

Abstract
Background
Hereditary elliptocytosis (HE) is a heterogeneous red blood cell membrane disorder characterized by the presence of elliptocytes on a peripheral blood smear. Clinical manifestations of HE vary widely from asymptomatic carriers to patients with severe transfusion‐dependent anemia. Most patients are asymptomatic or have mild anemia, which hinders diagnosis. The proband in this case had mild anemia and jaundice over a period of 4 years, the etiology of which was unclear. Hence, he was admitted to our hospital for further diagnosis.
Methods
Peripheral blood smears and routine blood tests were performed and biochemical parameters of the proband, and his family members were determined. To confirm the diagnosis, gene mutations were screened in the proband using next‐generation sequencing (NGS) and verified by Sanger sequencing in other family members.
Results
A novel mutation (c.1294delA, p.Ser432 fs) in exon 15 of the EPB41 gene was detected in the proband and his family members. This mutation results in a frameshift and a premature stop codon at position 455, encoding a truncated protein. The variant was likely pathogenic according to the criteria of the American College of Medical Genetics and Genomics. SWISS‐MODEL protein structure prediction indicated partial loss of the spectrin and actin binding and C‐terminal domains.
Conclusion
A heterozygous mutation 1294delA in exon 15 of the EPB41 gene was identified using NGS and Sanger sequencing in members of a Chinese family. This identification expands the spectrum of EPB41 mutations and contributes to the genetic diagnosis of families with HE.
Keywords: erythrocyte protein band 4.1 gene, hemolytic anemia, hereditary elliptocytosis, mutation
1. INTRODUCTION
Hereditary elliptocytosis (HE) is characterized by the presence of elliptical‐shaped erythrocytes (elliptocytes) on a peripheral blood smear. HE is associated with variable clinical manifestations. 1 The prevalence of HE is higher in malaria‐endemic regions, with probable association with protection against malarial infection. 2 HE has been reported in 1.6% of the native population in Benin, West Africa. 3 It has also been detected in populations in Mediterranean countries, the Middle East, Japan, Korea, the Indian subcontinent, and the Far East. 4 , 5 The true incidence of HE is unknown because many affected patients are asymptomatic. In China, HE is a relatively rare disease. No incidence has been reported, and only individual family cases or sporadic cases have been described. 6 , 7
The typical clinical characteristics of HE include splenomegaly, jaundice, and anemia. Most patients present with no anemia or hemolysis. The diagnosis of HE is made incidentally, after worsening of anemia due to infections or after diagnosis in symptomatic relatives. Severe anemia is observed only in rare cases. Mutations in several genes, including those that encode protein 4.1R and α‐ and β‐spectrin proteins, are associated with this condition. 8
In this study, we identified a novel EPB41 gene mutation responsible for HE in a Chinese family using next‐generation sequencing (NGS). We summarize the clinical features and laboratory results of the proband and his family, which can be used as a reference for the diagnosis of HE.
2. MATERIALS AND RESULTS
2.1. Patients
The proband was an 88‐year‐old male with mild anemia and jaundice for more than 4 years. The etiology was unclear, which prompted his visit to our hospital for further diagnosis. Abdominal ultrasonography revealed mild splenomegaly and gallstones. Hematological investigations showed only mild anemia (white blood cell count 3.88×109/L, red blood cell count 2.80×1012/L, hemoglobin 100 g/L, platelet count 102×109/L, and reticulocyte percentage of 2.5%). Elliptocytes were readily identifiable and accounted for 86% of the total erythrocyte population (Figure 1A). Biochemical tests indicated a slight elevation of bilirubin, mainly an indirect reaction (total bilirubin, 24.2 μmol/L; indirect bilirubin, 18.2 μmol/L). We ruled out iron deficiency or megaloblastic anemia based on laboratory findings of serum iron (96 µg/dl, reference range: 50–150 µg/dl), ferritin (256.91 ng/ml, reference range: 23.9–336.2 ng/ml), vitamin B12 (716.24 pmol/L, reference range 133–675 pmol/L), and folate (36.81 nmol/L, reference range: 7.0–45.1 nmol/L). Glucose‐6‐phosphate dehydrogenase (G‐6‐PD) screening test and Coombs test were negative. No abnormalities were detected by hemoglobin electrophoresis and thalassemia gene analyses. Bone marrow examination indicated erythroid hyperplasia with a G:E ratio of 0.77:1. Cytogenetic studies revealed a normal karyotype (46, XY). The clinical manifestations and laboratory results were highly suspicious of HE. This prompted examination of family members and genetic testing to confirm the diagnosis.
