

Abstract
We report the first Fe─CPh3 complex, and show that the long Fe─C bond can be disrupted by neutral π-acceptor ligands (benzophenone and phenylacetylene) to release the triphenylmethyl radical. The products are formally iron(I) complexes, but X-ray absorption spectroscopy coupled with density functional and multireference ab initio calculations indicates that the best description of all the complexes is iron(II). In the formally iron(I) complexes, this does not imply that the π-acceptor ligand has radical character, because the iron(II) description arises from doubly-occupied frontier molecular orbitals that are shared equitably by the iron and the π-acceptor ligand, and the unpaired electrons lie on the metal. Despite the lack of substantial radical character on the ligands, alkyne and ketone fragments can couple to form a high-spin iron(III) complex with a cyclized metalladihydrofuran core.
Graphical Abstract
INTRODUCTION
The assignment of ligands as "anionic" or "neutral" in the ionic electron counting formalism is typically done using rules about whether the free fragment has an all-octet Lewis structure. The assignment is more challenging to accomplish experimentally, because metal-ligand bonds have substantial covalency.1 With "anionic" alkyl ligands, the formal assignment of ligands as anionic is problematic because transition metals have electronegativities similar to carbon. With many "neutral" π-ligands, on the other hand, the assignment as neutral is also complicated because of π-backbonding as well as the potential for radical character on the ligand from electron transfer into low-lying π* orbitals of the ligand.2 It is challenging to find even-handed ways to compare formally "anionic" and "neutral" ligands to one another,3 particularly if one wants experimental comparisons that leave the metal in the same environment. For example, a ligand substitution reaction that exchanges a neutral ligand with alkyl− changes the overall charge on the complex, while exchanging a neutral ligand with alkyl• formally changes the oxidation state on the metal by one unit, and produces a radical that is typically unstable.
For almost two decades, we have studied the chemistry of iron complexes supported by bulky β-diketiminate ligands (Chart 1),4 which are ideal for addressing these questions because β-diketiminate complexes are often stable in a variety of formal oxidation levels with both odd- and even-electron counts.5 Some of the formally iron(I) complexes are stabilized through backbonding or electron transfer into unsaturated ligands such as nitriles,6 alkynes,7 N2,8 tetrazenes,9 hexazenes,10 and diazenes.11 In others, the β-diketiminate supporting ligand accepts excess charge, a tendency that is greatly increased when a formazanate supporting ligand is used in place of the β-diketiminate.12 This shift of electron density to the ligands can be understood through Pauling’s electroneutrality principle, which has been quantitatively re-emphasized in the recent manifesto of Wolczanski.13
Chart 1.
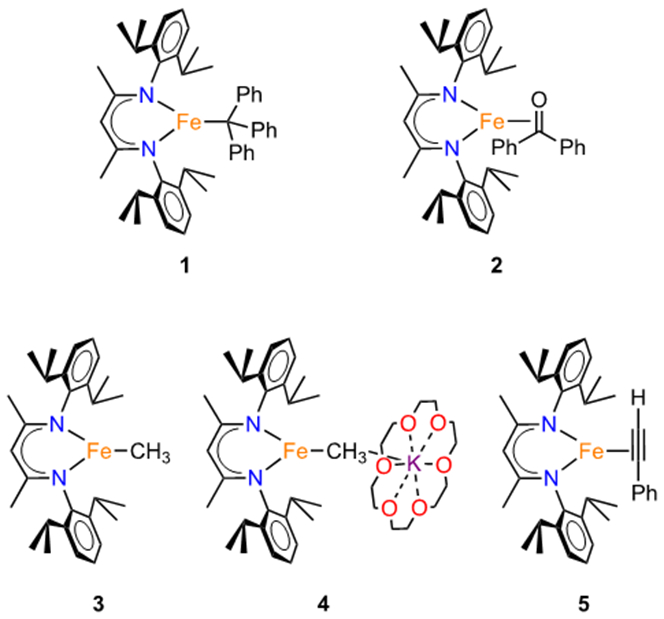
Iron species studied in this report.
In the context of these challenges, we note that homolytic dissociation of M─C bonds has an analogy to the dissociation of a formally neutral but redox-noninnocent ligand that has accepted unpaired electron density in the complex: both situations start as iron(II) but yield an identical iron(I) product. Taking advantage of this opportunity to directly compare alkyls with neutral ligands, we report here synthetic, spectroscopic, and computational comparisons of the formally iron(II) trityl complex LMeFeCPh3 and the formally iron(I) alkyne and benzophenone complexes LMeFe(PhCCH) and LMeFe(Ph2CO). X-ray absorption spectroscopy coupled to density functional theory (DFT) and ab initio multireference configuration interaction (MRCI) calculations enables an in-depth investigation of the electronic structures, quantitatively supporting the idea that the iron ions have similar electron densities in these complexes despite the differences in formal oxidation state.
RESULTS AND DISCUSSION
Synthesis and Characterization of LMeFe(CPh3) (1).
A diethyl ether solution of the known formally diiron(I) complex LMeFe(N2)FeLMe reacted with 0.5 equivalent of Gomberg’s dimer of the trityl radical14 to give the orange-red compound 1 (Scheme 1), which was characterized using X-ray crystallography (Figure 1). To our knowledge, this is the first Fe-trityl complex to be reported, and few metal-trityl complexes of any metal are known.15 In 1, the iron has a trigonal-planar geometry with Fe-N distances of 2.012(2) and 2.008(2) Å. The Fe─C distance of 2.124(3) Å is extremely long for an Fe─C bond (Fe─C bond lengths to three-coordinate iron in the Cambridge Structural Database have an average of 2.04 Å with a standard deviation of 0.04 Å).16 The only other triarylmethyl-iron complex has the Fe─C as part of a tetradentate tris(phosphine) ligand; it has a Fe─C distance of 2.081(3) Å in its iron(II) form and longer Fe─C bonds in reduced N2 complexes.17 Though the length of the Fe─C bond in 1 could be attributed to steric crowding from the bulky triphenylmethyl ligand, the ability of this compound to exchange Ph3C• (see below under Reactivity) suggests the possibility of an unconventional resonance structure with a triphenylmethyl radical, LFeI•••(•CPh3).
Scheme 1.
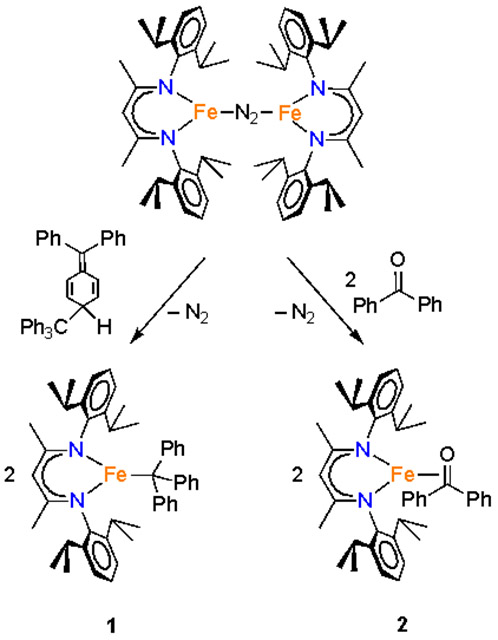
Synthesis of β-Diketiminatoiron Complexes 1 and 2
Figure 1.
