

Abstract
Background
Almonertinib Mesilate Tablets (HS-10296, Hansoh Pharma, Shanghai, China) is a novel and selective third-generation epidermal growth factor receptor tyrosine kinase inhibitor (EGFR-TKI). A phase I study of almonertinib in patients with non-small cell lung cancer (NSCLC) demonstrated a linear metabolic trend, a good tolerability/safety profile, and preliminary antitumor activity. However, the metabolism, excretion, and substance balance of almonertinib has not been clearly determined. Here, we investigated the pharmacokinetic characteristics and safety profile of almonertinib following a single oral dose (110 mg/50 µCi) in healthy Chinese male participants.
Methods
Total radioactivity (TRA) in whole blood, plasma, urine, and feces was measured by utilizing a liquid scintillation counter to obtain almonertinib substance balance data. The pharmacokinetic parameters of [14C]almonertinib and the parent drug almonertinib in whole blood and plasma were analyzed with noncompartmental analysis in the WinNonlin software (Pharsight Corp). The major metabolites in plasma, urine, and feces were analyzed by high-performance liquid chromatography (HPLC) coupled with an online or offline isotope detector. The safety of the drug was evaluated after administration.
Results
The safety and tolerability of a single oral dose of 110 mg/50 µCi [14C] almonertinib suspension were good in healthy Chinese male participants. There was no significant abnormality or special adverse reaction. TRA peaked quickly in plasma, with a Tmax of 4.0 h; however, TRA was cleared slowly in vivo, with a mean terminal elimination phase (half-life, T1/2) of up to 863 h. In addition to the parent drug, a total of 26 metabolites in blood, urine, and feces were analyzed. In plasma, parent drug was the major drug-related component, accounting for 69.97% of TRA, and M440 (almonertinib-M2 demethyl product) was the major metabolite, accounting for 5.08% of TRA; in urine, parent drug accounted for 0.48% of the dose administered and HAS-719 was the major metabolite, accounting for 1.20% of the administered dose; in feces, parent drug was about 8.61% of the dose administered and HAS-719 was the major metabolite, accounting for 12.33% of the administered dose, which was followed by M541a/M470a and M617/M575, accounting for 11.8% and 6.76% of the administered dose, respectively.
Conclusions
Almonertinib has a good safety profile, with parent drug as its main circulating component. almonertinib is extensively metabolized in vivo before excretion and is excreted as a parent drug and metabolites mainly via feces.
Trial registration
The trial registration number: CTR20192291.
Keywords: Almonertinib, HS-10296, 14C, excretion, metabolism, pharmacokinetics
Introduction
Tyrosine kinase inhibitors (TKI) play a role in suppressing tumor metastasis by competitively binding to the ATP site on the structural domain of intracellular tyrosine kinases, thereby inhibiting the tyrosine kinase activity and intracellular phosphorylation associated with the epidermal growth factor receptor (EGFR) and blocking the EGFR-induced growth of tumor cells and other processes (1,2). The first-generation epidermal growth factor receptor TKIs (EGFR-TKIs), gefitinib and erlotinib, are effective in treating advanced non-small cell lung cancer (NSCLC) harboring activating EGFR mutations (EGFRm+) (3,4); however, most patients develop acquired resistance (AR) after 9 to 14 months of treatment, leading to disease progression (5). Although the second-generation irreversible EGFR-TKI [e.g., afatinib (6) and dacomitinib (7,8)] monotherapy are capable of overcoming EGFR T790M-mediated resistance (9), their clinical use is limited due to dose-limiting toxicities associated with inhibition of wild-type EGFR (wtEGFR) (10). Therefore, there is an urgent need for third-generation EGFR-TKIs with high selectivity against EGFR T790M and with decreased toxicities caused by wtEGFR inhibition (11-13).
Almonertinib, a third-generation EGFR inhibitor independently developed by Hansoh Pharma, Shanghai, China, is a potent, selective irreversible inhibitor of mutant EGFR and is proposed for the treatment of advanced NSCLC (14,15). In vitro studies (14,15) have demonstrated the potent inhibition of EGFR T790M resistance mutations by almonertinib as almonertinib potently inhibited the growth of T790M-mutant NSCLC cell lines H1975 and LU1868 and the growth of NSCLC HCC827 cell line harboring EGFR-sensitive mutants. A phase I study on almonertinib for NSCLC (15) enrolled 26 patients in the dose-escalation cohort (including 4 dose groups of 55, 110, 220, and 260 mg). The results showed a linear trend of almonertinib metabolism, the drug was well tolerated at each single or multiple dose level (7 cycles of 21 consecutive days per cycle) and showed preliminary antitumor activity. However, the previous pharmacokinetic studies have not yet been able to clarify the metabolism, excretion, and substance balance of almonertinib. The aim of our current study was thus to identify the absorption, substance balance, and biotransformation pathways of almonertinib in the human body and to provide a reference for rational use of the drug.
We present the following article in accordance with the MDAR reporting checklist (available at http://dx.doi.org/10.21037/atm-21-1606).
Methods
Research design
This single-center, open-label, single-dose clinical trial was designed to investigate the total radioactivity (TRA) of [14C]almonertinib in whole blood and plasma in healthy Chinese male participants after administration of a single oral dose of 14C-labeled drug. Furthermore, the TRA of [14C]almonertinib in the excretions of healthy participants after oral administration of [14C]almonertinib was quantified to determine the substance balance and main excretion paths in the human body. The radiolabeled metabolite profiles in plasma, urine, and feces of healthy participants after oral administration of [14C]almonertinib were quantitatively analyzed and the major metabolites were identified, so as to determine the main metabolic pathways of almonertinib in the human body. A validated liquid chromatography-mass spectrometry (LC/MS) method was used to quantify the concentration of almonertinib in plasma and to obtain pharmacokinetic data of almonertinib in plasma. The safety of [14C]almonertinib in participants after a single dose was observed. The study was conducted in accordance with the Declaration of Helsinki (as revised in 2013). The study was reviewed and approved by the Drug Clinical Trial Ethics Committee of the Affiliated First Hospital of Nanjing Medical University (No. 2019-MD-049, the registration number of ethics board) and informed consent was taken from all individual participants.
Research methods
Participant selection
Four healthy male participants were enrolled in this study.
The main inclusion criteria included the following: (I) in good health; (II) with a body mass index (BMI) value of 19.0–26.0 kg/m2; (III) with regular bowel movements, and (IV) without abnormalities or abnormalities with clinical significance in vital signs or in the results of physical examination, 12-lead electrocardiography, or laboratory tests.
