

Introduction
Immunosuppressive therapy after solid organ transplant (SOT) is associated with increased risk of post-transplant lymphoproliferative disorder (PTLD).1 Plasma cell neoplasms rarely present after SOT and may adversely affect patient clinical outcomes.2,3 Plasmacytic post-transplantation lymphoproliferative disorder (plasmacytic-PTLD) traditionally has been described as a rare subtype of monomorphic B-cell post-transplant lymphoproliferation with features of plasmacytoma in the non-transplant population.2,3 It accounts for ∼4% of all PTLD after SOT.3 The salient clinical features of plasmacytic-PTLD described in the literature usually include an aggressive and rapid clinical presentation, often with extranodal and extramedullary manifestations, lack of osteolytic lesions, negative bone marrow involvement, lack of para-proteinemia, and positivity for Epstein Barr virus (EBV) in about 50% of cases.2,3 This traditional definition does not encompass systemic light chain (AL) amyloidosis occurring after SOT.
Plasmacytic-PTLD and other clonal plasma cell disorders are a rare and challenging group of disorders that presents difficulty in diagnosis and management in SOT recipients. Therapeutic strategies in this setting typically include anti-plasma cell therapy with a proteasome inhibitor such as bortezomib and or an immunomodulatory agent, such as lenalidomide, based on extrapolation from their effectiveness in treatment of multiple myeloma in patients without history of SOT. Currently, there are no data on effectiveness of immunotherapy with anti-plasma cell monoclonal antibody in patients with history of SOT and concomitant clonal plasma cell disorders.
Daratumumab (Dara) is an anti-CD38 monoclonal antibody that exhibits therapeutic activity against multiple myeloma, either as a monotherapy4 or in combination with immunomodulatory agents5 or proteasome inhibitors.6 Currently, there are limited reported experiences with Dara when used as a salvage therapy in relapsed/refractory patients with multiple myeloma, after allogeneic hematopoietic stem cell transplantation.7 Furthermore, the safety of Dara usage is completely unknown in patients with a history of SOT, who are also on immunosuppressive therapy and have concomitant clonal plasma cell neoplasms.
Targeted immunotherapy with Dara represents an attractive therapeutic modality as it may be better tolerated compared with conventional chemotherapy regimens. It may also be safely employed in frail patients with compromised organ functions. Efficacious management of clonal plasma cell neoplasms in the immunosuppressed SOT recipients still represents an unmet need. Therefore, we herein report on our experience with a Dara-based regimen for treatment of plasma cell dyscrasias in the setting of SOT and ongoing immunosuppressive therapy.
In this Institutional Review Board-approved, retrospective series, records were queried for all adult patients who had undergone SOT and were on immunosuppressive therapy. We included patients who: (1) had developed clonal plasma cell neoplasms (such as plasmacytoma or multiple myeloma or AL amyloidosis), and (2) had received a Dara-containing regimen, as salvage therapy, at any time during the disease course. Two patients with a history of AL amyloidosis preceding heart transplant, who subsequently received Dara combination therapy post-transplant, were also included in the analysis. A summary of clinical and pathologic data is presented in Table 1.
Table 1.
