

Abstract
Although there are exceptions and outliers, T cell functional responses generally correlate with the affinity of a TCR for a peptide/MHC complex. In one recently described outlier case, the most promising clinical candidate in a series of TCRs specific for the gp100209 melanoma antigen bound with the weakest solution affinity and produced the least amount of cytokine in vitro. Hypotheses for this outlier behavior included unusual cytokine expression patterns arising from an atypical TCR binding geometry. Studying this instance in more detail, we found here that outlier behavior is attributable not to unusual cytokine patterns or TCR binding, but the use of a position 2 anchor-modified peptide variant in in vitro experiments instead of the wild type antigen that is present in vivo. Although the anchor-modified variant has been widely used in basic and clinical immunology as a surrogate for the wild type peptide, prior work has shown that TCRs can clearly distinguish between the two. We show that when this differential recognition is accounted for, the functional properties of gp100209-specific TCRs track with their affinity towards the peptide/MHC complex. Beyond demonstrating the correlates with T cell function for a clinically relevant TCR, our results provide important considerations for selection of TCRs for immunotherapy and the use of modified peptides in immunology.
Keywords: T cell receptor, binding affinity, gp100, peptide, heteroclitic
Introduction
The properties which influence the functional responses of T cells are of considerable interest. This is particularly the case when selecting T cells or T cell receptors (TCR) for use in adoptive cell therapy or other forms of T cell-based immunotherapy. Although there are known exceptions and outliers (e.g., 1–4), in general T cell functional responses correlate well with the affinity of a TCR for a peptide/MHC complex5,6. Although early studies relating TCR affinity to T cell function were usually performed using in vitro studies, more recent work has confirmed the relationship in vivo. For example, the DMF4 and DMF5 TCRs specific for the MART-1 melanoma antigens have been examined in human clinical trials, with the higher-affinity DMF5 TCR eliciting superior responses but also evidence of on-target autoimmunity7,8. Studies in mouse models have shown similar results9,10.
Recently, Eby et al. studied a series of three TCRs specific for the gp100209 melanoma antigen (sequence ITDQVPFSV) presented by the common human class I MHC protein HLA-A*02:01 (referred to as HLA-A2)11. One of these TCRs, named SILv44, was obtained from a patient with the autoimmune condition vitiligo12. Vitiligo is of interest in melanoma immunotherapy due to a connection between spontaneous skin depigmentation and tumor rejection13–15. Compared to the other gp100209-specific TCRs, the SILv44 vitiligo TCR elicited the strongest antitumor immune responses in a murine model of human melanoma. Intriguingly though, of the three TCRs, SILv44 showed the weakest binding affinity when measured against the anchor-modified gp100209–2M peptide presented by HLA-A2 (sequence IMDQVPFSV). T cells transduced with SILv44 also produced the least amount of IFN-γ in response to gp100209–2M. The gp100209–2M peptide, which relies on a threonine to methionine substitution at position 2 to enhance binding to HLA-A2 resulting in more stable antigen presentation16–18, has been widely used as a surrogate for the wild type (WT) gp100209 melanoma antigen in basic and clinical immunology19–22. We previously showed that the static crystallographic structures of the WT and methionine modified peptides bound to HLA-A2 were identical within the limits of protein crystallography23. Because of the ensuing presumption that the WT and modified peptides are recognized identically by TCRs, Eby and colleagues proposed a mechanism through which the SILv44 TCR results in enhanced tumor control by eliciting greater amounts of the IL-17a cytokine, possibly in response to altered immune functions resulting from an atypical TCR binding geometry11.
Recently, however, we showed that, despite the structural identity of the two peptide/HLA-A2 complexes, multiple TCRs, including SILv44, do not perceive the WT and anchor-modified variants of the gp100209 peptide identically. Due to alterations in peptide/MHC motions that occur upon peptide anchor modification, different TCRs sense the gp100209 and gp100209–2M peptides presented by HLA-A2 with as much as a 40-fold difference in binding affinity24. Similar, albeit smaller, effects have been seen with TCRs that recognize the MART-1 and NY-ESO-1 tumor antigens25–28. These biochemical differences have been reflected in functional and clinical studies19,26,29–31. Thus, interpretation of the tumor rejection studies with the SILv44 TCR could be skewed by the widespread but incorrect presumption that the WT and anchor-modified variants of the gp100209 peptide are recognized the same regardless of TCR.
Here we explored how SILv44 and other gp100209-specific TCRs sense peptide anchor modification in greater detail. We found that of the three TCRs tested for tumor rejection, SILv44 indeed recognizes the WT gp100209 peptide with the strongest affinity and lowest EC50 in functional studies. Thus, when accounting for TCR sensitivity to peptide anchor modification, the greater potency of the SILv44 TCR towards melanoma tumors presenting the WT peptide can be rationalized based on established relationships between T cell function and binding affinity. The results are fully consistent with findings in other systems that relate the potency of TCR-transduced T cells to the affinity of the TCR towards its target antigen, without having to invoke mechanisms relying on differential cytokine expression patterns or altered signaling from unusual receptor binding properties that would further complicate selection of TCRs for immunotherapy.
Results
TCR binding affinity towards gp100209 peptide variants correlates with T cell function
We previously showed that the SILv44, T4H2, and R6C12 TCRs distinguish between WT and p2-modified variants of the gp100209 peptide presented by HLA-A224. Sensitivity to anchor modification was observed in both binding and functional experiments, with EC50 values for co-culture experiments correlating well with TCR binding affinities. These experiments were completed with the WT gp100209 peptide and its most studied anchor-modified variant, Thr2→Methionine (T2Met). To further explore the relationship between TCR affinity and function in this system, we performed functional measurements with two other p2-modified variants: Thr2→leucine (T2Leu) and Thr2→norleucine (T2Nle). These variants were previously selected to study how the chemistry of the p2 amino acid influences TCR binding24. As before, we measured EC50 values for IL-2 release from TCR-transduced CD8+ Jurkat cells (Fig. 1A–C). Using our previously published binding affinity values for each TCR towards each peptide variant, this expanded dataset confirmed a good correlation between binding affinity (expressed in terms of binding free energy) and functional avidity across all three TCRs (Fig. 1D; Table 1).
Figure 1. T cell potency correlates with TCR affinity for gp100209-specific TCRs.

