

Abstract
Achieving therapeutically efficacious levels of gene transfer in tumors has been a major obstacle for cancer gene therapy using replication-defective virus vectors. Recently, replicating viruses have emerged as attractive tools for cancer therapy, but generally achieve only transitory tumor regression. In contrast to other replicating virus systems, transduction by replication-competent retrovirus (RCR) vectors is efficient, tumor-selective, and persistent. Correlating with its efficient replicative spread, RCR vector expressing the yeast cytosine deaminase suicide gene exhibited remarkably enhanced cytotoxicity in vitro after administration of the prodrug 5-fluorocytosine. In vivo, RCR vectors replicated throughout preestablished primary gliomas without spread to adjacent normal brain, resulting in profound tumor inhibition after a single injection of virus and single cycle of prodrug administration. Furthermore, stable integration of the replicating vector resulted in persistent infection that achieved complete transduction of ectopic glioma foci that had migrated away from the primary tumor site. Thus, efficient and stable integration of suicide genes represents a unique property of the RCR vector that achieved multiple cycles of synchronous cell killing upon repeated prodrug administration, resulting in chronic suppression of tumor growth and prolonged survival.
Keywords: retrovirus, replication competent, gene therapy, suicide gene, yeast cytosine deaminase, prodrug, brain cancer, long-term survival
Introduction
Replicating forms of a number of different virus species, including adenovirus, herpesvirus, vaccinia virus, reovirus, poliovirus, and vesicular stomatitis virus, are now being exploited as oncolytic agents to treat tumors [1,2]. However, the basis for attenuation or tumor selectivity in many oncolytic viruses is not well characterized; furthermore, cytolytic effects achieved have often proven to be transient, followed by disease recurrence after viral clearance. Recently, we have developed a murine leukemia virus (MLV)-based replication-competent retrovirus (RCR) vector as a cancer therapeutic agent [3–5], and we propose that this strategy may have significant advantages for oncolytic therapy.
It is well established that the MLV nucleocapsid complex contains no nuclear localization signal for active nuclear uptake, hence the virus can infect only cells that are actively dividing [6], and this absolute selectivity for dividing cells allows highly efficient and tumor-selective replication and gene transfer. Indeed, this selectivity was precisely the rationale for the original clinical trials of retrovirus-mediated suicide gene therapy for glioblastoma; despite progressing to Phase III, this approach ultimately failed because the replication-defective retrovirus vectors employed simply could not achieve therapeutically adequate levels of gene transfer to diffusely infiltrating glioma cells despite mass reduction through surgical resection and multiple site injections around the entire tumor margin [7].
In contrast, we [3–5,8] and others [9,10] have already demonstrated that MLV-based RCR vectors containing transgene cassettes inserted precisely at the env–3′ UTR border can efficiently transduce and stably propagate over multiple infection cycles, thereby achieving a tremendous in situ amplification effect after initial administration of a small inoculum. As predicted from their robust replicative capabilities, intratumoral injection of RCR vectors was found to be capable of transmitting an inserted transgene throughout entire solid tumor masses in vivo, without detectable spread to extratumoral organs [3,5].
Although not intrinsically cytolytic, replicating MLV can achieve efficient delivery of suicide genes that permanently integrate into the target cell genome, stably expressing prodrug-converting enzymes, which would kill the infected cell through production of cytotoxic metabolites upon administration of the appropriate prodrug at any desired time. Since prodrug conversion is intracellular and confined to tumor cells, the adverse side effects associated with systemic administration of toxic chemotherapeutic agents can be avoided. Since gene transfer with MLV is stable and nonlytic, the prodrug can be administered at any appropriate time, e.g., upon recurrence of malignant cells that escaped initial surgical treatment; this also represents a mechanism through which the activity of the vector can be regulated. We previously reported initial results in an intracranial glioma model [5], demonstrating the therapeutic survival benefit achieved by an RCR vector encoding the yeast cytosine deaminase (CD) suicide gene [11], which results in conversion of the nontoxic prodrug 5-fluorocytosine (5-FC) to the highly toxic metabolite 5-fluorouracil (5-FU), one of the most active antineoplastic agents in conventional cancer chemotherapy [12].
