

Abstract
Post-translational modifications (PTMs) can affect the normal function and pathology of α-synuclein (αS), an amyloid fibril forming protein linked to Parkinson’s disease. Phosphorylation of αS Tyr39 has recently been found to display a dose-dependent effect on fibril formation kinetics and to alter the morphology of the fibrils. Existing methods to access site-specifically phosphorylated αS for biochemical studies include total or semi-synthesis by native chemical ligation (NCL) as well as chemoenzymatic methods to phosphorylate peptides, followed by NCL. Here we investigated a streamlined method to produce large quantities of phosphorylated αS by co-expressing a kinase with a protein fragment in E. coli. We also introduced the use of methyl thioglycolate (MTG) to enable one pot NCL and desulfurization. We compare our optimized methods to previous reports and show that we can achieve the highest yields of site-specifically phosphorylated protein through chemoenzymatic methods using MTG, and that our strategy is uniquely well-suited to producing 15N labeled, phosphorylated protein for NMR studies.
Keywords: α-synuclein, phosphorylation, native chemical ligation, semi-synthesis, NMR
Graphical Abstract

Several strategies for producing α-synuclein phosphorylated at Tyr39 (αS-pY39) are compared and the value of a chemoenzymatic strategy with one-pot ligation is demonstrated, in particular for producing 15N labeled protein for NMR studies.
Introduction
Post-translational modifications (PTMs) of proteins are implicated in a variety of human diseases.[1] In Parkinson’s disease (PD), there is substantial interest in studying the PTMs of α-synuclein (αS), a 140-residue, presynaptic protein. αS is an intrinsically disordered protein that can aggregate into amyloid fibrils,[2] which accumulate in the substantia nigra to form Lewy bodies (LBs), a hallmark feature of PD patient brains.[3] αS phosphorylated at tyrosine 39 (pTyr39 or pY39), a consequence of cAbl kinase activity, has been identified as playing a key role in PD.[4] Early studies found that PD patient brains display upregulated cAbl activity compared to healthy brains[4] and that activation of cAbl in mouse brain enhances neurodegeneration.[5] Other studies reported that αS-pY39 is characterized by slower fibrillization kinetics compared to wild type (WT) protein.[6] Reconciling these results, it has been reported recently that αS-pY39 alters aggregation kinetics in a dose-dependent manner, with lower phosphorylation accelerating fibril formation and higher proportions slowing the process.[7] Most recently, it has been reported that Tyr39 phosphorylation results in enhanced neuropathology in rat primary cortical neurons.[8] The same study showed that cryo-electron microscopy structures of αS-pY39 fibrils display altered morphology, with a stable fibril core formed by an electrostatic interaction network between pTyr39 and lysines in the αS N-terminus.[8] In order to assess such complex effects of this PTM on αS behavior and to uncover disease mechanisms, effective methods to site-specifically install the modification are indispensable and enable experiments like those in Figure 1.
Figure 1.
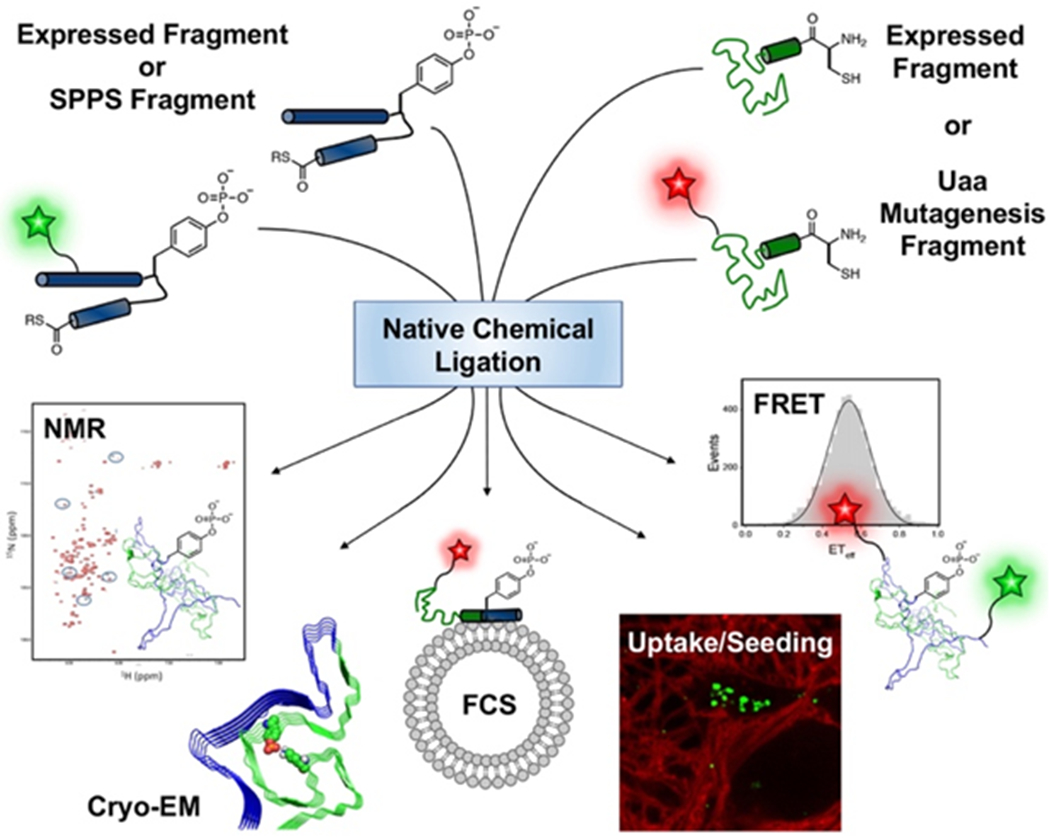
Experiments enabled by native chemical ligation (NCL). Top: αS fragments made by solid phase peptide synthesis (SPPS) or protein expression (including Uaa mutagenesis) can be ligated to form constructs with pY39 and fluorescent labels, enabling NMR experiments (this work),[6] cryo-EM structure determination,[8] fluorescence correlation spectroscopy (FCS) to study vesicle binding,[9] cell-based uptake and fibril seeding experiments,[8] or Förster resonance energy transfer (FRET) measurements of conformational ensembles.[7]
Several methods have been developed to site-specifically incorporate pTyr into proteins of interest. The most common approach is the use of a phosphomimetic, in which tyrosine is mutated to glutamate or aspartate to mimic the negative charge of phosphorylation. However, Glu/Asp does not bear the dianionic charge of the phosphoryl group[10] nor the steric bulk of the tyrosine side chain, which can lead to differences in properties between the authentically modified protein and the mimic protein.[7] Some studies have explored incorporating pTyr through unnatural amino acid (Uaa) mutagenesis, but this method has limitations as well. Fan et al. attempted to increase pTyr incorporation by engineering elongation factor Tu and reducing phosphatase activity, but this approach still suffers from low incorporation efficiency.[11] Luo et al. utilized a propeptide strategy to improve cell uptake and incorporation of pTyr or its non-hyrolyzable analog, but this method is limited when used to introduce multiple Uaas.[12]