A, B, C) Determination of EC50 values for IL-2 release from TCR-transduced CD8+ Jurkat T cells when recognizing the T2Leu and T2Nle gp100209 variants presented by HLA-A2+ T2 cells for the SILv44, R6C12, and T4H2 TCRs. These analyses complete a series that includes the WT and the T2Met peptides published previously, described in Table 124. Individual data points and error bars are averages and standard deviations of four measurements. Values and errors are from a nonlinear fit to all four datasets. D) Comparison of EC50 vs. TCR binding affinities, reported as binding free energies (ΔG° = RT ln KD), for the SILv44, T4H2, and R6C12 TCRs. Error bars are propagated error from panels A,B, C and values in ref.24.
Table 1.
Binding affinities and EC50 values for TCR recognition of gp100209 peptide and position 2 variants.
| gp100209 peptide variant | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT | T2Met | T2Leu | T2Nle | |||||||||
| Kda | ΔG°b | EC50c | Kda | ΔG°b | EC50c | Kda | ΔG°b | EC50c | Kda | ΔG°b | EC50c | |
| SILv44 | 65 ± 16 | −5.7 ± 0.1 | 2.1 ± 0.4 | 210 ± 65 | −5.0 ± 0.2 | 15 ± 3 | 100 ± 26 | −5.5 ± 0.2 | 1.4 ± 0.3 | 153 ± 24 | −5.2 ± 0.1 | 29 ± 6 |
| R6C12 | 62 ± 13 | −5.7 ± 0.1 | 6.5 ± 1.5 | 31 ± 7 | −6.2 ± 0.1 | 0.28 ± 0.08 | 128 ± 9 | −5.3 ± 0.1 | 11 ± 3 | 79 ± 14 | −5.6 ± 0.1 | 3.1 ± 0.9 |
| T4H2 | 72 ± 20 | −5.7 ± 0.2 | 26 ± 11 | 1.8 ± 0.5 | −7.9 ± 0.2 | 0.02 ± 0.01 | 68 ± 27 | −5.7 ± 0.2 | 0.3 ± 0.1 | 17 ± 6 | −6.6 ± 0.2 | 0.06 ± 0.02 |
KD values in μM are from ref. 24. Values are averages and standard deviations of at least three experiments, determined using SPR with the TCR amine coupled to the sensor surface.
Binding free energies in kcal/mol at 25 °C; calculated from KD values using ΔG° = RTlnKD.
EC50 values in μM. Values and errors are from simultaneous analysis of four measurements.
In addition to indicating a relationship between TCR affinity and signaling output, the functional data with cells expressing the three TCRs and panel of gp100209 peptides revealed a wide dispersion in EC50 values and a notable variation in the order of peptide sensitivity. The SILv44 TCR was most sensitive to the WT gp100209 peptide, whereas the T4H2 and R6C12 TCRs were most sensitive to the T2Met variant. Additionally, SILv44 was 3–5 fold more sensitive than T4H2 and R6C12 to the WT peptide. These results are consistent with the greater sensitivity of SILv44-transduced bulk PBMC to the WT peptide, as well as the superior control of melanoma tumors expressing the WT gp100 protein in mice by SILv44-expressing T cells11.
We next investigated the ability of peptides to activate IL-17a signaling in TCR-transduced Jurkat cells. Enhanced production of IL-17a by SILv44 at low peptide concentrations was previously proposed as a mechanism for tumor control, although SILv44 only showed enhanced production of IL-17a in one out of three co-culture experiments11. As Jurkat cells lack the ability to produce several cytokines, including IL-17a, we developed a CD8+ Jurkat cell line that produces firefly luciferase under the control of the IL-17a promotor. While less sensitive than a traditional ELISA, the approach still allowed us to examine if and how activation of the IL-17a pathway varied between the three TCRs and thus compare our results with the previously published data11. Activation appeared similar between the three TCRs in response to the WT peptide, although due to the low sensitivity of the reporter system we could not determine EC50 values (Fig. 2). Despite the low sensitivity of the assay, the IL-17a pathway was clearly activated more strongly for T4H2 recognition of the T2Met peptide, consistent with the substantially stronger binding affinity for this TCR-peptide pair and mirroring the low EC50 T4H2 has for T2Met when measuring IL-2 production. This observation is consistent with the known connection between IL-17a production and NFAT promoter activity, which generally scales well with TCR affinity32.
Figure 2. Activation of the IL-17a signaling pathway for the SILv44, R6C12, and T4H2 TCRs by the WT and T2Met peptides.
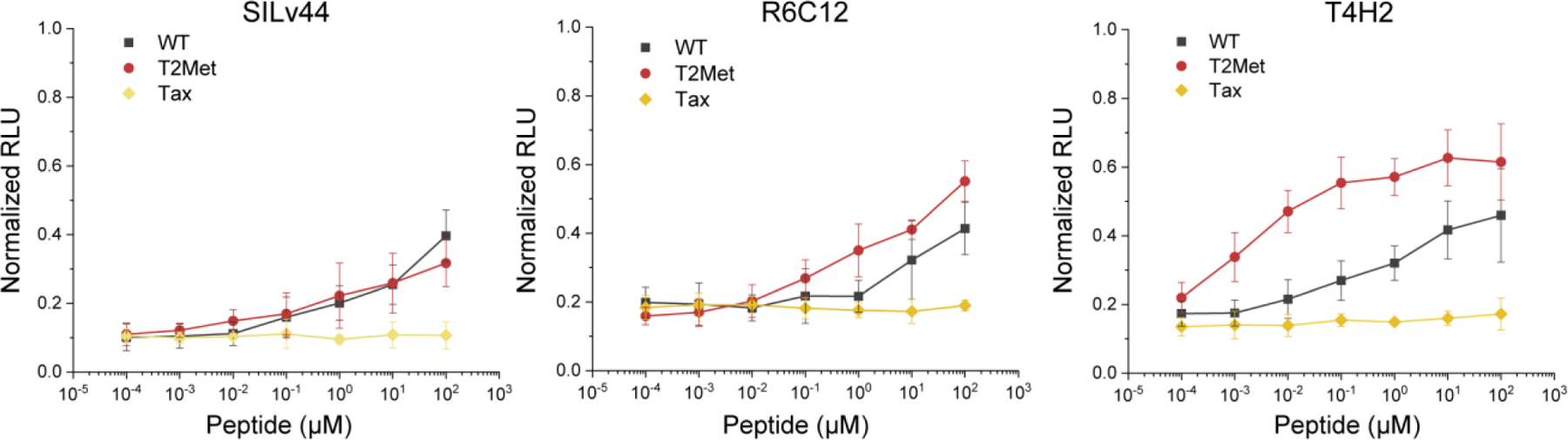
No significant difference is seen between the two peptides for the SILv44 TCR. In contrast, the T4H2 TCR, which has a 40-fold stronger binding affinity towards the T2Met peptide compared to the WT peptide, is more strongly activated by T2Met. Tax refers to an irrelevant control peptide (sequence LLFGYPVYV). RLU = relative light units. Individual data points and error bars are averages and standard deviations of six measurements.