Here, we present a definitive correlation of the contribution of replicative retroviral suicide gene vector spread, resulting in virtually complete and persistent tumor cell transduction, with highly efficient cell killing and significant improvements in survival. Also, this is the first morphological/immunohistochemical study demonstrating the highly tumor-selective nature of replicating retrovirus transduction within the CNS, without evidence of any significant spread to surrounding normal brain. Our results demonstrate that highly efficient and tumor-selective cell killing can be achieved both in culture and in vivo and furthermore that progressive and stable suicide gene transduction by the replicating retrovirus represents a unique property that confers the ability to achieve prolonged survival through multiple cycles of prodrug administration even after only a single intratumoral dose of virus supernatant.
Results
RCR Vectors Achieve Highly Efficient Transgene Delivery in Glioma Cells
The RCR vector ACE-GFP (Fig. 1A) contains the wild-type ecotropic Moloney MLV provirus in which the ecotropic envelope was replaced with the amphotropic envelope from 4070A [8]. We replaced the 5′ U3 region with the cytomegalovirus promoter and positioned an encephalomyocarditis virus (EMCV) internal ribosome entry site (IRES)–green fluorescent protein (GFP) cassette between the amphotropic envelope and the 3′ UTR [13]. To examine replicative spread and transduction efficiency of RCR vectors in glioma cells in culture, we infected U-7, A-172, and T-98G cells with the ACE-GFP virus at low multiplicity of infection. Flow-cytometric analysis showed that ACE-GFP could efficiently transduce each glioma cell line and spread throughout entire cell populations (Fig. 1B). We further studied the replication kinetics of ACE-GFP in U-87 by mixing infected and uninfected U-87 cells at varying percentages as indicated. When even as few as 0.1% of the cells initially expressed ACE-GFP, almost the entire U-87 population exhibited GFP fluorescence by postinoculation day 7 (Fig. 1C) with more rapid replication kinetics correlating with higher percentages of initial producer cells.
FIG. 1.

Replication kinetics of RCR vectors in glioma cells. (A) Structure of ACE-GFP, showing location of the IRES-GFP insert between env and 3′ UTR. CMV, cytomegalovirus promoter. (B) Glioma cells were inoculated with ACE-GFP at a multiplicity of infection of 0.05. At various time points after infection, the cells were analyzed by flow cytometry to determine the percentage of cells expressing GFP. The x axis shows days after virus inoculation. (C) ACE-GFP-transduced U-87 cells were mixed with their uninfected parental cells as indicated. At 4 and 7 days postmixing, the cells were analyzed for GFP expression. The x axis shows days after cell mixing.
Dose-Dependent Cell Killing with 5-FC Administration after RCR-Mediated Gene Delivery of Yeast CD
We have previously characterized RCR vectors containing various insertions at the env-3′ UTR boundary over multiple serial infection cycles in vitro and found that such inserts can be well tolerated as long as their size does not exceed approximately 1.3 kb or contain recombinogenic sequences [8]; therefore, we chose the relatively small yeast CD suicide gene (474 bp) as a therapeutic suicide gene for the current study. We first performed in vitro studies to examine the suicide gene function of RCR vector ACE-CD (Fig. 2A), which carries an EMCV IRES–yeast CD suicide gene cassette replacing the IRES-GFP cassette in ACE-GFP [5]. To determine the effective prodrug concentration, we first treated U-87 cells that had been fully transduced with ACE-CD with 5-FC at various concentrations ranging from 0.1 to 5 mM. To detect any potential nonspecific toxicity due to the prodrug itself, we also treated uninfected and ACE-GFP-transduced U-87 cells with corresponding concentrations of 5-FC. The prodrug showed no cytotoxicity at concentrations of 2 mM or less on uninfected and ACE-GFP-transduced cells (data not shown), and therefore, we designated 2 mM as the maximum tolerated concentration for further in vitro experiments. ACE-CD infection resulted in potent cell killing with 5-FC at all concentrations used, and killing efficiency was well correlated with increasing 5-FC dosage (Fig. 2B).
FIG. 2.

Cytotoxicity of ACE-CD/5-FC on tumor cells. (A) Structure of ACE-CD, showing location of IRES-CD insert between env and 3′ UTR. (B) ACE-CD-transduced U-87 cells were treated with 5-FC on day 0 at various concentrations ranging from 0 to 5 mM. Viability of cells was determined in a triplicate repeat with the MTS assay. The x axis shows days after 5-FC incubation. The y axis shows the viability percentage of the cells normalized to that of negative control, 0 mM. (C) The ACE-CD-transduced and untransduced U-87 cells were mixed at various initial ratios (0, 0.1, 1, 10, and 100% of ACE-CD-transduced cells) and seeded onto 96-well plates. The mixed cell populations were exposed to 2 mM 5-FC or to control medium without 5-FC, and viability of cells was determined with MTS assay. The values shown of viability percentage of the cells were normalized to that of negative control (0%/5-FC−). 5-FC−, without 5-FC incubation. 5-FC+, with 5-FC incubation.