FIGURE 1.
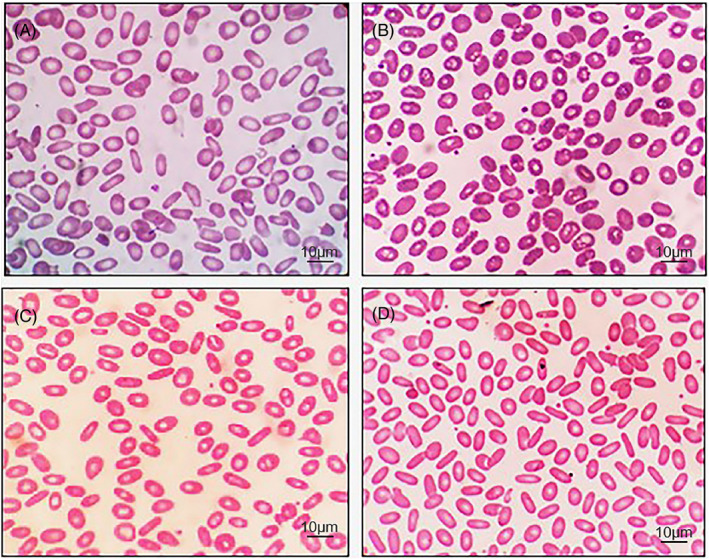
Peripheral blood smear of the proband and his family members (Wright‐Giemsa stain, original magnification ×1000): (A) proband, (B) proband's son, (C) proband's daughter, and (D) proband's granddaughter
In addition to the proband, three patients in the family were diagnosed with HE. The pedigree chart is shown in Figure 2. The proband's 55‐year‐old son (case II‐2) did not have anemia or jaundice but had gallstones. Elliptocytes accounted for 48% of the total erythrocyte population (Figure 1B). The proband's 53‐year‐old daughter (case II‐3) did not have anemia but had mild jaundice and gallstones. Elliptocytes accounted for 78% of the total erythrocyte population (Figure 1C). The proband's 30‐year‐old granddaughter (case III‐1) did not have anemia or jaundice but had gallstones. Elliptocytes accounted for 92% of the total erythrocyte population (Figure 1D). Laboratory test results of the proband and his family members are presented in Table 1.
FIGURE 2.
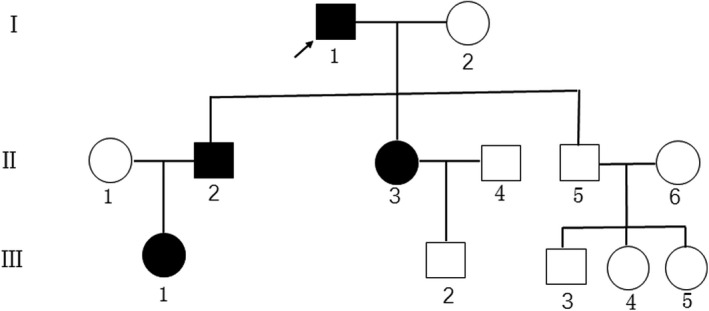
Pedigree of the family affected by hereditary elliptocytosis. The affected individuals are indicated by a black‐filled circle (females) or square (males). The proband (I‐1) is indicated by an arrow
TABLE 1.