The high functional avidity of SILv44-transduced T cells is associated with the strongest binding affinity towards the WT gp100209 peptide/HLA-A2 complex
When recognizing the WT peptide, SILv44 was the most sensitive among the three TCRs as measured by IL-2 and IFN-γ production, and SILv44 was the most effective at controlling tumors in mice11. However, in our prior work we found that SILv44’s binding affinity towards WT gp100209/HLA-A2 was indistinguishable from that of T4H2 and R6C12 (Table 1). Given the general correlation between affinity and functional avidity we have seen here and elsewhere24,33–35, we thought it important to examine the binding affinities of the TCR more closely.
The KD and ΔG° values in Table 1 were determined using SPR, with the TCRs immobilized on the sensor surface via amine coupling and purified peptide/HLA-A2 complexes injected as the analyte, as traditionally performed. As protein immobilization strategies can impact SPR-determined binding affinities36, we reversed the experimental setup and immobilized WT and T2Met gp100209 peptide/HLA-A2 complexes on separate sensor surfaces and used purified TCR as the analyte (as we could not produce sufficient amounts of purified R6C12, these experiments were only performed with the SILv44 and T4H2 TCRs). The values obtained in this reversed configuration for T4H2 were similar to those determined with the TCR coupled to the surface, as has been seen with other TCR systems37,38. However, for SILv44, the KD values determined with the reverse configuration were tighter than those reported previously: the KD values for SILv44 recognition of WT and T2Met in the reverse configuration were 8 μM and 19 μM, respectively, compared to 65 μM and 210 μM in the more traditional configuration (Fig. 3A, B). Importantly, the relative differences in affinity between the two TCRs for recognition of the WT and anchor-modified peptide was unchanged within error (Fig. 3C), confirming the ability of the two TCRs to sense peptide anchor modification. However, the results from these experiments indicating that SILv44 binds the WT peptide approximately 5-fold more tightly than T4H2 are consistent with the greater functional avidity of SILv44 with the WT peptide and its stronger efficacy in controlling gp100-expressing HLA-A2+ tumors.
Figure 3. Reversing the binding orientation in SPR experiments indicates SILv44 recognizes the WT peptide more strongly than the T4H2 TCR, consistent with the in vitro and in vivo functional data.
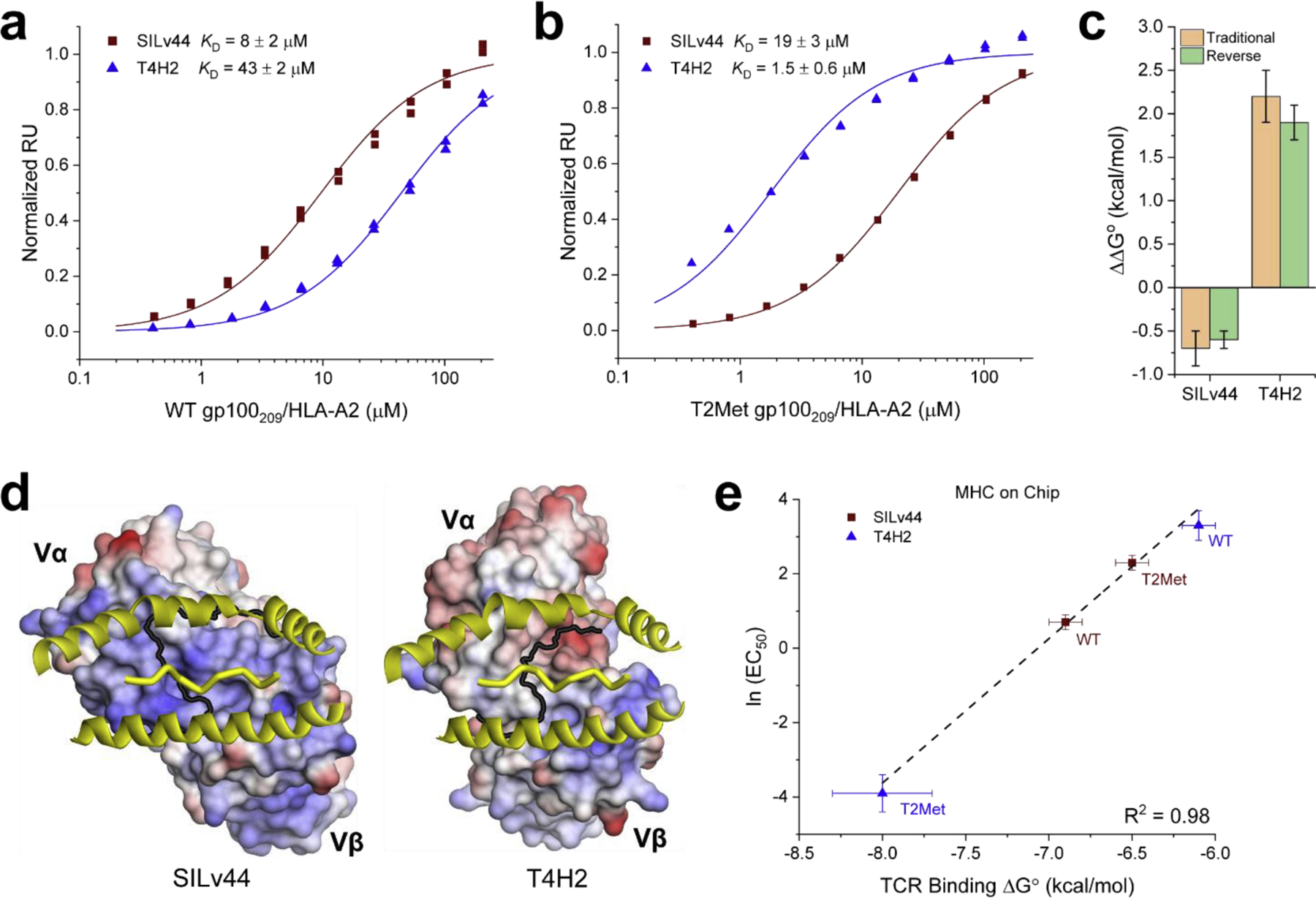
A) Reverse orientation (i.e., peptide/MHC complex on the sensor surface) SPR titration for SILv44 and T4H2 recognizing the WT gp100209 peptide. In this configuration, SILv44 binds more tightly than T4H2. B) Reverse orientation SPR titration for SILv44 and T4H2 recognizing the T2Met peptide. C) Although the reverse orientation experiments showed different affinities for the SILv44 TCR, the differences in affinity between the WT and T2Met peptide were the same within error as found with the traditional orientation experiments, confirming the TCR sensitivity to peptide anchor modification. D) Positive charges near and within the SILv44 binding site suggest a mechanism for artifactually weakening binding affinities when the TCR is coupled to the SPR sensor surface. The T4H2 TCR, whose binding affinity is only minimally impacted by coupling orientation, has a lower positive charge density. The view is through the HLA-A2 binding groove onto the surface of the TCR. Positively charged surface is blue, negatively charged surface is red. The black line demarcates the surfaces of the Vα and Vβ domains. E) Using affinities determined by reverse orientation, there is excellent agreement between IL-2 EC50 values and binding free energies for SILv44 and T4H2 recognition of the WT and T2Met peptides.
