

Summary
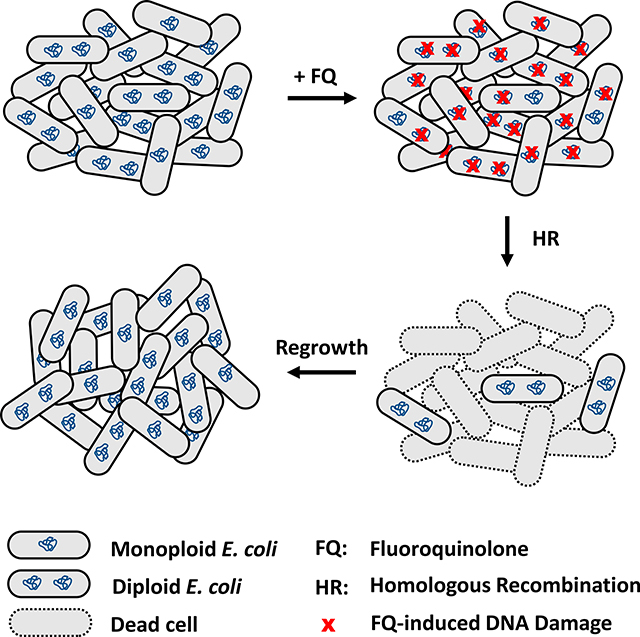
Genetic mutants have demonstrated the importance of homologous recombination (HR) to fluoroquinolone (FQ) persistence, which suggests that single-cell chromosome (Chr) abundance might be a phenotypic variable of importance to persisters. Here, we sorted stationary-phase E. coli based on ploidy and subjected the subpopulations to tolerance assays. Subpopulations sorted to contain diploid cells harbored up to ~40-fold more FQ persisters than those sorted to contain monoploid cells. This association was observed with distinct FQs, in independent environmental conditions, and with more than one strain of E. coli (MG1655, uropathogenic CFT073); but was abolished in HR-deficient strains (ΔrecA and ΔrecB). It was observed that the persister level of monoploid subpopulations exceeded those of ΔrecA and ΔrecB by 10-fold or more, and subsequent high-purity sorting confirmed that observation. Those data suggested the existence of distinct FQ persister subtypes; those that are and are not proficient with HR. Time-lapse microscopy revealed significant differences in initial size and growth dynamics during the post-antibiotic recovery period for persisters from monoploid- and diploid-enriched subpopulations. In addition, non-persisters in monoploid-enriched subpopulations elongated minimally following FQ treatment, resembling previous observations of HR-deficient strains; whereas, non-persisters in diploid-enriched subpopulations on average filamented extensively. Together, these results identify a phenotypic variable with a significant impact on FQ persistence, establish the existence of more than one type of persister to the same antibiotic in an isogenic culture, and reveal roles for RecA and RecB in FQ persistence even in the absence of homologous chromosomes.
Keywords: Persister, UPEC, Levofloxacin, Ciprofloxacin, Chromosome Number, Homologous Recombination, Stationary Phase
Graphical Abstract
eTOC Paragraph
Using cell sorting, antibiotic tolerance assays, genetics, and time-lapse microscopy, Murawski and Brynildsen demonstrate that nongrowing bacteria with two chromosomes are up to 40-fold more likely to be fluoroquinolone persisters than cells with one chromosome due to their ability to perform homologous recombination between sister chromosomes.
Introduction
Persisters are phenotypic variants in clonal populations that can transiently tolerate lethal doses of antibiotics1,2. Their presence is indicated on biphasic-killing curves, where normal cells are killed rapidly and persisters are killed more slowly by antibiotic treatment2,3. Persisters can arise from both growing and non-growing bacterial subpopulations4–6, and are particularly enriched in growth-inhibited cultures4,5,7, which may be of clinical concern since growth-inhibited bacteria are often the cause of chronic, relapsing infections that remain the most difficult to treat1,8. Further, recent work has shown that persisters can directly facilitate the development of antibiotic resistance9–11, which deepens concerns about their potential impact on global health.
Fluoroquinolones (FQs) are a class of antibiotics that has the capacity to kill both growing and non-growing bacteria1,8, yet the efficacy of FQs against non-growing populations wanes in comparison to growing populations4,7. FQs (e.g., levofloxacin (LEVO), ciprofloxacin (CIP)) function by trapping DNA-bound DNA gyrase and topoisomerase IV in a ternary complex along with the DNA, preventing the religation of the 5’ overhangs initially introduced by the topoisomerase12. These cleaved complexes can be converted into free single-stranded and double-stranded DNA breaks (DSBs) during replicational and transcriptional processes, compromising culturability if not repaired13–16. The main mechanism of DSB repair in Escherichia coli is homologous recombination (HR), which is mediated by the SOS response, involves a number proteins (e.g., recA, recB, ruvA, recG9,17,18), and requires the presence of a nearly identical genetic sequence for accurate repair19.
Persisters were previously thought to survive treatment due to the inactivity of antibiotic targets3,5,20; however, recent studies have shown that FQ persisters experience comparable amounts of DNA damage as their antibiotic-susceptible kin and form filaments during recovery from treatment before dividing into progeny that replicate normally6,7,9. A closer inspection of recovery periods revealed that the relative timing between DNA repair and growth-related processes may dictate survival from FQ treatment21. Furthermore, previous works from both growing and non-growing bacteria demonstrated that FQ persisters rely on the SOS response, including mediator lexA, and DNA repair proteins, such as recA, recB, recN, ruvA, and recG, for survival6,7,9,21–23.
Given the genetic evidence implicating HR in FQ persistence, we hypothesized that chromosome abundance may be a phenotypic variable of importance to persister survival. Conceivably, cells with a single chromosome (monoploid) should not be able to weather a DSB well, due to the lack of an additional genomic copy of the damaged locus required for HR. Although, it should be noted that partially homologous DNA can be used as a template, albeit at a lower efficiency24. Analogously, cells with more than two chromosomes should weather DSBs better than diploid cells, because their likelihood of receiving damage to all copies of a locus decreases as the genome abundance increases. However, monoploid, diploid, and polyploid cells differ in more than the copy number of their chromosome; such cells can be physiologically different25–28 and could exhibit varying levels of drug import, export, or topoisomerase activity, which could lead to different effects from the same FQ treatment. Importantly, though the significance of HR to FQ persistence has been established with genetic mutants, direct evidence supporting a role for ploidy in FQ persistence of wild-type populations has been lacking.
To probe the extent to which chromosome number matters to FQ persisters, we examined stationary-phase populations of E. coli, because previous work had shown them to exhibit subpopulations with well resolved chromosome numbers9,29. We used FACS to sort stationary-phase cells based on DNA content as determined by staining with Hoechst 33342 and observed that LEVO persisters arise at a higher frequency from cells harboring at least two genomes. Further, we found that the dependence of persistence on chromosome number is shared among distinct FQs and distinct environmental conditions; translates to uropathogenic E. coli (UPEC); and is abolished in HR-deficient mutants (ΔrecA and ΔrecB). An unresolved question from that data was why subpopulations of monoploid cells, which lack sister chromosomes to conduct HR, exhibited 10-fold or greater survival than ΔrecA and ΔrecB, which are HR-deficient mutants. We confirmed this difference with an origin-reporter strain30,31, which served as an independent method to count chromosomes, and high-purity sorting experiments, which suggested the existence of two distinct FQ persister subtypes; those that are HR-proficient and those that are HR-impaired, because they lack a homologous sister chromosome. To confirm the existence of distinct persister subtypes, we employed time-lapse microscopy. Untreated cells from monoploid-enriched and diploid-enriched subpopulations were observed to resume growth similarly. Following FQ treatment, non-persisters from monoploid-enriched subpopulations elongated minimally, which resembled previous observations on the post-FQ recovery of ΔrecA cells9; whereas, non-persisters from diploid-enriched subpopulations on average filamented extensively. Importantly, persisters from monoploid- and diploid-enriched subpopulations differed significantly in both their initial size and growth dynamics during recovery, with persisters from diploid-enriched subpopulations increasing in size faster and dividing earlier than those from monoploid-enriched subpopulations. Collectively, these results identify ploidy as a phenotypic variable with a significant impact on FQ persistence, establish the existence of functionally-distinct persister subtypes to the same antibiotic in an isogenic culture, and reveal that RecA and RecB are used to enhance FQ persistence even in the absence of a homologous sister chromosome. Further, these data suggest that agents that facilitate reductive division and induce monoploidy in bacteria could help reduce the threat posed by FQ persisters.
Results
LEVOFLOXACIN PERSISTERS DEMONSTRATE BIPHASIC KILLING
We examined stationary-phase populations of E. coli and elected to use LEVO as a representative FQ. Ofloxacin (OFL) is a common FQ used in persistence studies4,6,7,9,21,32,33, but it is a racemic mixture, and the biologically-active enantiomer of OFL is LEVO, which motivated our choice of LEVO for this study. Dose response curves for LEVO were comparable to OFL above 1 μg/mL (Figure S1A). Moreover, we used a concentration of 5 μg/mL (~ 250x the minimum inhibitory concentration (MIC), Figure S1B), which generated classic biphasic kill curves and ensured that only persisters remained after 5 h of treatment, as previously reported7,9 (Figure S1C). At concentrations that greatly exceed the MIC, DSBs are expected to be the predominant form of DNA damage14–16. We show that similar to OFL persisters9, LEVO persisters also require HR machinery, including recA, recB, ruvA, and recG, in order to survive, and complementation of these mutants restored persistence to wild-type levels (Figure S1D–F).
STAINING AND SORTING DO NOT ALTER FQ PERSISTENCE
Given the dependence of LEVO persistence on HR, we reasoned that cells harboring two or more chromosomes should be more likely to be persisters than cells harboring only one chromosome due to their ability to use a sister chromosome for HR to repair FQ-induced DNA damage. To test this hypothesis, we sought to use fluorescent, DNA-specific nucleic acid stains and fluorescence-activated cells sorting (FACS). Previously our lab used PicoGreen9,33, which is a cell-impermeant double-stranded DNA-specific (dsDNA) stain that requires fixation and permeabilization for DNA quantification, to demonstrate that single cells in stationary phase have unit numbers of chromosomes9. This is in contrast to exponentially-growing bacteria that often have partially-replicated chromosomes in addition to complete copies of the genome21,29. Because fixation is destructive and kills all of the cells, we identified Hoechst 33342 as a dsDNA-specific stain that could be used to quantify chromosomal content in single, living E. coli cells. As depicted in Figure S2A, staining with 5 μg/mL Hoechst 33342 recapitulates the chromosomal distribution observed using PicoGreen in both fixed and live cells. Figure 1A demonstrates that it does not interfere with the persistence phenotype, nor does it impact culturability of stationary-phase E. coli, as illustrated by the H2O-treated and Hoechst 33342-only controls.
Figure 1. Ploidy impacts persistence to fluoroquinolones in stationary-phase populations.

(A) Persistence to LEVO is not influenced by staining with 5 μg/mL Hoechst 33342 (n = 4). (B) Cell sorting does not influence the persistence phenotype, where unsorted cells were diluted to the same density as sorted cells (n = 7). (C) Representative DNA histogram and gating strategy indicating the number of chromosomes in a stationary-phase population of wild-type E. coli stained with 5 μg/mL Hoechst 33342. (D) Cells sorted to contain 2Chr (as determined by Hoechst 33342 staining) are > 16 times as likely to be LEVO persisters than cells sorted to contain 1Chr (n = 7). Untreated samples demonstrate that any survival differences observed can be attributed to treatment with 5 μg/mL LEVO (n = 7). Statistical significance was initially determined using a one-way ANOVA (F(3,24) = 12.0, p < 0.0001) at the 5 h timepoint, because at that time point, persisters are the only remaining CFUs. Tukey HSD post-hoc test showed a significant difference at 5 h when comparing the means of 1Chr vs. 2Chr (p = 0.0003), 1Chr vs. ≥2Chr (p = 0.0001), and 1Chr vs. Total Sort (p = 0.0007). 1Chr, 2Chr, and >2Chr indicate chromosome number as determined by experiments presented in Figure 2. See also Figures S1 and S2. Representative gating strategy is indicated by a, b, and c. Error bars portray ± SEM. All replicates were independent biological replicates. * indicates statistical significance.
