

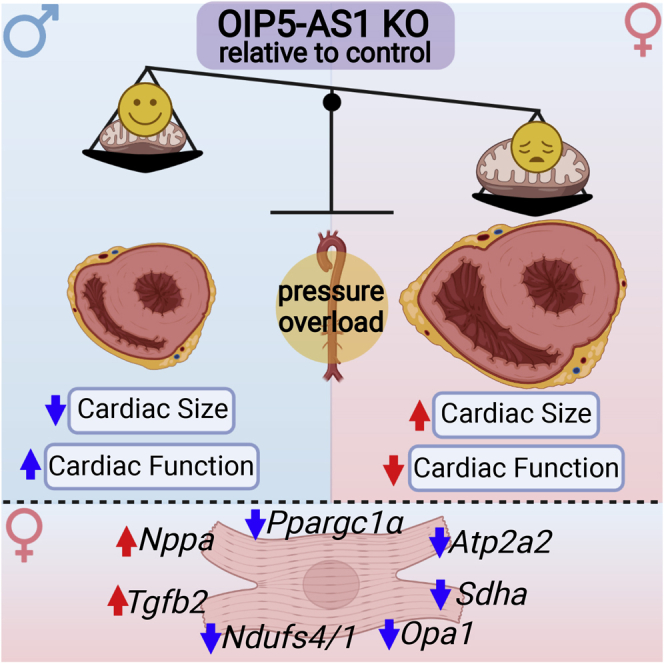
Summary
Long non-coding RNAs (lncRNAs) have been demonstrated to influence numerous biological processes, being strongly implicated in the maintenance and physiological function of various tissues including the heart. The lncRNA OIP5-AS1 (1700020I14Rik/Cyrano) has been studied in several settings; however its role in cardiac pathologies remains mostly uncharacterized. Using a series of in vitro and ex vivo methods, we demonstrate that OIP5-AS1 is regulated during cardiac development in rodent and human models and in disease settings in mice. Using CRISPR, we engineered a global OIP5-AS1 knockout (KO) mouse and demonstrated that female KO mice develop exacerbated heart failure following cardiac pressure overload (transverse aortic constriction [TAC]) but male mice do not. RNA-sequencing of wild-type and KO hearts suggest that OIP5-AS1 regulates pathways that impact mitochondrial function. Thus, these findings highlight OIP5-AS1 as a gene of interest in sex-specific differences in mitochondrial function and development of heart failure.
Subject areas: Cardiovascular medicine, Molecular physiology, Transcriptomics
Graphical abstract
Highlights
-
•
The lncRNA OIP5-AS1 is enriched in striated muscles in mice and humans.
-
•
OIP5-AS1 is regulated during heart development and in models of heart disease.
-
•
Global deletion of OIP5-AS1 exacerbates heart failure specifically in female mice.
-
•
Transcriptomics analysis suggests that loss OIP5-AS1 alters mitochondrial function.
Cardiovascular medicine; Molecular physiology; Transcriptomics
Introduction
Since the advent of deep-sequencing and genome mapping, it has become clear that genetic control of cellular function is evidently more complex than the classical model of DNA→RNA→protein. This dogma was initially re-examined upon the discovery of miRNAs (miRs), which were one of the first classes of non-coding (nc)RNAs demonstrated to regulate cellular disease pathways. Since then, the ncRNA field has expanded rapidly with the subsequent discovery of a wide range of ncRNA classes, including potentially thousands of predicted long ncRNAs (lncRNAs) (Cech and Steitz, 2014). LncRNAs are those described to be > 200 nucleotides long; however, most are several kilobases in length and are topographically similar in many ways to protein coding mRNAs (Kashi et al., 2016). Bona fide lncRNAs do not code for functional proteins and are therefore proposed to have a vast array of functions including acting as transcriptional regulators, structural scaffolds or RNA sponges (Mallory and Shkumatava, 2015; Ulitsky and Bartel, 2013). Although lncRNAs were discovered more recently, exciting studies have already emerged to suggest that they are likely promising targets for therapeutic and biomarker applications. Indeed, lncRNAs are often more cell-type specific and tightly regulated than protein coding RNAs (mRNA) (St Laurent et al., 2015).
Of particular interest to the current study, are several lncRNAs that have been linked with pathological conditions in cardiac muscle (McMullen and Drew, 2016). The molecular regulation of cardiac commitment and development has been an intense area of research for many years. The discovery of lncRNAs has thus pioneered a new area of investigation in cardiac biology, with several groups identifying cardiac specific lncRNAs that are involved in almost every facet of cardiac commitment, development, and function. The first detailed mechanistic description of a cardiac specific lncRNA was Braveheart (bvht) (Klattenhoff et al., 2013), which was shown to be important in cardiac lineage commitment through its actions on MesP1, the master regulator of cardiovascular lineage commitment. Since then, many lncRNAs have been implicated in cardiac commitment and differentiation including Fendrr, SRA and Novlnc6, all of which affect the activity of lineage specific transcriptional pathways (Colley and Leedman, 2011; Grote et al., 2013; Ounzain et al., 2015). Furthermore, the expression of some lncRNAs such as MIAT, LIPCAR, Mhrt, and CHRF are also strongly associated with cardiovascular disease (Han et al., 2014; Ishii et al., 2006; Kumarswamy et al., 2014; Wang et al., 2014). Specifically, MyHeart (Mhrt) and CHRF exhibit significantly altered expression in the setting of cardiac hypertrophy (Han et al., 2014; Wang et al., 2014). Importantly, mice with reconstitution of Mhrt expression in the setting of pathological hypertrophy, displayed less cardiac dysfunction than wild-type (WT) mice, providing evidence that lncRNAs have promising therapeutic potential in the heart (Han et al., 2014).
Here, we investigate a lncRNA known as OIP5-AS1 (also known as 1700020I14Rik and Cyrano), which has been reasonably well studied in the brain and embryonic stem (ES) cells, but there is limited data on its roles in cardiac pathologies such as heart failure. We reveal that OIP5-AS1 expression is enriched in striated muscle and differentiating cardiomyocytes (CMs), and its expression is reduced in the setting of heart failure. Furthermore, we show that global loss of OIP5-AS1 in mice leads to poorer outcomes following pressure-overload induced heart failure, but only in female mice. Thus, our studies demonstrate a previously unrecognized sex-specific function of OIP5-AS1 in the heart.
Results
OIP5-AS1 is enriched in striated muscles
OIP5-AS1 is located between the two protein coding genes Chp1 and Oip5 on chromosome 2 (Figure S1A) and is a bona fide lncRNA based on its absence in ribosomal transcriptome profiling (Ulitsky et al., 2011). It is called OIP5-AS1 because of its proximity and opposing topographical locality to the OIP5 gene, which is similar to that observed in the mouse (Kleaveland et al., 2018). OIP5-AS1 has been the topic of a number of studies in lab-based systems such as zebrafish and ES cells. Its function in these systems have been described to modulate the abundance of specific miRs to influence pathways important in proliferation (Kim et al., 2016), self-renewal (Smith et al., 2017), and differentiation (Zheng et al., 2019). However, functional genetic studies have failed to demonstrate a link between loss of OIP5-AS1 expression and robust phenotypes in animal systems, other than mild malformation of the neural tube and nasal placodes in zebrafish embryos – hence its alternative name, Cyrano (Ulitsky et al., 2011). Moreover, almost none of these phenotypes have translated across vertebrate species, with mild to no phenotype identified in knock-down or partial knockout (KO) models in higher order mammals (i.e. mice) (Kleaveland et al., 2018).
Using Genomic Evolutionary Rate Profiling data, we and others (Kleaveland et al., 2018; Ulitsky et al., 2011) demonstrate that unlike the majority of lncRNAs, OIP5-AS1 harbors several regions of high homology in its nucleotide sequence between mammalian species including humans. We also confirm previous findings that a small region of exon 3 is 100% conserved across vertebrates (Figure S1B; blue shaded area). This is uncommon for lncRNAs, which often exhibit poor sequence conservation between species (Diederichs, 2014).
In the current study, we were interested to know if OIP5-AS1 has functional relevance in the heart, especially given that deposited gene expression data from NCBI (Figure 1A) (Thorrez et al., 2008) and transcriptomics data from our lab (Data S1) (Ribas et al., 2016) and others (Kleaveland et al., 2018), demonstrate an enrichment for OIP5-AS1 in striated muscles (diaphragm, muscle, and heart), as well as in the brain. Indeed, our transcriptomics data (from skeletal muscle) demonstrated that OIP5-AS1 is expressed within the top 500 most abundant genes (of ∼7000 detected), together with several other previously annotated lncRNAs such as Malat1, H19 and Rhit1 (Nctc1) (Dey et al., 2014; Watts et al., 2013).
Figure 1.

OIP5-AS1 is enriched in developing cardiac muscle, and is altered during cardiac disease
(A) Expression of OIP5-AS1 across mouse tissues sourced from NCBI Gene Expression Omnibus (GEO, GSE24207), n = 4/group, mean ± SEM, ∗p < 0.001 from liver expression.
(B and C) (B) Expression of OIP5-AS1 as determined by RNA-sequencing in fibroblasts (Fb) and cardiomyocytes (CMs) digested from mouse hearts of neonate and 8-week old mice (n = 4/group, mean ± SEM, ∗p < 0.05 versus neonate Fb; #p < 0.05 versus neonate CM) from GEO deposited dataset GSE95764, and in (C) Fb and CM differentiated from human iPSCs (n = 3/group, mean ± SEM, ∗p < 0.05 versus Fb).
