

Summary
Single-molecule fluorescence in situ hybridization (smFISH) allows spatial mapping of gene expression. This protocol presents advances in smFISH fidelity and flexibility in intact murine sensory nervous system tissue. An approach using RNAscope probes allows multiplexing, enhanced target specificity, and immunohistochemistry compatibility. Computational strategies increase quantification accuracy of mRNA puncta with a point spread function for clustered transcripts in the dorsal root ganglion and 3D masking for intermingled sciatic nerve cell types. Approaches are validated for mRNAs of modest (Lin28a) and medium (Ppib) steady-state abundance in neurons.
Subject areas: Microscopy, Molecular Biology, In Situ Hybridization, Neuroscience, Molecular/Chemical Probes
Graphical Abstract
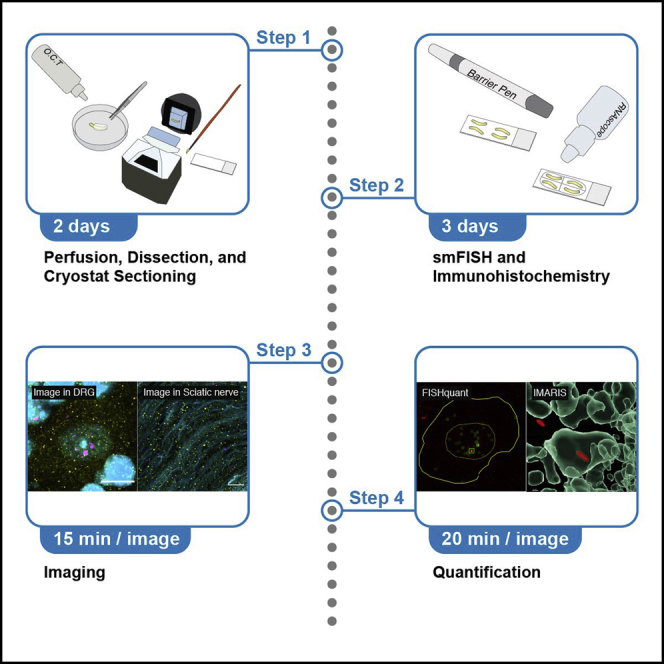
Highlights
-
•
Single mRNA spatial mapping with immunohistochemistry in murine peripheral nervous system
-
•
Point spread function (PSF) analysis approach for clustered transcript quantification
-
•
3D masking strategy for mRNA quantification in intermingled sciatic nerve cell types
Single-molecule fluorescence in situ hybridization (smFISH) allows spatial mapping of gene expression. This protocol presents advances in smFISH fidelity and flexibility in intact murine sensory nervous system tissue. An approach using RNAscope probes allows multiplexing, enhanced target specificity, and immunohistochemistry compatibility. Computational strategies increase quantification accuracy of mRNA puncta with a point spread function for clustered transcripts in the dorsal root ganglion and 3D masking for intermingled sciatic nerve cell types. Approaches are validated for mRNAs of modest (Lin28a) and medium (Ppib) steady-state abundance in neurons.
Before you begin
The protocol below describes the specific steps for single-molecule fluorescence in-situ hybridization (smFISH), image acquisition, and quantification approaches using RNAscope probes with Opal fluorophores in the dorsal root ganglion (DRG) and sciatic nerve compartments of the mouse sensory nervous system. Changes in transcript abundance and post-transcriptional regulation are important in sensory neuron plasticity. This protocol is based on an initial publication describing RNAscope (Shiers et al., 2020; Wang et al., 2012). Other smFISH protocols and tissues have not been tested, but it is likely that the presented customized RNA puncta quantification approaches could also be applied in a variety of settings. Before you begin the smFISH and image quantification approaches, you will need to complete the following preparatory steps.
Perfusion and fixation
Timing: 3 h
This step involves the perfusion-fixation and harvest of dorsal root ganglion (DRG) and sciatic nerve tissue from deeply anesthetized mice. For additional detail and visualization of the perfusion-fixation process see reference (Gage et al., 2012). Ensure that all procedures involving mice are preapproved by your institutional Animal Care and Use Committee.
-
1.
Deeply anesthetize the mouse with Anesthesia Solution delivered by intraperitoneal (IP) injection using a 25G needle. If using 20% urethane in sterile 0.9% NaCl, inject 200 μL per adult mouse. If using a mixture of ketamine and xylazine, adjust concentration of mixture to achieve target doses in a volume of 100 μl/10 g body weight. See preparation of Ketamine mixtures in materials and equipment.
Note: These doses work well for mice on a C57Bl/6 background. Mice on other genetic backgrounds may require adjustment of anesthetic dose or supplementation of ketamine and xylazine with acepromazine.
-
2.Monitor anesthesia.
-
a.Assess response to front paw pinch with forceps to test for sufficient depth of anesthesia. The mouse must show no response to firm paw pinch before any incision is made. Pinching the front paw will avoid production of acute changes in lumbar DRGs and sciatic nerve. Respiratory pattern should be slow and even.
-
b.If anesthesia is incomplete, inject 20–50 μL increments of 20% urethane or 10 ul increments of ketamine mixture and reassess at 1 min intervals.
-
a.
Note: It is important not to over-anesthetize the mouse. While one or two minutes of complete respiratory cessation may be compatible with good perfusion, prolonged cessation of respiration is associated with a state of circulatory stasis, intravascular coagulation, and thus poor perfusion.
-
3.
Rinse the perfusion line with distilled water, then fill with 1× RNase-free PBS.
-
4.
Affix mouse to dissection mat, using tape to immobilize both front paws and one hind paw. The remaining hind paw is left free to facilitate later visualization of fixative-induced movement.
-
5.
Make a transverse incision across the abdominal skin and musculature below the level of the ribcage using surgical scissors to expose the diaphragm, being careful not to cut the liver. Cut along the border of the diaphragm with the ribcage to expose the thoracic cavity. Use caution not to cut the heart or lungs. Then cut through the ribcage longitudinally on either side of the chest to expose the heart. Cut the connective tissue between the heart and chest wall to liberate the heart. The anterior ribcage may then be either removed or folded rostrally out of the way using hemostats to provide effective visualization prior to perfusion.
-
6.
Create an opening in the right auricle of the heart using scissors or a scalpel. Next, insert a 25G needle connected to the perfusion pump and filled with PBS into the apex of the left ventricle. Be sure to orient the needle longitudinally towards the left atrium to avoid puncturing the interventricular septum; do not insert the needle tip more than 5–7mm from the apex of the heart to avoid puncturing the atrioventricular septum. Ensure that there are no air bubbles in the tubing.
-
7.
Perfuse 1× RNase-free PBS (room temperature; 20°C–24°C) at 5 mL/min for 3–4 min or until the liquid exiting the heart through the right auricle is completely clear.
Note: A change in liver color from deep red to pale brown is indicative of successful perfusion. If color does not change within 20s of beginning perfusion, slightly adjust needle orientation and depth.
Note: The sharp tip of the needle can be slightly blunted before use by removing the point with a pair of needle-nosed pliers. This can decrease the risk of inadvertently penetrating the cardiac septa.
-
8.
Change perfusate from PBS to ice-cold 4% Paraformaldehyde (PFA). First, turn off the pump, then transfer the tubing into 4% PFA without allowing entry of air bubbles into the tubing. Restart pump and continue to perfuse with 4% PFA at 5mL/min for 10 min.
Note: Slow movement of the unbound leg or tail as paraformaldehyde reaches the central nervous system is indicative of successful perfusion.
CRITICAL: 4% PFA must be prepared fresh with RNase-free PBS on the same day as fixation.
-
9.
Clean the perfusion tubing with distilled water. Refill with PBS if a subsequent perfusion is to be performed.
Post-fixation and cryoprotection
-
10.Dissect required tissues from the sensory nervous system. Here, we describe the protocol using DRG and sciatic nerve tissues.
-
a.Dissect DRG tissue. With mouse in prone position, use blunt dissection to completely remove skin from the mouse back and the upper hind limbs. Using surgical scissors or a scalpel, remove muscular tissue surrounding the lumbosacral spinal column and overlying the iliac bones. Using scissors or small rongeurs, and beginning in the sacral vertebral column, gently remove the dorsal half of the vertebral column one vertebra at a time, progressing rostrally, to expose the cauda equina and lower spinal cord. Using small iris scissors, cut the spinal dorsal roots on both sides to liberate the caudal spinal cord and cauda equina and remove them from the vertebral canal to expose the dorsal root ganglia. Using sharp forceps, gently remove individual DRGs from their ventrolateral positions within the spinal canal and liberate them from their peripheral spinal nerves by cutting the nerves with iris scissors, putting as little tension on the ganglia as possible.Note: The sciatic nerve has contributions from the L3, L4, and L5 DRGs. The mouse L5 DRG is often located at approximately the level of the iliac crest, and is characteristically smaller than the more rostral L4 and L3 DRGs. A more definitive assignment can be made by dissecting distally from the spinal nerves emanating from the DRGs, to trace them to their merge to form the sciatic nerve. It is important to note, however, that sciatic nerve and lumbosacral DRG anatomy can vary among mouse strains(Rigaud et al., 2008).
-
b.Post-fix the excised DRGs in 1 mL ice cold 4% PFA 18–24 h at 4°C. We have not explored other fixation temperatures, but room temperature fixation for a longer time might be appropriate for some experiments. If sample is post-fixed only without perfusion-fixation, include gentle agitation in this step.
-
c.Bluntly dissect the musculature over the dorsolateral gluteal and upper leg regions to expose the sciatic nerve, which will appear as a white longitudinal band of tissue 1–2mm in diameter. Some muscle tissue may need to be cut with scissors to fully expose the nerve. Dissect distally to the trifurcation of the sciatic nerve into the sural, tibial, and common peroneal branches. Excise the desired length of nerve tissue and lay it on a 5mm × 7mm piece of filter paper to keep the tissue flat.