To demonstrate that FACS does not alter the persistence phenotype, we used controls that contained unstained or stained cells that were not sorted but were diluted to approximately the same density as sorted cells. Persistence levels from those samples were comparable and not statistically different than those from bacteria that had been stained and sorted (Total Sort) (Figure 1B). Moreover, as depicted in Figure S2B, pre-sort and post-sort controls further demonstrated that Hoechst 33342 staining and the time required for sorting do not alter LEVO persister levels. Together, these results show that Hoechst 33342 and using it in FACS experiments does not have a measurable impact on persistence to FQs in stationary-phase E. coli populations.
FQ PERSISTENCE DIFFERS IN SUBPOPULATIONS SORTED BASED ON CHROMOSOMAL ABUNDANCE
To assess the impact of chromosome number on persistence, MG1655 was grown to stationary phase in M9 minimal media with glucose as the sole carbon source. Cells were stained with 5 μg/mL Hoechst 33342 and diluted in preparation for FACS. A conservative gating strategy, as depicted in Figure 1C, was employed to reduce potential cell leakage between gates. Cells were collected in an equal volume of sterile-filtered spent media and treated with 5 μg/mL LEVO. Figure 1D shows that the b-gate subpopulation (2Chr) contained >16 times as many LEVO persisters than the a-gate subpopulation (1Chr). Moreover, the Total Sort and c-gate subpopulation (>2Chr) also demonstrated significant increases in persistence when compared to the a-gate population. Untreated controls demonstrated minimal survival differences among the sorted subpopulations (Figure 1D), which suggested that the survival differences observed in treated populations depended on the presence of LEVO.
PICOGREEN VERIFICATION OF HOECHST 33342-SORTED CELLS
To verify that the subpopulations segregated based on Hoechst 33342 staining represented cells with 1Chr (a-gate), 2Chr (b-gate), and >2Chr (c-gate), we restained the sorted subpopulations with PicoGreen. We observed excellent agreement at the single-cell level between Hoechst 33342 and PicoGreen staining with the a-gate and b-gate subpopulations (Figure 2, Sort a and b). The c-gate subpopulation was enriched in single cells harboring 4 Chr as compared to the Total population (19.0% compared to 4.6%); however, a substantial number of 1Chr and 2Chr cells were also observed in that subpopulation (Figure 2, Sort c and Total). We anticipate that some cell clumping occurred during the sorting of these samples due to the fixation buffer. Attempts to remedy this are detailed in the Methods; however, none were deemed sufficiently successful. Together, these data demonstrated that Hoechst 33342 staining recapitulates what would be observed with PicoGreen staining at the single-cell level for subpopulations corresponding to those with 1Chr and 2Chr. Due to the imprecision associated with c-gate subpopulations, we elected to focus on 1Chr- and 2Chr-enriched subpopulations (a- and b-gate) for the remainder of this study.
Figure 2. PicoGreen verification of sort fidelity.

Restaining Hoechst 33342-sorted cells with PicoGreen demonstrated good separation of 1Chr- and 2Chr-sorted samples, with minimal gate leakage. The subpopulation sorted to contain >2Chr (Sort c) had an enrichment in cells with four chromosomes; however, 1Chr and 2Chr populations remained abundant. Data represents the means of four biological replicates, where each replicate included the analysis of 50,000 cells.
PLOIDY DEPENDENCE EXTENDS TO CIP, UPEC, AND DIFFERENT GROWTH CONDITIONS
To assess the generality of our results to other FQs, we performed analogous experiments with CIP. As depicted in Figure 3A–B, CIP persisters displayed a similar dependence on chromosomal content, with a >6-fold reduction in persistence for the 1Chr-enriched subpopulation compared to the 2Chr-enriched subpopulation. Controls indicated that neither Hoechst 33342-staining nor cell sorting impacted CIP persistence assays (Figure S2F–J).
Figure 3. Ploidy dependence extends to ciprofloxacin, UPEC, and different growth conditions.

(A) Representative DNA histogram of stationary-phase wild-type E. coli stained with 5 μg/mL Hoechst 33342. (B) Cells that were FACS-sorted to contain 2Chr exhibit >6-fold higher survival when treated with 1 μg/mL CIP than cells harboring 1Chr (n = 5). Statistical significance was initially determined using a one-way ANOVA (F(2,12) = 16.0, p < 0.001) at the 5 h timepoint. Tukey HSD post-hoc test showed a significant difference at 5 h when comparing the means of 1Chr vs. 2Chr (p = 0.0008) and 1Chr vs. Total Sort (p = 0.0012). (C) Representative DNA histogram of uropathogenic E. coli strain CFT073 (UPEC) grown to stationary phase and stained with 5 μg/mL Hoechst 33342. (D) UPEC harboring 1Chr were >13-fold or >8-fold less likely to survive 5 μg/mL LEVO (n = 7) or 1 μg/mL CIP (n = 6) treatment, respectively, than cells harboring 2Chr. Statistical significance was initially determined using a one-way ANOVA (LEVO: F(2,18) = 4.4, p < 0.03; CIP: F(2,15) = 4.1, p < 0.04). Tukey HSD post-hoc test showed a significant difference when comparing the means of 1Chr vs. 2Chr for both the LEVO (p = 0.023) and CIP (p = 0.031) treatments. (E) Wild-type E. coli was grown to stationary phase in minimal media with 20 mM glycerol as the sole carbon source and stained with 5 μg/mL Hoechst 33342 to determine genomic content. (F) Cells grown in minimal glycerol media and sorted to have 2Chr were >6-fold more likely to survive treatment with 5 μg/mL LEVO than cells sorted to have 1Chr (n = 3). Statistical significance was initially determined using a one-way ANOVA (F(2,6) = 7.0, p < 0.03). Tukey HSD post-hoc test showed a significant difference when comparing the means of 1Chr vs. 2Chr (p = 0.024). Biphasic kill curves of UPEC treated with 5 μg/mL LEVO or 1 μg/mL CIP and wild-type E. coli grown in minimal glycerol media and treated with 5 μg/mL LEVO can be found in Figure S3. See also Figures S2 and S3. 1Chr and 2Chr indicate chromosome number as determined by experiments in Figure 2. Representative gating strategy is indicated by a and b. Error bars portray ± SEM. All replicates were independent biological replicates. * indicates statistical significance.
To test whether chromosome abundance influences FQ persistence in pathogenic bacteria, we performed experiments with UPEC. Strain CFT073 is quite distinct from that of MG1655; it’s genome is ~590,000 bp larger than MG1655 and it encodes 1623 unique proteins, four large pathogenicity islands, five cryptic prophages, and several toxins34. When we sorted UPEC based on chromosomal content, we found that persistence to LEVO or CIP in the 1Chr-enriched subpopulation were >13- or >8-fold lower, respectively, than cells in the 2Chr-enriched subpopulation (Figure 3C–D). Moreover, similar to MG1655, we demonstrated that staining with Hoechst 33342, cell sorting, and the time required for sorting did not alter the persistence phenotype of UPEC under the conditions used here (Figure S3A–G).
To assess the generality of this phenomenon to altered growth conditions, we performed experiments on E. coli grown to stationary phase in minimal glycerol media. Similar to E. coli grown in minimal glucose media, we observed a >6-fold enhancement in persisters arising from cells sorted to harbor 2Chr compared to 1Chr (Figure 3E–F). Further, we demonstrated that under these minimal glycerol conditions, cells exhibited biphasic killing when treated with LEVO, and the time required for sorting did not alter the persistence phenotype (Figure S3H–M). Collectively, these data demonstrate the generality of the impact of ploidy on persistence to FQs in growth-inhibited populations.
FQ PERSISTENCE OF HR-DEFICIENT STRAINS IS INDEPENDENT OF PLOIDY
To directly test our hypothesis that monoploid cells give rise to significantly fewer persisters than diploid cells due to their inability to conduct HR with a sister chromosome, we performed analogous experiments with HR-deficient mutants. We reasoned that in such mutants, we should not observe differences in persistence between 1Chr- and 2Chr-enriched subpopulations. When recA or recB were deleted, the persister levels were precipitously lower than wild-type and indistinguishable between 1Chr- and 2Chr-enriched subpopulations (Figure 4, Figure S4). These data demonstrated that HR was required to observe differences in survival between the 1Chr- and 2Chr-enriched subpopulations. We note that for ΔrecA, we sorted the peak that was less fluorescent than the a-gate subpopulation but did not observe any culturable cells when plated prior to treatment. This subpopulation has been observed previously, and represents cells without a nucleoid35.
Figure 4. Chromosomal association with FQ persistence eliminated in HR mutants.

(A) Representative DNA histogram of a stationary-phase population of MG1655 ΔrecA stained with 5 μg/mL Hoechst 33342. The small peak (arrow) that is less fluorescent than the 1Chr peak was not culturable after sorting and has been previously described as cells without nucleoids35. (B) When sorted and treated with 5 μg/mL LEVO, the association between chromosomal content and persistence was eliminated in a MG1655 ΔrecA mutant (n = 3). (C) Representative DNA histogram of a stationary-phase population of MG1655 ΔrecB stained with 5 μg/mL Hoechst 33342. (D) No association between chromosomal content and persistence was observed in a MG1655 ΔrecB mutant treated with 5 μg/mL LEVO (n = 3). Raw CFU/mL values and additional controls can be found in Figure S4. 1Chr and 2Chr indicate chromosome number as determined by experiments presented in Figure 2. Representative gating strategies are indicated by a and b. Error bars portray ± SEM. All replicates were independent biological replicates. No statistical significance was achieved as determined using a one-way ANOVA (ΔrecA: F(2,6) = 0.15, p = 0.86; ΔrecB: F(2,6) = 0.38, p = 0.70).
ORIGIN REPORTER AND HIGH-PURITY SORTING SUGGEST THE EXISTENCE OF FQ PERSISTER SUBTYPES
Although 1Chr-enriched subpopulations have significantly lower FQ persister levels than their 2Chr-counterparts and that difference is absent in HR-deficient mutants, we wondered why the survival of 1Chr-enriched subpopulations was significantly greater than that of HR-deficient mutants (Figures 1D and Figures 4B,D). We considered whether 1Chr-enriched subpopulations failed to more precisely phenocopy HR-deficient mutants because of the small abundance of 2Chr and >2Chr cells in that sorted subpopulation (~3% as measured with PicoGreen, Figure 2). Since PicoGreen and Hoechst 33342 are both nucleic acid stains, we sought to assess the purity of sorted subpopulations with an independent method. To accomplish that, we employed an origin-reporter strain based on the ParB-parS tagging system and fluorescence microscopy30,31,36. When supplied in trans, GFP-ParB binds to the genomically-integrated parS site (located near the origin of replication31) and polymerizes along the adjacent DNA, which produces a fluorescent foci at that site30,31. Because we are using stationary-phase populations where cells have unit chromosomes9,29 (Figure 1), the abundance of origin foci in a single cell should indicate the number of chromosomes. Controls demonstrated that the foci we observed required both a genomically-integrated parS site and GFP-ParB (Figure S5). We therefore grew this origin reporter to stationary phase, fixed the cells in paraformaldehyde (PFA), stained with 5 μg/mL Hoechst 33342, and sorted the cells with the gating strategy shown in Figure 5A. Using confocal fluorescence microscopy, we visualized each sorted subpopulation and quantified the number of foci within single cells. We found that approximately 90% of cells in the a-gate subpopulation contained 1Chr (Figure 5B, ‘Original Sorting’), whereas for the b-gate subpopulation, ~89% were 2Chr cells. In agreement with the PicoGreen experiments, the c-gate subpopulation contained a mix of cells containing 1–5 chromosomes, with an enrichment in cells containing >2 Chr when compared to a Total Sort population (Figure 2). Notably, the purities of the 1Chr- and 2Chr-enriched subpopulations were lower when assessed by the origin reporter than when assessed by PicoGreen.
Figure 5. Verification of sort fidelity using an origin reporter strain.

