

Abstract
Background & objectives:
Several phylogenetic classification systems have been devised to trace the viral lineages of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). However, inconsistency in the nomenclature limits uniformity in its epidemiological understanding. This study provides an integration of existing classifications and describes evolutionary trends of the SARS-CoV-2 strains circulating in India.
Methods:
The whole genomes of 330 SARS-CoV-2 samples were sequenced using next-generation sequencing (NGS). Phylogenetic and sequence analysis of a total of 3014 Indian SARS-CoV-2 sequences from 20 different States/Union Territories (January to September 2020) from the Global Initiative on Sharing All Influenza Data (GISAID) database was performed to observe the clustering of Nextstrain and Phylogenetic Assignment of Named Global Outbreak LINeages (Pangolin) lineages with the GISAID clades. The identification of mutational sites under selection pressure was performed using Mixed Effects Model of Evolution and Single-Likelihood Ancestor Counting methods available in the Datamonkey server.
Results:
Temporal data of the Indian SARS-CoV-2 genomes revealed that except for Uttarakhand, West Bengal and Haryana that showed the circulation of GISAID clade O even after July 2020, the rest of the States showed a complete switch to GR/GH clades. Pangolin lineages B.1.1.8 and B.1.113 identified within GR and GH clades, respectively, were noted to be indigenous evolutions. Sites identified to be under positive selection pressure within these clades were found to occur majorly in the non-structural proteins coded by ORF1a and ORF1b.
Interpretation & conclusions:
This study interpreted the geographical and temporal dominance of SARS-CoV-2 strains in India over a period of nine months based on the GISAID classification. An integration of the GISAID, Nextstrain and Pangolin classifications is also provided. The emergence of new lineages B.1.1.8 and B.1.113 was indicative of host-specific evolution of the SARS-CoV-2 strains in India. The hotspot mutations such as those driven by positive selection need to be further characterized.
Keywords: Clades, COVID-19- nucleotide substitution, India, SARS-CoV-2, selection pressure, whole genomes
Genome sequence analyses of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) strains aid in understanding of patterns and determinants of the global spread of the pandemic strain causing coronavirus disease 2019 (COVID-19)1. The phylogenetic analysis of the genome sequences showed that within a short span from the emergence of the SARS-CoV-2 virus, the genetic diversity expanded2,3. This resulted in the delineation of the viral strains into clades, lineages and sub-lineages. The Global Initiative on Sharing All Influenza Data (GISAID)4 database (https://www.gisaid.org/) in its earliest classification divided SARS-CoV-2 into two major lineages/clades 'L' and 'S' based on a mutation L84S in the ORF8 protein. Further, for the purpose of consistent reporting based on marker mutations, it identified three major clades denoted as G, V and O or an unclassified group. These clades evolved from 'L'. Further, the clade G was split into sub-clades GH and GR5. The GISAID clades are presently augmented with more detailed lineages assigned by the Phylogenetic Assignment of Named Global Outbreak LINeages (Pangolin tool (https://virological.org/t/pangolin-web-application-release/482)6. On the other hand, Nextstrain7 classified the SARS-CoV-2 initially into about nine clades referred to as A1a, A2, A2a, A3, A6, B, B1, B2 and B4. These are indicated in the form of ancestral nodes as 19A, 19B, 20A, 20B and 20C.
Thus, it can be noted that several phylogenetic classification systems based on different approaches have been devised to trace the viral lineages of the SARS-CoV-2 across the globe. Inconsistency in the nomenclature systems limits the uniformity in its epidemiological understanding. In this study, we describe the genetic lineages of the strains circulating in India as retrieved from GISAID and provide integration for the SARS-CoV-2 classification systems developed by GISAID, Nextstrain and Pangolin. This study also adds to the whole-genome sequences of SARS-CoV-2, majorly referred samples from different districts of Maharashtra during the period from March 9 to October 14, 2020. To further understand if adaptive evolution of the clades is being observed in the Indian context, selection pressure studies were undertaken.
Material & Methods
This study was conducted at the National Influenza Centre, ICMR-National Institute of Virology (NIV), Pune, India. The genomic analysis was based on samples from different States that were referred to the NIV and hence the approval for the study was obtained from the Institutional Ethics Committee.
RNA isolation, RT-PCR of clinical samples and next-generation sequencing (NGS): Throat and nasal swab samples of suspected cases fulfilling the case definition for SARS-CoV-2 were referred by the hospital authorities and COVID collection centers of State Health Services, Maharashtra, India, to ICMR-NIV, Pune, for diagnosis of SARS-CoV-2 during the period from March 9 to September 28, 2020. The detection of the SARS-CoV-2 was done by using the NIV reverse transcription-polymerase chain reaction kit as per the protocols described earlier8. Positive clinical samples were selected for whole-genome sequencing representing the geographical districts and disease severity.
In brief, 280 μl of each sample in duplicate was used for RNA extraction by Qiagen viral RNA extraction protocol. The extracted RNA was quantified using Qubit® Fluorometer (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA). A concentration of 10 ng of RNA was used for cDNA synthesis using the SuperScript™ VILO™ cDNA Synthesis Kit (Invitrogen, Carlsbad, CA, USA). Further, two-pool RNA panel libraries were prepared manually using the Ion AmpliSeq™ Library Kit Plus as per the manufacturer's instructions (Invitrogen, Carlsbad, CA, USA). The amplified amplicons were partially digested with FuPa reagent and were ligated with adaptors with Switch Solution and DNA Ligase. Purified libraries were quantified using the Qubit™ fluorometer or the Agilent™ 2100 Bioanalyzer™ instrument and diluted to 100 pM. The Ion Chef System was used for template preparation. Purified template beads were submitted to meta-transcriptome next-generation sequencing (NGS) in the Ion S5 platform (Thermo Fisher Scientific) using an Ion 540™ chip and the Ion Total RNA-Seq kit v2.0, as per the manufacturer's protocol (Thermo Fisher Scientific).
The Ion AmpliSeq SARS-CoV-2 Research Panel containing target region information was downloaded from Ion AmpliSeq designer (https://ampliseq.com/login/login.action) and utilized for analysis. Sequence data were processed using the Torrent Suite Software (TSS) v5.10.1 (Thermo Fisher Scientific, USA). Coverage analysis plugins were utilized to generate coverage analysis report for each of the samples. Reference-based reads gathering and assembly were performed for all the samples using Iterative Refinement Meta-Assembler (IRMA)9 developed by the Centers for Disease Control, USA incorporated within the TSS.
Phylogenetic analysis and classification: The whole-genome sequences from India available in GISAID as of October 14, 2020 with information of the sampling location (State information) (n=3014) were used as a starting dataset for this study. The selected sequences were aligned using MAFFT v.7.45010, and phylogenetic analysis was undertaken using MEGA v.611 based on the neighbour-joining approach with the composite likelihood as the substitution model. Further, the classification of the Indian sequences into the Nextstrain assigned new clades and the Pangolin nomenclature for clades/sub-clades was done using the respective tools directly. However, the GISAID nomenclature was assigned by the phylogeny and mutations noted.