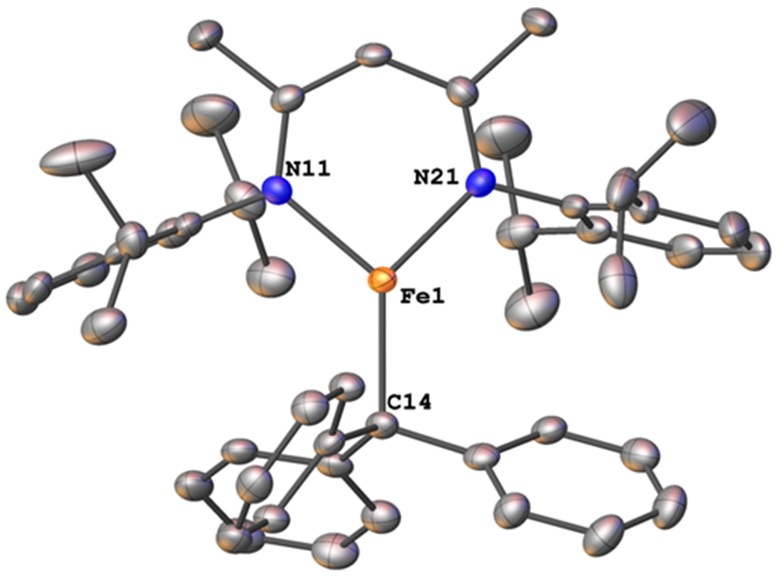
Molecular structure of 1 from X-ray crystallography. Thermal ellipsoids are displayed at the 50% probability level and hydrogens are omitted for clarity.
Magnetic and spectroscopic observations are relevant to this question. At 80 K, 1 exhibited a quadrupole doublet in its Mössbauer spectrum with δ = 0.58 mm s−1 and ∣ΔEQ∣ = 0.76 mm s−1. The isomer shift is within the range of other low-coordinate, high-spin iron(II) complexes that we have previously characterized.18 However, a number of diketiminate-supported iron(I) complexes have similar Mössbauer parameters as well,19 so these data do not readily distinguish the possibilities. The solution magnetic moment of 1 in C6D6 was 5.8(4) Bohr magnetons (BM), consistent with a high spin iron(II) system with significant spin-orbit coupling, and inconsistent with an iron(I)-radical complex unless there were extremely strong ferromagnetic coupling.
Synthesis and Characterization of LMeFe(Ph2CO) (2).
Addition of 2 equiv of benzophenone to LMeFe(N2)FeLMe in hexanes under an N2 atmosphere at room temperature produced a color change from red to brown/gold.20 X-ray crystallographic studies on the purple crystals gave compound 2 (Scheme 1), which has the structure that is displayed in Figure 2.
Figure 2.
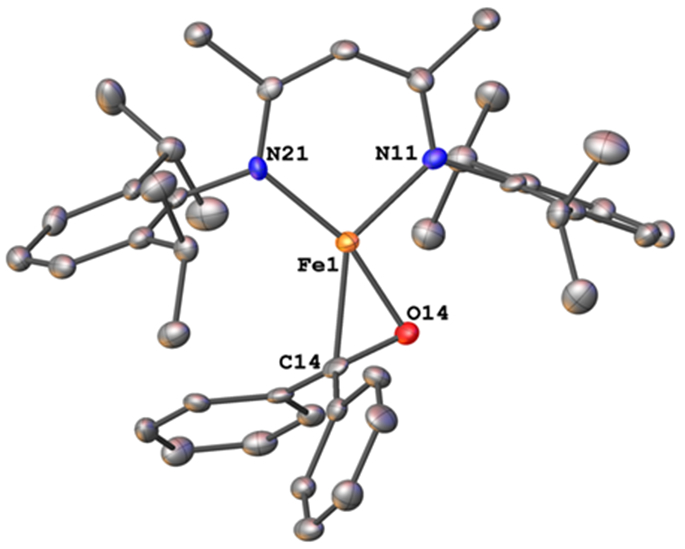
Molecular structure of 2, from X-ray diffraction studies. Thermal ellipsoids are displayed at the 50% probability level and hydrogens are omitted for clarity.
In the solid-state structure of 2, the geometry around iron is approximately square planar. The Fe─N distances of 1.944(4) and 2.001(4) Å lie in the expected range for iron β-diketiminate complexes.21 The Fe─C distance is 2.073(4) Å and the Fe─O distance is 1.836(3) Å. The C─O bond distance of the benzophenone carbonyl shows significant lengthening from 1.22(4) Å in free benzophenone to 1.358(5) Å in 2. Benzophenone ketyl radical anions have been crystallographically characterized, and have C─O bond lengths of 1.31(2) Å, closer to the observed distance in 2, while C─O single bonds are much longer, in the 1.43-1.46 Å range.17 Recent work using a phosphine-tethered ketone showed similarly that when bonded to iron(I), the C=O bond length of the ketone increases from 1.213(3) Å to 1.330(1) Å, which was attributed to a ketyl radical.22 The presence of radical character on a ketyl group with a C─O distance of 1.334(6) Å was documented thoroughly in a ketyl radical complex of uranium.23 While this uranium complex showed an intense visible absorption near 600 nm that is characteristic of a ketyl radical, 2 had no intense band in this region despite its similar C─O bond length.
The 1H NMR spectrum of 2 showed paramagnetically shifted peaks similar to those seen in other β-diketiminate iron(I) and iron(II) complexes. The observed solution magnetic moment24 of 4.2(2) BM was consistent with a high spin iron(I) system (S = 3/2) having a contribution from orbital angular momentum, which is common in diketiminate systems.25 However, another possible interpretation of the electronic structure of 2 is as a high-spin iron(II) ion with strong antiferromagnetic coupling to a ketyl radical on the benzophenone ligand, which would also provide an S = 3/2 ground state. The Mössbauer spectrum of 2 at 223 K displayed a doublet with δ = 0.46 mm s−1 and ∣ΔEQ∣ = 0.89 mm s−1. This isomer shift is within 0.02 mm s−1 that of the iron(II) complex LtBuFeCH3 and the iron(I) complex [LtBuFeH][K(18-crown-6],19,26 so Mössbauer spectroscopy again does not distinguish unambiguously between oxidation states. In order to resolve these ambiguities, 1 and 2 were examined using X-ray absorption spectroscopy and ab initio calculations.
X-ray absorption spectroscopy and computations.