Clinical Characteristics of Patients
| Clinical Characteristics | Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 |
|---|---|---|---|---|---|
| Age/gender | 57/M | 66/M | 72/M | 66/M | 59/M |
| Date and type of organ transplanted | 1/2017: deceased donor kidney transplantation for ESRD owing to diabetic and hypertensive nephropathy | 6/2009: OLT liver transplant for HCV cirrhosis | 8/2003: Heart transplant for IDCM | 9/2016: Heart transplantation for AL amyloid cardiomyopathy | 5/6/2019: Heart transplant for AL amyloid cardiomyopathy |
| Date of diagnosis and type of clonal plasma cell neoplasm | 10/2018: monomorphic post-transplant lymphoproliferative disorder, EBV-positive, extra-osseous plasmacytoma type | 4/2017: AL amyloidosis | 5/2017: AL amyloidosis | 7/2005: Multiple myeloma with AL amyloidosis | 4/2019: IgG lambda myeloma with Al amyloidosis |
| Clinical presentation leading to the diagnosis of clonal plasma cell disorder | Pelvic pain at site of transplanted kidney, hematuria, obstructive uropathy of transplanted kidney | Proteinuria 5 grams/24 hours | Congestive heart failure, proteinuria 10 grams/24 hours | Congestive heart failure, proteinuria | Non-ischemic cardiomyopathy |
| Tissue biopsy confirming the diagnosis | 8-cm pelvis mass encasing the transplanted kidney | Kidney and liver Congo Red-positive, kidney positive for amyloid by EM and MS | Endomyocardial biopsy, and bone marrow biopsy Congo Red-positive | Endomyocardial biopsy, fat pad biopsy AL amyloidosis affecting heart, liver and kidney | Bone marrow Fat pad Orthotopic heart |
| Bone marrow flow cytometry (CD38/CD138, cytoplasmic kappa/ lambda), IHC and FISH finding | Negative | 5%−10% lambda light chain restricted clonal plasma cells/monosomy 13, duplication of 1q, gain chromosome 5 | ∼10% lambda light chain restricted clonal plasma cells, 13q- and t(11;14) | 23% lambda light chain restricted clonal plasma cells; FISH not available | 15% lambda light chain restricted clonal plasma cells, normal FISH |
| Circulating monoclonal protein | Negative | IgG lambda monoclonal gammopathy | IgM lambda monoclonal gammopathy | IgG lambda monoclonal gammopathy | IgG lambda |
| Type of immunosuppressant | Tacrolimus, prednisone | Cyclosporine Prednisone Sirolimus | Cyclosporine, mycophenolate, prednisone | tacrolimus, prednisone | tacrolimus, prednisone Mycophenolate |
| Prophylactic antimicrobials | Valganciclovir Trimethoprimsulfamethoxazole | Trimethoprimsulfamethoxazole | Acyclovir | Valganciclovir Acyclovir | Valganciclovir Trimethoprimsulfamethoxazole |
| Complications during treatment with daratumumab | Right great toe acute osteomyelitis needing amputation | Mild acute cellular rejection of liver while on Bd | Influenza A Bacterial pneumonia | Disseminated nocardiosis CMV viremia EBV viremia | None |
| First line anti-plasma cell therapy (response) | CyBorD (PD) | CyBorD (PR) | CyBorD (VGPR) | MEL 140 (PR) | CyBorD (PD) |
| Second line anti-plasma cell therapy (response) | DPd (CR) | Bd (PR) | DRd (SD) | Td (CR) | DVd (VGPR) |
| Third line anti-plasma cell therapy (response) | NA | DVd plus POM (SD) | NA | Bd (SD) | NA |
| Fourth line anti-plasma cell therapy (response) | NA | NA | NA | Benda + dex (SD) | NA |
| Fifth line anti-plasma cell therapy (response) | NA | NA | NA | Did (VGPR) | NA |
| Best response to Dara-containing regimen/outcome | Hematologic CR with Dara containing regimen | Hematologic VGPR: progressive CKD | Hematologic VGPR, progressive CKD leading to ESRD | Hematologic VGPR to Dara containing regimen | Hematologic VGPR to Dara containing regimen |
Abbreviations: Bd= bortezomib plus dexamethasone; Benda + dex = bendamustine plus dexamethasone; CKD = chronic kidney disease; CMV = cytomegalovirus; CR = completeresponse; CyBorD = bortezomib, cyclophosphamide, and dexamethasone; Dara = daratumumab; Did = daratumumab plus ixazomib and dexamethasone; Dpd = daratumumab plus pomalidomide and dexamethasone; Drd = daratumumab plus lenalidomide and dexamethasone; Dvd = daratumumab plus bortezomib and dexamethasone; EBV = Epstein Barr virus; EM = electron microscopy; ESRD = end-stage renal disease; HCV = hepatitis C virus; IDCM = idiopathic dilated cardiomyopathy; IHC = immunohistochemistry; FISH = fluorescent in situ hybridization; M = male; MEL = melphalan; MS = mass spectrometry; NA = not applicable; OLT = orthotopic liver transplantation; PD = progressive disease; POM = pomalidomide; PR = partial response; SD = stable disease; Td = thalidomide plus dexamethasone; VGPR = very good partial response.
