

Abstract
Background
Tachycardia and heart rate irregularity are proposed triggers of premature ventricular contraction–induced cardiomyopathy (PVC-cardiomyopathy). Bigeminal premature atrial and ventricular contractions (PACs and PVCs) increase heart rate and result in rhythm irregularities but differ in their effects on ventricular synchrony. Comparing chronic bigeminal PACs with PVCs would provide insights into mechanisms of PVC-cardiomyopathy.
Objective
To compare the impact of chronic PACs and PVCs on ventricular hemodynamics, structure, and function.
Methods
Pacemakers were implanted in 27 canines to reproduce atrial (PACs, n = 7) or ventricular bigeminy (PVCs, n = 11) for 12 weeks, and compared to sham-operated animals (n = 9). Four additional animals were exposed to long-term bigeminal PVCs (48 weeks). Hemodynamic changes were assessed using a pressure-transducing catheter at baseline and 12 weeks. Cardiac remodeling was monitored by transthoracic echocardiography throughout the 12- and 48-week protocols in the respective groups.
Results
PVC group demonstrated a significant decrease in left ventricular (LV) ejection fraction and contractility (max dP/dt), impaired LV lusitropy (min dP/dt), and increase in LV dimensions and LV mass at 12 weeks without further deterioration beyond 16 weeks. Despite increased LV mass, relative wall thickness decreased, consistent with eccentric hypertrophy. No significant cardiac remodeling was noted in either sham or PAC groups at 12 weeks.
Conclusion
In contrast to bigeminal PACs, PVCs result in a cardiomyopathy characterized by reduced LV ejection fraction, LV dilation, and eccentric hypertrophy that plateaus between 12 and 16 weeks. The lack of remodeling in chronic PACs suggests that tachycardia and heart rate irregularity do not play a significant role on the development of PVC-cardiomyopathy.
Keywords: Cardiomyopathy, Eccentric hypertrophy, Heart rate irregularity, LV dyssynchrony, Premature atrial contraction, Premature ventricular contraction, Tachycardia
Graphical abstract
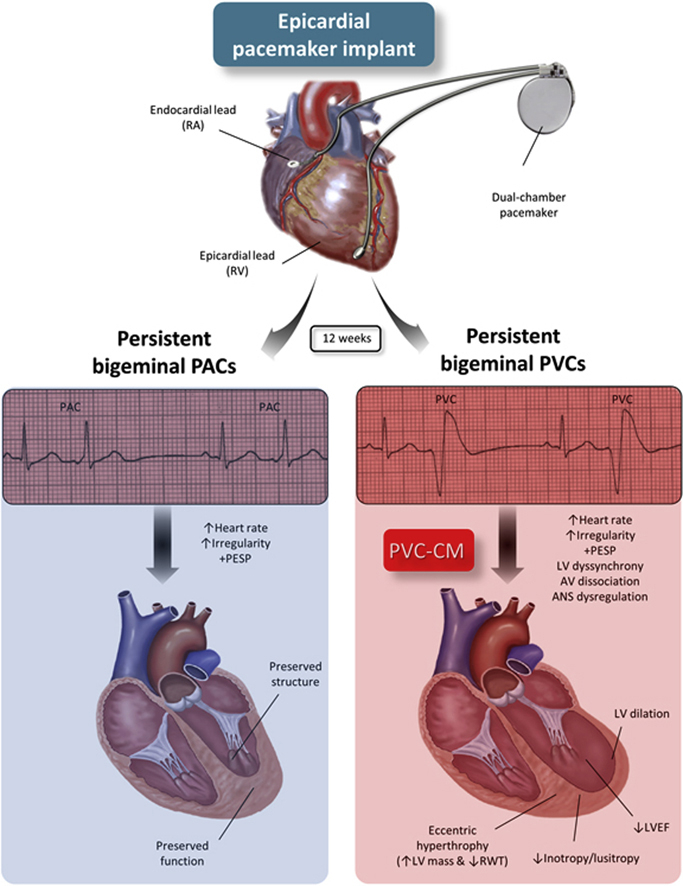
Key Findings.
-
▪
In contrast to frequent premature atrial contractions (PACs), frequent premature ventricular contractions (PVCs) can induce a cardiomyopathy characterized by reduced left ventricular (LV) ejection fraction, impaired contractility and relaxation (max and min dP/dt, respectively), and increase in systolic and diastolic chamber dimensions and LV mass index without relative wall thickening, indicative of eccentric hypertrophy.
-
▪
Frequent PVCs in a bigeminal pattern triggers LV cardiac remodeling that plateaus at week 12 without further deterioration beyond 16 weeks despite persistence of ventricular ectopy for nearly a year.
-
▪
In contrast to frequent PVCs, frequent PACs in a bigeminal pattern are unlikely to induce LV dysfunction despite similar tachycardia and heart rate irregularity. Thus, these phenomena do not seem to play a significant role in the development of PVC-induced cardiomyopathy.