Identification of synonymous/non-synonymous substitutions in dominant Pangolin lineages in India: The nucleotide substitutions were identified by comparing the alignment of all the Indian SARS-CoV-2 genomes against the reference human SARS-CoV-2 genome from Wuhan (NC_045512.2) using NUCmer version 3.112. The resulting list of nucleotide variations was translated into synonymous and non-synonymous amino acid changes using a previously developed R script13 and the updated list of gene features from NCBI RefSeq SARS-CoV-2 genome annotation (https://www.ncbi.nlm.nih.gov/sars-cov-2/). The substitutions which were present in more than 75 per cent of the sequences of only one lineage with a minimum of 10 representing genomes were considered as the substitutions characterizing the specific lineage.
Selection pressure analysis: Selection pressure analysis was performed using the Datamonkey adaptive evolution server14. The sequences were separated into different datasets based on the GISAID clades. For each clade, if the number of sequences was >500, then redundant (100% identical) sequences were removed. Further, if still the number of sequences exceeded 500, then random selection of 500 sequences was done. Stop codons were replaced by gaps. The individual codon sites under diversifying selection pressure were identified by employing two methods: Mixed Effects Model of Evolution15 method which detects episodic diversification by employing a mixed-effects maximum likelihood approach and Single-Likelihood Ancestor Counting16 that uses a combination of maximum likelihood and counting approaches to infer the non-synonymous and synonymous rates of substitution for each site.
The overall pipeline of work undertaken in this study is depicted in Figure 1.
Fig. 1.
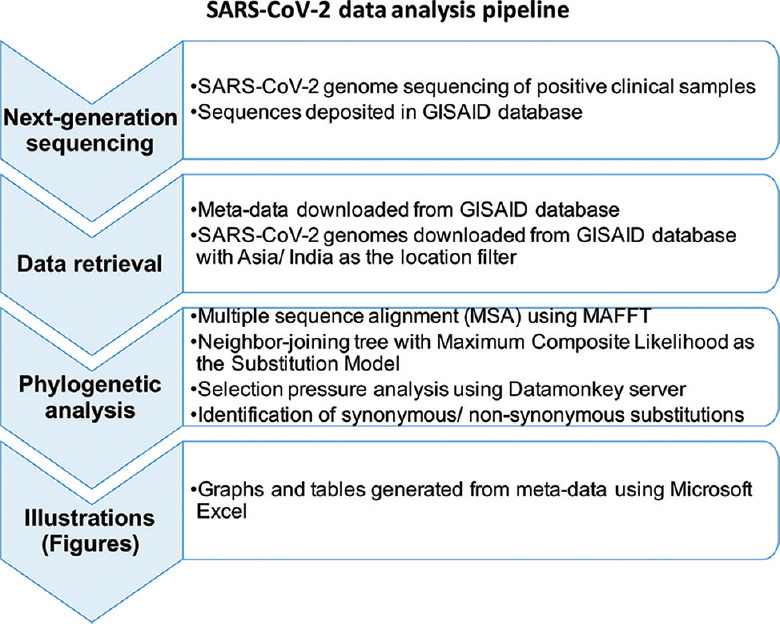
Workflow for SARS-CoV-2 data analysis.
Results
The whole-genome sequencing for 330 strains from Maharashtra (n=328) and Karnataka (n=2) was undertaken as a part of this study. The details of the study samples and the sequences obtained including the per cent of reads mapped, total reads and the per cent of genome coverage recovered are provided in (Supplementary Table I (466.9KB, pdf) (available from http://www.ijmr.org.in/articles/2021/153/1/images/IndianJMedRes_2021_153_1_166_311942_sm8.pdf).
Phylogenetic analysis (Supplementary Fig. 1 (423.2KB, pdf) (available from https://www.ijmr.org.in/articles/2021/153/1/images/IndianJMedRes_2021_153_1_166_311942_sm9.pdf)) revealed that the genomes from different parts of India (n=3014) could be classified under seven clades, viz. S, V, G, GR, GH, L and O, identified by the GISAID on the basis of the marker mutations as shown in Table I. The genetic make-up of the Indian sequences revealed that overall, the proportion of strains in clade G (including GH and GR) were found to be highest (74.98%) followed by strains in O (unclassified category) (21.53%) (Fig. 2A and Supplementary Table II (available from https://www.ijmr.org.in/articles/2021/153/1/images/IndianJMedRes_2021_153_1_166_311942_sm10.pdf)). Within the G clade, the highest proportion was noted in the GR clade. Fig. 2B represents the equivalence between the GISAID nomenclature and the Pangolin lineages for the Indian SARS-CoV-2 sequences. As per the Pangolin nomenclature, majority of the Indian sequences belonged to sub-lineages B.1.1.32, B.6, B.1, B.1.1, B.1.113 and B.1.1.8 (Fig. 2, Supplementary Fig. 1 (423.2KB, pdf) and Supplementary Table II).
Table I.
Establishing an equivalence between the Global Initiative on Sharing All Influenza Data (GISAID), Nextstrain and Phylogenetic Assignment of Named Global Outbreak LINeages (Pangolin) nomenclature systems with respect to the genome sequence data from India (n=3014)
| GISAID clades | Nextstrain | Dominant Pangolin lineages | Major marker mutations |
|---|---|---|---|
| G | 20A | B.1, B.1.80 | S: D614G |
| GR | 20B | B.1.1.32, B.1.1, B.1.1.8 | S: D614G + N: G204R |
| GH | 20C | B.1.113, B.1.36 | S: D614G + nsp3:Q57H |
| V | 19A | B.2.1 | nsp6:L37F + nsp3:G251V |
| L (Ref. seq. WIV04) | 19A | B | - |
| S | 19B | A | ORF8:L84S |
| O | 19A | B.6, B.4 | ORF1a: L3606F |
Fig. 2.
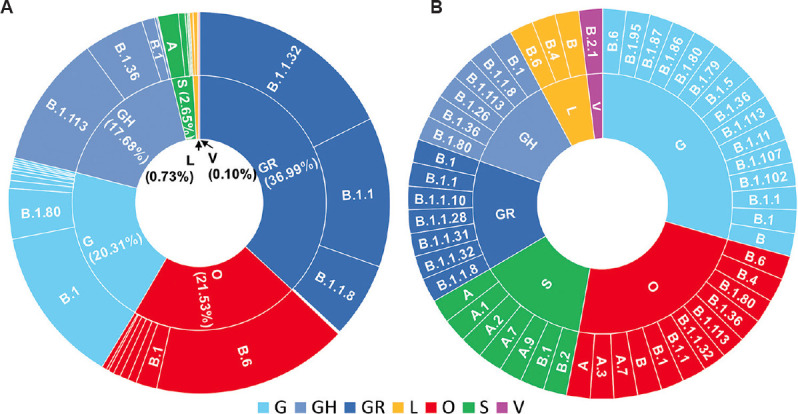
Sunburst diagrams coloured according to Global Initiative on Sharing All Influenza Data (GISAID) clades showing relationship between GISAID and Phylogenetic Assignment of Named Global Outbreak LINeages (Pangolin) annotations on the inner and outer circles, respectively for the Indian SARS-CoV-2 genomes (n=3014). (A) The proportionate chart showing dominant Pangolin corresponding to each of the GISAID clades (The count for individual clades/lineages is shown in Supplementary Table II). (B) The schematic representation of association between the GISAID clades and the Pangolin lineages.