(D) Network analysis of genes sets correlated with OIP5-AS1 expression as determined by RNA-sequencing in mouse cardiomyocytes versus fibroblasts from data sets in panel C. Enriched networks include cardiac contraction, muscle morphogenesis and mitochondrial pathways (assembly and electron transport chain (ETC)). For a description of each code shown within the nodes, see Table S2.
(E) Bar graph depicting most highly enriched pathways associated with OIP5-AS1 expression in mouse cardiomyocytes.
(F and G) (F) qPCR determined expression of OIP5-AS1, Atp2a2 (SERCA2) and Nppa (ANP) in hearts from WT or utrophin/dystrophin double KO (DKO) male mice (n = 8/group, mean ± SEM, ∗p < 0.05 versus WT) (Chen et al., 2017), and (G) WT male mice that have undergone sham or aortic constriction (AC); n = 4–6/group, mean ± SEM, ∗p < 0.05 versus sham). For A, B, C, F and G, a non-parametric one-way ANOVA with multiple comparisons correction (Dunnet's) was used.
OIP5-AS1 is regulated during development of cardiac muscle
To determine whether OIP5-AS1 expression is regulated in cardiac muscle, we examined its expression in various models and tissues. Firstly, we sought to investigate whether its expression was enriched in CMs relative to other cells types in the heart, and therefore investigated its expression profile in fibroblasts (Fbs) and CMs isolated from the murine heart. To do this, we examined RNA-seq data (GEO data set GSE95764) from mouse Fbs and CMs that were FACS sorted from digested neonatal and adolescent mouse hearts (Figure 1B) (Quaife-Ryan et al., 2017). These data demonstrated that OIP5-AS1 expression were higher in CMs of both neonates and adolescents, with an increasing expression observed in the adolescent (8-weeks) heart. We also investigated the expression of OIP5-AS1 in an in vitro human model of cardiomyogenesis. RNA-seq data from human Fbs and CMs (GEO data set GSE62913) induced from the same pluripotent stem cell (iPSCs) population, again demonstrated an increased expression of OIP5-AS1 in CM compared to Fb (Figure 1C). Collectively, these findings provide evidence that OIP5-AS1 expression is upregulated in CMs compared to Fb in both rodent and human cells.
Given that OIP5-AS1 appears to be regulated during cardiomyogenesis, we performed computational analyses in an attempt to predict the potential role that OIP5-AS1 might play in CMs. Using RNA-seq data from the mouse Fb and CM data sets described above, we demonstrated that OIP5-AS1 transcript associated with gene networks representative of ventricular contraction, muscle morphogenesis and mitochondrial function (Figure 1D). Enrichment analysis reveals a link to pathways that are consistent with alterations in respiration and heart contraction (Figure 1E), providing evidence of a link between OIP5-AS1 and cellular pathways involved in heart energetics.
OIP5-AS1 expression is regulated in the setting of disease
Given the results above, we considered whether OIP5-AS1 might also be altered in cardiac disease – a setting where developmental processes and cardiac energetics are often dysregulated. Alterations in the expression of lncRNAs have been observed in several models of cardiovascular disease such as cardiomyopathy and myocardial infarction (McMullen and Drew, 2016). Indeed, OIP5-AS1 expression was previously reported to be reduced in the hearts of rats that had suffered from a myocardial infarction (Niu et al., 2020). To demonstrate if OIP5-AS1 expression was specifically affected in heart failure, we chose to analyze tissues from two existing murine heart failure models. This included the mild model of heart failure, the utrophin/dystrophin double KO mouse (Kennedy et al., 2016) (Chen et al., 2017). Analysis of hearts from these mice demonstrated a reduction in SERCA2 (Atp2a2) and an increase in ANP (Nppa) mRNA expression compared to WT mice, consistent with molecular signatures of heart failure (Figure 1F). We also observed a reduced expression of OIP5-AS1, suggesting that OIP5-AS1 is downregulated in the setting of mild heart failure. Next, we studied hearts from mice that had undergone aortic constriction (AC) induced by cardiac pressure overload (McMullen et al., 2004, 2007), which is a severe model of heart failure that develops in less than 2 weeks (as opposed to other models that take 8-12 weeks, i.e. transverse aortic constriction [TAC]). In this model, we observed robust changes in the cardiac expression of SERCA2 and ANP, as well as a consistent reduction in OIP5-AS1 expression (Figure 1G). Collectively, these data provide evidence that OIP5-AS1 expression is downregulated in the setting of heart failure and cardiomyopathy in mice.
Generation of an OIP5-AS1 KO mouse
The above findings are mostly associative, and thus do not specifically demonstrate a causal role for OIP5-AS1 in cardiac pathologies. Therefore, to investigate whether OIP5-AS1 is causally linked to cardiac dysfunction and disease, we generated an OIP5-AS1 global KO mouse using CRISPR/Cas9 technology in C57BL/6J mice by deleting the OIP5-AS1 gene (Figure 2A). These mice were viable and subsequently bred with WT C57BL/6J mice for 5 generations before generating cohorts of WT and KO mice. Using qPCR analysis, we demonstrated complete ablation of OIP5-AS1 expression in all muscle tissues tested in homozygous null mice (KO) (Figure 2B), confirming successful generation of the model. Young adult (∼10 weeks of age) KO mice were phenotypically unremarkable and displayed no differences in body weight or organ weights compared to WT mice (Figure 2C). These findings are consistent with that from Kleaveland et al. who generated a partial exon 3 OIP5-AS1 KO mouse that also displayed no overt basal phenotype (Kleaveland et al., 2018). Furthermore, basal phenotyping of heart function in our model using echocardiography, demonstrated no difference in left ventricle (LV) dimensions or function between WT and KO male or female mice at ∼10 weeks of age (Table 1 - Baseline).
Figure 2.

Generation and basal phenotyping of an OIP5-AS1 KO mouse model
(A) Schematic outlining the CRISPR/Cas9 approach employed to delete the OIP5-AS1 gene (∼13.4kb) from the C57BL/6J genome. Two guide RNAs (gRNA1&2) were designed to bind at either end of the gene locus (red arrows) after which Cas9 induced removal of the entire gene by non-homologous repair mechanisms.
(B and C) (B) qPCR analysis of OIP5-AS1 gene expression from four different muscle tissues from 10-week old male wild type (WT) and OIP5-AS1 KO (KO) mice (n = 3–5/group, mean ± SEM, ∗p < 0.05 versus WT). Gastroc = Gastrocnemius (C) Body mass and organ weights of 10-week-old female WT and KO mice (n = 3–5/group, mean ± SEM). BW = body weight, LM = lean mass, FM = fat mass. For B and C, a non-parametric one-way ANOVA with multiple comparisons correction (Dunnet's) was used.
Table 1.
Echocardiography measurements in WT and KO male and female mice.
| All | Baseline |
8 Weeks post-procedure |
||||
|---|---|---|---|---|---|---|
| Sham | TAC | |||||
| Female | WT | KO | WT | KO | WT | KO |
| BW (g) | 21.6 ± 1.1 | 21.2 ± 1.0 | 26.7 ± 1.8 | 24.5 ± 1.1 | 24.1 ± 0.9† | 22.2 ± 0.8† |
| Heart rate (bpm) | 524 ± 19 | 499 ± 18 | 546 ± 8 | 525 ± 42 | 542 ± 17 | 559 ± 5 |
| AWd (mm) | 0.52 ± 0.03 | 0.54 ± 0.02 | 0.69 ± 0.07 | 0.68 ± 0.08 | 0.86 ± 0.02† | 0.82 ± 0.05† |
| LVEDD (mm) | 4.09 ± 0.10 | 4.15 ± 0.05 | 3.61 ± 0.21 | 3.97 ± 0.21 | 4.31 ± 0.25† | 4.84 ± 0.23† |
| PWd (mm) | 0.64 ± 0.02 | 0.58 ± 0.02 | 0.79 ± 0.09 | 0.72 ± 0.07 | 0.96 ± 0.05† | 0.93 ± 0.04† |
| LVESD (mm) | 2.91 ± 0.10 | 3.04 ± 0.06 | 2.51 ± 0.19 | 2.72 ± 0.19 | 3.51 ± 0.31† | 4.33 ± 0.21† |
| FS (%) | 29 ± 1.7 | 27 ± 0.9 | 31 ± 1.5 | 31 ± 4.5 | 19 ± 2.7† | 11 ± 1.1∗† |
| LV mass (mg) | 77.52 ± 5.94 | 79.59 ± 3.98 | 88.58 ± 5.51 | 97.20 ± 8.12 | 158.25 ± 9.70† | 198.04 ± 13.47∗† |
| Baseline |
8 Weeks post-procedure |
|||||
|---|---|---|---|---|---|---|
| Sham | TAC | |||||
| Male | WT | KO | WT | KO | WT | KO |
| BW (g) | 22.0 ± 1.0 | 23.1 ± 0.7 | 28.8 ± 1.2 | 30.8 ± 0.9 | 29.4 ± 0.4 | 29.9 ± 0.6 |
| Heart rate (bpm) | 536 ± 12 | 518 ± 18 | 545 ± 27 | 605 ± 34 | 602 ± 16 | 597 ± 12 |
| AWd (mm) | 0.53 ± 0.02 | 0.48 ± 0.01 | 0.61 ± 0.06 | 0.68 ± 0.03 | 0.87 ± 0.03† | 0.90 ± 0.02† |
| LVEDD (mm) | 4.06 ± 0.10 | 4.13 ± 0.10 | 4.25 ± 0.13 | 3.99 ± 0.05 | 4.84 ± 0.16† | 4.33 ± 0.15† |
| PWd (mm) | 0.60 ± 0.02 | 0.60 ± 0.01 | 0.70 ± 0.05 | 0.71 ± 0.05 | 0.97 ± 0.04† | 0.99 ± 0.03† |
| LVESD (mm) | 2.86 ± 0.10 | 2.94 ± 0.11 | 3.10 ± 0.16 | 2.59 ± 0.14 | 3.87 ± 0.16† | 3.25 ± 0.15∗† |
| FS (%) | 30 ± 1.0 | 29 ± 1.4 | 27 ± 1.7 | 35 ± 3.7 | 20 ± 1.3† | 25 ± 1.1∗† |
| LV Mass (mg) | 76.34 ± 3.51 | 75.64 ± 3.48 | 102.57 ± 15.00 | 97.14 ± 5.44 | 195.40 ± 11.73† | 172.54 ± 12.50† |
Echocardiography measurements performed in the same animal at baseline (pre-procedure) and 8-weeks after undergoing sham or TAC surgery. Group numbers (n = 9 for female, n = 15 for male) were influenced by whether the majority of data points were collected for a given animal at pre- and post-procedure (see STAR methods for exclusion and inclusion criteria). Overall group size was smaller for females due to more severe disease being observed at 8-weeks post-TAC. Bold text highlights values where there was a significant difference between WT and KO. Data are shown as mean ± SEM. ∗p < 0.05 vs WT TAC, †<0.05 vs sham of the same genotype, two-way ANOVA followed by Tukey post-hoc testing. Abbreviations: BW = body weight; (bpm) = beats per minute; AWd = anterior wall diameter; LVEDD = left ventricular end-diastolic dimensions; PWd = posterior wall diameter; LVESD = left ventricular end-systolic dimensions; FS% = fractional shortening percent; LV = left ventricle.
