

Abstract
The E3 ubiquitin ligase Cbl-b has been characterized as an intracellular checkpoint in T cells; however, the function of Cbl-b in primary human natural killer (NK) cells, an innate immune anti-tumor effector cell, is not well defined. Here we show that the expression of Cbl-b is significantly upregulated in primary human NK cells activated by IL-15, IL-2 and the human NK cell-sensitive tumor cell line K562 that lacks MHC class I expression. Pre-treatment with JAK or AKT inhibitors prior to IL-15 stimulation reversed Cbl-b upregulation. Downregulation of Cbl-b resulted in significant increases in granzyme B and perforin expression, IFN-γ production and cytotoxic activity against tumor cells. Collectively, we demonstrate upregulation of Cbl-b and its inhibitory effects in IL-15/IL-2/K562-activated human NK cells, suggesting that Cbl-b plays a negative feedback role in human NK cells.
Keywords: Human NK cells, Cbl-b, IL-15, checkpoint
Introduction
Natural killer (NK) cells are part of the innate immune system and act as the first line of defense-against infectious pathogens and tumors through cytotoxicity and cytokine production (1–3). NK cells were first described for their ability to spontaneously lyse target cells without any prior priming or restriction to target cells expressing major histocompatibility complex (MHC) molecules (4, 5). The genesis, survival, proliferation and activation of NK cells are regulated in large part by IL-15, which binds the IL15Rβγ expressed on NK cells or its precursors resulting in activation of JAK3 and the subsequent phosphorylation of STAT5 (6). NK cells express both activating and inhibitory receptors that receive their signals by engaging with target cell ligands, which then activate or inhibit NK cell function (7, 8). NK cells themselves can express non-MHC class I specific inhibitory receptors or “checkpoints” including programmed death-1 (PD-1), T cell immunoreceptor with Ig and ITIM domains (TIGIT), lymphocyte activation gene 3 protein (LAG3) and T cell immunoglobulin domain and mucin domain-3 (TIM-3) that can suppress NK cell function, and may represent new targets for checkpoint blockade-based NK cell immunotherapy (9).
The Casitas B-cell lymphoma (Cbl) protein family, which includes Cbl-b and c-Cbl in mammals, represents RING-finger domain-containing E3 ubiquitin ligases and provides critical inhibitory signaling for the proper regulation of protein tyrosine kinases (PTKs) (10, 11). Cbl-b also regulates CD28-dependent T cell activation by selectively suppressing TCR-mediated Vav activation (12). Additionally, Cbl-b regulates peripheral T cell tolerance, and the loss of Cbl-b results in the onset of autoimmunity (13). The Cbl family plays an important role in the induction of B-cell immune tolerance (14). Deleting Cbls in germinal center (GC) B cells abolishes antibody affinity maturation via the early exit of high-affinity antigen specific B cells from the GC (15).
NK cell activation is carefully titrated between activating and inhibitory signals in order to maintain effective immune surveillance and avoid auto-reactivity. In human NK cells expressing KIR2D1, following inhibitory receptor engagement, the expression level of Cbl-b is increased (16). In Cbl-b−/− mice, such activation results in significantly higher proliferation and interferon (IFN)-γ secretion when compared to NK cells from wild-type mice, yet there appears to be no difference in NK cell development. These data support the notion that Cbl-b is a negative regulator of murine NK cell proliferation and effector function (17). However, to our knowledge, the function of Cbl-b in human NK cells has not been explored. Our current study revealed that the expression of Cbl-b is significantly increased following activation of primary human NK cells. Knockdown of Cbl-b in resting and IL-15-activated NK cell enhances cytotoxicity and expression of granzyme B, perforin and IFN-γ. Our data suggest a negative feedback role of Cbl-b in activated human NK cells.
Materials and methods
Isolation of primary human NK cells
Blood leukopacks were obtained from City of Hope National Medical Center Blood Bank under the institutional review board approved protocols. NK cells were isolated by using the RosetteSep™ human NK cell enrichment cocktail (StemCell Technologies) and Ficoll-Paque (GE Healthcare). The purity of primary NK cells was confirmed with flow cytometry using anti-CD56 (Beckman Coulter, Cat# B46024) and anti-CD3 (Miltenyi Biotec, Cat# 130-113-134) antibodies. CD3−CD56bright, CD3−CD56dim NK cells and total NK cells co-cultured with tumor cells were sorted using an Aria Fusion sorter (BD Biosciences).
Cell culture
Molm-13, EOL-1 and MV4-11 cell lines were purchased from the American Type Culture Collection (ATCC). Molm-13 and EOL-1 cell lines were cultured in RPMI with 10% heat-inactivated FBS (Sigma-Aldrich), and MV4-11 in IMDM with 10% FBS. All cells were incubated at 37°C in a 5% CO2 humidified incubator.
Antibodies and other reagents
Antibodies to Cbl-b (#9498), Granzyme B (#17215), perforin (#62550) and Mertk (#4319) for immunoblotting analysis were purchased from Cell Signaling Technology (CST) and p-Mertk (#ab14921) from Abcam. An antibody to β-actin (#MAB1501R) for immunoblotting analysis was purchased from Millipore-Sigma. For flow cytometric analysis, the anti-hCD56-APC antibody (#B46024) was purchased from Beckman Coulter, anti-hCD3 (#130-113-134) and anti-hIFN-γ (#130-113-493) from Miltenyi Biotec, and anti-hTyro3 (FAB859P), anti-hAxl (#FAB154P) and anti-hMertk (FAB8912P) from R&D. The scrambled and Cbl-b-specific Accell siRNAs were purchased from Dharmacon. Recombinant human IL-2 and IL-15 proteins were obtained from National Institutes of Health (NIH). Recombinant human IL-7, IL-12, IL-18 and IL-21 were purchased from Miltenyi Biotec.
Transient transfection
Primary human NK cells were transfected with Accell siRNA by using the Accell delivery media from Dharmacon. Gene knockdown efficiency of siRNA was determined with immunoblotting analysis.
Reverse transcription-polymerase chain reaction
Total RNA was isolated from primary NK cells with the RNeasy Mini Kit (Qiagen). Complementary DNA (cDNA) was generated by Moloney murine leukemia virus (M-MLV) reverse transcriptase (Invitrogen) and amplified by qPCR with SYBR Green PCR Master Mix (Applied Biosystems) and gene-specific primers. Relative amplification values were normalized to the amplification of GAPDH or 18S rRNA.
Immunoblotting
Cells were harvested and suspended in RIPA lysis buffer (Thermo Fisher Scientific) on ice for 20 minutes. Equal amount of protein was resolved by 5–15% Criterion TGX gel (Bio-Rad) and then transferred onto a Nitrocellulose (NC) or PVDF membrane (Thermo Fisher Scientific). The membrane was incubated with a primary antibody at 4°C overnight and an IRDye secondary antibody (Li-COR Biosciences) for 1 hour at room temperature. The immunoblots were visualized with Odyssey CLx Imager (Li-COR Biosciences). Densitometric analysis was performed to quantify intensity of gel bands with Image J (National Institutes of Health).
ELISA
Primary NK cells were plated in an equivalent number of 100,000 cells/well in a 96-well plate in RPMI and supplemented with 10% FBS. Cells were treated with recombinant human IL-15 (10 ng/ml) for 24 h, and cell-free culture supernatant was collected and frozen at −80°C for later use. IFN-γ concentration was measured by ELISA using anti-IFN-γ antibody from Thermo Fisher Scientific. Granzyme B concentration was measured by ELISA kit purchased from R&D.
51Cr-release cytotoxicity assay
51Cr cytotoxicity assay was performed as described previously (18). Primary NK cells were transduced with siRNAs in Accell delivery media for 24 h, followed by being treated with or without IL-15 (2 ng/ml) for another 16 h. The treated NK cells were co-cultured with 51Cr labeled Molm-13, MV4-11 and EOL-1 cells in triplicates in a 96-well U-bottom plate at multiple ET ratios for 4 h at 37°C in a 5% CO2 incubator. The supernatant was harvested from each well and transferred into 96-well Luma plate and analyzed using a Microbeta scintillation counter (Wallac, PerkinElmer).
Flow cytometry
For flow cytometry to determine protein cell surface expression, cells were stained with monoclonal antibodies at room temperature for 20 minutes and washed with FACS buffer prior to analysis using a Fortessa X-20 flow cytometer (BD Biosciences). For IFN-γ intracellular flow cytometric analysis, 1 mg/ml GolgiPlug (BD Biosciences) was added for 4 h before cell harvest; cells were then permeabilized and fixed using a Cytofix/Cytoperm Fixation/Permeabilization Solution Kit (BD Biosciences), followed by analysis with a Fortessa X-20 flow cytometer. Intracellular flow cytometric analysis of Mertk was performed similarly except that GolgiPlug was not added. Data was analyzed by using Flowjo V10 software (Tree Star, Ashland, OR, USA).
Statistical analysis
For continuous, normally distributed data, student two-tailed t-tests or paired t-tests were used to compare two groups. Two-way ANOVA was used to compare two groups with different conditions. P value of 0.05 or less was considered statistically significant.
