

Abstract
Background:
The cystic fibrosis transmembrane conductance regulator (CFTR) potentiator, ivacaftor, was first approved for people with CF and the G551D CFTR mutation. This study describes the long-term clinical effectiveness of ivacaftor in this population.
Methods:
We conducted a multicenter, prospective, longitudinal, observational study of people with CF ages ≥6 years with at least one copy of the G551D CFTR mutation. Measurements of lung function, growth, quality of life, and sweat chloride were performed after ivacaftor initiation (baseline, 1 month, 3 months, 6 months, and annually thereafter until 5.5 years).
Results:
Ninety-six participants were enrolled, with 81% completing all study measures through 5.5 years. This cohort experienced significant improvements in percent predicted forced expiratory volume in 1 second (ppFEV1) of 4.8 [2.6, 7.1] (p<0.001) at 1.5 years, that diminished to 0.8 [−2.0, 3.6] (p=0.57) at 5.5 years. Adults experienced larger improvements in ppFEV1 (7.4 [3.6, 11.3], p<0.001 at 1.5 years and 4.3 [0.6, 8.1], p=0.02 at 5.5 years) than children (2.8 [0.1, 5.6], p=0.04 at 1.5 years and −2.0 [−5.9, 2.0], p=0.32 at 5.5 years). Rate of lung function decline for the overall study cohort from 1 month after ivacaftor initiation through 5.5 years was estimated to be −1.22 pp/year [−1.70, −0.73]. Significant improvements in growth, quality of life measures, sweat chloride, Pseudomonas aeruginosa detection, and pulmonary exacerbation rates requiring antimicrobial therapy persisted through five years of therapy.
Conclusions:
These findings demonstrate the long-term benefits and disease modifying effects of ivacaftor in children and adults with CF and the G551D mutation.
Keywords: ivacaftor, CFTR, lung function, sweat chloride, Pseudomonas aeruginosa, quality of life
1. Introduction
With the development of ivacaftor (1, 2), treatment targeted to the underlying protein defect became possible for people with cystic fibrosis (CF). Ivacaftor is a CF transmembrane conductance regulator (CFTR) potentiator, enhancing the open probability of the CFTR protein that reaches the cell surface but has defective gating.(3) Clinical trials demonstrated ivacaftor was highly efficacious for people with the G551D CFTR mutation.(1, 2) Ivacaftor was the first CFTR modulator approved for clinical use in 2012.
Following approval, a long-term, prospective observational study was launched in the United States to evaluate ivacaftor’s effectiveness in patients with at least one copy of the G551D allele (G551D observational study, GOAL). In addition to confirming the clinical benefits over a 6 month period in a real-world population, the GOAL study provided additional mechanistic insights into partial CFTR restoration.(4) This and other studies of ivacaftor have demonstrated reductions in Pseudomonas aeruginosa detection, and improvements in growth, gastrointestinal function, insulin secretion, and quality-of-life.(5–9) Retrospective and registry-based studies have reported on the long-term beneficial effects of ivacaftor.(10–17) The prospective design of the GOAL study provides the opportunity to expand upon these findings with five years of follow-up data and report on outcomes not captured in registries, including sweat chloride, quality of life (QOL), and inter-relationships between these measures.
We describe a prospective cohort study to examine the clinical effectiveness of ivacaftor in children (≥ 6 years) and adults (≥ 18 years) with CF and the G551D CFTR mutation for over 5 years, supported in part with data from the CF Foundation Patient Registry (CFFPR). Some of the results have been previously reported in the form of an abstract.(18)
2. Methods
2.1. Study design and participants
This was a longitudinal cohort study at 23 U.S. centers within the CF Therapeutics Development Network. The G551D Observational Study - Expanded to Additional Genotypes and Extended for Long Term Follow up (GOAL-e2) has been described previously (NCT01521338)(4, 6). CF participants ages 6 years and above with at least one G551D CFTR mutation and no prior exposure to ivacaftor were enrolled and initially followed for six months after initiation of ivacaftor.(4) These same study participants were approached to continue annual study visit assessments for five additional years. Written informed consent was obtained for each participant, according to each study site’s Institutional Review Board.
2.2. Procedures and outcome measures
For each subject, the following outcome measures were performed and recorded over the five year period: forced expiratory volume in one second (FEV1), percent predicted FEV1 (ppFEV1)(19), weight, height, body mass index (BMI), sweat chloride, and Cystic Fibrosis Questionnaire-Revised (CFQ-R). For participants under the age of 20 years, percentiles compared to reference populations were calculated for weight and BMI using the Centers for Disease Control formulas.(20, 21) For adolescents who had observations after their 20th birthday, the 20 year old percentile formulas continued to be used for continuity. For weight determinations, the CDC adult (absolute value) and pediatric (percentile) categories were applied to classify underweight (BMI<18.5 kg/m2; <5th percentile), normal (BMI 18.5 to <25 kg/m2; 5th to <85th percentile), overweight (BMI 25 - <30 kg/m2; 85th to < 95th percentile), and obese (BMI >30 kg/m2; ≥95th percentile) status at different time points. Data relating to pulmonary exacerbation rates (defined as any home IV or hospitalization care episode in which “pulmonary exacerbation” was listed as one of the care reasons, hospitalizations due to pulmonary exacerbations, and P. aeruginosa detection were captured from the CFFPR. Baseline was defined as the year prior to ivacaftor initiation.