Laboratory test results of the proband and his family members
| Test | Proband | Son | Daughter | Granddaughter | Normal range |
|---|---|---|---|---|---|
| WBC (×109/L) | 3.88 | 4.14 | 5.04 | 6.91 | 3.5–9.5 |
| RBC (×1012/L) | 2.8 | 5.11 | 4.11 | 4.32 | 4.3–5.8 |
| HGB (g/L) | 100 | 156 | 135 | 121 | 120–160 |
| MCV (fL) | 101.1 | 87.7 | 92.5 | 85.6 | 82–100 |
| MCH (pg) | 35.9 | 30.5 | 32.9 | 28.0 | 27–34 |
| MCHC (g/L) | 355 | 348 | 355 | 327 | 316–354 |
| PLT (×109/L) | 102 | 214 | 207 | 276 | 100–300 |
| RET (%) | 2.5 | 1.0 | 2.1 | 1.2 | 0.5–1.5 |
| Percentage of elliptocytes (%) | 86 | 48 | 78 | 92 | <2% |
| TBIL (µmol/L) | 24.20 | 13.00 | 21.92 | 12.30 | 1.7–20.5 |
| IBIL (µmol/L) | 18.20 | 8.20 | 18.26 | 8.60 | 0.00–14.0 |
Abbreviations: HGB, hemoglobin; IBIL, indirect bilirubin; MCH, mean corpuscular hemoglobin; MCHC, mean corpuscular hemoglobin concentration; MCV, mean corpuscular volume; PLT, platelets; RBC, red blood cells; RET, reticulocytes; TBIL, total bilirubin; WBC, white blood cells.
2.2. Genetic analysis
NGS was performed to detect all exons of 4811 genes, including ANK1, GYPC, EPB42, SPTB, SLC4A1, EPB41, and SPTA1. For this, 2 ml of peripheral blood of the proband was drawn into an EDTA‐K2 anticoagulated tube. Genomic DNA was extracted from whole blood using a QIAamp DNA Blood Mini Kit (Qiagen), and was fragmented to an average length of 180–280 bp using a Covaris crusher. After the ends were repaired and A tails were added, DNA fragments were ligated to the ends of the fragments to prepare DNA libraries. Exomes were captured using a TruSight One Sequencing Panel (Illumina Inc) according to the manufacturer's instructions. A Novaseq 6000 platform (Illumina) was used for genomic DNA sequencing to generate 150‐bp paired‐end reads. After sequencing, FASTQ data were obtained from an Illumina sequencing machine using bcl2fastq (Version 2.0.1). The acquired data were cleaned by processing using Trimmomatic (Version 0.36) to remove low‐quality reads, bases, trimming adaptors, and other data. The clean data were processed by the Burrows‐Wheeler Aligner using a reference sequence of hg19 and Genome Analysis Toolkit (GATK) for variant calling. Finally, the variant call format files were analyzed using ANNOVAR tools containing annotation databases, such as the 1000‐genome database, dbSNP database, ClinVar database, Polymorphism Phenotyping v2 database, and the Sorting Intolerant from the Tolerant database. Variations were screened using SIFT, Polyphen, Mutation Taster, and CADD software. Potentially deleterious variations were reserved if the score of more than half of these four software programs supported the harmfulness of the variation. To better predict the harmfulness of variation, the classification system of the American College of Medical Genetics and Genomics (ACMG) was used. The ACMG classifications of variations include pathogenic, likely pathogenic, uncertain significance, likely benign, and benign. Based on the patient's clinical history, variants in candidate genes linked to hyperbilirubinemia and HE were prioritized. Candidate mutations were identified. PCR primers were designed to amplify the suspected candidate mutation sites, and Sanger sequencing was performed for verification.
The NGS results met the quality control standard. Details are shown in Table 2. A heterozygous mutation (c.1294delA, p. Ser432 fs) in exon 15 of the EPB41 gene was identified in the proband. The variant was likely pathogenic according to the ACMG criteria. The mutation was further confirmed by Sanger sequencing. The same mutation was found in the proband's family members (cases II‐2 and II‐3). The other healthy members (cases II‐5 and III‐2) showed no variation at this site. The granddaughter of the proband (case III‐1) did not undergo genetic analysis due to personal reasons. The results of Sanger sequencing are shown in Figure 3.
TABLE 2.