To produce αS-pY39, semi-synthetic methods have been developed, involving solid phase peptide synthesis (SPPS), recombinant expression of protein fragments, and native chemical ligation (NCL).[6–8] Dikiy et al. reported the first semi-synthesis of αS-pY39 in 2016 along with NMR studies of the monomer structure, and Zhao et al. recently used a similar approach to make αS-pY39 for cryo-electron microscopy (cryo-EM) studies (Fig. 1 and Scheme 1, Route A).[6, 8] Even though semi-synthesis allows for site-specific incorporation of an authentic pTyr into a protein of interest, synthetic schemes involving multiple ligations can be laborious, and better scalability is ideally needed for biochemical studies.
Scheme 1.

The Semi-Synthetic Method for Producing αS-pY39. The N-terminal fragment was produced by SPPS (Route A) or by expression of an intein fusion (Route 1B). The appropriate central fragments for either route were synthesized by SPPS. The two fragments were ligated together. The resulting product was ligated with a recombinant C-terminal fragment. A final desulfurization step produced full-length αS-pY39.
Towards this goal, our laboratory has previously investigated a chemoenzymatic approach to obtain site-specifically phosphorylated αS-pY39.[7] This chemoenzymatic semi-synthesis involved the treatment of αS1-55 fragment thioester with purified cAbl kinase (Scheme 2, Top), followed by ligation with an αS56-140-C56 fragment and desulfurization (Scheme 2, Bottom). Although the cAbl kinase is not site-specific when acting on full-length αS, as a tyrosine kinase, it exclusively phosphorylates Tyr39, the only Tyr in the αS1-55 fragment. When compared to our 3-piece ligation strategy where we synthesized αS-pY39 by SPPS and NCL in 14% yield over all steps (Scheme 1B), we were able to incorporate pTyr as well as two fluorescent probes into αS using Uaas and our chemoenzymatic strategy to obtain triply modified αS in 63% yield over all steps, an extremely high yield for this type of multiply-modified protein.[13] Our high yields allowed for ample material for biophysical studies of the phosphorylated protein, including single molecule Förster resonance energy transfer (FRET) measurements of protein conformation ensembles[7] and fluorescence correlation spectroscopy (FCS) measurements of diffusion times, which can be used to determine vesicle binding affinity (Fig. 1). Labeled proteins can also be used in measurements of cell uptake and seeding to study fibril propagation between cells (Fig. 1).[8, 14]
Scheme 2.

The Chemoenzymatic Approach for Producing αS-pY39. αS1-55 fragment was expressed as an intein fusion and either phosphorylated in vitro or in E. coli cells by co-expression with cAbl kinase along with YopH1 phosphatase or added imatinib. The in cell procedure resulted in only very low levels of phosphorylation. For in vitro phosphorylation by purified cAbl kinase, the αS1-55 thioester resulting from intein cleavage was phosphorylated with cAbl kinase, converted to the MTG thioester, and then subjected to a one pot ligation/desulfurization procedure to produce αS-pY39.
Encouraged by these results, here we sought to investigate whether we could produce large quantities of modified and labeled αS by co-expression with cAbl kinase (Scheme 2, Middle), bypassing not only the synthesis of various αS fragments but also the separate-pot expression and purification of the enzyme. We expected such a strategy to allow for scalable semi-synthesis in high yield of full-length product bearing simultaneously a PTM and other modifications. Previous research has demonstrated the feasibility of enzyme co-expression to produce a post-translationally modified polypeptide: Large quantities of N-terminally acetylated proteins can be obtained from co-expressing a eukaryotic N-α-acetyltransferase B (NatB) complex in E. coli.[15] Motivated by these studies, we investigated whether a similar strategy can be used to produce tyrosine-phosphorylated αS. We also increased the efficiency of the route using a one-pot NCL and desulfurization strategy. We compared our route to others and found that the in vitro chemoenzymatic phosphorylation route offers the unique advantage of being easily amenable to the incorporation of 15N labels in αS-pY39. We examined the conformation of this site-specifically phosphorylated protein by heteronuclear single quantum coherence (HSQC) NMR spectroscopy and confirmed that the spectrum of the PTM-bearing αS is distinct from that of WT, consistent with the role that pTyr plays in altering the local environment of the protein.