(A) Representative DNA histogram and sort gates (indicated by a, b, and c) of an origin-reporter strain grown to stationary phase and stained with 5 μg/mL Hoechst 33342. This strain has fluorescent foci in single cells that are indicative of the number of chromosomes and was (B) used to quantify sorted sample purities by counting foci using confocal fluorescence microscopy. Quantification of foci sorted using two different Sort Precision Modes are portrayed (see Methods). Representative phase contrast, GFP, and merged images are displayed. Over 400 cells were quantified per sort gate using microscopy across three different sorts, where each was on a different biological replicate. 1Chr: number of cells with 1 foci; 2Chr: number of cells with 2 foci; >2Chr: number of cells with more than 2 foci.; U: undeterminable, which were cells having either diffuse green fluorescence or no fluorescence. We note that we did not sort the >2Chr population using the high-purity sort method. See Figure S5 for origin reporter controls. Scale bar represents 5 μM.
Because the origin reporter indicated purities of sorted subpopulations to be around 90%, we sought to adjust the sorting procedure to assess whether increased purity could be obtained. To accomplish this, we used a high-purity sorting method, which in general sacrifices yield and time to obtain higher purity (see Methods). For the a-gate, the purity increased to 98.3% 1Chr cells, whereas for the b-gate, purity increased to 91.5% 2Chr cells (Figure 5B, ‘High-Purity Sorting’). Importantly, when persistence to LEVO was quantified, the survival of the 1Chr-enriched subpopulation using the high-purity method did not differ significantly from the survival depicted in Figure 1D, despite the decrease in 2Chr and undetermined cells from 10.3% to 1.7% (Figure 6A). If considerable amounts of persisters in a-gate samples were not from 1Chr cells, then the persister level should have scaled with the 2Chr and undetermined cell populations and decreased by approximately 6-fold, which did not occur. Further, with survival in the 1Chr-enriched subpopulation at approximately 1%, nearly 60% of the 2Chr and undetermined cells in that sorted subpopulation would need to survive, which is an exceedingly high level considering that the highest survival we measured from the high-purity samples was 8% for the 2Chr-enriched subpopulation. These data suggest that persisters do arise from 1Chr cells at frequencies higher than ΔrecA and ΔrecB mutants, which occur at < 0.1% (Figure 6B). Further, the data point to the existence of functionally-distinct FQ persister subtypes: those that arise from cells with at least 2Chr and are HR-proficient, and others that arise from 1Chr cells and have compromised recombination capabilities due to the lack of a second chromosomal copy.
Figure 6. 1Chr persisters survive LEVO treatment at higher frequencies than ΔrecA and ΔrecB mutants.

(A) Five hour survival of wild-type E. coli stained with 5 μg/mL Hoechst 33342, sorted using original (n = 7) and high-purity sorting (n = 3) modes, and treated with 5 μg/mL LEVO demonstrates no significant difference in persistence levels, as determined using a one-way ANOVA (F(5,24) = 2.50, p > 0.05). (B) When compared to the survival of 1Chr-sorted ΔrecA (n = 3) and 1Chr-sorted ΔrecB mutants (n = 3), a significantly higher proportion of 1Chr-sorted wild-type cells (n = 7) survive treatment with 5 μg/mL LEVO. Statistical significance was initially determined using a one-way ANOVA (F(2,10) = 25.9, p = 0.0001). Tukey HSD post-hoc test showed a significant difference when comparing the means of 1Chr wild-type vs. 1Chr ΔrecA (p < 0.01) and vs. 1Chr ΔrecB (p <0.001). See also Figure S6. * indicates statistical significance.
TIME-LAPSE MICROSCOPY CONFIRMS DISTINCT FQ PERSISTER SUBTYPES
To investigate the existence of functionally-distinct FQ persister subtypes within the same bacterial population, we employed time-lapse microscopy to observe the recovery of 1Chr- and 2Chr-sorted subpopulations that had been treated with LEVO. Controls confirmed that LEVO treatment prior to sorting did not alter the Hoechst 33342 histograms appreciably, nor did it impact persister levels when compared to sorting prior to LEVO treatment (Figure S6). We attempted to observe the FQ recovery period of the origin reporter strain, so that we could observe the number of foci in persisters at the onset. However, we observed that fluorescent ParB-GFP foci become perturbed by DNA damage (Figure S7), which is consistent with previous results in Caulobacter crescentus37. Notably, when this was quantified for 1Chr- and 2Chr-sorted subpopulations, purities of approximately 90% dropped to approximately 15%, with large gains in the category of cells with undetermined chromosomal abundances. Although this prevented us from using the origin reporter to quantify the initial foci abundance in persisters as they recovered, it provided evidence that DNA damage occurred in the stationary-phase cells under investigation, regardless of whether they were monoploid or diploid, and that the DNA damage was being processed prior to exposure to fresh nutrients.
Using Hoechst 33342 staining alone as a metric for chromosomal content, we observed a total of 2,875 LEVO-treated 1Chr-sorted cells and 3,288 LEVO-treated 2Chr-sorted cells using time lapse microscopy (Videos S1–S2). Representative phase contrast images of the recovery of each sorted subpopulation are shown in Figure 7A–B. Non-persisters were identified as cells that failed to produce progeny that continued to grow within the 16 h timeframe of the experiment, whereas persisters were cells that produced daughter cells that continued to replicate normally. In all, six of the 1Chr-sorted cells became persisters (survival fraction = 0.002), whereas 262 of the 2Chr-sorted cells became persisters (survival fraction = 0.080), which represents a survival difference of approximately 40-fold.
Figure 7. Time-lapse microscopy of Hoechst-sorted monoploid and diploid E. coli.

Representative phase contrast images of recovering LEVO-treated cells sorted to contain (A) 1Chr or (B) 2Chr, as determined by staining with 5 μg/mL Hoechst 33342. Red arrows indicate persisters. In total, we observed a total of 2,875 LEVO-treated 1Chr-sorted cells and 3,288 LEVO-treated 2Chr-sorted cells. 1Chr and 2Chr persisters displayed significantly distinct (C) growth dynamics (Repeated Measures ANOVA with main effects of chromosome number and time, F(1,45) = 6.93, p <0.0001), (C panel inset) initial cell sizes (one-way ANOVA, F(1,55) = 25.4, p < 10−5), (D) rate of expansion through 240 min of recovery (one-way ANOVA, F(1,57) = 8.03, p < 0.01) and (E) time at which they divided during recovery from LEVO treatment (one-way ANOVA, F(1,55) = 8.2, p < 0.01), but (F) did not differ in the length at which they divided (one-way ANOVA, F(1,55) = 1.85, p = 0.18). (G) 1Chr and 2Chr non-persisters similarly displayed unique growth dynamics (Repeated Measures ANOVA with main effects of chromosome number and time, F(1,45) = 32.8, p <0.0001), and of the ~20% of each sorted subpopulation that lysed following treatment, (H) the length at which lysis occurred varied based on chromosomal content (one-way ANOVA, F(1,43) = 28.3, p < 10−5), but (I) the time of lysis during recovery did not (one-way ANOVA, F(1,43) = 1.31, p = 0.26). Untreated cells had similar (J) growth dynamics (Repeated Measures ANOVA with main effects of chromosome number and time, F(6,47) = 0.982, p = 0.45), (K) rate of expansion through 96 min of recovery (one-way ANOVA, F(1,65) = 0.15, p = 0.70), (L) time of cell division (one-way ANOVA, F(1,65) = 3.47, p = 0.07), and (M) length at cell division (one-way ANOVA, F(1,65) = 0.67, p = 0.42), regardless of chromosomal content. Most untreated 1Chr and 2Chr cells divided by three hours. See also Figures S6–S7 and Videos S1–S4. 1Chr and 2Chr indicate chromosome number as determined by staining with Hoechst 33342. Scale bar represents 5 μM. * indicates statistical significance.
We used MicrobeJ to quantify the growth of all six 1Chr persisters and 53 of the 2Chr persisters up until their first cell division (Figure 7C–F). Importantly, 1Chr and 2Chr persisters differed significantly in their initial size, rate of expansion, and growth dynamics during recovery, as well as the time of first cell division (Figures 7C–E), but did not differ in the length at which they divided (Figure 7F). For the non-persisters, 1Chr and 2Chr cells exhibited vastly different morphologies upon recovery from LEVO treatment (Figure 7G). 2Chr non-persisters (n = 111) displayed a variety of outcomes, ranging from lack of growth to filamentation up to >20x their original cell length. In contrast, the vast majority of 1Chr non-persisters (n = 108) failed to elongate much and resembled the recovery dynamics of ΔrecA non-persisters observed previously9. In the 2Chr subpopulation, non-persisters filamented on average up to 8 μM before lysing or ceasing to expand; whereas cells in the 1Chr subpopulation grew on average up to 2 μM before lysing or failing to expand. We also note that lysis occurred in 21.3% and 19.8% of the 1Chr and 2Chr non-persisters, respectively (Figures 7H–I). Interestingly, 1Chr and 2Chr untreated controls resumed growth comparably with similar times to and lengths at division, as well as indistinguishable rates of expansion (Videos S3–S4, Figures 7J–M). Notably, 1Chr persisters divided on average ~586 minutes into recovery at ~8 μM in length, whereas 1Chr untreated cells divided around ~152 minutes at ~3 μM in length, which suggests that 1Chr persisters were impacted by the LEVO treatment. Collectively, these data indicate that 1Chr and 2Chr persisters can be distinguished based on their responses to LEVO treatment during the recovery period, and that the majority of 1Chr cells that die exhibit physiology during the recovery period that resembles that of HR-deficient strains.
Discussion
Antibiotic treatment failure is of great concern to public health, and one of its potential drivers is antibiotic persistence, where subpopulations of bacteria that are genetically susceptible to the prescribed antibiotic are tolerant to treatments where drug concentrations exceed their MIC for extended periods38. More disheartening is that antibiotic persistence can lead to antibiotic resistance development9,10, exacerbating an internationally-recognized public health crisis39. Identifying mediators of persistence can allow for better understanding of persister survival and suggest avenues for therapeutic intervention3–5,32,33.
While some genetic mediators and phenotypic features impact persistence to more than one drug, others are specific to the antibiotic used. For instance, the TisB toxin was found to be important for CIP persisters in growing populations, but not impactful for AMP or streptomycin persisters in the same culture40. Studies of carbon source transitions revealed both shared and distinct mediators of AMP and OFL persister formation41,42. Additionally, persisters arising from exponentially-growing cultures treated with either AMP or OFL exhibited vastly different morphologies and physiologies: Goormaghtigh and Van Melderen6 showed that OFL persisters underwent massive filamentation after antibiotic exposure, whereas Balaban and colleagues3 demonstrated that AMP persisters appeared to undergo normal rounds of growth and division post-treatment. These studies suggest that the mechanism of action of each drug should be considered when understanding how persisters survive treatment.
Beyond the drug administered, the nutritional status of the environment also influences persistence. For example, the age of stationary-phase cultures and the subsequent inoculation of these cultures into different medias influenced persister levels43. While the TisB toxin was found to be important for CIP persister formation in exponentially-growing cultures, the same results were not observed in stationary-phase populations7,40. Moreover, starvation-assisted recovery after OFL treatment led to nearly 100% survival of persisters arising from nongrowing populations but not from growing cultures21. Thus, both the antibiotic and a bacterium’s environment are important features that can impact persister levels.
Here we focused on stationary-phase populations of E. coli that were treated with FQs. Previous works have discovered that persisters in non-growing bacterial populations were impacted by indole signaling44, GTPase Obg activity45, mutations in DNA repair7,9,23, and protein synthesis levels46. In addition, we found that FQ persisters in stationary-phase populations exhibited similar DNA damage responses when compared to their antibiotic-susceptible kin7,9. Further, the ability of persisters to repair DNA damage during the post-antibiotic recovery period was found to be critical to their survival7,21. While these findings contribute to a greater understanding of persistence in stationary phase, the defining feature of how or why FQ persisters survive has remained elusive.