Standard CD138 (anti-syndecan-1 antibody) immunostain was applied to the bone marrow core biopsy in order to estimate bone marrow plasmacytosis. Immunophenotyping by flow cytometry was performed on bone marrow aspirate (CD38, CD138, cytoplasmic kappa and lambda) to identify the clonal plasma cell population. Congo red staining of the tissue biopsy was used for the diagnosis of amyloidosis, and mass spectrometry, when available, was also used for proteomics analysis of the organ biopsy specimens.
Patient Series
From January 2017 to June 2019, 5 patients (n = 5) with history of SOT received a Dara-containing regimen for relapsed/refractory clonal plasma cell neoplasms, at the Cleveland Clinic Florida (Weston, FL) and Cleveland Clinic Main Campus (Cleveland, OH); one patient also received care both at Tufts Medical Center (Boston, MA) and Memorial Sloan Kettering Cancer Center (New York, NY). Three patients received heart transplants, and 1 patient each received a kidney and a liver transplant.
Four patients had AL amyloidosis, and 1 patient had EBV-positive monomorphic PTLD, extraosseous plasmacytoma type (PTLD-EP). All 5 patients were male. All patients were receiving immunosuppressive therapy with prednisone, tacrolimus or cyclosporine, and mycophenolate. Anti-rejection immunosuppressive therapy was continued as recommended by the SOT specialists before and during initiation of anti-plasma cell therapy in all cases except in case 1.
Patient 1
Patient 1 is a 57-year-old male who presented to the emergency room with sudden onset of lower right quadrant pelvic pain at the site of transplanted kidney, as well as oliguric acute renal failure, 22 months after renal transplantation for end-stage renal disease, owing to diabetic and hypertensive nephropathy. Computerized tomography showed obstructive nephropathy of transplanted kidney owing to extrinsic compression by a 6-cm mass enveloping the distal part of transplanted ureter, and a 5-cm mass at the right hemi-pelvis near pelvic wall. An emergency nephrostomy tube was placed to treat the obstructive uropathy. Histologic sections of core needle biopsies of both masses showed sheets of abnormal, mature plasma cells with cytologic atypia (Figure 1A). Immunohistochemical stains showed that the neoplastic plasma cells expressed CD138 (Figure 1B) and monotypic lambda immunoglobulin light chains. In addition, the Epstein-Barr encoding region (EBER) in situ hybridization was diffusely positive in the neoplastic plasma cells (Figure 1C). A skeletal survey revealed no lytic bone lesions. Bone marrow aspiration biopsy was normal and monoclonal gammopathy panels reported no monoclonal protein. An EBV DNA PCR in the blood reported an elevated level of 23480 IU/ml equivalent of EBV DNA (log IU/mL) of 4.37. Nuclear medicine positron emission tomography scan reported enlarging 6.8 × 4.6 × 7.8 cm intensely 18F-fluorodeoxyglucose-avid mass (standard uptake value, 16) in the right anterior pelvis abutting and possibly encasing the transplanted kidney and ureter (Figure 2A).
Figure 1.