We performed a more in-depth evaluation of the two above complexes, and compared them to iron(I) and iron(II) complexes of the σ-only CH3 ligand and the π-ligand phenylacetylene (Chart 1 above). Thus, our studies comprised 1 and 2, plus the iron(II) complex LMeFeCH3 (3),27 the new iron(I) methyl complex [LMeFeCH3][K(18-crown-6)] (4), and the formally iron(I) phenylacetylene complex LMeFe(HCCPh) (5). This acetylene complex was studied previously using Mössbauer spectroscopy, EPR, and DFT calculations.26
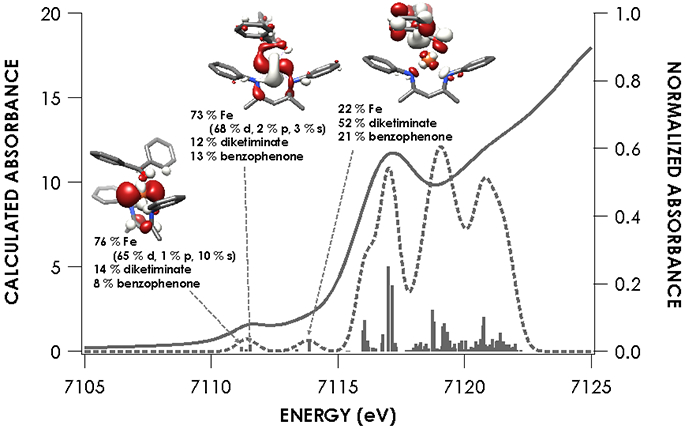
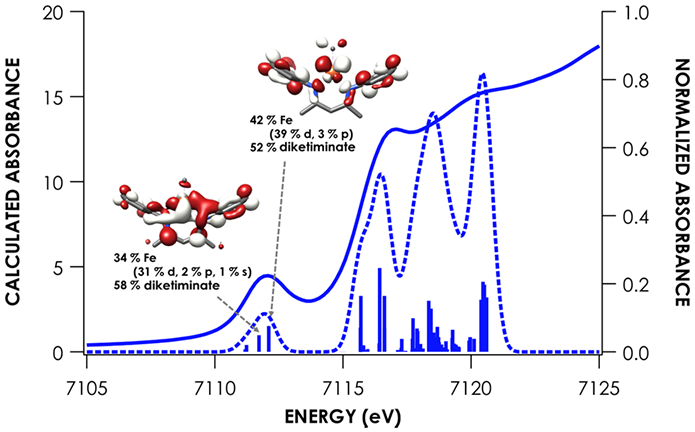
Because of its utility in establishing the oxidation state and electronic structure of the iron ion, we used X-ray absorption spectroscopy (XAS) at the iron K-edge.28 The experimental spectra (Figure 3) show little change in edge position between the five species investigated, which was an initial suggestion that there is no difference in oxidation state between species. Intense rising edge features were apparent for all species between 7114.2 and 7117.0 eV. All species showed a pre-edge feature centered at 7112.1 eV. These features were interpreted through the use of time-dependent density functional theory (TDDFT) computations, using optimized geometries based on the crystallographic structures. TDDFT calculations were based on unrestricted single point calculations and there was an excellent correlation between the computationally predicted and observed XAS features (Figure S-18). Previous work has validated the use of TDDFT to accurately predict metal and ligand K-edge XAS feature energies following linear correction to account for systematic errors in calculating absolute core (e.g. 1s) energies.29
Figure 3.
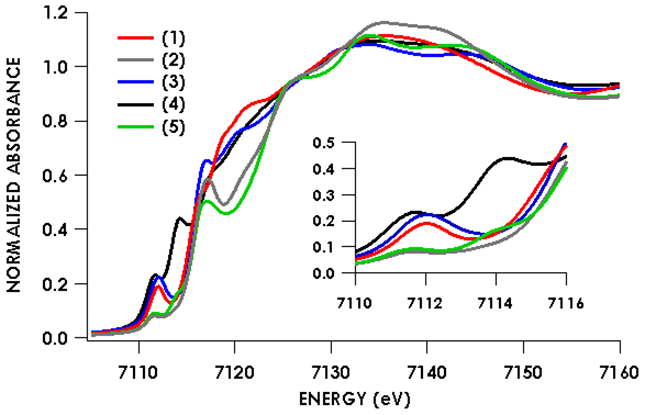
Fe K-edge absorption spectra of species 1-5 showing little to no pre-edge and rising-edge shifts between the formally iron(I) and formally iron(II) species.
Figures 4-8 show the individual pre-edge spectra for species 1-5, along with the orbitals to which core electrons are excited in the major absorption features. The lower energy feature in all cases is predicted to arise from excitation from Fe 1s into molecular orbitals featuring large contributions from Fe 3d, but exhibiting variable degrees of diketiminate ligand MO admixture. The diminished intensity of this feature in 2 and 5 can be attributed to decreased contributions from Fe p into the acceptor molecular orbitals. In addition, the small pre-edge features at 7113.9 eV in 2 and 5, which would be formally described as having neutral donors on iron(I), are predicted to involve unoccupied orbitals with substantial Fe character in both cases. The higher energy rising edge features for all species can be assigned to the dipole allowed 1s → 4p transition.
Figure 4.
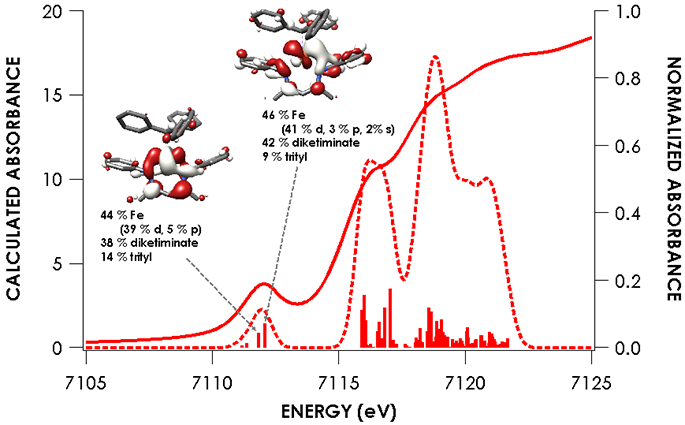
Overlay of experimental and TDDFT calculated spectra of 1 showing acceptor molecular orbitals for pre-edge transitions. Orbitals are plotted at an isovalue of 0.03 au. TDDFT calculations were initiated from B3LYP single point calculations employing the CP(PPP) basis set on Fe and ZORA-def2-TZVP(-f) basis set on all other atoms.
Figure 8.
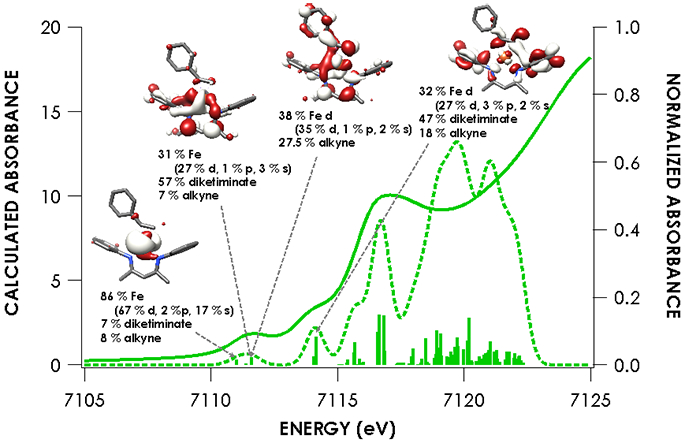
Overlay of experimental and TDDFT calculated spectra of 5 showing acceptor molecular orbitals for pre-edge transitions. Orbitals are plotted at an isovalue of 0.03 au. TDDFT calculations were initiated from B3LYP single point calculations employing the CP(PPP) basis set on Fe and ZORA-def2-TZVP(-f) basis set on all other atoms.