-
d.Post-fix the sciatic nerve tissue in 1 mL ice cold 4% PFA 18–24h at 4°C with gentle agitation. Tissue will stay attached on filter paper during agitation.
-
e.If using liver tissue as an unrelated tissue to define sample orientation (see Note in Preparation of Frozen Tissue Blocks), excise a wedge of liver lobe ∼ 2 X 4 mm and postfix as with DRG and sciatic nerve.
-
a.
-
11.
After the post-fix, rinse the tissues 3 times with 1×PBS, then transfer DRG tissue and sciatic nerve (along with its adhered filter paper) into pre-chilled cryoprotection solution (30% sucrose in 1× RNase-free PBS solution) at 4°C until the tissue sinks (24-48 hours).
Preparation of frozen tissue blocks
See Figure 1
-
12.
Prepare the required number of embedding molds by filling the molds with Optimal Cutting Temperature (OCT) Compound. Label embedding molds with a permanent marker.
-
13.
Prepare a dry ice/95% ethanol bath ∼2cm deep in a plastic box or ice bucket. Smashed dry ice is preferable.
-
14.
Remove tissue from the cryoprotection solution with a forceps. Blot excess dehydrating solution from the tissue by touching a corner of the tissue to a Kimwipe.
Note: Small tissues like DRG could be challenging to remove from the cryoprotection solution with forceps. Alternatively, the tissue may be removed by suction into a Pasteur pipette and subsequently dropped on an empty open Petri dish and retrieved with forceps or a silicone rubber probe.
-
15.
Drop a small portion of OCT Compound on an open Petri dish and use this drop to wrap a layer of OCT Compound around the tissue. Put the wrapped tissue into marked embedding molds filled with OCT Compound. Take care to lay the tissue at the bottom center of the mold (Figure 1A).
Note: If multiple pieces of tissue are to be embedded in the same block, be sure to submerge them to the same depth and embed them in an asymmetric pattern to avoid ambiguity of sample identity (Figure 1B). Ambiguity in sample identification may also arise from inadvertently flipping tissue sections during thaw-mounting onto slides. To facilitate unambiguous sample identification, mark the edges of the mold to define tissue arrangement, include of a piece of readily distinguishable tissue such as liver and keep a detailed record of sample organization.
-
16.
Place the mold containing the tissue into the dry ice/95% ethanol bath until the OCT Compound is completely opaque.
Note: You may temporarily store the opaque frozen OCT tissue blocks in a nest of smashed dry ice while making other tissue blocks.
-
17.
Store frozen molds at −80°C until ready for sectioning.
Pause point: Frozen blocks can be stored at −80°C for at least 9 months if RNAse-free; lengthy storage of OCT compound should be in an airtight container to avoid dessication. The blocks should never be allowed to thaw after freezing.
Figure 1.

Embed the tissue in OCT
General procedure for tissue embedding (A) and suggested arrangement of DRG specimens in OCT tissue block to avoid sample ambiguity (B).
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Microtubule-Associated Protein 2 (MAP2) rabbit polyclonal antibody; 1:500 | Millipore | Cat# AB5622 Lot# 3279314 |
| Anti-S-100 Protein, clone 15E2E2 mouse monoclonal antibody; 1:200 | Millipore | Cat# MAB079-1 Lot# 3308288 |
| Neu-N antigen chicken polyclonal anti-peptide antibody; 1:200 | Aves Labs | Cat# NUN Lot# NUN7807981 |
| Alexa Fluor® 488 AffiniPure Donkey Anti-Chicken IgY (IgG) (H+L); 1:500 | Jackson ImmunoResearch | Cat# 703-545-155 Lot# 147805 |
| Cy™2 AffiniPure Donkey Anti-Rabbit IgG (H+L); 1:500 | Jackson ImmunoResearch | Cat# 711-225-152 Lot# 146036 |
| DyLight™405 AffiniPure Goat Anti-Mouse IgG, Fcγ Subclass 1 Specific; 1:500 | Jackson ImmunoResearch | Cat# 115-475-205 Lot# 123839 |
| Biological samples | ||
| Mouse dorsal root ganglion tissue | Jackson Laboratory | C57BL/6, #000664 |
| Mouse sciatic nerve tissue | Jackson Laboratory | C57BL/6, #000664 |
| Chemicals, peptides, and recombinant proteins | ||
| Hoechst 33342 | Invitrogen | Cat# H3570 |
| Diethylpyrocarbonate (DEPC) | VWR | Cat# 97062-650 |
| 1× Phosphate-buffered saline | Corning | Cat# 21-040-CV |
| Potassium chloride (KCl) | Sigma-Aldrich | Cat# 60130 |
| Potassium dihydrogen phosphate (KH2PO4) | Fisher Biotech | Cat# AA1159430 |
| Sodium phosphate, dibasic, heptahydrate (Na2PO4) | Fisher Scientific | Cat# S373-500 |
| Sodium chloride (NaCl) | Sigma-Aldrich | Cat# 71380 |
| Sucrose | Sigma-Aldrich | Cat# S5016 |
| Paraformaldehyde | Sigma-Aldrich | Cat# P6148 |
| Urethane | Sigma | Cat# U2500 |
| Ketamine (Zetamine) | MWI | Cat# 501072 |
| Xylazine (AnaSed) | MWI | Cat# 003437 |
| Acepromazine maleate | MWI | Cat# 501075 |
| Triton-X100 | Fisher Scientific | Cat# BP151-100 |
| Ethyl Alcohol 200 Proof | Pharmco | Cat# 111000200 |
| Optimal cutting temperature (OCT) compound | Fisher Healthcare | Cat# 23-730-571 |
| ProlongTM Gold Antifade Mountant | Invitrogen | Cat# P36931 |
| Normal donkey serum (NDS) | Jackson ImmunoResearch | Cat# 017-000-121 |
| Opal 570 | Akoya Biosciences | Cat# FP1488001KT |
| Critical commercial assays | ||
| RNAscope® Multiplex Fluorescent Reagent Kit v2 | Advanced Cell Diagnostics | Cat# 323100 |
| 3-Plex Negative Control Probe | Advanced Cell Diagnostics | Cat# 320871 |
| Probe diluent | Advanced Cell Diagnostics | Cat# 300041 |
| RNAscope Probe, Ppib | Advanced Cell Diagnostics | Cat# 313911-C3 |
| RNAscope Probe, Lin28a | Advanced Cell Diagnostics | Cat# 437121 |
| Experimental models: Organisms/strains | ||
| C57BL/6J Mice 7–16 weeks, male or female | Jackson Laboratory | Stock# 000664 |
| Software and algorithms | ||
| Zeiss ZEN imaging software | Zeiss | N/A |
| FISHQuant v3 | Florian Mueller | https://bitbucket.org/muellerflorian/fish_quant/src/master/ |
| MATLAB R2019a | MathWorks | https://www.mathworks.com/ |
| Imaris | Oxford Instruments | https://imaris.oxinst.com/ |
| SortCellSize | Meffert Lab | https://github.com/Meffert-Lab/SortCellSize |
| Other | ||
| Confocal microscope | Zeiss | LSM 800 |
| Numerical aperture (NA) 1.4 objective, ×63 | Zeiss | N/A |
| Zeiss™ Immersol™ Immersion Oil | Zeiss | 12-624-66A |
| Microtome cryostat | Microm | HM505E |
| Edge-Rite Disposable Microtome Blades | Thermo Scientific | Cat# 4280L |
| Fine paint brush | N/A | N/A |
| Vacuum pump | N/A | N/A |
| Petri dish (100 mm × 15 mm) | Denville Scientific | Cat# 1156F15 |
| 0.2 Micron filter | Thermo Scientific | Cat# 565-0020 |
| Conical tubes, 15 mL | Greiner Bio-One | Cat# 188 271 |
| Conical tubes, 50 mL | VWR | Cat# 89039-656 |
| Microscope cover glass | Fisher Scientific | Cat# 12-545-E |
| Rotary shaker | N/A | N/A |
| Autoclave | N/A | N/A |
| Heat and stir plate | N/A | N/A |
| Incubator | VWR Scientific | Model 1565 |
| 25G ×1 needle | BD | Cat# 305125 |
| Perfusion peristaltic pump | N/A | N/A |
| Needle-nosed pliers | N/A | N/A |
| Tissue-embedding molds (22 mm × 22 mm), (Peel-A-Way Truncated T-12) | Polysciences | Cat# 18986 |
| Superfrost Plus microscopic slides | VWR | Cat# 48311-703 |
| Polysine adhesion slides | Electron Microscopy Sciences | Cat# 63412-01 |
| Steamer | Oster | N/A |
| Thermometer | N/A | N/A |
| Hydrophobic barrier pen | Vector Laboratories | Cat# H-4000 |
| Slide rack (staining jar) | Ted Pella | Cat# 432-1 |
| RapidFISH Slide Hybridization Oven (HybEZ Oven) | Boekel Scientific | Cat# 240200 |
| IHC dark humidity tray | Ted Pella | Cat# 21053 |
| Tissue wipes | Kimtech | Cat# 34120 |
| Glass Pasteur pipette | N/A | N/A |
| Dry ice | N/A | N/A |
Materials and equipment
-
•
Anesthesia solution (20 % Urethane in sterile saline (wt/vol)). For a total volume of 50 mL, add 20% (10 g, wt/vol) urethane to a 50 mL conical tube, add 0.9% NaCl to a final volume of 50 mL. Vortex the solution at room temperature (20°C–24°C) until all urethane is dissolved. The 20% urethane saline solution may be stored at 20°C–24°C for several weeks.