Supplementary Table II.
Distribution of the Indian sequences (n=3014) in the Global Initiative on Sharing All Influenza Data, Nextstrain and Phylogenetic Assignment of Named Global Outbreak LINeages clades
| GISAID clade | Count of sequences | Nextstrain new clades# | Pangolin |
|---|---|---|---|
| O | 649 | 19A=514 | B.6=501 |
| 19B=6 | B.1=52 | ||
| 20A=53 | B.1.1=24 | ||
| 20B=11 (65) | B.4=22 | ||
| B=18 | |||
| B.1.113=14 | |||
| A.7=6 | |||
| B.1.1.32=4 | |||
| A=3 | |||
| B.1.36=2 | |||
| B.1.80=2 | |||
| A.3=1 | |||
| L | 22 | 19A=19 (3) | B=10 |
| B.4=3 | |||
| B.6=9 | |||
| V | 3 | 19A=3 | B.2.1=3 |
| S | 80 | 19B=71 | A=51 |
| 19A=1 | A.7=17 | ||
| 20A=1(7) | A.9=6 | ||
| A.1=2 | |||
| B.1=2 | |||
| A.2=1 | |||
| B.2=1 | |||
| G | 612 | 20A=540 | B.1=404 |
| 19A=11 | B.1.80=130 | ||
| 20B=11 | B.1.113=21 | ||
| (50) | B.1.1=15 | ||
| B.1.95=11 | |||
| B.1.5=8 | |||
| B.1.87=6 | |||
| B=5 | |||
| B.1.36=5 | |||
| B.1.11=2 | |||
| B.1.102=1 | |||
| B.1.107=1 | |||
| B.1.79=1 | |||
| B.1.86=1 | |||
| B.6=1 | |||
| GH | 533 | 20A=460 | B.1.113=337 |
| 20C=18 | B.1.36=155 | ||
| 19A=6 (49) | B.1=32 | ||
| B.1.26=6 | |||
| B.1.80=2 | |||
| B.1.1.8=1 | |||
| GR | 1115 | 20B=877 | B.1.1.32=535 |
| 20A=4 | B.1.1=382 | ||
| 19A=2 (232) | B.1.1.8=192 | ||
| B.1.1.31=3 | |||
| B.1.1.28=1 | |||
| B.1=1 | |||
| B.1.1.10=1 | |||
| Total | 3014 | 3014 | 3014 |
Bold fonts indicate the major distribution. #Clade information not available as on October 14, 2020 for the number of strains depicted in bracket. GISAID, Global Initiative on Sharing All Influenza Data; PangoLIN, Phylogenetic Assignment of Named Global Outbreak LINeages
Other than the major globally circulating clades that possessed the marker mutations as shown in Table I, mutations specific to the dominant Indian Pangolin lineages were identified (Table II). As per the Pangolin lineage summaries (https://cov-lineages.org/lineages.html), some of the lineages most likely to have evolved in India are B.1.113 (n=372), B.1.1.8 (n=193), A.7 (n=23) and A.9 (n=6). Among these, the major lineage B.1.1.8 was found to possess unique mutations nsp3:S1285F and ORF3a:L46F, while B.1.113 possessed S194L in the N protein (Table II).
Table II.
Synonymous and non-synonymous substitutions characterizing the dominant Phylogenetic Assignment of Named Global Outbreak LINeages (Pangolin) in India
| Pangolin lineage | Synonymous substitution | Non-synonymous substitutions | Untranslated nucleotide change | ||
|---|---|---|---|---|---|
| Nucleotide variation | Gene (amino-acid change) | Nucleotide variation | Gene (amino-acid change) | ||
| B.1.113 | C22444T | S (D294D) | C28854T | N (S194L) | - |
| B.1.1.32 | C313T | NSP1 (L16L) | C5700A | nsp3 (A994D)/ORF1ab (A1812D) | - |
| B.1.1.8 | G4354A | NSP3 (E545E)/ORF1ab (E1363E) | C6573T | nsp3 (S1285F)/ORF1ab (S2103F) | - |
| C25528T | ORF3a (L46F) | ||||
| B.1.80 | C3634T | NSP3 (N305N)/ORF1ab (N1123N) | - | ||
| C15324T | NSP12b (N619N) | ||||
| B.4 | T28688C | N (A138A) | C884T | nsp2 (R27C)/ORF1ab (R207C) | 3’UTR (G29742T) |
| G1397A | nsp2 (V198I)/ORF1ab (V378I) | ||||
| G8653T | nsp4 (M33I)/ORF1ab (M2796I) | ||||
| G11083T | nsp6 (L37F)/ORF1ab (L3606F) | ||||
| B.6 | C13730T | nsp12b (A88V) | - | ||
| C28311T | N (P13L) | ||||
| C6312A | nsp3 (T1198K)/ORF1ab (T2016K) | ||||
| G11083T | nsp6 (L37F)/ORF1ab (L3606F) | ||||
On the basis of the new nomenclature by Nextstrain as per the ancestral nodes, majority of the sequences fell into the cluster having ancestral nodes as 20A and 20B and others fell into clusters with nodes as 19A, 19B and 20C (Supplementary Table II). The Nextstrain clade assignment was retrieved as on 14 October 2020. Extrapolating to the Nextstrain old nomenclature for classification, it could be seen that the Indian strains could be classified into clades A2a, A1a, A3, B, B4 and O (Supplementary Table III (available from https://www.ijmr.org.in/articles/2021/153/1/images/IndianJMedRes_2021_153_1_166_311942_sm11.pdf)).
Supplementary Table III.
Mutations representing the old Nextstrain clade and corresponding major new Nextstrain clade nomenclatures
| Nextstrain (old clades) | Mutation(s) defined for the clade | Major Nextstrain new clades (defined mutation) |
|---|---|---|
| A1a | ORF3a: G251V and ORF1a: L3606F | 19A (-) |
| A3 | ORF1a: L3606F and V378I | |
| A6 | nt: T514C | |
| A7 | ORF1a: A3220V | |
| B | ORF8: L84S | 19B (nt. C8782T) |
| B1 | ORF8: L84S, nt- C18060T | |
| B2 | ORF8: L84S, nt- C29095T | |
| B4 | ORF8: L84S, N: S202N | |
| A2 | S: D614G | 20A (ORF1b/nsp12b: P314L) |
| A2a | S: D614G, ORF1b: P314L | |
| A2a | S: D614G, ORF1b: P314L | 20B (N: R203K, G204R & ORF14: G50N) |
| - | - | 20C (ORF1a: T265I) |
The State-wise distribution of the SARS-CoV-2 genomes classified as per the different GISAID clades is shown in Fig. 3. A comparison of these genetic variants in the Indian States wherein sufficient sequence data were available (Supplementary Table IV (available from https://www.ijmr.org.in/articles/2021/153/1/images/IndianJMedRes_2021_153_1_166_311942_sm12.pdf)) was done. For States where a single clade was predominant, it was noted that clade O predominated in Delhi and Tamil Nadu while G predominated in West Bengal and Madhya Pradesh. Both clades GH and G were predominant in Gujarat. Clades GR and O predominated in Telangana; in Karnataka and Uttarakhand, GR and GH predominated; while in Haryana, O and GH were predominant. Clade S majorly circulated in Odisha along with GR, G, O and GH, and Maharashtra was also noted to have several clades in circulation including GR, G, O and S.