Introduction
Frequent premature ventricular contractions (PVCs) are implicated in the development of nonischemic cardiomyopathy referred to as PVC-cardiomyopathy.1,2 The mechanism of PVC-cardiomyopathy has not yet been determined. Persistent tachycardia and heart rate irregularity, as well as post-extrasystolic potentiation, have been implicated as potential mechanisms for PVC-cardiomyopathy.2,3 This has been supported by the observation of cardiomyopathy in subjects with atrial fibrillation despite adequate rate control and case reports of frequent premature atrial contractions (PACs) causing cardiomyopathy.4,5
The aim of this study was to better understand the role of tachycardia and irregularity on the mechanism of PVC-cardiomyopathy as well as the long-term (12-month period) effect of bigeminal PVCs in left ventricular (LV) remodeling. Using our established premature pacing algorithm,6,7 we examined the effects of chronic exposure (3-month period, mid-term) to premature ventricular and atrial beats on cardiac structure and function. While both PVCs and PACs can result in tachycardia, heart rate irregularity, and post-extrasystolic potentiation, they differ in the presence of LV synchrony, which is characteristic of PVCs.7, 8, 9 We hypothesized that if irregularity and post-extrasystolic potentiation were necessary triggers for PVC-cardiomyopathy, frequent PACs should induce a similar reversible cardiomyopathy. Moreover, comparing PACs and PVCs would provide mechanistic insights into the role of dyssynchrony on the development of cardiomyopathy.
Methods
Experimental protocol
Thirty-one mongrel female canines (>9 months old and 21 kg) were implanted with a dual-chamber epicardial pacemaker to deliver PACs or PVCs as a clinically relevant model of atrial or ventricular bigeminy. An epicardial right ventricle (RV) lead and pulse generator with premature pacing algorithm were surgically implanted as previously described.6,10 Through a left thoracotomy, a bipolar steroid-coated lead (Medtronic model 4968; Medtronic, Inc., Minneapolis, MN) was inserted through the right atrial appendage and positioned in the high right atrium, while a sutureless bipolar Myopore™ (Greatbatch Medical, Alden, NY) epicardial bipolar lead was attached to the RV apex for each animal.
Electrocardiography (without ectopy) and invasive hemodynamic evaluation were obtained at baseline and a 24-hour Holter monitor was performed. After a 2-week surgical recuperation, animals were randomized into 3 groups: (1) 50% burden of RV apical PVCs at 200 ms (n = 11), (2) right atrial PACs at 200 ms (n = 7), or (3) sham (no ectopy, n = 9), using our previously validated premature pacing algorithm.6,10 Thus, PAC and PVC groups were exposed to the respective arrhythmia for a total of 12 weeks, while the sham group had no ectopy during the same study period. Echocardiograms (without ectopy) were repeated at weeks 4, 8, 12. At the end of the 12-week study protocol, a final 24-hour Holter, echocardiogram, and hemodynamic assessment were repeated. Four additional animals were exposed to bigeminal PVCs for nearly 1 year (48-week protocol) to assess the long-term effects of frequent PVCs on LV function and structure. These 4 animals exposed to a 48-week protocol only underwent additional echocardiograms at weeks 32, 40, and 48. The experimental model design and protocols are summarized in Figure 1.
Figure 1.

Experimental design and study protocol. Animal randomization into sham, premature atrial contraction (PAC), and premature ventricular contraction (PVC) groups for 12-week period and 4 additional animals exposed to a 48-week PVC period. RA = right atrium; RV = right ventricle.
All procedures were approved by the McGuire Institutional Animal Care and Use Committee in accordance with the provisions of the USDA Animal Welfare Act Regulations and Standards, PHS Policy, the Guide for the Care and Use of Laboratory Animals, and VA Policy.
Ambulatory Holter monitors and echocardiography
Three-lead ambulatory Holter monitors were obtained for at least 24 hours (GE SEER light Holters; GE, Boston, MA; GE, Boston, MA) at baseline and prior to the end of protocol. Holter data were quantified for mean heart rate (R-R interval) and PAC and PVC burden (GE MARS ECG analysis software; GE, Boston, MA).