We hypothesized that the difference in SILv44 binding affinities between the two SPR configurations could be related to the heterogeneity in immobilized TCR orientations caused by a difference in surface charge between the SILv44 and T4H2 TCRs. With amine coupling, proteins are immobilized in random orientations on the sensor surface by covalent attachment to positive charges, usually the primary amines of lysine side chains. Because the distribution of surface lysines is heterogeneous, coupling can sometimes result in partial or full obstruction of the binding interface, impacting affinity measurements39–41. Although SILv44 and T4H2 use the same Vβ genes (TRBV 19), they use different Vα genes (TRAV 8–1 for SILv44, 12–2 for T4H2). Compared to TRAV 12–2, TRAV 8–1 has four additional lysines, two in the CDR2α loop and two in the HV4β loop near the antigen binding site. Additionally, SILv44 has two positively charged arginine residues at the tip of CDR3α which are absent in the T4H2 structure. Although arginine side chains are not typically cross-linked during amine coupling, their positive charges could still associate with the negatively charged carboxymethylated sensor surface and facilitate covalent linkage through the nearby lysines. Conversely, T4H2 has three additional aspartic/glutamic acids than SILv44, two of which are in or adjacent to CDR loops, which would lead to the opposite effect. The result of these sequence and structural differences is that the surface of the TCR binding site for SILv44 is more positively charged than that of T4H2, with multiple lysines present for coupling (Fig. 3D). As would be expected if the SILv44 affinity measurements were negatively impacted by coupling geometry when the TCR was on the sensor surface, the correlation between binding affinity and functional avidity was stronger for the experiments performed in this reverse configuration (Fig. 3E).
Titration calorimetry and tetramer staining confirm the stronger affinity of SILv44 for the WT gp100209 antigen
Given the configuration-dependent results with the SPR experiments, we sought independent ways to confirm that SILv44 possessed a stronger binding affinity for the WT gp100209 antigen. We first used isothermal titration calorimetry (ITC) to compare SILv44 and T4H2 binding to the WT gp100209 peptide/HLA-A2 complex, titrating solutions of TCR into a calorimeter cell containing the peptide/HLA-A2 complex (Fig. 4A). The values from ITC were slightly higher than those obtained by SPR as seen in other cases where ITC and SPR have been compared24,37,42, but the experiment nonetheless confirmed the stronger affinity of SILv44 over T4H2. Moreover, the relative differences in binding affinity between the SILv44 and T4H2 TCRs were the same within error as seen with SPR in the reverse configuration (a ΔΔG° of −1.0 ± 0.2 kcal/mol determined by reverse SPR compared to a ΔΔG° of −1.2 ± 0.2 kcal/mol determined by ITC).
Figure 4. Independent assessments confirm SILv44 binds the WT peptide more strongly compared to T4H2.
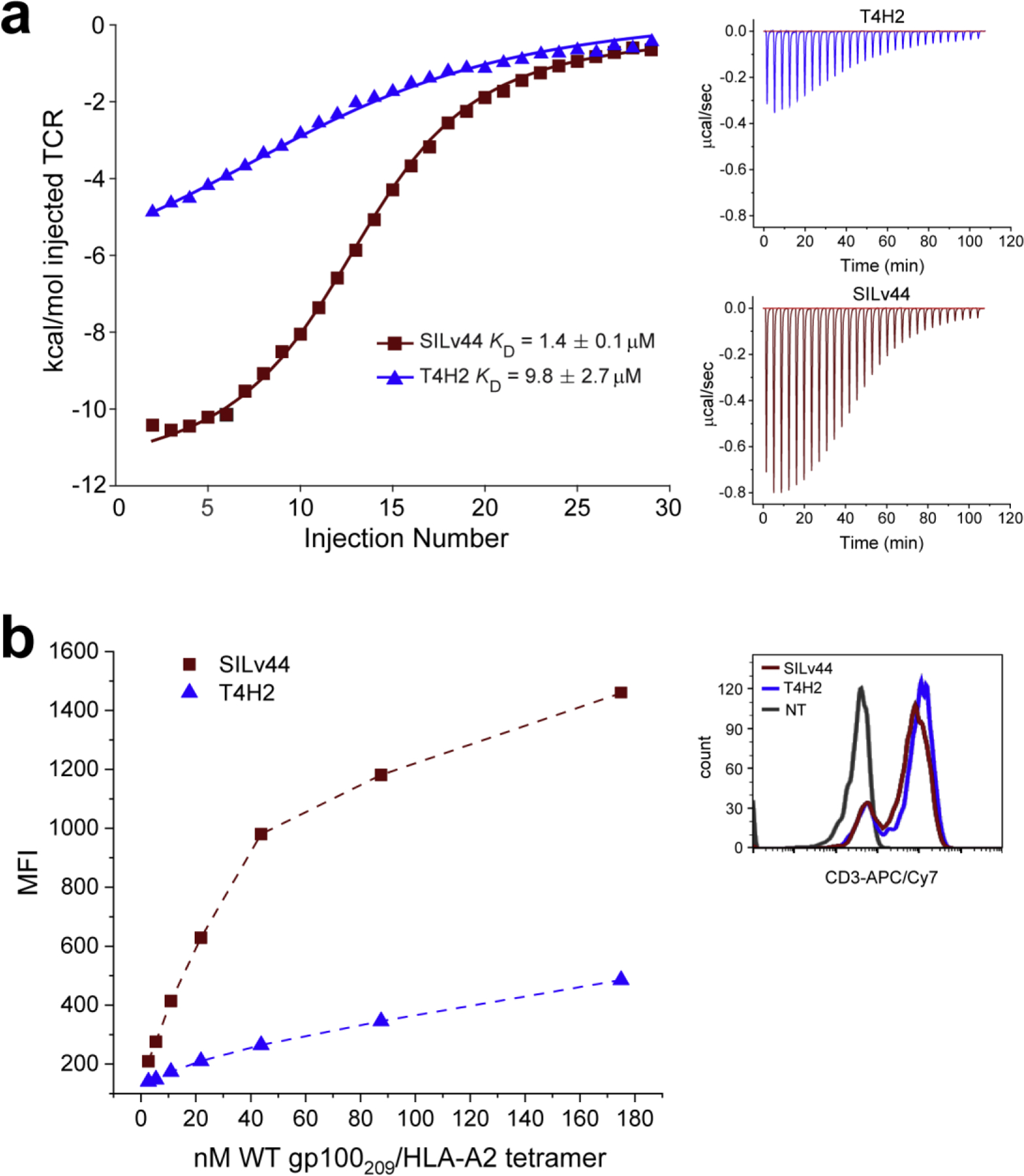
A) Isothermal titration calorimetry experiments confirm SILv44 binds the WT peptide/HLA-A2 complex more tightly than the T4H2 TCR. The panel on the left shows integrated heats fit to a single site binding isotherm. The two small panels on the right show individual titrations for the SILv44 and T4H2 TCRs. B) Staining of TCR-transduced Jurkat 76 cells with WT gp100209/HLA-A2 tetramers shows more efficient staining of SILv44 transduced cells compared to T4H2 transduced cells. The panel on the left shows the tetramer titrations. The small panel on the right indicates similar level of TCR expression for the TCR-transduced cell populations as indicated by anti-CD3 antibody staining. NT = non-transduced control.
We used tetramer staining as a final way to confirm the higher affinity of SILv44 towards the WT gp100209 peptide/HLA-A2 complex. Jurkat cells expressing the SILv44 and T4H2 TCRs were stained with WT gp100209/HLA-A2 tetramers at increasing concentrations and were evaluated by flow cytometry. Fully consistent with the binding affinity data, in these experiments SILv44-expressing Jurkat cells were most efficiently tetramer stained compared to those expressing T4H2 (Fig. 4B).
