

Summary
Recent advances in our understanding of tumour heterogeneity alongside studies investigating altered metabolism within transformed tissue have identified metabolic pathways critical to cancer cell survival. Leveraging this information presents a promising new avenue for the generation of cancer-specific therapeutics and improved patient outcomes.
Subject terms: Cancer metabolism, Drug development
Main
Traditional therapeutics, such as chemotherapy and radiation, induce a variety of undesirable effects in otherwise healthy tissues and many cancers ultimately become refractory to further treatment. Resistance is due in part to heterogeneity and metabolic adaptation within the stressed tumour microenvironment. Identification of tumour specific metabolic changes is therefore essential to improving targeted treatment of cancer and enhancing patient responses. One such cancer-specific metabolic adaptation is the opportunistic utilisation of acetate as an alternate carbon source for the generation of acetyl-CoA. Acetate-derived acetyl-CoA is primarily produced by the enzyme acetyl-CoA synthetase 2 (ACSS2) and is utilised by cancer cells to fuel a variety of processes associated with acetyl-CoA metabolism, including de novo fatty acid biosynthesis and protein acetylation (Fig. 1).1 Previous work from our lab has shown that ACSS2 is upregulated in response to nutrient and hypoxic stress, conditions that frequently arise in the harsh tumour microenvironment.2 ACSS2 expression promotes acetate uptake and utilisation by cancer cells thereby conferring a metabolic advantage to the cancer cell.3,4 Importantly, genetically targeting ACSS2 within tumour cells results in decreased growth of breast, melanoma, liver, prostate, myeloma, and glioblastoma cancers.2,4,5 The wide variety of cancers that appear susceptible to ACSS2 loss speaks to the universality of acetate metabolism in cancer.
Fig. 1. Targeting ACSS2 in human pathologies.
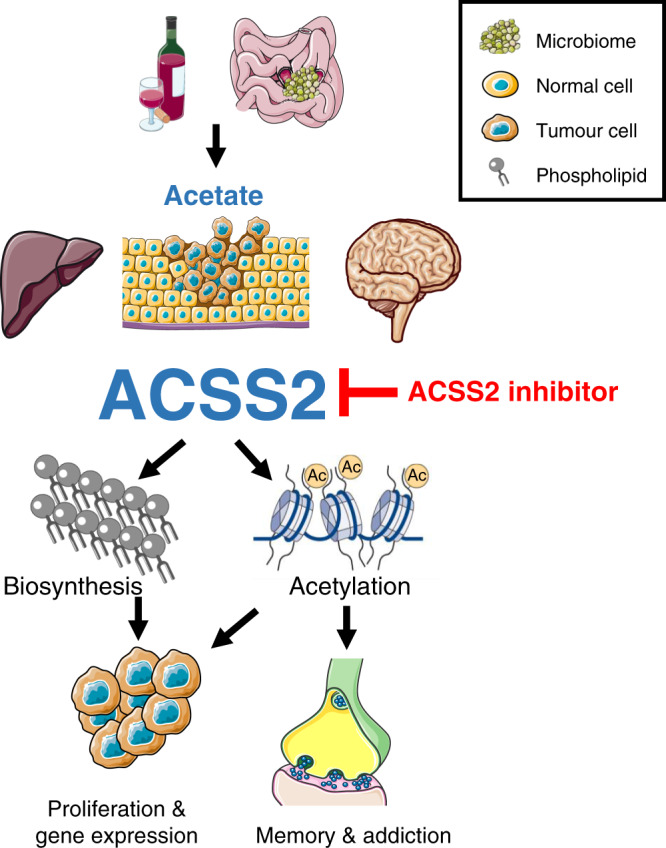
Acetate is primarily generated following the fermentation of carbohydrates by the host microbiome or detoxification of ethanol in the liver. Acetate is readily metabolised to acetyl-CoA by ACSS2 in tumour cells, the liver, and brain driving a variety of cellular processes including de novo lipogenesis, changes in gene expression, and memory formation in the hippocampus. Targeting ACSS2 genetically or with small molecules prevents the mobilisation of acetate by ACSS2 to fuel a variety of human pathologies resulting in improved patient outcomes. Figure generated and adapted from https://smart.servier.com.