The nearly constant rising edge energies (7115.5 eV) in the XANES obtained for the compounds studied, except for 4, suggest that these four compounds all remain physically d6 iron(II). While the iron(II) assignment is clear for 1, where the contribution to frontier orbitals is dominated by Fe, it necessitates that for 2 and 5 electron density is delocalized from the Fe center to the benzophenone or alkyne ligands, respectively, in accord with the observed elongation of the benzophenone C─O bond in 2 and the bending of the phenylacetylene in 5. While such a picture emerges from analysis of the MOs obtained from the single point DFT calculations (Figures S-19 through S-23), we favored spectroscopy-oriented configuration interaction (SORCI) calculations based on a CAS(15,11) reference as a means to scrutinize the nature of the bonding in these species. This analysis permits the possibility to address whether multiple ground state electronic configurations are operative. The SORCI results showed that both 2 and 5 are multiconfigurational. The SORCI results for 2 (abbreviated diagram in Figure 9) revealed two near-degenerate quartet states, both of which have leading electronic configurations accounting for ca. 60% of each state. In both of these low-lying quartet configurations, two electrons reside in an MO that is split between Fe (52%) and benzophenone π* (28%) parentage. The SORCI picture that emerges for 5 (Figure 10) is less complex, as the quartet ground state is effectively non-degenerate, and its leading configuration accounts for 85.5% of the state. As with 2, all configurations of the ground state of 5 feature two electrons in orbitals that are evenly distributed between Fe and the coordinated alkyne (46% Fe / 33% alkyne and 52% Fe / 38% alkyne). This leads us to propose sharing the two electrons evenly between the metal and ligand, resulting in six d electrons and a spectroscopic oxidation state of iron(II) in 2 and 5. This matches the formal (and physical) oxidation states of 1 and 3.
Figure 9.
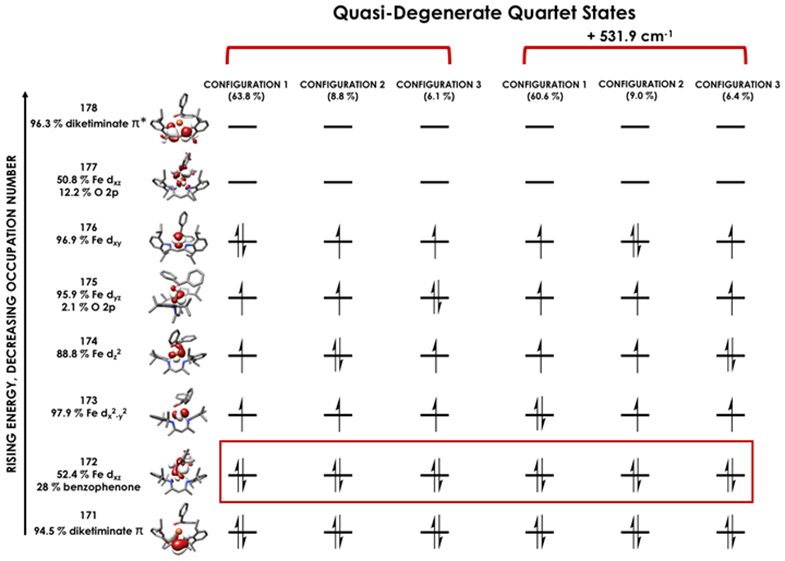
Leading configurations of the quartet ground states produced via the SORCI procedure carried out using a CAS(15,11) reference on compound 2. Electrons lying in orbitals with nearly equal parentage from Fe and benzophenone (2) are boxed in red. SORCI calculations employed the ZORA-def2-TZVPP basis set on Fe and ZORA-def2-SVP basis set on all other atoms.
Figure 10.
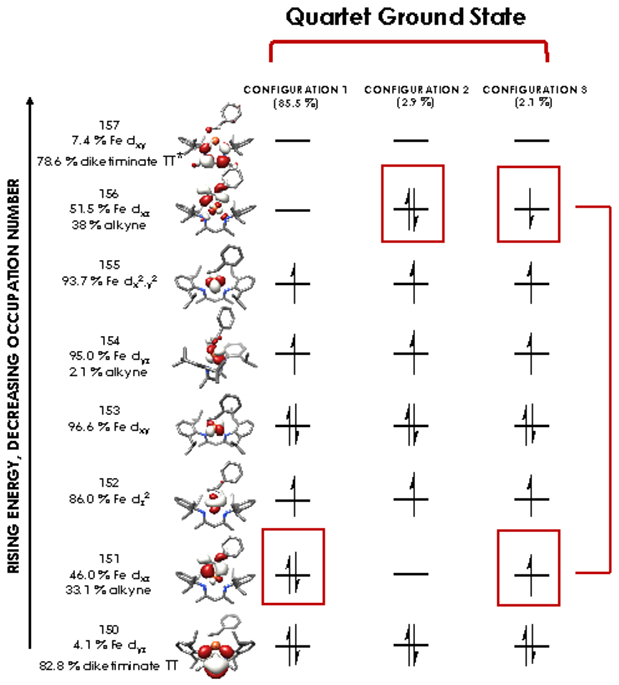
Leading configurations of the quartet ground states produced via the SORCI procedure carried out using a CAS(15,11) reference on compound 5. Electrons lying in orbitals with nearly equal parentage from Fe and alkyne (5) are boxed in red. SORCI calculations employed the ZORA-def2-TZVPP basis set on Fe and ZORA-def2-SVP basis set on all other atoms.
It is important to note that the iron(II) assignment does not imply that 2 and 5 have ligand radicals. Actually, the spectroscopically-validated calculations indicate that there is little population of configurations with unpaired electrons in ligand-localized orbitals. An alternative Dewar-Chatt-Duncanson formulation (resonance hybrid of iron(I) and iron(III) formal oxidation states) implies the presence of configurations with differing iron and ligand character, which is not evident in the SORCI results. Therefore, we favor the physical oxidation state description of iron(II).
Exchange of Ligands and Other Reactivity.
Exchange of the ligands between compounds 1, 2, and 5 occurred readily at room temperature, as shown by monitoring reactions using 1H NMR spectroscopy. For example, treating a C6D6 solution of compound 1 with 1 equiv of benzophenone gave rapid and quantitative formation of Gomberg’s dimer and 2.30 Likewise, treatment of 1 or 2 with 1 equiv of phenylacetylene gave 5. In each case, degradation of the mixtures over time prevented us from quantitatively determining the equilibrium constants as we had done previously for other formally iron(I) complexes in this system.8 However, the complete conversions in these ligand exchange reactions indicates that the relative binding affinities of the ligands are PhCCH > Ph2CO > •CPh3 > C6H6 > N2. The ability to handle the benzophenone and trityl complexes in benzene solution shows that the last two ligands in this series have weaker binding constants (their ordering has been discussed previously8). These experiments place the radical •CPh3 on a qualitative scale of binding energies for formally two-electron ligands in this formally iron(I) (but spectroscopically iron(II)) system. As shown above, though, the extent of electron transfer to the alkyne and benzophenone ligands is substantial, and it is likely that the induced polarity plays a role in strengthening these metal-ligand bonds.
These complexes have good thermal stability; notably, the trityl complex 1 showed no decomposition in benzene solution at 75 °C for several days, as judged by 1H NMR spectroscopy. However, mixtures of the benzophenone complex 2 with phenylacetylene, after the rapid formation of 5, showed slow formation of another product. Heating a benzene solution of 2 and phenylacetylene to 65 °C for 4 d formed the new green compound LMeFe(OCPh2CHCPh) (6), which could be isolated in 67% yield (Scheme 2). The X-ray crystal structure of 6 (Figure 11) showed the formation of a new C─C bond between the phenylacetylene and benzophenone fragments, to form a metalladihydrofuran ring with a C=C double-bond distance of 1.340(5) Å. The iron has a distorted tetrahedral geometry with an Fe─O bond length of 1.843(2) Å and an Fe─C bond length of 2.031(3) Å. The Fe─N bonds are again within the expected range, at 1.988(2) and 1.986(3) Å.