The main exclusion criteria included the following: (I) with a history of cardiac, respiratory, endocrine, renal, gastrointestinal, skin, infectious, hematological, neurological, or psychiatric diseases; (II) with a history of drug allergy; (III) with conditions including chronic constipation or diarrhea, irritable bowel syndrome, inflammatory bowel disease, and/or perianal disease; (IV) with factors affecting drug absorption, distribution, metabolism, and/or excretion; (V) previous participation in radiolabeled clinical trials or having exposure to significant radiation (e.g., more than 1 session of X-ray, computed tomography scan, or barium meal, and being involved in radiation-related occupations) within the past 12 months.
The endpoint for blood sampling was that the radioactivity concentrations at 2 consecutive time points were less than 2 times the plasma background value; collection of urine and feces samples were consecutively collected until the TRA in all excretions exceeded 80% of the dose administered and the radioactivity concentrations was below 1% of the dose administered for 2 consecutive days.
Drug dose, preparation, and administration
Based on the phase I/II trials using almonertinib tablets in Chinese patients with advanced NSCLC and a food effect study in healthy participants (15), almonertinib 110 mg was selected in our current study as it has shown a good safety profile. Meanwhile, the safe radioactive dose of [14C]almonertinib for oral administration in humans was estimated following the oral administration of [14C]almonertinib in a quantitative whole-body autoradiography (QWBA) study conducted in male rats. The radioactive dose of 50 µCi administered in our current study was safe and far below the radioactivity dose limit (30 mSv) in human radiopharmacological studies as prescribed by the US Food and Drug Administration (FDA) (16). The associated radiation exposure fell within International Commission on Radiological Protection Guidelines for Category IIa studies (0.1–1 mSv) (17). almonertinib was manufactured by Hansoh Pharmaceutical, Shanghai, China. [14C]almonertinib was synthesized by Chemdepo, Inc (USA). First, 110 mg/50 µCi [14C]almonertinib was isolated, purified, and prepared into solid powder by XenoBiotic Laboratories, Inc. (XBL-China, China) and stored at –70 °C. The [14C]almonertinib solid powder was prepared in a suspension at room temperature on the day of the test. The participants took the drug in the morning on an empty stomach, having fasted since 10 pm the previous night, and received lunch 4 hours after dosing.
Sample collection and processing
For blood sampling, 7 mL of blood samples were collected at 0 (before taking the drug) and 1, 2, 3, 4, 6, 8, 10, 12, 24, 48, 72, 96, 120, 168, 216, 312, 408, and 504 hours after dosing. The sample was first divided into 2 tubes of 1 ml whole blood sample. The remaining blood was centrifuged (3,000 rpm, 10 minutes) to obtain plasma samples. All the plasma and whole blood samples were stored temporarily in a refrigerator (–20 °C) for further tests. Blood sampling was terminated if blood sample radioactivity concentrations at 2 consecutive time points were less than 2 times the plasma background value.
For urine and feces sampling urine samples were collected before dosing and 0–4, 4–8, 8–12, and 12–24 hours after dosing, and then every 24 hours thereafter. Feces samples were collected before dosing and every 24 hours after dosing. The collection of urine and feces samples was terminated if the TRA in all excretions exceeded 80% of the dose administered and the radioactivity concentrations at 2 consecutive days were below 1% of the dose administered. The collected urine and fecal samples were weighed in designated containers and stored in a refrigerator at –10 to –30°C for further tests.
Safety evaluation
The types, intensity, and frequency of adverse events were described according to the National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE) Version 4.03 (18). The evaluation indicators included adverse drug reactions (ADRs), results of laboratory tests (including routine blood/urine/feces tests, blood biochemistry, and coagulation test) and vital signs, along with findings of physical examination, 12-lead electrocardiography, and other examinations/tests.
Analytical measurements
The radioactivity in the whole body, plasma, urine, and feces samples was measured by using a Tri-Carb model 3110 TR liquid scintillation counter (LSC) (Perkin Elmer, Wellesley, MA, USA). Radioactivities in plasma and urine samples were measured directly; in whole blood, a small amount of sample was combusted in an OX-501 oxidizer (R.J. Harvey, Tappan, NY, USA) before measurement. Fecal samples were added with an appropriate amount of solvent, followed by vortexing to produce a homogenized suspension; after a certain number of homogenized samples were combusted in an OX-501 oxidizer (R.J. Harvey), the CO2 resulting from burning was captured in a liquid scintillation cocktail; finally, the TRA was measured by LSC (Perkin Elmer). The drug excretion rate corresponding to each time interval was calculated based on the radioactivity measured in urinary or fecal samples at each time interval (excretion rate of drug = radioactivity excreted in urine and feces/percentage of actual radioactive dose administered). The cumulative excretion amount and the cumulative excretion rate of the drug were obtained by summing up the values at each time interval, and the results were used to obtain the substance balance data (i.e., total substance recovery rate) and the major excretion routes of almonertinib in the human body.
For metabolite analysis, plasma samples obtained at different time points were mixed, and a certain volume of 0.1% formic acid in methanol was added to each mixed plasma sample. After being thoroughly mixed by vortexing, the mixtures were then blown dry using nitrogen gas and then redissolved with 0.1% formic acid, and the redissolved samples were used for radiolabeled metabolite profiling. The urine samples of each subject were mixed at the same volume percentage according to the time intervals, and the supernatant was dried, concentrated, and centrifuged for radioisotope metabolite profiling. The feces samples were mixed at the same weight percentage according to the time intervals, and the homogenized samples were shaken, vortexed, and centrifuged with0.1% formic acid in methanol; then extracts were blown dry with nitrogen gas and redissolved. The redissolved samples were used for radiolabeled metabolite profiling. The above plasma, urine, and fecal samples were analyzed by high-performance liquid chromatograph (HPLC) coupled with a radioactivity detector or high-resolution mass spectrometer (LC-RAM/HRMS) for radioactive metabolite profiling. The radioisotope metabolite spectra were obtained by conversion and integration using ARC Convert and Evaluation software, and then the chromatograms of radioactive metabolite of each biological sample were integrated to obtain the peak area of each radioactive peak and the percentage of TRA of each peak in the sample. Finally, the proportions of the parent drug and its major metabolites over plasma TRA and the percentages of the parent drug and its metabolites over total drug administered were calculated. A validated liquid chromatography tandem spectroscopy (LC-MS/MS) method was used to identify the structures of metabolites in plasma [more than or close to 10% area under the curve (AUC)], urine, and feces (>5% of dose administered), and the main biotransformation pathway of [14C]almonertinib in the human body was obtained. The LC-MS/MS system was operated using a SCIEX API 5000 tandem MS (Applied Biosystems, CA, USA) coupled with either a 20 HPLC system or a 30-AD XR HPLC system (Shimadzu Co., Kyoto, Japan).
Statistical analysis
Pharmacokinetic parameters including maximum plasma concentration (Cmax), time to maximum plasma concentration (Tmax), area under the drug-time curve (AUC0-t and AUC0-∞), elimination half-life (T1/2), apparent oral clearance (CL/F), oral apparent volume of distribution (Vd/F), and mean residence time (MRT) were estimated using a noncompartmental model by WinNonlin software (Version 8.0 and above, Pharsight Corp.).