In this study, we add to the understanding of FQ persistence by characterizing ploidy as a phenotypic characteristic of importance to FQ persisters in non-growing populations. Populations enriched with bacteria containing 2Chr were up to ~40x more likely to survive LEVO treatment than populations enriched with 1Chr cells, and analogous results were seen with a different FQ, different E. coli strain, and cultures that had been grown on a different substrate (Figures 1 and 3). Using time-lapse microscopy (Figure 7, Videos S1–S2), we obtained images of persisters arising from both 1Chr- and 2Chr subpopulations, and observed significant differences in their growth dynamics during the post-antibiotic recovery period (Figure 7C–E). In addition, the physiologies of the non-persisters varied based on chromosomal content with heterogenous responses from 2Chr non-persisters post-treatment, whereas 1Chr non-persisters almost uniformly failed to expand appreciably (Figure 7G), which bore a striking resemblance to FQ-treated ΔrecA9. Indeed, our central hypothesis was that chromosomal content would influence FQ persistence due to the capacity of cells with at least 2Chr to perform HR, which was supported by experiments with HR-deficient mutants (ΔrecA, ΔrecB), where persistence was not influenced by ploidy (Figure 4). Collectively, these data suggest that an additional copy of the chromosome is important for persistence to FQs in E. coli, because it allows for a major DNA repair system to be efficiently used.
Initially, we postulated that cells harboring 1Chr would not be able to weather a FQ-induced DSB, the type of damage expected at the FQ concentrations administered here, any better than HR-deficient mutants. However, monoploid cell survival exceeded that of ΔrecA and ΔrecB by >10-fold (Figure 6B), which suggested that RecA and RecB can play a role in persister survival that does not involve using an undamaged sister chromosome as a template during HR (interchromosomal HR). Mechanisms that monoploid cells use to survive FQ treatment could involve alternate repair pathways or recombination on partially homologous or duplicated regions. RecA, for example, has been shown to help stabilize blocked replication forks in DNA-damaged bacteria, suggesting a role in DNA repair that does not involve interchromosomal HR, but rather involves uvr proteins47,48. Alternatively, repair could happen between duplicated genes, which occur at a rate of 10−2 – 10−4 within a population49,50, or semi-homologous chromosomal regions, including rRNA operons49,51, prophages52, or insertion sequences53–55. Such repair could happen in a recA-dependent or recA-independent manner, depending on the length of DNA homology56–58, but may result in genomic rearrangements including inversions or deletions52,59. The survival of 1Chr persisters could also be explained if the nature of the DNA damage is found to be singe-strand breaks, as recA plays a role in the repair of single-strand damage60. Delineating the mechanism responsible for how monoploid, HR-impaired bacteria survive FQ treatment better than HR-deficient mutants is an interesting topic for future work.
DNA content was previously assayed in exponentially-growing populations treated with OFL, yet it was observed that genomic copy number, as measured by HU-GFP fluorescence, did not alter FQ persistence6. An important distinction between non-growing and exponentially-growing cultures is that the former is starved long enough to yield cells with unit numbers of chromosomes, which will include monoploid cells9,29 (Figure 1). However, in exponentially-growing cultures, the majority of cells have more than one chromosome and overlapping cell cycles, leading to partially-replicated copies21,29. Therefore, it is understandable that Goormaghtigh and Van Melderen did not observe the chromosomal differences amongst persisters that we observed here. Our experimental conditions allowed us to compare the survival of a monoploid-rich, HR-impaired subpopulation to that of a diploid-rich, HR-proficient subpopulation to determine how harboring more than one copy of genomic DNA affects survival.
We speculate that ploidy could play a role in FQ persistence in any bacteria where HR is the dominant DNA repair method. For example, UPEC is a clinical pathogen that uses HR as its foremost means of DNA repair, and chromosomal content appears to influence its level of FQ persistence (Figure 3). As a clinical pathogen, UPEC is a leading cause of UTIs in women, potentially causing biofilm formation, pyelonephritis, and/or urosepsis if insufficiently treated, and FQs are one treatment strategy for drug-susceptible UPEC61. Moreover, approximately 25% of women who experience a UTI caused by UPEC will be plagued by a second UTI within 6 months62, and persisters have been found to be a cause of reinfection in these women63. Since FQs are a potential treatment for clinical UTIs caused by UPEC, identifying an adjuvant that fosters monoploidy could prove a useful strategy for eradicating recurrent UTIs.
Beyond persistence, chromosome abundance can impact microbial physiology in different ways. For example, Haloferax volcanii, an archaea, is polyploid and breaks down its extra copies of DNA for phosphate during starvation conditions64. The ploidy status of Deinococcus radiodurans65 and Thermus thermophilus66 may allow for protection against radiation or thermal stresses, respectively. Further, haploid Lactococcous lactis were found to be more sensitive to ultraviolet light when compared to a similar diploid strain67, and the ploidy status of yeast influences their ability to thrive under different environmental conditions25,26. Ploidy status is thus an important phenotypic variable in bacterial populations that is often under-appreciated, and additional research into how monoploid and diploid cells differ phenotypically is an interesting avenue for future research. We anticipate that its role in bacterial physiology will gain increasing recognition as its impact on important societal issues, such as antibiotic failure studied here, becomes better established.
This study has shown that harboring multiple chromosomes puts non-growing bacteria at a phenotypic advantage after exposure to FQs, which can be attributed to their capacity to perform HR (Figure 4), yet a duplicate genome still does not guarantee survival. Rather, the number of DSBs per genome or the location of DNA damage on the chromosome may contribute to a diploid cell’s demise. Whether bacteria with >2Chr have even greater survival than diploid cells remains to be determined due to the sorting imprecision we observed with the c-gate (Figures 2 and 5). In addition, what mechanisms enable HR-deficient mutants to yield FQ persisters and whether any of those are used by monoploid, diploid, or polyploid cells remains to be determined. Based on the data and analyses presented here, we postulate that finding an adjuvant that induces reductive division and drives the formation of monoploid cells in non-growing bacterial populations would increase the efficacy of FQs and decrease the number of FQ persisters.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Correspondence and requests for materials should be addressed to and will be fulfilled by the lead contact, Mark P. Brynildsen (mbrynild@princeton.edu).
Materials Availability
Data and materials that support the findings of this study are available from the corresponding author upon request.
Data and Code Availability
All data are included within the paper, and raw data can be requested from the Lead Contact.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Chemicals and Growth Media
All media components, antibiotics, and stains used in this study were purchased from Sigma Aldrich (St. Louis, MO) or Fisher Scientific (Waltham, MA) unless otherwise indicated. All growth media and agar were prepared in MilliQ water (resistivity 18.2 MΩ.cm) and autoclaved at 121°C for 30 min to achieve sterilization except where indicated. LB media (10 g/L tryptone, 5 g/L yeast extract, and 10 g/L NaCl) was prepared from individual components, and LB agar was prepared with Difco pre-mixed LB Miller broth and 15 g/L agar. M9 glucose media was prepared using autoclaved 5x M9 minimal salts (33.9 g/L Na2HPO4, 15 g/L KH2PO4, 5 g/L NH4Cl, 2.5 g/L NaCl), 0.1 mM CaCl2, 2 mM MgSO4, and 10 mM glucose as the sole carbon source. M9 glucose media was filter-sterilized using a 0.22 μm filter upon preparation. All media was used within one week. To obtain sterile spent media, ~15 mL of the same 20 h culture was centrifuged for 10 min at room temperature at 4,000 rpm, and then the supernatant was filtered (0.22 μm).
Stock solutions of AMP, KAN, and isopropyl β-D-q-thiogalactopyranoside (IPTG) were dissolved in autoclaved MilliQ water, LEVO was dissolved in 20 mM NaOH, and CIP was dissolved in 0.2 N HCl. All antibiotics were filter-sterilized with 0.22 μm filters prior to use. For mutant selection and or plasmid retention, AMP and KAN were used at final concentrations of 100 μg/mL and 50 μg/mL, respectively. 5 μg/mL LEVO and 1 μg/mL CIP were used for persistence assays. 1 mM IPTG was used for induction two hours prior to treatment. Hoechst 33342 (ThermoFisher) stock was diluted 1:10 in autoclaved MilliQ water and cells were stained at a final concentration of 5 μg/mL. PicoGreen stock was dissolved in dimethyl sulfoxide (DMSO) and diluted (1:100) in 25%/75%, DMSO/MilliQ water before being added to cells.
Bacterial strain construction
All strains and plasmids used in this study are listed in Table S1. AM01 was generated via P1 transduction of the parS site (located at position 3909402 near the origin of replication) from the oriC strain obtained from the lab of Frédéric Boccard, Université Paris-Saclay,31 to the wild-type strain used in this study, MG1655. Transductants were selected on 50 μg/mL KAN and colony PCR with primers listed in Table S1 was used to confirm the genetic modifications. MG1655 ΔruvA, ΔrecG, ΔrecA, ΔrecB, ΔruvAΔrecG, and pUA66-PrecA-recA, and pUA66-PrecB-recB were constructed as previously described9. pALA2705, harboring GFP-ParB under the control of a lac-repressible promoter, was obtained from the lab of Frédéric Boccard, Université Paris-Saclay,31 and was maintained in strains with 100 μg/mL AMP. Induction with IPTG was not necessary to observe foci, as determined previously31. Gibson assembly was performed to generate pALA2705ΔparB, pUA66-lacIq-PT5-ruvAB, and pUA66-lacIq-PT5-recG using the primers listed in Table S1 (NEB Gibson Assembly Cloning Kit). For complementation of recA9 and recB21, these genes were expressed under their native promoter on low copy plasmid pUA66, using KAN for plasmid retention. RuvAB and RecG (this study) did not complement when expressed from their native promoters on pUA66. We postulated that expression issues were to blame, and thus cloned ruvA and recG under a T5 promoter on a low copy plasmid, and found that induction with 1 mM IPTG two hours prior to treatment was sufficient to restore persistence to wild-type levels. Additionally, complementation of both ruvA and ruvB were required to restore ΔruvA persistence to wild-type levels. We expect that since ruvA is a part of an operon with ruvB, and insertional mutagenesis can potentially cause polar effects on neighboring genes68, P1 transduction of ΔruvA::kanR into the MG1655 genome may have disrupted ruvB expression. Similar observations have been described previously with ΔruvA69. Plasmid sequences were confirmed with PCR using the primers listed in Table S1 and sequencing (Genewiz, South Plainfield, NJ).
METHOD DETAILS
Antibiotic persistence assays
E. coli MG1655 or UPEC CFT073 strains were inoculated into 2 mL LB in a test tube (50 μg/mL KAN for plasmid retention if necessary) from a −80°C 25% glycerol stock and incubated at 37°C for 5 h with shaking at 250 rpm. Samples were then removed and centrifuged for 3 min at 15,000 rpm in sterile 1.5 mL microcentrifuge tubes. Supernatant was removed and cell pellets were resuspended in 300 μL of M9 media containing 10 mM glucose or 20 mM glycerol. Cells were then inoculated into 25 mL M9 media + 10 mM glucose or 20 mM glycerol in a 250 mL baffled flask to an OD600 ~ 0.01 for MG1655 or OD600 ~ 0.1 for UPEC (UPEC displayed a substantial lag in growth when initially inoculated to an OD600 ~ 0.01 in minimal media) and incubated at 37°C for 20 h with shaking (250 rpm). For complementation of RecG and RuvAB, cells were induced with 1 mM IPTG two hours prior to treatment. After 20 h of growth, 500 μL were removed from each flask, washed once in PBS, and serially diluted (five-fold dilutions) in PBS, and 10 μL of each dilution was plated onto LB agar to enumerate colony forming units at the beginning of the assay (CFU/mL). LEVO or CIP were then added to a final concentration of 5 μg/mL or 1 μg/mL, respectively, to each flask, and cultures were incubated for 5 h at 37°C with shaking (250 rpm). When the impact of Hoechst 33342 on persistence was assessed, Hoechst 33342 was diluted 1:10 in water and added to the 250 mL baffled flasks to a final concentration of 5 μg/mL. At various time points, 500 μL of cells were removed, washed twice in PBS (three times for ΔrecA, ΔrecB, ΔruvA, ΔrecG, and ΔruvAΔrecG mutants), serially diluted (five-fold dilutions) and spotted onto LB to enumerate CFU/mL. Untreated controls were inoculated with equivalent volumes of solvents. Plates were incubated at 37°C for 16–20 h before quantifying CFUs from dilutions that contained ~20–80 colonies.