Results
Cbl-b is upregulated when primary human NK cells are activated by IL-15, IL-2 or K562
We assessed the expression of Cbl-b protein in primary human NK cells enriched from the blood of healthy donors in response to 24-hour stimulation by different cytokines. We found that stimulation with either IL-15 or IL-2 induced an increase in the expression of Cbl-b, whereas stimulation with IL-7, IL-12, IL-18 and IL-21 showed no significant change compared to resting NK cells incubated without cytokines (Fig. 1A). Since certain tumor cells can trigger NK cell activation, we co-cultured NK cells for 24 h with different tumor cell lines in order to assess this effect on the expression of Cbl-b. Our results showed a 3-fold increase in the expression of Cbl-b when fresh human NK cells were co-cultured with the NK-sensitive K562 myeloid leukemia cell line that lacks MHC class I (MHC I−) (Fig. 1B), whereas the more NK cell-resistant cell lines, Molm-13 and MV4-11 cells, which express MHC class I (MHC I+) (19), did not induce a significant upregulation of Cbl-b in resting NK cells (S Fig.1A). Moreover, primary human NK cells incubated with an NKG2D activating antibody (clone 1D11) overnight showed no significant change of Cbl-b expression compared to those incubated with mouse IgG (S Fig.1B). When incubating NK cells with IL-15, we did not find evidence of a dose response change (Fig. 1C), nor did we find a difference in Cbl-b upregulation between CD56bright and CD56dim NK cells incubated in IL-15 (Fig. 1D). Then Cbl-b transcript was quantified at different time points in NK cells following incubation with IL-15 or IL-2; Cbl-b mRNA increased within 2 h and peaked following 4 h of stimulation with IL-15 or IL-2 compared to unstimulated NK cells (S Fig. 1C). The upregulation of Cbl-b mRNA by IL-15 or IL-2 or K562 stimulation was also observed to be sustained at the longer time point, 24 h post stimulation (S Fig. 1D and 1E).
Fig. 1. Cbl-b is upregulated when primary human NK cells are activated by IL-15, IL-2 or K562.
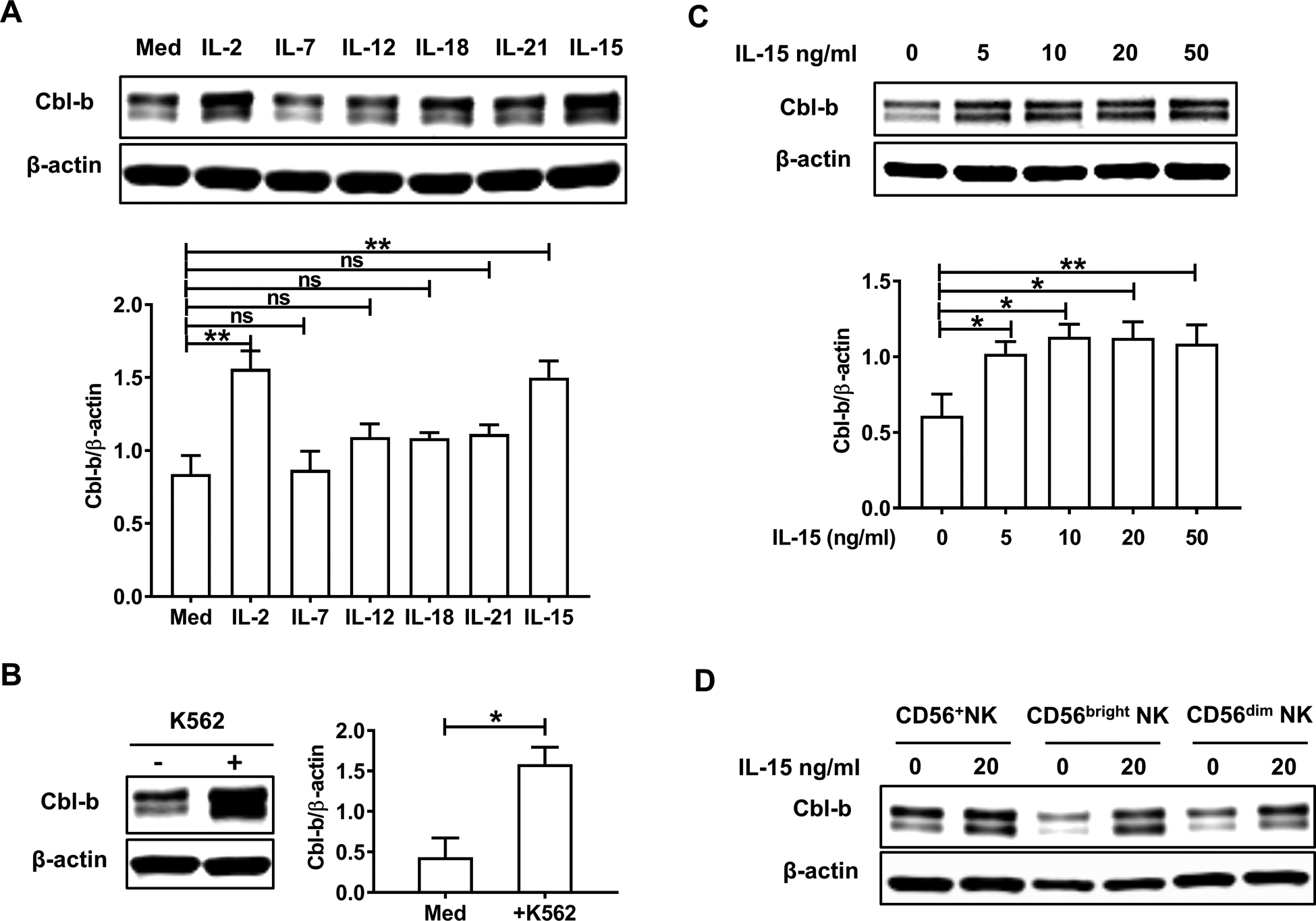
(A) Enriched primary human NK cells were treated with IL-2 (150 IU/ml), IL-7 (50 ng/ml), IL-12 (10 ng/ml), IL-18 (10 ng/ml), IL-21 (20 ng/ml) or IL-15 (20 ng/ml) for 24 h followed by immunoblot analysis (n=5 donors). Densitometric quantification from five independent experiments shows the ratio of Cbl-b protein to β-actin protein. (B) Enriched primary human NK cells were co-cultured with the K562 myeloid leukemia cell line (E/T ratio=10:1) for 24 h followed by immunoblot analysis. Summary data are from three independent experiments. (C) Primary human NK cells were treated with different concentrations of IL-15 for 24 h followed by immunoblot analysis (n=5 donors). (D) Equal numbers of purified CD56+ human NK cells, CD56bright NK cells and CD56dim NK cells were sorted from peripheral blood mononuclear cells isolated by Ficoll, cultured and stimulated with IL-15 (20 ng/ml for 24 h) followed by immunoblot analysis. The data are representative of three independent experiments. Med, medium only; *P<0.05, **P<0.01 by two-tailed unpaired t-test or One-way ANOVA; ns, not significant. Data are presented as mean ± SEM.
JAK-STAT and AKT pathways mediate upregulation of Cbl-b in NK cells
We next investigated the underlying mechanism by which IL-15 or IL-2 up-regulated the expression of Cbl-b protein. Despite the lack of homology in the amino acid sequence between IL-15 and IL-2, both proteins can bind to the IL-2/15Rβγ heterodimer, activating the intracellular signal leading to cell activation (20, 21). Since IL-15 utilizes select JAK-STAT proteins to initiate cellular activation, we pre-treated primary NK cells with the JAK3 inhibitor CP-690550 (22) or the JAK1/2 inhibitor AZD1480 (23) in increasing concentrations prior to IL-15 stimulation then Cbl-b expression levels were evaluated. As shown in Fig. 2A–C, blockade of the JAK-STAT pathway with pre-treatment of either JAK inhibitor followed by stimulation of primary NK cells with IL-15 resulted in a significant decrease in the expression of Cbl-b protein compared with IL-15-stimulated NK cells in the presence of vehicle control. Furthermore, we observed nearly identical results in primary NK cells pre-treated with the AKT1/2/3 inhibitor Afuresertib (24) prior to activation by IL-15 or IL-2 stimulation (Fig. 2D and E). As K562 cells were also able to induce expression of Cbl-b in primary human NK cells (Fig. 1B), we repeated the inhibitor experiments under the K562 cell stimulation. Blockade of the JAK-STAT or AKT pathway in NK cells showed that only the AKT1/2/3 inhibitor Afuresertib rather than either of the two JAK inhibitors (CP-690550 and AZD1480) resulted in a significant downregulation of Cbl-b expression compared to NK cells co-cultured with K562 cells in the presence of vehicle control (S Fig. 2A and B).
Fig. 2. JAK-STAT and AKT pathways mediate upregulation of Cbl-b in NK cells.