In our recent study, we characterised a highly potent and selective ACSS2 inhibitor, called VY-3-135, that displays excellent pharmacokinetic properties in mice.6 VY-3-135 as a single agent significantly impedes in vivo tumour growth of triple-negative breast cancers (TNBC) that have high ACSS2 expression.6 Importantly, VY-3-135 has little to no inhibitory activity against other acetyl-CoA synthetase family members, such as ACSS1 and ACSS3, and caused no further reduction in growth of ACSS2 knockout tumours, suggesting that tumour growth inhibition is probably due to the on-target activity of the inhibitor. The on-target activity of VY-3-135 was further confirmed by metabolomic analysis of tumours from mice administered deuterated or carbon-13-labelled acetate.
Since ACSS2 is a nucleo-cytosolic enzyme, it can generate acetyl-CoA in the nucleus and cytosol which can then be used by acetyltransferases for protein and histone acetylation. Indeed, there are reports on the ability of acetate metabolism to modulate gene expression. One recent study showed that ACSS2 provides acetyl-CoA for the acetylation and stabilisation of the IRF4 transcription factor which then drives oncogenic gene expression in diet-induced mouse models of myeloma.5 The authors showed that this could be reversed by ACSS2 inhibitors or ACSS2 knockdown. Cytosolic ACSS2 also generates acetyl-CoA for de novo fatty acid synthesis and acetylation of metabolites such as UDP-N-acetyl-glucosamine (UDP-GlcNAc). We showed that ACSS2 inhibitors caused no significant changes to gene expression patterns in tumours but we did observe significant reductions in de novo fatty acid synthesis and UDP-GlcNAC synthesis from acetate.6 In light of our results, it seems that the mechanism by which ACSS2 fuels cancer growth appears to be context dependent. It is this multifaceted role of acetate metabolism in cancer that makes drugging ACSS2 an appealing cancer therapeutic.
The idea of acetate metabolism as a cancer-specific metabolic vulnerability with a favourable therapeutic index is perhaps best demonstrated by the fact that ACSS2 knockout (Acss2−/−) mice are phenotypically normal. Acss2−/− mice are viable, fertile, and display no overt signs of pathology when fed standard chow diet.4,7 Interestingly, Acss2−/− mice fed a high fat diet were resistant to fat accumulation and weight gain suggesting a potential role for acetate metabolism in pathological liver disease, such as non-alcoholic fatty liver disease.7 A key source of diet-derived acetate is generated by the host microbiome following fermentation of carbohydrates to acetate. Indeed, microbiome-derived acetate has been suggested to be one way by which high fructose diets induce hepatic lipogenesis and fat deposition.8 Furthermore, the end product of ethanol detoxification in the liver is acetate. Alcohol consumption leads to substantial amounts of acetate being generated in the liver that could be then be converted into acetyl-CoA in an ACSS2-dependent manner. Acetyl-CoA is the precursor for all de novo fatty acid and cholesterol biosynthesis and therefore could be associated with the enhanced lipid deposition and steatosis observed in chronic alcoholics. Ethanol-derived acetate has also been shown to regulate hippocampal memory in an ACSS2-dependent manner indicating a role of ACSS2 in memory formation and addiction.9 Exactly how differing diets directly influence changes to microbiome-derived acetate is an active area of research and will likely reveal novel situations in which acetate metabolism drives disease.