MIC testing
E. coli MG1655, MG1655 ΔrecB, MG1655 ΔrecA, or UPEC CFT073 were inoculated into 2 mL LB in a test tube from a −80°C 25% glycerol stock and incubated for 16 h. One mL samples were removed, centrifuged for 3 min at 15,000 rpm, and resuspended in 1 mL 0.85% NaCl. Cells were then diluted to an OD600 ~ 0.2 in 0.85% NaCl. Using a sterile cotton swab, cells were inoculated onto a Mueller-Hinton agar plate by swiping repeatedly along the entire surface of the plate three times, rotating the plate 60° each time. After plates dried, a LEVO Etest strip (bioMérieux, Marcy-l’Étoile, France) was aseptically placed in the center of the plate. Plates were incubated at 37°C for 20 h. MIC was determined by reading the value at which the growth inhibition intersected with the Etest strip.
PicoGreen staining
E. coli strains were grown to stationary phase as described above. At the completion of 20 h of growth, three 500 μL samples were taken from each flask, centrifuged at 15,000 rpm for 3 min, and supernatant was removed. One sample was resuspended in 500 μL PBS (live sample for Hoechst 33342 staining). A second sample was resuspended and fixed in 500 μL 4% paraformaldehyde (PFA; in PBS) at room temperature for 30 min, before being centrifuged and resuspended in 500 μL PBS (fixed sample for Hoechst 33342 staining). These two samples were diluted to an OD600 ~ 0.025 – 0.03 in PBS and stained with 5 μg/mL Hoechst 33342 for flow cytometric analysis.
The third sample (permeabilized samples for PicoGreen staining) was prepared as described by Ferullo and colleagues70. Briefly, 500 μL cells were added to 4.5 mL 70% cold ethanol and stored at 4°C for at least three hours. The day prior to flow cytometric analysis, cells were spun at 4,000 rpm for 10 min at 4°C. Four mL of supernatant was removed, the cells were resuspended in the remaining 1 mL and then transferred to a microcentrifuge tube. Cells were subsequently spun at 15,000 rpm for 3 minutes. The remaining supernatant was removed, and tubes were left open and covered with low-lint tissue overnight to allow residual ethanol to evaporate. The following morning, dried cell pellets were resuspended in 1 mL PBS, diluted to an OD600 = 0.4 in 500 μL PBS, and stained with 100 μL of 1:100 dilution of PicoGreen dissolved in 25% DMSO in PBS at room temperature in the dark for ~3 hours. Stained cells were then transferred to a flow cytometry tube, and further diluted with 1 mL 1:1000 dilution PicoGreen in PBS (total volume 1.6 mL).
Single-cell flow cytometric data were acquired using an LSRII SORP flow cytometer (BD Biosciences, San Jose, CA) and analyzed using FACSDiva version 8.0 software. E. coli were initially identified using forward scatter (FSC) and side scatter (SSC). To exclude doublets, cells were subsequently gated based on SSC-H and SSC-W, followed by gating based on staining with either Hoechst 33342 (as determined by Hoechst 33342-W and Hoechst 33342-A) or PicoGreen (as determined by PicoGreen-W and PicoGreen-A). Single-cell Hoechst 33342 fluorescence values were acquired by exciting each cell with a 355 nm laser run at 60 mW power (Coherent, Santa Clara, CA) and collected using a 410 nm long pass filter and a 450/50 nm bandpass filter. PicoGreen was excited using a 488 nm laser run at 100 mW power (Coherent, Santa Clara, CA) and fluorescence intensities were collected using a 505 nm long pass filter and a 525/50 band pass filter. FCS Express Software version 6 Research Use Only (De Novo Software, Glendale, CA) was used to analyze samples.
FACS: Sample Preparation
E. coli MG1655, MG1655 ΔrecA, MG1655 ΔrecB, or UPEC CFT073 strains were grown to stationary phase in M9 media + 10 mM glucose or 20 mM glycerol as described above. At the completion of 20 h of growth, two 1 mL samples from each flask were removed and centrifuged at 15,000 rpm for 3 min. The supernatants were removed and the cell pellets were resuspended in 1 mL PBS (for live-cell analysis) or 1 mL PFA for 30 min at room temperature (for fixed cell analysis). The fixed cells were subsequently centrifuged for 3 min at 15,000 rpm and resuspended in 1 mL PBS. Cells were then diluted to an OD600 ~ 0.025–0.03 in 5 mL PBS in a 15 mL conical falcon tube. The cells were stained with a final concentration of 5 μg/mL Hoechst 33342 (diluted 1:10 in water from the 10 mg/mL stock solution) at 37°C in the dark for 30 minutes. An equivalent volume of water was added to the unstained sample and treated likewise. Cells were then transferred to flow cytometry tubes.
FACS: Sorting Parameters
A BD Biosciences FACSAria Fusion Special Order Research Product (SORP) (San Jose, CA) was used to sort single cells based on Hoechst 33342 fluorescence intensity as determined by FACSDiva version 8.0 software. For details on sorter set-up, readers are referred to a recent methods article36. For cell gating, E. coli were initially identified using FSC and SSC by comparing to an unstained control. To exclude cell doublets, subsequently gate cells based on on SSC-H and SSC-W, followed by gating with Hoechst33342-W and Hoechst33342-A. Single-cell Hoechst 33342 fluorescence values were acquired by exciting each cell with a 355 nm laser run at 60 mW power (Coherent, Santa Clara, CA) and collected using a 410 nm long pass filter and a 450/50 nm bandpass filter. FCS Express Software version 6 Research Use Only (De Novo Software, Glendale, CA) was used to analyze samples.
Cells were sorted either using Four-Way Purity Mode (original sorting method) or Single-Cell Mode (high-purity sorting method). These sorting modes vary based on how the software determines which cells are sorted. Briefly, the sort stream starts off solid and is broken up into droplets. The software interrogates all droplets and evaluates the leading and trailing drops surrounding each droplet in which it observes a target cell. Four-way purity mode (original mode) sacrifices a target cell if a nearby droplet also has a non-target cell in it, but sorts that droplet if a nearby droplet also has a target cell in it. Single-Cell mode (high-purity) only sorts a droplet if there are no cells in the neighboring droplets, regardless of whether they are target cells or non-target cells.
FACS: Persister assay after sorting
For MG1655 and UPEC, one million Hoechst-stained single cells (original sorting method) or 500,000 wild-type Hoechst-stained single cells (high-purity sorting method) were sorted at 37 °C for each gate, as depicted in Figures 1, 3, and 5, as well as a total population sort, and eluted from the sorter in ~1 mL or ~500 mL PBS, respectively and collected into an equal volume of sterile-filtered spent media. The volume of the cells sorted using the high-purity mode was adjusted with an additional 1 mL of 50% PBS / 50% sterile spent media, bringing the total volume to 2 mL. Prior to treatment, each 2 mL culture was divided into two equal subcultures (1 mL each for untreated and for FQ-treated) in 15 mL polystyrene tubes, and 100 μL of cells was removed from each tube to determine initial CFUs. Cultures were then treated with LEVO or CIP (untreated controls were inoculated with equal volume of solvent) and incubated at 37°C with shaking (250 rpm) for 5 h. For untreated samples, 100 μL of cells from each sample were removed at various time points, added to a 1.5 mL microcentrifuge tube, and 900 μL PBS was added to bring the total volume to 1 mL. Cells were then centrifuged at 15,000 rpm for 3 min and 900 μL of supernatant was removed. 900 μL of fresh PBS was added again to each tube, and cells were resuspended, spun, and supernatant removed, leaving a final volume of 100 μL. 20 μL of cells were then added to 80 μL PBS in a 96-well round-bottom plate, serially diluted (five-fold dilutions) in PBS, and 10 μL of each dilution for the first six wells was spotted onto LB agar. Treated samples were washed in PBS and serially diluted as described for untreated samples, then 50 μL of the undiluted, 5-fold diluted, 25-fold diluted, and 125-fold diluted samples were plated onto LB agar and allowed to dry. For UPEC and high purity sorting, only 0 h and 5 h time points were assessed because low survivability following treatment prevented the intermediate time points from being collected. For these samples, at the final time point, the remaining 800 μL of the treated samples were also centrifuged in a 1.5 mL microcentrifuge tube for 3 min at 15,000 rpm, and 700 μL supernatant was removed, and 900 μL fresh PBS was added. Cells were washed twice as described (removing 900 μL supernatant and adding 900 μL fresh PBS), and the remaining 100 μL was plated onto LB agar using a sterile glass cell spreader, which was soaked in ethanol and flamed beforehand. All plates were incubated at 37°C for 16–20 h for CFU enumeration. We note that untreated controls demonstrated a ~2-fold increase in CFUs after 5 h of treatment. This trend was only observed in experiments where cells were at low density and were in media that was 50% PBS and 50% sterile-filtered spent media, which were the treatment conditions for sorted samples and their associated controls (e.g., not sorted-stained, not sorted-unstained). We suspect that the change in environment produced trace levels of division or culturability gain, because analogous experiments in 100% spent media and full culture density did not produce that trend (e.g., Figure 1A).
FACS: ΔrecA and ΔrecB persister assays
For MG1655 ΔrecA and ΔrecB, four million Hoechst-stained single cells were sorted at 37 °C for each gate, as well as a total population sort, and eluted from the sorter in ~4 mL PBS. We note that this was done because ΔrecA and ΔrecB cells did not survive sorting well, as we observed only 50,000–300,000 cells per 4 million sorted, as determined by initial CFU counts. Each sample was then centrifuged for 10 min at 4,000 rpm in 15 mL conical tubes, and supernatant was removed so that 1 mL remained. One mL filter-sterilized spent media was added to the tube, the cells were resuspended, and each 2 mL culture was divided into two equal subcultures (1 mL each for untreated and for FQ-treated) in 15 mL polystyrene tubes. Prior to treatment with LEVO, 100 μL of cells from each sample were removed, washed, and plated as described above. At t = 5, the untreated samples were processed as described above. The remaining 900 μL of the treated samples were centrifuged and 800 μL supernatant was removed. Cells were washed three times in PBS as described above. After the final wash, 900 μL supernatant was removed, the cells were resuspended, and all 100 μL cells were plated onto LB agar and allowed to dry. For both ΔrecA and ΔrecB, data is depicted only after 5 h of treatment due to low survival of the mutants, which precluded the collection of intermediate time points because it would have reduced the measurements to be below the limit of detection. MG1655 ΔrecA and ΔrecB persisters were enumerated after 36 hours because it was difficult to count colonies at earlier time points. For ΔrecA, we sorted 12 million total cells from the nucleoid-less peak over multiple replicates and never observed a colony. We note that all ΔrecA and ΔrecB persisters were cPCR verified for the absence of recA and recB, respectively.
FACS: Controls
Pre-sort and post-sort controls were performed as previously described4,7 to assess whether the duration of sorting alters the persistence phenotype (Figures S2B, S2I, S3F, S3L, S4D, S4I). For the pre-sort control, the stained and unstained tubes from which cells were to be sorted were diluted to an OD600 < 0.001 in 2 mL 50–50% PBS/sterile spent media (approximating the OD600 of the sorted cells in 50%−50% PBS/sterile spent media prior to antibiotic treatment). Prior to sorting, each 2 mL pre-sort culture was divided into two equal subcultures (1 mL each for untreated and for FQ-treated) in 15 mL polystyrene tubes, and persistence assays were conducted on these diluted cells as described above by taking time points at 0 h and 5 h. For the post-sort controls, after sorting had occurred, the stained and unstained tubes from which cells were sorted were diluted to an OD600 < 0.001 in 2 mL 50–50% PBS/sterile spent media and split into two equal subcultures for persistence assays, with samples taken at 0 h and 5 h. To demonstrate that sorting itself does not influence the persister phenotype (Figures 1B, S2D, S2F), cells that had been stained and prepared for FACS were diluted to an OD600 < 0.001 in 2 mL 50–50% PBS/sterile spent media, split into two equal subcultures for a time-course persistence assay, and compared to a persistence assay of the total sort population. To demonstrate that MG1655 grown in minimal glycerol and UPEC exhibited biphasic killing kinetics in an environment similar to those of sorted populations, cells that had been stained and prepared for FACS were diluted to an OD600 < 0.001 in 2 mL 50–50% PBS/sterile spent media and then split into two equal subcultures for a time-course persistence assay (Figures S3C and S3K).