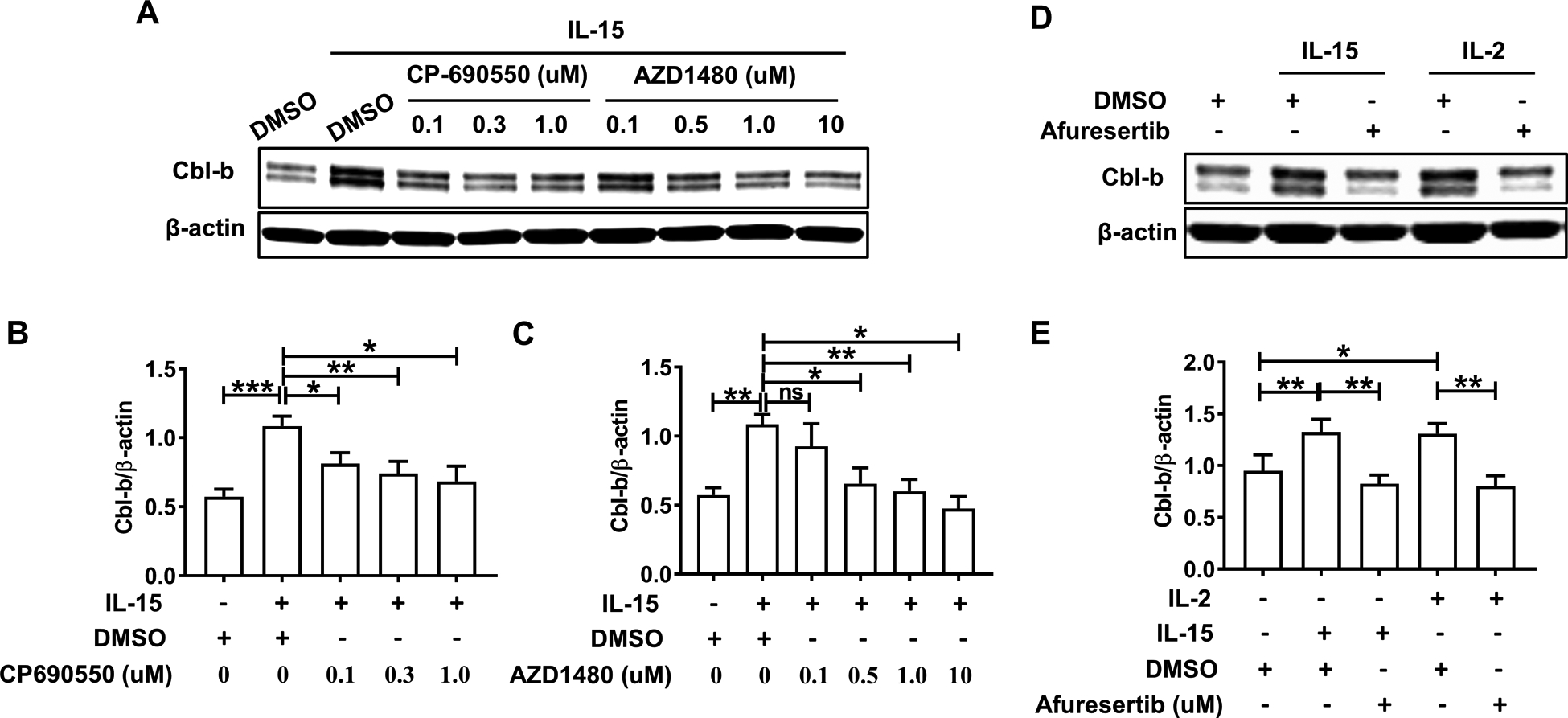
(A) Primary human NK cells were pretreated with the JAK3 inhibitor CP-690550 and the JAK1/2 inhibitor AZD1480 at various concentrations for 90 min prior to a 24 h activation by IL15 (10 ng/ml). NK cells were then harvested for quantification of Cbl-b protein expression by immunoblot analysis. Data are representative of 6 donors performed in a similar fashion. Densitometric quantification assessing the ratio of the Cbl-b protein to β-actin protein levels for 6 donors is summarized in (B) and (C). (D) Primary NK cells were pretreated with the AKT1/2/3 inhibitor Afuresertib (10 uM) for 90 min prior to a 24 h activation by IL-15 (10 ng/ml) or IL-2 (150 IU/ml). Data are representative of 5 donors performed in a similar fashion. Densitometric quantification assessing the ratio of the Cbl-b protein levels to β-actin protein levels for 5 donors is summarized in (E). *P<0.05, **P<0.01, ***P<0.001 by One-way ANOVA or Student’s two-tailed paired t-test. Data are presented as mean ± SEM.
Downregulation of Cbl-b enhances the cytotoxicity of primary human NK cells
We transduced primary human NK cells with Cbl-b-specific siRNA (Cbl-b siRNA) or scrambled siRNA (Scr siRNA) for 48 h and confirmed a significant decrease in the expression of Cbl-b in the experimental group (S Fig. 3A). Both groups were next incubated without or with IL-15 and co-cultured with 51Cr labeled MV4-11, Molm-13 or EOL-1 leukemia cell lines, followed by assessment of specific lysis measured after 4 h of co-culture. In the absence of IL-15 stimulation, NK cells transduced with Cbl-b siRNA showed a significant increase in cytotoxicity against three of the four AML cell lines at the highest E/T ratio of 40:1, compared to NK cells transduced with Scr siRNA (Fig. 3A). When co-incubated with a low concentration of IL-15 (2 ng/ml), NK cells transduced with Cbl-b siRNA also showed a significant increase in cytotoxicity against all four AML cell lines at all three E/T ratios compared to NK cells transduced with Scr siRNA (Fig. 3B). Granzyme B mRNA was significantly increased in NK cells transduced with Cbl-b siRNA compared to NK cells transduced with Scr siRNA, either without or with IL-15 stimulation (Fig. 3C), while perforin mRNA trended toward upregulation in NK cells transduced with Cbl-b siRNA but the upregulation did not reach a statistical significance (S Fig. 3B). The secretion of soluble granzyme B protein was moderately but significantly higher in resting NK cells transduced with Cbl-b siRNA compared to resting NK cells transduced with Scr siRNA (Fig. 3D), whereas with IL-15 stimulation, the secretion of soluble granzyme B protein from the NK cells transduced with Cbl-b siRNA was 2.6 times higher compared to NK cells transduced with Scr siRNA (Fig. 3D). Immunoblot analysis demonstrated an increase of total GZMB protein accompanied with the knockdown of Cbl-b by siRNA (Fig. 3E). In addition, the total protein level of perforin was also significantly increased in NK cells transduced with Cbl-b siRNA compared to NK cells transduced with Scr siRNA with IL-15 stimulation (S Fig. 3C).
Fig. 3. Downregulation of Cbl-b enhances the cytotoxicity of primary human NK cells.
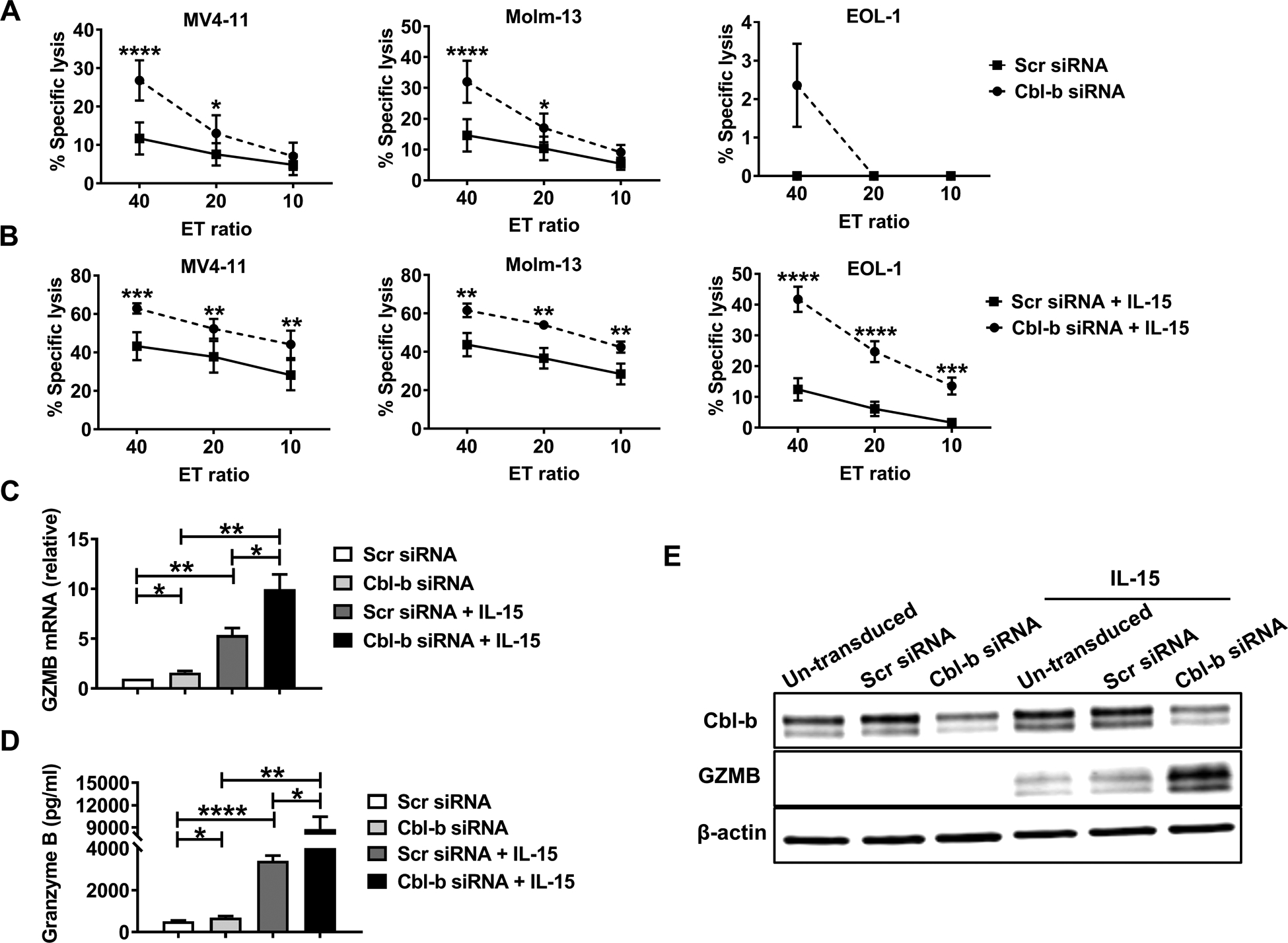
(A and B) Primary human NK cells transduced with Cbl-b siRNA or Scr siRNA for 24h were stimulated without (A) or with (B) a low concentration of IL-15 (2 ng/ml) for 16h, then co-cultured with 51Cr labeled Molm-13, MV4-11 or EOL-1 leukemia cell lines for 4 h, followed by quantification of specific tumor cell lysis. Each experiment was repeated with 6 different normal donors. (C) qRT-PCR was performed to quantify the granzyme B (GZMB) mRNA levels (n=8 donors) in siRNA- and Scr siRNA-transduced primary human NK cells incubated without or with IL-15 (10 ng/ml) for 16 h. (D) The concentration of the granzyme B protein in the supernatants of Cbl-b siRNA- and Scr siRNA-transduced primary human NK cells incubated without (688.1 pg/ml vs 525.4 pg/ml; P=0.0232) or with IL-15 (10 ng/ml) for 24 h (8789 pg/ml vs 3397 pg/ml; P=0.0232) was measured by ELISA (n=8 donors). (E) The level of granzyme B protein in Cbl-b siRNA- and Scr siRNA-transduced primary human NK cells incubated without or with IL-15 (10 ng/ml) for 24 h was measured by immunoblot analysis. The data are representative of three experiments. Scr-scrambled; *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 by One-way or Two-way ANOVA; ns, not significant. Data presented as mean ± SEM.