Female OIP5-AS1 KO mice develop exacerbated disease in a pressure overload induced model of heart failure
Since there were no basal alterations in cardiac function in OIP5-AS1 KO mice, we sought to investigate whether inducing cardiac stress might reveal a phenotypic difference between the genotypes. Given that we observed reductions in expression of OIP5-AS1 in the setting of AC induced pressure overload in WT mouse hearts as shown in Figure 1G, we chose to subject OIP5-AS1 KO mice to a similar pressure overload procedure. Pressure overload also represents an appropriate model to test the effect of OIP5-AS1 deletion because it induces high-energy demand on the heart, thus directly impacting on the energetic pathways predicted to be associated with OIP5-AS1 function. For these studies, we performed TAC, which progressively induces heart failure over an approximately 8-week period. We performed TAC and sham surgery on both male and female weight matched OIP5-AS1 WT and KO mice at 7-10 weeks of age, and the phenotype was monitored over the ensuing 8 weeks (Figure 3A). Because the mice were weight matched, the aortic diameter and thus constriction at the time of surgery were assumed to be equivalent. Indeed, intra-cardiac and aortic catheter pressure analysis confirmed an equivalent pressure increase in a subset of WT and KO animals (Figure S2A). Consistent with our previous studies (Bernardo et al., 2012, 2014; Gao et al., 2005; Kiriazis et al., 2008), we demonstrated that the TAC procedure promoted significant pathological cardiac hypertrophy in WT mice following 8 weeks of TAC in both male and female mice compared to sham operated animals (light gray bars, Figures 3B–3F). Specifically, we demonstrated that TAC operated WT mice (light gray bars) demonstrated an increase in total heart, LV, right ventricle (RV), atria and lung weights compared to sham operated animals (white bars), consistent with cardiomyopathy and congestive heart failure. We also demonstrated that TAC leads to the expected changes in the transcription of genes in the heart such as SERCA2 (Atp2a2) and ANP (Nppa) in both male and female animals, as measured by qPCR that are consistent with heart failure (Figures S2B and S2C). With regards to OIP5-AS1 deletion, we demonstrated that female OIP5-AS1 KO mice (dark gray bars) had a more severe pathological hypertrophy and heart failure phenotype post-TAC than female WT mice, as indicated by significantly (p < 0.05) increased heart weight, LV and RV weight, atrial weight and substantially heavier lung weight (Figures 3B–3F), the latter representing advanced cardiomyopathy and congestive heart failure. Interestingly, the same differences between genotypes were not observed in male mice, or were even opposing, despite male WT mice displaying significant pathological cardiac hypertrophy (increased heart, LV and atria weights) following the TAC procedure. This was consistent with qPCR data from hearts of male WT and KO mice, where ANP expression trended to be substantially lower in KO TAC hearts (indicating a lessened heart failure phenotype) (Figure S2D), compared to female KO TAC hearts which demonstrated equivalent ANP expression but even lower SERCA2 expression (Figure S2E).
Figure 3.

Female OIP5-AS1 mice demonstrated a worsened heart failure phenotype following TAC surgery
(A–G) (A) Schematic of experimental (transverse aortic constriction [TAC] or Sham surgery) protocol performed on male and female wild type (WT) and knock out (KO) mice, which were followed for a further 8 weeks before cull. Tissue weights from cull were normalized to tibia length for (B) whole heart weights, (C) left ventricle (LV), (D) right ventricle (RV), (E) atria, and (F) whole lung (♀ = female, ♂ = male, female n = 7–14/group, male n = 9–19, mean ± SEM, ∗p < 0.05 versus sham, #p < 0.05 versus WT-TAC of the same sex) (G) Percent fractional shortening (FS %) as determined by echocardiography in male and female WT and KO Sham and TAC mice at 8 weeks post-procedure (n = 3–5/group for sham, n = 6–11/group for TAC, mean ± SEM, ∗p < 0.05 versus sham for each sex.
(H) Representative photomicrographs of whole hearts from female and male WT and KO mice 8-weeks post procedure.
(I) Representative images of picrosirius red stained LV sections from female WT and KO mice post-TAC procedure, 400 x magnification, scale bar = 20m.
(J) Quantification of percent area picrosirius red staining (fibrosis) of entire LV section from WT and KO female mice undergoing TAC surgery (n = 6/group). For panels B-G and panel J, a non-parametric one-way ANOVA with multiple comparisons correction (Dunnet's) was used to test for significance, ∗p < 0.05.
Echocardiography analysis demonstrated significant (p < 0.05) alterations in LV wall thickness, dimensions, and function in mice that had undergone TAC, compared to sham surgery mice, in both female and male animals (Table 1). Female OIP5-AS1 KO mice demonstrated a more pronounced decline in fractional shortening (FS%) following TAC compared to WT mice (11 ± 1.1% vs 19 ± 2.7%, respectively) (Figure 3G), indicating a worsened left ventricular function in female animals. Male OIP5-AS1 KO mice did not show a further decline in LV function in comparison to male WT mice post-TAC as demonstrated by FS% (Table 1 and Figure 3G). Photomicrographs of the hearts from male and female mice confirmed the increase in heart size in TAC-induced mice and demonstrated the significant enlargement in both the ventricles and atria of KO mice, particularly in female mice (top row, Figure 3H). These pathological effects in the heart were associated with a greater incidence of clinical phenotypes of organ congestion, as indicated by an increase in lung weights in female KO mice in Figure 3F, and a greater percentage of female KO mice exhibiting thrombi in their atria compared to WT mice (55% vs 0%; shown visually in Figure 3H). Liver and kidney weights were equivalent between WT and KO male and female mice (Figures S2F and S2G), whilst spleen weights were significantly larger in female KO TAC treated mice, potentially reflecting a general loss of health in these animals (Figure S2H). We did not observe any alterations in fibrosis between WT and KO female mice as determined by picrosirius red staining of LV sections, which demonstrated no significant difference in percent area of fibrosis between WT and KO female mice following TAC surgery (Figures 3I and 3J).
Collectively, these data demonstrated that female but not male OIP5-AS1 KO mice displayed a more severe heart failure phenotype following pressure-induced cardiac-overload compared to WT mice.
Loss of OIP5-AS1 leads to transcriptional changes in gene networks associated with cardiomyopathy and mitochondrial function
In light of the exacerbated heart failure phenotype observed in female KO mice, we investigated transcriptional pathways that might be altered in OIP5-AS1 male and female mice. We did not observe differences in the basal expression of OIP5-AS1 in LVs between male and female WT mice, although the KO was equivalent between sexes (Figure 4A). We also observed no difference in OIP5-AS1 abundance between male and female WT mice following TAC (Figure 4B). These results suggest that sex specific factors such as circulating hormones are most likely not influencing the expression of OIP5-AS1 in the hearts of WT mice either basally or after TAC, and thus more detailed analyses were necessary to tease out the potential causal pathways.
Figure 4.

Expression of OIP5-AS1 and its neighboring genes are not influenced by sex or cis-specific transcriptional mechanisms
(A) Abundance of OIP5-AS1 transcripts as determined by qPCR in left ventricle (LV) of male and female wild type (WT) and OIP5-AS1 KO (KO) mice (n = 9–10/group, mean ± SEM, ∗p < 0.05 versus WT, NS = not significant between male and female WT mice).
(B–D) (B) Abundance of OIP5-AS1 transcripts as determined by RNA-seq in female and male WT LVs following TAC. Expression of OIP5-AS1 neighboring genes (Oip5, Chp1, Nusap1 and Exd1) in sham treated mice in (C) female and (D) male WT and OIP5-AS1 KO hearts as determined by qPCR (n = 7–10/group, mean ± SEM). Non-parametric one-way ANOVA with multiple comparisons correction (Dunnet's) was used to test for significance, ∗p < 0.05.