Assessing Sort Fidelity: PicoGreen staining
To evaluate the fidelity of chromosome sorting based on Hoechst 33342 with PicoGreen, MG1655 was incubated for 20 h in M9 media with 10 mM glucose as previously described. After 20 h, 1 mL samples were removed, centrifuged for 3 min at 15,000 rpm in microcentrifuge tubes, and fixed in 4% PFA for 30 minutes in the dark at room temperature. Cells were then centrifuged again for 3 min at 15,000 rpm, resuspended in PBS, and stored at 4°C. On the day of the sort, cells were stained with Hoechst 33342 to a final concentration of 5 μg/mL as described above. One million single cells were sorted per gate based on Hoechst 33342 fluorescence using the BD FACSAria Fusion SORP, as described above. Sorted cells were subsequently fixed with 9 mL 70% ethanol (which is the permeabilization procedure for PicoGreen) in a 15 mL conical tube for at least three hours at 4°C and up to two weeks. The day prior to flow cytometric analysis, cells were spun at 4,000 rpm for 10 min at 4°C. 9 mL supernatant was removed, and the cells were resuspended in the remaining 1 mL and transferred to a microcentrifuge tube. Cells were subsequently spun at 15,000 rpm for 3 minutes. The remaining supernatant was removed, and tubes were left open and covered with low-lint tissue overnight to allow residual ethanol to evaporate. The following morning, dried cell pellets were resuspended in 500 μL PBS, and stained with 100 μL of 1:100 dilution of PicoGreen dissolved in 25% DMSO in PBS at room temperature in the dark for ~3 hours. Stained cells were then centrifuged at 15,000 rpm for 3 minutes, 500 μL of supernatant was removed, and the cell pellet was resuspended in the remaining 100 μL. The cells were then transferred to de-capped PCR tubes, which were then placed into a flow cytometry tube for single cell analysis.
We observed that fixed cells sorted to contain more than two chromosomes had an enrichment in cells harboring three and four chromosomes, but also observed significant levels of cells with one or two chromosomes in that subpopulation (Figure 2). Several procedures were attempted to remedy this situation, such as the use of 1 mM EDTA, 0.1% Tween-80, vigorous vortexing, and alternative gating strategies; however, none were deemed sufficiently successful and subsequent analyses focused on one- and two-chromosome sorted subpopulations, because those were well resolved.
Single-cell flow cytometric data were acquired using an LSRII SORP flow cytometer (BD Biosciences, San Jose, CA) and analyzed using FACSDiva version 8.0 software. E. coli were initially identified using FSC and SSC. No gates were applied to the sort-check samples. PicoGreen was excited using a 488 nm laser run at 100 mW power (Coherent, Santa Clara, CA) and fluorescence intensities were collected using a 505 nm long pass filter and a 525/50 band pass filter.
Assessing Sort Fidelity: Origin Reporter
To verify the accuracy of sorting for chromosome abundance based on Hoechst 33342 with an independent methodology (not a nucleic acid stain), AM01 with pALA2705 was grown for 20 h in M9 media with 10 mM glucose and 100 μg/mL AMP for plasmid retention as previously described above. After 20 hr, 500 μL of cultures were centrifuged at 15,000 rpm for 3 min. Supernatant was removed and cells were fixed in 500 μL 4% PFA in the dark at room temperature for ~30 min. After fixation, cells were centrifuged at 15,000 rpm for 3 min. Supernatant was removed, and cells were stored in 500 μL PBS in the dark at 4 °C for up to two weeks. On the days FACS fidelity was measured, fixed cells were diluted to an OD600 ~ 0.025 – 0.03 in 2 mL PBS, stained with 5 μg/mL Hoechst 33342 as previously described, followed by transfer to a flow cytometry tube. Using the FACSAria Fusion SORP (parameters and thresholds as described above), 2 million cells were sorted per gate, as depicted in Figure 5A, as well as a total sort. Eluted cells were concentrated to a final volume of ~30 μL in PBS and stored in the dark at 4 °C. For controls, MG1655 with pALA2705 (no parS binding site) and AM01 with pALA2705ΔparB (contains parS binding site but lacks ParB) were fixed identically to AM01 harboring pALA2705, before being centrifuged and resuspended in PBS for microscopic analysis (Figure S5).
For microscopic imaging, 0.1 g agarose was dissolved in 10 mL autoclaved MilliQ water by microwaving for ~1 min in a 50 mL falcon tube. The solution was allowed to cool slightly before pipetting 1 mL onto plain microscopy slides that had been cleaned with 200-proof ethanol (Fisher Scientific). A second plain microscopy slide was laid cross-wise over top (supported by adjacent double-stacked plain microscopy slides laid parallel to and on both sides of the original slide). The solution was allowed to harden at room temperature for ~1 h. The top slide was carefully removed, and the agarose pad was cut into a ~2 cm x ~2 cm square. 2 μL of the fixed, sorted, and concentrated AM01 with pALA2705 samples were pipetted onto the agarose pad and allowed to air dry. To increase the density of cells for microscopy, an additional 2 μL of the same sample was pipetted immediately on top of the original sample on the agar and allowed to dry. This step was repeated one more time. For MG1655 with pALA2705 and AM01 with pALA2705ΔparB, samples were diluted 1:12 in PBS, and 2 μL were spotted onto the agarose pad and allowed to air dry. Cover glass was overlaid and sealed with clear nail polish.
A Nikon Ti-E equipped with Yokogawa spinning disc (CSU-21) and quad dichroic accommodating 405, 488, 561, and 647 lasers was used for microscopy. NIS-Elements, V4.5 (Nikon Instruments, Melville, NY) was used to control the microscope. A Hammamatsu ImagEm back-thinned EMCCD was the spinning disc detector employed, and an Agilent laser launch with a 488 laser was used for fluorescence imaging. The system was outfitted with a piezo stage with 100 μm range, which had the capacity to make 100 steps/second, and a CF160 Plan Fluor Phase Contrast DLL 100x oil objective with 1.3NA was used. For fixed cell analysis, GFP Z-stacks were taken every 0.2 μM steps for 15 steps. 300 ms exposure was used for phase contrast, and 1 s exposure at 50% laser power was used for fluorescence imaging. MicrobeJ was used to count foci within single cells. Three sorting replicates allowed for the total classification of at least 400 cells per sort gate. Since foci clarity in single cells varied based on location in the microscopic image, foci were visually quantified by manually adjusting the min- and max- GFP fluorescence. % Unknown (U) represents cells that lacked discernible foci.
We note that in Figure 5, the origin reporter was used to quantify sorting precision in E. coli K12 cultures grow to stationary phase in minimal glucose media. Additional samples, such as E. coli K12 cultures grown to stationary phase in minimal glycerol media and UPEC cultures, were not assayed for sorting precision with the origin reporter. This was because they exhibited comparable DNA histograms to those of E. coli K12 grown to stationary phase in minimal glucose media, consisting of unimodal peaks at similar fluorescence intensities, and conservative gating methodologies were used while sorting. We expect that the sorting precision for those samples is comparable to what is presented in Figure 5. In addition, we note that the cultures used in this study (stationary phase, where PicoGreen staining confirmed highly resolved DNA peaks), the conservative gating strategy employed, and the agreement observed between two independent chromosome counting methods (Hoechst 33342, origin reporter), suggest that the bacterial populations examined contained cells with fully-replicated chromosomes. Under different conditions, such as exponential-phase, where partially-replicated chromosomes are prevalent21, the use of an additional, independent reporter of ter abundance would be advisable to distinguish between cells with fully-replicated and partially-replicated chromosomes.
FACS with time lapse microscopy
AM01 + pALA2705 was grown to stationary in M9 minimal glucose media in the presence of 100 μg/mL AMP as described above. After 20 h, 1 mL was removed, centrifuged at 15,000 rpm for three minutes, and the pellet was resuspended in 1 mL PBS. The cells were diluted to an OD600 ~ 0.025 – 0.03 in 5 mL 50% PBS / 50% sterile-filtered spent media in a 15 mL conical falcon tube in the presence of 100 μg/mL AMP. Two 15 mL polystyrene tubes were then filled with 1 mL of these diluted cells. Both samples were then treated with either 5 μg/mL LEVO or equal volume water and incubated at 37°C with shaking (250 rpm) for 5 h. 100 μL of the remaining diluted cells in the 15 mL conical falcon tube were used for CFU measurements.
After 4.5 hours of treatment, one of the two cell samples were stained with a final concentration of 5 μg/mL Hoechst 33342 (diluted 1:10 in water from the 10 mg/mL stock solution) at 37°C with shaking (250 rpm) in the dark for 30 minutes. The other cell sample remained unstained. Cells were then transferred to flow cytometry tubes. One million of the Hoechst-determined 1Chr and 2Chr cells (see Gates a and b, respectively, in Figure 1C) were sorted at 37°C as described above, eluted from the sorter in ~1 mL PBS, and collected into an equal volume of sterile-filtered spent media containing 200 μg/mL AMP. If cells had also been treated with LEVO, the sterile-filtered spent media collection tube also contained 10 μg/mL LEVO (double the concentration of the treatment condition since the final volume of sorted cells + collection media was 2 mL). The sorted cells were centrifuged for 3 minutes at 15,000 rpm, and all but 100 μL of supernatant was removed. These cells were then washed a total of three times in 900 μL of PBS, removing 900 μL of supernatant each time. Upon the final wash, 100 μL of cells were removed for CFU measurements. The cells were centrifuged a final time, ~880 μL of supernatant were removed, and the cell pellet was resuspended in the residual ~20 μL.
For the control images shown in Figure S7, AM01 + pALA2705 was grown to stationary phase in M9 minimal glucose in the presence of 100 μg/mL AMP as described above. After 20 h, 2 × 1 mL were removed and centrifuged at 15,000 rpm for three minutes. One pellet was resuspended in 1 mL 4% PFA for 30 min (‘Stationary phase Untreated and Unsorted’ sample), and the other pellet was resuspended in 1 mL PBS. The live cells were diluted to an OD600 ~ 0.025 – 0.03 in 5 mL 50% PBS / 50% sterile-filtered spent media in a 15 mL conical falcon tube in the presence of 100 μg/mL AMP. Two 15 mL polystyrene tubes were then filled with 1 mL of these diluted cells. One sample was treated with 5 μg/mL LEVO and the other with equal volume water, and both were incubated at 37°C with shaking (250 rpm) for 5 h. After 5 h, both samples were centrifuged for 3 min at 15,000 rpm in microcentrifuge tubes, nearly all of the supernatant was removed, and the cells were resuspended in 4% PFA for 30 min. After 30 min in PFA, samples were centrifuged for 3 min at 15,000 rpm, and resuspended in 1 mL PBS.
Microscopy of sorted cells
To prepare for live-cell microscopy of the sorted cells, we adapted protocols from Barrett and colleagues9 and Young and colleagues71. Briefly, coverslips, spatulas, and stir bars used for LB pad preparation were autoclaved at 121°C for 30 min. To prepare the agar pads, 0.2 g Certified Molecular Biology agarose (Bio-Rad, Hercules, CA) was dissolved in 20 mL of LB media in a sterile flask and microwaved at power level 4 for ~2.5 minutes. The solution was cooled with stirring using a sterilized stir bar for ~5 minutes, AMP was added for plasmid retention, 500 μL was pipetted onto a 10.5 × 35 mm rectangular coverslip (Chemglass Life Sciences, Vineland, NJ), and another coverslip was place on top. The approximate thickness of the agarose pad was ~1 mm (the thickness of a microscopy slide). The agar pad was allowed to cool at room temperature for ~ 1 h before the top coverslip was removed and the agar pad was cut into three equally-sized pads using a sterile spatula. Two microliters of resuspended sorted cells were pipetted onto the center agar pad and allowed to dry. This was repeated until all ~20 μL of cells were pipetted onto the pad in order to increase cell density for microscopy. This exact process was repeated for the fixed cell microscopy controls (Figure S7), except that the 20 h stationary-phase fixed cells were first diluted 1:12 in PBS and 2 μL were then pipetted onto the center of the agar pad only one time. Once the pad was completely dry, the coverslip containing the pads was placed cell-side down into a Lab-Tek chambered #1.5 coverglass (Thermo Fisher Scientific, Waltham, MA). The coverslip was sealed to the chambered coverglass using valap (1:1:1[w/w/w] of petroleum jelly, lanolin, and paraffin wax).