Highly Efficient and Progressive Suicide Gene-Mediated Cytotoxicity Correlates with ACE-CD Infectivity and Replication Kinetics
We treated ACE-CD-transduced U-87 cells mixed with uninfected cells at varying percentages with prodrug 5-FC at 2 mM concentration and normalized cell viability over time to that of a negative control composed of uninfected cells without prodrug (0%/5-FC−) (Fig. 2C). The 5-FC prodrug alone (0%/5-FC+) and the ACE-CD virus alone without addition of prodrug (100%/5-FC−) were both confirmed to be nontoxic. However, when initially even as few as 0.1% of the tumor cells were producing ACE-CD, we observed substantial cell killing over time after incubation with 5-FC (Fig. 2C); this killing effect could not have been achieved by a bystander effect alone [14] and indicates amplification of the transduced population by replicative spread of the vector. Higher percentages of initially transduced cells in the culture plates achieved correspondingly faster clearance of the tumor cell population and correlated with the kinetics of viral replication observed previously.
It should be noted that, at prodrug concentrations that were nontoxic to uninfected cells, cytotoxicity induced by intracellular conversion of 5-FC to 5-FU required some time to exert its maximum effect. In our cell culture studies, even in the fully transduced control group in which 100% of the cells expressed CD from the outset, it still required 10 to 12 days to achieve 100% cell killing with 2 mM 5-FC (Fig. 2C). However, this time delay may be advantageous for sustaining viral replication: during prodrug administration cycles, virus-transduced cells are concurrently being killed over time by converted 5-FU while in the process of producing more ACE-CD virus. Thus, fewer ACE-CD viruses may be produced from these dying cells, resulting in a slower time course of RCR spread; this may be partially offset by the prolonged time course of cell killing.
ACE-CD Significantly Prolongs Survival of Athymic Mice Implanted with U-87 Intracranial Gliomas after a Single Cycle of Treatment with 5-FC
To determine whether the high level of transduction efficiency achieved by replicating retrovirus vectors could lead to therapeutic benefit, we then tested ACE-CD in an intracerebral U-87 glioma model. After stereotactic intratumoral injection of ACE-CD or saline vehicle control (PBS) into preestablished gliomas, we performed a single cycle of intraperitoneal 5-FC or PBS administration for 15 consecutive days. The mice treated with ACE-CD and 5-FC showed a doubling of median survival time over a follow-up period of more than 70 days (Fig. 3A), compared to three control groups.
FIG. 3.

Survival and histological analyses after single-cycle RCR suicide gene therapy. (A) Survival analysis of athymic mice bearing intracerebral U-87 glioma. ACE-CD or PBS was stereotactically injected into intracerebral U-87 xenografts 7 days after tumor inoculation. Eight days after injection, mice received daily intraperitoneal injections of 5-FC or PBS for 15 consecutive days. Survival curves were constructed for four treatment groups as indicated. ACE-CD/5-FC vs ACE-CD/PBS, ACE-CD/5-FC vs PBS/5-FC, and ACE-CD/5-FC vs PBS/PBS, all show significance at P < 0.0001. (B) Brain sections and immunohistochemical analysis. Top shows low-magnification views of whole brain sections at primary tumor inoculation site. Bottom shows immunohistochemistry using antiviral envelope antibody at primary tumor site (PBS/5-FC, ACE-CD/PBS, ACE-CD/5-FC) or ectopic tumor site (ACE-CD/5-FC). Red/brown staining indicates positive immunohistochemical signal. Original magnifications: bottom left three, ×200; bottom right, ×100. (C) Immunohistochemical staining of periventricular region in ACE-CD/PBS-treated animals. Sections in which the tumor mass is encroaching upon (left) or penetrating (right) the ventricle were stained using antiviral envelope antibody, as above. Original magnification: ×400.