Downregulation of Cbl-b increases IFN-γ secretion in IL-15-activated primary human NK cells
To further explore how Cbl-b regulates the functions of primary human NK cells, we performed RNA-seq analyses using enriched primary NK cells from healthy donors transduced with Cbl-b siRNA or Scr siRNA without or with IL-15 treatment for 16 h (n=4 in each group). After normalizing the level of each transcript in Cbl-b downregulated NK cells transduced with Cbl-b siRNA to their corresponding controls, we drew a heatmap quantifying the differentially expressed genes noted between NK cells transduced with Cbl-b siRNA or Scr siRNA and co-cultured without or with IL-15 (Fig. 4A). The RNA-seq data revealed that the transcriptional level of granzyme B was increased in human NK cells transduced with Cbl-b siRNA compared to those transduced with Scr siRNA either without or with IL-15 stimulation, which is consistent with our above results (Fig. 3C). We detected IFN-γ mRNA expression levels in IL-15-activated primary human NK cells, and the result was consistent with our RNA-seq data, with IFN-γ mRNA increased dramatically in the Cbl-b siRNA group compared to that in the Scr siRNA group (Fig. 4B). Using intracellular flow cytometry, we demonstrated that IL-15-activated primary human NK cells transduced with Cbl-b siRNA expressed 2.5-fold higher levels of IFN-γ compared to NK cells transduced with Scr siRNA (S Fig. 4A and Fig. 4C). The intracellular results were supported by assessment IFN-γ secretion into the supernatant, in that IL-15-activated primary human NK cells transduced with Cbl-b siRNA expressed 10-fold higher levels of IFN-γ compared to NK cells transduced with Scr siRNA (Fig. 4D). Additionally, the percentage of IFN-γ+ fraction in IL-18-stimulated primary human NK cells transduced with Cbl-b siRNA was 2.5-fold higher than that in NK cells transduced with Scr siRNA (S Fig. 4B), while there was no significant difference in IL-12-stimulated or IL-12 and IL-18 co-stimulated primary human NK cells (S Fig. 4C and D).
Fig. 4. Downregulation of Cbl-b increases IFN-γ secretion in IL-15-activated primary human NK cells.
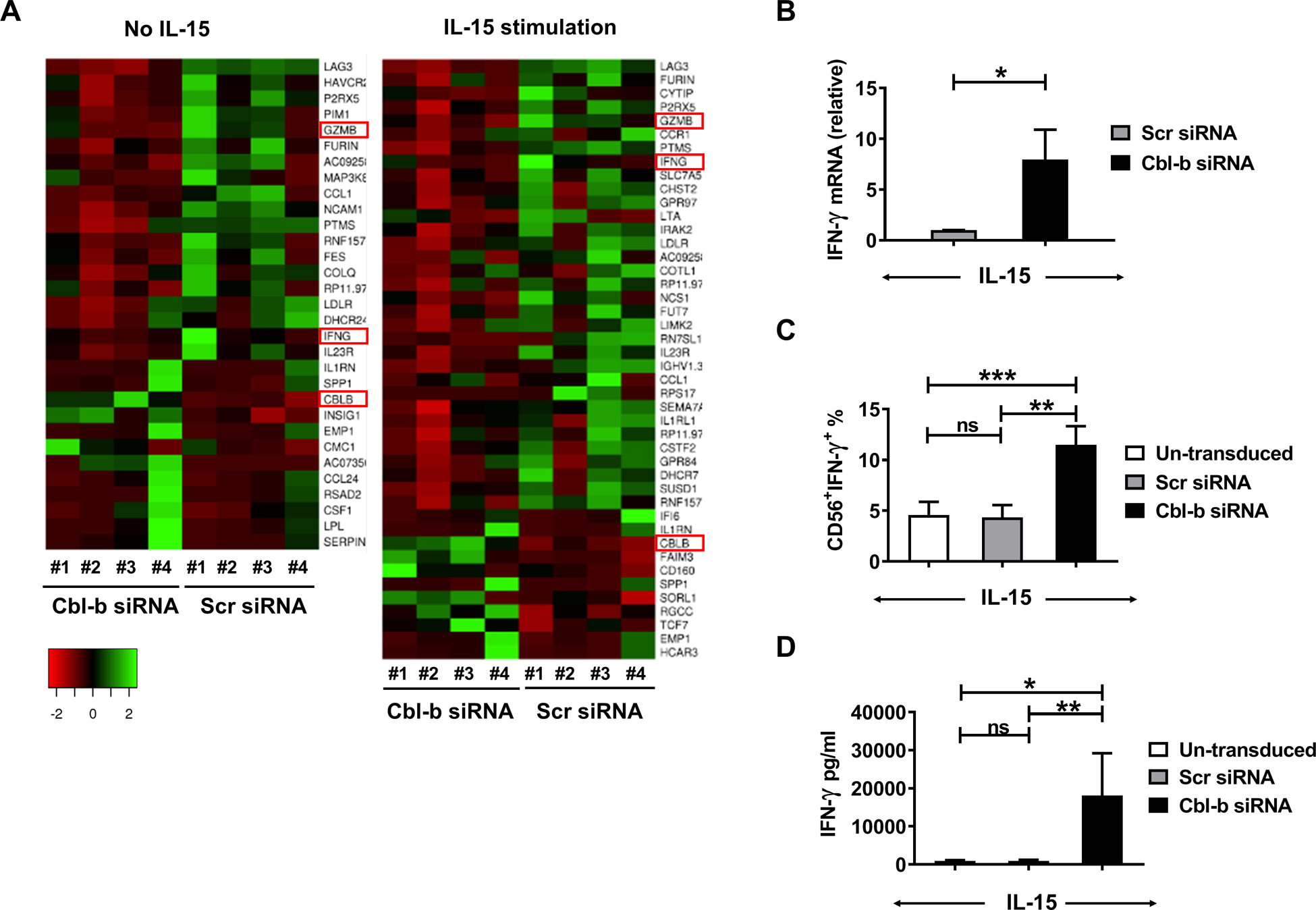
(A) RNAseq was conducted using primary human NK cells transduced with either Cbl-b siRNA or Scr siRNA without or with incubation in IL-15 (10 ng/ml) for 16 h (4 donors for each group). The heatmap illustrates significantly up-regulated (red) or downregulated (green) gene expression. (B) qRT-PCR was performed to quantify the mRNA levels of IFN-γ (n=6 donors) after IL-15 (10 ng/ml) 6 h stimulation. (C) The percentage of intracellular IFN-γ+ cells in un-transduced NK cells or those transduced with Cbl-b siRNA or Scr siRNA, each activated with IL-15 (10 ng/ml) for 24 h (11.5% vs 4.338%; P=0.0008; n=5 donors). (D) Supernatants were collected after IL-15 (10 ng/ml) for 24 h and IFN-γ concentrations measured by ELISA (18115 pg/ml vs 882.5 pg/ml; P=0.0099; n=7 donors). Scr-scrambled; *P<0.05, **P<0.01, ***P<0.001 by Student’s two-tailed paired t-test or One-way ANOVA; ns, not significant. Data presented as mean ± SEM.
