 2010;9(4):705–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dammer EB, Fallini C, Gozal YM, Duong DM, Rossoll W, Xu P, et al. Coaggregation of RNA-binding proteins in a model of TDP-43 proteinopathy with selective RGG motif methylation and a role for RRM1 ubiquitination. PLoS One. 2012;7(6):e38658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hans F, Eckert M, von Zweydorf F, Gloeckner CJ, Kahle PJ. Identification and characterization of ubiquitinylation sites in TAR DNA-binding protein of 43 kDa (TDP-43). J Biol Chem. 2018;293(41):16083–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell. 2011;44(2):325–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wagner SA, Beli P, Weinert BT, Nielsen ML, Cox J, Mann M, et al. A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol Cell Proteomics. 2011;10(10):M111 013284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kametani F, Obi T, Shishido T, Akatsu H, Murayama S, Saito Y, et al. Mass spectrometric analysis of accumulated TDP-43 in amyotrophic lateral sclerosis brains. Sci Rep. 2016;6:23281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cohen TJ, Hwang AW, Restrepo CR, Yuan CX, Trojanowski JQ, Lee VM. An acetylation switch controls TDP-43 function and aggregation propensity. Nat Commun. 2015;6:5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cohen TJ, Hwang AW, Unger T, Trojanowski JQ, Lee VM. Redox signalling directly regulates TDP-43 via cysteine oxidation and disulphide cross-linking. EMBO J. 2012;31(5):1241–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Aguzzi A, Heikenwalder M, Polymenidou M. Insights into prion strains and neurotoxicity. Nat Rev Mol Cell Biol. 2007;8(7):552–61. [DOI] [PubMed] [Google Scholar]
- 84.Kawakami I, Arai T, Hasegawa M. The basis of clinicopathological heterogeneity in TDP-43 proteinopathy. Acta Neuropathol. 2019;138(5):751–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hasegawa M, Nonaka T, Tsuji H, Tamaoka A, Yamashita M, Kametani F, et al. Molecular dissection of TDP-43 proteinopathies. J Mol Neurosci. 2011;45(3):480–5. [DOI] [PubMed] [Google Scholar]
- 86.Nonaka T, Masuda-Suzukake M, Arai T, Hasegawa Y, Akatsu H, Obi T, et al. Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep. 2013;4(1):124–34. [DOI] [PubMed] [Google Scholar]
- 87.Porta S, Xu Y, Restrepo CR, Kwong LK, Zhang B, Brown HJ, et al. Patient-derived frontotemporal lobar degeneration brain extracts induce formation and spreading of TDP-43 pathology in vivo. Nat Commun. 2018;9(1):4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Davidson YS, Robinson AC, Flood L, Rollinson S, Benson BC, Asi YT, et al. Heterogeneous ribonuclear protein E2 (hnRNP E2) is associated with TDP-43-immunoreactive neurites in Semantic Dementia but not with other TDP-43 pathological subtypes of Frontotemporal Lobar Degeneration. Acta Neuropathol Commun. 2017;5(1):54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kattuah W, Rogelj B, King A, Shaw CE, Hortobagyi T, Troakes C. Heterogeneous Nuclear Ribonucleoprotein E2 (hnRNP E2) Is a Component of TDP-43 Aggregates Specifically in the A and C Pathological Subtypes of Frontotemporal Lobar Degeneration. Front Neurosci. 2019;13:551. [DOI] [PMC free article] [PubMed] [Google Scholar]