Some lncRNAs are proposed to function via a cis acting mechanism, whereby active transcription at the locus of an lncRNA promotes opening of local chromatin and recruits transcriptional machinery to facilitate expression of neighboring genes. However, we did not observe any difference between WT and KO female or male mice in the expression of the four neighboring genes of OIP5-AS1 (two proximal, two distal; Oip5, Chp1, Nusap1, and Exd1) (Figures 4C and 4D), suggesting that the OIP5-AS1 phenotype is unlikely to be secondary to dysregulation of cis acting mechanisms. Thus, given that none of these archetypical pathways appeared to explain the mechanism by which OIP5-AS1 was functioning, we performed transcriptomic analysis on the WT and KO hearts in order to obtain a global overview of transcriptional differences between the genotypes.
For these analyses, we performed RNA-sequencing on LVs from male and female WT and KO mice, all of which had undergone TAC surgery (n = 6/group). Initial quality control analyses including PCA demonstrated segregation of the four groups (Figure S3A). Importantly, we demonstrate from this RNA-seq analysis that OIP5-AS1 expression is similar to that of other known lncRNAs such as Xist and Malat1 and was indeed more highly expressed than the well characterized Bvht (Figure S3B). Consistent with in vivo phenotyping data, the greatest changes in differential gene expression were observed in female KO mice, whether it be compared to female WT mice (Figure S3C) or male KO mice (Figure 5A). The number of genes significantly (q < 0.1) altered between female WT and female KO hearts was 66, whereas between female KO and male KO hearts it was ∼1700 genes (q < 0.1) (Figure 5A). This latter finding may not seem that surprising, given that it is a comparison between male and female mice. However, when we compared the transcriptomes between male and female WT hearts, there were only 8 genes that were significantly (q < 0.1) different, including known sex specific transcripts such as Xist, Kdm5c, and Ddx3x (Figure S3D). Thus, this indicated that the large transcriptional effect of OIP5-AS1 deletion between KO female and KO male hearts (∼1700 genes) was not purely due to sex effects alone but was a likely interaction between sex and the loss of OIP5-AS1 expression. Only one gene was significantly (q < 0.1) altered between male WT and KO mice and that was OIP5-AS1 itself (Figure S3E). Thus, these data provide evidence that the loss of OIP5-AS1 results in specific alterations in cardiac gene expression post-TAC only in female mice.
Figure 5.

Transcriptomic and integrative analysis of OIP5-AS1 KO hearts reveals alterations in pathways associated with cardiomyopathy and mitochondrial metabolism
(A) Volcano plot of LV gene expression analyzed by RNA-seq that were differentially regulated between female (F) and male (M) KO mice following TAC (n = 5–6/group), gray dots = non-significant, magenta = >0.25 log2FC, yellow dots = FDR<0.01, blue/red dots = >0.25log2FC and FDR<0.01. Off-scale points are specifically annotated with log2FC values at the individual data point.
(B) Functional map of genes altered in OIP5-AS1 KO female mice compared to WT, in LV following TAC. Colors indicate the association to either a positive (red = higher in KO) or negative (blue = lower in KO) enrichment. Node size is proportional to the total number of genes in each set. Edge thickness represents the similarity coefficient between gene/pathway sets (circle). Significance (FDR q-value as a percentage) of the enrichment is represented as a color gradient, where a fuller color is more significant. The major functional groups are highlighted by the shaded background bubbles - enriched in KO (red); reduced in KO (blue).
(C–E) (C) Abundance of transcripts in WT and OIP5-AS1 KO female mice as determined by RNA-sequencing in LV following TAC for genes related to heart failure (HF) (Nppa, Tgfb2 and Atp2a2) and (D) Related to mitochondrial function (Ndufs1, Ndufs4, Sdha, Ppargc1a, and Esrrg), (data in C-D are mean ± SEM, n = 5–6) (E) Table of proteins and genes that were shown to interact with OIP5-AS1 in a previously published data set (Smith et al., 2018) and are also significant DEGs in our RNA-seq analysis. Targets were cross-reference against MitoCarta 2.0 to identify those that are mitochondrial enriched (indicated by a “Y”). Fold change is represented with a gradient (blue = decreased in KO, red = increased in KO), where mitochondrial proteins show a strong trend to be decreased in OIP5-AS1 KO hearts.
(F) Gene expression of mitochondrial related genes as determined by qPCR analysis in left ventricles from female WT and KO sham operated mice (n = 3/group).
(G and H) (G) Oxygen consumption rate (OCR) as performed by Seahorse in mitochondria isolated from basal WT and OIP5-AS1 KO female hearts to test ETC Complex activity and to test (H) Maximal respiratory capacity using the mitochondrial stress test (Rot = rotenone, Complex I inhibitor; Succ = Succinate, Complex II substrate; Anti-A = antimycin A, Complex III inhibitor; L-Asc/TMPD = L-ascorbate and Tetramethyl-p-phenylenediamine, Complex IV substrate; ADP = adenosine diphosphate; Oligo = Oligomycin, Complex V inhibitor; FCCP = Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone, mitochondrial uncoupler) (n = 7/group). For panels C and D, a non-parametric one-way ANOVA with multiple comparisons correction (Dunnet's) was used to test for significance, ∗p < 0.05.
To gain a better understanding of the pathways that were altered by OIP5-AS1 deletion, we performed pathway enrichment and network analysis on the gene sets altered between male and female KO mice. These studies demonstrated a notable enrichment (red networks) in genes associated with extracellular matrix (ECM)/cytoskeleton, and hypertrophic cardiomyopathy in female KO mice following TAC (Figure 5B). Moreover, we observed a substantial depletion (blue networks) in the expression of genes associated with cardiac metabolism in KO female hearts including electron transport chain (ETC) respiration, amino acid catabolism and fatty acid metabolism (blue networks, Figure 5B). Alterations in similar networks were also observed in the comparison of WT v KO female hearts, with changes in classic heart failure related genes such as Nppa (ANP), Tgfb2 and Atp2a2 (SERCA2) (Figure 5C). Moreover, changes in core components of complex I and II of the ETC were also observed including reductions in Ndufs1, Ndufs4, and Sdha (Figure 5D). These findings were reassuring because gene correlation analysis performed in Figure 1D suggested that OIP5-AS1 was likely associated with cardiomyopathy and mitochondrial/oxidative gene networks, predictions that appear to be supported by these analyses in KO mouse hearts. Moreover, our initial data in Figures 3I and 3J demonstrating that fibrosis pathways were not markedly different between WT and KO mice, was also supported by our RNA-seq data, with the majority of genes coding for collagens not substantially increased in female or male KO versus WT mice (Figures S3F and S3G).
Regarding specific mediators of this phenotype, it is interesting to note that two bona fide mitochondria related transcription factors were significantly decreased in OIP5-AS1 KO female hearts versus WT hearts. Specifically, we observed ∼30% reduction in expression of Ppargc1a (PGC1alpha), a co-activator and regulator of genes important for mitochondrial biogenesis (Mootha et al., 2003), and a ∼25% reduction in Esrrg (ERRgamma) (Figure 5D). ERRgamma is a transcriptional co-activator that regulates transcription of genes important for oxidative capacity and mitochondrial function (Alaynick et al., 2007) and has also been shown to influence neonatal to adult transition in CMs (Quaife-Ryan et al., 2017), an important time in development where we observed substantial increases in OIP5-AS1 expression (Figure 1).
These observed alterations in mitochondrial networks were of potential mechanistic interest. However, because we have no direct evidence for how OIP5-AS1 functions in CMs, we instead mined previously published datasets in an attempt to gain further mechanistic insights. One such dataset was a protein interaction data set, which used pull down approaches to identify proteins that interacted with OIP5-AS1 in ES cells (Smith et al., 2018). By comparing their list of OIP5-AS1 interacting proteins with our differentially expressed gene set, we identified 18 genes/proteins that were consistent between the two datasets (Figure 5E and Table S3). There were a mix of genes upregulated (red shading) and downregulated (blue shading) in the KO heart; however it was apparent that the majority of the down regulated genes (blue shading) were known mitochondrial associated proteins as indicated by their presence in the MitoCarta 2.0 database(Calvo et al., 2016) (indicated by a “Y” in the table). Thus, we hypothesize that OIP5-AS1 may interact with critical proteins from the mitochondria (e.g. Ndufa4, Idh1, Hadh, Etfa), and that loss of OIP5-AS1 leads to a disruption of this function. This effect is likely to be stress specific, such as that induced by TAC, because we also demonstrate that the expression of these mitochondrial genes were not different between WT and KO female hearts in the basal (i.e. sham) setting (Figure 5F), nor was mitochondrial function different in the basal setting as measured by Seahorse (Figures 5G and 5H). Thus, OIP5-AS1 loss potentially alters mitochondrial networks in the setting of stress in the female heart, yet loss of OIP5-AS1 in a basal, unstressed setting does not impact mitochondrial function.
Collectively, our findings provide evidence that disruption OIP5-AS1 leads to an exacerbated heart failure progression following stress in female mice, a phenotype that may be linked to specific components of the mitochondrial network.