2.3. Statistical Analyses
Summary statistics and 95% confidence intervals (CI) [in brackets] are reported. Paired t tests were used to estimate changes from baseline in clinical outcomes overall and by age and baseline ppFEV1 categories. The Stuart-Maxwell test was applied to assess differences in BMI category distribution. Mixed effect models with a random participant effect were fit to estimate longitudinal trends and annual rate of decline in ppFEV1 for adults and children separately. Lung function data from 1-month post baseline through 5.5 years were used to avoid the incident change that occurred immediately after initiating ivacaftor. Rates of pulmonary exacerbations and P. aeruginosa detection were modeled as any/none within each period, using McNemar’s test to assess differences from baseline. CFF Registry data were used to examine use of common CF medications in the year prior to starting ivacaftor, the period 0.5–1.5 years after starting ivacaftor (Period 1), and the period 4.5–5.5 years after starting ivacaftor (Period 5). Counts and percentages of participants using each medication type were used to determine odds ratios and p-values with McNemar’s exact test. Correlations between sweat chloride and clinical outcomes were analyzed using Pearson correlation coefficients. All reported p-values are two-sided and no corrections for multiplicity or sub-group analyses were performed. SAS version 9.4 and R version 3.6.1 were used for analyses.
3. Results
3.1. Study population
Of the 96 participants who enrolled in the extension study, 82 (85%) completed study visits through 18 months and 78 (81%) completed all study measures through 5.5 years of follow-up. At baseline (pre-ivacaftor), the overall cohort (n=96) had a mean age of 19.8 years (SD=11.5), a mean ppFEV1 of 82.0 (SD=25.0), and a mean BMI of 20.4 (SD=4.6). Demographic and baseline clinical characteristics of the pediatric and adult cohorts are reported in Table 1.
Table 1:
Participant demographics and clinical characteristics by age group at baseline.
| Variable | Age <18 (N=52) | Age >=18 (N=44) | All (N=96) | |
|---|---|---|---|---|
| Sex, n (%) | Female | 26 (50%) | 17 (39%) | 43 (45%) |
| Male | 26 (50%) | 27 (61%) | 53 (55%) | |
| Enrollment Age, mean (SD) | 11.6 (3.6) | 29.5 (10.0) | 19.8 (11.5) | |
| Enrollment Age, n (%) | 6–11 | 29 (56%) | -- | 29 (30%) |
| 12–17 | 23 (44%) | -- | 23 (24%) | |
| 18–29 | -- | 26 (59%) | 26 (27%) | |
| 30+ | -- | 18 (41%) | 18 (19%) | |
| Genotype Class of the second allele, n (%) | Class I | 4 (8%) | 4 (9%) | 8 (8%) |
| Class II | 44 (85%) | 28 (64%) | 72 (75%) | |
| Class III | 1 (2%) | 1 (2%) | 2 (2%) | |
| Class IV | -- | 1 (2%) | 1 (1%) | |
| Class V | 2 (4%) | 3 (7%) | 5 (5%) | |
| Unknown | 1 (2%) | 7 (16%) | 8 (8%) | |
| Race, n (%) | Black/African-American | 1 (2%) | 2 (5%) | 3 (3%) |
| White | 51 (98%) | 42 (95%) | 93 (97%) | |
| FEV1 % Predicted, mean (SD) | 94.7 (19.3) | 67.0 (22.7) | 82.0 (25.0) | |
| FVC % Predicted, mean (SD) | 99.8 (16.5) | 81.8 (17.1) | 91.6 (19.0) | |
| BMI kg/m2, mean (SD) | 17.9 (2.7) | 23.4 (4.6) | 20.4 (4.6) |
3.2. Effect of ivacaftor on lung function
The overall cohort experienced a significant improvement in ppFEV1 of 4.8 [2.6, 7.1] (p<0.001) at 1.5 years, reverting towards baseline over time to 0.8 [−2.0, 3.6] (p=0.57) at 5.5 years (Figure 1A, Table 2). The pediatric population experienced an increase of 2.8 [0.1, 5.6] (p=0.045) at 18 months and a change of −2.0 [−5.9, 2.0] (p=0.32) at 5.5 years that was not statistically significant. Notably, the pediatric cohort maintained mean ppFEV1 above 90 throughout the entire 5.5 year observational period. In comparison, adult participants experienced a significant increase in ppFEV1 at 18 months (7.4 [3.6, 11.3], p<0.001) and at 5.5 years (4.3 [0.6, 8.1] p=0.024) (Supplemental Table 1).
Figure 1. Clinical outcomes over 5.5 years.

Each individual participant’s results for each visit are represented by a single line. A linear spline, with a knot at 30 days,is represented by a bolded line; the solid line represents all participants, the dotted line adult participants only, and the dashed pediatric participants only. A. ppFEV1; B. BMI; C.CFQ-R Respiratory Domain score; D. Sweat chloride.