Quality control results of next‐generation sequencing
| Data category | Results |
|---|---|
| Raw data (G) | 17.27 |
| Raw depth (X) | 285.66 |
| Q20 (%) | 97.38 |
| Q30 (%) | 93.36 |
| Initial bases on target (n) | 60456963 |
| Base covered on target (n) | 60234106 |
| Coverage of target region (%) | 99.6 |
| Effective bases on target (Mb) | 12512.09 |
| Fraction of effective bases on target (%) | 73.9 |
| Average sequencing depth on target | 206.96 |
| Fraction of target covered with at least 20× (%) | 95.4 |
FIGURE 3.

DNA sequence analysis of the EPB41 gene. A novel mutation in exon 15 (c.1294delA) of the EPB41 gene was found in the proband and his family members. Arrows indicate point mutations. (A) Reference sequence, (B) proband, (C) proband's son, and (D) proband's daughter
2.3. Structure prediction of the mutant protein
The SWISS‐MODEL homology modeling program (http://swissmodel.expasy.org) was used to develop an appropriate model to mimic the effects of the mutated region. Sequence alignment revealed that the c.1294delA mutation resulted in a substitution of amino acid 432, changing it from serine to valine, and converting the codon 445 to a stop codon, resulting in a truncated protein (Figure 4A). Structure prediction of the truncated protein demonstrated a lack of part of the spectrin and actin binding (SAB) domain and C‐terminal domain (CTD) (Figure 4B).
FIGURE 4.

The mutant protein structure. (A) The wild‐type protein consists of 588 amino acids. The c.1294delA mutation causes a substitution of the 432nd amino acid, changing it from serine to valine, and converting codon 445 to a stop codon, resulting in a truncated protein. (B) The mutant protein (p.Ser432fs) was predicted to result in partial loss of the SAB domain and CTD using SWISS‐MODEL online software compared to the wild type
3. DISCUSSION
Clinical manifestations of HE vary widely from asymptomatic carriers to patients with severe transfusion‐dependent anemia. HE is nearly always asymptomatic, however, approximately 10% of patients have moderate to severe anemia, including a few reported cases of hydrops fetalis anemia. Typically, individuals with heterozygosity for an elliptocytic variant are asymptomatic. Individuals with homozygosity or compound heterozygosity for HE variants experience mild to severe anemia, including severe variant hereditary pyropoikilocytosis. 9 , 10 HE is typically diagnosed incidentally during testing for unrelated conditions. The erythrocyte life span is normal in most patients. Symptomatology may vary between members of the same family and is attributed to modifier alleles that influence the expression of spectrin. In the present study, the proband displayed only mild anemia and jaundice. The other three family members had compensated for anemia. This can be related to their genetic types and heterozygous mutations. All patients had gallstones, suggesting chronic hemolysis. The family investigation revealed HE in all three generations, with both sexes affected. These findings are consistent with HE as a genetically and phenotypically heterogeneous hemolytic anemia.
The mechanistic basis for decreased membrane mechanical stability in HE is weakened “horizontal” linkages in the membrane skeleton because of either defective spectrin dimer‐dimer interactions or a defective spectrin‐actin‐protein 4.1R junctional complex. 1 Spectrin is a primary red blood cell (RBC) cytoskeleton protein composed of α‐β heterodimers assembled in an antiparallel fashion into flexible rods that self‐associate head‐to‐head to form tetramers. 11 , 12 Binding of the spectrin tetramer with actin at the junctional complex is mediated by 4.1R. Binding is essential for the stability of membranes of RBCs. 13 Defects in the spectrin‐protein 4.1R‐actin complex weaken the horizontal cytoskeletal associations, causing decreased mechanical stability and deformability of erythrocytes. 14 HE is caused by mutations in the SPTA1 and SPTB genes, which produce qualitative defects of α‐ and β‐spectrin, respectively, and by a mutation in the EPB41 gene, causing quantitative or qualitative defects of protein 4.1R.