Echocardiography was performed with a 5MHz probe using a commercial system (Vivid-7; GE, Boston, MA) and data were analyzed offline with EchoPAC software (EchoPAC v201; GE, Boston, MA). To assess chronic effects of frequent PVCs and PACs on LV function and structure, we performed echocardiograms during sinus rhythm at least 15 minutes after disabling the pacing algorithm. All parameters were obtained following the American Society of Echocardiography guidelines.11 LV systolic function was quantified by calculating LV ejection fraction (LVEF) using by the biplane method of disks (modified Simpson method). The LV relative wall thickness (RWT) was calculated as RWT = 2 ∗ posterior wall thickness / LVEDD, where the parameter LVEDD is left ventricular end-diastolic dimension. LV mass was calculated by 2 methods: (1) linear method (Cube formula), LV mass = 0.8 ∗ 1.04 (IVSEDD + LVEDD + PWEDD)3 − LVEDD3 + 0.6 g,12 where the parameter IVSEDD is interventricular septum end-diastolic dimension and PWEDD is posterior wall end-diastolic dimension; and (2) area-length method, LV mass = 1.05 ∗ 5/6 (A1 ∗ [LV length + t] − A2 ∗ LV length), where t = √(A1/π) − √(A2/π) represents the mean wall thickness, and A1 and A2 are epicardial and endocardial cross-sectional areas, respectively, obtained in short-axis view at the papillary muscle level (with the papillary muscles considered part of the LV cavity).11 LV cavity length is measured in an apical 4-chamber view as the distance from apex to the midmitral annulus plane (Supplemental Figure 1). LV mass index was normalized for variances in animal size (LV mass/kg body weight). Individual RWT versus LV mass index was plotted in the classic “LV remodeling diagram” to better describe the LV geometric remodeling phenotype associated with our model of PVC-Cardiomyopathy13. Doppler-ultrasound techniques, such as pulsed-wave and color flow imaging, were performed to measure the mitral inflow velocities (e.g., E/A ratio) and to assess mitral regurgitation (Supplemental Figure 1). Holter and echocardiogram data were reviewed by cardiologists blinded to the randomization arm.
Hemodynamic assessment
Percutaneous hemodynamic assessment was performed during survival and final surgeries under general anesthesia with isofluorane 1.5%–2% (Figure 1). An impedance-based multipolar catheter (Ventricath 507 5F; Millar Inc, Houston, TX) was introduced into the left ventricle through a carotid artery cutdown to assess LV diastolic and systolic pressures, contractility, and lusitropy (ie, max and min dP/dt, respectively) during sinus rhythm and after disabling premature pacing algorithm for at least 5 minutes in those animals randomized to PACs or PVCs.
Data and statistical analysis
Statistical analyses were performed using GraphPad Prism Software 8.0.2 (GraphPad, San Diego, CA). Normality of distributions was tested using the Shapiro-Wilk method and assessment of histograms and Q-Q plots. Normally distributed variables are presented as mean ± standard error of the mean and otherwise as median with interquartile range.
Groups of normally distributed data with equal variances were compared using 1-way analysis of variance with Bonferroni post hoc test. When examining the influence of 2 different categorical independent variables on a continuous dependent variable we performed 2-way analysis of variance for repeated measures with the Tukey comparison test to adjust for multiple comparisons within the model. Group means of continuous variables with unequal variances across groups or non-normally distributed data were compared using the Friedman test with Dunn’s multiple comparison test comparing the mean rank of each time point with the mean rank of baseline or between different groups. The relationship between different calculations of LV mass was tested using linear regression with Pearson correlation. Unadjusted 2-tailed P values < .05 were considered statistically significant.
Results
Ambulatory electrocardiogram Holter data
Ambulatory electrocardiogram Holter data of all groups are shown in Supplemental Table 1. As expected, animals with persistent atrial and ventricular bigeminy demonstrated a higher mean heart rate (HR) (119.1 ± 3.2 beats per minute [bpm] and 122.5 ± 4.4 bpm, respectively) compared to sham animals (99 ± 1.4 bpm, P = .0001). There was no difference in mean HR between PAC and PVC groups (P = .6). PAC burden was significantly higher in the PAC group vs PVC or sham groups (40.1%, 0.5%, and 3.5%, respectively; P < .001). Similarly, PVC burden was significantly higher in the PVC group compared to PAC or sham groups (49.8%, 0.4%, and 1.0%, respectively; P < .001).
Echocardiographic data
Figure 2, Figure 3, Figure 4 summarize all functional and structural parameters in sham, PAC, and PVC groups obtained throughout the 12- and 48-week protocols. Detailed echocardiographic data are presented in Supplemental Tables 2 and 3.
Figure 2.

Differences in left ventricular ejection fraction (LVEF) and geometry between study groups during the 12-week protocol. Progression of (A) LVEF, (B) LV end-diastolic volume (LVEDV), (C) LV end-systolic volume (LVESV), (D) relative wall thickness (RWT), and (E) LV mass index during 12 weeks of follow-up in sham, premature atrial contraction (PAC), and premature ventricular contraction (PVC) groups (mean ± standard error of the mean). Boxes next to individual values represent the median + interquartile range; P < .05 (∗) vs sham, (†) vs PAC, (‡) vs baseline, (§) vs week 4.
Figure 3.