ACE-CD Achieves Complete and Highly Selective Transduction of Intracranial Gliomas in Vivo with Profound Inhibition of Primary Tumor Growth after a Single Cycle of 5-FC Treatment
Examination of whole brain sections from the control groups revealed sizable U-87 gliomas at the primary tumor inoculation site, with disturbance of the normal brain architecture (Fig. 3B, PBS/5-FC and ACE-CD/PBS). Immunohistochemical staining of the primary tumors using an antiviral envelope antibody showed no staining of gliomas treated with 5-FC alone (Fig. 3B, PBS/5-FC), but robust staining of gliomas that had been injected with the replicating ACE-CD vector (Fig. 3B, ACE-CD/PBS). Upon higher magnification, it was observed that the anti-MLV signal was completely confined to the tumor cells without spread to surrounding normal brain, with a sharp demarcation between tumor and peritumoral brain tissues, suggesting that viral replication is highly selective for the rapidly dividing glioma cells.
At high magnification, we also observed masses of tumor cells that were encroaching upon or penetrating into the ventricles, which showed distinct immunohistochemical staining with the viral envelope-specific antibody in ACE-CD-transduced animals. Notably, however, adjacent normal-appearing areas underlying the ependymal cell layer lining the ventricle did not show significant staining (Fig. 3C).
In contrast to the large tumor masses observed in the control groups, ACE-CD vector plus 5-FC-treated animals showed only small lesions remaining at the primary inoculation site, which appeared to consist primarily of residual scar tissue with little evidence of any viable tumor (Fig. 3B, ACE-CD/5-FC). Thus, even a single injection of a relatively low dose of RCR vector supernatant was able to achieve highly efficient delivery of the suicide gene throughout the primary lesion, resulting in profound inhibition of tumor growth.
Eventually, despite successful treatment of the primary lesion and doubling of the mean survival time (Fig. 3A), we observed poor mobility and reduced feeding behavior, lethargy, and death of ACE-CD/5-FC-treated animals, associated with migratory spread of glioma cells to ectopic intracranial sites. Interestingly, however, anti-MLV immunohistochemistry demonstrated viral persistence in all glioma foci examined; that is, not only the primary tumor inoculation site but also ectopic sites to which the tumor cells had migrated were completely positive for virus staining (Fig. 3B, ACE-CD/PBS), including lesions that had apparently escaped the single cycle prodrug treatment (Fig. 3B, ACE-CD/5-FC). This suggested that the permanent integration and stable transduction achieved by the RCR vector in tumor cells could be exploited for ongoing destruction of migrating tumor foci and that additional cycles of 5-FC treatment might achieve further therapeutic benefit even after only a single injection of RCR vector supernatant.
Multiple Cycles of 5-FC Administration after a Single Treatment with ACE-CD Further Improve Long-Term Survival
As before, we performed a single intratumoral injection of ACE-CD vector or saline vehicle after prior establishment of intracerebral U-87 gliomas; subsequently, we performed multiple cycles of 5-FC prodrug or PBS administration by intraperitoneal injection for 8 consecutive days at 3-week intervals. Both control groups (Fig. 4A, ACE-CD/PBS, PBS/5-FC) showed similar results with a median survival time of approximately 30 days, and none of the mice in the control groups survived longer than 38 days. In contrast, the group treated with ACE-CD plus multiple cycles of 5-FC showed 100% survival for more than 100 days (Fig. 4A). Interestingly, toward the end of each 3-week rest interval during which prodrug administration was interrupted, we observed that the animals repeatedly became moribund and exhibited reduced mobility and poor feeding and grooming; strikingly, however, with commencement of the next cycle of 5-FC administration, the animals regained normal mobility and feeding behavior, suggesting that significant suppression of tumor regrowth associated with symptomatic relief was achieved in each cycle.
FIG. 4.

Survival and histological analyses after multiple cycles of 5-FC administration. (A) Survival analysis after multicycle 5-FC treatments. Survival curves were constructed for three treatment groups as indicated. ACE-CD/5-FC vs ACE-CD/PBS and ACE-CD/5-FC vs PBS/5-FC both show significance at P < 0.0001. (B) Histological analysis after multicycle 5-FC treatments. Control groups PBS/5-FC and ACE-CD/PBS showed extensive growth of several large tumors, indicated by numbers, and in contrast, ACE-CD/5-FC group showed a small tumor confined within the primary inoculation site. Anti-MLV immunohistochemistry confirmed viral persistence in glioma foci (ACE-CD/PBS, ACE-CD/5-FC).