Scheme 2.
Coupling of Benzophenone and Phenylacetylene
Figure 11.
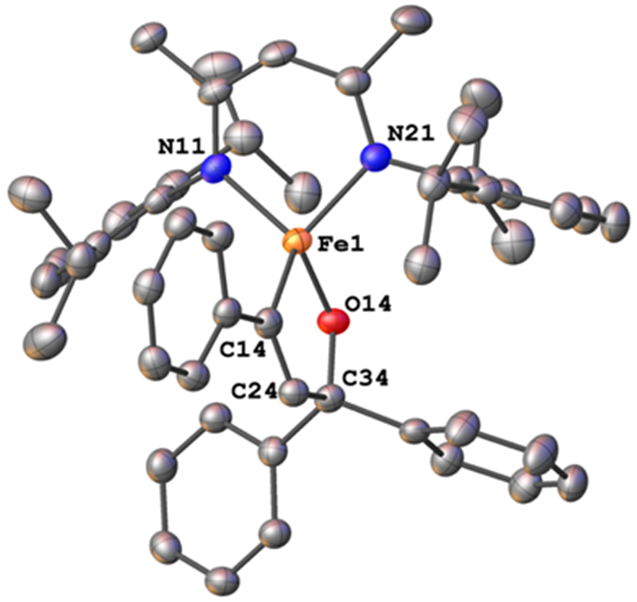
Molecular structure of 6. Thermal ellipsoids are displayed at the 50% probability level and hydrogens are omitted for clarity.
The magnetic moment of 5.6(3) BM for 6 agrees with the spin-only value of 5.92 BM expected for high-spin iron(III). The observed Mössbauer parameters at 173 K (δ = 0.10 mm s−1; ∣ΔEQ∣ = 1.66 mm s−1) are much lower than those in high-spin iron(I) or iron(II) complexes, and therefore support the assignment of this complex as having an iron(III) center. The 1H NMR spectrum of 6 was broad, as is common for high-spin iron(III) complexes, and combined with the magnetic moment, the preponderance of the evidence supports a high-spin electronic configuration for 6.
The reductive coupling of ketone and alkyne ligands to form metalladihydrofuran rings is best known starting from titanium(II) complexes which are oxidized to titanium(IV) in the process.31 A related series of alkyne-aldehyde coupling reactions have been studied in the context of nickel-catalyzed cyclizations.32 Mechanistic studies on the nickel systems indicate that bond formation between coordinated alkyne and carbonyl fragments is concurrent with oxidation of the metal,33 and we presume that the low coordination number of the iron is conducive to the coupling to form the new C─C bond. Note that this bond formation would often be attributed to radical character on coordinated benzophenone and/or alkyne, but the computational studies above indicate that the radical character on the ligands is not large in the ground state. We speculate that low-lying excited states with radical character can be accessed during the reaction pathway to form the C─C bond, an idea that can be tested with future computations.
CONCLUSIONS
Using the highly unsaturated and electron-rich environment of formally iron(I) species supported by β-diketiminate ligands, it has been possible to isolate a number of unusual species. One is the first example of an iron complex of CPh3, which can exchange the CPh3 group as a radical (stabilized by dimerization to Gomberg’s dimer). Though the loss of •CPh3 might suggest iron(I)–trityl radical character, spectroscopic characterization strongly indicates that an iron(II) formulation is most appropriate. The spectroscopic oxidation state of iron(II) is also shared by other formally iron(I) complexes with benzophenone and phenylacetylene. The iron(II) spectroscopic oxidation state formally implies radical character on the ligand, but the mixing of metal and ligand orbitals is so extensive that little radical character is present on the ligand. This serves as a warning about using purely valence-bond pictures to rationalize reactivity (such as the C─C bond formation between benzophenone and alkyne observed here) through ligand radicals.
EXPERIMENTAL SECTION
General.
All manipulations were performed under a nitrogen atmosphere, by Schlenk techniques or in an M. Braun or Vigor glovebox maintained at or below 1 ppm of O2. Glassware was oven-dried at 150 °C for at least 12 h before use. Graphite, Celite, and 3 & 4 Å molecular sieves were dried at 200 °C under vacuum for at least 12 h. Pentane, hexane, benzene, and diethyl ether were purified by passage through activated alumina and Q5 columns from Glass Contour Co, under Ar. All solvents were stored over 4 Å molecular sieves and passed through a plug of activated alumina immediately prior to use. Benzene-d6 was dried over activated alumina and stored over 4 Å molecular sieves. Potassium on graphite was prepared by heating stoichiometric amounts of potassium and graphite at 140 °C under an argon atmosphere.34 Warning: Alkali metals and their graphite intercalates KC8 is a powerful reductant, which ignites on contact with air and moisture. Therefore, extreme care must be taken when synthesizing and handling alkali graphite reductants! Benzophenone (99%) was obtained from ACROS Organics, and phenylacetylene (98%) was obtained from Alfa Aesar. LMeFeNNFeLMe was prepared according to the previously reported procedure.21 Gomberg’s dimer was prepared according to literature methods.35 NMR data were collected on an Agilent 400 or 500 MHz spectrometer. Chemical shifts in 1H NMR spectra are referenced to external SiMe4 using the residual protiated solvent peaks as internal standards: C6D5H (δ 7.16 ppm) and C7D7H (δ 2.34 ppm). Solution magnetic susceptibilities were determined by the Evans method.25 Elemental analyses were performed at the CENTC Elemental Analysis Facility at the University of Rochester. IR spectra were collected on an Alpha Platinum ATR IR Spectrometer. UV-vis spectra were recorded on a Cary 50 spectrometer using Schlenk-adapted quartz cuvettes with a 1 mm path length. Mössbauer samples were packed in Delrin sample cups and loaded into the spectrometer at 77 K. Mössbauer measurements were performed using a SEE Co. MS4 Mössbauer spectrometer integrated with a Janis SVT-400T He/N2 cryostat for measurements at 80 K and 223 K with a 0.07 T applied magnetic field. Isomer shifts were determined relative to α-iron at 298 K. All Mössbauer spectra were fit using the program WMoss (SEE Co.), using Lorentzian doublets.
Synthesis and characterization
LMeFe(CPh3) (1).