Discussion
In the current study, we have described and characterized the lncRNA OIP5-AS1 to be enriched and functional in cardiac muscle. OIP5-AS1 demonstrates several regions of high conservation between mouse and human in its nucleotide sequence, and thus its function and roles are also likely to be conserved. Upon generating and studying an OIP5-AS1 KO mouse model, we demonstrated that loss of OIP5-AS1 renders female mice more prone to cardiac overload induced heart failure, a phenotype not observed in male mice. Transcriptomic and integrative analysis indicated that OIP5-AS1 modulates molecular regulators of mitochondrial function, exemplified by alterations in key mitochondrial gene sets both in CMs isolated from mouse hearts, and in OIP5-AS1 KO heart tissue. Interestingly, we did not observe alterations in mitochondrial gene expression or mitochondrial function in non-stressed hearts, suggesting that the changes observed in stressed KO hearts were a maladaptive response. These data are consistent with the phenotype being primarily driven by changes in metabolism of CMs in female OIP5-AS1 KO mice, rather than ECM remodeling such as scarring and fibrosis. We believe this phenotype is most likely driven by loss of OIP5-AS1 in CMs based on the fact that its expression is highest in this cell type, and that there were no transcriptional signatures reminiscent of other phenotypes such as endothelial or immune cell dysfunction. The phenotype is also reminiscent of congenital defects of mitochondrial dysfunction that often precipitates as cardiomyopathy and heart failure in the absence of significant fibrosis.
Our transcriptional analyses identified alterations in two mitochondrial transcription factors (ERRgamma and PGC1alpha) in the female TAC KO heart. This may represent one mechanism by which OIP5-AS1 KO mice exhibit a worsening heart failure phenotype in a setting of pressure overload. ERRgamma was specifically downregulated in female OIP5-AS1 KO hearts following chronic cardiac pressure overload, a pathological setting that increases energy demand and thus aids in revealing a contractile dysfunction phenotype. ERRgamma is a transcriptional regulator of genes important for energy production including mitochondrial activity (Alaynick et al., 2007). Evidently, others have demonstrated that ERRgamma is an essential co-ordinator of cardiac metabolism and function (Wang et al., 2015).
Consistent with an effect on mitochondrial dysfunction, we also observed a significant reduction in PGC1alpha expression, a critical regulator of mitochondrial biogenesis pathways (Karkkainen et al., 2019). Given the significant loss of both PGC1alpha and ERRgamma, it is likely that the hearts of female OIP5-AS1 KO mice would have an impaired ability to generate energy and thus deprive the failing heart of ATP, driving a more severe phenotype in the absence of fibrosis. Indeed, this is important given that other studies have proposed that a 30-40% loss of PGC1alpha is sufficient to drive exacerbated heart failure (Jia et al., 2016; Chang et al., 2016; Wang et al., 2015). Moreover, similar to our findings, studies by Chang et al. (Chang et al., 2016) and Warren et al. (Warren et al., 2018) demonstrated that dystrophic- and Smyd1-driven cardiomyopathy respectively, were characterized by a dysregulation of mitochondrial biogenesis and function, exemplified by a downregulation of PGC1alpha.
Previous studies have demonstrated that lncRNAs can directly influence the expression of nuclear and mitochondrial encoded genes, with loss of lncRNAs leading to reduced mitochondrial activity and pathological phenotypes (Vendramin et al., 2018; Noh et al., 2016; Long et al., 2016). Thus, OIP5-AS1 may interact with transcriptional complexes harboring PGC1alpha or ERRgamma that directly regulate the expression of these genes. Our findings are consistent and supportive of the work recently published by Niu et al. who demonstrated that overexpression of OIP5-AS1 in rat hearts and cardiac cell cultures was protective against ischemia reperfusion injury resulting from transient myocardial infarction (Niu et al., 2020). They proposed that this protection was mediated by changes in mitochondrial function driven in part by alterations in the PGC1alpha axis. Thus these findings, combined with our new data generated in the KO mouse model, provide strong evidence for OIP5-AS1 regulating a conserved cardiac energetics program in the heart.
Regarding the specific mechanisms of OIP5-AS1 function, work by Kleaveland et al. (Kleaveland et al., 2018) demonstrated that OIP5-AS1 in the brain was predominantly localized to the cytoplasm (73%), where OIP5-AS1 (Cyrano) acted as a sponge for miRNA-7, subsequently decreasing the nuclear abundance of a circular RNA called Cdr1-AS. Others have demonstrated that OIP5-AS1 interacts with the competing endogenous RNA (ceRNA), HuR (Elavl1) in HeLa cells, alters the abundance of HuR (Elavl1) complex (Kim et al., 2016), or binds and effects the activity of miR-29a (Niu et al., 2020). Whether these mechanisms are active in our model in the mouse heart is not known; however our RNA-seq analysis demonstrates that miRNA-7, miR-29a, or Elavl1 were not altered in hearts of female KO mice. These discrepancies might be explained by divergent actions of OIP5-AS1 in striated muscle compared to other tissues, or that our transcriptome data were not optimized to capture miRs efficiently. It may also be related to the method in which OIP5-AS1 was deleted/silenced, or that our KO studies were performed in a pathological setting and the majority of other studies were not.
Regarding the mouse models, Kleaveland et al generated mice that deleted a specific region of exon 3 of Cyrano (OIP5-AS1) through cre-lox recombination, whereas we used CRISPR/Cas9 to delete the entire coding region of the gene. Kleaveland et al also demonstrated mild subclinical cis-effects in the brain of their KO model, namely a small but significant increase in the expression of the neighboring gene Nusap1, whereas we did not observe any change in Nusap1 expression, at least not in the heart. Each of these models comes with their own limitations. For example, in our model there is the possibility we have removed other transcripts present in the region; however given that no other genes or ncRNAs are annotated to the locus, this is unlikely. We may also have inadvertently removed important sites that are necessary for chromatin looping, or other long range chromatin interactions (Mumbach et al., 2017); however given that 91% of these interactions were found to be mediated by a promoter or enhancer, which are intact in our KO model, this is also not likely. With regard to the model by Kleaveland et al., many regions of Cyrano have been shown to be important for its function. Specifically, Smith et al. demonstrated that exon 3 of OIP5-AS1 is important for protein binding at multiple sites, and therefore removing only a small fraction of exon 3 is likely to impact very specific OIP5-AS1 mechanisms (which was likely the desired goal), which may not induce an overt phenotype. Similarly, the in vivo data presented by Niu et al. in the setting of MI in rats remains somewhat inconclusive because the authors use AAVs to overexpress OIP5-AS1, which have a well-accepted packaging limit of ∼5-6kb. Given that the major transcript of OIP5-AS1 (accounting for >90% of total OIP5-AS1 in the heart) is well above this limit at ∼9.5kb, this suggests that a truncated version or some other approach was engaged to overexpress OIP5-AS1 in their studies, clouding the interpretation of their findings.
Nevertheless, with each model there remain specific advantages and disadvantages and thus each of these approaches contribute unique knowledge to the field. An alternative approach to studying lncRNAs has been to insert loxP sites that flank the promoter region of the lncRNA, which would remove its ability to be transcriptionally regulated; however, no studies have yet been published using such an approach to study OIP5-AS1.
Another important distinction between our study and those from other groups is that we investigated both sexes and observed a phenotype only in female mice. We also studied these animals in a chronic disease setting that directly increases load on the heart, similar to that which occurs with hypertension and other cardiovascular conditions, which may be required to reveal specific phenotypes. Although not explicitly stated, it is assumed that previous groups only studied male OIP5-AS1 KO mice, possibly explaining in part the discrepancies observed in molecular phenotypes between our study and others.
In conclusion, the major findings from this study include (1) use of experimental and bioinformatic analyses of cardiac tissue and cells from both murine and human origin to confirm OIP5-AS1 as a cardiac muscle enriched lncRNA, (2) generation and characterization of a global mouse model with deletion of OIP5-AS1, (3) uncovering a critical role of OIP5-AS1 in the female heart in a setting of cardiac pressure overload, and (4) in-depth transcriptional analyses highlighting a dysregulation of mitochondrial genes in the female heart in the setting of pressure overload.
In summary, our study sheds light on our current understanding of sexual dimorphism observed in heart disease by demonstrating the female-sex restricted regulation of cardiac maladaptation by lncRNA OIP5-AS1. This is of particular importance considering the clinical observation that women are up to 4-fold more likely to develop heart failure in some settings than men, with a poor understanding of the molecular underpinnings of this observation. Our data suggest that future studies into sex disparities in heart failure progression in humans, might consider analyzing mitochondrial function and lncRNAs such as OIP5-AS1 as being potential regulators.
Limitations of the study
The current study investigates the impact of global deletion of OIP5-AS1 on heart function, and thus a limitation to this study is the inability to definitively demonstrate that CMs are the causal cell type. Separately, the current study was unable to directly measure mitochondrial function after TAC in either WT or KO animals, which would aid in understanding whether changes in mitochondrial transcriptional networks were indeed reflective of reduced mitochondrial activity. Last, although we provide insights into the mechanism by which OIP5-AS1 may influence heart function, we still have a poor understanding as to how OIP5-AS1 alters these pathways, and what proteins or RNAs interact with OIP5-AS1 to elicit such a response.
STAR★METHODS
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| ketamine hydrochloride | Ceva Animal Health | CAS 1867-66-9 |
| xylazine hydrochloride | Troy Laboratories | CAS 23076-35-9 |
| atropine sulfate monohydrate | Pfizer | CAS 5908-99-6 |
| atipamezole hydrochloride | Jurox | CAS 104075-48-1 |
| Isoflurane | Abbvie | CAS 26675-46-7 |
| Deposited Data | ||
| Collaborator's RNA-sequencing data: mouse (deposited) | NCBI Gene Expression Omnibus | GSE95764 |
| Collaborator's RNA-sequencing data: human (deposited) | NCBI Gene Expression Omnibus | GSE62913 |
| LV RNA-Seq gene counts generated in this publication | Figshare | DOI: https://doi.org/10.6084/m9.figshare.14538537 |
| Experimental Models: Organisms/Strains | ||
| Mouse: OIP5-AS1 KO; C57BL/6J | Baker Heart & Diabetes Institute | N/A |
| Oligonucleotides | ||
| qPCR primers | See Table S1 | N/A |
| Software and Algorithms | ||
| STAR Software | GitHub | V2.7.1 |
| Featurecounts | Bioconductor | N/A |
| edgeR and DESeq2 | Bioconductor | N/A |
| DAVID | NIH | v6.8 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Brian Drew (brian.drew@baker.edu.au)
Materials availability
The OIP5-AS1 KO mouse line generated in this study is available on request. This study did not generate any other new or unique reagents.