Histological examination again revealed that control groups already showed extensive growth of multiple large tumors throughout the CNS (Fig. 4B, PBS/5-FC) by the time of death b38 days post-tumor cell implantation. Immunohistochemistry using a viral envelope-specific antibody again confirmed that, within this time frame, the RCR vector achieved highly efficient spread within every observable glioma lesion throughout the CNS (Fig. 4B, ACE-CD/PBS). In contrast, in the group receiving ACE-CD followed by multiple cycles of 5-FC administration, at the time of termination N100 days post-tumor implantation the majority of animals showed no evidence of significant ectopic foci apart from the primary inoculation site, which again showed profound inhibition of tumor growth (Fig. 4B, ACE-CD/5-FC).
Genetic Stability and Biodistribution of ACE-CD during Prolonged Replication in Vivo
Stably integrated ACE-CD sequences in brain tumor specimens after prolonged periods of replication in vivo were examined by PCR analysis of tumor genomic DNA. As expected, ACE-CD could be readily detected in the transduced gliomas by PCR amplification of MLV sequence flanking the IRES-CD insert (Fig. 5A). In rare cases, we also detected fainter signals corresponding to mutants with partial deletions of the IRES-CD sequence after long-term propagation in brain tumors; however, the full-length IRES-CD sequence was the predominant species amplified in all ACE-CD-infected tumor samples examined and represented the only species detected in the majority of these specimens even after 5-FC prodrug administration.
FIG. 5.
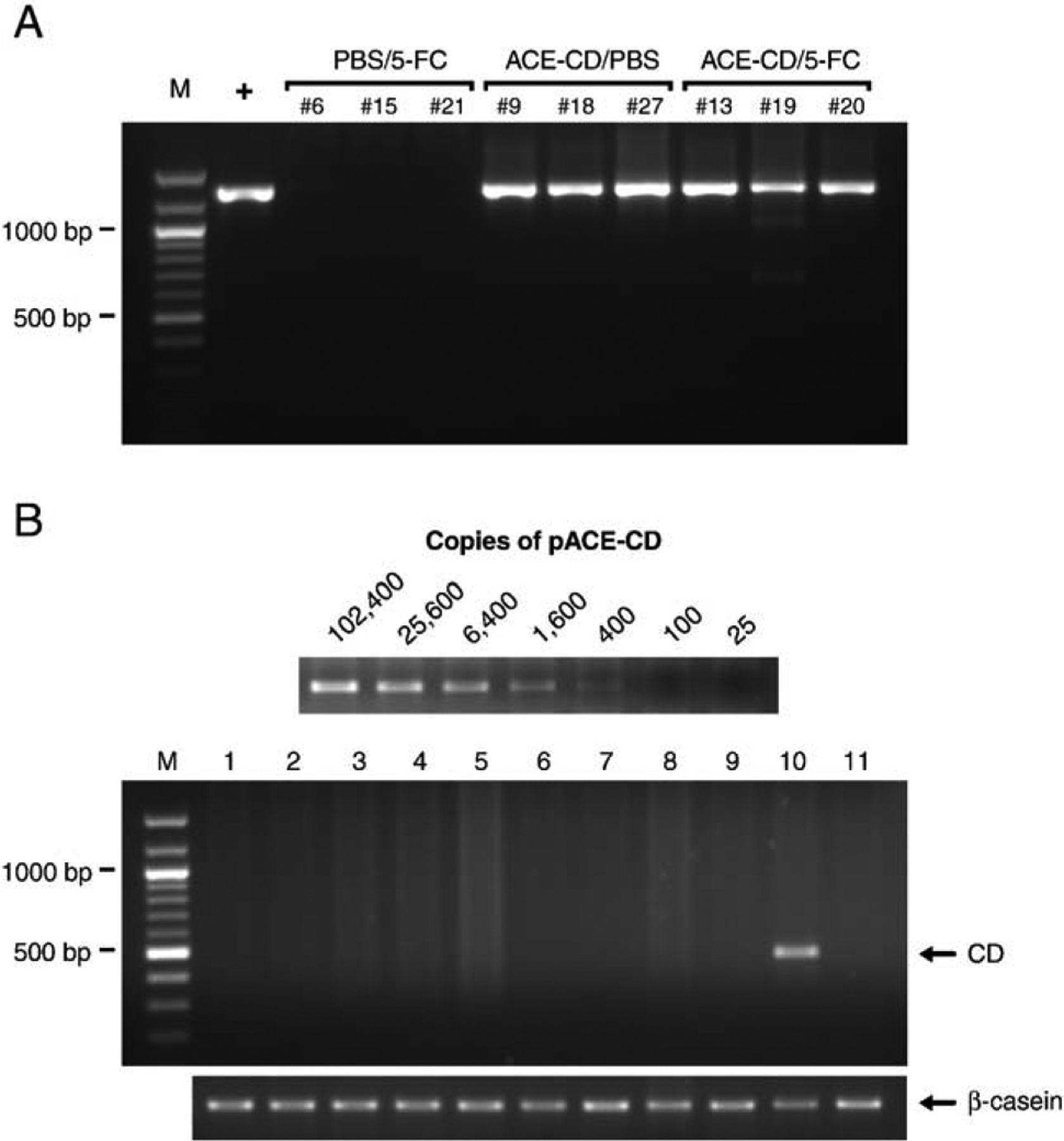
PCR analyses of virus stability and spread. (A) Genetic stability of ACE-CD during prolonged replication in tumors. Genomic DNA of intracranial gliomas was analyzed by PCR using primers in the MLV sequence flanking the IRES-CD insert. The expected size of the full-length PCR product is approximately 1300 bp. Three tumors from each group were assayed, and only the virus-injected tumors (ACE-CD/PBS, ACE-CD/5-FC) show a detectable IRES-CD signal. M, 100-bp DNA marker. +, pACE-CD plasmid DNA. (B) Biodistribution of ACE-CD during prolonged replication in vivo. The sensitivity of the PCR assay was determined by amplification of the CD gene from serially diluted pACE-CD plasmid mixed with untransduced mouse tissue genomic DNA (top); above each lane is indicated the number of copies of the CD gene per approximately 