Effect of long-term premature ventricular contractions (PVCs) on left ventricular ejection fraction (LVEF) and geometry. Progression of (A) LVEF, (B) LV end-diastolic (LVEDV) and (C) end-systolic volumes (LVESV), (D) LV mass index, and (E) relative wall thickness (RWT) after a long-term exposure to bigeminal PVCs (n = 4, 48-week follow-up). Solid lines represent each individual animal. Red dashed line represents mean value. (‡) P < .05 vs baseline.
Figure 4.

Left ventricular (LV) mass index and classic “LV remodeling diagram.” A: Progression of LV mass index throughout the 12-week protocol in sham, premature atrial contraction (PAC), and premature ventricular contraction (PVC) groups (mean ± standard error of the mean). B: Individual relative wall thickness (RWT) vs LV mass index is plotted in “LV remodeling diagram” to better describe the LV geometric hypertrophic phenotype associated with our model of PVC-induced cardiomyopathy. Dashed lines represent the 95th percentile threshold (P95) for each variable calculated from baseline values for all animals (n = 31), indicating 2 different thresholds (in the y and x axes) for RWT and LV mass index, respectively.13P < .05 (∗) vs sham, (†) vs PAC, (‡) vs baseline, (§) vs week 4. Abbreviations as in Figures 1 and 3.
LV ejection fraction
In contrast to both sham (58.8% ± 0.9%) and PAC (56.1% ± 1.9%) groups, LVEF significantly decreased in the PVC group (44.2% ± 1.7%, P < .001) at 12 weeks, while no significant difference was observed between sham and PAC groups (Figure 2A, Supplemental Table 2).
The decline in LVEF in the PVC group was significant at 4 weeks and continued to decrease during the following 12 weeks. However, the 48-week PVC protocol demonstrated that LVEF plateaus after 8 weeks without further deterioration beyond 16 weeks (Figure 3).
LV end-diastolic dimension and volume / LV end-systolic dimension and volume
After 12 weeks, the PVC group had a significant increase in LVEDD, LV end-diastolic volume (LVEDV), LV end-systolic dimension (LVESD), and LV end-systolic volume (LVESV) compared to PAC and sham groups (Figure 2B and 2C, Supplemental Figure 2 and Supplemental Table 2). LVEDD, LVEDV, LVESD, and LVESV followed a progression similar to LVEF, reaching significance at 4 weeks and increasing throughout the 12-week follow-up, reaching a plateau between weeks 8 and 16 (Figures 2 and 3).
LV wall thickness and mass index
No significant changes in either interventricular septal (IVSEDD) or posterior-wall thickness (PWEDD) were observed within or between groups (Supplemental Figure 2A and 2B, Supplemental Table 2). However, the RWT, a proportional ratio of wall thickness to LVEDD, was significantly decreased in the PVC group after 12 weeks of ventricular bigeminy (Figure 2D). Despite a decrease in RWT, LV mass index increased in the PVC group (P < .0001) compared to sham and PAC groups throughout the 12-week follow-up (Figure 4A), reaching an equilibrium between 8 and 16 weeks as noted on the 48-week PVC protocol (Figure 3D and 3E).
The increase in LV mass index in the PVC group was corroborated using both linear and area-length methods (R2 = 0.72, P < .0001, Supplemental Figure 3). This pattern of increased LV mass without increased RWT is consistent with eccentric hypertrophy (Figure 4B). None of the sham or PAC animals satisfied these criteria, and on average, they remained within normal (≤95th percentile threshold) values.
Pulsed-wave and color flow imaging
No significant change in E/A ratio in sham, PAC, or PVC groups were seen throughout the 12 weeks. However, the PVC group had lower E/A ratio at week 12 but only when compared to the PAC group (Supplemental Table 2). No significant mitral regurgitation was observed among the sham, PAC, and PVC groups.
Hemodynamic data
While LV end-diastolic and end-systolic pressures did not change significantly in sham, PAC, or PVC groups over the course of the 12-week study (Figure 5), the rates of pressure change (max and min dP/dt) were significantly decreased in the PVC group, suggestive of impaired contractility (max dP/dt 1355 ± 208 mm Hg/ms at baseline to 733 ± 59 mm Hg/ms, P < .0002) and lusitropy (min dP/dt from -1449 ± 83 mm Hg/ms at baseline to -921 ± 89 mm Hg/ms, P < .001).
Figure 5.

Changes in left ventricular (LV) pressure by different study groups. Hemodynamic changes in (A) LV end-diastolic pressure (LVEDP), (B) LV end-systolic pressure (LVESP), (C) LV contractility (max dP/dt), and (D) lusitropy (min dP/dt) in sham, premature atrial contraction (PAC), and premature ventricular contraction (PVC) groups at baseline and after 12 weeks. Maximum LV dP/dt decreased over the 12-week period in the PVC group, indicating decreased contractility during isovolumetric contraction (inotropy), while minimum LV dP/dt significantly decreased from baseline in the PVC group, suggestive of impaired relaxation (lusitropy). Boxes next to individual values represent the median + interquartile range. P < .05 (‡) vs baseline. Abbreviations as in Figure 1.