Data and code availability
The raw RNA-seq gene counts from LVs of WT and KO OIP5-AS1 mice are available at Figshare (https://doi.org/10.6084/m9.figshare.14538537). The NCBI Expression Omnibus accession codes for data from mouse and human cardiomyogenesis experiments are: GSE95764 and GSE95764.
Experimental models and subject details
Animals
OIP5-AS1 KO mice were generated by the Australian Phenomics Network with deletion being achieved by engaging CRISPR/Cas9 technology in single cell embryos, using gRNAs targeted to either end of the genomic sequence of the 1700020I14Rik (OIP5-AS1) gene. Edited C57BL/6J embryos were implanted into pseudo-pregnant C57BL/6J female mice, and founder offspring were sequenced to confirm genetic modification. Founders were subsequently mated to confirm germline editing and bred for >5 generations to C57BL/6J mice that had not been genetically manipulated. All animals were housed at 22°C on a 12hr light/dark cycle with ad libitum access to food (standard rat and mouse chow, Specialty feeds, Australia) and water, with cages changed weekly. Experiments were approved by the Alfred Research Alliance (ARA) Animal Ethics Committee (E/1769/2017/B), and performed in accordance with the NH&MRC of Australia guidelines for the care and use of laboratory animals.
Method details
Animal experiments
At four weeks of age, wild type (WT) and KO male and female mice were weaned from the Dam and placed in group housing conditions. Mice were housed until they reached an age of 7-10 weeks, at which time they were selected to achieve a weight matched comparison before undergoing surgery. Because male mice are commonly bigger than female mice, male mice were generally 1-2 weeks younger than females at time of surgery.
Transverse aortic constriction
Transverse restriction of aortic outflow was performed on 7-10 week old animals as previously described (Gao et al., 2005; Kiriazis et al., 2008). Briefly, animals were anesthetized with a mixture of ketamine, xylazine, and atropine (10, 2, and 0.12 mg/100 g, respectively, ip), intubated via the oral cavity, and ventilated. Following a sternotomy, the transverse aorta between the right innominate and left carotid arteries was dissected and banded with a 26-gauge blunt needle using a 5-0 silk suture. The probe was then removed allowing for the suture to remain in place and thus permit chronic constriction of the aorta. With the use of this diameter needle, aortic diameter was predicted to be reduced by 50–55%, which leads to an approximately 70% reduction in cross-sectional area. This procedure was performed similarly on both male and female mice between the ages of 7-10 weeks, which allowed us to weight match the animals and thus assume a mouse of a similar weight would have a similar sized aorta and percent constriction. Previously, other studies have used mice of the same age when comparing sex differences, meaning that the bigger male mice would likely receive a more severe aortic restriction in age-matched animals, resulting in more severe disease. Sham-operated mice underwent the same surgical procedure as TAC treated mice, but no suture was place around the aorta to restrict flow. Mice were recovered with antisedan and then followed for 8 weeks to monitor the development of HF.
Intra-aortic and LV pressure analysis
Blood pressure and intra-cardiac pressure was assessed by a catheter placed into the right carotid artery (proximal to the stenotic site) and advanced into the LV as previously described (Du et al., 2000). Mice were anesthetized using isoflurane at 2-4% and placed in the supine position on a heating pad, and the right main carotid artery was dissected. A micro-tipped transducer catheter (1.4F, Millar Instrument Co) was inserted into the artery and measurements including aortic blood pressure and LV pressures were recorded.
Echocardiography
Echocardiography was performed on mice anesthetized with isoflurane (1.75%) at baseline and at the end of the 8-week study using a 15-MHz linear transducer L15-7io with a Philips iE33 Ultrasound Machine (North Ryde, NSW, Australia). Data were analyzed and verified by two independent researchers according to QC procedures and validation measures as outlined previously (Donner et al., 2018).
Quantitative PCR (qPCR)
RNA for qPCR analysis was isolated from tissues as previously described (Bond et al., 2019a, 2019b). Briefly, tissues were homogenized in RNAzol reagent and precipitated using isopropanol. cDNA was generated from 1μg of RNA using MMLV reverse transcriptase (Invitrogen) according to the manufacturer's instructions. qPCR was performed on 10ng of cDNA using the SYBR-green method on a QuantStudio 7 Flex (ThermoFisher Scientific) using gene specific primers. Quantification of a given gene was expressed by the relative mRNA level compared with control, which was calculated after normalization to the housekeeping gene 36B4 (Rplp0) using the delta-CT method. Primers were designed to span exon-exon junctions where possible and were tested for specificity using BLAST (Basic Local Alignment Search Tool; National Centre for Biotechnology Information) (see Table S1 for Primer Details). In order to account for the two main variants of OIP5-AS1 expressed in most cell types, including striated muscles, we designed 4 sets of primers. Two of these primer sets recognized all variants, whilst another two recognized only the most abundant, and longest variant of OIP5-AS1 (known as Oip5os1-202 in mus musculus). Amplification of a single amplicon was estimated from melt curve analysis, ensuring only a single peak and an expected temperature dissociation profile were observed.
RNA-sequencing analysis
RNA was isolated from LV tissue using RNAzol reagent and purified using columns according to the manufacturer's instructions (Zymo Research). RNA integrity was evaluated using the Agilent Tape Station 2200 according to the manufacturer's instructions (Agilent). RNA libraries were prepared using Kapa Stranded RNA-seq kits on samples with a RIN>0.8 according to the manufacturer's instructions (Roche). Library quantities were determined using QUBIT (ThermoFisher) and equal amounts of all 24 libraries were pooled and run across 2 lanes using an Illumina HiSeq 2500 Sequencer. FASTQ sequencing data were demultiplexed and aligned to the Mus Musculus mm10 genome using STAR Software (V2.7.1) with default parameters. Mapped reads were counted using Featurecounts against an mm10 reference file. QC and batch effect analyses were performed using edgeR and DEseq2 bioconductor packages in R.
Pathway and network analysis
Enrichment analysis and gene ontology was performed using the Database for Annotation, Visualization and Integrated Discovery (DAVID v6.8) hosted by the National Institute of Allergy and Infectious Diseases (NIAID), NIH, USA (Huang Da et al., 2009). Cluster analysis from human iPSC-derived cardiomyocyte RNA-sequencing was derived by analyzing gene sets correlated with OIP5-AS1 expression. Cluster analysis from RNA-sequencing data was derived from genes sets significantly altered between WT and KO animals. These data sets were separately analyzed using GSEA (v3.0) (Mootha et al., 2003; Subramanian et al., 2005) and functional enrichment analysis was mapped using g:Profiler (v95_eg42_p13_f6e58b9) with g:SCS multiple testing correction method applied with a significance threshold of p < 0.05 (Raudvere et al., 2019; Reimand et al., 2007). Enrichment and cluster analysis were mapped to a network of the curated MSigDB C5 gene set collection (nodes) (Subramanian et al., 2005).
Histology
LV tissues were fixed in paraformaldehyde overnight before being placed in 70% ethanol. Tissues were subsequently embedded in paraffin blocks and 5μm sections were cut and mounted on glass slides. After dewaxing and hydration, sections were stained using picrosirius red (Sigma) to visualize cell collagen and fibrosis abundance. Slides images were captured using Olympus Slide scanner VS120 (Olympus, Japan) and viewed in the supplied program (OlyVIA Build 13771, Olympus, Japan). Whole tissue slides were quantified based on threshold analysis in Fiji (Schindelin et al., 2012).
Primary endpoints and data inclusion/exclusion criteria
Our primary end point was to test if OIP5-AS1 KO affected heart function in either male or female mice, compared to their relative WT controls. For in vitro and basal animal phenotyping data, individual data points were excluded if they were technically implausible or a methodological error had resulted in a spurious outcome. Analyses from animals following TAC surgery were excluded if animals were found dead from acute heart failure overnight (samples compromised), did not recover from surgery (2 female KO mice post-TAC) or technical/analytical problems were identified (compromised RNA, failed analysis, improper tissue collection, equipment failure). For echocardiography, data were excluded if heart rates were outside of 450-650bpm or animals were too ill to undergo anesthesia. Echocardiography outputs were only included in final analysis if we had a full pre- and post-surgery data set, meaning that numbers were lower in groups where TAC surgery induced a more severe heart failure (i.e. female KO mice).
Quantification and statistical analysis
Our primary endpoint was to identify if there were differences between WT and KO mice within each sex (not specifically between sexes), therefore sample size was determined accordingly. All animal and laboratory data underwent blinding and randomization at time of collection and during technical analysis. Data were expressed as mean ± standard error of the mean (SEM), unless otherwise stated. All statistical analyses of animal and laboratory based experiments were performed using PRISM7 software. Normally distributed data in cell culture experiments were compared by paired student’s t-test whilst animal studies were analyzed by one-way and two-way ANOVA with testing for multiple comparisons (Dunnet’s) between WT and KO animals of the same sex. As our primary endpoint was not to determine if male and females were statistically different from each other, we did not perform multiple comparison testing with all four groups. In these analyses, a p-value of p < 0.05 was considered statistically significant.