Alternatives: A mixture of ketamine (80 mg/kg) and xylazine (8 mg/kg) in sterile saline delivered intraperitoneally using a 25G needle in a volume of 100 μl/10 g body weight. See Perfusion and Fixation section above.
-
•
Diethyl pyrocarbonate-treated water (DEPC-H2O): For a total volume of 900 mL, add .05% (vol/vol) DEPC (450 μL) to distilled water (900 mL). Cap and shake vigorously. Shake on rotary shaker for at least 2 hours (recommend 100 revolutions per minute (rpm)). Autoclave with a single 50-min liquid cycle achieving 121°C to decompose DEPC. Cool DEPC-H2O to room temperature. DEPC-treated H2O may be stored at 20°C–24°C or 4°C for several months.
-
•
1× RNase-free PBS: For a total volume of 1L, dissolve 8 g NaCl, 0.2 g KCl, 1.44 g Na2HPO4, 0.24 g KH2PO4 in 800 mL DEPC-H2O. Adjust the pH to 7.4 with HCl or NaOH, then add DEPC-H2O to a final volume of 1L. Sterilze the PBS by filtering with a 0.2-micron filter. RNase-free PBS may be stored at 20°C–24°C for several months.
| Reagent | Final concentration | Amount |
|---|---|---|
| NaCl | 137 mM | 8 g |
| KCl | 2.7 mM | 0.2 g |
| Na2HPO4 | 10 mM | 1.44 g |
| KH2PO4 | 1.8 mM | 0.24 g |
| DEPC-H2O | - | Up to 1L |
-
•
4% Paraformaldehyde (4% PFA (wt/vol)): For a total volume of 200 mL, heat RNase-free PBS on hot plate stirrer with temperature set to 78°C. Add a stir bar and begin stirring. Add 4% Paraformaldehyde (8 g, wt/vol) to PBS (200 mL). Continue to stir until fully dissolved. Cool 4% Paraformaldehyde solution on ice and filter with a 0.2-micron filter. Make fresh 4% PFA solution prior to each use.
-
•
Cryoprotection solution (30% Sucrose (wt/vol)): For a total volume of 50 mL, add 15g sucrose to a 50 mL conical tube, then add RNase-free PBS to a final volume of 50 mL. Vortex the solution at room temperature until all sucrose is dissolved. The 30% sucrose-PBS solution may be stored at 4°C for up to one week.
-
•RNAscope® Multiplex Fluorescent Reagent Kit v2 includes:
- RNAscope Hydrogen Peroxide
- RNAscope Protease III
- RNAscope 10× Target Retrieval
- RNAscope Multiplex FL v2 AMP 1
- RNAscope Multiplex FL v2 AMP 2
- RNAscope Multiplex FL v2 AMP 3
- RNAscope Multiplex FL v2 HRP-C1
- RNAscope Multiplex FL v2 HRP-C2
- RNAscope Multiplex FL v2 HRP-C3
- RNAscope Multiplex FL v2 HRP blocker
- RNAscope Multiplex FL v2 TSA Buffer
- RNAscope 50× Wash Buffer
Note: Store solutions in the RNAscope® Multiplex Fluorescent Reagent Kit v2 at 2°C–8°C for up to one year except RNAscope 10× Target Retrieval and RNAscope 50× Wash Buffer. Store RNAscope 10× Target Retrieval and RNAscope 50× Wash Buffer at 20°C–24°C for up to one year.
-
•
1× RNAscope wash buffer: For a total volume of 1 L, warm RNAscope 50× wash buffer to 37°C. Add 2% 50× wash buffer (20 mL) to DEPC-H2O (980 mL). Mix well. Buffer may be stored at 20°C–24°C for up to one month.
-
•
RNAscope 1× Target Retrieval Reagent: For a total volume of 200 mL, Add 10% RNAscope 10× Target Retrieval reagent (20 mL) to DEPC-H2O (180 mL). Store the buffer at 20°C–24°C for up to one month.
Note: If white flocculent matter is observed in the RNAscope 50× wash buffer or in the 10× Target Retrieval buffer, pre-warm either buffer in a 37°C incubator (or water bath) for 5–10 min until dissolved prior to use.
-
•
RNAscope Probes: Order the desired specific probes for target RNA of interest from ACDBio. Target sequence regions of interest can be indicated by the investigator. Typically, RNAscope probes consist of 20 double Z pairs spanning 1000 bp of unique sequence in the target mRNA, with each double Z pair together spanning 36–50 bp of RNA; details of the probe design are described in (Shiers et al., 2020; Wang et al., 2012) and at ACDBio (https://acdbio.com/science/how-it-works). Probes are also designed to have compatible melting temperatures for optimal hybridization. While the use of other smFISH probes should be compatible with the analysis strategies presented here, the RNAscope system offers advantages of signal amplification which are beneficial particularly for the sciatic nerve compartment, as well as specificity. RNAscope pre-amplifier molecules can bind only when two RNAscope probes are associated contiguously with the target mRNA which makes it highly unlikely for off-target hybridization events to generate non-specific signal amplification (Shiers et al., 2020; Wang et al., 2012).
Note: Store probes and probe diluent at 4°C.
Note: If you plan to hybridize multiple RNAscope probes on a single section, ensure that the probes are ordered in different visualization channels (e.g., C1, C2, or C3). RNAscope probes, preamplifiers, amplifiers are all assigned to specific channels. Although any dye can be used with any channel, the specific channel for a given probe is selected at the time the probe is purchased so prior consideration should be given to which probes need to be co-visualized in the same tissue section, as described in the protocol below.
Note: When using multiple probes in different channels and/or more than one IHC target on one section, ensure adequate separation of emission/excitation spectra in your experimental design. For visualization, a wide variety of Opal dyes with non-overlapping imaging spectra are available (excitation/emission spectra here: http://microscopynotes.com/880/opals/index.html) and can be multiplexed with different RNAscope probes by ordering the probes (complete with corresponding pre-amplifier and amplifier components) in up to 4 channels. Staining tissues with positive control probes using different individual dyes can provide an independent means of empirically confirming lack of channel cross-talk in your imaging system
Note: Species non-specific negative control probes should be employed alongside probes of interest. Negative control probes test for background staining related to the RNAscope assay or technical issues of tissue specimen preparation. When using RNAscope Multiplex Fluorescent Reagent Kit with target RNA probes in channels 1, 2 or 3, run a negative control probe in the same channel using the same preamplifier, amplifier, and detection dye as used for the target probe (e.g., run a negative control probe in channel 1 with detection by opal 570 on a control slide with a target RNA probe in channel 1 with Opal 570 detection on a sample slide).
Note: Species-specific positive control probes are recommended to permit assessment of sample quality and assay technical performance. Suitable positive control probes may be selected according to expression level and cell-type specificity of the target RNAs.
-
•
Opal Dye working solution: Dilute Opal dye (1 vol:750 vol) with RNAscope MultiplexTSA buffer. Freshly prepare this solution just before use.
-
•
PBST.1 Wash Buffer (vol/vol): For a total volume of 1L, add 0.1% Triton-X100 solution (1 mL) to 1× RNase-free PBS (1000 mL). Store the buffer at 4°C for up to one month.
-
•
PBST.3 (vol/vol): For a total volume of 50 mL, add 0.3% Triton-X100 solution (150 μL) to 1× RNase-free PBS Buffer (50 mL). Store the buffer at 4°C for up to one month.
-
•
Blocking Reagent (10% NDS (vol/vol)): For a total volume of 2 mL, add 200 μL normal donkey serum (NDS) to 1.8 mL PBST.3 Buffer. Freshly prepare this solution just before use.
-
•
Antibody Diluent Buffer (1% NDS (vol/vol)): For a total volume of 2 mL, add 20 μL NDS to 2 mL PBST.3 Buffer. Freshly prepare this solution just before use.
-
•
Hoechst 500× stock solution: Dissolve the Hoechst in DEPC-H2O to a concentration of 10 mg/mL, pipette solution to mix and divide the solution into 20 μL aliquots. Store the aliquots at −20°C for up to one year.
-
•
Cryostat preparation: Set the temperature of the cryostat chamber between −22°C and −24°C in preparation for cryosectioning.
-
•
Imaging System: We performed RNAscope imaging on a confocal laser scanning microscope (LSM800, Zeiss) equipped with laser lines 405nm, 488nm, 561nm, and 640nm, and an Airyscan super-resolution module. A 63× magnification oil immersion objective with high-NA (1.4), as well as a 25× magnification/NA (0.8) oil/water immersion objective was used. Airyscan is a detector module consisting of a Gallium Arsenide Phosphide (GaAsP) photomultiplier tube (PMT) array of 32 elements arranged in a compound eye fashion.
Step-by-step method details
Tissue sectioning
This step describes how to section and thaw-mount tissue onto slides in preparation for RNAscope-based in situ hybridization and immunohistochemistry.
See Figure 2.
-
1.
Set the cryostat to −23°C and place tissue blocks frozen in OCT inside the cryostat. Allow 1 h for the blocks to reach −23°C; this acclimatization step helps achieve high quality tissue slices by reducing slice curling or damage during subsequent cryosectioning.
-
2.
Mount the blocks onto the cryostat chuck, keeping track of any specific orientation dictated by the tissue embedding pattern. Align tissue block with cryostat blade to ensure that sectioning occurs parallel to the bottom of the block, and thus keep all embedded tissues in the same plane of section.
-
3.
Cryosection the blocks by cutting sections of 12 μm in thickness, using fine paint brushes to prevent the tissue sections from curling or to gently uncurl prior to mounting. Thaw-mount tissue sections onto room temperature (20°C–24°C) microscope slides; the frozen sections will melt on the slide due to the temperature difference.