Discussion
The properties which influence the potency of T cell responses have been of interest for many years, with increasing scrutiny developing with the advent of gene engineered T cell therapy. Typically, T cell potency correlates with the affinity of a TCR towards its ligand5,6. However, there have been many outliers and exceptions. Exceptions could arise due to relatively simple mechanisms, such as differences in the affinity of a peptide to a presenting MHC protein, which influences the amount of available ligand and thus amount of TCR-peptide/MHC complex formed in a cell-cell interface16,17. Or, exceptions could be more complex, such as modulation of the lifetime of the TCR-peptide/MHC complex through catch bonds that occur across TCR interfaces, with longer lifetimes leading to sustained T cell signaling43–46. The architecture of the TCR-peptide/MHC complex has also been proposed as a variable, with different TCR binding geometries potentially influencing the construction and function of a multi-component signaling system involving the αβ TCR, coreceptor, CD3 modules, and any higher order oligomers47,48. The potency of T cell responses may also be differentially regulated by different cytokines, whose output may scale differently with TCR affinity49. Given these complexities, instances in which T cell potency does not correlate with affinity are of considerable interest.
One such instance was described recently with TCRs specific for the gp100209 tumor antigen presented by HLA-A2. Intriguingly, in a panel of three TCRs, the receptor which was most effective in controlling gp100+ tumors in a murine model was the least potent in in vitro assays and possessed the weakest TCR binding affinity11. Not only was this outcome of interest given the general correlation between affinity and function, it stood in contrast to similar experiments in the gp100209 system, which found a strong correlation between TCR affinity and the potency of gene-engineered T cells10.
An important point in the prior studies was that the in vitro experiments relied predominantly on an anchor-modified variant of the gp100209 peptide, in which the suboptimal threonine at position 2 was replaced with methionine. This variant, which binds more tightly to HLA-A2, has been widely used as a surrogate for the WT peptide, both in vitro and in vivo, including human clinical trials19–22. Yet, despite similar recognition by many T cell clones, and despite nearly identical structures of the peptide/HLA-A2 complexes23, differential recognition of the anchor-modified and WT peptides is known, as is the case for other shared tumor antigens19,26,29–31. We recently showed that different TCRs can perceive the two peptides markedly differently, owing to how peptide anchor modifications tune the physical properties of the peptide/HLA-A2 complex24. Given this finding and the potential clinical significance as TCRs are translated from the laboratory to the clinic, we thought it prudent to investigate T cell responses in the gp100209 system in greater detail. Importantly, the particular TCR of interest, SILv44, was derived from a vitiligo patient and is under consideration as a clinical candidate for T cell immunotherapy of melanoma12.
When focusing on the WT gp100209 peptide, our studies showed clearly that SILv44 possesses the strongest affinity towards the WT gp100209 peptide and indeed is the most sensitive towards the WT peptide in in vitro assays. These results are fully consistent with SILv44’s greater potency in mice, as well as its activity in a variety of other functional studies, to include not only TCR-transduced Jurkat cells, but primary T cells11. Thus, earlier hypotheses to explain SILv44’s enhanced activity, such as an unusual TCR binding geometry that overcomes or compensates for weak binding, are not supported. Indeed, we recently determined the crystallographic structure of the SILv44 TCR bound to gp100209/HLA-A224. Although the binding topology does differ from how another gp100209-specific TCR binds, the complex still displays features common to other TCR-peptide/HLA-A2 complexes, with no unusual or outlying structural parameters50,51.