Mutations in α‐spectrin are the most common cause of HE, accounting for approximately 65% of cases. Missense mutations most frequently occur in the amino‐terminal region of α‐spectrin that is involved in spectrin dimer‐dimer interactions. Mutations in other parts of the α‐spectrin that are not directly involved in spectrin self‐association have also been identified. Mutations in β‐spectrin account for 30% of HE cases. Point mutations and truncations in the carboxyl‐terminus of β‐spectrin, which impair spectrin self‐association, have been identified. Heterozygous mutations in this region of β‐spectrin are associated with variable clinical severity, whereas in the homozygous state, they are fatal or near‐fatal. Importantly, in almost all cases of spectrin mutations associated with HE, the degree of spectrin self‐association disruption correlates with clinical severity. The larger the degree of disruption, the more severe the clinical phenotype. Quantitative deficiency of protein 4.1R and qualitative defects in protein 4.1R account for 5% of HE cases. In all these instances, defects in protein 4.1R lead to a weakened spectrin‐actin junctional complex, which in turn leads to decreased membrane mechanical stability. 15 , 16 Black populations have a higher incidence of spectrin defects, whereas protein 4.1R abnormalities are prevalent among Caucasians. Patients with HE having spectrin defects tend to present a more severe clinical phenotype than those with protein 4.1R deficiency. 17
Protein 4.1R consists of 588 amino acid residues, including the B41, FERM domain, FERM adjacent (FA), SAB domain, and CTD. It is a product of the human gene EPB41. This gene is located on chromosome 1p35.3 and contains 28 exons. Fifteen pathogenic mutations in the EPB41 gene are included in the Human Gene Mutation Database. Five of these are missense mutations, one is a splicing mutation, one is a regulatory mutation, three are small deletions, four are gross deletions, and one is a gross insertion. In our study, the heterozygous mutation 1294delA in exon 15 of the EPB41 gene was found in the proband and his family members. The mutation was not evident in the ESP6500, 1000 genome, Human Genome, and dbSNP databases, suggesting that it could be a novel mutation. This mutation causes a frameshift and converts the codon 445 to a stop codon, resulting in a protein that is truncated from 588 to 445 amino acids. An important SAB functional domain and CTD are missing. These domains bind to both spectrin and actin. Truncated proteins are highly unstable and can be rapidly degraded. Therefore, the frameshift mutation is expected to have a significant impact on the structure and function of 4.1R.
Generally, most individuals with single heterozygous EPB41 gene mutations are asymptomatic or have mild forms of anemia, such as the proband and his family members. Only homozygotes or compound heterozygotes for HE variants exhibit moderate to severe hemolysis and require blood transfusion. Lacy and colleagues 18 presented a case of a 3‐year‐old male with lifelong and severe transfusion‐dependent anemia of unclear etiology, despite extensive clinical workup. Given the difficulty in making the diagnosis and the potential side effects of performing interventions in patients with congenital anemia of unknown etiology, the researchers opted to perform whole‐exome sequencing on the patient and his parents. The sequencing identified a homozygous loss‐of‐function mutation in the EPB41 gene (c.444_450delGAATCAG, p.Asn149fs), which caused a rare and severe form of HE in the patient.
4. CONCLUSIONS
NGS revealed a novel mutation in the EPB41 gene in a Chinese family. This is the first clinical case to confirm the diagnosis of HE using molecular analysis in the Chaoshan region of southern China. This identification expands the spectrum of EPB41 mutations and contributes to the genetic diagnosis of families with HE. More data need to be collected from HE patients to fully understand the genetic distributions and genotype‐phenotype correlations.
CONFLICT OF INTEREST
The authors declare that there are no conflicts of interest regarding the publication of this paper.
AUTHOR CONTRIBUTIONS
Zhanqin Huang and Manxiong Cao contributed equally to this work. Zhanqin Huang and Dongqing Zhang designed the study, analyzed the data, and drafted the manuscript. Manxiong Cao, Huanbing Zhou, Jinghua Lin, and Dongqing Zhang were involved in collecting patient information and analyzing the data.
ACKNOWLEDGMENTS
This work was supported in part by research grants from the National Natural Science Foundation of China (No. 30901810).
Manxiong Cao and Zhanqin Huang contributed equally.