Results and Discussion
Co-expression of αS and cAbl kinase does not produce αS-pY39
Restriction sites were respectively introduced into αS and cAbl kinase plasmids in order to clone both proteins into pETDuet-1, a plasmid harboring two cloning sites for co-expression of proteins. After constructing the plasmid, we wished to test whether co-expression of the kinase and our protein of interest in E. coli would yield phosphorylated protein that we could then purify. αS contains four tyrosines: at sites 39, 125, 133, and 136. Since our goal was to obtain site-specifically phosphorylated αS at Tyr39, we aimed to eventually generate recombinant peptide constructs singly phosphorylated at this site. Such a peptide would allow for NCL with partner peptide fragments to afford full-length, site-specifically modified αS-pY39. Nevertheless, as a preliminary investigation, we sought to test whether cAbl kinase could phosphorylate αS at all when co-expressed and to examine the degree of specificity if any. Previous studies attempting to use purified cAbl to phosphorylate full-length WT αS in vitro found that the modification was not site-specific, affording at least two phosphorylation sites in the protein.[4, 7] In our co-expression strategy, we transformed the pETDuet plasmid bearing an αS C-terminal intein fusion and cAbl into E. coli BL21 cells and grew the cells in media supplemented with ampicillin. Upon lysis and cleavage of the αS-intein with β-mercaptoethanol (βME) to generate the C-terminal carboxylic acid, we purified the protein by Nickel-NTA affinity chromatography. Product was analyzed by matrix-assisted laser desorption ionization mass spectrometry (MALDI MS). We found no mass corresponding to any number of phosphorylations on αS (Supporting Information, SI, Fig. S1).
Since co-expression of αS with cAbl alone did not result in phosphorylated protein, we explored additional co-expression with the phosphatase YopH. In previous methods to obtain purified cAbl, it has been established that co-expression with YopH prevents kinase self-phosphorylation and reduces toxicity to cells, thereby improving yields in cAbl expression.[16] YopH plasmid and our pETDuet vector were therefore co-transformed into E. coli BL21 cells by heat shock and grown in media supplemented with ampicillin and streptomycin. MALDI MS of the intein-cleaved, Ni-NTA purified αS showed peaks corresponding to a +116 mass adduct (observed [M+H]+ = 14574) in addition to unmodified αS (SI, Fig. S2). The additional mass did not correspond to mono- or diphosphorylated αS.
Co-expressed cAbl phosphorylates αS fragment for NCL
Despite these results, we decided to attempt the phosphorylation of the αS fragment consisting of residues 1-55, containing the one Tyr of interest at position 39 (Scheme 2, Middle). It is known from previous studies that full-length αS translocates to the periplasm when expressed in E. coli cells, and that the C-terminal residues 99 to 140 play a signal-like role in this process.[17] On the other hand, since cAbl is most likely expressed in the cytoplasm, we thought that the lack of co-localization between cAbl and full-length αS may be at least partially responsible for the absence of phosphorylation. Therefore, we hypothesized that the N-terminal αS1-55 fragment, which lacks the segments needed for translocation, would be better phosphorylated by cAbl.
Deletion PCR was performed on our pETDuet plasmid, resulting in an αS fragment consisting of residues 1-55. This could be used in NCL with a partner fragment consisting of residues 56-140 with a Cys ligation handle at 56. The pETDuet vector containing cAbl and αS1-55 was transformed into YopH competent E. coli cells and grown as before. After Ni-NTA purification, αS1-55 was dialyzed into buffer and cleaved with sodium 2-mercaptoethanesulfonate (MESNa) to generate a C-terminal thioester for NCL. Low levels of phosphorylated αS1-55-pY39 were observed by MALDI MS, showing that the modification did not go to completion (Fig. 2).
Figure 2.

MALDI MS of αS1-55-MES co-expressed with cAbl and YopH. *Unmodified αS1-55-MES calculated [M+H]+ = 5715.7, observed [M+H]+ = 5719.3. ‡Phosphorylated αS1-55-pY39 calculated [M+H]+ = 5795.7, observed [M+H]+ = 5799.2.
Overall, several reasons may account for the lower levels of phosphorylation in the co-expression approach. First, although αS and cAbl were cloned into the same pETDuet vector to allow for use of a single antibiotic, the burden on the E. coli cells may still have been significant, as the process involved the simultaneous production of an intrinsically disordered protein and a ~33kDa kinase. Second, although YopH is necessary to keep cAbl from phosphorylating itself and to reduce toxicity to the cells, the phosphatase is also possibly playing a role in removing the PTM from αS. Whereas for in vitro phosphorylation the YopH can be separated from kinase, this separation is not possible when phosphorylation is taking place in cells. Third, the slightly different growth conditions, induction conditions,[18] etc. required by αS, cAbl, and YopH make controlling the levels of each component challenging. To this end, we also investigated co-expression of αS1-55 and cAbl using various concentrations of the kinase inhibitor imatinib to buffer cAbl activity,[18a] but observed decreased αS1-55 expression and no phosphorylation (see SI). In general, despite literature precedence in co-expressing enzymes with proteins of interest,[15a] the enzyme co-expression strategy may not be compatible with every protein of interest. On the other hand, cAbl is rather promiscuous and indifferent to sequence and has been demonstrated to phosphorylate many substrates in vitro.[19] Finally, unlike phosphorylation of peptides using purified cAbl in vitro, co-expression is not conducive to controlling stoichiometry of enzyme to substrate or reaction time. While the subtleties of these conditions may be important for improving yield, co-expression does not lend itself to these optimizations.