Note: Use SuperFrost Plus slides for DRG tissue and poly-lysine adhesion coated slides for sciatic nerve tissue to avoid tissue detachment from the slide during processing.
Note: If multiple tissues are mounted in a block in a specific pattern, maintain consistency in the orientation in which multiple sections are mounted on the slides, keeping in mind that the tissue orientation may be inverted during the cutting and mounting process.
Note: Representing sections from all conditions upon one slide will minimize and allow detection of potential batch effects in staining and subsequent analysis; example diagram in Figure 2.
-
4.
Air dry slides at room temperature for 60 - 120 min for DRG tissue, air dry the slides at room temperature overnight (12 - 16 h) for sciatic nerve tissue. Maintain RNAse-free.
Note: The drying time is important to securely mount the tissue sample onto the slides; insufficient drying may lead to tissue detachment during subsequent steps. Longer drying has empirically been found to help retain sciatic nerve sections.
Figure 2.

Frozen section and mount slices on slides
Distribute all conditions on a single slide whenever possible, including ipsilateral (ipsi) or contralateral (contra) to an experimental manipulation such as nerve injury.
Sample permeabilization
In this step, tissue that has been thaw-mounted onto slides is permeabilized and a hydrophobic barrier is subsequently created between individual tissue sections in preparation for in-situ hybridization with RNAscope and immunohistochemistry.
-
5.
Acclimate the slides (stored at −80°C) for 30–60 min in a 37°C incubator. Sciatic nerve tissue requires a subsequent air-drying step at 20°C–24°C for 12–16 h to improve tissue adherence to the slide before the following steps.
-
6.
Wash the slides with 1× RNase-free PBS in a slide rack at room temperature (20°C–24°C ; RT) for 5 min on a rotary shaker with gentle rotation (recommend: 30–40 rpm).
-
7.
Lay the slides on the bench and remove excess PBS by suction using a vacuum line fitted with a glass Pasteur pipette. Quench endogenous peroxidases by adding 4–6 drops of RNAscope Hydrogen Peroxide (∼ 1 drop per section) to cover all the sections. Allow to sit for 10 min exactly.
-
8.
Remove the RNAscope Hydrogen Peroxide solution by gently tapping the bottom edge of the slide against an absorbent towel. Excess droplets of RNAscope Hydrogen Peroxide solution will be absorbed by the towel. Immediately wash the slides with DEPC-H2O in a slide rack at RT for 2 min on a rotary shaker (30 - 40 rpm).
-
9.
Wash again with DEPC-H2O for 2 min at RT on a rotary shaker (30–40 rpm).
-
10.Optional step: Target RetrievalCRITICAL: Performing target retrieval steps for sciatic nerve tissue is not recommended as it may cause this particular tissue to detach from the slide; alternative approaches such as using gelatin-coated slides could be assayed to improve retention of sciatic nerve sections on slides during target retrieval. We found that target retrieval was beneficial for all probes that we tested in DRG tissue, although the utility of target retrieval could change if an experimenter varied the fixation conditions or the tissue type.
-
a.Fill the Steamer water reservoir with cold tap water, and place a clear Steaming Bowl onto the base. Place two slide racks into the bowl, fill one with 200 mL of RNAscope 1× Target Retrieval Reagent, and fill the other with 200 mL of DEPC-H2O.
-
b.Turn on the Steamer. Block all vent holes (covering with tape will suffice) to prevent escape of steam. Allow temperature to rise to at least 97°C.Note: Insert a thermometer into the container containing RNA scope 1× Target Retrieval Reagent to check temperature.Note: Be sure to use a steamer with a bowl tall enough to accommodate the slide racks
-
c.Add slides to the slide rack containing DEPC-H2O for 10 seconds to allow them to acclimate.
-
d.Transfer slides into the slide rack containing RNAscope 1× Target Retrieval Reagent. Block any vent holes to prevent escape of steam and treat for 5 min.
-
e.Remove slides from the steamer and transfer immediately to a separate rinse slide rack filled with DEPC-H2O. Rinse for 15 sec.
-
f.Transfer slides to a slide rack filled with RNase-free 100% ethanol for 3 min.
-
g.Dry slides at RT.
-
a.
-
11.
Draw a barrier of 20mm × 20mm around each section on the slide with the ImmEdge hydrophobic barrier pen (Figure 3). Draw each outline 2–4 times to ensure a continuous barrier.
-
12.
Turn on the HybEZ Oven and set its temperature to 40°C.
-
13.
Place the dried slides into the HybEZ slide tray and permeabilize the tissue by adding ∼ one drop per section of the RNAscope Protease III, to entirely cover each tissue section.
-
14.
Load the slide tray into the oven and incubate for 30 min at 40°C.
Note: Ensure that the oven is already at 40°C before loading in the slide rack.
-
15.
Remove the RNAscope Protease III solution tapping the bottom edge of the slide against an absorbent towel. Droplets of RNAscope Protease III solution will fall downwards and become absorbed by the towel. Immediately wash the slides with DEPC-H2O in a slide rack at RT for 2 min on a rotary shaker (30 - 40 rpm).
-
16.
Wash the slides again with fresh DEPC-H2O for 2 min at RT on a rotary shaker (30 - 40 rpm).
Figure 3.

Draw hydrophobic barrier
Outline tissue on slides with the hydrophobic barrier pen, right image.
Probe hybridization
This step involves the hybridizing of RNAscope Double-Z probes to target mRNAs.
See Figure 4
Note: RNAscope probes and Solution are stored at 4°C. Pre-warm the RNAscope Probe Solution for 10 min in a 37°C incubator or water bath checking that white precipitate is dissolved, then cool to room temperature ( 20°C - 24°C ).
Note: If the probe requires dilution, dilute the probe with RNAscope Probe Dilution Buffer (1 vol : 50 vol). If hybridizing multiple probes on one section, dilute all the probes together with probe dilution buffer (1 vol : 50 vol) to make a probe master mix. If hybridizing multiple probes on one section and one of probes is more dilute from the manufacturer (e.g control and C1 RNAscope probes), dilute all other probes into the manufacturer- diluted probe solution (1 vol : 50 vol) to make the probe master mix. Diluted probes may be stored at 2°C- 8°C for up to 6 months.
-
17.
Remove any excess liquid by suctioning the liquid with a vacuum pump fitted with a glass Pasteur pipette to maintain gentle suction. Place the slides into the HybEZ slide tray and add about one drop (∼50 μl) per 20 mm × 20 mm section of the diluted probes to entirely cover the tissue.
-
18.
Load the slide tray into the oven and incubate for 2 h at 40°C.
-
19.
Wash the slides with1× RNAscope Wash Buffer in a slide rack for 2 min at RT on a rotary shaker (30 - 40 rpm).
-
20.
Wash again with fresh 1× Wash Buffer for 2 min at RT on a rotary shaker (30–40 rpm).
Figure 4.

Hybridize probe and amplifiers
Amplification of probe signal
This step involves the binding of amplifiers to amplify the signal of a single double Z probe pair used for smFISH; components are included in RNAscope® Multiplex Fluorescent Reagent Kit v2.
See Figure 4
Note: Solutions are stored in 4°C. Pre-warm the RNAscope Multiplex FL v2 AMP 1 solution (included in RNAscope® Multiplex Fluorescent Reagent Kit v2) for 10 min in a 37°C incubator (or water bath), then cool to RT.
-
21.Hybridize AMP 1.
-
a.Remove excess liquid from the slides by suctioning the liquid with a vacuum pump fitted with a glass Pasteur pipette to maintain gentle suction.
-
b.Place the slides into the slide tray and add one drop per section of RNAscope Multiplex FL v2 AMP 1 solution to entirely cover the tissue.
-
c.Load the slide tray into the oven and incubate for 30 min at 40°C.
-
a.
-
22.
Wash the slides with 1× Wash Buffer in a slide rack for 2 min at RT on a rotary shaker (30 - 40 rpm).
-
23.
Wash again with fresh 1× Wash Buffer for 2 min at RT on a rotary shaker (30 - 40 rpm).
Note: Pre-warm the RNAscope Multiplex FL v2 AMP 2 solution for 10 min in a 37°C incubator (or water bath), then cool to 20°C –24°C .
-
24.Hybridize AMP 2.
-
a.Remove excess liquid from the slides by suctioning the liquid using a vacuum line fitted with a glass Pasteur pipette.
-
b.Place the slides into the slide tray and add one drop per section of RNAscope Multiplex FL v2 AMP 2 solution to entirely cover the tissue.
-
c.Load the slide tray into the oven and incubate for 30 min at 40°C.
-
a.
-
25.
Wash the slides with 1× Wash Buffer in a slide rack for 2 min at 20°C –24°C on a rotary shaker (30–40 rpm).
-
26.
Wash again with fresh 1× Wash Buffer for 2 min at 20°C–24°C on a rotary shaker (30–40 rpm).
Note: Pre-warm the RNAscope Multiplex FL v2 AMP 3 solution for 10 min in a 37°C incubator (or water bath), then cool to 20°C–24°C.
-
27.Hybridize AMP 3.
-
a.Remove excess liquid from the slides by suctioning the liquid with a vacuum pump fitted with a glass Pasteur pipette.
-
b.Place the slides into the slide tray and add one drop per section of RNAscope Multiplex FL v2 AMP 3 solution to entirely cover the tissue.
-
c.Load the slide tray into the oven and incubate for 30 min at 40°C.
-
a.
-
28.
Wash the slides with 1× Wash Buffer in a slide rack for 2 min at 20°C–24°C on a rotary shaker (30–40 rpm).
-
29.
Wash again with fresh 1× Wash Buffer for 2 min at 20°C–24°C on a rotary shaker (30 - 40 rpm).