Table 2:
Change from baseline in clinical outcomes after initiation of ivacaftor
| Absolute Change | |||||||
|---|---|---|---|---|---|---|---|
| Variable | Visit | N | Mean (SD) | N | Mean | 95% C.I. | P-value |
| ppFEV1 (GLI) | Baseline | 96 | 82 (25) | - | - | - | - |
| 1 month | 96 | 89.4 (23.9) | 96 | 7.5 | (5.4, 9.5) | <.0001 | |
| 3 month | 93 | 88.5 (23.8) | 93 | 6.1 | (4.5, 7.7) | <.0001 | |
| 6 month | 90 | 89.8 (23.3) | 90 | 7.9 | (5.8, 10.1) | <.0001 | |
| 1.5 years | 82 | 87.4 (23.1) | 82 | 4.8 | (2.6, 7.1) | <.0001 | |
| 5.5 years | 78 | 84.7 (23.3) | 78 | 0.8 | (−2.0, 3.6) | 0.5739 | |
| Weight, kg | Baseline | 96 | 52.1 (20.4) | - | - | - | - |
| 1 month | 96 | 53.4 (20.5) | 96 | 1.3 | (1.0, 1.6) | <.0001 | |
| 3 month | 94 | 54.2 (20.8) | 94 | 2.1 | (1.6, 2.5) | <.0001 | |
| 6 month | 92 | 54.3 (19.8) | 92 | 2.9 | (2.2, 3.6) | <.0001 | |
| 1.5 years | 82 | 58.7 (18.9) | 82 | 6.6 | (5.3, 7.9) | <.0001 | |
| 5.5 years | 78 | 64.4 (15.4) | 78 | 12.8 | (10.1, 15.5) | <.0001 | |
| BMI, kg/m2 | Baseline | 96 | 20.4 (4.6) | - | - | - | - |
| 1 month | 96 | 20.9 (4.5) | 96 | 0.4 | (0.3, 0.6) | <.0001 | |
| 3 month | 94 | 21.1 (4.6) | 94 | 0.7 | (0.5, 0.9) | <.0001 | |
| 6 month | 92 | 21.2 (4.7) | 92 | 0.9 | (0.7, 1.1) | <.0001 | |
| 1.5 years | 82 | 21.9 (4.5) | 82 | 1.6 | (1.2, 2.0) | <.0001 | |
| 5.5 years | 78 | 23 (4.7) | 78 | 2.5 | (2.0, 3.1) | <.0001 | |
| CFQR Respiratory | Baseline | 96 | 71 (20.1) | - | - | - | - |
| 1 month | 95 | 81.8 (13.2) | 95 | 10.4 | (7.2, 13.5) | <.0001 | |
| 3 month | 93 | 83.7 (15.4) | 93 | 11.8 | (8.6, 15.0) | <.0001 | |
| 6 month | 92 | 80.3 (16.5) | 92 | 8.8 | (4.8, 12.8) | <.0001 | |
| 1.5 years | 81 | 80.1 (16.5) | 81 | 9 | (4.7, 13.3) | <.0001 | |
| 5.5 years | 78 | 79 (16.8) | 78 | 6.7 | (2.5, 10.9) | 0.0020 | |
| Sweat chloride, mEq/L | Baseline | 93 | 103.9 (11.3) | - | - | - | - |
| 1 month | 88 | 55.2 (22.3) | 86 | −48.9 | (−53.3, −44.4) | <.0001 | |
| 3 month | 85 | 49.4 (20.3) | 84 | −55 | (−59.2, −50.8) | <.0001 | |
| 6 month | 89 | 47.3 (19.9) | 87 | −56.4 | (−60.5, −52.2) | <.0001 | |
| 1.5 years | 79 | 48.9 (22.2) | 79 | −55.3 | (−60.5, −50.1) | <.0001 | |
| 5.5 years | 76 | 54.5 (24) | 74 | −49.5 | (−55.0, −44.1) | <.0001 | |
To determine whether the discordance in ivacaftor response between the pediatric and adult cohorts was related in part to baseline lung function, participants were divided into those with a baseline ppFEV1 <90 (n=57) or ≥90 (n=39) (Supplemental Table 2). Among participants with baseline ppFEV1 <90, 19 (33%) were in the pediatric cohort and 38 (67%) were in the adult cohort. In the group of participants with baseline ppFEV1 ≥90, 33 (85%) and 6 (15%) were in the pediatric and adult cohorts, respectively. Participants with lower baseline lung function experienced significant improvements in ppFEV1 at 1.5 years and at 5.5 years, whereas those with higher baseline lung function did not show statistically significant changes in ppFEV1 at 1.5 and 5.5 years.
Rate of decline in ppFEV1 for the overall cohort from the one-month post-ivacaftor visit through 5.5 years was estimated to be −1.22 pp/year [−1.70, −0.73] (Supplemental Table 3). The rate of decline in pediatric participants was −1.68 pp/year [−2.31, −1.04], and in adult participants was −0.63 pp/year [−1.35, 0.09] (Figure 2). The difference in rate of decline between the pediatric and adult cohorts was statistically significant (p=0.03).
Figure 2. Rate of change over time of ppFEV1.