The microscope described above (CSU-21) was used for time-lapse microscopy of the sorted samples. Samples were placed into an environmental control chamber in a fully-motorized XY stage and held at 37°C throughout the course of the time lapse experiment. Images were taken every 12 minutes for 16 hours. At each time point, a 2×2 tile of phase contrast images was taken at each XY position. Exposure was set to 300 ms. At the end of the experiment, sterility was assessed by imaging an adjacent agarose pad that had not been inoculated, and no growth was observed. Brightness and contrast were adjusted using ImageJ software, and 2×2 tiles were stitched together using the Stitching plugin of ImageJ72.
MicrobeJ was used to quantify the post-treatment growth of sorted cells observed using time lapse microscopy. Default settings were used, except for the following: Algorithm: MaxEntropy; Mode of Detection: Medial Axis; Area: 0.15-max; Length: 0.3-max; Width: 0–0.65. Using this MicrobeJ algorithm, approximately 98% of all data points were readily calculated without manual length correction. The remaining 2% of data points were listed as NaN in the data sets. Hoechst-determined 1Chr and 2Chr persister lengths were quantified up until the time they divided. 1Chr and 2Chr non-persister lengths were quantified up until the time they lysed or stop growing, or until crowding of cells hindered analysis by the software. Untreated controls were quantified up until the first cell division. Time 0 h was considered to be the first images obtained from the microscope.
To calculate the rates of volume expansion (r), we assumed that the increases in volumes of individual cells could be described by exponential functions of the form: , where V is cellular volume and t is time. We note that this equation mirrors the exponential growth equation. Rearrangement of the equation yields, , which is plotted in Figure 7. Solving the differential equation for V = Vo at t = 0, yields, V = Vo ∗ exp(rt). Then taking the natural logarithm of both sides yields, ln V = rt + ln Vo. To calculate r for each cell, the natural logarithms of the lengths were plotted as a function of time and straight lines were fit to the data. We note that we approximated cells as cylinders with fixed diameters for this analysis, which allows lengths data to be processed in this manner to calculate r. From those linear fits, r can be extracted as the slope of the line. The time periods for calculating those linear fits for each cell were selected based on the times when the first cell in each sample set divided. We note that the average R2 for the linear fits for the four different samples were 0.945 (1Chr persisters), 0.955 (2Chr persisters), 0.913 (1Chr untreated), and 0.984 (2Chr untreated), which suggested that the exponential function used was justified.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details of each experiment can be found in the figure legends. All replicates were independent biological replicates (n). Error bars represent SEM. Significance was assessed using an omnibus one-way analysis of variance (ANOVA) followed by Tukey’s post-hoc test on log-transformed values and a p-value threshold of 0.05. For growth dynamics (Figure 7), significance was assessed using a repeated Measures ANOVA with main effects of chromosome number and time on log-transformed values and a p-value threshold of 0.05. For Figure 1D and Figure 3B, statistical significance was assessed only at the 5 h timepoint, because at that time point persisters are the only remaining CFUs, as we previously reported7.
Supplementary Material
Video S1. Recovery of LEVO-treated Hoechst-sorted 1Chr cells, related to Figure 7.
Time-lapse microscopy of LEVO-treated Hoechst-sorted 1Chr cells. Phase contrast images were taken every 12 min. The video includes three distinct time courses. This video is representative of data obtained from five biological replicates.
Video S2. Recovery of LEVO-treated Hoechst-sorted 2Chr cells, related to Figure 7.
Time-lapse microscopy of LEVO-treated Hoechst-sorted 2Chr cells. Phase contrast images were taken every 12 min. The video includes three distinct time courses. This video is representative of data obtained from three biological replicates.
Video S3. Recovery of untreated Hoechst-sorted 1Chr cells, related to Figure 7.
Time-lapse microscopy of untreated Hoechst-sorted 1Chr cells. Phase contrast images were taken every 12 min. The video includes three distinct time courses. This video is representative of data obtained from two biological replicates.
Video S4. Recovery of untreated Hoechst-sorted 2Chr cells, related to Figure 7.
Time-lapse microscopy of untreated Hoechst-sorted 2Chr cells. Phase contrast images were taken every 12 min. The video includes three distinct time courses. This video is representative of data obtained from two biological replicates.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Escherichia coli MG1655; F−λ− ilvG−rfb-50 rph-1 | ATCC | Strain Number 700926 |
| Uropathogenic E. coli CFT073 | ATCC, GenBank | Strain Number 700928 (ATCC); Strain Number AE014075 (GenBank) |
| Origin reporter strain AM01: MG1655 oriC-parS + pALA2705 | This paper; P1 transduction from strain received from the lab of Frédéric Boccard, Université Paris-Saclay31 | N/A |
| For MG1655 recombination mutants, see Table S1 | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Levofloxacin | Fisher/Alfa Aesar | Catalog Number AA6694303 |
| Ciprofloxacin | Fisher/Acros | Catalog Number AC449620050 |
| Ampicillin | Fisher/Acros | Catalog Number AC611770050 |
| Kanamycin | Fisher Scientific | Catalog Number 11815032 |
| Hoechst 33342 | Thermofisher | Catalog Number H3570 |
| Quant-iT PicoGreen dsDNA reagent | ThermoFisher | Catalog Number P7581 |
| Paraformaldehyde, 4% in PBS | Fisher | Catalog Number AAJ61899AK |
| Certified Molecular Biology agarose | Bio-Rad | Catalog Number 161–3100EDU |
| 10.5 × 35 mm rectangular coverslip | VWR | Catalog Number 100493–658 |
| Lab-Tek chambered #1.5 coverglass | Fisher | Catalog Number 12–565-335 |
| coverglass | ||
| Oligonucleotides | ||
| Primers to confirm genetic mutants and plasmids: See Table S1 | This paper | N/A |
| Primers for plasmid construction: See Table S1 | This paper | N/A |
| Recombinant DNA | ||
| Plasmid pALA2705 | Frédéric Boccard Lab, Université Paris-Saclay31 | N/A |
| Plasmid pALA2705ΔparB | This paper | N/A |
| For empty vector and complementation plasmids, see Table S1 | This paper | N/A |
| Software and algorithms | ||
| FCS Express Software version 6 Research Use Only | De Novo Software | RRID: SCR_016431 |
| FACSDiva version 8.0 software | BD Biosciences | RRID: SCR_001456 |
| NIS-Elements, V4.5 | Nikon Instruments | RRID: SCR_014329 |
| ImageJ | 67 | https://imagej.nih.gov/ij/; RRID: SCR_003070 |
| Stitching plugin of ImageJ | 68 | RRID: SCR_016568 |
| MicrobeJ | 69 | https://www.microbej.com/ |
Highlights.
FQ persisters arise less frequently from E. coli harboring only one chromosome
HR-deficient mutants demonstrate no association between ploidy and FQ persistence
RecA and RecB enable survival of both monoploid and diploid FQ persisters
Monoploid and diploid FQ persisters demonstrate distinct recovery dynamics
Acknowledgments
We thank Christina DeCoste and Katherine Rittenbach (Princeton Flow Cytometry Resource Center), Gary Laevsky (Princeton Confocal Imaging Facility), Thomas Silhavy, and Zemer Gitai for their assistance and suggestions. We thank the Frédéric Boccard lab, Université Paris-Saclay, for their generous donation of MG1655 oriC-parS strain and pALA270531. We also acknowledge the National BioResource Project (NIG, Japan) for distribution of the Keio Collection. This work was supported by the NIAID of the NIH (A.M.M: F30AI140697; M.P.B.: R01AI130293). This work is the responsibility of the authors and the funders has no role in the design or implementation of the experiments or the decision to publish.
Footnotes
Declaration of interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lewis K (2010). Persister Cells. Annu. Rev. Microbiol 64, 357–372. [DOI] [PubMed] [Google Scholar]
- 2.Balaban NQ, Helaine S, Lewis K, Ackermann M, Aldridge B, Andersson DI, Brynildsen MP, Bumann D, Camilli A, Collins JJ, et al. (2019). Definitions and guidelines for research on antibiotic persistence. Nat. Rev. Microbiol 17, 441–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balaban NQ, Merrin J, Chait R, Kowalik L, and Leibler S (2004). Bacterial Persistence as a Phenotypic Switch. Science 305, 1622–1625. [DOI] [PubMed] [Google Scholar]
- 4.Orman MA, and Brynildsen MP (2013). Dormancy Is Not Necessary or Sufficient for Bacterial Persistence. Antimicrob. Agents Chemother 57, 3230–3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shah D, Zhang Z, Khodursky AB, Kaldalu N, Kurg K, and Lewis K (2006). Persisters: a distinct physiological state of E. coli. BMC Microbiol. 6, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goormaghtigh F, and Melderen LV (2019). Single-cell imaging and characterization of Escherichia coli persister cells to ofloxacin in exponential cultures. Sci. Adv 5, eaav9462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Völzing KG, and Brynildsen MP (2015). Stationary-Phase Persisters to Ofloxacin Sustain DNA Damage and Require Repair Systems Only during Recovery. mBio 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eng RH, Padberg FT, Smith SM, Tan EN, and Cherubin CE (1991). Bactericidal effects of antibiotics on slowly growing and nongrowing bacteria. Antimicrob. Agents Chemother 35, 1824–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barrett TC, Mok WWK, Murawski AM, and Brynildsen MP (2019). Enhanced antibiotic resistance development from fluoroquinolone persisters after a single exposure to antibiotic. Nat. Commun 10, 1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levin-Reisman I, Ronin I, Gefen O, Braniss I, Shoresh N, and Balaban NQ (2017). Antibiotic tolerance facilitates the evolution of resistance. Science 355, 826–830. [DOI] [PubMed] [Google Scholar]
- 11.Windels EM, Michiels JE, Fauvart M, Wenseleers T, Bergh BV den, and Michiels J (2019). Bacterial persistence promotes the evolution of antibiotic resistance by increasing survival and mutation rates. ISME J. 13, 1239–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koster DA, Crut A, Shuman S, Bjornsti M-A, and Dekker NH (2010). Cellular Strategies for Regulating DNA Supercoiling: A Single-Molecule Perspective. Cell 142, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pohlhaus JR, and Kreuzer KN (2005). Norfloxacin-induced DNA gyrase cleavage complexes block Escherichia coli replication forks, causing double-stranded breaks in vivo. Mol. Microbiol 56, 1416–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drlica K, Malik M, Kerns RJ, and Zhao X (2008). Quinolone-Mediated Bacterial Death. Antimicrob. Agents Chemother 52, 385–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drlica K, and Zhao X (1997). DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol. Mol. Biol. Rev 61, 377–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tamayo M, Santiso R, Gosalvez J, Bou G, and Fernández JL (2009). Rapid assessment of the effect of ciprofloxacin on chromosomal DNA from Escherichia coli using an in situ DNA fragmentation assay. BMC Microbiol. 9, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lesterlin C, Ball G, Schermelleh L, and Sherratt DJ (2014). RecA bundles mediate homology pairing between distant sisters during DNA break repair. Nature 506, 249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lloyd RG, and Sharples GJ (1993). Processing of recombination intermediates by the RecG and RuvAB proteins of Escherichia coli. Nucleic Acids Res. 21, 1719–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krasin F, and Hutchinson F (1977). Repair of DNA double-strand breaks in Escherichia coli, which requires recA function and the presence of a duplicate genome. J. Mol. Biol 116, 81–98. [DOI] [PubMed] [Google Scholar]