A red solution of LMeFe(N2)FeLMe (150.5 mg, 0.154 mmol) in diethyl ether (8 mL) was treated with a solution of C2Ph6 (“Gomberg’s dimer,” 88.7 mg, 0.167 mmol) in diethyl ether (8 mL). The mixture was stirred for 90 min. Volatile materials were removed under vacuum to give an orange residue. The solid was extracted with hexanes (18 mL), filtered through Celite, concentrated under vacuum to 10 mL, and placed in a −35 °C freezer overnight to give red crystals of 1 (214 mg, 97%). 1H NMR (500 MHz, C6D6) δ 107 (1H, backbone C─H), 49 (6H, backbone CH3), 16 (6H, o-H trityl aryl), 0 (12H, CH(CH3)2), −12 (6H, m-H trityl aryl), 31 (4H, CH(CH3)2), 54 (3H, p-H trityl aryl), 75 (12H, CH(CH3)2). μeff (Evans, C6D6, 298 K) = 5.8(4) BM. IR (ATR, neat): 3055 (w), 2960 (m), 2926 (m), 2867 (w), 1617 (w), 1588 (w), 1530 (m), 1518 (m), 1491 (w), 1481 (w), 1462 (m), 1434 (m), 1367 (s), 1310 (s), 1255 (s), 1199 (w), 1175 (w), 1152 (w), 1138 (w), 1100 (w), 1079(w), 1056 (w), 1032 (m), 932 (w), 910 (w), 882 (w), 853 (w), 836 (w), 794 (s), 780 (m), 759 (m), 756 (m), 721 (w), 700 (s), 657 (w), 610 (w), 522 (w), 510 (w), 504 (w), 480 (m), 457 (w) cm1. Anal. Calcd for C48H56N2Fe: C, 80.43; H, 7.79; N, 3.91. Found: C, 80.08; H, 8.02; N, 3.55. Mössbauer (80 K, solid): δ = 0.58(2) mm s1, ∣∆EQ∣ = 0.76(2) mm s1.
LMeFe (Ph2CO) (2).
A red solution of LMeFeNNFeLMe (107.9 mg, 0.111 mmol) in hexanes (8 mL) was treated with a solution of benzophenone (38.8 mg, 0.213 mmol) in hexanes (8 mL). The reaction mixture was stirred for 90 min and a change to a golden brown color was observed. Volatile materials were removed under vacuum yielding a brown residue. The solid was extracted with hexanes (15 mL), filtered through Celite, concentrated under vacuum to 3 mL and placed in a 35 °C freezer overnight to give purple crystals of 2 (120 mg, 82%). 1H NMR (500 MHz, C6D6): δ 87 (4H, m-H benzophenone aryl), 78 (2H, p-H benzophenone), 60 (6H, backbone CH3), 35 (1H, backbone CH), 3 (12H, CH(CH3)2), 16 (2H, p-H DIPP), 35 (4H, CH(CH3)2), 39 (12H, CH(CH3)2), 81 (4H, m-H DIPP). The above assignments do not account for the ortho protons of the benzophenone, which may be too broad and shifted to observe. μeff (Evans, C6D6, 298 K) = 4.2(2) BM. IR (ATR, neat): 3055 (w), 3029 (w), 2954 (m), 2925(m), 2865 (m), 1593 (w), 1539 (w), 1520 (m), 1488 (w), 1458 (m), 1436 (m), 1382 (s), 1377 (s), 1315 (s), 1280 (w), 1260 (m), 1252 (m), 1232 (w), 1175 (m), 1155, w), 1102(w), 1076 (w), 1053 (w), 1026 (m), 933 (m), 900 (w), 863 (w), 853 (w), 794 (s), 757 (s), 718 (w), 693(s), 646 (m), 619 (m), 552 (w), 541 (w), 519 (w), 447 (w) cm1. Anal. Calcd for C42H51N2OFe: C, 76.93; H, 7.84; N, 4.27. Found: C, 77.25; H, 8.04; N, 4.00. Mössbauer (223 K, solid): δ = 0.46(2) mm s1, ∣∆EQ∣ = 0.89(2) mm s1.
[K(18-crown-6)][LMeFeCH3] (3).
LMeFeCH3 (187.3 mg, 0.383 mmol) and 18-crown-6 (116.0 mg, 0.439 mmol) were dissolved in toluene (6 mL) to give a yellow solution. KC8 (62.0 mg, 0.459 mmol) was added, and the mixture turned dark green. After stirring for 2 h, the mixture was filtered through Celite, then the filter pad was rinsed with 20 mL of toluene. The green filtrate was concentrated to 15 mL under vacuum and cooled to −40 °C. After 3 days, crystals of 3 (269.3 mg, 79.6%) were isolated and dried under vacuum. The 1H NMR and elemental analysis results are consistent with one toluene of crystallization. 1H NMR (400 MHz, C6D6) δ 34 (6H, backbone CH3), 22 (4H, CH(CH3)2 or m-HL), 7 (5H, toluene), 3.0 (12H, CH(CH3)2), 2.5 (24 H, 18-c-6), 2.1 (3H, toluene), 3 (12H, CH(CH3)2), 35 (4H, CH(CH3)2 or m-HL), 147 (1H, backbone CH). μeff (Evans, C6D6, 298 K) = 4.1(2) BM. IR (ATR, neat): 3045 (w), 2953 (m), 2889 (m), 2861 (m), 1582 (w), 1458 (w), 1428 (m), 1376 (m), 1350 (m), 1318 (m), 1250 (m), 1103 (s), 960 (m), 836 (m), 754 (m), 731 (m), 695 (w), 464 (w) cm1. Anal. Calcd for C42H68O6N2FeK•C7H8: C, 66.57; H, 8.66; N, 3.17. Found: C, 66.41; H, 8.67; N, 3.01.
LMeFe(OCPh2CHCPh) (6).
Phenylacetylene (11.8 μL, 0.107 mmol) was added to a red/brown solution of LMeFe(Ph2CO) (1) (70.6 mg, 0.101 mmol) in benzene (15 mL), and the reaction mixture was heated at 65 °C for 4 d. Volatile materials were removed under vacuum to give a green residue. The solid was extracted with hexanes (15 mL), filtered through Celite, concentrated under vacuum to 2 mL, and placed in a 35 °C freezer overnight to give green crystals of 6 (51 mg, 67%). Peaks in the 1H NMR spectrum were broad and overlapped, and so the integrations are not reliable. Figure S-8 shows the spectrum, in which the clearest chemical shifts lie at 37, 35, 34, 24, 17, 8, 5, 4, 2, and 40 ppm. μeff (Evans, C6D6, 298 K) = 5.6(3) BM. IR (ATR, neat): 3060 (w), 2962 (m), 2938 (m), 2928 (m), 2860 (m), 1517 (s), 1486 (m), 1459 (m), 1438 (s), 1370 (s), 1361 (s, sh), 1315 (s), 1284 (m), 1259 (s), 1252 (s), 1198 (w), 1172 (m), 1100 (m), 1057 (m), 1019 (m), 1003 (w), 985 (m), 934 (m), 894 (w), 862 (w), 796 (m), 753 (s), 694 (s), 652 (w), 632 (m), 625 (m), 610 (m), 546 (w), 525 (m), 501 (w) cm1. Anal. Calcd for C50H57N2Fe: C, 79.24; H, 7.58; N, 3.70. Found: C, 79.10; H, 7.68; N, 3.49. Mössbauer (173 K, solid): δ = 0.10(2) mm s1, ∣∆EQ∣ = 1.66(2) mm s1.
Supplementary Material
Figure 5.
Overlay of experimental and TDDFT calculated spectra of 2 showing acceptor molecular orbitals for pre-edge transitions. Orbitals are plotted at an isovalue of 0.03 au. TDDFT calculations were initiated from B3LYP single point calculations employing the CP(PPP) basis set on Fe and ZORA-def2-TZVP(-f) basis set on all other atoms.
Figure 6.
Overlay of experimental and TDDFT calculated spectra of 3 showing acceptor molecular orbitals for pre-edge transitions. Orbitals are plotted at an isovalue of 0.03 au. TDDFT calculations were initiated from B3LYP single point calculations employing the CP(PPP) basis set on Fe and ZORA-def2-TZVP(-f) basis set on all other atoms.
Figure 7.