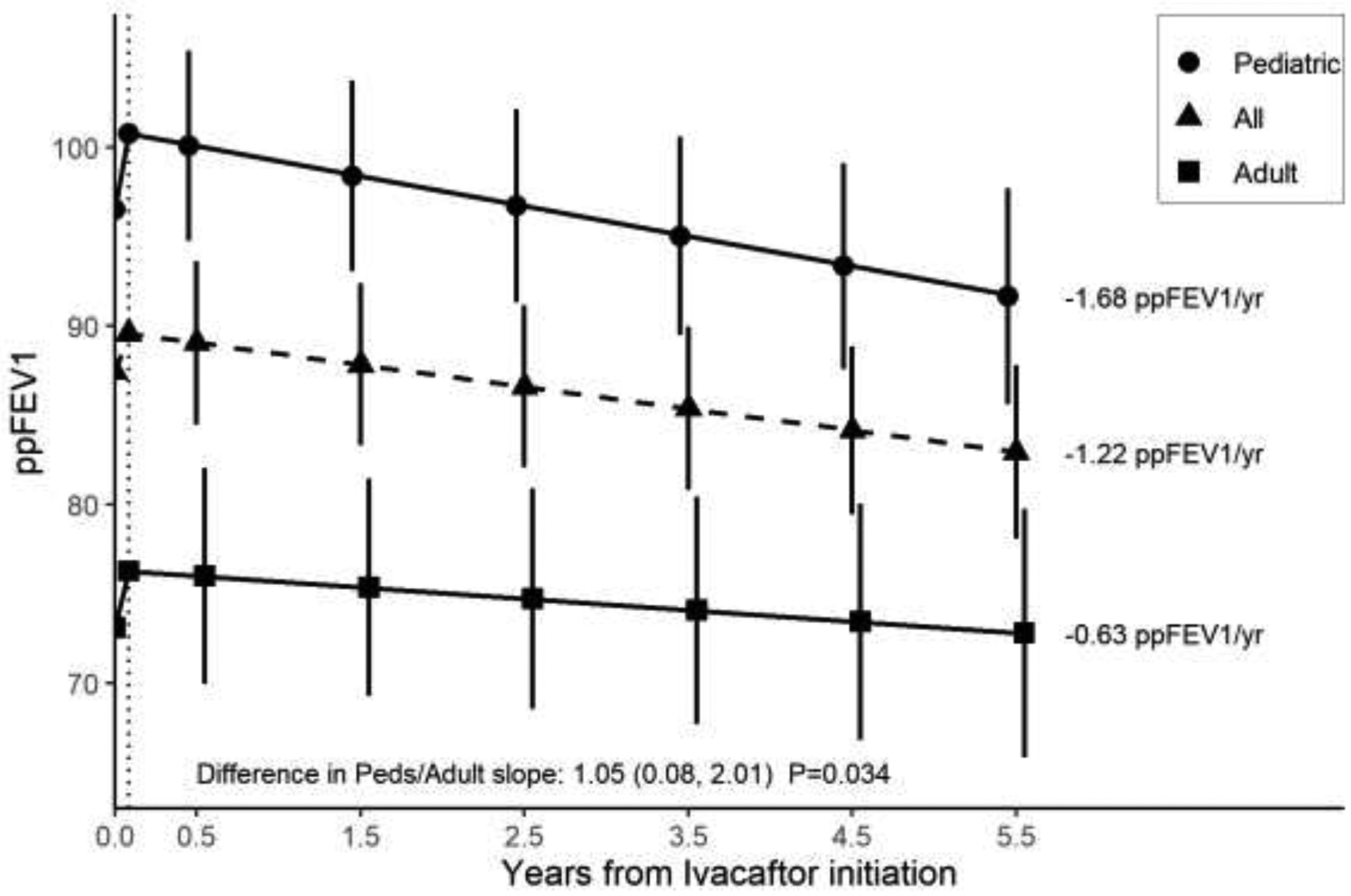
The annualized rate of decline was stratified by age cohort. The rate of change was calculated from the initial 1-month change (vertical dotted line) through 5.5 years. The rate of change for each group is indicated by the annotations to the right of each line.
3.3. Effect of ivacaftor on nutritional status
The overall cohort experienced significant improvements in weight and BMI throughout the study period, with increases in weight of 12.8 kg [10.1, 15.5] and BMI of 2.5 kg/m2 [2.0, 3.1] at 5.5 years (p<0.001 for both) (Figure 1b, Table 2). Changes in weight and BMI in the pediatric and adult cohorts are displayed in Supplemental Table 1. Patients were categorized as underweight, normal weight, overweight, and obese using CDC definitions for both adult and pediatric groups. There were no significant changes from baseline to 5.5 years in the distribution of weight categories (p=0.23 for adult and p=0.30 for pediatric), though there was a notable trend with increased numbers shifting to the overweight category in both groups at 5.5 years (Figure 3).
Figure 3.

Distribution of CDC weight categories at baseline and 5.5 years.
3.4. Effect of ivacaftor on patient-reported quality-of-life
For the overall cohort, there was a significant improvement in CFQ-R respiratory domain at 1.5 years that was sustained at 5.5 years (Figure 1c, Table 2). Both adults and children experienced significant increases in CFQ-R at 1.5 years, but only adults showed significant improvement at 5.5 years (Supplemental Table 1). Participants with ppFEV1 <90 had lower CFQ-R scores at baseline and experienced significant improvements in CFQ-R scores at both 1.5 and 5.5 years. Those with ppFEV1 ≥90 experienced a significant increase in CFQ-R at 1.5 years, while the changes were no longer significantly different from baseline at 5.5 years (Supplemental Table 2).
3.5. Effect of ivacaftor on P. aeruginosa detection, pulmonary exacerbations, and medication use (CFFPR data)
A reduction in P. aeruginosa detection, which was observed through 6 months of ivacaftor treatment,(6) continued through 5.5 years. As shown in Table 3, there was a significant reduction in P. aeruginosa detection from 62.4% at baseline to 44.1% in the 0.5–1.5 years after ivacaftor initiation to 38.9% in the 4.5–5.5 years after ivacaftor initiation (p<0.001). Significant reductions in positivity rates were seen in both adults and children (Supplemental tables 4–5).
Table 3:
Clinical outcomes captured through the U.S. CF Foundation National Patient Registry
| Variable | Period* | N | Experienced outcome (%) | OR | 95% CI | p value |
|---|---|---|---|---|---|---|
| P. aeruginosa positive | Baseline | 93 | 58 (62.4) | - | - | - |
| 0.5 – 1.5 years | 93 | 41 (44.1) | 0.095 | (0.011, 0.390) | <0.001 | |
| 4.5 – 5.5 years | 90 | 35 (38.9) | 0.045 | (0.001, 0.281) | <0.001 | |
| Any pulmonary exacerbation | Baseline | 94 | 34 (36.2) | - | - | - |
| 0.5 – 1.5 years | 94 | 15 (16.0) | 0.136 | (0.026, 0.454) | <0.001 | |
| 4.5 – 5.5 years | 90 | 15 (16.7) | 0.250 | (0.084, 0.628) | 0.001 | |
| Pulmonary exacerbation with hospitalization | Baseline | 94 | 32 (34.0) | - | - | |
| 0.5 – 1.5 years | 94 | 15 (16.0) | 0.190 | (0.048, 0.565) | 0.001 | |
| 4.5 – 5.5 years | 90 | 15 (16.7) | 0.273 | (0.090, 0.694) | 0.004 |
This data comes exclusively from the CFFPR. Patients are seen as clinically indicated at not at specified intervals as they were for all other data in this study, therefore intervals were used instead of precise time points. Baseline period is the one-year period before ivacaftor initiation. The other two periods are the number of years after ivacaftor initiation.