Beyond firmly establishing the correlates with SILv44 in vivo potency, our results provide an important consideration for the use of anchor-modified peptides (sometimes referred to as heteroclitic peptides or mimotopes) in basic and clinical immunology. Often, the assumption is made that such peptides are recognized similarly by T cells, e.g., the only effect of an anchor substitution is to enhance binding to presenting MHC proteins. As demonstrated here and hinted at in in many other cases26,29–31, this assumption is likely wrong. Lastly, our studies provide a foundation for further exploration of the clinical utility of the SILv44 TCR.
Methods
Proteins and peptides
Recombinant, soluble TCRs and peptide/MHC complexes were generated as previously described35. Briefly, TCR α and β chains, the HLA-A2 heavy chain, and β2m were expressed as inclusion bodies in E. coli and dissolved in 8 M urea following purification. TCR α and β chains with the α chain in 20% molar excess were rapidly diluted into 50 mM Tris-HCl (pH 8.3), 2.5 M urea, 6.3 mM cysteamine, 3.7 mM cystamine, 2 mM EDTA, and 0.2 mM PMSF with the addition of 400 mM ʟ-arginine for SILv44 and R6C12. HLA-A2 heavy chain and β2m in a 1:3 molar ratio with excess peptide were rapidly diluted into 100 mM Tris-HCl (pH 8.3), 400 mM ʟ-arginine, 6.3 mM cysteamine, 3.7 mM cystamine, 2 mM EDTA, and 0.2 mM PMSF. TCR and peptide/MHC were incubated in refold buffers for 12 hours at 4 °C followed by dialysis against 10 mM Tris-HCL (pH 8.3) for 48 hours. TCRs and peptide/MHCs were purified by anion exchange followed by size-exclusion chromatography. Peptides were commercially synthesized by AAPTEC or Genscript.
Cells and functional assays
Cells and functional assays were produced, maintained, and performed as described previously24. Briefly, Jurkat 76 cells stably transduced with the CD8 co-receptor and T2 cells were maintained in RPMI-1640 media supplemented with 10% FBS, 100 units/mL penicillin, and 100ug/. Jurkat cells were electroporated using the Neon Transfection System (Thermo Fisher) with the pCMV(CAT)T7-SB100 plasmid and the pSBbi-Neo Sleeping Beauty vector bearing either the SILv44, R6C12, or T4H2 TCR α and β chains separated by the P2A self-cleaving peptide as previously described35. Stable transformants were positively selected by culturing cells in complete media containing 1.2 mg/mL of G418. hIL-17A Firefly luciferase reporter cell lines were similarly generated with the Sleeping Beauty system by cloning the hIL-17A-Luciferase construct from the pGL3E-hIL-17prom(−1125)-Luc vector (Addgene #2567) into the pSBtet-Puro vector and growing transformants in media containing 0.6mg/mL of puromycin. After selection of hIL-17A-Luc transformants, reporter cells were electroporated with a TCR-bearing vector as described above. Prior to co-culture experiments, SILv44, R6C12, or T4H2 TCR expression was confirmed via flow cytometry by staining cells with anti-human CD3 PE-conjugated antibody (BioLegend). Co-culture experiments were conducted with TCR-expressing cells and T2 antigen-presenting cells pulsed with peptide for two hours at the indicated concentrations, then incubated at a 1:1 ratio at 37 °C for 18–20 hours. T cell cytokine release was determined by measuring IL-2 cytokine concentration from co-culture supernatants via ELISA (BioLegend) and luciferase activity was measured by firefly luciferase assay (Promega). Cytokine responses were measured in two independent experiments, each consisting of two biological replicates, with all four datasets analyzed simultaneously for determination of EC50 values. hIL-17-luciferase reporter co-culture assays were completed in two independent experiments consisting of three biological replicates. Luciferase activity was normalized to maximum responses attained by culturing TCR positive cells with 2.5 μM of ionomycin for 18–20 hours. pSBbi-Neo was a gift from Eric Kowarz, obtained from Addgene (Addgene plasmid #60525). pCMV(CAT)T7-SB100 was a gift from Zsuzsanna Izsvak, obtained from Addgene (Addgene plasmid #34879). Jurkat 76 and T2 cells were a gift from Michael Nishimura (Loyola University Stritch School of Medicine).
Tetramer preparation and staining
Monomeric peptide/HLA-A2 complexes with the heavy chain bearing a C-terminal BSP tag (LHHILDAQKMVWNHR) were generated and purified as described above. After purification, peptide/MHC was biotinylated as previously described52, with the extent of biotinylation determined using a gel-shift SDS-PAGE assay. Biotinylated peptide/MHCs were then incubated at a 1:1 ratio with streptavidin, R-phycoerythrin conjugate (ThermoFisher Scientific) as recommended by the manufacturer to generate peptide/MHC tetramers. Cells were stained with PE-conjugated tetramers by incubating samples at the indicated concentrations at 4 °C for 30 minutes, washed with PBS and loaded onto an FC500 (Beckman-Coulter) flow cytometer instrument. Mean fluorescence intensity (MFI) values were determined from analysis on FlowJo (v10.4) software.
Surface plasmon resonance
Surface plasmon resonance (SPR) experiments were performed using a Biacore T200 instrument in 10 mM HEPES (pH 7.4), 150 mM NaCl, 3 mM EDTA and 0.005% surfactant P-20 at 25 °C as previously described24, modified for reverse orientation. Peptide/MHCs were coupled to a Biacore CM5 sensor chip using EDC-NHS amine coupling to approximately 700–1500 response units. Increasing concentrations of TCR spanning 0.8–200 μM were flowed over immobilized peptide/MHC at a rate of 5 μL/min. Each concentration was injected in duplicate for each experiment. For determining KD values, steady-state responses (RU) were determined by averaging the final 10 seconds of each injection and subtracting the response values from identical injections over a blank surface. To enhance accuracy and precision, we used a previously described global analysis approach where each experiment consisted of injecting SILv44 and T4H2 TCRs over the same peptide/MHC sensor surface53. This data, consisting of RU vs. concentration from duplicate injections of different TCRs over the same peptide/MHC surface, was then globally fit to a 1:1 binding model in OriginPro 2018 with local KD values for each individual TCR series and a shared maximum response. KD values for each TCR experiment were averaged with errors reported as the standard deviation of three independent experiments.