Discussion
PVC-cardiomyopathy, like any cardiomyopathy, leads to heart failure admissions and implantation of defibrillators and resynchronization devices.2 Recently, a secondary analysis of the CHF-STAT study suggests that PVC-cardiomyopathy is likely associated with increased mortality.14 This cardiomyopathy is characterized by a mild-to-moderate LV systolic dysfunction and LV dilatation, while limited data support the presence of LV diastolic dysfunction, mild mitral regurgitation, and left atrial enlargement.2,6,15 The diagnosis of PVC-cardiomyopathy can only be confirmed with resolution or improvement of LV dysfunction after elimination of PVCs.
The main contributions of this translational study are as follows: (1) PVC-cardiomyopathy is characterized by an increase in LV mass index and a decrease in RWT, consistent with a pattern of eccentric hypertrophy; (2) LV structural remodeling in PVC-cardiomyopathy reaches its plateau around 12–16 weeks in bigeminal PVCs; (3) chronic bigeminal PACs are not associated with cardiomyopathy; (4) tachycardia and irregularity are not major triggers of PVC-cardiomyopathy, since high-burden PACs (similar tachycardia and irregularity without LV dyssynchrony) do not induce cardiomyopathy (Graphical Abstract).
LV remodeling in PVC-cardiomyopathy
Frequent PVCs in a bigeminal pattern triggered LV systolic dysfunction with a specific structural LV remodeling (eccentric hypertrophy) in our canine model, which plateaus at week 12 without further changes beyond 16 weeks despite persistence of ventricular ectopy for nearly a year. This finding is in contrast to clinical studies where the development of PVC-cardiomyopathy is thought to take several months and up to years to develop.16,17 However, the timeline to develop PVC-cardiomyopathy in these clinical studies is based on the initiation of symptoms, which has its own limitations. One possible explanation of an earlier development of PVC-cardiomyopathy in our animal model is that our premature pacing algorithm exposes animals to a persistent uninterrupted ventricular bigeminy, while patients have intermittent and significantly lower PVC burden (10%–33%) and therefore may require longer exposure to develop PVC-cardiomyopathy. Thus, future animal studies will address the progression and time to develop PVC-cardiomyopathy with intermittent and lower PVC burden more representative of the clinical setting.
No prior clinical or preclinical studies have assessed changes in LV mass in PVC-cardiomyopathy, limiting their assessment to LV function and dimensions. An increase in LV mass found in our PVC-cardiomyopathy model was not surprising, since LV internal dimensions increased notably in association with LV systolic dysfunction without significant changes in LV wall thickness. The increase in LV mass was corroborated by a second and independent echocardiographic method (Supplemental Figure 3).11
To identify the LV remodeling pattern associated with PVC-cardiomyopathy, the classic “LV remodeling diagram”13,18 was used to integrate the information of LV volume, mass, and relative wall thickness. In this context, we found that more than 50% of animals (6/11) in the PVC group develop an eccentric hypertrophy pattern (Figure 4B). This hypertrophy pattern is supported by our previous findings in isolated cardiomyocytes of PVC-cardiomyopathy, as they had larger length-to-width ratios and higher cell capacitance than sham (225 ± 6 pF vs 189 ± 5 pF; P < .001), consistent with cellular enlargement.10 This suggests that cardiomyocytes undergo hypertrophy as a response to chronic stress of ventricular bigeminy. Although we found a significant diastolic impairment evaluated by min dP/dt and modestly but significant changes in E/A ratio in the PVC group, we did not observe significant changes in max LV end-diastolic pressure after 12 weeks of ventricular bigeminy.