Pulmonary exacerbations decreased significantly as well. In the year prior to starting ivacaftor, 36.2% of patients experienced a pulmonary exacerbation, which decreased to 16.0% in the 0.5–1.5 year interval and to 16.7% in the 4.5–5.5 year interval (p<0.001). Pulmonary exacerbations requiring hospitalizations also declined significantly during both follow-up time intervals (Table 3). When separated by age, reductions were seen in both adults and children, although changes in the adult group were not statistically significant (Supplemental tables 4–5).
To assess changes in CF-specific medication use, we analyzed CFFPR data to examine use of common CF medications at baseline, at 1.5 years, and at 5.5 years. Among the CF medication categories summarized, reductions were seen in inhaled tobramycin, other inhaled antibiotics, and dornase alfa use in the 4.5–5.5 year period compared to the baseline pre-ivacaftor period (Supplemental Table 6). There were no significant changes in CF medication use in the 0.5–1.5 year period.
3.6. Effect of ivacaftor on sweat chloride
The overall cohort experienced significant reductions in sweat chloride of −55.3 mEq/L at 1.5 years and −49.5 mEq/L at 5.5 years (Figure 1d, Table 2). Changes in sweat chloride were not significantly different between children and adults (Supplemental Table 1) or between those with lower lung function versus those with higher lung function (Supplemental Table 2).
In the overall cohort, there were no significant correlations between changes in ppFEV1 and changes in sweat chloride at any of the time points including 5.5 years (Supplemental Figure 1). However, in adults, the 5.5 year change in ppFEV1 correlated significantly with a decrease in sweat chloride with a correlation of −0.48 [−0.71, −0.16], (p=0.006). To explore the impact of outliers in this sub-group, we conducted both Spearman rank and Pearson correlations, and in both cases the correlations were significant with negligible impact from outliers. There were no significant correlations between changes in ppFEV1 and sweat chloride in the pediatric cohort at any time-point. There were no significant correlations between changes in BMI and sweat chloride at 5.5 years in the pediatric and adult cohorts.
4. Discussion
We describe a real-world, 5.5 year, prospective observational study of ivacaftor treatment in people with CF and the G551D mutation. The overall cohort experienced clinical improvements in ppFEV1, pulmonary exacerbations, quality of life, growth, and P. aeruginosa detection. We observed differences in the degree of benefit based on age and lung function. Adults and those with lower baseline lung function experienced greater improvements in ppFEV1 at 1.5 years and at 5.5 years, compared to children and those with higher baseline lung function. Significant reductions in P. aeruginosa detection and pulmonary exacerbation rates were sustained through 5 years of treatment and mirror previous long-term, real-world studies. (12, 17) We observed significant long-term improvements in growth and quality of life, noting the effect on quality of life was more prominent and sustained in adults who had lower baseline quality of life scores. Sweat chloride was significantly reduced over the 5 years, with significant correlations seen between changes in ppFEV1 and sweat chloride in adults at 5.5 years.
The overall cohort maintained an average lung function above the pre-ivacaftor level throughout the study; but participants continued to experience lung function decline, with children experiencing a greater rate of decline than adults. The data further suggest that baseline lung function is an important determinant of the response to ivacaftor particularly as measured by lung function. These results are similar to other investigations, which have reported significant declines in relevant comparator groups in comparison to ivacaftor-treated patients, for example, annualized rates of decline in ppFEV1 of −1.72% among matched F508del homozygous patients compared to −0.91% for ivacaftor-treated patients with G551D CFTR.(13) Other studies have shown that CF patients with similar genetic background and disease severity have rates of decline in ppFEV1 ranging from −1.38 to −2.29% per year when not treated with modulators. (22–24) Direct comparisons with prior studies are difficult because the calculation of lung function decline may differ and methodology has evolved over time. Comparisons with natural history studies of healthy adults can provide some context, as one key goal is to bring lung function decline for patients with CF closer to that of the healthy, non-CF population. In healthy adults, lung function declines approximately 1–2% per year after age 25. (25, 26) The annualized rate of decline for children under 18 years of age in our study is improved compared to rates seen in two prior studies (~−2–3%/year) conducted in children without modulator treatment(27, 28) The disease-modifying effect of ivacaftor may contribute to a reduced rate of change in adults and children with CF, but additional treatment strategies are needed to eliminate lung function decline, particularly evident in children with CF.
Participants experienced significant decreases in sweat chloride regardless of age or baseline lung function. Reductions in sweat chloride correlated with increases in ppFEV1 at 5.5 years among adult participants, a finding that contrasts with previously published studies reporting a lack of correlation between changes in sweat chloride and improvements in FEV1(4, 29) within any single genetic group. This finding may be due to the longer study period, allowing for differences in lung disease progression to become evident, as in natural history studies that show a relationship between lung function and sweat chloride. (30, 31) This provides additional evidence that sweat chloride is a valuable marker for monitoring long-term health of individuals with CF.