Develop HRP channels with fluorescent signals
This step involves the sequential development of HRP channels and binding of fluorophores for probes in different channels, allowing individual mRNAs to be subsequently visualized by confocal microscopy.
Note: Solutions are stored at 4°C. Pre-warm the RNAscope Multiplex FL v2 HRP solutions for 10 min in a 37°C incubator (or water bath), then cool to 20°C–24°C prior to use.
-
30.
Remove excess liquid from the slides by suctioning the liquid with a vacuum pump fitted with a glass Pasteur pipette. Place the slides into the slide tray and add one drop per section of RNAscope Multiplex FL v2 HRP solution to entirely cover the tissue.
Note: Choose the appropriate HRP channel to develop depending on the probe channel (e.g., Develop HRP channel 1 with RNAscope Multiplex FL v2 HRP-C1 solution for C1 probes). Load the slide tray into the oven and incubate for 15 min at 40°C.
-
31.
Wash the slides with 1× Wash Buffer in a slide rack for 2 min at 20°C–24°C on a rotary shaker (30–40 rpm).
-
32.
Wash again with fresh 1× Wash Buffer for 2 min at 20°C–24°C on a rotary shaker (30–40 rpm).
-
33.
Remove excess liquid from the slides by suctioning the liquid with a vacuum pump fitted with a glass dropper pipette. Place the slides into the slide tray and add ∼50 μL Opal Dye working solution per section to entirely cover the tissue.
Note: See preparation for Opal Dye working solution in materials and equipment.
-
34.
Load the slide tray into the oven and incubate for 30 min at 40°C.
-
35.
Wash the slides with 1× Wash Buffer in a slide rack for 2 min at 20°C–24°C on a rotary shaker (30–40 rpm).
-
36.
Wash again with fresh 1× Wash Buffer for 2 min at 20°C–24°C on a rotary shaker (30–40 rpm).
Note: Pre-warm the RNAscope Multiplex FL v2 HRP blocker solution for 10 min in a 37°C incubator (or water bath), then cool to 20°C–24°C.
-
37.
Remove excess liquid from the slides by suctioning the liquid with a vacuum pump fitted with a glass Pasteur pipette. Place the slides into the slide tray and add one drop per section of the RNAscope Multiplex FL v2 HRP blocker solution to entirely cover the tissue.
-
38.
Load the slide tray into the oven and incubate for 15 min at 40°C.
-
39.
Wash the slides with 1× Wash Buffer in a slide rack for 2 min at 20°C–24°C on a rotary shaker (30 - 40 rpm).
-
40.
Wash again with fresh 1× Wash Buffer for 2 min at 20°C–24°C on a rotary shaker (30 - 40 rpm).
Note: If hybridizing multiple RNAscope probes assigned to different channels (e.g., C1,C2, or C3) on a single section, ensure that the probes are in different channels (e.g., C1, C2, or C3), repeat the above steps (30–40) to develop fluorescent signals for each channel, using a distinct dye for each channel until all channels have been developed. When using multiple probes in different channels and/or combining smFISH with IHC on one section, nsure adequate separation of emission/excitation spectra in your experimental design, as described in the probe-ordering section of this protocol.
Immunohistochemistry
This step identifies proteins present within the fixed tissue with primary and fluorescently-labeled secondary antibodies so that their abundance, spatial localization and relationship to target mRNAs may be observed by microscopy.
Note: Maintain RNAse-free conditions. We have had consistent results conducting immunohistochemistry (IHC) steps after the in-situ hybridization (ISH), as presented in this protocol. ACDBio recently released an ancillary kit in which IHC is instead conducted prior to ISH with the RNAscope probes.
-
41.
Prepare the IHC dark humidity tray by filling up to 1 mL distilled water into each well between the rails of the tray to keep the tray humidified and prevent slides from drying during staining. Carefully place the slides on rails of the IHC tray.
Note: If not using a commercial IHC humidity tray, a humidified tray can be made by soaking several paper towels in distilled water without wringing, rolling the towels into cylinders, and then using them to line the internal walls of an opaque and flat plastic container.
-
42.
Remove excess liquid from the slides by gently suctioning the liquid using a vacuum line fitted with a glass Pasteur pipette.
-
43.
Lay slides in dark humidity tray and drop ∼100 μL PBST.1 wash buffer per section onto tissue. Wash for 10 min and suction the liquid with a vacuum pump fitted with a glass Pasteur pipette. Wash the slides three times at room temperature (20°C–24°C) in the dark.
-
44.
Add ∼50 μL per section of the Blocking Reagent to entirely cover the tissue. Incubate for 30 - 60 min in dark humidity tray at 20°C–24°C.
-
45.
Dilute primary antibody in Antibody Diluent Buffer.
-
46.
Remove the blocking reagent by suctioning the liquid with a vacuum pump fitted with a glass Pasteur pipette. Add ∼50 μL per section of the diluted primary antibody to entirely cover the tissue. Incubate the slides in dark humidity tray 16–18 h at 4°C.
-
47.
Wash the slides three times in PBST.1 Wash Buffer for 10 min per wash at 20°C–24°C in the dark; an opaque box can be placed over the slides and removed only briefly during addition/suction of liquid.
-
48.
Dilute fluorophore-conjugated secondary antibody in Antibody Diluent Buffer, as required. Our antibody dilutions are listed in the Resource Table.
-
49.
Add ∼50 μL per section of the diluted secondary antibody to entirely cover the tissue. Incubate the slides for 1–2 h in dark humidified tray at 20°C–24°C.
-
50.
Wash the slides three times in RNase-free PBS for 10 min per wash at 20°C–24°C in the dark.
Counterstaining and mounting slides
This step prepares the tissue that has undergone RNAscope and IHC for microscopy.
-
51.Optional step: Nuclear Staining
-
a.Dilute the Hoechst 500× stock solution with RNase-free PBS (1 vol : 500 vol).
-
b.Remove excess liquid on the slides by suctioning the liquid with a vacuum pump fitted with a glass dropper pipette.
-
c.Add ∼50 μL per section of the diluted Hoechst solution onto the slide to entirely cover the tissue. Incubate the slides for 10 min in the dark in the humidified tray at 20°C–24°C.
-
d.Remove the excess liquid by suction the liquid with a vacuum pump fitted with a glass Pasteur pipette to maintain gentle suction.
-
a.
Note: We have not found a wash step to be required at this Hoechst dilution, however a wash step could be inserted if the background is found not to be negative when imaged.
-
52.
Remove excess liquid from the slides and add 1 small drop (∼5 μL) of Prolong Gold antifade mounting medium on top of each section (∼ 20 μL per slide).
-
53.
Carefully place a 24mm × 50mm glass coverslip over the tissue sections, avoiding production of air bubbles.
Note: Moving or lifting the coverslip after placing it on top of the tissue may cause tissue damage.
-
54.
Dry slides for at least 24 h to cure in the dark before proceeding to imaging.
Imaging slides
This step discusses approaches to image the slides of fixed tissue that has undergone RNAscope and immunohistochemistry preparation for microscopy.
We performed super-resolution RNAscope imaging on a confocal laser scanning microscope (LSM800, Zeiss) equipped with an Airyscan super-resolution module (producing an ∼1.7 fold increase in resolution in all XYZ spatial dimensions; Application Note (Huff, 2015)) and a 63× magnification oil immersion objective with high-NA (1.4) (see “materials and equipment”).
Note: A 63× super-resolution imaging approach is not required for RNA puncta counting in DRG samples for low to medium expression RNAs, but may be beneficial to quantify the increased puncta density of high abundance RNAs. The imaging of smFISH in DRG and effective quantification by the approach presented here may also be done with a 25× oil objective or equivalent. An aside of relevance to some investigators is that application of the Airyscan super-resolution module can yield improvements over non-Airyscan even if the objective (e.g., a lower magnification 25×) is not optimized for Airyscan.
Note: A high resolution imaging approach is imperative to allow correct assignment of RNA puncta in sciatic nerve sample with small structures and cellular heterogeneity. As an example in this protocol, we obtained reliable results using a 63× objective and imaging with an Airyscan super-resolution module.
-
55.
Select an objective and find the fields to be imaged:
Note: While lower magnification objectives (25×) can capture a broader field of view, higher magnification (63×) may be needed to resolve small numerous puncta and cellular complexity. An image tiling feature, if available, may be used to combine higher magnification with a broader field.
Note: If an oil objective (25× or 63×) is needed, the area of interest for imaging can be efficiently found by first scanning the image with a lower magnification dry objective.
-
56.
Set the image acquisition parameters for each channel: Resolution, scan speed, and frame averaging should be set optimally taking into consideration that the parameters should be held constant across samples intended for quantitative comparison. Excitation line and emission collection range will be determined by the excitation and emission spectra for the fluorescence dyes selected for a particular channel. Excitation power, master gain and digital offset should be adjusted for maximal resolution and signal:noise ratio; pinhole is set to optimal based on other imaging parameters.
Note: Higher resolution, slower scan speed and more averaging can be used to decrease noise if observed, but are more time-consuming and could result in bleaching.
Note: We use the Zeiss Airyscan super-resolution module for the RNAscope channel to improve the resolving power for individual mRNA puncta when imaging DRG, and for all channels when imaging sciatic nerve to improve spatial resolution.
Note: A recommended starting power for the excitation line in our system or similar is between 0.2%–0.3%. To determine your optimal laser power and scan settings, check for image saturation in orthogonal projections as well as single slices before proceeding, also check for visualization of signal above the negative control slide. The optimal excitation power will generate no saturation in orthogonal projections and no signal in appropriate negative control slides.
Note: Lower master gain may enhance the signal:noise ratio (SNR).
-
57.