100,000 cell genomes. Six hundred nanograms of genomic DNA isolated from intracranial tumor as well as various extratumoral tissues from the same ACE-CD-injected animal was analyzed by PCR using a primer flanking the CD transgene. The expected size of the full-length PCR product is 458 bp. A 525-bp fragment of the α-casein gene was also amplified from the same genomic DNA sample as an internal control for the PCR procedure. M, 100-bp DNA marker. Lanes 1, lung; 2, liver; 3, esophagus and stomach; 4, intestine; 5, spleen; 6, kidney; 7, skin; 8, bone marrow; 9, contralateral normal brain; 10, intracranial tumor; 11, negative control tumor (no virus injection).
We analyzed the biodistribution of ACE-CD by PCR of genomic DNA from various extratumoral organs, using primers specific for the yeast CD sequence. We determined the detection sensitivity of the PCR assay by amplification of the CD gene from serially diluted pACE-CD plasmid mixed with untransduced mouse tissue genomic DNA. As shown in Fig. 5B, this assay could detect down to 400 copies of the CD gene per approximately 100,000 cell genomes (600 ng of genomic DNA), representing a transduction level of approximately 0.4%. The RCR sequence could be readily detected in the transduced glioma tissue, but extratumoral spread of RCR vector was not detected in any of the normal tissues examined (Fig. 5B). We also tested MLV sequence-specific primers for biodistribution analysis; however, PCR of various extratumoral mouse tissues resulted in multiple bands even in samples from uninfected negative controls, presumably due to the presence of endogenous murine retrovirus-like sequences in the mouse genome (data not shown). Thus, detection of virus revertants that might have lost the CD transgene is not possible with this assay, and we cannot formally rule out the possibility of systemic dissemination of wild type RCR. However, as noted above, because the tumor samples consisted predominantly of human glioma cells, which lack endogenous murine retrovirus-like sequences, we were able to employ MLV-specific primers flanking the IRES-CD cassette in the PCR assay for RCR genomic stability. Therefore, wild-type RCR revertants could be detected as shorter amplification products in this assay, and it is encouraging that, in the majority of cases, only the full-length RCR vector was detected without any evidence of deletions in the CD suicide gene even after prodrug administration (Fig. 5A).