No prior studies have reported changes in quality of life with ivacaftor beyond two years. We observed sustained improvements in quality of life scores over five years of ivacaftor use. While both CFQ-R and ppFEV1 overall remained above baseline, in certain subgroups these measures were not fully concordant. Prior studies have shown an inconsistent, modest correlation between these two measures, which suggest that CFQ-R scores and lung function provide different, complementary information. (32–35) Furthermore, while improvements in growth have been reported by other investigators (12), we identified a modest increase in the percentage of patients in CDC-defined overweight and obese weight categories. While not statistically significant, this finding raises the possibility that highly effective modulator treatment, may necessitate a reduction in caloric intake in patients on long-term ivacaftor treatment. Although further study is needed, closer monitoring of weight trends, promotion of healthier eating, increased physical activity, and devising weight-loss strategies may be required in patients.
This study lacked a control group, limiting conclusions that can be drawn from this longitudinal cohort. We did not prospectively track medication adherence, side effects, drug tolerance, or adverse events. There have been other long-term longitudinal studies of ivacaftor response (10–13), including one of comparable length.(12) The cohort included in this study was very similar to the original GOAL cohort in terms of demographics and clinical characteristics and broadly representative of the U.S. G551D population treated with ivacaftor. (13) Since that time, ivacaftor has been expanded to patients with additional genotypes, who were not included in this study. This multicenter prospective study of children and adults adds to what has been reported by retrospective and registry-based real-world studies of ivacaftor. We monitored sweat chloride results to assess biologic response in addition to other clinical outcomes, as well as stratified analyses by age and baseline lung function.
Taken as a whole, this five-year prospective investigation provides additional evidence of the disease-modifying effects of ivacaftor and estimates of clinical benefits people with CF and the G551D mutation experience over time on this therapy. These data further support improvements in clinical status and reduction in healthcare utilization with the use of ivacaftor, which others have shown contributes to cost-effectiveness(36, 37). Ivacaftor, the first highly effective modulator therapy in CF, appears to restore 40%–50% of CFTR activity. (3) The recently approved triple CFTR modulator combination may restore over 60% of CFTR activity. (38) This study provides a detailed picture of what patients taking highly effective modulator therapy might expect after long-term use, which would likely reflect the disease-modulating effects described above. It also serves as a reminder that while CFTR modulator therapy is changing the course of disease for many patients, our data also show there is additional need to improve care for people with CF. Particularly for younger individuals with high baseline lung function, there is the possibility of more rapid lung function decline that needs to be attenuated. Further optimization of treatment, which includes personalized strategies and the introduction of novel therapeutics tailored to individual patients, are needed to further advance CF care.
Supplementary Material
Highlights.
Ivacaftor led to long-term benefits in adults and children with the G551D mutation.
Adults and those with lower lung function had larger improvements in lung function.
Rate of lung function decline was higher in children compared with adults.
Ivacaftor resulted in sustained reductions in sweat chloride over 5 years.
Improvements in growth, quality of life, P. aeruginosa, and exacerbations were seen.
ACKNOWLEDGEMENTS
We thank all the volunteers and their families who consented to participate in this study and made this work possible. We thank all site investigators and research coordinators who were involved in this study: Peter Mogayzel, MD, Pamela Zeitlin, MD, Karen Callahan, Britany Zeglin, Ahmet Uluer, MD, Robert Fowler, Carla Frederick, MD, Jameelah Ali, Christine Roach,Churee Pardee, Mary Cross, Ibrahim Abdulhamid, MD, Catherine Van Wagnen, Debra Driscoll, Peter Hiatt, MD, Nicoline Schaap, Ami Patel, Joanne Billings, MD, Denise Stacklie, Brooke Noren, Mary Lynch, Ronald Rubenstein, MD, Erin Donnelly, Christina Kubrak, Joseph Pilewski, MD, Elizabeth Hartigan, Rose Lanzo, Michael Powers, MD, Kim Simmons, Jenna Bucher, Erika Simeon, Ginger Reeves, Heather Hathorne, Alvin Singh, MD, Philip Black, MD, Candy Schmoll, April Williams, Scott Donaldson, MD, Nadia Shive, Robin Johnson, Rose Cunnion, Theodore Liou, MD, Kristyn Packer, Jessica Francis Susan Millard, MD, Thomas Symington, Cynthia Gile, Patrick Flume,MD, Isabel Virella-Lowell, MD, Ashley Warden, Gary McPhail, MD,John Clancy, MD, Delana Terrill, Emily Bell, Sharon Kadon, Carlos Milla, MD, Zoe Davies, Colleen Dunn, Sean Ryan, Isabel Neuringer, MD, Leonard Sicilian, MD, Grace Park, Kieu-Tram Bach, Caitlin Doolittle, Rebekah Brown, MD, Pamela Berry, Brijesh Patel, Gregory Montgomery, MD, Lori Shively, Raksha Jain, MD, Ashley Keller, Lynn Fernandez, Edward Naureckas, MD, Spring Holland
SOURCES OF FUNDING
The authors acknowledge funding support for this work from #GOAL13K2 from CFF. S.D.S is supported by the CFF (#GOAL13K2) and by NIH/NCATS Colorado CTSA Grant Number UL1 TR002535. J.S.G. and S.M.R. are supported by the NIH (DK072482 and R35HL35816 to S.M.R., K23 HL143167-01A1 to J.S.G.) and CFF (ROWE19R0, ROWE17A0 and GOAL13K1 to S.M.R.); GUIMB18A0-Q to J.S.G.). S.L.H. is supported by the NIH (DK089507 to S.L.H.)