Acknowledgments
We acknowledge funding support from the Victorian State Government OIS program to Baker Heart & Diabetes Institute (BHDI). These studies were supported in part by funding from the National Health & Medical Research Council (NHMRC) of Australia to B.G.D. and J.R.M. (APP1127336). B.G.D. and A.C.C. received funding from the National Heart Foundation of Australia, via the Future Leader Fellowship Scheme (101789 and 100067, respectively). J.R.M. and P.G. are Research Fellows of the NHMRC (APP1078985 and APP1117835, respectively). We acknowledge the use of facilities and technical assistance from the Monash Histology Platform, Department of Anatomy and Developmental Biology, Monash University, and use of the gene set enrichment analysis (GSEA) software, and Molecular Signature Database (MSigDB) (Subramanian et al., 2005). We thank all members of the Cardiac Hypertrophy, MMA, and LMCD laboratories at BHDI for their ongoing contributions.
Author contributions
B.G.D. and J.R.M. conceived the study and wrote the manuscript. B.G.D., J.R.M., A.C.C., and X.J.D. designed the mouse studies and directed experimental analysis. B.G.D., J.R.M., A.C.C., A.Z., S.L., Y.L., S.T.B., S.C.M., E.A.M.G., and S.R.C. analyzed data and interpreted findings. B.G.D., S.L., J.R.M., H.K., Y.L., A.Z., D.G.D., X.M.G., and X.J.D. performed animal experiments and analyzed data. B.G.D., J.R.M., A.C.C., T.D.C., T.Q.A.V., E.J.T., J.E.H., G.Q.R., E.P., P.G., and M.I. provided resources, data analysis, experimental guidance, and reagents. All authors had the opportunity to read, edit, and identify points for clarification before submission.
Declaration of interests
The authors declare they have no conflict of interest.
Published: June 25, 202
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102537.
Contributor Information
Julie R. McMullen, Email: julie.mcmullen@baker.edu.au.
Brian G. Drew, Email: brian.drew@baker.edu.au.
Supplemental information
Nucleotide sequence (5’ – 3′) of the primers used for quantitative PCR analysis using the SYBR green method.
References
- Alaynick W.A., Kondo R.P., Xie W., He W., Dufour C.R., Downes M., Jonker J.W., Giles W., Naviaux R.K., Giguere V., Evans R.M. ERRgamma directs and maintains the transition to oxidative metabolism in the postnatal heart. Cell Metab. 2007;6:13–24. doi: 10.1016/j.cmet.2007.06.007. [DOI] [PubMed] [Google Scholar]
- Bernardo B.C., Gao X.M., Winbanks C.E., Boey E.J., Tham Y.K., Kiriazis H., Gregorevic P., Obad S., Kauppinen S., Du X.J. Therapeutic inhibition of the miR-34 family attenuates pathological cardiac remodeling and improves heart function. Proc. Natl. Acad. Sci. U S A. 2012;109:17615–17620. doi: 10.1073/pnas.1206432109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardo B.C., Nguyen S.S., Winbanks C.E., Gao X.M., Boey E.J., Tham Y.K., Kiriazis H., Ooi J.Y., Porrello E.R., Igoor S. Therapeutic silencing of miR-652 restores heart function and attenuates adverse remodeling in a setting of established pathological hypertrophy. FASEB J. 2014;28:5097–5110. doi: 10.1096/fj.14-253856. [DOI] [PubMed] [Google Scholar]
- Bond S.T., Kim J., Calkin A.C., Drew B.G. The antioxidant moiety of MitoQ imparts minimal metabolic effects in adipose tissue of high fat fed mice. Front. Physiol. 2019;10:543. doi: 10.3389/fphys.2019.00543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond S.T., Moody S.C., Liu Y., Civelek M., Villanueva C.J., Gregorevic P., Kingwell B.A., Hevener A.L., Lusis A.J., Henstridge D.C. The E3 ligase MARCH5 is a PPARgamma target gene that regulates mitochondria and metabolism in adipocytes. Am. J. Physiol. Endocrinol. Metab. 2019;316:E293–E304. doi: 10.1152/ajpendo.00394.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo S.E., Clauser K.R., Mootha V.K. MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016;44:D1251–D1257. doi: 10.1093/nar/gkv1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cech T.R., Steitz J.A. The noncoding RNA revolution-trashing old rules to forge new ones. Cell. 2014;157:77–94. doi: 10.1016/j.cell.2014.03.008. [DOI] [PubMed] [Google Scholar]
- Chang A.C., Ong S.G., Lagory E.L., Kraft P.E., Giaccia A.J., Wu J.C., Blau H.M. Telomere shortening and metabolic compromise underlie dystrophic cardiomyopathy. Proc. Natl. Acad. Sci. U S A. 2016;113:13120–13125. doi: 10.1073/pnas.1615340113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.L., Walton K.L., Hagg A., Colgan T.D., Johnson K., Qian H., Gregorevic P., Harrison C.A. Specific targeting of TGF-beta family ligands demonstrates distinct roles in the regulation of muscle mass in health and disease. Proc. Natl. Acad. Sci. U S A. 2017;114:E5266–E5275. doi: 10.1073/pnas.1620013114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colley S.M., Leedman P.J. Steroid Receptor RNA Activator - a nuclear receptor coregulator with multiple partners: insights and challenges. Biochimie. 2011;93:1966–1972. doi: 10.1016/j.biochi.2011.07.004. [DOI] [PubMed] [Google Scholar]
- Dey B.K., Pfeifer K., Dutta A. The H19 long noncoding RNA gives rise to microRNAs miR-675-3p and miR-675-5p to promote skeletal muscle differentiation and regeneration. Genes Dev. 2014;28:491–501. doi: 10.1101/gad.234419.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diederichs S. The four dimensions of noncoding RNA conservation. Trends Genet. 2014;30:121–123. doi: 10.1016/j.tig.2014.01.004. [DOI] [PubMed] [Google Scholar]
- Donner D.G., Kiriazis H., Du X.J., Marwick T.H., Mcmullen J.R. Improving the quality of preclinical research echocardiography: observations, training, and guidelines for measurement. Am. J. Physiol. Heart Circ. Physiol. 2018;315:H58–H70. doi: 10.1152/ajpheart.00157.2018. [DOI] [PubMed] [Google Scholar]
- Du X.J., Autelitano D.J., Dilley R.J., Wang B., Dart A.M., Woodcock E.A. beta(2)-adrenergic receptor overexpression exacerbates development of heart failure after aortic stenosis. Circulation. 2000;101:71–77. doi: 10.1161/01.cir.101.1.71. [DOI] [PubMed] [Google Scholar]
- Gao X.M., Kiriazis H., Moore X.L., Feng X.H., Sheppard K., Dart A., Du X.J. Regression of pressure overload-induced left ventricular hypertrophy in mice. Am. J. Physiol. Heart Circ. Physiol. 2005;288:H2702–H2707. doi: 10.1152/ajpheart.00836.2004. [DOI] [PubMed] [Google Scholar]
- Grote P., Wittler L., Hendrix D., Koch F., Wahrisch S., Beisaw A., Macura K., Blass G., Kellis M., Werber M., Herrmann B.G. The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev. Cell. 2013;24:206–214. doi: 10.1016/j.devcel.2012.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han P., Li W., Lin C.H., Yang J., Shang C., Nurnberg S.T., Jin K.K., Xu W., Lin C.Y., Lin C.J. A long noncoding RNA protects the heart from pathological hypertrophy. Nature. 2014;514:102–106. doi: 10.1038/nature13596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Da W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Ishii N., Ozaki K., Sato H., Mizuno H., Saito S., Takahashi A., Miyamoto Y., Ikegawa S., Kamatani N., Hori M. Identification of a novel non-coding RNA, MIAT, that confers risk of myocardial infarction. J. Hum. Genet. 2006;51:1087–1099. doi: 10.1007/s10038-006-0070-9. [DOI] [PubMed] [Google Scholar]
- Jia Y., Chang H.C., Schipma M.J., Liu J., Shete V., Liu N., Sato T., Thorp E.B., Barger P.M., Zhu Y.J. Cardiomyocyte-specific ablation of Med1 subunit of the mediator complex causes lethal dilated cardiomyopathy in mice. PLoS One. 2016;11:e0160755. doi: 10.1371/journal.pone.0160755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karkkainen O., Tuomainen T., Mutikainen M., Lehtonen M., Ruas J.L., Hanhineva K., Tavi P. Heart specific PGC-1alpha deletion identifies metabolome of cardiac restricted metabolic heart failure. Cardiovasc. Res. 2019;115:107–118. doi: 10.1093/cvr/cvy155. [DOI] [PubMed] [Google Scholar]
- Kashi K., Henderson L., Bonetti A., Carninci P. Discovery and functional analysis of lncRNAs: methodologies to investigate an uncharacterized transcriptome. Biochim. Biophys. Acta. 2016;1859:3–15. doi: 10.1016/j.bbagrm.2015.10.010. [DOI] [PubMed] [Google Scholar]
- Kennedy T.L., Swiderski K., Murphy K.T., Gehrig S.M., Curl C.L., Chandramouli C., Febbraio M.A., Delbridge L.M., Koopman R., Lynch G.S. BGP-15 improves aspects of the dystrophic pathology in mdx and dko mice with differing efficacies in heart and skeletal muscle. Am. J. Pathol. 2016;186:3246–3260. doi: 10.1016/j.ajpath.2016.08.008. [DOI] [PubMed] [Google Scholar]