Data are represented as mean ± standard deviation () and were analyzed using SAS software (SAS Institute).
Results
Demographic data
All 4 participants in this study were Han Chinese males, with a mean age of 33±4 years (range, 29–37 years), a mean height of 169.8 (±4.9) cm, a mean weight of 66.9 (±7.9) kg, and a mean BMI of 23.1 (±1.7) kg/m2 (range, 20.6–24.5 kg/m2). All the participants weighed more than 50 kg, and all of them completed the trial as scheduled. During the study, the participants complied with the requirements on general conditions (e.g., avoid strenuous activity or exercise and avoid prolonged sitting or lying) and dietary restrictions (e.g., abstain from tea, coffee, and other coffee-containing or alcoholic beverages) described in the protocol.
Safety results
All 4 participants completed the trial as scheduled, two participants (50%) had 3 cases of ADRs possibly related to the test drug, including 2 cases of ADRs (perioral dermatitis and positive urinary occult blood; both grade 1) in 002 participant and 1 case of ADR (rash; grade 2) in 003 participant. No serious adverse events occurred throughout the trial, and no participant withdrew from the trial due to adverse events. Participant 2 spontaneously recovered from the adverse reactions, and participant 3 returned to normal after antiallergy treatment. No clinically significant abnormalities in vital signs, electrocardiography, or physical examination were noted in this study.
Actual administered radioactivity and actual amount of drug administered
The actual TRA administered to each subject was obtained by deducting the total amount of radioactivity contained in each vial of the preparation (measured before administration) with the amount of residual radioactivity in the vial; subsequently, the actual dose of the test drug administered to each subject was obtained by dividing the actual TRA administered with the specific activity of the administered preparation. The 4 participants were given 108.9–113.3 mg (mean: 111.2 mg) of almonertinib, and the actual administered radioactivity was 49.5–1.5 µCi (mean: 50.6 µCi).
Pharmacokinetic results
Plasma pharmacokinetic results of the unlabeled drug almonertinib and its metabolite HAS-719
A validated LC-MS/MS method was applied to analyze the concentrations of almonertinib and its metabolite HAS-719 in plasma samples. The pharmacokinetic parameters of almonertinib and HAS-719 in participants after dosing are shown in Table 1, while the mean blood concentration-time curves are shown in Figure 1.
Table 1. Pharmacokinetic parameters of HS-10296, HAS-719, and total radioactivity after dosing.
| Metabolite name | Tmax (h) | Cmax (ng/mL) | AUC0-t (h·ng/mL) | AUC0-∞ (h·ng/mL) | CL/F (L/h) | Vd/F (L) | t½ (h) |
|---|---|---|---|---|---|---|---|
| HS-10296 | 5.25±3.20 | 119±27.7 | 2,904±727 | 3,158±794 | 36.8±10.8 | 1,267±219 | 25.0±6.41 |
| HAS-719 | 6.50±2.52 | 22.2±3.94 | 1,027±121 | 1,117±103 | 99.1±8.92 | 5,965±724 | 41.8±3.93 |
| Total radioactivity | 4.00 | 479±43.1a | 76,718±46,938b | 235,209±194,880a | – | – | 863±930 |
a, ng Eq/g; b, h.ng Eq/g. Tmax, time of maximum concentration; Cmax, maximum concentration; AUC, area under curve; CL/F, apparent clearance; Vd/F, apparent volume of distribution; T1/2, half-life.
Figure 1.
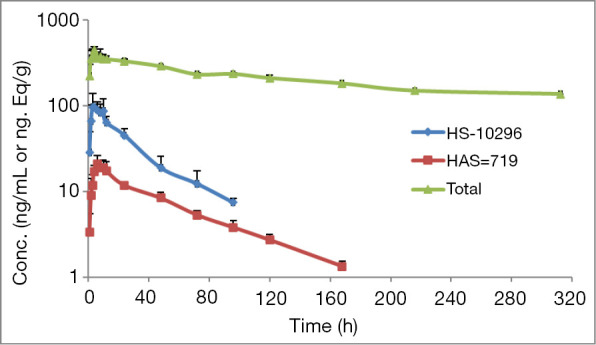
Plasma total radioactivity (total), plasma non-labeled prototype drug (almonertinib) and its metabolite (HAS-719) mean concentration-time curve (mean ± SD).
Results of pharmacokinetic analysis of the TRA of plasma
Limited by the detection sensitivity, complete elimination phase data were not available in 2 participants, in whom the elimination phase parameters were not available. Thus, the total plasma radioactivity pharmacokinetic results were calculated and analyzed only with data from the remaining 2 participants. The concentration-time curve of plasma TRA in 2 healthy males after a single oral dose of [14C]almonertinib is shown in Figure 1. The pharmacokinetic parameters of plasma TRA in 2 healthy male participants after a single oral dose of [14C]almonertinib on an empty stomach are shown in Table 1.
TRA in whole blood
Following a single oral dose of 110 mg/50 uCi [14C] almonertinib to four subjects,TRA concentration in the whole blood was only detected at three time pints in two subjects (4 and 24 h post dose in subject 1 and 8 h post dose in subject 2), the remaining samples were detected below the limit of detection. These data indicated that there was a low possible trend in the association of the radioactivity to blood cells.
Substance balance and excretion results
The mean cumulative excretion rates of participants at different time intervals are shown in Figure 2. The cumulative excretion rates of drug TRA were 90.19%±4.18% within 0–504 hours of drug administration. The cumulative excretion rate was 84.75%±3.35% in feces and 5.44%±0.83% in urine, suggesting that almonertinib was mainly excreted via feces.
Figure 2.
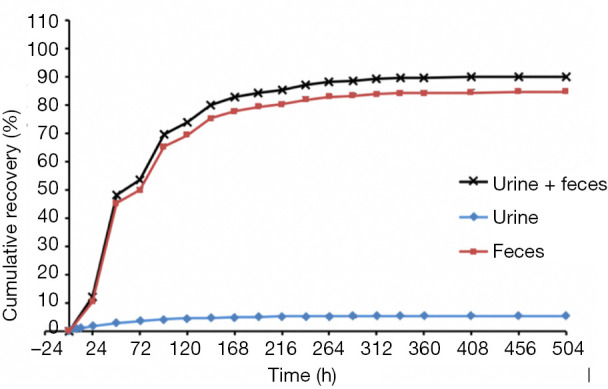
Average cumulative excretion rate of urine and stool samples within 0–504 hours.