Cbl-b regulates phosphorylation of Mertk in primary human NK cells
The TAM receptor family consists of Tyro3, Axl and Mertk, all of which are expressed in mature mouse NK cells (25) and are molecular substrates for Cbl-b ubiquitylation both in vitro and in vivo (17). To investigate the possible relationship between Cbl-b and the TAM receptor family in primary human NK cells, we detected the expression of the three TAM receptor members in NK cells. Intracellular flow cytometric analysis showed that Mertk could be detected in primary human NK cells while the other two-family members Tyro3 and Axl were undetectable (Fig. 5A). Immunoblotting analysis showed that the phosphorylation level of Mertk (p-Mertk) was significantly increased in NK cells transduced with Cbl-b siRNA compared to those transduced with Scr siRNA (Fig. 5B and S Fig. 4E). Previous studies showed that the TAM family receptors are the molecular substrates for Cbl-b-mediated ubiquitylation in murine NK cells (17). However, the level of total Mertk is low in human NK cells (S Fig. 4F), preventing us to characterize the mechanism associated with the p-Mertk regulation by Cbl-b. Next, primary human NK cells were pre-treated with or without Mertk inhibitors (UNC2550 or UNC2881), followed by IL-15 stimulation. We found that in the presence of IL-15, primary human NK cells treated with Mertk inhibitors expressed approximately 3-fold lower levels of IFN-γ compared to control with no treatment of the inhibitors (Fig. 5C). Immunoblotting analysis showed that the level of p-STAT5 decreased in IL-15-acitvated primary human NK cells treated with Mertk inhibitors compared to control with no treatment of the inhibitors (Fig. 5D). Our data suggest that Cbl-b regulates Mertk phosphorylation in primary human NK cells, leading to a suppressive role in activated NK cells.
Fig. 5. Cbl-b regulates phosphorylation of Mertk in primary human NK cells.
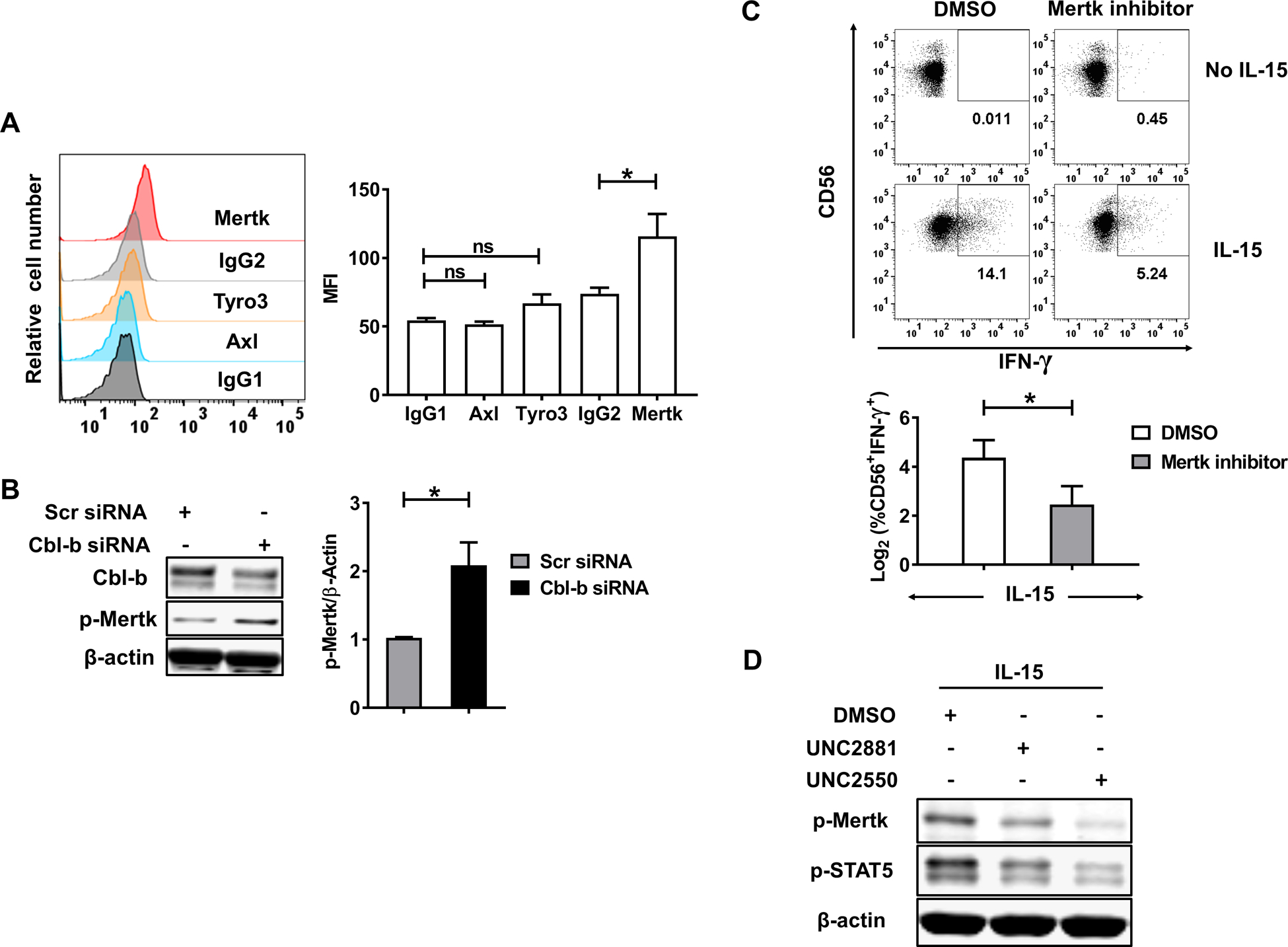
(A) Representative intracellular flow cytometric analysis of the TAM family receptors expression in primary human NK cells. Summarized data are shown from 6 donors. IgG1 is the isotype for Tyro3 and Axl, while IgG2 for Mertk. (B) The levels of Mertk phosphorylation and Cbl-b in Cbl-b siRNA- or Scr siRNA-transduced primary human NK were measured by immunoblot analysis. Data presented are representative of five independent experiments. (C) Primary NK cells were pre-treated with the Mertk inhibitors UNC2550 or UNC2881 (5 uM) for 90min prior to a 24 h activation by IL-15 (10 ng/ml), then the percentage of intracellular IFN-γ+ cells in NK cells was detected (20.64 % vs 5.49 %; P=0.0228). Data presented are representative of 4 donors performed in a similar fashion and are summarized in the panel below the representative flow dot plots. (D) Primary human NK cells were pretreated with the Mertk inhibitors UNC2550 or UNC2881 (5 uM) for 90 min prior to 30 min activation by IL-15 (10 ng/ml), followed by immunoblot analysis for phospho-Mertk. Data are representative of 4 donors with similar results. Scr-scrambled; *P<0.05; ns, not significant. Data presented as mean ± SEM.
Discussion
A multitude of immune checkpoint molecules have been discovered that help to keep immune cells from auto-reactivity, and yet when engaged can also prevent such cells from effectively surveying against malignant transformation. Examples of these checkpoint molecules include killer inhibitory receptors (KIRs) (26, 27), T cell immunoglobulin- and mucin-domain-containing molecule 3 (TIM-3) (28, 29), programmed cell death protein 1 (PD-1) (30, 31) and lymphocyte activation gene 3 (LAG-3) (32, 33). Each of these has been considered as a potentially druggable target in order to promote effective tumor immune surveillance and therapy. During the past two decades, several studies revealed that Cbl-b negatively regulates activation pathways such as T-cell receptor (TCR) signaling in T cells (34, 35), CD40 signaling in B cells (36) and FcεR1 signaling in mast cells (37). However, the role of Cbl-b in regulating human NK cell activation via any signaling pathway is largely unknown.
Stimulation of NK cells by cytokines can prime them by lowering the threshold for further activation and inducing expression of effector molecules (38, 39). IL-2 and IL-15 transduce their activation signal via a heterodimeric receptor consisting of IL-2/15β and the γc chain that are expressed on NK cells, with subsequent activation of the JAK-STAT5 pathway (40, 41), the MAPK pathway (42) and the PI3K pathway (43), followed by the enhancement of their cytolytic potential and anti-tumor responses. When the heterodimeric cytokine IL-12 binds its receptor expressed on NK cells, activation is triggered via the JAK-STAT4 pathway (44). IL-15 has been shown to be the critical endogenous cytokine for human NK cell development (18, 45), survival (46, 47) and activation (48). IL-21, another γc cytokine, is not required for the development of NK cells in that IL-21R−/− mice demonstrate normal NK cell development (49). IL-21 alone does not induce IFN-γ expression in human NK cells (39). IL-18 is a member of the pro-inflammatory IL-1 family, and is usually recognized as a NK cell co-stimulatory cytokine with IL-12 and/or IL-15 (50, 51). IL-7 selectively promotes the survival of the CD56bright NK subset but does not enhance NK cell cytotoxicity or IFN-γ secretion (52). We observed that IL-15, IL-2 and IL-18 significantly upregulate Cbl-b but not IL-7, IL-12 or IL-21, suggesting that upregulation of Cbl-b in activated NK cells is cytokine-dependent.