Support: The study was funded by Cystic Fibrosis Foundation Therapeutics (GOAL13K1, GOAL13K2, ROWE19R0) and the NIH (UL1TR003096, P30DK072482, R35HL135816, UL1TR002535).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICTS OF INTEREST
SMR and MJ serve as investigators and/or consultants on the design and conduct of CF clinical trials to Vertex Pharmaceuticals, the manufacturer of ivacaftor. A.P. declares a previous (within 36 months) equity interest in AbbVie, Inc. All other authors declare no conflicts of interest.
References cited.
- 1.Accurso FJ, Rowe SM, Clancy JP, Boyle MP, Dunitz JM, Durie PR, et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. The New England journal of medicine. 2010;363(21):1991–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Drevinek P, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. The New England journal of medicine. 2011;365(18):1663–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Goor F, Hadida S, Grootenhuis PD, Burton B, Cao D, Neuberger T, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(44):18825–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rowe SM, Heltshe SL, Gonska T, Donaldson SH, Borowitz D, Gelfond D, et al. Clinical Mechanism of the Cystic Fibrosis Transmembrane Conductance Regulator Potentiator Ivacaftor in G551D-mediated Cystic Fibrosis. American journal of respiratory and critical care medicine. 2014;190(2):175–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gelfond D, Heltshe S, Ma C, Rowe SM, Frederick C, Uluer A, et al. Impact of CFTR Modulation on Intestinal pH, Motility, and Clinical Outcomes in Patients With Cystic Fibrosis and the G551D Mutation. Clin Transl Gastroenterol. 2017;8(3):e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heltshe SL, Mayer-Hamblett N, Burns JL, Khan U, Baines A, Ramsey BW, et al. Pseudomonas aeruginosa in cystic fibrosis patients with G551D-CFTR treated with ivacaftor. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2015;60(5):703–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stalvey MS, Pace J, Niknian M, Higgins MN, Tarn V, Davis J, et al. Growth in Prepubertal Children With Cystic Fibrosis Treated With Ivacaftor. Pediatrics. 2017;139(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bellin MD, Laguna T, Leschyshyn J, Regelmann W, Dunitz J, Billings J, et al. Insulin secretion improves in cystic fibrosis following ivacaftor correction of CFTR: a small pilot study. Pediatric diabetes. 2013;14(6):417–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bell SC, Mainz JG, MacGregor G, Madge S, Macey J, Fridman M, et al. Patient-reported outcomes in patients with cystic fibrosis with a G551D mutation on ivacaftor treatment: results from a cross-sectional study. BMC pulmonary medicine. 2019;19(1):146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hubert D, Dehillotte C, Munck A, David V, Baek J, Mely L, et al. Retrospective observational study of French patients with cystic fibrosis and a Gly551Asp-CFTR mutation after 1 and 2years of treatment with ivacaftor in a real-world setting. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society. 2018;17(1):89–95. [DOI] [PubMed] [Google Scholar]
- 11.Bessonova L, Volkova N, Higgins M, Bengtsson L, Tian S, Simard C, et al. Data from the US and UK cystic fibrosis registries support disease modification by CFTR modulation with ivacaftor. Thorax. 2018;73(8):731–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Volkova N, Moy K, Evans J, Campbell D, Tian S, Simard C, et al. Disease progression in patients with cystic fibrosis treated with ivacaftor: Data from national US and UK registries. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society. 2019. [DOI] [PubMed] [Google Scholar]
- 13.Sawicki GS, McKone EF, Pasta DJ, Millar SJ, Wagener JS, Johnson CA, et al. Sustained Benefit from ivacaftor demonstrated by combining clinical trial and cystic fibrosis patient registry data. American journal of respiratory and critical care medicine. 2015;192(7):836–42. [DOI] [PubMed] [Google Scholar]
- 14.Chassagnon G, Hubert D, Fajac I, Burgel PR, Revel MP, investigators. Long-term computed tomographic changes in cystic fibrosis patients treated with ivacaftor. The European respiratory journal. 2016;48(1):249–52. [DOI] [PubMed] [Google Scholar]
- 15.Rosenfeld M, Cunningham S, Harris WT, Lapey A, Regelmann WE, Sawicki GS, et al. An open-label extension study of ivacaftor in children with CF and a CFTR gating mutation initiating treatment at age 2–5years (KLIMB). Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heltshe SL, Rowe SM, Skalland M, Baines A, Jain M, Network GIotCFFTD. Ivacaftor-treated Patients with Cystic Fibrosis Derive Long-Term Benefit Despite No Short-Term Clinical Improvement. American journal of respiratory and critical care medicine. 2018;197(11):1483–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frost FJ, Nazareth DS, Charman SC, Winstanley C, Walshaw MJ. Ivacaftor Is Associated with Reduced Lung Infection by Key Cystic Fibrosis Pathogens: A Cohort Study Using National Registry Data. Annals of the American Thoracic Society. 2019. [DOI] [PubMed] [Google Scholar]
- 18.Guimbellot J, Baines A, Khan U, Heltshe SL, VanDalfsen J, Jain M, Rowe SM, Sagel S. Long Term Effects of Ivacaftor in G551D Patients: Five Year Follow-up Data in GOAL-E2. The 32nd Annual North American Cystic Fibrosis Conference; October 18–20, 2018; Denver, Colorado. Pediatric Pulmonology: Pediatric Pulmonology; 2018. p. S1–S481. [Google Scholar]
- 19.Quanjer PH, Stanojevic S, Cole TJ, Baur X, Hall GL, Culver BH, et al. Multi-ethnic reference values for spirometry for the 3–95-yr age range: the global lung function 2012 equations. The European respiratory journal. 2012;40(6):1324–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Control CfD. Overweight and Obesity: Centers for Disease Control; 2020. [Available from: https://www.cdc.gov/obesity/index.html.