- 20.Roostalu J, Jõers A, Luidalepp H, Kaldalu N, and Tenson T (2008). Cell division in Escherichia coli cultures monitored at single cell resolution. BMC Microbiol. 8, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mok WWK, and Brynildsen MP (2018). Timing of DNA damage responses impacts persistence to fluoroquinolones. Proc. Natl. Acad. Sci 115, E6301–E6309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dörr T, Lewis K, and Vulić M (2009). SOS Response Induces Persistence to Fluoroquinolones in Escherichia coli. PLOS Genet. 5, e1000760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Theodore A, Lewis K, and Vulić M (2013). Tolerance of Escherichia coli to Fluoroquinolone Antibiotics Depends on Specific Components of the SOS Response Pathway. Genetics 195, 1265–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Watt VM, Ingles CJ, Urdea MS, and Rutter WJ (1985). Homology requirements for recombination in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A 82, 4768–4772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ding M-Z, Li B-Z, Cheng J-S, and Yuan Y-J (2010). Metabolome Analysis of Differential Responses of Diploid and Haploid Yeast to Ethanol Stress. OMICS J. Integr. Biol 14, 553–561. [DOI] [PubMed] [Google Scholar]
- 26.Zörgö E, Chwialkowska K, Gjuvsland AB, Garré E, Sunnerhagen P, Liti G, Blomberg A, Omholt SW, and Warringer J (2013). Ancient Evolutionary Trade-Offs between Yeast Ploidy States. PLoS Genet. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Soppa J (2017). Polyploidy and community structure. Nat. Microbiol 2, 1–2. [DOI] [PubMed] [Google Scholar]
- 28.Soppa J (2015). Polyploidy in Archaea and Bacteria: About Desiccation Resistance, Giant Cell Size, Long-Term Survival, Enforcement by a Eukaryotic Host and Additional Aspects. J. Mol. Microbiol. Biotechnol 172730 24, 409–419. [DOI] [PubMed] [Google Scholar]
- 29.Akerlund T, Nordström K, and Bernander R (1995). Analysis of cell size and DNA content in exponentially growing and stationary-phase batch cultures of Escherichia coli. J. Bacteriol 177, 6791–6797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Y, and Austin S (2002). The P1 plasmid is segregated to daughter cells by a ‘capture and ejection’ mechanism coordinated with Escherichia coli cell division. Mol. Microbiol 46, 63–74. [DOI] [PubMed] [Google Scholar]
- 31.Espeli O, Mercier R, and Boccard F (2008). DNA dynamics vary according to macrodomain topography in the E. coli chromosome. Mol. Microbiol 68, 1418–1427. [DOI] [PubMed] [Google Scholar]
- 32.Orman MA, and Brynildsen MP (2015). Inhibition of stationary phase respiration impairs persister formation in E. coli. Nat. Commun 6, ncomms8983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aedo SJ, Orman MA, and Brynildsen MP (2019). Stationary phase persister formation in Escherichia coli can be suppressed by piperacillin and PBP3 inhibition. BMC Microbiol. 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Welch RA, Burland V, Plunkett G, Redford P, Roesch P, Rasko D, Buckles EL, Liou S-R, Boutin A, Hackett J, et al. (2002). Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc. Natl. Acad. Sci 99, 17020–17024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zyskind JW, Svitil AL, Stine WB, Biery MC, and Smith DW (1992). RecA protein of Escherichia coli and chromosome partitioning. Mol. Microbiol 6, 2525–2537. [DOI] [PubMed] [Google Scholar]
- 36.Murawski AM, Rittenbach K, DeCoste CJ, Laevsky G, and Brynildsen MP Counting chromosomes in individual bacteria to quantify their impacts on persistence. In Methods in Mol Biol, p. Accepted. [DOI] [PubMed] [Google Scholar]
- 37.Badrinarayanan A, Le TBK, and Laub MT (2015). Rapid pairing and resegregation of distant homologous loci enables double-strand break repair in bacteria. J. Cell Biol 210, 385–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Michiels JE, Van den Bergh B, Verstraeten N, and Michiels J (2016). Molecular mechanisms and clinical implications of bacterial persistence. Drug Resist. Updat 29, 76–89. [DOI] [PubMed] [Google Scholar]
- 39.WHO | No Time to Wait: Securing the future from drug-resistant infections WHO. http://www.who.int/antimicrobial-resistance/interagency-coordination-group/final-report/en/.
- 40.Dörr T, Vulić M, and Lewis K (2010). Ciprofloxacin Causes Persister Formation by Inducing the TisB toxin in Escherichia coli. PLOS Biol. 8, e1000317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Amato SM, and Brynildsen MP (2015). Persister Heterogeneity Arising from a Single Metabolic Stress. Curr. Biol 25, 2090–2098. [DOI] [PubMed] [Google Scholar]
- 42.Amato SM, Orman MA, and Brynildsen MP (2013). Metabolic Control of Persister Formation in Escherichia coli. Mol. Cell 50, 475–487. [DOI] [PubMed] [Google Scholar]
- 43.Luidalepp H, Jõers A, Kaldalu N, and Tenson T (2011). Age of Inoculum Strongly Influences Persister Frequency and Can Mask Effects of Mutations Implicated in Altered Persistence. J. Bacteriol 193, 3598–3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vega NM, Allison KR, Khalil AS, and Collins JJ (2012). Signaling-Mediated Bacterial Persister Formation. Nat. Chem. Biol 8, 431–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Verstraeten N, Knapen WJ, Kint CI, Liebens V, Van den Bergh B, Dewachter L, Michiels JE, Fu Q, David CC, Fierro AC, et al. (2015). Obg and Membrane Depolarization Are Part of a Microbial Bet-Hedging Strategy that Leads to Antibiotic Tolerance. Mol. Cell 59, 9–21. [DOI] [PubMed] [Google Scholar]
- 46.Henry TC, and Brynildsen MP (2016). Development of Persister-FACSeq: a method to massively parallelize quantification of persister physiology and its heterogeneity. Sci. Rep 6, srep25100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Courcelle J, and Hanawalt PC (2001). Participation of recombination proteins in rescue of arrested replication forks in UV-irradiated Escherichia coli need not involve recombination. Proc. Natl. Acad. Sci. U. S. A 98, 8196–8202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Courcelle J, Donaldson JR, Chow K-H, and Courcelle CT (2003). DNA Damage-Induced Replication Fork Regression and Processing in Escherichia coli. Science 299, 1064–1067. [DOI] [PubMed] [Google Scholar]
- 49.Anderson P, and Roth J (1981). Spontaneous tandem genetic duplications in Salmonella typhimurium arise by unequal recombination between rRNA (rrn) cistrons. Proc. Natl. Acad. Sci 78, 3113–3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Langridge J (1969). Mutations conferring quantitative and qualitative increases in β-galactosidase activity in Escherichia coli. Mol. Gen. Genet. MGG 105, 74–83. [DOI] [PubMed] [Google Scholar]
- 51.Reams AB, and Roth JR (2015). Mechanisms of Gene Duplication and Amplification. Cold Spring Harb. Perspect. Biol 7, a016592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Iguchi A, Iyoda S, Terajima J, Watanabe H, and Osawa R (2006). Spontaneous recombination between homologous prophage regions causes large-scale inversions within the Escherichia coli O157:H7 chromosome. Gene 372, 199–207. [DOI] [PubMed] [Google Scholar]
- 53.Schneider D, Duperchy E, Coursange E, Lenski RE, and Blot M (2000). Long-Term Experimental Evolution in Escherichia coli. IX. Characterization of Insertion Sequence-Mediated Mutations and Rearrangements. Genetics 156, 477–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cooper VS, Schneider D, Blot M, and Lenski RE (2001). Mechanisms Causing Rapid and Parallel Losses of Ribose Catabolism in Evolving Populations of Escherichia coli B. J. Bacteriol 183, 2834–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Long H, Miller SF, Strauss C, Zhao C, Cheng L, Ye Z, Griffin K, Te R, Lee H, Chen C-C, et al. (2016). Antibiotic treatment enhances the genome-wide mutation rate of target cells. Proc. Natl. Acad. Sci 113, E2498–E2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morag AS, Saveson CJ, and Lovett ST (1999). Expansion of DNA repeats in Escherichia coli: effects of recombination and replication functions11Edited by J. H. Miller. J. Mol. Biol 289, 21–27. [DOI] [PubMed] [Google Scholar]
- 57.Lovett ST, Gluckman TJ, Simon PJ, Sutera VA, and Drapkin PT (1994). Recombination between repeats in Escherichia coli by a recA-independent, proximity-sensitive mechanism. Mol. Gen. Genet. MGG 245, 294–300. [DOI] [PubMed] [Google Scholar]
- 58.Bi X, and Liu LF (1994). recA-independent and recA-dependent Intramolecular Plasmid Recombination: Differential Homology Requirement and Distance Effect. J. Mol. Biol 235, 414–423. [DOI] [PubMed] [Google Scholar]
- 59.Hill CW, and Harnish BW (1981). Inversions between ribosomal RNA genes of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A 78, 7069–7072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miguel AG, and Tyrrell RM (1986). Repair of near-ultraviolet (365 nm)-induced strand breaks in Escherichia coli DNA. The role of the polA and recA gene products. Biophys. J 49, 485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Naber KG (2001). Which fluoroquinolones are suitable for the treatment of urinary tract infections? Int. J. Antimicrob. Agents 17, 331–341. [DOI] [PubMed] [Google Scholar]
- 62.Foxman B (1990). Recurring urinary tract infection: incidence and risk factors. Am. J. Public Health 80, 331–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Skjøt-Rasmussen L, Hammerum AM, Jakobsen L, Lester CH, Larsen P, and Frimodt-Møller N (2011). Persisting clones of Escherichia coli isolates from recurrent urinary tract infection in men and women. J. Med. Microbiol 60, 550–554. [DOI] [PubMed] [Google Scholar]
- 64.Zerulla K, Chimileski S, Näther D, Gophna U, Papke RT, and Soppa J (2014). DNA as a Phosphate Storage Polymer and the Alternative Advantages of Polyploidy for Growth or Survival. PLOS ONE 9, e94819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Krisko A, and Radman M (2013). Biology of Extreme Radiation Resistance: The Way of Deinococcus radiodurans. Cold Spring Harb. Perspect. Biol 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ohtani N, Tomita M, and Itaya M (2010). An Extreme Thermophile, Thermus thermophilus, Is a Polyploid Bacterium. J. Bacteriol 192, 5499–5505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Michelsen O, Hansen FG, Albrechtsen B, and Jensen PR (2010). The MG1363 and IL1403 Laboratory Strains of Lactococcus lactis and Several Dairy Strains Are Diploid. J. Bacteriol 192, 1058–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, and Mori H (2006). Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol 2, 2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sutherland JH, and Tse-Dinh Y-C (2010). Analysis of RuvABC and RecG Involvement in the Escherichia coli Response to the Covalent Topoisomerase-DNA Complex. J. Bacteriol 192, 4445–4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ferullo DJ, Cooper DL, Moore HR, and Lovett ST (2009). Cell cycle synchronization of E. coli using the stringent response, with fluorescence labeling assays for DNA content and replication. Methods San Diego Calif 48, 8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Young JW, Locke JCW, Altinok A, Rosenfeld N, Bacarian T, Swain PS, Mjolsness E, and Elowitz MB (2012). Measuring single-cell gene expression dynamics in bacteria using fluorescence time-lapse microscopy. Nat. Protoc 7, 80–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Preibisch S, Saalfeld S, and Tomancak P (2009). Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics 25, 1463–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Recovery of LEVO-treated Hoechst-sorted 1Chr cells, related to Figure 7.
Time-lapse microscopy of LEVO-treated Hoechst-sorted 1Chr cells. Phase contrast images were taken every 12 min. The video includes three distinct time courses. This video is representative of data obtained from five biological replicates.
Video S2. Recovery of LEVO-treated Hoechst-sorted 2Chr cells, related to Figure 7.
Time-lapse microscopy of LEVO-treated Hoechst-sorted 2Chr cells. Phase contrast images were taken every 12 min. The video includes three distinct time courses. This video is representative of data obtained from three biological replicates.
Video S3. Recovery of untreated Hoechst-sorted 1Chr cells, related to Figure 7.
Time-lapse microscopy of untreated Hoechst-sorted 1Chr cells. Phase contrast images were taken every 12 min. The video includes three distinct time courses. This video is representative of data obtained from two biological replicates.
Video S4. Recovery of untreated Hoechst-sorted 2Chr cells, related to Figure 7.
Time-lapse microscopy of untreated Hoechst-sorted 2Chr cells. Phase contrast images were taken every 12 min. The video includes three distinct time courses. This video is representative of data obtained from two biological replicates.
Data Availability Statement
All data are included within the paper, and raw data can be requested from the Lead Contact.