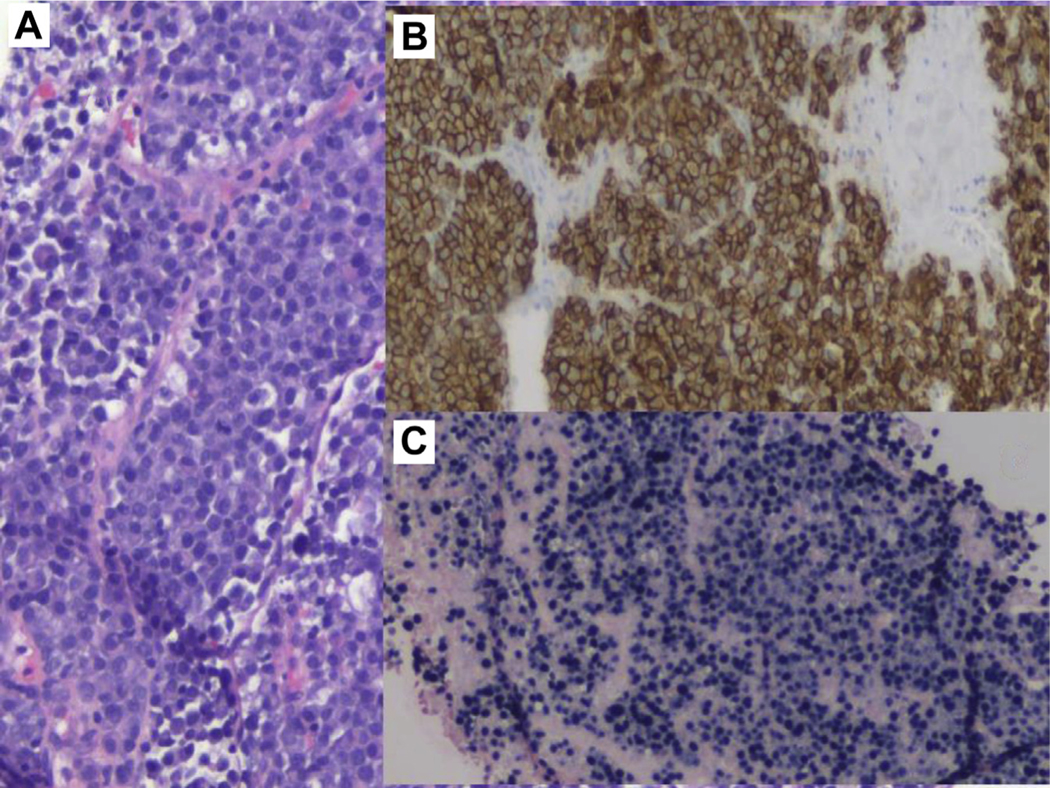
Abdominal Mass Biopsy. A, Sheets of Atypical Plasma Cells, Hematoxylin and Eosin 20×. B, CD138 Immunostain Confirming that the Mass Is Composed of Plasma Cells. CD138 Immunohistochemistry, 10×. C, Epstein-Barr Encoding Region (EBER) Chromogenic in Situ Hybridization (CISH) 10×. Small Encoded RNA (EBER) by CISH Shows the Presence of Epstein Barr Virus (EBV), in This EBV-driven Monomorphic Plasmacytic Post-transplant Lymphoproliferative Disorder
Figure 2.

Positron Emission Tomography Scan Image at Diagnosis, Prior to Initiation of Treatment, Reported Intensely 18F- Fluorodeoxyglucose Avid Mass (A) in the Right Anterior Pelvis Abutting and Encasing the Distal Aspect of the Transplanted Kidney and Ureter (Blue Arrow Pointing to the Ureteric Stent and Red Arrow Pointing to the Mass). The Mass Measures ∼7.8 cm in Maximum Dimension. Post Treatment Positron Emission Tomography Scan Showed Normal Transplanted Right Pelvic Kidney With Interval Resolution of the Previously Seen Right Anterior Pelvic Mass and the Ureteric Stent Has Been Removed (B)
Tacrolimus and mycophenolate were both discontinued, and the patient was maintained on prednisone, 10 mg daily, for immunosuppression. A diagnosis of extramedullary multiple myeloma was made, and the patient started on bortezomib, cyclophosphamide, and dexamethasone and completed 1 cycle of therapy. On the day the patient was scheduled to begin the second cycle of therapy, he presented to the emergency room with a large amount of gross hematuria and hemorrhagic shock. Computerized tomography scans showed enlargement of right pelvic mass (now 10 cm) invading both the urinary bladder and anterior abdominal wall. Radiation therapy was discussed but was not employed owing to the concern for radiation-induced kidney injury (and graft loss). In addition, the radiation field was estimated to include the entire pelvis with potential for damage to the pelvic organs, particularly the colon, as the whole pelvic radiation was required for curative intention. He was immediately started on salvage anti-myeloma therapy with Dara, pomalidomide, and dexamethasone, and achieved complete radiologic response based on a positron emission tomography scan completed after 2 cycles of therapy (Figure 2B). EBV viremia subsequently resolved. A clinical decision was made to administer the patient 2 more cycles of Dara, pomalidomide, and dexamethasone therapy for consolidation and to consider Dara as an ongoing regimen for maintenance. During the third cycle of therapy, the patient was admitted for a right, lower extremity, diabetic foot ulcer owing to osteomyelitis of the right great toe. He completed a 6-week intravenous antibiotic course, without improvement, and eventually required amputation of the great toe for osteomyelitis. A clinical decision was made to discontinue further therapy with Dara, pomalidomide, and dexamethasone, given the infectious complication. The patient remains in complete radiologic remission, 11 months post-completion of Dara-based treatment.