As in the case with every activation signal in the immune system, there needs to be a counter-regulatory mechanism to prevent autoimmunity or other toxicities that can result from excessive immune stimulation. Based on our data, it seems that activation by different cytokines is associated with different counter-regulatory mechanisms. In animal models, chronic stimulation of NK cells with an excess of endogenous IL-15 can result in malignant transformation resulting in large granular lymphocytic leukemia (53–55) or cutaneous T cell lymphoma (56), both incurable cancers in man. Thus, counter-regulation of IL-15-induced NK cell activation, which appears to be mediated at least in part by Cbl-b in our study, is likely critical for the proper regulation of NK cells in vivo.
The Cbl protein family are multifunctional adaptor molecules with ubiquitin ligase activity. The phospholipase Cγ (PLCγ) family, which is crucial for NK cell effector functions, is recruited to the contact site of the NK cell and its target mediated by the linker for activation of T cells (LAT) (57–59). Matalon et al. determined that LAT is ubiquitylated by c-Cbl and Cbl-b, leading to the abrogation of NK cell cytotoxicity (16). The hematopoietic-specific Rho-family GTP exchange factor Vav-1 is not only a regulator in B and T cells, but also forms part of signaling pathways required for human NK cell cytotoxicity (60). In human γδT cells, Cbl-b inhibits their cytotoxicity by decreasing the level of phosphorylated Vav-1 (61). The current study demonstrated that downregulation of Cbl-b in primary human NK cells resulted in higher cytotoxicity and IFN-γ production, especially in the presence of low, near physiologic concentrations of IL-15. Moreover, the results here are consistent with data from Cbl-b−/− mice, which showed an increase in NK cell perforin expression, cytotoxicity and secretion of IFN-γ when activated with NKG2D antibody (17). However, in human NK cell, triggered by NKG2D and 2B4 co-engagement, the enhancement of NK cell function was observed with knockdown of c-Cbl while similar enhancement of NK cell function was not observed with knockdown of Cbl-b (62). Collectively, the past and current studies suggest that Cbl-b in murine and human NK cells is complicate, which can be species- and context-dependent.
NK cells represent the first line of defense against tumor cells and likely play a critical role in tumor immune surveillance in human. Tumors lacking MHC class I antigens are highly susceptible to elimination by NK cells (63). NK cells are able to launch killing of additional target cells after the first one is eradicated, a process termed serial killing (64). A previous study reveals that NK cells undergo inactivation and lose their cytotoxic function with decreased levels of their perforin and granzyme B after being exposed to target cells (65). The K562 cell line, which is a leukemic cell line susceptible to NK cell cytotoxicity lacking MHC class I expression, can activate primary human NK cells, and our data show that the activation results in upregulation of Cbl-b, which is a negative regulator that controls NK cell effector functions. These data suggest that Cbl-b may accumulate in NK cells, leading to downregulation of effector functions, after serial killing of each target. This serial killing process is continuous and may eventually result in the complete loss of NK cell cytotoxicity against target cells.
The roles of and mechanisms by which TAM receptors regulate innate immune cell function are complex and cell-type dependent. In macrophages, IFN-α is able to up-regulate Axl, which in turn inhibits TNF-α secretion (66). Mertk binding of apoptotic cells in dendritic cells (DCs) also regulates NF-κB-dependent transcription (67). T cells were originally thought not to express TAM receptors but the expression of Mertk was later found to be expressed by activated human CD4+ T cells, in which the receptor plays a negative role (68). However, Marlies et al. revealed that Mertk serves as a late co-stimulatory signal for CD8+ T cells (69). Different from murine NK cells in which all three TAM receptors are expressed (17), our current study showed that primary human NK cells do not have expression of the Tyro3 and Axl receptors and only intracellular Mertk could be detected at very low level. Recently, Paul et al. reported that the surface expression of TAM receptors could not be detected in either resting or IL-15-activated primary human NK cells (70). To our knowledge, prior to our current study, intracellular expression of Mertk in primary human NK cells has not been explored. In the current study, we found that downregulation of Cbl-b increases the phosphorylation levels of Mertk and blocking Mertk phosphorylation inhibits the function of activated human NK cells. Paolino M et al. uncovered that Cbl-b negatively regulates NK cell function via increasing ubiquitination and subsequently degrading TAM receptors in a murine model (17). However, due to the limitation of the low level of total Mertk expression in human NK cells, we were unable to confirm the same mechanism by which Mertk is regulated by Cbl-b. Collectively, our data suggest Cbl-b plays a negative regulatory role in activated primary human NK cells and it remains to be characterized whether the ubiquitin ligase function of Cbl-b regulates Mertk expression and/or activity.
In conclusion, our study demonstrates that Cbl-b is an intracellular checkpoint in human NK cells, negatively regulating cytokine- or target-induced NK cell cytotoxicity and cytokine production. Targeting the Cbl-b in NK cells may therefore result in enhanced innate anti-tumor cytolytic activity.
Supplementary Material
Key points:
Cbl-b is induced in IL-15/IL-2/K562-activated primary human NK cells.
Abrogation of Cbl-b enhances cytotoxicity of primary human NK cells.
Funding
This work was supported by grants from the NIH (CA210087, CA068458 and CA163205 to M.A. Caligiuri). M.J. Yu and N. Brooks were supported by City of Hope’s Eugene and Ruth Roberts Summer Student Academy.
Footnotes
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Sun JC, and Lanier LL. 2011. NK cell development, homeostasis and function: parallels with CD8(+) T cells. Nat Rev Immunol 11: 645–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vivier E, Tomasello E, Baratin M, Walzer T, and Ugolini S. 2008. Functions of natural killer cells. Nat Immunol 9: 503–510. [DOI] [PubMed] [Google Scholar]
- 3.Spits H, Lanier LL, and Phillips JH. 1995. Development of human T and natural killer cells. Blood 85: 2654–2670. [PubMed] [Google Scholar]
- 4.Herberman RB, Nunn ME, Holden HT, and Lavrin DH. 1975. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumors. II. Characterization of effector cells. Int J Cancer 16: 230–239. [DOI] [PubMed] [Google Scholar]
- 5.Lanier LL, Phillips JH, Hackett J Jr., Tutt M, and Kumar V. 1986. Natural killer cells: definition of a cell type rather than a function. J Immunol 137: 2735–2739. [PubMed] [Google Scholar]
- 6.Muntasell A, Ochoa MC, Cordeiro L, Berraondo P, Lopez-Diaz de Cerio A, Cabo M, Lopez-Botet M, and Melero I. 2017. Targeting NK-cell checkpoints for cancer immunotherapy. Current opinion in immunology 45: 73–81. [DOI] [PubMed] [Google Scholar]
- 7.Morvan MG, and Lanier LL. 2016. NK cells and cancer: you can teach innate cells new tricks. Nat Rev Cancer 16: 7–19. [DOI] [PubMed] [Google Scholar]
- 8.Vivier E, Nunes JA, and Vely F. 2004. Natural killer cell signaling pathways. Science 306: 1517–1519. [DOI] [PubMed] [Google Scholar]
- 9.Kim N, and Kim HS. 2018. Targeting Checkpoint Receptors and Molecules for Therapeutic Modulation of Natural Killer Cells. Front Immunol 9: 2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duan L, Reddi AL, Ghosh A, Dimri M, and Band H. 2004. The Cbl family and other ubiquitin ligases: destructive forces in control of antigen receptor signaling. Immunity 21: 7–17. [DOI] [PubMed] [Google Scholar]
- 11.Liu YC 2004. Ubiquitin ligases and the immune response. Annu Rev Immunol 22: 81–127. [DOI] [PubMed] [Google Scholar]
- 12.Chiang YJ, Kole HK, Brown K, Naramura M, Fukuhara S, Hu RJ, Jang IK, Gutkind JS, Shevach E, and Gu H. 2000. Cbl-b regulates the CD28 dependence of T-cell activation. Nature 403: 216–220. [DOI] [PubMed] [Google Scholar]