Overlay of experimental and TDDFT calculated spectra of 4 showing acceptor molecular orbitals for pre-edge transitions. Orbitals are plotted at an isovalue of 0.03 au. TDDFT calculations were initiated from B3LYP single point calculations employing the CP(PPP) basis set on Fe and ZORA-def2-TZVP(-f) basis set on all other atoms.
SYNOPSIS.
An isolable iron(II) trityl complex exchanges the trityl radical with neutral π-acceptor ligands to form formally iron(I) complexes, but all of these are best described as iron(II).
ACKNOWLEDGMENT
The authors acknowledge funding from the National Institutes of Health (GM065313 to P.L.H.), the National Science Foundation (CHE-1454455 to K.M.L.), the Alfred P. Sloan Foundation (Research Fellowship to K.M.L.) and Saint Francis University (E.P.Z. sabbatical leave). XAS data were obtained at SSRL, which is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the Department of Energy’s Office of Biological and Environmental Research, and by NIH/NIGMS (including P41GM103393).
Funding Sources
National Institutes of Health (GM065313)
Footnotes
Supporting Information. The following files are available free of charge. Spectroscopy, calculations (PDF) Crystallographic information files (CIF)
REFERENCES
- 1.Orgel LE An Introduction to Transition-Metal Chemistry: Ligand Field Theory, Wiley: New York, 1966. [Google Scholar]
- 2(a).Chirik PJ; Wieghardt K Radical Ligands Confer Nobility on Base-Metal Catalysts. Science 2010, 327, 794–795. [DOI] [PubMed] [Google Scholar]; (b) Dzik WI; van der Vlugt JI; Reek JNH; de Bruin B Ligands that Store and Release Electrons during Catalysis. Angew. Chem., Int. Ed 2011, 50, 3356–3358. [DOI] [PubMed] [Google Scholar]; (c) Eisenberg R; Gray HB Noninnocence in Metal Complexes: A Dithiolene Dawn. Inorg. Chem 2011, 50, 9741–9751. [DOI] [PubMed] [Google Scholar]
- 3(a).Green MLH A new approach to the formal classification of covalent compounds of the elements. J. Organomet. Chem 1995, 500, 127–148. [Google Scholar]; (b) Green MLH; Parkin G Application of the Covalent Bond Classification Method for the Teaching of Inorganic Chemistry. J. Chem. Ed 2014, 91, 807–816. [Google Scholar]
- 4.Chen C; Bellows SM; Holland PL Tuning steric and electronic effects in transition-metal β-diketiminate complexes. Dalton Trans. 2015, 44, 16654–16670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holland PL Electronic Structure and Reactivity of Three-Coordinate Iron Complexes. Acc. Chem. Res 2008, 41, 905–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cowley RE; Christian GJ; Brennessel WW; Neese F; Holland PL A Reduced (β-Diketiminato)iron Complex with End-On and Side-On Nitriles: Strong Backbonding or Ligand Non-Innocence? Eur. J. Inorg. Chem 2012, 479–483. [Google Scholar]
- 7.Yu Y; Smith JM; Flaschenriem CJ; Holland PL Binding Affinity of Alkynes and Alkenes to Low-Coordinate Iron. Inorg. Chem 2006, 45, 5742–5751. [DOI] [PubMed] [Google Scholar]
- 8.Stoian SA; Vela J; Smith JM; Sadique AR; Holland PL; Münck E; Bominaar EL Mössbauer and Computational Study of an N2-Bridged Diiron Diketiminate Complex: Parallel Alignment of the Iron Spins by Direct Antiferromagnetic Exchange with Activated Dinitrogen. J. Am. Chem. Soc 2006, 128, 10181–10192. [DOI] [PubMed] [Google Scholar]
- 9.Cowley RE; Bill E; Neese F; Brennessel WW; Holland PL Iron(II) Complexes with Redox-Active Tetrazene (RNNNNR) Ligands. Inorg. Chem 2009, 48, 4828–4836. [DOI] [PubMed] [Google Scholar]
- 10.Cowley RE; Elhaik J; Eckert NA; Brennessel WW; Bill E; Holland PL A Bridging Hexazene (RNNNNNNR) Ligand from Reductive Coupling of Azides. J. Am. Chem. Soc 2008, 130, 6074–6075. [DOI] [PubMed] [Google Scholar]
- 11.Sadique AR; Gregory EA; Brennessel WW; Holland PL Mechanistic Insight into N:N Cleavage by a Low-Coordinate Iron(II) Hydride Complex. J. Am. Chem. Soc 2007, 129, 8112–8121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12(a).Broere DLJ; Mercado BQ; Lukens JT; Vilbert AC; Banerjee G; Lant HMC; Lee SH; Bill E; Sproules S; Lancaster KM; Holland PL Reversible Ligand-Centered Reduction in Low-Coordinate Iron Formazanate Complexes. Chem. Eur. J 2018, 24, 9417–9425. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Broere DLJ; Mercado BQ; Bill E; Lancaster KM; Sproules S; Holland PL Alkali Cation Effects on Redox-Active Formazanate Ligands in Iron Chemistry. Inorg. Chem 2018, 57, 9580–9591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wolczanski PT Flipping the Oxidation State Formalism: Charge Distribution in Organometallic Complexes As Reported by Carbon Monoxide. Organometallics 2017, 36, 622–631. [Google Scholar]
- 14.Neumann WP; Peneory A; Stewen U; Lehnig M Sterically Hindered Free Radicals. Stabilization of Free Radicals by Substituents as Studied by Using Triphenylmethyls. J. Am. Chem. Soc 1989, 111, 5845–5851. [Google Scholar]
- 15(a).Sonoda A; Bailey PM; Maitlis PM Crystal and molecular structures of pentane-2,4-dionato-(α,1,2-ɳ-triphenylmethyl)-palladium and platinum, J. Chem. Soc., Dalton Trans 1979, 346–350. [Google Scholar]; (b) Hitchcock PB; Khvostov AV; Lappert MF; Protchenko AV Heteroleptic ytterbium(II) complexes supported by a bulky β-diketiminato ligand, Dalton Trans. 2009, 2383–2391. [DOI] [PubMed] [Google Scholar]; (c) Bartless RA; Dias HVR; Power PP Isolation and X-ray crystal structures of the organolithium etherate complexes, [Li(Et2O)2(CPh3)] and [{Li(Et2O)(2,4,6-(CHMe2)3C6H2)}2], J. Organomet. Chem 1988, 341, 1–9. [Google Scholar]; (d) Brooks JJ; Stucky GD π-Groups in Ion Pair Bonding. Triphenylmethyllithium Tetramethylethylenediamine, J. Am. Chem. Soc 1972, 94, 7333–7338. [Google Scholar]; (e) Gielen M; Vanden Eynde I Synthesis and X-ray Crystal Structure of Two Triorganostannylcobalt complexes, Bull. Soc. Chim. Belg 1980, 89, 915–927. [Google Scholar]; (f) Englehardt LM; Harvey S; Raston CL; White AH Organomagnesium reagents: the crystal structures of [Mg(anthracene)(THF)3] and [Mg(triphenylmethyl)Br(OEt2)2], J. Organomet. Chem 1988, 341, 39–51. [Google Scholar]; (g) Xiong Y; Szilvasi T; Yao S; Tan G; Driess M Synthesis and Unexpected Reactivity of Germyliumylidene Hydride Stabilized by a Bis(N-heterocyclic carbene)borate Ligand. ? 2014, 136, 11300–11303. [DOI] [PubMed] [Google Scholar]; (h) Hoffmann D; Bauer W; von Rague Schleyer P; Pieper U; Stalke D Cation-induced structural altrerations in the organo alkali metal derivatives of triphenylmethane: a combined X-ray and NMR study of the potassium-cesium salts, Organometallics 1993, 12, 1193–1200. [Google Scholar]; (i) Viebrock H; Panther T; Behrens U; Weiss E Über Metallalkyl- and Arylverbindungen: Neue Triphenylmethyl-Verbindungen des Kaliums, J. Organomet. Chem 1995, 491, 19–25. [Google Scholar]