Fig. 3.
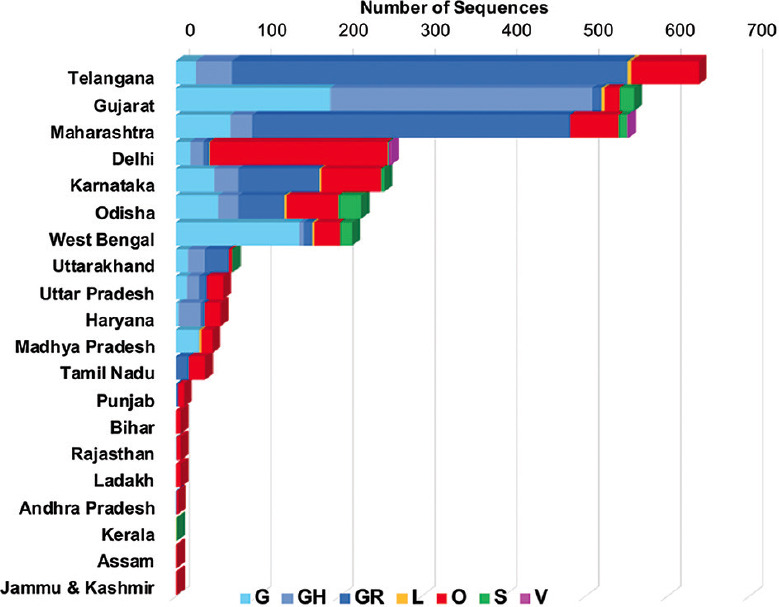
State-wise distribution of total number of SARS-CoV-2 sequences deposited from India to Global Initiative on Sharing All Influenza Data (GISAID) from January to September 2020. The colours on the graph denote the GISAID clades.
Supplementary Table IV.
State-wise list with clade information
| Clade | Nextstrain | Lineage | Andhra Pradesh | Assam | Bihar | Delhi | Gujarat | Haryana | Jammu and Kashmir | Karnataka | Kerala | Ladakh |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| G | 19A | B | 1 | 2 | ||||||||
| B.1 | 2 | 3 | ||||||||||
| B.1.1 | ||||||||||||
| B.1.113 | 5 | |||||||||||
| B.1.36 | 1 | |||||||||||
| B.1.80 | ||||||||||||
| B.6 | ||||||||||||
| 20A | B | |||||||||||
| B.1 | 2 | 134 | 2 | 13 | ||||||||
| B.1.1 | 2 | 1 | ||||||||||
| B.1.102 | 1 | |||||||||||
| B.1.107 | ||||||||||||
| B.1.11 | ||||||||||||
| B.1.113 | 6 | 7 | 1 | |||||||||
| B.1.36 | 3 | |||||||||||
| B.1.5 | ||||||||||||
| B.1.79 | ||||||||||||
| B.1.80 | 34 | 21 | ||||||||||
| B.1.86 | ||||||||||||
| B.1.87 | 2 | |||||||||||
| B.1.95 | ||||||||||||
| 20B | B.1 | |||||||||||
| B.1.1 | 1 | |||||||||||
| B.1.113 | 2 | |||||||||||
| Blank | B.1 | 3 | ||||||||||
| B.1.1 | 2 | |||||||||||
| B.1.5 | 1 | |||||||||||
| B.1.80 | 6 | |||||||||||
| GH | 20A | B.1 | 2 | 12 | 1 | |||||||
| B.1.113 | 11 | 219 | 26 | 16 | ||||||||
| B.1.36 | 1 | 85 | 4 | |||||||||
| B.1.80 | 2 | |||||||||||
| 20C | B.1 | 2 | 2 | 1 | ||||||||
| B.1.1.8 | ||||||||||||
| B.1.26 | ||||||||||||
| Blank | B.1 | 4 | ||||||||||
| B.1.113 | 2 | |||||||||||
| B.1.36 | 3 | |||||||||||
| GR | 20B | B.1 | ||||||||||
| B.1.1 | 7 | 4 | 4 | 74 | ||||||||
| B.1.1.28 | ||||||||||||
| B.1.1.31 | 1 | |||||||||||
| B.1.1.32 | 7 | 1 | 3 | |||||||||
| B.1.1.8 | 1 | |||||||||||
| Blank | B.1.1 | 17 | ||||||||||
| B.1.1.10 | 1 | |||||||||||
| B.1.1.32 | 3 | |||||||||||
| L | 19A | B | 1 | |||||||||
| B.4 | 3 | |||||||||||
| B.6 | 1 | 1 | 2 | |||||||||
| O | 19A | A.3 | 1 | |||||||||
| B | 1 | 1 | ||||||||||
| B.1 | 9 | 1 | 6 | |||||||||
| B.1.1 | 2 | 2 | ||||||||||
| B.1.113 | 5 | |||||||||||
| B.4 | 1 | 9 | 6 | |||||||||
| B.6 | 2 | 2 | 6 | 195 | 11 | 11 | 1 | 48 | ||||
| 19B | A | |||||||||||
| A.7 | ||||||||||||
| B.1 | ||||||||||||
| B.1.1 | ||||||||||||
| 20A | B | 1 | ||||||||||
| B.1 | 1 | 3 | 1 | |||||||||
| B.1.113 | 2 | 3 | 2 | |||||||||
| B.1.36 | 1 | 1 | ||||||||||
| 20B | B.1 | |||||||||||
| B.1.1 | 2 | 1 | ||||||||||
| B.1.1.32 | ||||||||||||
| Blank | B.1 | 1 | ||||||||||
| B.1.1 | 2 | |||||||||||
| B.1.80 | 2 | |||||||||||
| B.6 | 1 | 1 | ||||||||||
| S | 19B | A | 17 | 3 | 1 | |||||||
| A.1 | 1 | |||||||||||
| A.2 | 1 | |||||||||||
| A.7 | ||||||||||||
| A.9 | ||||||||||||
| B.1 | 1 | 1 | ||||||||||
| B.2 | ||||||||||||
| V | 19A | B.2.1 | 2 | |||||||||
| Total | 3 | 2 | 6 | 263 | 560 | 55 | 2 | 255 | 2 | 6 | ||
| Clade | Madhya Pradesh | Maharashtra | Odisha | Punjab | Rajasthan | Tamil Nadu | Telangana | Uttar Pradesh | Uttarakhand | West Bengal | Total | |