Patient 2
Patient 2 is a 66-year-old man who presented with nephrotic range proteinuria and worsening creatinine levels, 8 years following liver transplantation. He was found to have IgG lambda monoclonal protein in both urine and serum samples. A renal biopsy confirmed amyloid deposition in the glomeruli and interstitium on Congo red stain. Electron microscopy revealed characteristic 11-nm amyloid fibrils within the glomeruli. Amyloid typing by mass spectrometry identified predominance of lambda light chains. Given his comorbidities, the patient was felt not to be a candidate for autologous peripheral blood stem cell transplantation, and thus began treatment with CyBorD (cyclophosphamide, bortezomib, and dexamethasone) as previously described.8 He completed 11 cycles of therapy, leading to very good partial response (VGPR) based on previously published consensus criteria.9
Cycle 2 of CyBorD therapy was complicated by elevated liver enzymes and hyperbilirubinemia – a liver biopsy demonstrated mild acute cellular rejection and focal deposition of amyloid by Congo red (Figure 3A-C); a previous liver biopsy, performed 2 years earlier, showed no amyloidosis. After 6 cycles of therapy, he achieved VGPR, and he continued therapy with stable disease for 1 year, but he showed renal, cardiac, and hepatic progression after 11 cycles of CyBorD as manifested with increasing creatinine, progressive elevation of NTproBNP and troponin T, and elevation of alkaline phosphatase levels. An echocardiogram was reported to show a granular speckled pattern consistent with cardiac amyloidosis. Patient started salvage therapy with Dara-Vd (Dara, bortezomib, and dexamethasone) as previously described1–3 without further increase in cardiac biomarkers (troponin T and NTproBNP) and without meaningful further hematologic response, despite 20 dosages of Dara-Vd. Pomalidomide was added to the regimen and was tolerated.
Figure 3.

Liver Biopsy. A, Hepatic Arterioles With Thickened Walls Owing to Acellular Eosinophilic Deposits. Hematoxylin and Eosin, 20x. B, Congo Red Stain Showing Waxy Acellular, Salmon Pink Material on Liver Arteriole Walls. Congo Red Stain, Pre-polarization 20x. C, Hepatic Arterioles With Amyloid Deposition on the Walls After Light Polarization Demonstrating Typical Apple-Green Birefringence. Congo Red Stain, 20x
The patient has received 24 dosages of Dara-Vd thus far, with 2 cycles of pomalidomide. Despite the quadrupled therapy, he continues to exhibit cardiac, hepatic, and renal disease progression, indicating a resistant nature of AL amyloidosis in post-SOT patients. The presence of duplication of 1q by fluorescence in situ hybridization analysis in the bone marrow aspirate of this patient may explain the resistance to therapy and progressive disease. This particular fluorescence in situ hybridization finding is known to be a poor prognostic feature in patients with multiple myeloma.