Alterations in LV structure and geometry are seen in a variety of conditions and develop in a pattern that is unique to the “inciting overload.”18 Distinct basic patterns of cardiac hypertrophy occur in response to hemodynamic overload, depending on whether it is volume or pressure overload. An eccentric hypertrophic remodeling of the LV appears to be the usual adaptation mechanism to volume overload imposed either by physiologic (eg, exercise, normal pregnancy) or pathophysiological circumstances (eg, mitral or aortic regurgitation).18, 19, 20 Under volume overload conditions, increased diastolic wall stress leads to lengthening of cardiac myocytes with the addition of sarcomeres in series, thereby engendering increased LV dilatation.21 This remodeling pattern clearly contrasts with concentric hypertrophy owing to an increase in systolic wall stress caused by pressure overload (eg, with aortic stenosis or hypertension), which results in an increment in ventricular mass, high RWT, and LV wall thickening, with cardiomyocytes expansion by synthesis of new contractile proteins and the assembly of new sarcomeres in parallel.21
To the best of our knowledge, this is the first time that the PVC-cardiomyopathy-specific remodeling phenotype has been described. We can only speculate that the reduced stroke volume caused by PVC contraction (owing to lack of LV filling caused by prematurity) together with the compensatory pause after PVC leads to increased diastolic time and subsequent volume overload. The reduced cardiac output during ventricular bigeminy9 could also lead to incomplete LV emptying and volume overload despite subsequent increased contractility (post-extrasystolic potentiation and Frank-Starling mechanism in a lesser degree).8 Moreover, atrial contraction during mitral valve closure owing to PVC has been shown to cause a significant increase in pulmonary venous regurgitation flow.22 Consequently, this hemodynamic impairment can lead to the activation of renin-angiotensin-aldosterone and sympathetic nervous systems with known detrimental long-term effects on LV structure and function23,24 that could also trigger eccentric remodeling. These could be part of the substrate that contributes to the potential increase in morbidity and mortality associated with PVC-cardiomyopathy.14,25
Triggers and mechanism of PVC-cardiomyopathy
Various triggers have been implicated in PVC-cardiomyopathy, including (1) heart rate irregularity and/or higher mean heart rate2,6 with similar pathophysiology responsible for atrial fibrillation– and tachycardia-induced cardiomyopathy, respectively; (2) abnormal LV mechanics and dyssynchrony with subsequent disruption of LV wall motion synergy7,22,26,27; (3) increased intracellular calcium concentration and myocardial oxygen consumption associated with post-extrasystolic potentiation8; and (4) hemodynamic alterations with remodeling of the autonomic nervous system.23,28 Our premature pacing algorithm6,15 has allowed us to systematically study and compare the chronic effects of PACs and PVCs on LV function and dimensions.
Our study found a significant difference in LVEF, LV dimensions, RWT, and LV mass index between PVC- and PAC-treated subjects after 12 weeks despite similar mean HR, both of which were significantly higher compared to the sham-operated group. This is consistent with a recent clinical retrospective study and a preclinical study in a swine model where frequent PACs were not associated with cardiomyopathy.27,29 These findings are contradictory with the results of Pacchia and colleagues,30 where 4 of 5 animals exposed to a 50% PAC burden (using a dual-chamber pacemaker) developed LV dysfunction (LVEF <50%). Furthermore, the PVC-swine model demonstrated that bigeminal PVCs led to a significantly lower LVEF when compared to chronic RV apical pacing at 140 bpm. They went on to propose significant differences in QRS duration and LV dyssynchrony during PVC and confirmed molecular changes of calcium handling proteins, including upregulation of phosphorylated ryanodine receptor 2, sodium-calcium exchanger-1, Ca2+/calmodulin-dependent protein kinase II, and phospholamban, with downregulation of SERCA2a.10,27,31 Finally, our prior study with escalating PVC burdens demonstrated that PVC-cardiomyopathy was induced in half of the animals when exposed to 33% PVC burden despite a lack of tachycardia (mean HR was 80 ± 14 bpm).15 Thus, the evidence suggests that there are other mechanisms at play that trigger PVC-cardiomyopathy that cannot be explained by tachycardia and heart rate irregularity alone.
Limitations
This study simulates PVCs originating from the RV apex only. Both atrial and ventricular ectopy is persistent and uninterrupted, which could explain an earlier development of PVC-cardiomyopathy compared to clinical literature. Even though the premature pacing algorithm delivers effectively atrial bigeminy, not all PACs result in atrioventricular nodal conduction, which in part explains the lower than expected PAC reported by ambulatory Holter monitors. GE SEERS software can only assess PACs based on an R-R variability owing to the low amplitude of the P wave. Based on our experience, echocardiography is not a reliable tool to evaluate detailed RV function in canines. Thus, we solely evaluated LV function/mechanics as demonstrated in the clinical setting and other animal models.
Conclusion
In contrast to bigeminal PACs, PVCs result in a cardiomyopathy characterized by reduced LVEF, impaired contractility and relaxation (max and min dP/dt, respectively), and diastolic and systolic LV dilatation without relative wall thickening, consistent with eccentric hypertrophy that plateaus after 12 weeks without further deterioration beyond 16 weeks. The lack of cardiac remodeling in an animal model of bigeminal PACs suggests that tachycardia and heart rate irregularity are unlikely to play a significant role on the development of PVC-cardiomyopathy. Further studies are warranted to determine if these findings are reproducible in the clinical setting and reversible after elimination of frequent ventricular ectopy.
Funding Sources
NIH/NHLBI 1R01HL139874-01 (PI: Huizar), NIH/NHLBI 1R56HL133182-01 (PI: Huizar), AHA SDG 16SDG3128001 (PI: Tan), Research grant from Abbott, Inc. (PI: Huizar).