Isothermal titration calorimetry
Isothermal titration calorimetry was performed using a Microcal VP-ITC instrument in 10 mM HEPES (pH 7.4) and 150 mM NaCl at 25 °C as previously described24. Briefly, titrations were performed with the peptide/MHC in the calorimeter cell at a concentration of 25 μM and TCR in the syringe at a concentration of 250 μM. Injection volumes of 10 μL were injected over 20 seconds spaced 220 seconds apart for a total of 30 injections. Data were processed and integrated using Origin 7.0 distributed with the instrument. The first data point was removed due to diffusion across the syringe tip during equilibration as is standard. The c values for the for the titrations were 17 and 2.5 for SILv44 and T4H2, respectively. While these values are within the accepted range of 1–100054, to improve accuracy at the low value we used a global analysis approach similar to that used with SPR where a single experiment consisted of titrating different TCR (SILv44 and T4H2) into a fresh peptide/MHC solution from the same stock. These datasets were then fit globally to a single-site binding model using MATLAB R2019a, with ΔH°, KD, and a baseline offset as local parameters and the stoichiometry as a shared, global parameter. This approach allowed us to improve accuracy with the low affinity, low c titrations, as the stoichiometry was constrained by the higher affinity SILv44 titration55,56. Errors were propagated from standard fitting error using standard error propagation methods.
Highlights:
T cell receptors distinguish between the wild type and anchor-modified gp100209 melanoma antigens.
The clinical candidate SILv44 TCR recognizes the wild-type antigen with higher affinity and greater potency than the anchor-modified antigen.
Higher affinity for the wild type antigen explains the SILv44’s superior tumor control in mouse models of melanoma.
Anchor modified peptides should be used cautiously, as the presumption that they are wild type surrogates may be wrong.
Acknowledgements
We thank Dr. Lance Hellman for technical support. Supported by grants R35GM118166 and S10OD028553 from the National Institutes of Health. JAA was supported by a fellowship from the I-CTSI, supported in part by grant TL1TR001107 from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sibener LV et al. Isolation of a Structural Mechanism for Uncoupling T Cell Receptor Signaling from Peptide-MHC Binding. Cell 174, 672–687.e627 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kalergis AM et al. Efficient T cell activation requires an optimal dwell-time of interaction between the TCR and the pMHC complex. Nat Immunol 2, 229–234. (2001). [DOI] [PubMed] [Google Scholar]
- 3.Kersh GJ, Kersh EN, Fremont DH & Allen PM High- and low-potency ligands with similar affinities for the TCR: the importance of kinetics in TCR signaling. Immunity 9, 817–826 (1998). [DOI] [PubMed] [Google Scholar]
- 4.Huang J et al. The kinetics of two-dimensional TCR and pMHC interactions determine T-cell responsiveness. Nature 464, 932–936 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stone JD & Kranz D Role of T cell receptor affinity in the efficacy and specificity of adoptive T cell therapies. Frontiers in Immunology 4, 1–16 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hebeisen M et al. Molecular Insights for Optimizing T Cell Receptor Specificity Against Cancer. Frontiers in Immunology 4 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson LA et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 114, 535–546 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Borbulevych OY, Santhanagopolan SM, Hossain M & Baker BM TCRs Used in Cancer Gene Therapy Cross-React with MART-1/Melan-A Tumor Antigens via Distinct Mechanisms. J Immunol 187, 2453–2463 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller AM, Bahmanof M, Zehn D, Cohen EEW & Schoenberger SP Leveraging TCR Affinity in Adoptive Immunotherapy against Shared Tumor/Self-Antigens. Cancer Immunol Res 7, 40–49 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhong S et al. T-cell receptor affinity and avidity defines antitumor response and autoimmunity in T-cell immunotherapy. Proceedings of the National Academy of Sciences 110, 6973–6978 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eby JM et al. Molecular properties of gp100-reactive T-cell receptors drive the cytokine profile and antitumor efficacy of transgenic host T cells. Pigment Cell & Melanoma Research 32, 68–78 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klarquist J et al. Functional cloning of a gp100-reactive T cell receptor from vitiligo patient skin. Pigment Cell Melanoma Res (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oyarbide-Valencia K et al. Therapeutic implications of autoimmune vitiligo T cells. Autoimmun Rev 5, 486–492 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Palermo B et al. Qualitative difference between the cytotoxic T lymphocyte responses to melanocyte antigens in melanoma and vitiligo. Eur J Immunol 35, 3153–3162 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Moore T et al. Clinical and immunologic evaluation of three metastatic melanoma patients treated with autologous melanoma-reactive TCR-transduced T cells. Cancer Immunology, Immunotherapy 67, 311–325 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parkhurst M et al. Improved induction of melanoma-reactive CTL with peptides from the melanoma antigen gp100 modified at HLA-A*0201-binding residues. J Immunol 157, 2539–2548 (1996). [PubMed] [Google Scholar]
- 17.Bakker AB et al. Analogues of CTL epitopes with improved MHC class-I binding capacity elicit anti-melanoma CTL recognizing the wild-type epitope. Int J Cancer 70, 302–309 (1997). [DOI] [PubMed] [Google Scholar]
- 18.Yu Z et al. Poor immunogenicity of a self/tumor antigen derives from peptide-MHC-I instability and is independent of tolerance. J Clin Invest 114, 551–559 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clay TM et al. Changes in the fine specificity of gp100(209–217)-reactive T cells in patients following vaccination with a peptide modified at an HLA-A2.1 anchor residue. J Immunol 162, 1749–1755 (1999). [PubMed] [Google Scholar]
- 20.Rosenberg SA et al. Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nat Med 4, 321–327 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwartzentruber DJ et al. gp100 Peptide Vaccine and Interleukin-2 in Patients with Advanced Melanoma. New England Journal of Medicine 364, 2119–2127 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee P et al. Effects of interleukin-12 on the immune response to a multipeptide vaccine for resected metastatic melanoma. J Clin Oncol 19, 3836–3847 (2001). [DOI] [PubMed] [Google Scholar]