- 21.Kuczmarski RJ, Ogden CL, Grummer-Strawn LM, Flegal KM, Guo SS, Wei R, et al. CDC growth charts: United States. Adv Data. 2000(314):1–27. [PubMed] [Google Scholar]
- 22.Konstan MW, McKone EF, Moss RB, Marigowda G, Tian S, Waltz D, et al. Assessment of safety and efficacy of long-term treatment with combination lumacaftor and ivacaftor therapy in patients with cystic fibrosis homozygous for the F508del-CFTR mutation (PROGRESS): a phase 3, extension study. The lancet Respiratory medicine. 2017;5(2):107–18. [DOI] [PubMed] [Google Scholar]
- 23.Sawicki GS, McKone EF, Millar SJ, Pasta DJ, Konstan MW, Lubarsky B, et al. Patients with Cystic Fibrosis and a G551D or Homozygous F508del Mutation: Similar Lung Function Decline. American journal of respiratory and critical care medicine. 2017;195(12):1673–6. [DOI] [PubMed] [Google Scholar]
- 24.Caley L, Smith L, White H, Peckham DG. Average rate of lung function decline in adults with cystic fibrosis in the United Kingdom: Data from the UK CF registry. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society. 2020. [DOI] [PubMed] [Google Scholar]
- 25.Association AL. Lung Capacity and Aging American Lung Association: Americal Lung Association; 2020. [updated March 11, 2020. Available from: https://www.lung.org/lung-health-diseases/how-lungs-work/lung-capacity-and-aging.
- 26.Thomas ET, Guppy M, Straus SE, Bell KJL, Glasziou P. Rate of normal lung function decline in ageing adults: a systematic review of prospective cohort studies. BMJ Open. 2019;9(6):e028150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Konstan MW, Morgan WJ, Butler SM, Pasta DJ, Craib ML, Silva SJ, et al. Risk factors for rate of decline in forced expiratory volume in one second in children and adolescents with cystic fibrosis. The Journal of pediatrics. 2007;151(2):134–9, 9 e1. [DOI] [PubMed] [Google Scholar]
- 28.Szczesniak RD, Li D, Su W, Brokamp C, Pestian J, Seid M, et al. Phenotypes of Rapid Cystic Fibrosis Lung Disease Progression during Adolescence and Young Adulthood. American journal of respiratory and critical care medicine. 2017;196(4):471–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Durmowicz AG, Witzmann KA, Rosebraugh CJ, Chowdhury BA. Change in sweat chloride as a clinical end point in cystic fibrosis clinical trials: the ivacaftor experience. Chest. 2013;143(1):14–8. [DOI] [PubMed] [Google Scholar]
- 30.Caudri D, Zitter D, Bronsveld I, Tiddens H. Is sweat chloride predictive of severity of cystic fibrosis lung disease assessed by chest computed tomography? Pediatric pulmonology. 2017;52(9):1135–41. [DOI] [PubMed] [Google Scholar]
- 31.McCague AF, Raraigh KS, Pellicore MJ, Davis-Marcisak EF, Evans TA, Han ST, et al. Correlating Cystic Fibrosis Transmembrane Conductance Regulator Function with Clinical Features to Inform Precision Treatment of Cystic Fibrosis. American journal of respiratory and critical care medicine. 2019;199(9):1116–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DiMango E, Spielman DB, Overdevest J, Keating C, Francis SF, Dansky D, et al. Effect of highly effective modulator therapy on quality of life in adults with cystic fibrosis. International forum of allergy & rhinology. 2020. [DOI] [PubMed] [Google Scholar]
- 33.Quittner AL, Sawicki GS, McMullen A, Rasouliyan L, Pasta DJ, Yegin A, et al. Psychometric evaluation of the Cystic Fibrosis Questionnaire-Revised in a national sample. Qual Life Res. 2012;21(7):1267–78. [DOI] [PubMed] [Google Scholar]
- 34.Sawicki GS, Rasouliyan L, McMullen AH, Wagener JS, McColley SA, Pasta DJ, et al. Longitudinal assessment of health-related quality of life in an observational cohort of patients with cystic fibrosis. Pediatric pulmonology. 2011;46(1):36–44. [DOI] [PubMed] [Google Scholar]
- 35.Cheney J, Vidmar S, Gailer N, Wainwright C, Douglas TA, Australasian Cystic Fibrosis Bronchoalveolar Lavage study g. Health-related quality-of-life in children with cystic fibrosis aged 5-years and associations with health outcomes. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society. 2020;19(3):483–91. [DOI] [PubMed] [Google Scholar]
- 36.Dilokthornsakul P, Hansen RN, Campbell JD. Forecasting US ivacaftor outcomes and cost in cystic fibrosis patients with the G551D mutation. The European respiratory journal. 2016;47(6):1697–705. [DOI] [PubMed] [Google Scholar]
- 37.Feng LB, Grosse SD, Green RF, Fink AK, Sawicki GS. Precision Medicine In Action: The Impact Of Ivacaftor On Cystic Fibrosis-Related Hospitalizations. Health Aff (Millwood). 2018;37(5):773–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Veit G, Roldan A, Hancock MA, Da Fonte DF, Xu H, Hussein M, et al. Allosteric folding correction of F508del and rare CFTR mutants by elexacaftor-tezacaftor-ivacaftor (Trikafta) combination. JCI Insight. 2020;5(18). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.