Set Z stacks for the image: The upper and lower limit of the stack may be set based on the RNAscope or, preferably, the IHC channel. Set the optical inter-slice interval to optimal based on acquisition and imaging parameters. The experimenter collecting images should be blinded to the sample condition or identity to reduce the potential for bias.
Note: Keep the inter-slice interval of the Z stack images constant when imaging the same batch of samples. Oversampling by halving the ‘optimal’ inter-slice interval can be used in occasional settings requiring high axial resolution and improved SNR if photo-bleaching is not an issue.
-
58.
Obtain the images: mRNAs in RNAscope channel with Airyscan module are detected as discrete or compound (clustered) puncta. The mRNA puncta will be quantified within co-stained markers using the Z stack images.
Expected outcomes
Files from confocal imaging can be saved for analysis as image Z-stacks, projections and videos, as shown in Figures 5 and 6, Methods videos S1, S2, S3, and S4.
Figure 5.

Lin28a mRNA smFISH in mouse dorsal root ganglia (DRG) tissue combined with immunostaining for neuronal cell bodies and nuclei
(A and I) RNAscope channel for Lin28a mRNAs; probes were visualized using opal 570 dye [ex/em: 561 / 570–650 nm; laser intensity: 0.25%; pinhole: 1 airy unit; detector: Airyscan; detector gain: 750 V].
(B and J) RNAscope channel for negative control DapB mRNAs probe detected with opal 570 and the same confocal settings as in (A).
(C, D, K, and L) Immunohistochemistry for NeuN to mark neuron cell bodies visualized with Alexa Fluor 488 [ex/em: 488 / 494–550 nm; laser intensity: 0.25%; pinhole: 1 airy unit; detector: Airyscan; detector gain: 700V].
(E, F, M, and N) Hoechst 33342 nuclear staining [ex/em 405 nm/ 400–480 nm; laser intensity: 0.5%; pinhole: 1 airy unit; detector: GaAsP; detector gain: 700V]. Hoechst 33342 staining intensity is typically weaker in neuronal nuclei, compared with satellite glial nuclei. The Hoechst channel is therefore intentionally semi-saturated to enhance the visibility of neuronal nuclei.
(G, H, O, and P) Merged images for all channels: Lin28a mRNA/opal 570 (red); NeuN/Alexa Fluor 488 (green); Hoechst 33342 (blue). NeuN transparency is increased for visualization purposes in merged images. Scale bars: 10 μm.
Figure 6.

Ppib smFISH in mouse sciatic nerve tissue combined with immunostaining for neuronal processes and Schwann cells
(A) RNAscope channel for Ppib mRNAs; probes were visualized using opal 570 dye [ex/em: 561 / 565–630 nm; laser intensity: 0.25%; pinhole: 1 airy unit; detector: Airyscan; detector gain: 700V].
(B) RNAscope channel for negative control DapB mRNAs probe detected with opal 570 and the same confocal settings as in (A).
(C and D) IHC for MAP2 to mark neuronal processes visualized with Cy2 [ex/em: 488 / 494–550 nm; laser intensity:0.3; pinhole: 1 airy unit; detector: Airyscan; detector gain: 680V].
(E and F) IHC for S100 to mark Schwann cells visualized with Dylight 405 [ex/em 405 nm/ 400–480 nm; laser intensity: 0.4%; pinhole: 1 airy unit; detector: GaAsP; detector gain:680V].
(G and H) Merged images for all channels: Ppib mRNA/opal 570 (red); MAP2/Cy2 (green); S100/Dylight 405 (blue). Scale bars: 10 μm.
In Figure 5 (also see Methods videos S1 and S2), cryosections (12 μm) of an intact mouse DRG (8-week-old, male, C57Bl6) were subject to smFISH with RNAscope probes (magenta) for Lin28a mRNA, combined with immunochemistry for neuron cells (anti-NeuN, yellow) and nucleus (Hoechst 33342, cyan). Lin28a, which encodes an RNA binding protein, is used as an example of a modest abundance transcript. Lin28a mRNA can be regulated by nuclear retention through inverted Alu elements in its 3’UTR (Chen and Carmichael, 2009; Elbarbary et al., 2013). Larger nuclear puncta may represent Transcription Start Sites, as previously demonstrated using RNAscope for dystrophin mRNA(Hildyard et al., 2020). LSM800 Airyscan superresolution confocal Z-stacks were collected with 63× magnification oil immersion objective with high-NA (1.4). Images shown are orthogonal projections of confocal Z-stacks.
In Figure 6 (also see Methods videos S3 and S4), cryosections (12 μm) of an intact mouse sciatic nerve (8-week-old, male, C57BL/6) were subject to smFISH with RNAscope probes (magenta) for Ppib mRNA, combining with immunochemistry for neuronal processes (anti-MAP2, yellow) and Schwann cells (anti-S100, cyan). LSM800 Airyscan superresolution confocal stacks were collected with 63× magnification oil immersion objective with high-NA (1.4).
Quantification and statistical analysis
Quantification of mRNA puncta in DRG neurons by a batch semi-automated approach
A strategy using FISH-quant, a freeware Matlab-based toolbox (Mueller et al., 2013) is applied to quantify the number of discrete and compound mRNA puncta in DRG cell bodies using 3D confocal stacks obtained by high resolution 63× objective Airyscan imaging.
Note: The FISH-quant toolbox and detailed general use tutorial may be downloaded here: https://bitbucket.org/muellerflorian/fish_quant/src/master/Documentation/
-
1.
Begin by inputting the experimental parameters for each analysis session including pixel size for XY plane and Z axis of the images, refractive index of medium and numeric aperture (NA) of objective used, excitation and emission wavelength of fluorophore, and type of microscope ('confocal’, ‘nipkow’, ‘widefield’ are supported).
Note: These values are used to calculate the theoretical point spread function (PSF) in XY and Z(Zhang et al., 2007)
-
2.
Define cell and nucleus outlines manually in the “Define outlines” GUI(e.g., using images with NeuN and Hoechst channels, or as distinguished by cell-selective markers). Outlines can be saved as a text file containing XY position information of the outlines (Figure 7B-1).
Note: This “Define outlines” GUI converts 3D image stacks into 2D projections. Avoid drawing outlines where cells overlap in the 2D projection of an image stack.
-
3.Discrete mRNA puncta numbers in the nuclear and cytoplasmic compartments are analyzed using our customized FISH-quant mature mRNA counting protocol (Mueller et al., 2013).
-
a.Apply two-step Gaussian filtering to the image with default Gaussian kernels. The first filter with a large kernel (default = 5 pixels for XY and Z) is for background rejection and the second filter with a small kernel (default = 1 pixel for XY and Z) is to increase the signal-to-noise ratio (SNR) (Figure 7A).
-
b.Import the cell & nuclear outlines to the RNAscope channel image (FQ menu > Load > Outline from their respective channel) (Figure 7B-2).Note: The purpose of these dual filters is to increase the fidelity of automatic spot counting, which depends significantly on background rejection and signal intensity. If the image background is too high, the software may call spots in the background. If the signal of the RNA puncta is too low due to exaggerated background rejection, puncta may be missed during automatic spot counting. The kernels may be adjusted as necessary. If RNA puncta are highly clustered, an XY kernel less than 1 pixel may help automatic spot counting, and a smaller Z kernel may be desired for an image with fewer Z-stacks as described in the FISH-quant v3 user manual (https://bitbucket.org/muellerflorian/fish_quant/src/master/Documentation/).
-
c.Apply local maximum spot detection to the filtered images from step ‘a’. The parameters for detection (including size of the detection region, intensity threshold and quality scores for puncta calling) are manipulated in test images by assessing program calling of puncta boundaries through direct visualization and sample condition-blinded experimenter/program automated counting agreement (Figures 7C and 7D).Note: A ‘detection region’ is defined around each detected punctum; this sub-region will be extracted for the calculation of the quality scores and fitting of 3D Gaussian in later steps. The size of the detection region in pixels is set in XY and Z and is usually twice the size of theoretical PSF of a spot by default. The region should be large enough to capture the entire signal but not so large that background signal becomes dominant. Values can be adjusted: larger values are desirable if not enough of the signal is detected, smaller values are desirable if too much background or neighboring puncta are detected (Figure 7C-1).Note: Intensity threshold is the minimum value a signal must have to be detected as a discrete punctum. Adjust the intensity threshold by assessing program calling: higher threshold gives less detected puncta (Figure 7C-2).Note: Quality scores are determined by user-selection of either: (1) standard deviation of all the pixel intensities in the defined sub-region around each detected punctum or (2) 3D curvature of signal intensity (suitable if FISH image is bright). Thresholds may also be adjusted for the quality score: larger quality scores give brighter spots (Figure 7C-3).
-
d.Fit detected spots with 3D Gaussian and exclude spots that are false-positives. Experimenter/program counting agreement can be improved if needed by manipulating the upper and lower threshold for the fitting parameters including the size of the spot in XY, the size of the spot in Z, and the amplitude (Figure 7C-4).Note: The FISH-quant developers recommend adjusting the detection threshold in step c to a setting that avoids false-positives as much as possible.Note: Background nonspecific spots often have a large spot size and small amplitude.
-
e.Settings for single mRNA puncta detection can be saved as a txt file and load back into FISH-quant program for batch processing for the same batch of sample.Note: Settings may be held constant across a single batch of collectively stained/imaged slides, but may need to be varied slightly between experimental batches. For this reason, we advise test monitoring the experimenter/program automated counting agreement with distinct slide batches.
-
a.
-
4.Compound puncta (puncta that represent more than one discrete RNA punctum but are incompletely spatially separated) are quantified by application of a script originally developed for quantification of transcript number at transcription start sites (TXsites scripts) in FISH-quant (Figure 8).
-
a.Outlines for compound puncta are manually defined (along with cell/nucleus outlines) in the “Define outlines” GUI for “transcription start sites”.