| G | 1 | 4 | ||||||||||
| 3 | 8 | |||||||||||
| 1 | 1 | |||||||||||
| 5 | ||||||||||||
| 1 | ||||||||||||
| 2 | 2 | |||||||||||
| 1 | 1 | |||||||||||
| 1 | 1 | |||||||||||
| 22 | 24 | 33 | 1 | 1 | 19 | 10 | 6 | 124 | 391 | |||
| 1 | 4 | |||||||||||
| 1 | ||||||||||||
| 1 | 1 | |||||||||||
| 1 | 1 | 2 | ||||||||||
| 14 | ||||||||||||
| 1 | 4 | |||||||||||
| 3 | 1 | 3 | 7 | |||||||||
| 1 | 1 | |||||||||||
| 2 | 34 | 9 | 6 | 1 | 9 | 6 | 122 | |||||
| 1 | 1 | |||||||||||
| 4 | 6 | |||||||||||
| 11 | 11 | |||||||||||
| 2 | 2 | |||||||||||
| 1 | 1 | 5 | 8 | |||||||||
| 2 | ||||||||||||
| 3 | ||||||||||||
| 2 | ||||||||||||
| 1 | ||||||||||||
| 6 | ||||||||||||
| GH | 1 | 3 | 2 | 2 | 23 | |||||||
| 12 | 7 | 11 | 13 | 18 | 2 | 335 | ||||||
| 1 | 14 | 14 | 1 | 27 | 1 | 4 | 152 | |||||
| 2 | ||||||||||||
| 5 | ||||||||||||
| 1 | 1 | |||||||||||
| 4 | 2 | 6 | ||||||||||
| 4 | ||||||||||||
| 2 | ||||||||||||
| 3 | ||||||||||||
| GR | 1 | 1 | ||||||||||
| 63 | 40 | 2 | 15 | 124 | 9 | 20 | 3 | 365 | ||||
| 1 | 1 | |||||||||||
| 2 | 3 | |||||||||||
| 321 | 16 | 168 | 9 | 7 | 532 | |||||||
| 191 | 192 | |||||||||||
| 17 | ||||||||||||
| 1 | ||||||||||||
| 3 | ||||||||||||
| L | 2 | 2 | 2 | 3 | 10 | |||||||
| 3 | ||||||||||||
| 1 | 1 | 3 | 9 | |||||||||
| O | 1 | |||||||||||
| 1 | 1 | 1 | 1 | 1 | 1 | 1 | 8 | 17 | ||||
| 2 | 1 | 6 | 1 | 5 | 31 | |||||||
| 4 | 5 | 13 | ||||||||||
| 5 | ||||||||||||
| 5 | 1 | 22 | ||||||||||
| 8 | 42 | 31 | 6 | 4 | 20 | 80 | 19 | 2 | 11 | 499 | ||
| 3 | 3 | |||||||||||
| 6 | 6 | |||||||||||
| 1 | 1 | |||||||||||
| 1 | 1 | |||||||||||
| 1 | ||||||||||||
| 2 | 5 | 6 | 18 | |||||||||
| 1 | 1 | 9 | ||||||||||
| 2 | ||||||||||||
| 1 | 1 | |||||||||||
| 1 | 3 | 1 | 8 | |||||||||
| 4 | 4 | |||||||||||
| 1 | ||||||||||||
| 2 | ||||||||||||
| 2 | ||||||||||||
| 2 | ||||||||||||
| S | 3 | 14 | 1 | 12 | 51 | |||||||
| 1 | 2 | |||||||||||
| 1 | ||||||||||||
| 6 | 11 | 17 | ||||||||||
| 3 | 3 | 6 | ||||||||||
| 2 | ||||||||||||
| 1 | 1 | |||||||||||
| V | 1 | 3 | ||||||||||
| Total | 45 | 553 | 227 | 10 | 6 | 36 | 639 | 58 | 70 | 216 | 3014 | |
State-wise temporal data (March to August 2020) are shown in Fig. 4 and Supplementary Table V (available from https://www.ijmr.org.in/articles/2021/153/1/images/IndianJMedRes_2021_153_1_166_311942_sm13.pdf). Several clades were noted to be circulating in many of the States between March and May. Beyond this, a switch to majorly GR/GH was observed. The temporal distribution in Maharashtra was analyzed based on the sequences generated as a part of this study. The clades during March were majorly O, S and G. The proportion of strains of clade O was noted to decrease gradually, and a replacement to GR strains was noted consistently during May to September.
Fig. 4.
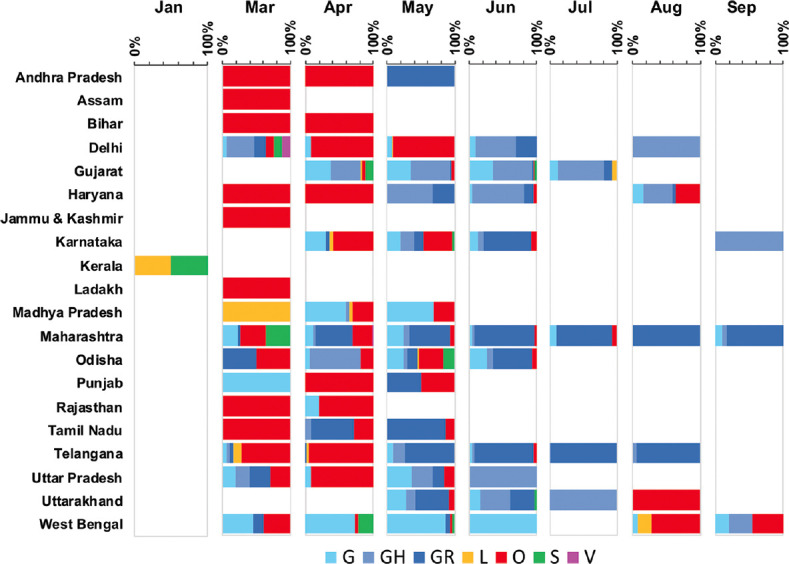
Temporal distribution of SARS-CoV-2 sequences from different States of India. The number of SARS-CoV-2 sequences belonging to distinct GISAID clades is represented as a percentage plot of the clades for each month.
Supplementary Table V.