Results of human metabolism tests
Results of plasma sample metabolism tests
After equal volumes of plasma samples obtained from these 4 participants were mixed, the redissolved solutions were directly used for the radiolabeled metabolite profiling, and the obtained liquid phase radioactivity chromatograms are shown in Figure 3. The percentage (%TRA) and concentration of [14C]almonertinib (ng Eq./g) and its radioactive metabolite spectrum peaks over the TRA of the plasma samples are shown in Table 2. The pharmacokinetic parameters of the major metabolites in plasma are shown in Table 3. In addition to the parent drug, a total of 12 metabolites were detected in the plasma samples. The parent drug was the major circulating substance in the plasma samples, accounting for approximately 69.97% of the mixed plasma exposure within 0–48 hours (AUC0–48 h). M440 was the major metabolite in the plasma samples, accounting for about 5.08% of the 0–48-hour mixed plasma exposure (AUC0–48 h).
Figure 3.

Representative radiochromatograms of human samples from four subjects after oral dosing of 110 mg/50 µCi [14C] almonertinib: (A) pooled human plasma 6 hours, (B) pooled urine samples 0–96 hours, and (C) pooled feces samples 0–240 hours, respectively.
Table 2. Intensity percentage (%HPLC) and levels (almonertinibng Eq./g) of [14C]almonertinib and its metabolites over the total radioactivity in plasma samples.
| Metabolic peak, no. | Liquid phase time, Rt (min) | % HPLCa | Level (almonertinib ng Eq./g)a | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 h | 4 h | 6 h | 10 h | 24 h | 48 h | 2 h | 4 h | 6 h | 10 h | 24h | 48 h | |||
| 1 | 2.6–3.9 | 5.41 | ND | ND | 3.45 | ND | ND | 17.8 | ND | ND | 12.5 | ND | ND | |
| 3 | 10.1–10.6 | 1.35 | ND | ND | ND | ND | ND | 4.44 | ND | ND | ND | ND | ND | |
| 5 | 12.1–13.1 | ND | ND | ND | 3.45 | ND | ND | ND | ND | ND | 12.5 | ND | ND | |
| 7 | 14.1–15.4 | 1.35 | ND | ND | ND | ND | ND | 4.44 | ND | ND | ND | ND | ND | |
| 9 | 16.9–17.9 | ND | ND | ND | 3.45 | ND | ND | ND | ND | ND | 12.5 | ND | ND | |
| 10 (M717) | 18.3–18.9 | 1.35 | ND | 4.08 | ND | ND | ND | 4.44 | ND | 15.2 | ND | ND | ND | |
| 17 | 27.1–29.6 | ND | 1.67 | ND | ND | ND | ND | ND | 7.75 | ND | ND | ND | ND | |
| 23 (M617 and M575) | 41.1–42.6 | 1.35 | ND | ND | ND | ND | ND | 4.44 | ND | ND | ND | ND | ND | |
| 24 (M541a and M470a) | 43.4–44.4 | 1.35 | ND | ND | ND | ND | ND | 4.44 | ND | ND | ND | ND | ND | |
| 29 (almonertinib-M30) | 49.6–50.6 | 1.35 | 10 | 4.08 | 10.35 | ND | ND | 4.44 | 46.4 | 15.2 | 37.2 | ND | ND | |
| 32 (M511a) | 54.1–54.9 | ND | ND | 2.04 | ND | ND | ND | ND | ND | 7.61 | ND | ND | ND | |
| 33 (M470e) | 55.4–56.1 | 2.7 | 1.67 | ND | ND | ND | ND | 8.88 | 7.75 | ND | ND | ND | ND | |
| 34 (M440) | 56.4–56.9 | 10.81 | 21.67 | 16.33 | 3.45 | 9.68 | ND | 35.5 | 101 | 60.8 | 12.5 | 31.5 | ND | |
| 35 (M497b) | 56.9–57.4 | 1.35 | 1.67 | ND | ND | ND | ND | 4.44 | 7.75 | ND | ND | ND | ND | |
| 36 (HAS-719) | 57.9–58.4 | 2.7 | ND | 8.16 | 3.45 | 16.13 | ND | 8.88 | ND | 30.4 | 12.5 | 52.3 | ND | |
| 37 (almonertinib-M2) | 58.6–59.4 | 8.11 | 1.67 | 2.04 | 3.45 | 3.23 | 25 | 26.7 | 7.75 | 7.61 | 12.5 | 10.5 | 70.5 | |
| 38 ([14C]almonertinib) | 59.6–60.1 | 60.81 | 61.67 | 63.27 | 68.97 | 70.97 | 75 | 200 | 286 | 236 | 249 | 231 | 212 | |
a, the percentages of [14C]almonertinib and its metabolites over plasma extract samples can be seen in ARC1340N: 9a, 9b, 10a, 10b, 11a, 11b, 12a, 12b, 13a, 13b, 14a, and 14b. Due to the low radioactivity in the 96-h plasma sample, no radioactive metabolites were detected.
Table 3. Pharmacokinetic parameters of [14C]almonertinib and its metabolites in mixed plasma of participants.
| Metabolite No. | T1/2 (h) | tmax (h) | Cmax (ng Eq/g) | AUC0–48 h (h ng Eq/g) | MRT0–48 h (h) | %AUCa (%) |
|---|---|---|---|---|---|---|
| Total plasma | 105 | 4 | 464 | 15,513 | 22.9 | 100.00 |
| [14C]almonertinib | 167 | 4 | 286 | 10,854 | 23.7 | 69.97 |
| M440 | 1.95* | 4 | 101 | 788 | 10.7 | 5.08 |
The half-life (T1/2) of M440 was calculated using the data at 4, 6, and 10 h. %AUC = the plasma exposure of the parent drug or metabolite within 0–48 h (AUC0-48 h) (ng×h/mL) ÷ the total plasma exposure within 0–48 h (AUC0-48 h) (ng×h/mL). MRT, mean residence time.
Results of urine sample metabolism tests
After equal volumes of urine samples obtained from these 4 participants were mixed, the redissolved solutions were directly used for the radiolabeled metabolite profiling; the obtained liquid phase radioactivity chromatograms are shown in Figure 3. The %TRA and the percentage of drug dose delivered (%Dose) of the parent drug and its radioactive metabolite spectrum peaks over the TRA of the urine samples are shown in Table 4. The results of the relative quantification of the parent drug and its major metabolites in urine samples are shown in Table 5. Thirteen metabolites were found in 0–96 hours mixed urine samples. The parent drug was about 0.48% of the dose administered. HAS-719 was the major metabolite, accounting for 1.20% of the administered dose, which was followed by M497a, which accounted for 0.21% of the dose administered. The other metabolites were trace metabolites.