-
b.An “average punctum” value with quantified fluorescence intensity and volume is collected by averaging the intensity at each pixel for all identified discrete puncta from all outlined cells in the image. This “average punctum” can be saved as a TIFF image (Figure 8A).CRITICAL: It is critical to obtain a high-quality image of the “average punctum” that can be used for compound puncta quantification. The image quality will improve if more puncta are averaged. Average all discrete mRNAs from the same batch of images to obtain a high-quality image of the individual mRNA molecules.Note: When defining size of average region, it depends on the size of detection region: puncta are only considered in the averaging processes when the entire sub-region can be extracted. A smaller averaging area has to be specified if too many puncta are not considered.
-
c.Define the background of the “average punctum” image using Fiji (open source imaging software); the scalar value of background will be subtracted from each pixel during compound puncta analysis.
-
d.Point spread function is then employed in both the XY plane and the Z axis to quantify compound puncta by defining local minimum and maximum intensities based on peak calling using the average single puncta value. The number of the peaks is the number of single puncta quantified within a compound puncta (Figure 8B).Note: This approach is highly suitable for in situ with RNAscope probes since each of the single puncta are of fairly uniform size and intensity.
-
e.Settings for compound mRNA puncta detection can be saved as a txt file and loaded back into the FISH-quant program for batch processing with the same batch of samples.
-
a.
-
5.The total number of mRNA puncta in each cell is obtained by adding the number of discrete puncta as well as compound puncta (if present).Note: We suggest enhancing rigor using comparative quantification of RNA puncta in cells in a yoked manner: quantify puncta number in cells for different condition or cell size/type on the same image. Data collection parameters may be set prospectively. For example, to maintain even sample representation, we typically analyze no more than 4 paired cells of different conditions per region of interest (ROI 75 x 75 um2), no more than 4 ROI per section and no more than 4 sections per mouse for data collection in which all data points will be represented. If per condition (same condition, same mouse) averages are to be plotted, additional sampling may be ideal to account for biological heterogeneity. Optimal sampling density will be influenced by the expression heterogeneity of the assayed RNA and the specific biological question. Representative sampling of rare neuronal subpopulations can be facilitated through cell-type-specific IHC co-labeling and/or retrograde labeling from specific target tissues. Regardless of the sampling density used, quantification of similar numbers of neurons and sections across mice will prevent imbalanced representation.Optional: Separate Analyses for Cells of Different Diameter (e.g., large and small diameter DRG neuronal subtypes)Note: The SortCellSize script can be used to separate cells by their size into FISH-quant outline files and folders so they may be analyzed separately. This custom script may be downloaded from GitHub at the following address: https://github.com/Meffert-Lab/SortCellSize
-
a.Find the directory where SortCellSize is located in Finder.
-
b.Launch Terminal.
-
c.Navigate to the location of SortCellSize within Terminal.
-
d.Once you have successfully navigated to the folder containing SortCellSize, run SortCellSize.
-
i.SortCellSize must first be compiled. This is done by typing the following:
-
ii.javac SortCellSize.java
-
i.
-
e.SortCellSize can then be run after compiling. This is done by typing the following:
-
i.java SortCellSize.java
-
i.
-
f.SortCellSize will prompt for the directory where outlines are located. Enter this address.
- Example input (OSX): /Users/yourname/Documents/Outlines or /Volumes/NAME_OF_DRIVE/Subfolder/Outlines
-
g.SortCellSize will attempt to locate the outlines and will produce 3 folders containing small, medium, and large sized cells.
-
i.The default size threshold is 700 square microns, separating small and medium sized cells into one folder and large sized cells >700 into another.
Note: this default parameter can be adjusted in the source code. The conversion used in these experiments was 1 sq. um = 57.3049 px based on the resolution and frame size of our microscope images. Any threshold entered within the source should be in pixels. -
i.
-
a.
Figure 7.

Quantification of discrete mRNA puncta in DRG neurons using FISH-quant
(A) Gaussian filtering of RNAscope channel image (red). (1) Unfiltered Lin28a RNAscope channel image. (2) Lin28a RNAscope image with two-step Gaussian filtering with default Gaussian kernels. First filter: kernel = 5 (pixel) for XY and Z; second filter: kernel = 1 (pixel) for XY and Z.
(B) Define cell, nucleus and compound puncta outlines. (1) Manually delineate the outlines for neuronal cells using the NeuN immunohistochemistry channel (copper) and the nuclear outlines using Hoechst 333542 staining (bone). Define cell and nuclear outlines with polygons (yellow). Save outlines as a text file containing XY position information to a specified directory. (2) Import the cell & nuclear outline text file from the respective channel to function as an overlay in the RNAscope channel image (FQ menu > Load > Outline from other channel), and manually draw the outlines for compound puncta (if present) on RNAscope channel images. Compound puncta outlines may be defined with rectangles (green).
(C) Flow chart of puncta detection. (1) Define size of detection region (sub-region) around puncta. The region should be large enough to capture entire signal but not so large that background gets dominant. (2) Define intensity threshold for local maximum detection of puncta. (3) Define quality score threshold for detected puncta, quality scores are determined by standard deviation of all pixel intensities in the sub-region around each detected punctum. (4) Adjust 3D Gaussian fitting threshold to exclude false-positive puncta.
(D) Assessing program calling of puncta boundaries through visualization. (1) Lin28a puncta calling (green) with optimal parameters for detection [size of detection region: XY=2, Z=3; intensity threshold = 1; quality score threshold = 0.2; Gaussian fitting threshold: sigma-XY (size in XY) = 18.68–199.11; sigma-Z = 34.70–1749.72; amplitude = 2.31–403.93]. (2) DapB (negative control probe) puncta calling with the same detection settings as in (1). (3) An example appearance of inappropriate puncta calling when intensity threshold is assigned too high, missing puncta are marked with arrowheads (blue). [size of detection region: XY=2, Z=3; intensity threshold = 1.7; quality score threshold = 0; Gaussian fitting threshold: sigma-XY = 19.15–106.82; sigma-Z = 62.26–1211.90; amplitude = 4.06–204.88]. (4) An example appearance of inappropriate excess puncta calling when detection region around puncta and intensity threshold is assigned too low [size of detection region: XY=2, Z=2; intensity threshold = 0.5; quality score threshold = 0; Gaussian fitting threshold: sigma-XY = 14.92–216.18; sigma-Z = 25.44–1991; amplitude = 0–705844.00]. Scale bars: 10 μm.
Figure 8.

Quantification of compound mRNA puncta in DRG neurons using FISH-quant
(A) Obtain the image of “average punctum” by averaging all discrete mRNA puncta from the same batch of images. Image quality improves as more puncta are averaged. Left: Images of the ‘average puncta’ with maximum intensity projections along the three axes. Right: Images of the ‘average puncta’ subjected to Gaussian fit along the XYZ axes. Size of the averaging region in this example: XY = 9 (pixel), Z = 9 (pixel).
(B) An estimate calling of discrete puncta number quantified within a compound puncta by PSF superposition approach along the three axis with the ‘average puncta’. Using the PSF superposition approach, the compound punctum is reconstructed. The software trials placement of ‘average puncta’ until the best description of the compound puncta is obtained.
Quantification for mRNA puncta in sciatic nerves (3D, IMARIS-based approach)
See Figure 9
Figure 9.

Quantification of mRNA puncta in sciatic nerves using IMARIS
(A) Generate 3D virtual puncta for Ppib mRNA using the 'Spot' image analysis module of Imaris and 3D virtual surfaces for neuronal processes and Schwann cells using the 'Surface' module of Imaris. (1) - (3) Generate 3D virtual puncta for Ppib mRNA with Ppib RNAscope channel image (red). [Diameters for creation: XY = 0.25 μm, Z = 0.9 μm; filter: ‘Quality > 2.5’]. (1) Ppib RNAscope channel raw image. (2) Virtual puncta for Ppib mRNA (yellow). (3) Merged images of Ppib RNAscope channel and virtual Ppib puncta. (4) – (6) Generate 3D virtual surfaces for neuronal processes with MAP2 immunohistochemistry channel (green). [Enable smooth = true; surface grain size = 0.45 μm; enable eliminate background = true; diameter of largest sphere = 4 μm; intensity threshold = 0.29–43.22; filter: “Number of voxels > 100” ]. (4) MAP2 immunohistochemistry channel raw image. (5) Virtual surfaces for neuronal processes. (6) Merge of MAP2 immunohistochemistry channel and virtual neuronal processes surfaces. (7) – (9) Generate 3D virtual surfaces for Schwann cells with S100 immunohistochemistry channel (blue). [Enable smooth = true; surface grain size = 0.4 μm; enable eliminate background = true; diameter of largest sphere = 2.5 μm; intensity threshold = 3 – 52.6187; filter: “Number of voxels > 100”]. (7) S100 immunohistochemistry channel raw image. (8) Virtual surfaces for Schwann cells. (9) Merge of S100 immunohistochemistry channel and virtual Schwann cell surfaces. (10) Merged images for all channels: Ppib mRNA/opal 570 (red); MAP2/Cy2 (green); S100/Dylight 405 (blue). (11) Merged images for all virtual puncta and surfaces: Ppib puncta (yellow); neuronal processes (green); Schwann cells (blue). (12) Merged images for all channels and virtual masks: Ppib puncta/Ppib RNAscope (red); neuronal processes/MAP2 (green); Schwann cells/S100 (blue).
(B) Ppib mRNA punctum (red) colocalized with neuronal processes (green) in 3D is marked with a yellow arrowhead. Ppib mRNA punctum not colocalized with neuronal processes is marked with a blue arrowhead.