State-wise clade information with temporal distribution
| State | Clade | January | February | March | April | May | June | July | August | September | NA | Total |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Andhra Pradesh | GR | 1 | 1 | |||||||||
| O | 1 | 1 | 2 | |||||||||
| Assam | O | 2 | 2 | |||||||||
| Bihar | O | 3 | 3 | 6 | ||||||||
| Delhi | G | 1 | 2 | 14 | 1 | 18 | ||||||
| GH | 7 | 6 | 3 | 16 | ||||||||
| GR | 3 | 1 | 3 | 7 | ||||||||
| L | 1 | 1 | ||||||||||
| O | 2 | 25 | 185 | 5 | 217 | |||||||
| S | 2 | 2 | ||||||||||
| V | 2 | 2 | ||||||||||
| Gujarat | G | 27 | 45 | 112 | 5 | 189 | ||||||
| GH | 33 | 74 | 184 | 29 | 320 | |||||||
| GR | 2 | 4 | 5 | 11 | ||||||||
| L | 1 | 3 | 4 | |||||||||
| O | 4 | 8 | 6 | 18 | ||||||||
| S | 8 | 10 | 18 | |||||||||
| Haryana | G | 1 | 3 | 4 | ||||||||
| GH | 2 | 17 | 8 | 27 | ||||||||
| GR | 1 | 3 | 1 | 5 | ||||||||
| O | 1 | 10 | 1 | 7 | 19 | |||||||
| Jammu and Kashmir | O | 2 | 2 | |||||||||
| Karnataka | G | 14 | 16 | 13 | 4 | 47 | ||||||
| GH | 17 | 8 | 2 | 3 | 30 | |||||||
| GR | 3 | 13 | 72 | 11 | 99 | |||||||
| L | 2 | 2 | ||||||||||
| O | 28 | 35 | 8 | 2 | 73 | |||||||
| S | 4 | 4 | ||||||||||
| Kerala | L | 1 | 1 | |||||||||
| S | 1 | 1 | ||||||||||
| Ladakh | O | 6 | 6 | |||||||||
| Madhya Pradesh | G | 12 | 16 | 28 | ||||||||
| GH | 1 | 1 | ||||||||||
| L | 2 | 1 | 3 | |||||||||
| O | 6 | 7 | 13 | |||||||||
| Maharashtra | G | 7 | 12 | 31 | 6 | 3 | 8 | 67 | ||||
| GH | 3 | 12 | 5 | 7 | 27 | |||||||
| GR | 1 | 59 | 79 | 128 | 23 | 23 | 75 | 388 | ||||
| O | 12 | 32 | 9 | 4 | 2 | 59 | ||||||
| S | 11 | 11 | ||||||||||
| V | 1 | 1 | ||||||||||
| Odisha | G | 1 | 38 | 13 | 52 | |||||||
| GH | 13 | 8 | 4 | 25 | ||||||||
| GR | 1 | 26 | 29 | 56 | ||||||||
| L | 3 | 3 | ||||||||||
| O | 1 | 3 | 56 | 3 | 63 | |||||||
| S | 28 | 28 | ||||||||||
| Punjab | G | 1 | 1 | |||||||||
| GR | 2 | 2 | ||||||||||
| O | 5 | 2 | 7 | |||||||||
| Rajasthan | G | 1 | 1 | |||||||||
| O | 1 | 4 | 5 | |||||||||
| Tamil Nadu | GH | 1 | 1 | |||||||||
| GR | 9 | 6 | 15 | |||||||||
| O | 15 | 4 | 1 | 20 | ||||||||
| Telangana | G | 1 | 15 | 8 | 1 | 25 | ||||||
| GH | 1 | 29 | 6 | 1 | 7 | 44 | ||||||
| GR | 1 | 1 | 123 | 159 | 47 | 152 | 483 | |||||
| L | 2 | 2 | 1 | 5 | ||||||||
| O | 13 | 61 | 8 | 82 | ||||||||
| Uttar Pradesh | G | 2 | 1 | 11 | 14 | |||||||
| GH | 2 | 9 | 4 | 15 | ||||||||
| GR | 3 | 5 | 1 | 9 | ||||||||
| O | 3 | 12 | 5 | 20 | ||||||||
| Uttarakhand | G | 10 | 5 | 15 | ||||||||
| GH | 5 | 13 | 3 | 21 | ||||||||
| GR | 18 | 11 | 29 | |||||||||
| O | 3 | 1 | 4 | |||||||||
| S | 1 | 1 | ||||||||||
| West Bengal | G | 8 | 34 | 93 | 12 | 1 | 3 | 151 | ||||
| GH | 1 | 5 | 6 | |||||||||
| GR | 3 | 7 | 10 | |||||||||
| L | 3 | 3 | ||||||||||
| O | 7 | 3 | 3 | 11 | 7 | 31 | ||||||
| S | 10 | 5 | 15 | |||||||||
| Total | 2 | 130 | 453 | 1086 | 868 | 121 | 221 | 107 | 26 | 3014 |
NA, not available
In addition, as the information of the outcome of the infection in terms of fatality was available for Maharashtra (n=41 of 328 sequences, Supplementary Table I (466.9KB, pdf) ), the proportion of fatal cases were estimated in the clade G (including GR as none of the sequences belonged to GH clade). It was observed that 14.38 per cent (41 of 285) of cases which possessed the D614G mutation resulted in fatal outcomes, while the rest of the cases that possessed the mutation were mild.
Nextstrain inference (Supplementary Fig. 2 (1.5MB, tif) (available from https://www.ijmr.org.in/articles/2021/153/1/images/IndianJMedRes_2021_153_1_166_311942_sm14.pdf)) of the most likely transmission events (https://nextstrain.org/ncov/asia?c=clade_membership&f_country=India&f_region=Asia) revealed that the dominant clade B.6 (GISAID O) that emerged from 19A was introduced into India from China, Europe, South-East Asia and Middle-East while B.1 (G) and B.1.36 (GH) that emerged from 20A had their origins from Europe, Middle-East and Africa. The B.1.1 (GR) clade that emerged from 20B was introduced from the Europe, Middle-East and Far-East. Selection pressure analysis revealed that site nsp3:994A/D was identified to be under positive selection pressure in both clades G and GR, nsp6:37 L/F and nsp12:323 L/P in both G and GH and nsp16:298N/L/I in GR and GH (Table III).
Table III.
Selection pressure analysis based on the whole-genome sequences using the methods Mixed Effects Model of Evolution and Single-Likelihood Ancestor Counting, available in the Datamonkey server
| Clade | Gene | Site | Variable amino acid residues | P MEME | P SLAC |
|---|---|---|---|---|---|
| G | nsp3 | 994 | A/D | 0.01 | 0.042 |
| nsp6 | 37 | L/F | 0.01 | 0.036 | |
| nsp12 | 323 | L/P | 0.01 | 0.008 | |
| GR | nsp3 | 994 | D/A | 0.01 | 0.05 |
| nsp3 | 1103 | P/L/S | 0.02 | 0.085 | |
| nsp4 | 380 | A/V | 0.05 | 0.088 | |
| nsp7 | 54 | S/L/P | 0.07 | 0.066 | |
| nsp16 | 298 | N/L/I | 0 | 0.037 | |
| ORF3a | 46 | L/F | 0.01 | 0.06 | |
| GH | nsp6 | 37 | L/F | 0.02 | 0.04 |
| nsp12 | 323 | L/P | 0.03 | 0.059 | |
| nsp14 | 372 | T/I | 0.03 | 0.062 | |
| nsp16 | 298 | N/L/H/I | 0 | 0.062 | |
| S | - | - | - | - | - |
| O | nsp3 | 1197 | S/R/K | 0 | 0.021 |
| nsp3 | 1198 | K/T | 0.07 | 0.022 | |
| nsp3 | 1768 | V/G | 0 | 0.004 | |
| nsp6 | 37 | F/L | 0.02 | 0.009 |
Sites were identified as showing evidence of positive selection as per the statistical significance level (P<0.1) by both the methods. MEME, Mixed Effects Model of Evolution; SLAC, Single-Likelihood Ancestor Counting
Discussion
A dynamic nomenclature for SARS-CoV-2 proposed by Rambaut et al5 initially identified two lineages (A and B) at the root of the phylogeny based on the sharing of two nucleotides at positions 8782 in ORF1ab and 28144 in ORF817,18. Subsequently, descendent lineages were assigned a numerical value provided; these satisfied certain criteria of nucleotide substitutions within and between lineages. Several lineages and sub-lineages were thus identified. On the other hand, Nextstrain is based on a maximum likelihood approach as implemented in TreeTime19. Considering temporal dating of ancestral nodes and discrete trait geographic reconstruction based on the SARS-CoV-2 sequences, Nextstrain identified five nodes that were labelled as 19A, 19B, 20A, 20B and 20C.