DATA AVAILABILITY STATEMENT
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Gallagher PG. Hereditary elliptocytosis: spectrin and protein 4.1R. Semin Hematol. 2004;41(2):142‐164. [DOI] [PubMed] [Google Scholar]
- 2. Dhermy D, Schrevel J, Lecomte MC. Spectrin‐based skeleton in red blood cells and malaria. Curr Opin Hematol. 2007;14(3):198‐202. [DOI] [PubMed] [Google Scholar]
- 3. Glele‐Kakai C, Garbarz M, Lecomte MC, et al. Epidemiological studies of spectrin mutations related to hereditary elliptocytosis and spectrin polymorphisms in Benin. Br J Haematol. 1996;95(1):57‐66. [DOI] [PubMed] [Google Scholar]
- 4. Yawata Y. Characteristics of red cell membrane disorders in the Japanese population. Rinsho Byori. 1997;45(4):367‐376. [PubMed] [Google Scholar]
- 5. Park ES, Jung HL, Kim HJ, et al. Hereditary hemolytic anemia in Korea from 2007 to 2011: A study by the Korean Hereditary Hemolytic Anemia Working Party of the Korean Society of Hematology. Blood Res. 2013;48(3):211‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ma S, Qin J, Wei A, et al. Novel compound heterozygous SPTA1 mutations in a patient with hereditary elliptocytosis. Mol Med Rep. 2018;17(4):5903‐5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang X, Liu A, Huang M, Shen N, Lu Y, Hu Q. Hereditary elliptocytosis with variable expression and incomplete penetrance in a Chinese family. Br J Haematol. 2019;186(5):e159‐e162. [DOI] [PubMed] [Google Scholar]
- 8. Gaetani M, Mootien S, Harper S, Gallagher PG, Speicher DW. Structural and functional effects of hereditary hemolytic anemia‐associated point mutations in the alpha spectrin tetramer site. Blood. 2008;111(12):5712‐5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Baklouti F, Moriniere M, Haj‐Khelil A, et al. Homozygous deletion of EPB41 genuine AUG‐containing exons results in mRNA splicing defects, NMD activation and protein 4.1R complete deficiency in hereditary elliptocytosis. Blood Cells Mol Dis. 2011;47(3):158‐165. [DOI] [PubMed] [Google Scholar]
- 10. Niss O, Chonat S, Dagaonkar N, et al. Genotype‐phenotype correlations in hereditary elliptocytosis and hereditary pyropoikilocytosis. Blood Cells Mol Dis. 2016;61:4‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cherry L, Menhart N, Fung LW. Interactions of the alpha‐spectrin N‐terminal region with beta‐spectrin. Implications for the spectrin tetramerization reaction. J Biol Chem. 1999;274(4):2077‐2084. [DOI] [PubMed] [Google Scholar]
- 12. Ipsaro JJ, Harper SL, Messick TE, Marmorstein R, Mondragon A, Speicher DW. Crystal structure and functional interpretation of the erythrocyte spectrin tetramerization domain complex. Blood. 2010;115(23):4843‐4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Da Costa L, Galimand J, Fenneteau O, Mohandas N. Hereditary spherocytosis, elliptocytosis, and other red cell membrane disorders. Blood Rev. 2013;27(4):167‐178. [DOI] [PubMed] [Google Scholar]
- 14. Iolascon A, King MJ, Robertson S, et al. A genomic deletion causes truncation of alpha‐spectrin and ellipto‐poikilocytosis. Blood Cells Mol Dis. 2011;46(3):195‐200. [DOI] [PubMed] [Google Scholar]
- 15. An X, Mohandas N. Disorders of red cell membrane. Br J Haematol. 2008;141(3):367‐375. [DOI] [PubMed] [Google Scholar]
- 16. Andolfo I, Russo R, Gambale A, Iolascon A. New insights on hereditary erythrocyte membrane defects. Haematologica. 2016;101(11):1284‐1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Delaunay J, Dhermy D. Mutations involving the spectrin heterodimer contact site: clinical expression and alterations in specific function. Semin Hematol. 1993;30(1):21‐33. [PubMed] [Google Scholar]
- 18. Lacy JN, Ulirsch JC, Grace RF, et al. Exome sequencing results in successful diagnosis and treatment of a severe congenital anemia. Cold Spring Harb Mol Case Stud. 2016;2(4):a000885. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.