In vitro phosphorylation of αS fragment by cAbl kinase
In order to directly compare the results from enzyme co-expression with the chemoenzymatic method, we performed in vitro phosphorylation of αS1-55-MES with cAbl kinase and monitored the phosphorylation rate at each time point. The analytical HPLC peak integration showed 53% phosphorylation of αS1-55-MES after 2 h and 74% phosphorylation after 5 h, similar to previously reported phosphorylation yields using this method (SI, Fig. S3).[7] MALDI MS confirmed the existence of these phosphorylated αS fragments. Interestingly, after 31 h, the level of phosphorylated αS1-55-MES was very low, indicating that phosphorylation could be a reversible process under certain conditions. Nevertheless, several hours of treatment with cAbl in vitro allows sufficient amounts of αS1-55-pY39-MES to be purified for NCL with a partner fragment. Such comparison demonstrates that the in vitro chemoenzymatic method is ideal for producing large quantities of phosphorylated αS.
Comparison of methods to generate αS-pY39
It is important to compare strategies for generating phosphorylated αS and determine how each affects scalability and incorporation efficiency. In vitro phosphorylation with WT αS is scalable but has poor specificity, due to the target protein having multiple tyrosine residues. pTyr incorporation through Uaa mutagenesis is limited by low incorporation and low yield. Due to poor uptake of pTyr into cells and endogenous phosphatase activity, the incorporation of tyrosine has been observed using the model Green Fluorescent Protein (GFP), and the expression yield of pTyr-containing protein has been reported to be 5% that of WT. [11] Semisynthetic methods have been reported with higher yields of phosphorylated final product. The three-fragment ligation scheme of Dikiy et al. afforded αS-pY39 in 21.7% overall yield (5.3 mg, 0.3 μmols).[6] Our laboratories previously utilized a similar three-fragment ligation approach involving the synthesis of peptides αS1-29-NHNH2 and αS30-55-C30pY39-NHNH2 by SPPS, affording the final product αS-pY39 in 11% yield over all steps (Scheme 1A).[7]
In attempts to efficiently produce larger quantities of αS peptides for ligation, we reported an alternative semi-synthetic scheme with the ligation site at V37 (Scheme 1B.[7] By increasing the length of the N-terminal fragment, we could express the αS1-36 fragment in bacteria with a high yield (9.2 mg per L of culture), allowing for scalability by increasing culture size. This fragment underwent NCL with αS37-55-V*37pY39-SR (where V* is penicillamine). The intermediate product underwent another ligation with αS56-140-C56 followed by desulfurization, resulting in full-length αS-pY39 in 14% yield over all steps. Given these synthetic yields involving SPPS and three-piece ligations, it is clear that scalability of bacterial culture can be further capitalized upon to increase yield.
An advantage of producing ligation fragments by expression is the ability to decrease the number of fragments that need to be ligated to produce full-length αS. The chemoenzymatic approach previously developed in our lab, which bypasses SPPS, involved bacterially expressing both the αS1-55-MES thioester fragment and the αS56-140-C56 fragment for ligation.[7] Since αS1-55 only has one Tyr residue at position 39, in vitro phosphorylation of Y39 could be achieved enzymatically with cAbl kinase (Scheme 2, Top). This method produced 8.0 mg/L of αS1-55-MES starting material and phosphorylated αS1-55-pY39-MES thioester in 80% yield, similar to the 74% phosphorylation yield reported in this study. Over all steps, our chemoenzymatic semi-synthesis produced triply modified αS-pY39 bearing two fluorophores in 63% yield.
In this study, we explored an enzyme co-expression approach in which the pETDuet vector was used to simultaneously express αS1-55 and cAbl kinase (Scheme 2, Middle). We expected that large quantities of the phosphorylated fragment would be produced without the need for separate-pot expression and purification of the enzyme. However, these experiments resulted in very little phosphorylated αS1-55 fragments that were difficult to quantify and isolate. Instead, 1.59 mg/mL of unmodified αS1-55 was obtained after HPLC purification, suggesting that co-expression with enzyme was not as scalable compared to expression of αS alone. Comparing all these methods, we find that the in vitro chemoenzymatic semi-synthesis is the most efficient and scalable method to produce αS-pY39.
15N αS-pY39 semi-synthesis
Given its efficiency and scalability, we took advantage of the in vitro chemoenzymatic semi-synthesis method to produce isotopically labeled αS-pY39 for studies to characterize the effect of the PTM on the structure of the αS monomer. To semi-synthesize 15N-labeled αS-pY39, we expressed αS1-55 and αS56-140-C56 fragments in media supplemented with heavy ammonium chloride (15NH4Cl). αS1-55-Mxe-His6 and αS56-140-Mxe-His6 plasmids were each transformed into BL21 cells, and cultures were grown in 15NH4Cl media. After transthioesterification with MESNa and HPLC purification, 12.6 mg of 15N αS1-55-MES was obtained per L of culture (SI, Fig. S4), comparable to amounts obtained from unlabeled cultures (8 mg per L of culture). In vitro phosphorylation with purified cAbl afforded 15N αS1-55-pY39-MES in 67.7% yield (SI, Fig. S5). The partner fragment, 15N αS56-140-C56, was obtained after methoxyamine-mediated thiazolidine deprotection with an isolated yield of 9 mg per L of culture (SI, Fig. S6–S7). The incorporation efficiency of heavy nitrogen label into both fragments was quantitative, as there was total absence of unlabeled species.