Table 4. Intensity percentage (%HPLC) and percentage of dose administered (%Dose) of [14C] almonertinib and its metabolites over the total radioactivity in urine samples.
| Metabolic peak, No. | Liquid phase time, Rt (min) | 0–48 h (2.97%Dose) | 0–96 h (4.16%Dose) | |||
|---|---|---|---|---|---|---|
| % HPLCa | %Doseb | % HPLCa | %Doseb | |||
| 1 | 3.4–3.9 | 0.78 | 0.02 | 3.81 | 0.16 | |
| 2 | 6.4–6.6 | 2.75 | 0.08 | ND | ND | |
| 3 | 10.1 | 5.10 | 0.15 | 5.93 | 0.25 | |
| 4 | 10.9 | 10.20 | 0.30 | 12.29 | 0.51 | |
| 5 | 11.4 | 3.14 | 0.09 | 5.09 | 0.21 | |
| 6 | 12.6 | 3.92 | 0.12 | 2.97 | 0.12 | |
| 7 | 13.4 | 0.78 | 0.02 | 2.54 | 0.11 | |
| 8 | 14.4–14.6 | 2.35 | 0.07 | 4.24 | 0.18 | |
| 9 | 15.6–15.9 | 4.71 | 0.14 | 3.81 | 0.16 | |
| 10 (M717) | 17.4 | 3.53 | 0.10 | 0.85 | 0.04 | |
| 11 | 18.4–18.6 | ND | ND | 0.85 | 0.04 | |
| 12 (M607) | 20.1 | 1.57 | 0.05 | ND | ND | |
| 13 (M621) | 21.4 | ND | ND | 0.42 | 0.02 | |
| 14 (M621a) | 22.1–22.4 | 0.78 | 0.02 | ND | ND | |
| 15 | 23.1–23.6 | 0.78 | 0.02 | ND | ND | |
| 17 | 27.6 | ND | ND | 0.42 | 0.02 | |
| 20 | 34.6 | 0.78 | 0.02 | ND | ND | |
| 21 (M527b) | 36.9 | ND | ND | 0.42 | 0.02 | |
| 22 (M687) | 40.1 | 2.35 | 0.07 | 1.27 | 0.05 | |
| 23 (M617 and M575) | 41.4–41.6 | 3.53 | 0.10 | 1.70 | 0.07 | |
| 24 (M541a and M470a) | 43.4 | 2.35 | 0.07 | 3.39 | 0.14 | |
| 25 (M632b) | 44.1–44.4 | 0.78 | 0.02 | ND | ND | |
| 27 (M688) | 45.6–45.9 | 0.39 | 0.01 | ND | ND | |
| 28 (M470b and M541c) | 47.4–48.1 | 0.39 | 0.01 | ND | ND | |
| 29 (almonertinib-M30 and M470c) | 49.4 | ND | ND | 0.42 | 0.02 | |
| 31 (M497a) | 52.9 | 5.88 | 0.17 | 5.09 | 0.21 | |
| 36 (HAS-719) | 56.4 | 26.67 | 0.79 | 28.81 | 1.20 | |
| 37(almonertinib-M2) | 57.6 | ND | ND | 1.70 | 0.07 | |
| 38 ([14C]almonertinib) | 58.4–59.1 | 13.33 | 0.40 | 11.44 | 0.48 | |
| 39c | 71.6–71.9 | 3.14 | 0.09 | 2.54 | 0.11 | |
| Others | ND | ND | <0.01 | <0.01 | ||
a, the intensity percentages of [14C]almonertinib and its metabolites over the TRA in urine samples. For specific data, see ARC1340N: 2a, 2b, 3a, and 3b; b, the dose percentages of [14C]almonertinib and its metabolites (%Dose) = the total radioactivity in urine sample over the dose administered (RTC01289) × the intensity percentages of [14C]almonertinib and its metabolites over the TRA in urine sample; c, contains multiple <0.1% metabolites. ND, not detected.
Table 5. Relative quantitative analysis of the parent drug and its major metabolites in urine and feces samples.
| Metabolite no. | Urine (0–96 h), 4.16%Dose | Feces (0–240 h), 81.99%Dose | Urine + feces, 86.15%Dose | ||||
|---|---|---|---|---|---|---|---|
| %TRA | %Dose | %TRA | %Dose | %Dose | |||
| Sum of parent drug and major metabolites | 55.51 | 2.32 | 84.48 | 69.23 | 71.55 | ||
| M717 | 0.85 | 0.04 | 1.05 | 0.86 | 0.90 | ||
| M607 | ND | ND | 2.87 | 2.35 | 2.35 | ||
| M621 | 0.42 | 0.02 | 1.34 | 1.10 | 1.12 | ||
| M621a | ND | ND | 1.92 | 1.57 | 1.57 | ||
| M527a | ND | ND | 0.48 | 0.39 | 0.39 | ||
| M527b | 0.42 | 0.02 | 3.45 | 2.83 | 2.85 | ||
| M687 | 1.27 | 0.05 | 1.53 | 1.25 | 1.30 | ||
| M617 | 1.70 | 0.07 | 8.24 | 6.76 | 6.83 | ||
| M575 | |||||||
| M541a | 3.39 | 0.14 | 14.37 | 11.78 | 11.92 | ||
| M470a | |||||||
| M632b | ND | ND | 2.78 | 2.28 | 2.28 | ||
| M646 | ND | ND | 2.30 | 1.89 | 1.89 | ||
| M688 | ND | ND | 2.49 | 2.04 | 2.04 | ||
| M470b | ND | ND | 3.54 | 2.90 | 2.90 | ||
| M541c | |||||||
| Almonertinib-M30 | 0.42 | 0.02 | 1.15 | 0.94 | 0.96 | ||
| M470c | |||||||
| M470d | ND | ND | 1.25 | 1.03 | 1.03 | ||
| M497a | 5.09 | 0.21 | 0.38 | 0.31 | 0.52 | ||
| M511a | ND | ND | 3.64 | 2.98 | 2.98 | ||
| M470e | ND | ND | 1.34 | 1.10 | 1.10 | ||
| M440 | ND | ND | 2.68 | 2.20 | 2.20 | ||
| M497b | ND | ND | 0.96 | 0.79 | 0.79 | ||
| HAS-719 (M511b) | 28.81 | 1.20 | 15.04 | 12.33 | 13.53 | ||
| almonertinib-M2 | 1.70 | 0.07 | 1.15 | 0.94 | 1.01 | ||
| [14C]almonertinib | 11.44 | 0.48 | 10.54 | 8.61 | 9.09 | ||
ND, not detected.
Results of fecal sample metabolism tests
After equal volume of fecal samples obtained from these 4 participants were mixed, the redissolved solutions were directly used for the radiolabeled metabolite profiling; the obtained liquid phase radioactivity chromatograms are shown in Figure 3. The %TRA and %Dose of the parent drug and its radioactive metabolites in fecal samples at different time intervals are shown in Table 6. The results of the relative quantification of the parent drug and its major metabolites in fecal samples are shown in Table 5. In addition to the parent drug, a total of 26 metabolites were detected in mixed fecal samples within 0–240 hours. The parent drug was the main component in the feces, accounting for about 8.61% of the dose administered. HAS-719 was the major metabolite in feces samples, accounting for 12.33% of the drug dose administered. The M541a/M470a and M617/M575 metabolites were less common, accounting for 11.8% and 6.76% of the administered dose, respectively. The other metabolites were trace metabolites.