Patient 3
Patient 3 is a 72-year-old man who presented with systemic AL amyloidosis, 14 years after heart transplantation for idiopathic cardiomyopathy. The AL amyloidosis was diagnosed following prominent gastrointestinal symptoms (nausea, diarrhea), tubular dysfunction, and proteinuria of 8 g/24 hours and IgM lambda monoclonal gammopathy. An endomyocardial biopsy reported no rejection but confirmed cardiac amyloidosis. Bone marrow aspiration and biopsy reported 10% lambda light chain restricted plasma cells. He began treatment with CyBorD, which was poorly tolerated with worsening of diarrhea, nausea, and vomiting.
Despite hematologic VGPR to 6 cycles of CyBorD, renal disease progressed during the therapy. He was initiated on Dara and lenalidomide as rescue therapy to prevent renal progression. He had severe diarrhea and nausea from lenalidomide and was discontinued after 2 months; daratumumab was continued for the next 6 months. The patient, however, progressed to end-stage renal disease after 6 months of Dara therapy and was started on hemodialysis. Owing to comorbidities and frailty, he is being managed conservatively without further anti-plasma cell-directed therapy. He had frequent exacerbation of heart failure owing to amyloid cardiomyopathy.
Patient 4
Patient 4 is a 66-year-old man with history of heart transplantation for AL amyloid cardiomyopathy, which was performed 11 years after initial diagnosis of AL amyloidosis, with cardiac and renal involvement. He started with risk-adapted autologous hematopoietic stem cell transplantation, with melphalan 140 mg/m2 conditioning, and had received several lines of therapy (Table 1) for treatment of AL amyloidosis, with complete hematologic response for 3 years prior to receiving heart transplantation. He was monitored expectantly with hematologic relapse of AL amyloidosis, documented 2 years post-heart transplant, and he was started on salvage combination therapy with Dara, ixazomib, and dexamethasone.
Three months after starting Dara-based therapy, the patient presented with fever, malaise, subcutaneous nodules, and perinephric fluid collections. Blood cultures, as well as cultures from one of the abdominal subcutaneous nodules, were positive for Nocardia species consistent with disseminated nocardiosis. He was also found to have concomitant cytomegalovirus viremia (treated with valganciclovir) and EBV viremia (monitored with lowering immunosuppressive medications). The disseminated nocardiosis was treated initially with sulfamethoxazole trimethoprim, linezolid, and imipenem – linezolid and sulfamethoxazole trimethoprim were discontinued owing to bone marrow suppression. The patient was successfully treated with a combination of imipenem and amoxicillin clavulanic acid. He resumed Dara-based therapy and continues to show hematologic response. He has not had recurrence of any of his infections. One could infer that the anti-plasma cell therapy in this case may have predisposed to nocardiosis and opportunistic viral infections.
Patient 5
Patient 5 was a 59-year-old male with no significant past medical history, who presented with complaints of shortness of breath and anasarca and was diagnosed with non-ischemic cardiomyopathy. He subsequently had a ventricular fibrillation arrest outside of the hospital and was hospitalized after successful resuscitation. Echo-cardiographic findings were felt to be suggestive for amyloid cardiomyopathy. Patient was found to have an elevated lambda serum free light chain level. A bone marrow biopsy showed 15% clonal plasma cells, and Congo red stained positive for amyloid. A fat pad biopsy was positive for amyloidosis.
On cycle 1, day 4 of CyBorD, he had a second episode of in-hospital, ventricular fibrillation arrest and remained dependent on intra-aortic balloon pump and inotropic support to maintain perfusion. On day 15 of cycle 1 of CyBorD, there was evidence of a rising level of free lambda light chain, indicating a primary refractory AL amyloidosis. He underwent heart transplant, 4 weeks after initiation of CyBorD therapy. The pathology of the recipient heart revealed cardiomegaly (548 grams), and both the heart and the aorta were positive for AL amyloidosis, confirmed by Congo red staining and mass spectrometry. Two months following heart transplant, serum free lambda light chain levels continued to increase. Therefore, he was initiated on salvage Dara-Vd therapy approximately 3 months following heart transplant.