Based on the equivalence between the GISAID clade nomenclature, the new Nextstrain clades and the Pangolin sub-lineages, initially, Nextstrain clade names were ad hoc letter number combinations that were never intended to be a permanent naming system. At least ten clades (B, B1, B2, B4, A3, A6, A7, A1a, A2 and A2a) based on specific marker mutations were identified. The marker mutations specific to these clades are shown in Supplementary Table III. The clades A1a, A3, A6 and A7 emerged from the node labelled 19A, while clades B, B1, B2 and B4 emerged from the node 19B. The strains belonging to clade A2 correlated to strains having ancestral nodes 20A, while the A2a strains could be traced back to nodes 20A, 20B and 20C. Thus, the old Nextstrain clade nomenclature was found to be undefined and did not reflect on the time scale of evolution. We further analyzed the predominance of the strains in different Indian States based on the Pangolin and GISAID clade nomenclatures (Supplementary Table IV) in association with their emergence times as per the Nextstrain new clades classification nomenclature.
The earliest Indian cases2,3 of SARS-CoV-2 were based on laboratory confirmation of suspected cases of persons with international travel history20. Since March 2020, the reported cases saw an increase in different States of the country. Genome sequencing efforts in India resulted in generation of whole-genome sequence data representing 20 different States/Union Territories (UTs). Good representation was noted from the States of Telangana, Gujarat, Maharashtra, Delhi, Karnataka, Odisha, West Bengal, Uttarakhand, Uttar Pradesh and Haryana (Supplementary Table IV and Fig. 2). In the other 16 States/UTs, though cases of SARS-CoV-2 were reported, no genome data were deposited.
The genetic make-up of the Indian sequences revealed that the predominant clades (Pangolin/GISAID) circulating in India are the B.1.1.32/GR, B.6/O, B.1/G, B.1.1/GR, B.1.113/GH and B.1.1.8/GR. Thus, as also observed in other studies21,22, the G clade (including GR and GH) is seen to have established itself in India as well as the world over13,23 (Supplementary Fig. 3 (608.8KB, tif) (available from https://www.ijmr.org.in/articles/2021/153/1/images/IndianJMedRes_2021_153_1_166_311942_sm15.pdf)). Temporal data of the Indian SARS-CoV-2 genomes revealed that except for Uttarakhand, West Bengal and Haryana that showed the circulation of O clade even after July, other States showed a complete switch to GR/GH. The dominant clades were noted to have emerged from nodes 19A, 20A, 20B and 20C. The same Nextstrain clades/Pangolin lineages were found to occur in multiple GISAID clades. Hence, the GISAID nomenclature system that is specifically based on amino acid substitutions can be considered more robust than the other two nomenclatures.
The State-wise distribution of the prevalence of the different clades was observed. Within clade GR, a sub-group (Pangolin B.1.1.32 lineage) showed the combinations of strains from Maharashtra interspersed with strains from Telangana. Another sub-group (B.1.1) showed strains mainly from Telangana along with strains from Karnataka, Odisha and Tamil Nadu. The lineage B.1.1.8 which was identified as an indigenous lineage of India could most likely be attributed to evolution within Telangana. On the other hand, within the clade G, groups with mixing of strains from Gujarat, Madhya Pradesh, West Bengal, Odisha, Karnataka or Gujarat, Karnataka and Maharashtra were evident. These may be associated with the inter-State movements of migrant workers, tourists, students and professionals before or following the lock down imposed in the country. Another indigenous lineage (B.1.113), a major component of the clade GH, was noted to have emerged in Gujarat. Within clade O, two prominent sub-groups were noted. In one of these sub-groups (Pangolin B.6 lineage), Delhi strains were noted to be interspersed with strains from several States all over the country including Odisha, Maharashtra, Karnataka, Telangana, Madhya Pradesh, Andhra Pradesh, Haryana, Uttar Pradesh, Bihar, Tamil Nadu, West Bengal, Telangana and Rajasthan. The other sub-group (B.4) involved mainly Karnataka, Maharashtra, Ladakh, Telangana and Gujarat. The O clade was prevalent across several States in the country in March and April, suggesting their expansion due to introductions before the lockdown on March 19, 2020 (Fig. 4).
It was noted that the sites putatively identified to be under positive selection pressure within the GISAID clades were found to occur majorly in the non-structural proteins coded by ORF1a and ORF1b. A few of the sites were found to be common to clades G/GH/GR. This was a reflection of the evolution within the dominant clade G. It remains to be observed whether these and the other sites would be future hotspots of evolution. Such sites need to be further characterized to understand if the virus is adapting further towards enhanced human transmissions24,25,26,27. The clade G/GH/GR strains possess the mutation D614G in the spike protein S. It has been demonstrated that this mutation increases infectivity, resulting in potentially more transmissible SARS-CoV-228,29,30. Insight into the associated mechanism was obtained from cryo-EM studies of the SARS-CoV-2 S protein trimer which revealed that D614G shifted the S conformation toward an ACE2 binding-competent state28. Further, considering that a lower proportion of the clade G cases resulted in fatality, if could be difficult to attribute the outcome of infection solely to the D614G marker mutation. It is necessary to focus on the viral genomic variations arising from rapid local expansions of the GISAID or Pangolin lineages.
In summary, this study revealed the genetic variants circulating in India during the period from March to September 2020. The increased prevalence of the GH and GR clades from May 2020 onwards was noted to parallel the global trend. The observation of emergence of new lineages B.1.1.8 and B.1.113 was indicative of host-specific evolution of the SARS-CoV-2 strains within GR and GH clades, respectively, in India. To conclude on the robustness of the existing classification nomenclatures, there would be need to continue observing the global evolutionary trends and delineation of the strains. The study had limitations due to the non-availability or less sequence data at uniform time intervals from many parts of the country and also the lack of clinical information. This would benefit in exploring the establishment of the clades, molecular clock, transmissions within the country and further evidence of indigenous evolution. It may also help infer the potential association of SARS-CoV-2 lineages and mortality, as well as identify possible ethnic and genetic correlations.
Strains of this study including details of their sequenced genome
Neighbour-joining tree of the Indian genome sequences, with the taxa colour coded state-wise.
Global transmissions as captured from Nextstrain analyses.
Graphical representation of the temporal distribution of severe acute respiratory syndrome coronavirus 2 sequences from different continents of the world (n=140,560).
Acknowledgment:
Authors acknowledge the support of Thermo Fisher Scientific India field applications specialist (FAS), Laboratory, Bioinformatics and Technical Sales Specialist (TSS) team. The authors acknowledge Dr G.B. Shantala, Bangalore Medical College Research Institute, Bengaluru, Karnataka, for sharing clinical samples.
Footnotes
Supplementary material available from https://www.ijmr.org.in/article.asp?issn=0971-5916;year=2021;volume=153;issue=1;spage=166;epage=174;aulast=Potdar
Financial support & sponsorship: None.
Conflicts of Interest: None.
References
- 1.WHO Coronavirus Disease (COVID-19) Dashboard. [accessed on February 4, 2021]. Available from: https://covid19.who.int .