- 13.Jeon MS, Atfield A, Venuprasad K, Krawczyk C, Sarao R, Elly C, Yang C, Arya S, Bachmaier K, Su L, Bouchard D, Jones R, Gronski M, Ohashi P, Wada T, Bloom D, Fathman CG, Liu YC, and Penninger JM. 2004. Essential role of the E3 ubiquitin ligase Cbl-b in T cell anergy induction. Immunity 21: 167–177. [DOI] [PubMed] [Google Scholar]
- 14.Kitaura Y, Jang IK, Wang Y, Han YC, Inazu T, Cadera EJ, Schlissel M, Hardy RR, and Gu H. 2007. Control of the B cell-intrinsic tolerance programs by ubiquitin ligases Cbl and Cbl-b. Immunity 26: 567–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li X, Gadzinsky A, Gong L, Tong H, Calderon V, Li Y, Kitamura D, Klein U, Langdon WY, Hou F, Zou YR, and Gu H. 2018. Cbl Ubiquitin Ligases Control B Cell Exit from the Germinal-Center Reaction. Immunity 48: 530–541 e536. [DOI] [PubMed] [Google Scholar]
- 16.Matalon O, and Barda-Saad M. 2016. Cbl ubiquitin ligases mediate the inhibition of natural killer cell activity. Commun Integr Biol 9: e1216739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paolino M, Choidas A, Wallner S, Pranjic B, Uribesalgo I, Loeser S, Jamieson AM, Langdon WY, Ikeda F, Fededa JP, Cronin SJ, Nitsch R, Schultz-Fademrecht C, Eickhoff J, Menninger S, Unger A, Torka R, Gruber T, Hinterleitner R, Baier G, Wolf D, Ullrich A, Klebl BM, and Penninger JM. 2014. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature 507: 508–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mrozek E, Anderson P, and Caligiuri MA. 1996. Role of interleukin-15 in the development of human CD56+ natural killer cells from CD34+ hematopoietic progenitor cells. Blood 87: 2632–2640. [PubMed] [Google Scholar]
- 19.Dong W, Wu X, Ma S, Wang Y, Nalin AP, Zhu Z, Zhang J, Benson DM, He K, Caligiuri MA, and Yu J. 2019. The Mechanism of Anti-PD-L1 Antibody Efficacy against PD-L1-Negative Tumors Identifies NK Cells Expressing PD-L1 as a Cytolytic Effector. Cancer Discov 9: 1422–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giri JG, Ahdieh M, Eisenman J, Shanebeck K, Grabstein K, Kumaki S, Namen A, Park LS, Cosman D, and Anderson D. 1994. Utilization of the beta and gamma chains of the IL-2 receptor by the novel cytokine IL-15. EMBO J 13: 2822–2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waldmann T, Tagaya Y, and Bamford R. 1998. Interleukin-2, interleukin-15, and their receptors. Int Rev Immunol 16: 205–226. [DOI] [PubMed] [Google Scholar]
- 22.Kudlacz E, Perry B, Sawyer P, Conklyn M, McCurdy S, Brissette W, Flanagan, and Changelian P. 2004. The novel JAK-3 inhibitor CP-690550 is a potent immunosuppressive agent in various murine models. Am J Transplant 4: 51–57. [DOI] [PubMed] [Google Scholar]
- 23.Plimack ER, Lorusso PM, McCoon P, Tang W, Krebs AD, Curt G, and Eckhardt SG. 2013. AZD1480: a phase I study of a novel JAK2 inhibitor in solid tumors. Oncologist 18: 819–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spencer A, Yoon SS, Harrison SJ, Morris SR, Smith DA, Brigandi RA, Gauvin J, Kumar R, Opalinska JB, and Chen C. 2014. The novel AKT inhibitor afuresertib shows favorable safety, pharmacokinetics, and clinical activity in multiple myeloma. Blood 124: 2190–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Caraux A, Lu Q, Fernandez N, Riou S, Di Santo JP, Raulet DH, Lemke G, and Roth C. 2006. Natural killer cell differentiation driven by Tyro3 receptor tyrosine kinases. Nat Immunol 7: 747–754. [DOI] [PubMed] [Google Scholar]
- 26.Kohrt HE, Thielens A, Marabelle A, Sagiv-Barfi I, Sola C, Chanuc F, Fuseri N, Bonnafous C, Czerwinski D, Rajapaksa A, Waller E, Ugolini S, Vivier E, Romagne F, Levy R, Blery M, and Andre P. 2014. Anti-KIR antibody enhancement of anti-lymphoma activity of natural killer cells as monotherapy and in combination with anti-CD20 antibodies. Blood 123: 678–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benson DM Jr., Hofmeister CC, Padmanabhan S, Suvannasankha A, Jagannath S, Abonour R, Bakan C, Andre P, Efebera Y, Tiollier J, Caligiuri MA, and Farag SS. 2012. A phase 1 trial of the anti-KIR antibody IPH2101 in patients with relapsed/refractory multiple myeloma. Blood 120: 4324–4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tan S, Xu Y, Wang Z, Wang T, Du X, Song X, Guo X, Peng J, Zhang J, Liang Y, Lu J, Peng J, Gao C, Wu Z, Li C, Li N, Gao L, Liang X, and Ma C. 2019. Tim-3 hampers tumor surveillance of liver resident and conventional NK cells by disrupting PI3K signaling. Cancer research. [DOI] [PubMed] [Google Scholar]
- 29.Xu L, Huang Y, Tan L, Yu W, Chen D, Lu C, He J, Wu G, Liu X, and Zhang Y. 2015. Increased Tim-3 expression in peripheral NK cells predicts a poorer prognosis and Tim-3 blockade improves NK cell-mediated cytotoxicity in human lung adenocarcinoma. International immunopharmacology 29: 635–641. [DOI] [PubMed] [Google Scholar]
- 30.Vari F, Arpon D, Keane C, Hertzberg MS, Talaulikar D, Jain S, Cui Q, Han E, Tobin J, Bird R, Cross D, Hernandez A, Gould C, Birch S, and Gandhi MK. 2018. Immune evasion via PD-1/PD-L1 on NK cells and monocyte/macrophages is more prominent in Hodgkin lymphoma than DLBCL. Blood 131: 1809–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hsu J, Hodgins JJ, Marathe M, Nicolai CJ, Bourgeois-Daigneault MC, Trevino TN, Azimi CS, Scheer AK, Randolph HE, Thompson TW, Zhang L, Iannello A, Mathur N, Jardine KE, Kirn GA, Bell JC, McBurney MW, Raulet DH, and Ardolino M. 2018. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. The Journal of clinical investigation 128: 4654–4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lino AC, Dang VD, Lampropoulou V, Welle A, Joedicke J, Pohar J, Simon Q, Thalmensi J, Baures A, Fluhler V, Sakwa I, Stervbo U, Ries S, Jouneau L, Boudinot P, Tsubata T, Adachi T, Hutloff A, Dorner T, Zimber-Strobl U, de Vos AF, Dahlke K, Loh G, Korniotis S, Goosmann C, Weill JC, Reynaud CA, Kaufmann SHE, Walter J, and Fillatreau S. 2018. LAG-3 Inhibitory Receptor Expression Identifies Immunosuppressive Natural Regulatory Plasma Cells. Immunity 49: 120–133 e129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williams JB, Horton BL, Zheng Y, Duan Y, Powell JD, and Gajewski TF. 2017. The EGR2 targets LAG-3 and 4–1BB describe and regulate dysfunctional antigen-specific CD8+ T cells in the tumor microenvironment. The Journal of experimental medicine 214: 381–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shamim M, Nanjappa SG, Singh A, Plisch EH, LeBlanc SE, Walent J, Svaren J, Seroogy C, and Suresh M. 2007. Cbl-b regulates antigen-induced TCR down-regulation and IFN-gamma production by effector CD8 T cells without affecting functional avidity. J Immunol 179: 7233–7243. [DOI] [PubMed] [Google Scholar]
- 35.Zhang W, Shao Y, Fang D, Huang J, Jeon MS, and Liu YC. 2003. Negative regulation of T cell antigen receptor-mediated Crk-L-C3G signaling and cell adhesion by Cbl-b. J Biol Chem 278: 23978–23983. [DOI] [PubMed] [Google Scholar]
- 36.Qiao G, Lei M, Li Z, Sun Y, Minto A, Fu YX, Ying H, Quigg RJ, and Zhang J. 2007. Negative regulation of CD40-mediated B cell responses by E3 ubiquitin ligase Casitas-B-lineage lymphoma protein-B. J Immunol 179: 4473–4479. [DOI] [PubMed] [Google Scholar]
- 37.Qu X, Sada K, Kyo S, Maeno K, Miah SM, and Yamamura H. 2004. Negative regulation of FcepsilonRI-mediated mast cell activation by a ubiquitin-protein ligase Cbl-b. Blood 103: 1779–1786. [DOI] [PubMed] [Google Scholar]
- 38.Konjevic GM, Vuletic AM, Mirjacic Martinovic KM, Larsen AK, and Jurisic VB. 2019. The role of cytokines in the regulation of NK cells in the tumor environment. Cytokine 117: 30–40. [DOI] [PubMed] [Google Scholar]
- 39.Strengell M, Matikainen S, Siren J, Lehtonen A, Foster D, Julkunen I, and Sareneva T. 2003. IL-21 in synergy with IL-15 or IL-18 enhances IFN-gamma production in human NK and T cells. J Immunol 170: 5464–5469. [DOI] [PubMed] [Google Scholar]
- 40.Park SY, Saijo K, Takahashi T, Osawa M, Arase H, Hirayama N, Miyake K, Nakauchi H, Shirasawa T, and Saito T. 1995. Developmental defects of lymphoid cells in Jak3 kinase-deficient mice. Immunity 3: 771–782. [DOI] [PubMed] [Google Scholar]
- 41.Teglund S, McKay C, Schuetz E, van Deursen JM, Stravopodis D, Wang D, Brown M, Bodner S, Grosveld G, and Ihle JN. 1998. Stat5a and Stat5b proteins have essential and nonessential, or redundant, roles in cytokine responses. Cell 93: 841–850. [DOI] [PubMed] [Google Scholar]