Discussion
A wide range of strategies have been developed for delivery of chemotherapeutic agents for glioma treatment, including infusion pumps, surfactant-coated nanoparticles, implantable polymeric carriers, and osmotic disruption of the blood–brain barrier [15]. Systemic administration of 5-FU is ineffective against brain tumors, due to its poor diffusion across the blood–brain barrier when administered at physiologically tolerable doses [16]. Direct intratumoral administration of 5-FU has been explored for local chemotherapy in patients with malignant gliomas [17], but due to its short half-life and cell cycle phase-specific activity, this approach requires a sustained-release polymer carrier system to achieve any significant therapeutic effect [18,19]. Even this approach, however, is restricted by the limited diffusion of released drug from the polymer implantation site, and median survival was prolonged by less than 1.5-fold [18]. While biodegradable carmustine-impregnated chips for sustained release in the postresection tumor cavity are now in clinical use as Gliadel wafers [20], Phase III trials showed a median improvement in survival of only 2 months [21].
In contrast, replication-competent retroviruses represent a stably integrating, naturally persistent mechanism for sustained release of intracellularly generated chemotoxin, triggered at will by high-dose systemic administration of a nontoxic and well-tolerated prodrug. This represents the first comprehensive report describing the therapeutic application of such a therapeutic RCR vector, which displayed highly efficient tumor cell-selective replication and highly effective suicide gene transduction and cell killing of glioma cells in vitro and in vivo. In vitro cytotoxicity studies showed highly efficient cell killing by ACE-CD even with an initial inoculum as low as 0.1% of the target cell population, indicating that the high efficiency of gene transfer by progressive replicative spread greatly augmented suicide gene toxicity. In vivo, our current results confirm and extend the findings of our previous study [5] demonstrating that a single dose of cell-free vector supernatant followed by a single cycle of 5-FC administration was able to achieve profound inhibition of preestablished primary gliomas without discernable damage to adjacent normal brain tissue, resulting in a doubling of the median survival time compared to controls. These findings are also consistent with and extend previous studies demonstrating that in situ production of retroviral vectors in glioma models can achieve higher transduction levels for longer periods of time due to stable transgene inheritance in progeny tumor cells, without notable transgene expression in normal brain cells [22].
Notably, we observed complete transduction of tumor foci that developed at multiple ectopic sites extending down to the brain stem; thus infection by the RCR vector was sufficiently persistent to follow the tumor cells even as they migrated away from the primary lesion, and continuing multiple cycles of 5-FC administration was found to achieve further long-term survival benefit. The prolonged therapeutic effectiveness of this multiple-cycle treatment regimen represents a novel finding and was possible due to the ability of replicating MLV to integrate stably into the genome of tumor cells and spread even to metastatic lesions after only a single injection; such persistence is unique among oncolytic viruses, which generally achieve only transitory benefit.
Certainly the U-87 tumor model is different from many human gliomas, which generally show more diffuse infiltration into surrounding normal brain. Furthermore, U-87 and other transplantable glioma models contain a high percentage of dividing cells, whereas in human glioblastoma multiforme only a small fraction of tumor cells are dividing at any one time. Thus, optimization of the dosage and timing of RCR and prodrug injections will certainly be required to sustain log-phase growth of the virus and achieve comparable results in human brain tumors.
A major advantage of this multicycle strategy is that the 5-FC prodrug is amenable to oral administration and is well-tolerated at doses 10-fold higher than those typical of systemic 5-FU treatment, resulting in good CNS penetration despite the blood–brain barrier [23]; thus, multiple cycles of systemic 5-FC administration can be performed without the toxicity associated with conventional chemotherapy. Again, further optimization of RCR vector dosage, duration of vector spread prior to initiation of prodrug administration, timing and interval of prodrug administration cycles, etc., may yield further improvements in long-term survival.
In contrast to ganciclovir and its derivatives [24–26], 5-FC and 5-FU can diffuse across cell membranes in the absence of cellular gap junctions, thereby achieving more potent bystander effects on neighboring tumor cells [14]. However, immune responses have also been reported to contribute to bystander effects [27,28]; therefore to assess purely the therapeutic efficacy of RCR-mediated suicide gene therapy in the absence of immune responses, these in vivo studies were performed in athymic mice. Studies using immunocompetent rats bearing intracerebral gliomas derived from syngeneic cells are now under way to examine whether immune responses will affect RCR spread and antitumor effects; from preliminary studies (data not shown) it appears that immunological responses have little impact on RCR spread in the immunosuppressive microenvironment of tumors within the CNS, itself an immunoprivileged site.