Disclosures
K Kaszala – Research support from Boston Scientific Corp (BSX) and St. Jude Medical (SJM); A Tan – Research support from BSX, Medtronic (MDT), and Biotronik, Inc. (BTK); KA Ellenbogen – Research support from BSX, Biosense Webster (BSW), MDT, SJM, NIH, Consultant for BSX, SJM, Atricure, Medtronic, Honoraria from MDT, BSX, BTK, BSW, and Atricure; JF Huizar – Research support from Abbott. The remaining co-authors do not have anything to disclose.
Authorship
All authors attest they meet the current ICMJE criteria for authorship.
Ethics statement
All procedures were approved by the McGuire Institutional Animal Care and Use Committee in accordance with the provisions of the USDA Animal Welfare Act Regulations and Standards, PHS Policy, the Guide for the Care and Use of Laboratory Animals, and VA Policy.
Footnotes
Supplementary data associated with this article can be found in the online version at https://doi.org/10.1016/j.hroo.2020.12.021.
Appendix. Supplementary data
References
- 1.Bozkurt B., Colvin M., Cook J. Current diagnostic and treatment strategies for specific dilated cardiomyopathies: a scientific statement from the American Heart Association. Circulation. 2016;134:e579–e646. doi: 10.1161/CIR.0000000000000455. [DOI] [PubMed] [Google Scholar]
- 2.Huizar J.F., Ellenbogen K.A., Tan A.Y., Kaszala K. Arrhythmia-induced cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol. 2019;73:2328–2344. doi: 10.1016/j.jacc.2019.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kowlgi G.N., Ramirez R.J., Kaszala K. Post-extrasystolic potentiation as a predictor of premature ventricular contraction-cardiomyopathy in an animal model. Europace. 2020;22:813–820. doi: 10.1093/europace/euaa025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cha Y.M., Redfield M.M., Shen W.K., Gersh B.J. Atrial fibrillation and ventricular dysfunction: a vicious electromechanical cycle. Circulation. 2004;109:2839–2843. doi: 10.1161/01.CIR.0000132470.78896.A8. [DOI] [PubMed] [Google Scholar]
- 5.Liuba I., Schaller R.D., Frankel D.S. Premature atrial complex-induced cardiomyopathy: Case report and literature review. HeartRhythm Case Rep. 2020;6:191–193. doi: 10.1016/j.hrcr.2019.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huizar J.F., Kaszala K., Potfay J. Left ventricular systolic dysfunction induced by ventricular ectopy: a novel model for premature ventricular contraction-induced cardiomyopathy. Circ Arrhythm Electrophysiol. 2011;4:543–549. doi: 10.1161/CIRCEP.111.962381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Potfay J., Kaszala K., Tan A.Y. Abnormal left ventricular mechanics of ventricular ectopic beats: insights into origin and coupling interval in premature ventricular contraction-induced cardiomyopathy. Circ Arrhythm Electrophysiol. 2015;8:1194–1200. doi: 10.1161/CIRCEP.115.003047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cooper M.W. Postextrasystolic potentiation. Do we really know what it means and how to use it? Circulation. 1993;88:2962–2971. doi: 10.1161/01.cir.88.6.2962. [DOI] [PubMed] [Google Scholar]
- 9.Takada H., Takeuchi S., Ando K., Kaito A., Yoshida S. Experimental studies on myocardial contractility and hemodynamics in extrasystoles. Jpn Circ J. 1970;34:419–430. doi: 10.1253/jcj.34.419. [DOI] [PubMed] [Google Scholar]
- 10.Wang Y., Eltit J.M., Kaszala K. Cellular mechanism of premature ventricular contraction-induced cardiomyopathy. Heart Rhythm. 2014;11:2064–2072. doi: 10.1016/j.hrthm.2014.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lang R.M., Badano L.P., Mor-Avi V. Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J Am Soc Echocardiogr. 2015;28:1–39. doi: 10.1016/j.echo.2014.10.003. e14. [DOI] [PubMed] [Google Scholar]
- 12.Devereux R.B., Alonso D.R., Lutas E.M. Echocardiographic assessment of left ventricular hypertrophy: comparison to necropsy findings. Am J Cardiol. 1986;57:450–458. doi: 10.1016/0002-9149(86)90771-x. [DOI] [PubMed] [Google Scholar]
- 13.Ganau A., Devereux R.B., Roman M.J. Patterns of left ventricular hypertrophy and geometric remodeling in essential hypertension. J Am Coll Cardiol. 1992;19:1550–1558. doi: 10.1016/0735-1097(92)90617-v. [DOI] [PubMed] [Google Scholar]
- 14.Huizar JF, Fisher SG, Ramsey FV, et al. Outcomes of premature ventricular contraction–cardiomyopathy in the veteran population: a secondary analysis of the CHF-STAT Study. J Am Coll Cardiol Clin Electrophysiol. https://doi.org/10.1016/j.jacep.2020.08.028 2020. 2020.11.25. [DOI] [PMC free article] [PubMed]