Table 6. Intensity percentage (%HPLC) and percentage of dose administered (%Dose) of [14C] almonertinib and its metabolites over the total radioactivity in feces samples at different time intervals in 4 participants.
| Metabolic peak No. | Liquid phase time, Rt (min) | 0–48 h (45.32%Dose) | 48–96 h (20.06%Dose) | 96–240 h (16.61%Dose) | |||||
|---|---|---|---|---|---|---|---|---|---|
| % HPLCa | % Doseb | %HPLCa | % Doseb | %HPLCa | % Doseb | ||||
| 1 | 2.9–3.1 | ND | ND | 0.42 | 0.08 | ND | ND | ||
| 3 | 9.9–10.4 | 0.29 | 0.13 | 0.69 | 0.14 | ND | ND | ||
| 4 | 10.6–11.1 | 0.57 | 0.26 | 1.25 | 0.25 | ND | ND | ||
| 5 | 11.6–12.1 | 0.90 | 0.41 | 0.63 | 0.13 | 0.45 | 0.07 | ||
| 6 | 12.6–13.4 | 0.98 | 0.44 | 1.11 | 0.22 | 0.45 | 0.07 | ||
| 7 | 13.6–13.9 | 0.41 | 0.19 | 0.56 | 0.11 | ND | ND | ||
| 8 | 15.1–15.4 | 2.86 | 1.30 | 1.74 | 0.35 | ND | ND | ||
| 9 | 16.60 | 1.60 | 0.72 | 1.67 | 0.34 | 1.35 | 0.22 | ||
| 10 (M717) | 17.4–17.9 | 0.74 | 0.34 | 1.11 | 0.22 | 0.45 | 0.07 | ||
| 11 | 18.6–19.4 | 2.54 | 1.15 | 2.36 | 0.47 | 2.69 | 0.45 | ||
| 12 (M607) | 19.9–20.6 | 3.11 | 1.41 | 2.64 | 0.53 | 2.24 | 0.37 | ||
| 13 (M621) | 21.1–21.9 | 1.68 | 0.76 | 1.94 | 0.39 | 0.45 | 0.07 | ||
| 14 (M621a) | 23.1–23.4 | 1.76 | 0.80 | 1.67 | 0.34 | 2.69 | 0.45 | ||
| 15 | 23.9–24.3 | 0.82 | 0.37 | 1.11 | 0.22 | 0.90 | 0.15 | ||
| 16 | 24.90 | 1.06 | 0.48 | 1.25 | 0.25 | 0.45 | 0.07 | ||
| 17 | 26.1–27.6 | 2.50 | 1.13 | 2.50 | 0.50 | 0.90 | 0.15 | ||
| 18 (M527a) | 29.9–30.3 | 0.74 | 0.34 | 0.42 | 0.08 | ND | ND | ||
| 19 | 31.1–31.6 | 0.98 | 0.44 | 0.83 | 0.17 | 0.45 | 0.07 | ||
| 20 | 32.9–34.4 | 2.37 | 1.07 | 1.94 | 0.39 | 0.45 | 0.07 | ||
| 21 (M527b) | 36.9–37.4 | 4.74 | 2.15 | 3.12 | 0.63 | 0.90 | 0.15 | ||
| 22 (M687) | 37.9–38.9 | 1.47 | 0.67 | 2.92 | 0.59 | 2.69 | 0.45 | ||
| 23 (M617 and M575) | 41.1–41.5 | 8.55 | 3.87 | 7.56 | 1.52 | 8.97 | 1.49 | ||
| 24 (M541a and M470a) | 42.6–42.9 | 13.25 | 6.02 | 12.98 | 2.61 | 21.53 | 3.57 | ||
| 25 (M632b) | 44.1–44.4 | 1.51 | 0.68 | 1.60 | 0.32 | 0.90 | 0.15 | ||
| 26 (M646) | 45.3–45.6 | 1.64 | 0.74 | 1.94 | 0.39 | 4.48 | 0.74 | ||
| 27 (M688) | 46.1–46.4 | 2.45 | 1.11 | 3.05 | 0.61 | 2.69 | 0.45 | ||
| 28 (M470b and M541c) | 47.90 | 2.78 | 1.26 | 3.12 | 0.63 | 2.24 | 0.37 | ||
| 29 (almonertinib-M30 and M470c) | 48.9–49.8 | 1.19 | 0.54 | 0.97 | 0.19 | ND | ND | ||
| 30 (M470d) | 50.6–51.6 | 1.47 | 0.67 | 1.60 | 0.32 | 0.90 | 0.15 | ||
| 31 (M497a) | 52.8–53.4 | 0.16 | 0.07 | 0.28 | 0.06 | ND | ND | ||
| 32 (M511a) | 54.1–54.5 | 2.95 | 1.34 | 3.05 | 0.61 | 1.35 | 0.22 | ||
| 33 (M470e) | 54.90 | 1.55 | 0.70 | 2.08 | 0.42 | 1.79 | 0.30 | ||
| 34 (M440) | 55.6–55.9 | 2.45 | 1.11 | 1.80 | 0.36 | 4.48 | 0.74 | ||
| 35 (M497b) | 56.10 | 1.06 | 0.48 | 1.25 | 0.25 | 0.90 | 0.15 | ||
| 36 (HAS-719) | 56.9–57.1 | 12.31 | 5.57 | 17.07 | 3.44 | 22.87 | 3.80 | ||
| 37 (almonertinib-M2) | 57.8–58.1 | 0.74 | 0.34 | 0.83 | 0.17 | 1.35 | 0.22 | ||
| 38 ([14C]almonertinib) | 58.9–59.1 | 11.94 | 5.39 | 7.56 | 1.52 | 7.18 | 1.19 | ||
| 39c | 60.1–69.9 | 1.88 | 0.85 | 1.39 | 0.28 | 0.90 | 0.15 | ||
| Othersc | <0.01 | ND | <0.01 | ND | <0.01 | ND | |||
a, the intensity percentages of [14C]almonertinib and its metabolites over the TRA in urine samples. For specific data, see ARC1340N: 2a, 2b, 3a, and 3b; b, the dose percentages of [14C]almonertinib and its metabolites (%Dose) = the total radioactivity in urine sample over the dose administered (RTC01289) × the intensity percentages of [14C]almonertinib and its metabolites over the TRA in urine sample; c, contains multiple <0.1% metabolites. ND, not detected.