Since our chemoenzymatic semi-synthesis scheme reduces the number of steps to one in vitro phosphorylation reaction and a single NCL between recombinant partner fragments, we employed methyl thioglycolate (MTG),[20] an alkyl thiol additive compatible with one-pot desulfurization (Scheme 2, Bottom). We also considered the use of trifluoroethanethiol (TFET),[21] which has very efficient reactivity, but opted for MTG for its lower volatility (b.p. 152 °C vs. TFET, 37 °C) and little malodor.[20] 15N αS1-55-pY39-MES was reacted with 2 equiv 15N αS56-140-C56, 100 equiv MTG, and 40 mM TCEP at 37 °C. Even though the MTG-thioester is expected to display slower reactivity compared to the more commonly used aryl thiol mercaptophenyl acetic acid (MPAA) thioester, and the Val55 thioester under attack is sterically hindered, the reaction proceeded to completion after overnight incubation at 37 °C (Fig. 3 and SI, Fig. S8). One-potdesulfurization using radical initiator VA-044, t-butylthiol, and TCEP afforded the product 15N αS-pY39 in 77.4% isolated yield, an improvement over traditional MPAA-mediated NCL and separate desulfurization described in our earlier report.[7]
Figure 3.
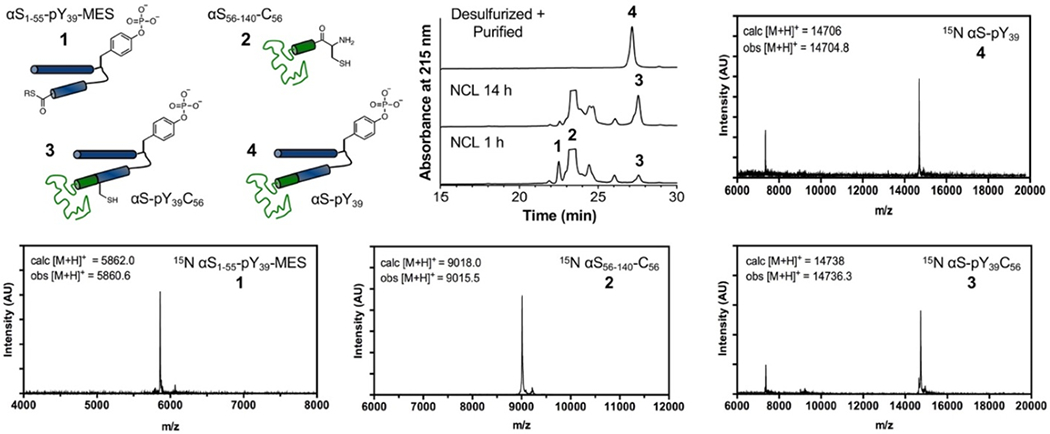
Incorporation of Isotopic Labels by NCL of in vitro Phosphorylated N-terminal Fragment with C-terminal Fragment. Clockwise from top left: Schematic representations of NCL reactants and product. Analytical HPLC trace (10-50% B over 30 min) of NCL reaction; 15N αS1-55-pY39-MES (1), 15N αS56-140-C56 (2), ligated product 15N αS-pY39C56 (3), and desulfurized product 15N αS-pY39 (4) (Retention time 27.2 min). MALDI MS of reacting partners, ligated product, and desulfurized product.
NMR characterization of αS-pY39
To characterize our final product, we acquired proton-nitrogen correlation spectra (1H-15N HSQC) for αS-pY39 and compared them with spectra obtained for WT αS (Fig. 4). Changes in the spectrum of αS-pY39 can be observed for signals from residues near the phosphorylation site, confirming a local change in the electrochemical environment. Furthermore, these changes corresponded exactly to changes previously observed for the same signals in αS-pY39F125F131 (SI, Fig. S9), confirming successful phosphorylation of residue 39. Unlike the spectrum of the phosphorylated double Tyr-to-Phe mutant, there were no changes for signals from residues near Tyr125 and Tyr131 for our product compared to WT αS (Figs. 4 and S9). The clustering of peaks observed here is common for intrinsically disordered proteins, as the electrochemical environment of each residue is degenerate, consisting mostly of solvent molecules, leading to similar backbone NH chemical shifts for similar chemical groups.
Figure 4.