- 42.Zhu X, Suen KL, Barbacid M, Bolen JB, and Fargnoli J. 1994. Interleukin-2-induced tyrosine phosphorylation of Shc proteins correlates with factor-dependent T cell proliferation. J Biol Chem 269: 5518–5522. [PubMed] [Google Scholar]
- 43.Gu H, Maeda H, Moon JJ, Lord JD, Yoakim M, Nelson BH, and Neel BG. 2000. New role for Shc in activation of the phosphatidylinositol 3-kinase/Akt pathway. Molecular and cellular biology 20: 7109–7120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang KS, Frank DA, and Ritz J. 2000. Interleukin-2 enhances the response of natural killer cells to interleukin-12 through up-regulation of the interleukin-12 receptor and STAT4. Blood 95: 3183–3190. [PubMed] [Google Scholar]
- 45.Kennedy MK, Glaccum M, Brown SN, Butz EA, Viney JL, Embers M, Matsuki N, Charrier K, Sedger L, Willis CR, Brasel K, Morrissey PJ, Stocking K, Schuh JC, Joyce S, and Peschon JJ. 2000. Reversible defects in natural killer and memory CD8 T cell lineages in interleukin 15-deficient mice. The Journal of experimental medicine 191: 771–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koka R, Burkett PR, Chien M, Chai S, Chan F, Lodolce JP, Boone DL, and Ma A. 2003. Interleukin (IL)-15R[alpha]-deficient natural killer cells survive in normal but not IL-15R[alpha]-deficient mice. The Journal of experimental medicine 197: 977–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carson WE, Fehniger TA, Haldar S, Eckhert K, Lindemann MJ, Lai CF, Croce CM, Baumann H, and Caligiuri MA. 1997. A potential role for interleukin-15 in the regulation of human natural killer cell survival. The Journal of clinical investigation 99: 937–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carson WE, Ross ME, Baiocchi RA, Marien MJ, Boiani N, Grabstein K, and Caligiuri MA. 1995. Endogenous production of interleukin 15 by activated human monocytes is critical for optimal production of interferon-gamma by natural killer cells in vitro. The Journal of clinical investigation 96: 2578–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kasaian MT, Whitters MJ, Carter LL, Lowe LD, Jussif JM, Deng B, Johnson KA, Witek JS, Senices M, Konz RF, Wurster AL, Donaldson DD, Collins M, Young DA, and Grusby MJ. 2002. IL-21 limits NK cell responses and promotes antigen-specific T cell activation: a mediator of the transition from innate to adaptive immunity. Immunity 16: 559–569. [DOI] [PubMed] [Google Scholar]
- 50.Fehniger TA, Shah MH, Turner MJ, VanDeusen JB, Whitman SP, Cooper MA, Suzuki K, Wechser M, Goodsaid F, and Caligiuri MA. 1999. Differential cytokine and chemokine gene expression by human NK cells following activation with IL-18 or IL-15 in combination with IL-12: implications for the innate immune response. J Immunol 162: 4511–4520. [PubMed] [Google Scholar]
- 51.French AR, Holroyd EB, Yang L, Kim S, and Yokoyama WM. 2006. IL-18 acts synergistically with IL-15 in stimulating natural killer cell proliferation. Cytokine 35: 229–234. [DOI] [PubMed] [Google Scholar]
- 52.Michaud A, Dardari R, Charrier E, Cordeiro P, Herblot S, and Duval M. 2010. IL-7 enhances survival of human CD56bright NK cells. J Immunother 33: 382–390. [DOI] [PubMed] [Google Scholar]
- 53.Yokohama A, Mishra A, Mitsui T, Becknell B, Johns J, Curphey D, Blaser BW, Vandeusen JB, Mao H, Yu J, and Caligiuri MA. 2010. A novel mouse model for the aggressive variant of NK cell and T cell large granular lymphocyte leukemia. Leukemia research 34: 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fehniger TA, Suzuki K, Ponnappan A, VanDeusen JB, Cooper MA, Florea SM, Freud AG, Robinson ML, Durbin J, and Caligiuri MA. 2001. Fatal leukemia in interleukin 15 transgenic mice follows early expansions in natural killer and memory phenotype CD8+ T cells. The Journal of experimental medicine 193: 219–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sato N, Sabzevari H, Fu S, Ju W, Petrus MN, Bamford RN, Waldmann TA, and Tagaya Y. 2011. Development of an IL-15-autocrine CD8 T-cell leukemia in IL-15-transgenic mice requires the cis expression of IL-15Ralpha. Blood 117: 4032–4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mishra A, La Perle K, Kwiatkowski S, Sullivan LA, Sams GH, Johns J, Curphey DP, Wen J, McConnell K, Qi J, Wong H, Russo G, Zhang J, Marcucci G, Bradner JE, Porcu P, and Caligiuri MA. 2016. Mechanism, Consequences, and Therapeutic Targeting of Abnormal IL15 Signaling in Cutaneous T-cell Lymphoma. Cancer Discov 6: 986–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Braiman A, Barda-Saad M, Sommers CL, and Samelson LE. 2006. Recruitment and activation of PLCgamma1 in T cells: a new insight into old domains. EMBO J 25: 774–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Caraux A, Kim N, Bell SE, Zompi S, Ranson T, Lesjean-Pottier S, Garcia-Ojeda ME, Turner M, and Colucci F. 2006. Phospholipase C-gamma2 is essential for NK cell cytotoxicity and innate immunity to malignant and virally infected cells. Blood 107: 994–1002. [DOI] [PubMed] [Google Scholar]
- 59.Upshaw JL, Schoon RA, Dick CJ, Billadeau DD, and Leibson PJ. 2005. The isoforms of phospholipase C-gamma are differentially used by distinct human NK activating receptors. J Immunol 175: 213–218. [DOI] [PubMed] [Google Scholar]
- 60.Chan G, Hanke T, and Fischer KD. 2001. Vav-1 regulates NK T cell development and NK cell cytotoxicity. European journal of immunology 31: 2403–2410. [DOI] [PubMed] [Google Scholar]
- 61.Yin S, Zhang J, Mao Y, Hu Y, Cui L, Kang N, and He W. 2013. Vav1-phospholipase C-gamma1 (Vav1-PLC-gamma1) pathway initiated by T cell antigen receptor (TCRgammadelta) activation is required to overcome inhibition by ubiquitin ligase Cbl-b during gammadeltaT cell cytotoxicity. J Biol Chem 288: 26448–26462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim HS, Das A, Gross CC, Bryceson YT, and Long EO. 2010. Synergistic signals for natural cytotoxicity are required to overcome inhibition by c-Cbl ubiquitin ligase. Immunity 32: 175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Correa I, Corral L, and Raulet DH. 1994. Multiple natural killer cell-activating signals are inhibited by major histocompatibility complex class I expression in target cells. European journal of immunology 24: 1323–1331. [DOI] [PubMed] [Google Scholar]
- 64.Bhat R, and Watzl C. 2007. Serial killing of tumor cells by human natural killer cells--enhancement by therapeutic antibodies. PLoS One 2: e326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jewett A, and Bonavida B. 1995. Target-induced anergy of natural killer cytotoxic function is restricted to the NK-target conjugate subset. Cell Immunol 160: 91–97. [DOI] [PubMed] [Google Scholar]
- 66.Sharif MN, Sosic D, Rothlin CV, Kelly E, Lemke G, Olson EN, and Ivashkiv LB. 2006. Twist mediates suppression of inflammation by type I IFNs and Axl. The Journal of experimental medicine 203: 1891–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sen P, Wallet MA, Yi Z, Huang Y, Henderson M, Mathews CE, Earp HS, Matsushima G, Baldwin AS Jr., and Tisch RM. 2007. Apoptotic cells induce Mer tyrosine kinase-dependent blockade of NF-kappaB activation in dendritic cells. Blood 109: 653–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cabezon R, Carrera-Silva EA, Florez-Grau G, Errasti AE, Calderon-Gomez E, Lozano JJ, Espana C, Ricart E, Panes J, Rothlin CV, and Benitez-Ribas D. 2015. MERTK as negative regulator of human T cell activation. J Leukoc Biol 97: 751–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Peeters MJW, Dulkeviciute D, Draghi A, Ritter C, Rahbech A, Skadborg SK, Seremet T, Carnaz Simoes AM, Martinenaite E, Halldorsdottir HR, Andersen MH, Olofsson GH, Svane IM, Rasmussen LJ, Met O, Becker JC, Donia M, Desler C, and Thor Straten P. 2019. MERTK Acts as a Costimulatory Receptor on Human CD8(+) T Cells. Cancer Immunol Res 7: 1472–1484. [DOI] [PubMed] [Google Scholar]
- 70.Giroud P, Renaudineau S, Gudefin L, Calcei A, Menguy T, Rozan C, Mizrahi J, Caux C, Duong V, and Valladeau-Guilemond J. 2020. Expression of TAM-R in Human Immune Cells and Unique Regulatory Function of MerTK in IL-10 Production by Tolerogenic DC. Front Immunol 11: 564133. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.