Structures of metabolites
Besides the prodrug, 26 metabolites including HS-10296-M2, HS-10296-M30, HAS-719 (M511b), M440, M470a/b/c/d/e, M497a/b, M511a, M527a/b, M541a/c, M575, M607, M617, M621/M621a, M632b, M646, M687 M687, M688, and M717 were analyzed for their structures. Among them, HS-10296-M30 was the N-dealkylation product of the prodrug aminopyridine, HS-10296-M2 was the N-dedimethylethylaminization product of the prodrug, M440 was the demethylation product of HS-10296-M2, M470a/b/c/d/e were the mono-oxidation products of HS-10296-M2, M497a/b were the didemethylation products of the prodrug, M511a/M511b (HAS-719) were the demethylation products of prodrug, M527a/b were the mono-oxidation products of the demethylation product of the prodrug, M541a/c were the mono-oxidation product of the prodrug, M575 was the cysteine-binding product of HS-10296-M2, M607 was the sulfation product of the mono-oxidation product of the demethylation product of the prodrug, M617 was the acetylated cysteine-binding product of HS-10296-M2, M621 and M621a were the sulfated binding products of monooxidation products of prodrug, M632b was the cysteine-binding product of demethylation product of the prodrug, M646 was the cysteine-binding product of the prodrug, M687 was the glucuronide-binding product of demethylation product of the prodrug, M688 was the acetylated cysteine-binding product of the prodrug, and M717 was the glucuronide-binding product of demethylation product of the prodrug.
Metabolic pathways of HS-10296 in the human body
Based on the results of the metabolite identification of [14C]HS-10296, it was assumed that the major metabolic pathway of HS-10296 in the human body was: (I) N-dedimethylethylaminization to form HS-10296-M2; and (II) demethylation to form M511a or M511b (HAS-719). The secondary metabolic pathway was oxidation to form M541a and M541c. Other metabolic pathways might include N-dealkylation to form HS-10296-M30 or binding to cysteine to form M646. Figure 4 shows the metabolic pathways of HS-10296 in the human body.
Figure 4.

Metabolic pathways of almonertinib in healthy participants.
Clearance pathways of HS-10296 in the human body
Based on the substance balance data and the main metabolic pathways of [14C]HS-10296 in the human body, it is presumed that HS-10296 is secreted into bile and eliminated through feces, mainly as prodrug and metabolites, and the main clearance pathways are: (I) N-dedimethylethylaminization to form HS-10296-M2, followed by oxidation, cysteine-binding, acetylated cysteine-binding, or demethylation before elimination through the liver; and (II) demethylation of the prodrug to form M511a or M511b (HAS-719), followed by demethylation, oxidation, cysteine-binding, glucuronide-binding, or sulfation before direct elimination by the liver. The secondary clearance pathway is the oxidation of the prodrug to form M541a or M541c, followed by glucuronide-binding or sulfation binding before direct elimination by the liver. The liver may also eliminate HS-10296 via one of the following pathways: (I) N-dealkylation of the prodrug to form HS-10296-M30; (II) direct cysteine-binding; or (III) acetylated cysteine-binding.
Discussion
This study's main aim was to investigate the pharmacokinetic characteristics and the major metabolic and clearance pathways of orally administered [14C]HS-10296 in healthy Chinese males. The pharmacokinetic results showed that, after a single oral dose of 110 mg/50 µCi [14C]HS-10296 suspension, the total plasma radioactivity peaked rapidly, with a Tmax of 4 h. These results were comparable to that of the non-labeled prodrug HS-10296 (5.25±3.2 h); the terminal elimination half-life (T1/2) of plasma total radioactivity was 863±930 h, which was much higher than the half-lives of the prodrug HS-10296 (25.0 h) and its metabolite HAS-719 (41.8 h). Also, the total radioactivity concentrations in whole blood were almost all below the lower detection limit, suggesting HS-10296 and its metabolites were not distributed into blood cells.
The errors of each metabolite’s measured mass spectrometry signals were within 5 ppm, showing that the experimental data had high accuracy and credibility. The isotope peaks of [14C]HS-10296-M30 had a peak-to-peak deviation of 13.6884 ppm because of this metabolite’s low content and small molecular weight, retention time, and mass spectral information were consistent with HS-10296-M30 standard, so the radioactive peak was identified as [14C]HS-10296-M30. During the metabolic analysis, 26 metabolites including HS-10296-M2, HS-10296-M30, HAS-719(M511b), M440, M470a/b/c/d/e, M497a/b, M511a, M527a/b, M541a/c, M575, M607, M617, M621/M621a, M632b, M646, M687 M687, M688, and M717 were identified in blood, urine, and feces. Among them, 12 metabolites had not been found in rat metabolism studies, namely HS-10296-M30, HS-10296-M2, M440, M470a/b/c/d/e, M575, M617, M687, and M688. In plasma, the major metabolite was M440 (accounting for 5.08% of total plasma radioactivity) in humans and M511a, M511b, and M541b in rats. In urine, the major metabolite was HAS-719 (accounting for 1.2% of drug administered; followed by M497a) in humans and M511a and M689b in rats. In feces, the major metabolite was HAS-719 (accounting for 12.33% of drug administered; followed by M541a/M470a and M617/M575) in humans and M632b, M646, and M541a in rats. Therefore, the major metabolic pathways in humans and rats are species-specific.
It is further assumed that the major metabolic pathways of HS-10296 in the human body are the N-dedimethylethylaminization to form HS-10296-M2 and the demethylation to form M511a or M511b (HAS-719), and the minor metabolic pathway is the oxidation to form M541a and M541c. Other metabolic pathways may include N-dealkylation to form HS-10296-M30 or binding to cysteine to form M646. In contrast, the main metabolic pathways of HS-10296 in SD rats are O-demethylation, N-demethylation, and oxidation; other pathways may include: phase II glucuronide-binding, sulfate-binding, glutathione-binding, and binding to glutathione followed by hydrolysis to form cysteine-binding or cysteine-and-glycine-binding products.
In summary, HS-10296 is extensively metabolized in the human body, producing a variety of metabolites. Although 12 metabolites are absent or not found in animals, they are relatively small in amount.
The most commonly known adverse effects of third-generation EGFRTKI on humans include rash, nausea, vomiting, and fatigue. For HS-10296, a Phase I dose-escalation study in patients with advanced NSCLC has been completed, with four doses (55 mg, 110 mg, 220 mg, and 260 mg) being safely tolerated by patients and showing preliminary antitumor activity. The common adverse events (AEs) of HS-10296 were rash, malaise, blurred vision, decreased white blood cells, gastrointestinal symptoms (constipation, diarrhea, and nausea), and elevated glutamic-pyruvic transaminase; however, these symptoms were mild or non-clinical and could resolve spontaneously. Our current study showed the good safety and tolerability of a single oral dose of 110 mg/50 µCi [14C]HS-10296 suspension in healthy Chinese males. No obvious or special adverse reaction was noted during the observation.