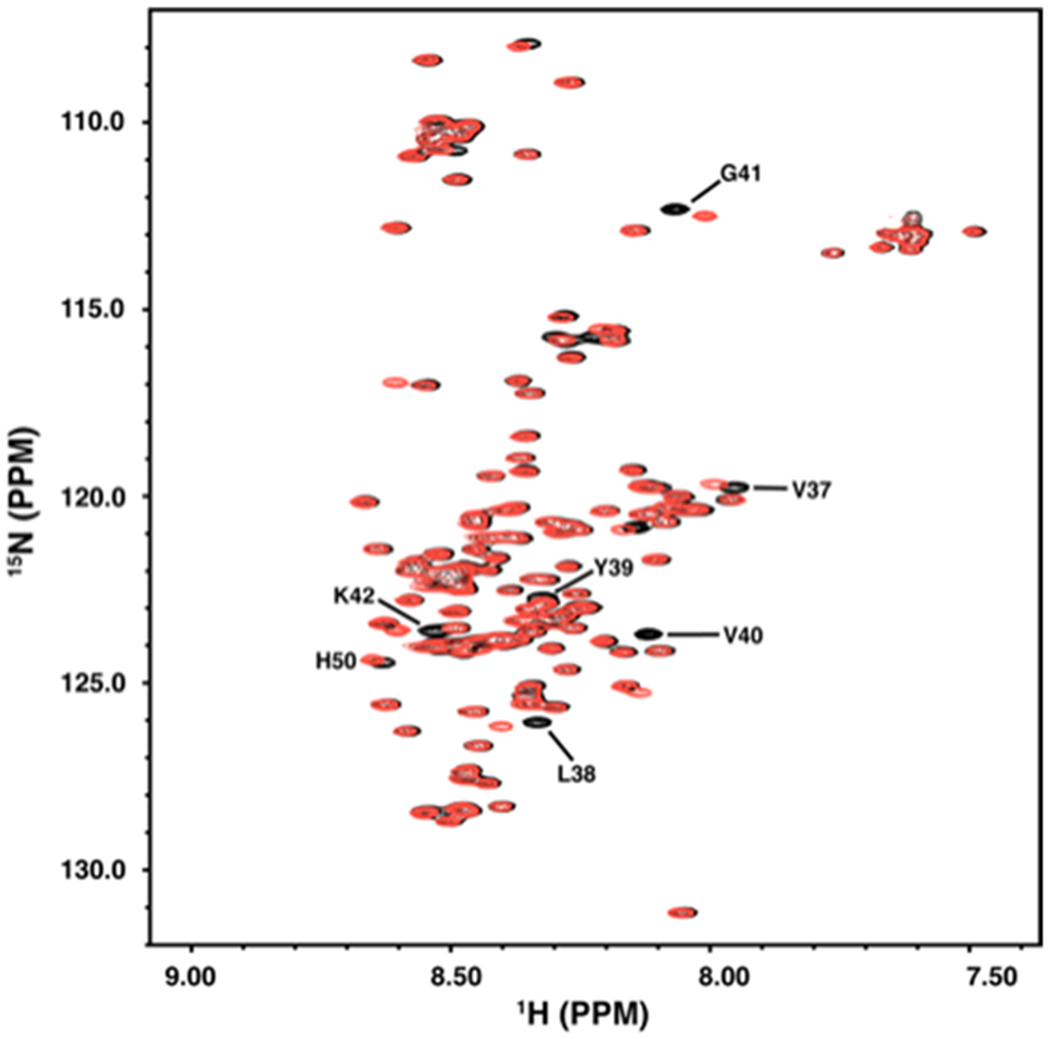
NMR 2D proton-nitrogen correlation spectrum (1H-15N HSQC) of αS-pY39. The 1H-15N spectrum of site-specifically modified semi-synthetic 15N αS-pY39 (red) is compared to that of the unmodified protein (black). Signals near residue Tyr39 that exhibit chemical shift changes as a result of the altered local electrochemical environment are indicated. Residue His50 is also indicated as this resonance is exquisitely sensitive to pH shifts, indicating a small difference in the pH of the samples. Resonance assignments are as in Eliezer et al.[22]
Conclusion
We constructed a vector consisting of αS and cAbl in an attempt to develop a strategy for the generation of proteins and peptides bearing pTyr by co-expression. Despite some success in obtaining phosphorylated αS1-55 when co-expressed with cAbl and YopH, the levels of phosphorylation were not as high compared to previous strategies taking advantage of in vitro phosphorylation: Up to 80% isolated yield of phosphorylated peptide was attained using separately purified cAbl. Several factors may affect the success of producing a modified peptide or protein of interest via the co-expression method: the burden on E. coli cells of expressing certain types of proteins, the compatibility of growth conditions required by each protein, the extent of co-localization of enzyme and substrate in the cell, and the difficulty of controlling stoichiometry compared to in vitro reactions. Thus, the feasibility of producing modified peptides by co-expression with enzyme may vary depending on the enzyme and substrate in question. Nevertheless, the strategy is worthy of further exploration with a variety of other modifications and would be a valuable additional tool in the semi-synthesis of modified proteins.
In this work, we presented an evaluation of all methods – established, novel, and potential – for the semi-synthesis of αS-pY39. Comparing the various methods that we and others have established for producing αS-pY39, we found that in vitro chemoenzymatic semi-synthesis is the most efficient and scalable. Importantly, it is the most amenable to the incorporation of heavy isotopic labels. Compared to incorporation of 15N by SPPS, the in vitro chemoenzymatic method obviates the need for expensive labeled, protected amino acids. With this chemoenzymatic semi-synthesis, combined with strategic use of MTG, which allowed for one-pot desulfurization, we obtained 15N αS-pY39 in superior yield to all other synthetic routes. It is worth noting that other methods may still be worth exploring, such as trans-splicing using split inteins,[23] which would avoid the need to cleave each intein separately and can be used for segmental isotope labelling.[24] Other thiol additives like TFET could also be useful, although we have found MTG to be quite favourable here as well as in our recent execution of an efficient synthesis of αS bearing multiple arginylation modifications using MTG for one-pot desulfurization.[25] Finally, HSQC NMR of αS-pY39 allowed us to confirm the effects of this site-specific PTM on the local environment of the protein in the absence of additional mutations and validated the utility of our scalable, efficient, and facile semi-synthesis of phosphorylated αS.
Experimental Section
Construction of pETDuet-αS-cAbl plasmid.
Site-directed mutagenesis was carried out by polymerase chain reaction (PCR) using Pfu Turbo polymerase on cAbl plasmid to introduce SacI/HindIII restriction sites and on αS to introduce the XhoI restriction site. The corresponding restriction enzymes were used to excise the DNA sequence of interest to subclone the cAbl gene into the pETDuet vector multiple cloning site 1 (MCS1) and the sequence for αS-Mxe-His6 gene into MCS2. Full-length αS fused to a polyhistidine-tagged GyrA intein from Mycobacterium xenopi (Mxe) in pTXB plasmid was used as a starting point for αS cloning.[14] The XhoI restriction site was introduced at the end of the sequence of interest to be used in combination with an existing NdeI site at the beginning. For the kinase, the restriction sites SacI and HindIII were introduced into a plasmid consisting of ABL1_HUMAN_D0 in a pET 2B-T-10 backbone.[16] Using the corresponding enzyme for these two sites, cAbl kinase was inserted into pETDuet multiple cloning site 2 (MCS2). Similarly, αS was inserted into MCS1 of pETDuet.