Following completion of cycle 1 of the Dara-based regimen, his serum free lambda light chain had dropped from 170 mg/L to 32 mg/L with improvement in the kappa/lambda ratio. As of his last visit, at the time of writing of this report, he is clinically responding well and tolerating treatment.
Discussion
Immunosuppressive therapy after SOT, or after allogeneic hematopoietic stem cell transplantation, is associated with an increased risk of PTLD as a result of immune dysregulation and EBV reactivation.1 Emerging data indicates that the frequency of systemic AL amyloidosis in the general population is increasing, probably as a result of heightened awareness of this rare disease among clinicians.10,11 One registry study suggests that the frequency of AL amyloidosis may be elevated in the post-SOT setting, although the evidence is correlative and retrospective without definitive clinical-translational support.8 We analyzed the effectiveness of a Dara-based regimen for the treatment of clonal plasma cell neoplasm in SOT recipients. Our results indicate variable success among the 2 evaluated types of plasma cell dyscrasias: (1) systemic AL amyloidosis, and (2) PTLD-EP.
In all patients, the first line of therapy was a bortezomib-based regimen. Immunomodulatory drugs (including lenalidomide and pomalidomide) were employed at the time of relapse, or after failure of the bortezomib-based regimen. In 1 patient with PTLD-EP and another patient with heart transplant for AL amyloid cardiomyopathy who progressed to a first-line bortezomib-based regimen, the Dara-containing regimen was the second-line therapy, but in the 4 remaining patients, Dara was employed as a third-line setting or after.
A Dara-based regimen, used as second-line therapy in combination with pomalidomide and dexamethasone, elicited a complete response in the patient with PTLD-EP. This favorable response was observed following aggressive progression of disease, not stymied by the withdrawal of immunosuppressive therapy and the first-line bortezomib therapy (administered with dexamethasone and cyclo-phosphamide). The Dara-based regimen was also very effective as second-line treatment, after failure of a bortezomib-based regimen in 1 patient who had heart transplant for AL amyloid cardiomyopathy.
All patients with AL amyloidosis had at least a partial response, and 3 patients exhibited VGPR with a Dara-based regimen in the relapsed/refractory setting when measured by previously published criteria.12 Three patients had infectious complications while receiving immunotherapy with Dara.
Although rare, clinicians should be aware of the clinical presentation of clonal plasma cell neoplasms in the recipients of SOT. It should also be noted that a patient with a clonal plasma cell neoplasm such as AL amyloidosis can undergo a solid organ transplant and will need to continue both the antirejection immunosuppression and the anti-plasma cell therapy. Our limited observations show that Dara-based salvage regimens were not associated with any organ dysfunction including the graft, and were well-tolerated, aside from an increased risk of infectious complications. As such, a heightened level of clinical vigilance is needed for infections, and appropriate antimicrobial prophylaxis should be considered. In our series, immunosuppression was not held or reduced except in case 1. In patients with PTLD, the first therapeutic step is usually the reduction in the immunosuppression therapy, but whether a similar approach can be considered in AL amyloidosis developing after SOT is unknown. It is possible that the concomitant use of immunosuppressive therapy and Dara-based regimen may have increased the risk of infections is some of our patients. The Dara-based regimen, when used for second-line therapy, was extremely efficacious in 1 patient with PTLD-EP achieving a complete response to therapy, and in another patient observed to have VGPR after 4 dosages. This observation may indicate that early use of a Dara-based regimen, either as a first-line or second-line therapy, may help prevent progressive organ damage in the clonal plasma cell neoplasm in this setting.
Two patients with AL amyloidosis also had renal progression, despite achieving very good partial hematologic response and improved proteinuria. Progression of renal disease, despite achieving hematologic response on the underlying plasma cell neoplasm, indicates that the progression of nephropathy in this setting could be owing to calcineurin inhibitor therapy- related nephrotoxicity. Our study is limited by its retrospective design and low power; therefore, it is difficult to draw a definitive conclusion regarding the treatment of clonal plasma cell neoplasms in the SOT setting. A multicenter registry study and or a multicenter prospective clinical study, further examining this rare group of disorders, may provide additional insight regarding proper diagnosis and management.