Conclusions
Our current study showed the good safety and tolerability of a single oral dose of 110 mg/50 µCi [14C]HS-10296 suspension in healthy Chinese males. No obvious or special adverse reaction was noted during the observation. TRA peaked quickly in plasma, with a Tmax of 4.0 h; however, TRA was cleared slowly in vivo, with a mean terminal elimination phase (half-life, T1/2) of up to 863 h, suggesting HS-10296 and its metabolites were not distributed into blood cells. Besides the prodrug, 26 metabolites in blood, urine, and feces were analyzed. Twelve metabolites were found in plasma, 13 in urine, and 26 in feces. In plasma, prodrug was the major drug-related component, accounting for 69.97% of TRA, and M440 (HS-10296-M2 demethylation product) was the major metabolite, accounting for 5.08% of TRA; in urine, the prodrug was about 0.48% of the dose administered. HAS-719 was the major metabolite, accounting for 1.20% of the administered dose; M497a was the minor metabolite, accounting for 0.21% of the dose administered. In feces, prodrug was about 8.61% of the dose administered. HAS-719 was the major metabolite, accounting for 12.33% of the administered dose, followed by M541a/M470a and M617/M575, accounting for 11.8% and 6.76% of the administered dose, respectively. The major metabolic pathways of HS-10296 in the human body were N-demethylacetylamination and demethylation, and oxidation was a minor pathway involved in HS-10296 metabolism. Other pathways such as N-dealkylation or binding to cysteine may also play a role. It is assumed that the main route of HS-10296 clearance in the human body is the excretion of the prodrug and its metabolites via bile from feces.
Supplementary
The article’s supplementary files as
Acknowledgments
Funding: This study was supported by funding from the 12th Five-Year Plan New Drug Creation Program-Clinical Evaluation Research Technology Platform for New Drugs for Respiratory Diseases (no. 2011ZX09302-003-02) and the 13th Five-Year Plan New Drug Creation Program - Isotope Tracer Technology Platform for Clinical Evaluation of Innovative Drugs (no. 2017ZX09304032).
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. The study was conducted in accordance with the Declaration of Helsinki (as revised in 2013). The study was reviewed and approved by the Drug Clinical Trial Ethics Committee of the Affiliated First Hospital of Nanjing Medical University (No. 2019-MD-049, the registration number of ethics board) and informed consent was taken from all individual participants.
Footnotes
Reporting Checklist: The authors have completed the MDAR reporting checklist. Available at http://dx.doi.org/10.21037/atm-21-1606
Data Sharing Statement: Available at http://dx.doi.org/10.21037/atm-21-1606
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/atm-21-1606). The authors have no conflicts of interest to declare.
References
- 1.Hojjat-Farsangi M. Small-molecule inhibitors of the receptor tyrosine kinases: promising tools for targeted cancer therapies. Int J Mol Sci 2014;15:13768-801. 10.3390/ijms150813768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morin MJ. From oncogene to drug: development of small molecule tyrosine kinase inhibitors as anti-tumor and anti-angiogenic agents. Oncogene 2000;19:6574-83. 10.1038/sj.onc.1204102 [DOI] [PubMed] [Google Scholar]
- 3.Jackman D, Pao W, Riely GJ, et al. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J Clin Oncol 2010;28:357-60. 10.1200/JCO.2009.24.7049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gahr S, Stoehr R, Geissinger E, et al. EGFR mutational status in a large series of Caucasian European NSCLC patients: data from daily practice. Br J Cancer 2013;109:1821-8. 10.1038/bjc.2013.511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med 2010;362:2380-8. 10.1056/NEJMoa0909530 [DOI] [PubMed] [Google Scholar]
- 6.Li D, Ambrogio L, Shimamura T, et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 2008;27:4702-11. 10.1038/onc.2008.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Engelman JA, Zejnullahu K, Gale CM, et al. PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res 2007;67:11924-32. 10.1158/0008-5472.CAN-07-1885 [DOI] [PubMed] [Google Scholar]
- 8.Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007;316:1039-43. 10.1126/science.1141478 [DOI] [PubMed] [Google Scholar]
- 9.Ramalingam SS, Blackhall F, Krzakowski M, et al. Randomized phase II study of dacomitinib (PF-00299804), an irreversible pan-human epidermal growth factor receptor inhibitor, versus erlotinib in patients with advanced non-small-cell lung cancer. J Clin Oncol 2012;30:3337-44. 10.1200/JCO.2011.40.9433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eskens FA, Mom CH, Planting AS, et al. A phase I dose escalation study of BIBW 2992, an irreversible dual inhibitor of epidermal growth factor receptor 1 (EGFR) and 2 (HER2) tyrosine kinase in a 2-week on, 2-week off schedule in patients with advanced solid tumours. Br J Cancer 2008;98:80-5. 10.1038/sj.bjc.6604108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou W, Ercan D, Chen L, et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 2009; 462:1070-4. 10.1038/nature08622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walter AO, Sjin RT, Haringsma HJ, et al. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov 2013;3:1404-15. 10.1158/2159-8290.CD-13-0314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dong WK, Sang WK, Tae MK, et al. Phase I study of HM61713, a novel epidermal growth factor receptor (EGFR) mutant selective inhibitor, in non-small cell lung cancer (NSCLC) patients having an activating EGFR mutation but failed to prior EGFR tyrosine kinase inhibitor (TKI) therapy. WCLC (HM61713) 2013:Abstract #1048.
- 14.Ge X, Zhou Q, Zhang Y, et al. EGFR tyrosine kinase inhibitor almonertinib induces autophagy and apoptosis in triplenegative breast cancer MDA-MB-231 cells. Nan Fang Yi Ke Da Xue Xue Bao 2020;40:981-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang JC, Camidge DR, Yang CT, et al. Safety, Efficacy, and Pharmacokinetics of Almonertinib (almonertinib) in Pretreated Patients With EGFR-Mutated Advanced NSCLC: A Multicenter, Open-label, Phase 1 Trial. J Thorac Oncol 2020;15:1907-18. 10.1016/j.jtho.2020.09.001 [DOI] [PubMed] [Google Scholar]
- 16.U.S Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research. The Radioactive Drug Research Committee: Human Research Without An Investigational New Drug Application [DB/OL]. Silver Spring: CDER, 2009 [2009-06]. Available online: http://www.fda.gov/downloads/Drugs/Guidances/UCM163892.pdf.
- 17.Protection Radiological. in Biomedical Research. A report of Committee 3 adopted by the International Commission on Radiological Protection. Ann ICRP 1991;22:1-28, v-xxiv. [PubMed] [Google Scholar]
- 18.Common Terminology Criteria for Adverse Events (CTCAE), Version 4.03, June 14, 2010. US Department of Health and Human Services. National Institutes of Health National Cancer Institute. Available online: https://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdf
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The article’s supplementary files as