- 23.Borbulevych OY, Baxter TK, Yu Z, Restifo NP & Baker BM Increased Immunogenicity of an Anchor-Modified Tumor-Associated Antigen Is Due to the Enhanced Stability of the Peptide/MHC Complex: Implications for Vaccine Design. J Immunol 174, 4812–4820 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith AR et al. Structurally silent peptide anchor modifications allosterically modulate T cell recognition in a receptor-dependent manner. Proceedings of the National Academy of Sciences 118, e2018125118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Madura F et al. Structural basis for ineffective T-cell responses to MHC anchor residue-improved “heteroclitic” peptides. European Journal of Immunology 45, 584–591 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cole DK et al. Modification of MHC anchor residues generates heteroclitic peptides that alter TCR binding and T cell recognition. J Immunol 185, 2600–2610 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen J-L et al. Structural and kinetic basis for heightened immunogenicity of T cell vaccines. J. Exp. Med 201, 1243–1255 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Romero P et al. CD8+ T-cell response to NY-ESO-1: relative antigenicity and in vitro immunogenicity of natural and analogue sequences. Clin Cancer Res 7, 766s–772s (2001). [PubMed] [Google Scholar]
- 29.Wieckowski S et al. Fine Structural Variations of αβTCRs Selected by Vaccination with Natural versus Altered Self-Antigen in Melanoma Patients. J Immunol 183, 5397–5406 (2009). [DOI] [PubMed] [Google Scholar]
- 30.Speiser DE et al. Unmodified self antigen triggers human CD8 T cells with stronger tumor reactivity than altered antigen. Proceedings of the National Academy of Sciences 105, 3849–3854 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Webb AI et al. Functional and Structural Characteristics of NY-ESO-1-related HLA A2-restricted Epitopes and the Design of a Novel Immunogenic Analogue. J. Biol. Chem 279, 23438–23446 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Gomez-Rodriguez J et al. Differential Expression of Interleukin-17A and −17F Is Coupled to T Cell Receptor Signaling via Inducible T Cell Kinase. Immunity 31, 587–597 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hellman LM et al. Improving T Cell Receptor On-Target Specificity via Structure-Guided Design. Molecular Therapy 27, 300–313 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Riley TP et al. T cell receptor cross-reactivity expanded by dramatic peptide–MHC adaptability. Nature Chemical Biology 14, 934–942 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Devlin JR et al. Structural dissimilarity from self drives neoepitope escape from immune tolerance. Nat Chem Biol 16, 1269–1276 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Myszka DG Improving biosensor analysis. J Mol Recognit 12, 279–284 (1999). [DOI] [PubMed] [Google Scholar]
- 37.Davis-Harrison RL, Armstrong KM & Baker BM Two Different T Cell Receptors use Different Thermodynamic Strategies to Recognize the Same Peptide/MHC Ligand. Journal of Molecular Biology 346, 533–550 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Baker BM & Wiley DC alpha beta T cell receptor ligand-specific oligomerization revisited. Immunity 14, 681–692. (2001). [DOI] [PubMed] [Google Scholar]
- 39.Trilling AK, Harmsen MM, Ruigrok VJ, Zuilhof H & Beekwilder J The effect of uniform capture molecule orientation on biosensor sensitivity: dependence on analyte properties. Biosens Bioelectron 40, 219–226 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Wang X et al. Impact of SPR biosensor assay configuration on antibody: Neonatal Fc receptor binding data. MAbs 9, 319–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang J et al. Comparison of binding characteristics and in vitro activities of three inhibitors of vascular endothelial growth factor A. Mol Pharm 11, 3421–3430 (2014). [DOI] [PubMed] [Google Scholar]
- 42.Armstrong KM & Baker BM A Comprehensive Calorimetric Investigation of an Entropically Driven T Cell Receptor-Peptide/Major Histocompatibility Complex Interaction. Biophys. J 93, 597–609 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu B, Chen W, Evavold, Brian D. & Zhu, C. Accumulation of Dynamic Catch Bonds between TCR and Agonist Peptide-MHC Triggers T Cell Signaling. Cell 157, 357–368 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hong J et al. Force-Regulated In Situ TCR–Peptide-Bound MHC Class II Kinetics Determine Functions of CD4+ T Cells. The Journal of Immunology 195, 3557–3564 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hong J et al. A TCR mechanotransduction signaling loop induces negative selection in the thymus. Nature Immunology 19, 1379–1390 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Das DK et al. Force-dependent transition in the T-cell receptor β-subunit allosterically regulates peptide discrimination and pMHC bond lifetime. Proceedings of the National Academy of Sciences 112, 1517–1522 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li Y & Mariuzza R Structural and Biophysical Insights into the Role of CD4 and CD8 in T Cell Activation. Frontiers in Immunology 4 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Birnbaum ME et al. Molecular architecture of the αβ T cell receptor–CD3 complex. Proceedings of the National Academy of Sciences 111, 17576–17581 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spear TT et al. Altered Peptide Ligands Impact the Diversity of Polyfunctional Phenotypes in T Cell Receptor Gene-Modified T Cells. Molecular Therapy 26, 996–1007 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Blevins SJ et al. How structural adaptability exists alongside HLA-A2 bias in the human αβ TCR repertoire. Proceedings of the National Academy of Sciences 113, E1276–E1285 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Szeto C, Lobos CA, Nguyen AT & Gras S TCR Recognition of Peptide-MHC-I: Rule Makers and Breakers. Int J Mol Sci 22 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fairhead M & Howarth M Site-specific biotinylation of purified proteins using BirA. Methods Mol Biol 1266, 171–184 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Blevins SJ & Baker BM Using Global Analysis to Extend the Accuracy and Precision of Binding Measurements with T cell Receptors and Their Peptide/MHC Ligands. Frontiers in Molecular Biosciences 4, 1–9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wiseman T, Williston S, Brandts JF & Lin L-N Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Analytical Biochemistry 179, 131–137 (1989). [DOI] [PubMed] [Google Scholar]
- 55.Beechem JM Global analysis of biochemical and biophysical data. Methods Enzymol 210, 37–54 (1992). [DOI] [PubMed] [Google Scholar]
- 56.Turnbull WB & Daranas AH On the Value of c: Can Low Affinity Systems Be Studied by Isothermal Titration Calorimetry? J. Am. Chem. Soc 125, 14859–14866 (2003). [DOI] [PubMed] [Google Scholar]