- Kim J., Abdelmohsen K., Yang X., De S., Grammatikakis I., Noh J.H., Gorospe M. LncRNA OIP5-AS1/cyrano sponges RNA-binding protein HuR. Nucleic Acids Res. 2016;44:2378–2392. doi: 10.1093/nar/gkw017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiriazis H., Wang K., Xu Q., Gao X.M., Ming Z., Su Y., Moore X.L., Lambert G., Gibbs M.E., Dart A.M., Du X.J. Knockout of beta(1)- and beta(2)-adrenoceptors attenuates pressure overload-induced cardiac hypertrophy and fibrosis. Br. J. Pharmacol. 2008;153:684–692. doi: 10.1038/sj.bjp.0707622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klattenhoff C.A., Scheuermann J.C., Surface L.E., Bradley R.K., Fields P.A., Steinhauser M.L., Ding H., Butty V.L., Torrey L., Haas S. Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell. 2013;152:570–583. doi: 10.1016/j.cell.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleaveland B., Shi C.Y., Stefano J., Bartel D.P. A network of noncoding regulatory RNAs acts in the mammalian brain. Cell. 2018;174:350–362.e17. doi: 10.1016/j.cell.2018.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumarswamy R., Bauters C., Volkmann I., Maury F., Fetisch J., Holzmann A., Lemesle G., De Groote P., Pinet F., Thum T. Circulating long noncoding RNA, LIPCAR, predicts survival in patients with heart failure. Circ. Res. 2014;114:1569–1575. doi: 10.1161/CIRCRESAHA.114.303915. [DOI] [PubMed] [Google Scholar]
- Long J., Badal S.S., Ye Z., Wang Y., Ayanga B.A., Galvan D.L., Green N.H., Chang B.H., Overbeek P.A., Danesh F.R. Long noncoding RNA Tug1 regulates mitochondrial bioenergetics in diabetic nephropathy. J. Clin. Invest. 2016;126:4205–4218. doi: 10.1172/JCI87927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallory A.C., Shkumatava A. LncRNAs in vertebrates: advances and challenges. Biochimie. 2015;117:3–14. doi: 10.1016/j.biochi.2015.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcmullen J.R., Amirahmadi F., Woodcock E.A., Schinke-Braun M., Bouwman R.D., Hewitt K.A., Mollica J.P., Zhang L., Zhang Y., Shioi T. Protective effects of exercise and phosphoinositide 3-kinase(p110alpha) signaling in dilated and hypertrophic cardiomyopathy. Proc. Natl. Acad. Sci. U S A. 2007;104:612–617. doi: 10.1073/pnas.0606663104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMullen J.R., Drew B.G. Long non-coding RNAs (lncRNAs) in skeletal and cardiac muscle: potential therapeutic and diagnostic targets? Clin. Sci. (Lond) 2016;130:2245–2256. doi: 10.1042/CS20160244. [DOI] [PubMed] [Google Scholar]
- McMullen J.R., Sherwood M.C., Tarnavski O., Zhang L., Dorfman A.L., Shioi T., Izumo S. Inhibition of mTOR signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circulation. 2004;109:3050–3055. doi: 10.1161/01.CIR.0000130641.08705.45. [DOI] [PubMed] [Google Scholar]
- Mootha V.K., Lindgren C.M., Eriksson K.F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstrale M., Laurila E. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Mumbach M.R., Satpathy A.T., Boyle E.A., Dai C., Gowen B.G., Cho S.W., Nguyen M.L., Rubin A.J., Granja J.M., Kazane K.R. Enhancer connectome in primary human cells identifies target genes of disease-associated DNA elements. Nat. Genet. 2017;49:1602–1612. doi: 10.1038/ng.3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu X., Pu S., Ling C., Xu J., Wang J., Sun S., Yao Y., Zhang Z. lncRNA Oip5-as1 attenuates myocardial ischaemia/reperfusion injury by sponging miR-29a to activate the SIRT1/AMPK/PGC1alpha pathway. Cell Prolif. 2020;53:e12818. doi: 10.1111/cpr.12818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noh J.H., Kim K.M., Abdelmohsen K., Yoon J.H., Panda A.C., Munk R., Kim J., Curtis J., Moad C.A., Wohler C.M. HuR and GRSF1 modulate the nuclear export and mitochondrial localization of the lncRNA RMRP. Genes Dev. 2016;30:1224–1239. doi: 10.1101/gad.276022.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ounzain S., Micheletti R., Beckmann T., Schroen B., Alexanian M., Pezzuto I., Crippa S., Nemir M., Sarre A., Johnson R. Genome-wide profiling of the cardiac transcriptome after myocardial infarction identifies novel heart-specific long non-coding RNAs. Eur. Heart J. 2015;36:353–368a. doi: 10.1093/eurheartj/ehu180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quaife-Ryan G.A., Sim C.B., Ziemann M., Kaspi A., Rafehi H., Ramialison M., El-Osta A., Hudson J.E., Porrello E.R. Multicellular transcriptional analysis of mammalian heart regeneration. Circulation. 2017;136:1123–1139. doi: 10.1161/CIRCULATIONAHA.117.028252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raudvere U., Kolberg L., Kuzmin I., Arak T., Adler P., Peterson H., Vilo J. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update) Nucleic Acids Res. 2019;47:W191–W198. doi: 10.1093/nar/gkz369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimand J., Kull M., Peterson H., Hansen J., Vilo J. g:Profiler--a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res. 2007;35:W193–W200. doi: 10.1093/nar/gkm226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas V., Drew B.G., Zhou Z., Phun J., Kalajian N.Y., Soleymani T., Daraei P., Widjaja K., Wanagat J., De Aguiar Vallim T.Q. Skeletal muscle action of estrogen receptor alpha is critical for the maintenance of mitochondrial function and metabolic homeostasis in females. Sci. Transl. Med. 2016;8:334ra54. doi: 10.1126/scitranslmed.aad3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith K.N., Starmer J., Magnuson T. Interactome determination of a long noncoding RNA implicated in embryonic stem cell self-renewal. Sci. Rep. 2018;8:17568. doi: 10.1038/s41598-018-34864-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith K.N., Starmer J., Miller S.C., Sethupathy P., Magnuson T. Long noncoding RNA moderates MicroRNA activity to maintain self-renewal in embryonic stem cells. Stem Cell Rep. 2017;9:108–121. doi: 10.1016/j.stemcr.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Laurent G., Wahlestedt C., Kapranov P. The Landscape of long noncoding RNA classification. Trends Genet. 2015;31:239–251. doi: 10.1016/j.tig.2015.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorrez L., Van Deun K., Tranchevent L.C., Van Lommel L., Engelen K., Marchal K., Moreau Y., Van Mechelen I., Schuit F. Using ribosomal protein genes as reference: a tale of caution. PLoS One. 2008;3:e1854. doi: 10.1371/journal.pone.0001854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitsky I., Bartel D.P. lincRNAs: genomics, evolution, and mechanisms. Cell. 2013;154:26–46. doi: 10.1016/j.cell.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitsky I., Shkumatava A., Jan C.H., Sive H., Bartel D.P. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell. 2011;147:1537–1550. doi: 10.1016/j.cell.2011.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vendramin R., Verheyden Y., Ishikawa H., Goedert L., Nicolas E., Saraf K., Armaos A., Delli Ponti R., Izumikawa K., Mestdagh P. SAMMSON fosters cancer cell fitness by concertedly enhancing mitochondrial and cytosolic translation. Nat. Struct. Mol. Biol. 2018;25:1035–1046. doi: 10.1038/s41594-018-0143-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K., Liu F., Zhou L.Y., Long B., Yuan S.M., Wang Y., Liu C.Y., Sun T., Zhang X.J., Li P.F. The long noncoding RNA CHRF regulates cardiac hypertrophy by targeting miR-489. Circ. Res. 2014;114:1377–1388. doi: 10.1161/CIRCRESAHA.114.302476. [DOI] [PubMed] [Google Scholar]
- Wang T., Mcdonald C., Petrenko N.B., Leblanc M., Wang T., Giguere V., Evans R.M., Patel V.V., Pei L. Estrogen-related receptor alpha (ERRalpha) and ERRgamma are essential coordinators of cardiac metabolism and function. Mol. Cell. Biol. 2015;35:1281–1298. doi: 10.1128/MCB.01156-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren J.S., Tracy C.M., Miller M.R., Makaju A., Szulik M.W., Oka S.I., Yuzyuk T.N., Cox J.E., Kumar A., Lozier B.K. Histone methyltransferase Smyd1 regulates mitochondrial energetics in the heart. Proc. Natl. Acad. Sci. U S A. 2018;115:E7871–E7880. doi: 10.1073/pnas.1800680115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts R., Johnsen V.L., Shearer J., Hittel D.S. Myostatin-induced inhibition of the long noncoding RNA Malat1 is associated with decreased myogenesis. Am. J. Physiol. Cell Physiol. 2013;304:C995–C1001. doi: 10.1152/ajpcell.00392.2012. [DOI] [PubMed] [Google Scholar]
- Zheng D., Wang B., Zhu X., Hu J., Sun J., Xuan J., Ge Z. LncRNA OIP5-AS1 inhibits osteoblast differentiation of valve interstitial cells via miR-137/TWIST11 axis. Biochem. Biophys. Res. Commun. 2019;511:826–832. doi: 10.1016/j.bbrc.2019.02.109. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Nucleotide sequence (5’ – 3′) of the primers used for quantitative PCR analysis using the SYBR green method.
Data Availability Statement
The raw RNA-seq gene counts from LVs of WT and KO OIP5-AS1 mice are available at Figshare (https://doi.org/10.6084/m9.figshare.14538537). The NCBI Expression Omnibus accession codes for data from mouse and human cardiomyogenesis experiments are: GSE95764 and GSE95764.