Recombinant production of αS and cAbl from pETDuet.
For expression of αS and cAbl only, pETDuet plasmid containing αS and cAbl was transformed into BL21 DE3 competent cells by heat shocking at 42 °C. Cells plated on ampicillin (Amp) plates were grown at 37 °C overnight. Primary cultures in 5 mL LB media supplemented with 0.1 mg/mL Amp were grown from single colonies and used to inoculate secondary cultures. These were incubated at 37 °C with shaking at 250 rpm until optical density (OD) reached ~0.6. isopropyl-β-D-1-thiogalactopyranoside (IPTG) was added to final concentration 1 mM to induce expression of the genes of interest. Cells were then incubated with shaking at 18 °C overnight. Protein was harvested by centrifuging the culture (5000 rpm, 20 min, 4 °C), resuspending cell pellets in buffer (20 mM Tris pH 8.3, 1 Roche protease inhibitor tablet) and sonicating the sample in a cup on ice (5 min, 1 s ON, 1 s OFF). The resulting lysate was centrifuged (14,000 rpm, 25 min, 4 °C), and supernatant containing the protein of interest (POI) was purified over a Ni-NTA affinity column. To generate full-length αS with a C-terminal carboxylate, intein cleavage was carried out by incubation with 200 mM β-mercaptoethanol (βME) on a rotisserie over night at room temperature.[26]
For expression of αS and cAbl with YopH using competent cells without pre-transformed plasmid, pETDuet and protein tyrosine phosphatase (YopH) plasmids were co-transformed into BL21 cells by heat shocking, and cells were grown as above except using media supplemented with Amp and streptomycin (Str). For expression of αS and cAbl using pre-transformed YopH competent cells, the pETDuet plasmid was transformed by heat shocking into pre-prepared competent cells bearing the YopH plasmid, and expression was again carried out as described. Existence of phosphorylated protein was checked by MALDI MS.
Semi-synthesis of αS1-55 fragment.
Deletion PCR was performed to generate the αS1-55-intein construct in the pETDuet plasmid containing cAbl and full-length αS. PCR was carried out using Q5 polymerase and Master Mix. Reaction mixture was purified using a DNA extraction kit. DNA was incubated with DpnI and simultaneously circularized by incubating with T4 DNA ligase and T4 polynucleotide kinase (PNK) at 37 °C for 1 h. Plasmids were then directly transformed into DH5α cells and sequenced to verify identity.
After expressing the proteins as described, intein cleavage was carried out to obtain the αS1-55 fragment bearing a C-terminal thioester. Excess imidazole was removed by dialysis before transthioesterification with 200 mM sodium 2-mercaptoethanesulfonate (MESNa) overnight at 4 °C on a stir plate. Cleaved POI was dialyzed into 20 mM Tris, pH 8. Protein products were purified by reverse-phase high-performance liquid chromatography (HPLC) using a C4 column.
In Vitro phosphorylation of αS1-55.
Purified αS1-55-MES was dissolved in buffer (50 mM Tris, 150 mM NaCl,pH 7.5) and incubated with 0.1 equiv cAbl, 2 mM Mg-ATP, and 5 mM MgCl2 in a water bath at 30 °C. The reaction was supplemented with cAbl, Mg-ATP, and MgCl2 as necessary. Samples collected at each time point were analyzed by analytical HPLC to monitor the extent of phosphorylation. Product identity was confirmed by MALDI MS.
Native Chemical Ligation (NCL).
15N αS1-55-pY39-MES was re-dissolved in NCL buffer (6M Gdn•HCl, 200 mM Na2HPO4, pH 7.0) to 2 mM final concentration. 2 equiv 15N αS56-140-C56 ligation partner was added as a powder, and reaction was initiated by adding 100 equiv MTG from a stock dilution in NCL buffer pH 7.0. The reaction was supplemented with tris(2-carboxyethyl) phosphine (TCEP) to a 40 mM final concentration, adjusted to pH 6.8-7.0, and incubated at 37 °C with agitation at 500 rpm. After overnight incubation, NCL reaction mixture was diluted ~5-fold with NCL buffer pH 7.0. One-pot desulfurization was carried out by adding 20 mM radical initiator VA-044, 10% (v/v) t-BuSH, and 250 mM TCEP. The reaction tube was purged with argon and incubated overnight at 37 °C. The desulfurized product was purified by RP-HPLC using a C4 column.
NMR.
Proton-nitrogen correlation (1H-15N HSQC) spectra of unmodified and αS-pY39 were obtained on a 600 MHz Bruker Avance instrument equipped with a cryogenic probe using methods similar to those previously reported for αS-pY39F125F131 in Dikiy et al.[6]
Supplementary Material
Acknowledgements
This research was supported by the National Institutes of Health (NIH NS103873 to E.J.P.; NS079955 to E.R.; GM136686 and AG019391 to D.E.; S10OD016320 to the Weill Cornell NMR Core Facility). Instruments supported by the National Science Foundation include a matrix-assisted laser desorption ionization mass spectrometer (NSF MRI-0820996). B.P. thanks the University of Pennsylvania for support through a Dissertation Completion Fellowship.
Footnotes
Supporting information (SI), including detailed protocols for cloning, protein expression, ligation and purification, mass spectra and NMR spectra, is given via a link at the end of the document.