(C) Ppib mRNA punctum (red) colocalized with Schwann cells (blue) in 3D is marked with a yellow arrowhead. Ppib mRNA puncta not colocalized with Schwann cells are marked with blue arrowheads. Scale bar: 10 μm.
Bitplane: IMARIS, a 3D image analysis software, is used to quantify the number of discrete RNA puncta in neuronal processes and surrounding Schwann cells. A 3D analysis approach is required for resolving the fine processes and the intermingled axons and Schwann cells in nerve tissue.
-
6.
Virtual spots are generated for each mRNA puncta by using 3D puncta diameters defined in the XYZ axis (the software uses the largest puncta diameter in the XYZ plane). Assess the puncta calling quality and adjust spot ‘Quality’ filter (intensity detected at center of the spot) if needed, by comparing agreement of automated puncta boundaries with direct visualization by an experimenter blinded to experimental condition; parameter settings should be held constant throughout images to be compared (Figure 9A 1–3).
-
7.
Virtual surfaces are generated separately for MAP2 (neuronal processes) (Figure 9A 4–6) and S100 (Schwann cell) (Figure 9A 7–9; Methods video S5) staining using the determined masking thresholds. Thresholds are first adjusted to fit the compartment boundaries by visualization of an experimenter blinded to condition.
-
8.
Spots located with neuronal processes are quantified by applying a “smallest distance to MAP2 below zero” and a “smallest distance to S100 above zero” filter to the spots (Figure 9B).
-
9.
Spots located with Schwann cells are quantified by applying a “smallest distance to S100 below zero” and a “smallest distance to MAP2 above zero” filter to the spots (Figure 9C).
-
10.
Spot density in neuronal process or Schwann cells is generated based on puncta number per MAP2 or S100 volume in a region of interest.
Note: Enhance rigor by implementing data collection guidelines at the initiation of quantification, e.g., we analyze no more than 4 regions of interest (ROI 75×75 μm2) per section and no more than 4 sections per mouse tissue to reduce bias of data from any one particular sample. Optimal sampling density for a given experiment will need to be established based upon variables such as the prevalence of the RNA and specific cell type(s) to be analyzed and the homogeneity of the biological process under study.
Limitations
This protocol using may not be suitable for counts of mRNA abundance if the target mRNA is highly aggregated (or imaging resolution is low) since highly overlapping RNA puncta signals decrease the fidelity with which the Fish-quant software can generate an accurate count. In this scenario we suggest an alternative intensity-based approach for quantitation, such as measuring the mean gray value of an ROI or cell, using ImageJ/Fiji.
63× Airyscan super-resolution imaging approach can be time-consuming and may not be suitable as a high-throughput approach. We suggest imaging with a lower magnification objective if puncta assignment accuracy is not affected.
Puncta colocalization assignment with IMARIS may be resolution-limited in distinguishing puncta located at the contacting border of two intermixed cell markers. We suggest either discounting the indeterminate puncta during analysis, or increase the resolution of imaging for better discrimination of puncta localization with intermingled cell types.
Troubleshooting
Problem 1
Background fluorescence is too high to be distinguished from target-specific fluorescence signals (step 58).
Potential solution
One common cause of high background fluorescence is insufficient washing during IHC staining which can allow retention of non-specific antibody binding on tissue sections. One potential solution is to switch from dropping PBST.1 wash buffer onto sections, to washing the slides in a larger volume by placing slides into a slide rack in a staining jar filled with PBST.1 wash buffer on a rotary shaker (30–40 rpm) (steps 41–50).
Alternatively, if high background is observed only at the edge of sections, desiccation of the sections during staining may be a cause. Do not allow the slides to dry during staining steps, leave liquid on the tissue sections until it is suctioned off to be immediately replaced by the solution in the subsequent step (steps 41–50).
Switching to a distinct Opal dye may also reduce background, particularly if the background source is the tissue itself. In our setting, Opal 570 tends to give less background than Opal 690 and so is preferable for use with lower abundance targets (step 33).
Problem 2
Tissue detaches from microscope slides during RNAscope and IHC washing steps (step 6).
Potential solution
Dry slides completely before initiating staining, place slides in an incubator with circulating ventilation to help dehydrate tissue sections. Use poly-lysine coated slides (Electron microscopy sciences, cat# 63412-01) instead of SuperFrost slides. Additional general suggestions (not specifically tested here), include the use of gelatin-coated slides, and incubation of slides in a vacuum chamber prior to staining (step 4).
Problem 3
Direct experimenter visualization does not agree with automated calling of puncta boundaries by Fish-quant (step 3 in quantification and statistical analysis).
Potential solution
Adjust parameters for detection, including: i) size of the detection region (area of each detected spot); too small if not enough of the signal is detected, too large if too much background is detected); ii) intensity threshold (minimum intensity value for discrete puncta calling); too low if excess puncta are called, too high if puncta are missed; and iii) quality scores for puncta calling (intensity at the center of the spot relative to standard deviation of pixel intensities in the surrounding sub-region, and the 3D curvature of local maxima) values may be too large if dimmer puncta are not detected. False-positive puncta may be avoided by adjusting the threshold of fitting parameters with the 3D Gaussian. 'Fitting parameters' include the size of the spot in XY, the size of the spot in Z, and the intensity amplitude. Background non-specific spots often have a large spot size but small amplitude. Ensure that no puncta are called by the selected FISH-quant settings on images with negative control probes.
Problem 4
Puncta are absent, dim or less frequent than expected, based on expression level of target RNA (step 58)
Potential solution
If you have not already done so, try incorporating the optional Target Retrieval step, as described above. This may be especially important if tissue is over-fixed. We have found that target retrieval improves RNAscope signal from some probes, but not others. Additionally, confirm that your probe is designed for the species from which your tissue was derived and consider the possibility that splice variants of your RNA of interest might not contain the full segment used to design the probe.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Mollie K. Meffert (mkm@jhmi.edu)
Materials availability
This study did not generate new unique reagents.
Data and code availability
The SortCellSize script developed for this study is available on GitHub:
https://github.com/Meffert-Lab/SortCellSize
The FISHquant MATLAB toolbox used in this study is available on BitBucket https://bitbucket.org/muellerflorian/fish_quant.
The IMARIS software used in this study is available for licensing by Oxford Instruments at https://imaris.oxinst.com/.
Acknowledgments
We thank members of the Meffert and Caterina laboratories for helpful discussions and critical feedback on the manuscript and Julie Gohkale for assistance with pilot RNAScope experiments. This work was funded by NINDS R01NS103974 and a Discovery Fund Synergy Award to M.J.C. and M.K.M.; a Braude Foundation award to M.K.M.; NIH R25GM109941 to G.M.; and NINDS NS050274 to the Hopkins Neuroscience MPI core.
Author contributions
All authors contributed to the protocol design. X.L., S.E., S.J., G.M., M.J.C., and M.K.M. contributed to data collection; X.L., S.E., M.J.C., and M.K.M. contributed to data analysis and interpretation. All authors contributed to the drafting, revision, and final approval of the article.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2021.100555.
References
- Chen L.L., Carmichael G.G. Altered nuclear retention of mRNAs containing inverted repeats in human embryonic stem cells: functional role of a nuclear noncoding RNA. Mol. Cell. 2009;35:467–478. doi: 10.1016/j.molcel.2009.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbarbary R.A., Li W., Tian B., Maquat L.E. STAU1 binding 3' UTR IRAlus complements nuclear retention to protect cells from PKR-mediated translational shutdown. Genes Dev. 2013;27:1495–1510. doi: 10.1101/gad.220962.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage G.J., Kipke D.R., Shain W. Whole animal perfusion fixation for rodents. J. Vis. Exp. 2012;65:3564. doi: 10.3791/3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildyard J.C.W., Rawson F., Wells D.J., Piercy R.J. Multiplex in situ hybridization within a single transcript: RNAscope reveals dystrophin mRNA dynamics. PLoS One. 2020;15:e0239467. doi: 10.1371/journal.pone.0239467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huff J. The Airyscan detector from ZEISS: confocal imaging with improved signal-to-noise ratio and super-resolution. Nat. Methods. 2015;12 i–ii. [Google Scholar]
- Mueller F., Senecal A., Tantale K., Marie-Nelly H., Ly N., Collin O., Basyuk E., Bertrand E., Darzacq X., Zimmer C. FISH-quant: automatic counting of transcripts in 3D FISH images. Nat. Methods. 2013;10:277–278. doi: 10.1038/nmeth.2406. [DOI] [PubMed] [Google Scholar]
- Rigaud M., Gemes G., Barabas M.E., Chernoff D.I., Abram S.E., Stucky C.L., Hogan Q.H. Species and strain differences in rodent sciatic nerve anatomy: implications for studies of neuropathic pain. Pain. 2008;136:188–201. doi: 10.1016/j.pain.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiers S., Klein R.M., Price T.J. Quantitative differences in neuronal subpopulations between mouse and human dorsal root ganglia demonstrated with RNAscope in situ hybridization. Pain. 2020;161:2410–2424. doi: 10.1097/j.pain.0000000000001973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F., Flanagan J., Su N., Wang L.C., Bui S., Nielson A., Wu X., Vo H.T., Ma X.J., Luo Y. RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J. Mol. Diagn. 2012;14:22–29. doi: 10.1016/j.jmoldx.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B., Zerubia J., Olivo-Marin J.C. Gaussian approximations of fluorescence microscope point-spread function models. Appl. Opt. 2007;46:1819–1829. doi: 10.1364/ao.46.001819. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The SortCellSize script developed for this study is available on GitHub:
https://github.com/Meffert-Lab/SortCellSize
The FISHquant MATLAB toolbox used in this study is available on BitBucket https://bitbucket.org/muellerflorian/fish_quant.
The IMARIS software used in this study is available for licensing by Oxford Instruments at https://imaris.oxinst.com/.