- 16.Groom CR; Bruno IJ; Lightfoot MP; Ward SC The Cambridge Structural Database. Acta Cryst. B 2016, 72, 171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Creutz SE; Peters JC Catalytic Reduction of N2 to NH3 by an Fe─N2 Complex Featuring a C-Atom Anchor. J. Am. Chem. Soc 2014, 136, 1105–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Andres H; Bominaar EM; Smith JM; Eckert NA; Holland PL; Münck E Planar Three-Coordinate High Spin Fe(II) Complexes with Large Orbital Angular Momentum: Mössbauer, Electron Paramagnetic Resonance, and Electronic Structure Studies. J. Am. Chem. Soc 2002, 124, 3012–3025. [DOI] [PubMed] [Google Scholar]
- 19.McWilliams SF; Brennan-Wydra E; MacLeod KC; Holland PL Density Functional Calculations for Prediction of 57Fe Mössbauer Isomer Shifts and Quadrupole Splittings in β-Diketiminate Complexes. ACS Omega 2017, 2, 2594–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith JM; Sadique AR; Cundari TR; Rodgers KR; Lukat-Rodgers G; Lachiocotte RJ; Flaschenreim CJ; Vela J; Holland PL Studies of Low-Coordinate Iron Dinitrogen Complexes J. Am. Chem. Soc 2006, 128, 756–769. [DOI] [PubMed] [Google Scholar]
- 21.Lewis RA; MacLeod KC; Mercado BQ; Holland PL Geometric and redox flexibility of pyridine as a redox-active ligand that can reversibly accept one or two electrons Chem. Commun 2014, 50, 11114–11117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Verhoeven DGA van Wiggen MAC Kwakernakk J; Lutz M; Gebbink RJMK; Moret M-C Periodic Trends in the Binding of a Phosphine-Tethered Ketone Ligand to Fe, Co, Ni, and Cu. Chem. Eur. J 2018, 24, 5163 – 5172 [DOI] [PubMed] [Google Scholar]
- 23.Lam OP; Anthon C; Heinemann FW; O’Connor JM; Meyer K Structural and Spectroscopic Characterization of a Charge-Separated Uranium Benzophenone Ketyl Radical Complex. J. Am. Chem. Soc 2008, 130, 6567–6576. [DOI] [PubMed] [Google Scholar]
- 24.Schubert EM Utilizing the Evans Method with a Superconducting NMR Spectrometer in the Undergraduate Laboratory, J. Chem. Ed 1992, 69, 62. [Google Scholar]
- 25.Stoian S; Yu Y; Smith JM; Holland PL; Bominaar EL; Münck E Mössbauer, EPR, and Crystallographic Characterization of a High-Spin Fe(I) Diketiminate Complex with Orbital Degeneracy. Inorg. Chem 2005, 44, 4915–4922. [DOI] [PubMed] [Google Scholar]
- 26.Chiang KP; Scarborough CC; Horitani M; Lees NS; Ding K; Dugan TR; Brennessel WW; Bill E; Hoffman BM; Holland PL Characterization of the Fe-H Bond in a Three-Coordinate Terminal Hydride Complex of Iron(I). Angew. Chem., Int. Ed 2012, 51, 3658–3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vela J; Vaddadi S; Cundari TR; Smith JM; Gregory EA; Lachicotte RJ; Flaschenriem CJ; Holland PL Reversible Beta-Hydrogen Elimination of Three-Coordinate Iron(II) Alkyl Complexes: Mechanistic and Thermodynamic Studies. Organometallics 2004, 23, 5226–5239. [Google Scholar]
- 28.Penner-Hahn JE X-ray absorption spectroscopy in coordination chemistry. Coord. Chem. Rev 1999, 190–192, 1101–1123. [Google Scholar]
- 29(a).DeBeer George S; Petrenko T; Neese F Prediction of Iron K-Edge Absorption Spectra Using Time-Dependent Density Functional Theory. J. Phys. Chem. A 2008, 112, 12936–12943. [DOI] [PubMed] [Google Scholar]; (b) DeBeer George S; Petrenko T; Neese F Time-dependent density functional calculations of ligand K-edge X-ray absorption spectra. Inorg. Chim. Acta 2008, 361, 965–972. [Google Scholar]; (c) DeBeer George S; Neese F Calibration of Scalar Relativistic Density Functional Theory for the Calculation of Sulfur K-Edge X-ray Absorption Spectra. Inorg. Chem. 2010, 49, 1849–1853. [DOI] [PubMed] [Google Scholar]
- 30.The equilibrium constant for the dissociation of Gomberg’s dimer in toluene at 20 °C is K = 3.08 x 10−4 M. Colle TH; Glaspie PS; Lewis ES The triphenylmethyl radical: equilibrium measurements and the reaction with thiophenol. J. Org. Chem 1978, 43, 2722–2725. [Google Scholar]
- 31(a).Hewlett DF; Whitby RJ Intramolecular cocyclisation of carbonyl groups and unactivated alkenes or alkynes induced by a low-valent titanium complex. J. Chem. Soc., Chem. Commun 1990, 1684–1686. [Google Scholar]; (b) Harada K; Urabe H; Sato F Generation and reactions of low-valent titanium alkoxide-acetylene complexes. A practical preparation of allyl alcohols. Tetrahedron Lett. 1995, 36, 3203–3206. [Google Scholar]
- 32(a).Ogoshi S; Arai T; Ohashi M; Kurosawa H Nickeladihydrofuran. Key intermediate for nickel-catalyzed reaction of alkyne and aldehyde. Chem. Commun 2008, 1347–1349. [DOI] [PubMed] [Google Scholar]; (b) Oblinger E; Montgomery J A New Stereoselective Method for the Preparation of Allylic Alcohols. J. Am. Chem. Soc 1997, 119, 9065–9066. [Google Scholar]
- 33(a).Liu P; McCarren P; Cheong PH-Y; Jamison TF; Houk KN Origins of Regioselectivity and Alkene-Directing Effects in Nickel-Catalyzed Reductive Couplings of Alkynes and Aldehydes. J. Am. Chem. Soc 2010, 132, 2050–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Baxter RD; Montgomery J Mechanistic Study of Nickel-Catalyzed Ynal Reductive Cyclizations through Kinetic Analysis. J. Am. Chem. Soc 2011, 133, 5728–5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dresselhaus MS; Dresselhaus G Intercalation compounds of graphite. Adv. Phys 1981, 30, 139–326. [Google Scholar]
- 35.Neumann WP; Penenory A; Stewen U; Lehnig M Sterically hindered free radicals. 18. Stabilization of free radicals by substituents as studied by using triphenylmethyls. J. Am. Chem. Soc 1989, 111, 5845–5851. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.