Institute and/or researcher Twitter usernames: @EJPLab
References
- [1].a) Hunter T, Curr. Opin. Cell Biol 2009, 21, 140–146; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Blume-Jensen P, Hunter T, Nature 2001, 411, 355–365; [DOI] [PubMed] [Google Scholar]; c) Tan CSH, Bodenmiller B, Pasculescu A, Jovanovic M, Hengartner MO, Jorgensen C, Bader GD, Aebersold R, Pawson T, Linding R, Sci. Signal 2009, 2, 13; [DOI] [PubMed] [Google Scholar]; d) Krause DS, Van Etten RA, N. Engl. J. Med 2005, 353, 172–187. [DOI] [PubMed] [Google Scholar]
- [2].Spillantini MG, Schmidt ML, Lee VMY, Trojanowski JQ, Jakes R, Goedert M, Nature 1997, 388, 839–840. [DOI] [PubMed] [Google Scholar]
- [3].Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M, Proc. Natl. Acad. Sci. U. S. A 1998, 95, 6469–6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Mahul-Mellier AL, Fauvet B, Gysbers A, Dikiy I, Oueslati A, Georgeon S, Lamontanara AJ, Bisquertt A, Eliezer D, Masliah E, Halliday G, Hantschel O, Lashuel HA, Human Molecular Genetics 2014, 23, 2858–2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Brahmachari S, Ge P, Lee SH, Kim D, Karuppagounder SS, Kumar M, Mao XB, Shin JH, Lee Y, Pletnikova O, Troncoso JC, Dawson VL, Dawson TM, Ko HS, J. Clin. Invest 2016, 126, 2970–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Dikiy I, Fauvet B, Jovičić A, Mahul-Mellier A-L, Desobry C, El-Turk F, Gitler AD, Lashuel HA, Eliezer D, ACS Chem. Biol 2016, 11, 2428–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Pan B, Rhoades E, Petersson EJ, ACS Chem. Biol 2020, 15, 640–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhao K, Lim Y-J, Liu Z, Long H, Sun Y, Hu J-J, Zhao C, Tao Y, Zhang X, Li D, Li Y-M, Liu C, Proc. Natl. Acad. Sci. U. S. A 2020, 117, 20305–20315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Middleton ER, Rhoades E, Biophys. J 2010, 99, 2279–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wojciechowski M, Grycuk T, Antosiewicz JM, Lesyng B, Biophys. J 2003, 84, 750–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Fan CG, Ip K, Soll D, FEBS Lett. 2016, 590, 3040–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Luo XZ, Fu GS, Wang RSE, Zhu XY, Zambaldo C, Liu RH, Liu T, Lyu XX, Du JT, Xuan WM, Yao AZ, Reed SA, Kang MC, Zhang YH, Guo H, Huang CH, Yang PY, Wilson IA, Schultz PG, Wang F, Nat. Chem. Biol 2017, 13, 845-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Haney CM, Wissner RF, Petersson EJ, Curr. Opin. Chem. Biol 2015, 28, 123–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Haney CM, Wissner RF, Warner JB, Wang YXJ, Ferrie JJ, Covell DJ, Karpowicz RJ, Lee VMY, Petersson EJ, Org. Biomol. Chem 2016, 14, 1584–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Johnson M, Coulton AT, Geeves MA, Mulvihill DP, PLoS One 2010, 5, 5; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Trexler AJ, Rhoades E, Protein Sci. 2012, 21, 601–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Albanese SK, Parton DL, Isik M, Rodriguez-Laureano L, Hanson SM, Behr JM, Gradia S, Jeans C, Levinson NM, Seeliger MA, Chodera JD, Biochemistry 2018, 57, 4675–4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ren GP, Wang X, Hao SF, Hu HY, Wang CC, J. Bacteriol 2007, 189, 2777–2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].a) Xu R, Liu DS, Cowburn D, Mol. Biosyst 2012, 8, 1878–1885; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Seeliger MA, Young M, Henderson MN, Pellicena P, King DS, Falick AM, Kuriyan J, Protein Sci. 2005, 14, 3135–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wu JJ, Afar DEH, Phan H, Witte ON, Lam KS, Comb. Chem. High Throughput Screen 2002, 5, 83–91. [DOI] [PubMed] [Google Scholar]
- [20].Huang Y-C, Chen C-C, Gao S, Wang Y-H, Xiao H, Wang F, Tian C-L, Li Y-M, Chem. Eur. J 2016, 22, 7623–7628. [DOI] [PubMed] [Google Scholar]
- [21].Thompson RE, Liu X, Alonso-García N, Pereira PJB, Jolliffe KA, Payne RJ, J. Am. Chem. Soc 2014, 136, 8161–8164. [DOI] [PubMed] [Google Scholar]
- [22].Eliezer D, Kutluay E, Bussell R, Browne G, J. Mol. Biol 2001, 307, 1061–1073. [DOI] [PubMed] [Google Scholar]
- [23].Vila-Perelló M, Liu Z, Shah NH, Willis JA, Idoyaga J, Muir TW, J. Am. Chem. Soc 2013, 135, 286–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yamazaki T, Otomo T, Oda N, Kyogoku Y, Uegaki K, Ito N, Ishino Y, Nakamura H, J. Am. Chem. Soc 1998, 120, 5591–5592. [Google Scholar]
- [25].Pan B, Kamo N, Shimogawa M, Huang Y, Kashina A, Rhoades E, Petersson EJ, Submitted 2020. [DOI] [PMC free article] [PubMed]
- [26].Batjargal S, Walters CR, Petersson EJ, J. Am. Chem. Soc 2015, 137, 1734–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.