Heterogeneity within the tumour microenvironment presents one of the largest challenges in successfully eradicating cancerous tissue. ACSS2 inhibitors show promise in the treatment of TNBC and myeloma and this will likely extend to other cancers and disease states as the field progresses. However, no studies have explored how ACSS2 inhibition could synergise with simple standard-of-care chemotherapies or immunotherapies. The efficacy and tolerability of ACSS2 inhibitors in preclinical models shows great promise and future studies are warranted to test the synergy between ACSS2 inhibition and other targeted treatments. Considering the range of diseases acetate metabolism and ACSS2 may impact, it comes as no surprise that multiple groups, including our own, have mobilised to develop small molecule inhibitors against ACSS2 (patents WO/2019/097515; WO/2015/175845; WO/2020/252407). Further enhancing the pharmacokinetic properties of these compounds while verifying on-target effects in preclinical models will generate further enthusiasm for transition to the clinic. Importantly 11C-acetate PET imaging has shown that acetate is avidly consumed by both human and mouse tumours, with some tumours showing better signal from 11C-acetate as opposed to the canonically used fluorodeoxyglucose (FDG) probe.4,10 11C-acetate PET imaging could be leveraged as a powerful, clinically relevant, biomarker to single out patients likely to respond to ACSS2 inhibitor therapy while also allowing for a means to demonstrate on target therapy in human patients. At present, targeting ACSS2 appears to fulfil the lofty goals of an ideal treatment: eradicating transformed cells with minimal toxicity to normal healthy tissues. Achilles should be worried.
Author contributions
K.D.M. and Z.T.S. generated the figures and wrote the manuscript.
Ethics approval and consent to participate
Not applicable.
Consent to publish
Not applicable.
Data availability
Not applicable.
Competing interests
The authors declare no competing interests.
Funding information
This work was supported by grants from NIH NCI DP2 CA249950-01 (Z.T.S.), NIH NCI P01 CA114046-12, Susan G. Komen® CCR19608782 (Z.T.S.), the V Foundation for Cancer Research (Z.T.S.) and NIH NCI T32 CA009171-44 (K.D.M.).
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Schug ZT, Vande Voorde J, Gottlieb E. The metabolic fate of acetate in cancer. Nat. Rev. Cancer. 2016;16:708–717. doi: 10.1038/nrc.2016.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schug ZT, Peck B, Jones DT, Zhang Q, Grosskurth S, Alam IS, et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell. 2015;27:57–71. doi: 10.1016/j.ccell.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bulusu V, Tumanov S, Michalopoulou E, van den Broek NJ, MacKay G, Nixon C, et al. Acetate recapturing by nuclear Acetyl-CoA synthetase 2 prevents loss of histone acetylation during oxygen and serum limitation. Cell Rep. 2017;18:647–658. doi: 10.1016/j.celrep.2016.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Comerford SA, Huang Z, Du X, Wang Y, Cai L, Witkiewicz AK, et al. Acetate dependence of tumors. Cell. 2014;159:1591–1602. doi: 10.1016/j.cell.2014.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li Z, Liu H, He J, Wang Z, Yin Z, You G, et al. Acetyl-CoA synthetase 2: a critical linkage in obesity-induced tumorigenesis in myeloma. Cell Metab. 2021;33:78–93 e7. doi: 10.1016/j.cmet.2020.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miller, K. D., Pniewski, K., Perry, C. E., Papp, S. B., Shaffer, J. D., Velasco-Silva, J. N. et al. Targeting ACSS2 with a transition state mimetic inhibits triple-negative breast cancer growth. Cancer Res.81, 1252–1264 (2021). [DOI] [PMC free article] [PubMed]
- 7.Huang Z, Zhang M, Plec AA, Estill SJ, Cai L, Repa JJ, et al. ACSS2 promotes systemic fat storage and utilization through selective regulation of genes involved in lipid metabolism. Proc. Natl Acad. Sci. USA. 2018;115:E9499–E9506. doi: 10.1073/pnas.1806635115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao S, Jang C, Liu J, Uehara K, Gilbert M, Izzo L, et al. Dietary fructose feeds hepatic lipogenesis via microbiota-derived acetate. Nature. 2020;579:586–591. doi: 10.1038/s41586-020-2101-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mews P, Egervari G, Nativio R, Sidoli S, Donahue G, Lombroso SI, et al. Alcohol metabolism contributes to brain histone acetylation. Nature. 2019;574:717–721. doi: 10.1038/s41586-019-1700-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oyama N, Okazawa H, Kusukawa N, Kaneda T, Miwa Y, Akino H, et al. 11C-acetate PET imaging for renal cell carcinoma. Eur. J. Nucl. Med. Mol. Imaging. 2009;36:422–427. doi: 10.1007/s00259-008-0981-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.