- 2.Yadav PD, Potdar VA, Choudhary ML, Nyayanit DA, Agrawal M, Jadhav SM, et al. Full-genome sequences of the first two SARS-CoV-2 viruses from India. Indian J Med Res. 2020;151:200–9. doi: 10.4103/ijmr.IJMR_663_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Potdar V, Cherian SS, Deshpande GR, Ullas PT, Yadav PD, Choudhary ML, et al. Genomic analysis of SARS-CoV-2 strains among Indians returning from Italy, Iran & China, & Italian tourists in India. Indian J Med Res. 2020;151:255–60. doi: 10.4103/ijmr.IJMR_1058_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elbe S, Buckland-Merrett G. Data, disease and diplomacy: GISAID's innovative contribution to global health. Glob Chall. 2017;1:33–46. doi: 10.1002/gch2.1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alm E, Broberg EK, Connor T, Hodcroft EB, Komissarov AB, Maurer-Stroh S, et al. Geographical and temporal distribution of SARS-CoV-2 clades in the WHO European Region, January to June 2020. Euro Surveill. 2020;25:2001410. doi: 10.2807/1560-7917.ES.2020.25.32.2001410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rambaut A, Holmes EC, O'Toole Á Hill V, McCrone JT, Ruis C, et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat Microbiol. 2020;5:1403–7. doi: 10.1038/s41564-020-0770-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hadfield J, Megill C, Bell SM, Huddleston J, Potter B, Callender C, et al. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics. 2018;34:4121–3. doi: 10.1093/bioinformatics/bty407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choudhary ML, Vipat V, Jadhav S, Basu A, Cherian S, Abraham P, et al. Development of in vitro transcribed RNA as positive control for laboratory diagnosis of SARS-CoV-2 in India. Indian J Med Res. 2020;151:251–4. doi: 10.4103/ijmr.IJMR_671_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shepard SS, Meno S, Bahl J, Wilson MM, Barnes J, Neuhaus E. Viral deep sequencing needs an adaptive approach: IRMA, the Iterative Refinement Meta-Assembler. BMC Genomics. 2016;17:708. doi: 10.1186/s12864-016-3030-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–66. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–9. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Delcher AL, Phillippy A, Carlton J, Salzberg SL. Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res. 2002;30:2478–83. doi: 10.1093/nar/30.11.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mercatelli D, Giorgi FM. Geographic and genomic distribution of SARS-CoV-2 mutations. Front Microbiol. 2020;11:1800. doi: 10.3389/fmicb.2020.01800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weaver S, Shank SD, Spielman SJ, Li M, Muse SV, Kosakovsky Pond SL. Datamonkey 2.0: A modern web application for characterizing selective and other evolutionary processes. Mol Biol Evol. 2018;35:773–7. doi: 10.1093/molbev/msx335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murrell B, Wertheim JO, Moola S, Weighill T, Scheffler K, Kosakovsky Pond SL. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012;8:e1002764. doi: 10.1371/journal.pgen.1002764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kosakovsky Pond SL, Frost SD. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol Biol Evol. 2005;22:1208–22. doi: 10.1093/molbev/msi105. [DOI] [PubMed] [Google Scholar]
- 17.Tang X, Wu C, Li X, Song Y, Yao X, Wu X, et al. On the origin and continuing evolution of SARS-CoV-2. Natl Sci Rev. 2020;7:1012–23. doi: 10.1093/nsr/nwaa036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khailany RA, Safdar M, Ozaslan M. Genomic characterization of a novel SARS-CoV-2. Gene Rep. 2020;19:100682. doi: 10.1016/j.genrep.2020.100682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sagulenko P, Puller V, Neher RA. TreeTime: Maximum-likelihood phylodynamic analysis. Virus Evol. 2018;4:vex042. doi: 10.1093/ve/vex042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ministry of Health and Family Welfare, Government of India. COVID-19. [accessed on February 4, 2021]. Available from: https://www.mohfw.gov.in/
- 21.Kumar P, Pandey R, Sharma P, Dhar MS, Vivekanand A, Uppili B, et al. Integrated genomic view of SARS-CoV-2 in India. Wellcome Open Res. 2020;5:184. doi: 10.12688/wellcomeopenres.16119.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singh H, Singh J, Khubaib M, Jamal S, Sheikh JA, Kohli S, et al. Mapping the genomic landscape & diversity of COVID-19 based on >3950 clinical isolates of SARS-CoV-2: Likely origin & transmission dynamics of isolates sequenced in India. Indian J Med Res. 2020;151:474–8. doi: 10.4103/ijmr.IJMR_1253_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Islam MR, Hoque MN, Rahman MS, Alam AS, Akther M, Puspo JA, et al. Genome-wide analysis of SARS-CoV-2 virus strains circulating worldwide implicates heterogeneity. Sci Rep. 2020;10:14004. doi: 10.1038/s41598-020-70812-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Callaway E. Making sense of coronavirus mutations. Nature. 2020;585:174–7. doi: 10.1038/d41586-020-02544-6. [DOI] [PubMed] [Google Scholar]
- 25.Hodcroft EB, Zuber M, Nadeau S, Crawford KH, Bloom JD, Veesler D, et al. Emergence and spread of a SARS-CoV-2 variant through Europe in the summer of 2020. Medrxiv. 2020 doi: 10.1038/s41586-021-03677-y. doi: 10.1101/2020.10.25.20219063. [DOI] [PubMed] [Google Scholar]
- 26.Sardar R, Satish D, Birla S, Gupta D. Comparative analyses of SAR-CoV2 genomes from different geographical locations and other coronavirus family genomes reveals unique features potentially consequential to host-virus interaction and pathogenesis. bioRxiv. 2020 doi: 10.1016/j.heliyon.2020.e04658. doi:10.1101/2020.03.21.001586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lo Presti A, Rezza G, Stefanelli P. Selective pressure on SARS-CoV-2 protein coding genes and glycosylation site prediction. Heliyon. 2020;6:e05001. doi: 10.1016/j.heliyon.2020.e05001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang L, Jackson CB, Mou H, Ojha A, Rangarajan ES, Izard T, et al. The D614G mutation in the SARS-CoV-2 spike protein reduces S1 shedding and increases infectivity. bioRxiv. 2020 doi: 10.1038/s41467-020-19808-4. doi: 10.1101/2020.06.12.148726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yurkovetskiy L, Wang X, Pascal KE, Tomkins-Tinch C, Nyalile TP, Wang Y, et al. Structural and functional analysis of the D614G SARS-CoV-2 spike protein variant. Cell. 2020;183:739–51. doi: 10.1016/j.cell.2020.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Starr TN, Greaney AJ, Hilton SK, Ellis D, Crawford KH, Dingens AS, et al. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell. 2020;182:1295–310.e20. doi: 10.1016/j.cell.2020.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Strains of this study including details of their sequenced genome
Neighbour-joining tree of the Indian genome sequences, with the taxa colour coded state-wise.
Global transmissions as captured from Nextstrain analyses.
Graphical representation of the temporal distribution of severe acute respiratory syndrome coronavirus 2 sequences from different continents of the world (n=140,560).