Clinical Practice Points.
Clonal plasma cell neoplasm in solid organ transplant recipients can manifest in the form of AL amyloidosis or post-transplant lymphoproliferative disorder extraosceous plasmacytoma type.
The typical treatment in clinical practice includes multiple myeloma type anti-plasma cell therapy, but there is no consensus on how to best treat this rare group of disorders.
In our series (n = 5), all patients received anti-CD38 monoclonal antibody treatment with daratumumab in combination with other agents as salvage therapy with variable clinical response.
Daratumumab-based therapy was safe and well-tolerated but was associated with increased infectious complications and did not prevent progressive kidney damage, despite achieving significant anti-plasma cell response.
Acknowledgments
The authors want to thank Felipe Martinez, MD (Department of Radiology, Cleveland Clinic Florida) who provided positron emission tomography scan image (Figure 2). Funding support for this publication was provided by Malignant Hematology Research Fund of Maroone Cancer Center, Cleveland Clinic Florida.
Footnotes
Disclosure
The authors have stated that they have no conflicts of interest.
References
- 1.Dierickx D, Tousseyn T, Gheysens O. How I treat posttransplant lymphoproliferative disorders. Blood 2015; 126:2274–83. [DOI] [PubMed] [Google Scholar]
- 2.Trappe R, Zimmermann H, Fink S, et al. Plasmacytoma-like post-transplant lymphoproliferative disorder, a rare subtype of monomorphic B-cell post-transplant lymphoproliferation, is associated with a favorable outcome in localized as well as in advanced disease: a prospective analysis of 8 cases. Haematologica 2011; 96:1067–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karuturi M, Shah N, Frank D, et al. Plasmacytic post-transplant lymphoproliferative disorder: a case series of nine patients. Transpl Int 2013; 26:616–22. [DOI] [PubMed] [Google Scholar]
- 4.Usmani SZ, Weiss BM, Plesner T, et al. Clinical efficacy of daratumumab monotherapy in patients with heavily pretreated elapsed or refractory multiple myeloma. Blood 2016; 128:37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chari A, Suvannasankha A, Fay JW, et al. CASTOR Investigators. Daratumumab plus pomalidomide and dexamethasone in relapsed and/or refractory multiple myeloma. Blood 2017; 130:974–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Palumbo A, Chanan-Khan A, Weisel K, et al. CASTOR Investigators. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med 2016; 375:754–66. [DOI] [PubMed] [Google Scholar]
- 7.Klyuchnikov E, von Pein UM, Ayuk F, et al. Daratumumab is an effective and safe salvage therapy in relapsed/refractory patients with multiple myeloma after allogeneic stem cell transplantation. Blood 2016; 128:3437. [Google Scholar]
- 8.Sharpley FA, Fontana M, Gilbertson JA, et al. Amyloidosis diagnosed in solid-organ transplant recipients. Transplantation 2019; 3. 10.1097/TP.0000000000002813. [DOI] [PubMed] [Google Scholar]
- 9.Mikhael JR, Schuster SR, Jimenez-Zepeda VH, et al. Cyclophosphamide-borte-zomib-dexamethasone (CyBorD) produces rapid and complete hematologic response in patients with AL amyloidosis. Blood 2012; 119:4391–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duhamel S, Mohty D, Magne J, et al. Incidence and prevalence of light chain amyloidosis: a population-based study. Blood 2017; 130:5577. [Google Scholar]
- 11.Quock TP, Yan T, Chang E, Guthrie S, Broder MS. Epidemiology of AL amyloidosis: a real-world study using US claims data. Blood Adv 2018; 2:1046–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Comenzo RL, Reece D, Palladini G, et al. Consensus guidelines for the conduct and reporting of clinical trials in systemic light-chain amyloidosis. Leukemia 2012; 26:2317–25. [DOI] [PubMed] [Google Scholar]