- 15.Tan A.Y., Hu Y.L., Potfay J. Impact of ventricular ectopic burden in a premature ventricular contraction-induced cardiomyopathy animal model. Heart Rhythm. 2016;13:755–761. doi: 10.1016/j.hrthm.2015.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ban J.E., Park H.C., Park J.S. Electrocardiographic and electrophysiological characteristics of premature ventricular complexes associated with left ventricular dysfunction in patients without structural heart disease. Europace. 2013;15:735–741. doi: 10.1093/europace/eus371. [DOI] [PubMed] [Google Scholar]
- 17.Carballeira Pol L., Deyell M.W., Frankel D.S. Ventricular premature depolarization QRS duration as a new marker of risk for the development of ventricular premature depolarization-induced cardiomyopathy. Heart Rhythm. 2014;11:299–306. doi: 10.1016/j.hrthm.2013.10.055. [DOI] [PubMed] [Google Scholar]
- 18.Gaasch W.H., Zile M.R. Left ventricular structural remodeling in health and disease: with special emphasis on volume, mass, and geometry. J Am Coll Cardiol. 2011;58:1733–1740. doi: 10.1016/j.jacc.2011.07.022. [DOI] [PubMed] [Google Scholar]
- 19.Konstam M.A., Kramer D.G., Patel A.R., Maron M.S., Udelson J.E. Left ventricular remodeling in heart failure: current concepts in clinical significance and assessment. JACC Cardiovasc Imaging. 2011;4:98–108. doi: 10.1016/j.jcmg.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 20.Mihl C., Dassen W.R., Kuipers H. Cardiac remodelling: concentric versus eccentric hypertrophy in strength and endurance athletes. Neth Heart J. 2008;16:129–133. doi: 10.1007/BF03086131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grossman W., Paulus W.J. Myocardial stress and hypertrophy: a complex interface between biophysics and cardiac remodeling. J Clin Invest. 2013;123:3701–3703. doi: 10.1172/JCI69830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuroki K., Tada H., Seo Y. Prediction and mechanism of frequent ventricular premature contractions related to haemodynamic deterioration. Eur J Heart Fail. 2012;14:1112–1120. doi: 10.1093/eurjhf/hfs095. [DOI] [PubMed] [Google Scholar]
- 23.Tan A.Y., Elharrif K., Cardona-Guarache R. Persistent proarrhythmic neural remodeling despite recovery from premature ventricular contraction-induced cardiomyopathy. J Am Coll Cardiol. 2020;75:1–13. doi: 10.1016/j.jacc.2019.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mann D.L. Left ventricular size and shape: determinants of mechanical signal transduction pathways. Heart Fail Rev. 2005;10:95–100. doi: 10.1007/s10741-005-4636-y. [DOI] [PubMed] [Google Scholar]
- 25.Huizar J.F., Ellenbogen K.A. Is PVC-induced cardiomyopathy truly reversible?: A deep dive into questions that remain unanswered. JACC Clin Electrophysiol. 2020;6:1377–1380. doi: 10.1016/j.jacep.2020.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kanei Y., Friedman M., Ogawa N., Hanon S., Lam P., Schweitzer P. Frequent premature ventricular complexes originating from the right ventricular outflow tract are associated with left ventricular dysfunction. Ann Noninvasive Electrocardiol. 2008;13:81–85. doi: 10.1111/j.1542-474X.2007.00204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walters T.E., Rahmutula D., Szilagyi J. Left ventricular dyssynchrony predicts the cardiomyopathy associated with premature ventricular contractions. J Am Coll Cardiol. 2018;72:2870–2882. doi: 10.1016/j.jacc.2018.09.059. [DOI] [PubMed] [Google Scholar]
- 28.Hamon D., Rajendran P.S., Chui R.W. Premature ventricular contraction coupling interval variability destabilizes cardiac neuronal and electrophysiological control: insights from simultaneous cardioneural mapping. Circ Arrhythm Electrophysiol. 2017;10 doi: 10.1161/CIRCEP.116.004937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gunda S., Akyeampong D., Gomez-Arroyo J. Consequences of chronic frequent premature atrial contractions: Association with cardiac arrhythmias and cardiac structural changes. J Cardiovasc Electrophysiol. 2019;30:1952–1959. doi: 10.1111/jce.14067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pacchia C.F., Akoum N.W., Wasmund S., Hamdan M.H. Atrial bigeminy results in decreased left ventricular function: an insight into the mechanism of PVC-induced cardiomyopathy. Pacing Clin Electrophysiol. 2012;35:1232–1235. doi: 10.1111/j.1540-8159.2012.03466.x. [DOI] [PubMed] [Google Scholar]
- 31.Jiang M., Zhang M., Howren M. JPH-2 interacts with Cai-handling proteins and ion channels in dyads: Contribution to premature ventricular contraction-induced cardiomyopathy. Heart Rhythm. 2016;13:743–752. doi: 10.1016/j.hrthm.2015.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.