The immunohistochemical studies presented here also represent the first direct correlation between replicative transduction efficiency and therapeutic effect of RCR vectors, and we also observed a striking demarcation between transduced tumor tissue and untransduced adjacent brain tissue in the context of the adult CNS. In particular, immunohistochemical staining revealed no evidence of infection by the replicating ACE-CD vector in ependymal cells lining the ventricles or in the subventricular zone, considered to be one of the principal sources of adult neural stem cells in human as well as rodent brain [29]. Certainly, however, this is not definitive, and the possibility that the RCR vectors might transduce noncancerous but actively replicating cells in the peritumoral normal brain requires further careful examination. While wild-type MLV infection in the murine CNS has been reported to cause spongiform neuropathies, these occur only in neonatal mice and in particular mouse strains [30–32] and appear to involve recombination of variant envelope proteins from endogenous murine retroviral elements [33–35]. Thus, the neuropathogenic potential of MLV is unlikely to be a relevant safety concern in humans, as endogenous murine retrovirus-like sequences do not naturally exist in the human genome. In any case, the incorporation of suicide genes into the RCR vector ensures that inadvertently transduced normal cells will have a high probability of eventually being eliminated and that the spread of the vector will be inherently self-limited.
Of course, the potential for adverse events due to uncontrolled systemic spread of replication-competent revertants has been a foremost concern of investigators in the field of retroviral gene therapy [36], and certainly it is now clear that even replication-defective vectors have the potential to cause insertional mutagenesis events that can contribute to the development of malignancies in humans [37]. However, a number of considerations mitigate this concern when contemplating the use of RCR vectors expressing suicide genes for application as a cancer therapeutic agent, particularly for poor-prognosis malignancies such as glioblastoma. The primary setting in which retroviral insertional mutagenesis has been demonstrated to contribute to clonal proliferation has been in the course of gene replacement therapy, when hematopoietic progenitor cells have been directly transduced ex vivo under heavy cytokine stimulation with genes that confer a selective growth advantage [36–39]. In contrast, as observed in current and previous studies [5], due to their absolute requirement for cell mitosis to achieve successful infection, MLV-based vectors showed an inherent tumor selectivity, transducing only actively dividing glioma cells and associated neovasculature [3], but not quiescent normal brain or other extratumoral tissues.
It should be noted, however, that in this nude mouse model, there are few circulating T cells, which represent the natural host cells of this murine leukemia virus. Thus, it is possible that potential routes of cell-borne spread and biodistribution were reduced in this athymic host. Nonetheless, even in this situation, the majority of previous safety studies examining the effect of systemically administering wild-type MLV to primates showed no evidence of any retrovirus-induced pathology [40,41]. Furthermore, as an additional safety mechanism, a variety of retrovirus inhibitors such as 3′-azido-3′-deoxythymidine (AZT) could be used effectively to block retrovirus replication and dissemination [42], and again, the incorporation of a suicide gene results in an inherent self-destruct mechanism that would help to eliminate inadvertently transduced cells.
In these studies, RCR vectors were introduced at extremely low dosages (on the order of 104 infectious units total dose) directly into tumor tissues in vivo. Thus, the potential risk of environmental transmission is likely to be low, particularly since retroviruses are enveloped viruses, which cannot survive for long periods of time in the environment and are not stable as aerosols. As with other enveloped retroviruses such as HIV, the primary route of potential horizontal transmission would be through mucosal contact or direct inoculation into the bloodstream. However, in immunocompetent primates and humans, it has been reported that MLV retrovirus in the bloodstream is rapidly cleared by the immune response [40,41,43].
In conclusion, in the case of poor prognosis malignancies such as glioblastoma, in which conventional surgery, chemotherapy, and radiation regimens constitute little more than palliative measures, the use of replicating viruses as a promising experimental treatment strategy may be warranted, particularly if the therapeutic benefits justify the potential risk. In this context, MLV-based RCR vectors exhibit significant advantages for virotherapy, including inherent tumor selectivity due to an absolute inability to infect quiescent normal cells; a relatively small genome size, which makes it easy to manipulate; and an ability to transduce tumor cells efficiently and stably in vivo. In this experimental glioma model, we have demonstrated that stable integration represents a unique property of RCR vectors that can allow long-term persistence and further viral spread, which allows the virus to follow the tumor cells regardless of their pattern of dissemination. Thus, RCR vectors may be able to achieve sufficiently persistent transgene expression for long-term survival benefit, thereby converting highly aggressive cancers to more indolent chronic disease, and ultimately fulfill the promise of retroviral gene therapy for cancer.
Materials and Methods
Cell lines and virus vector production.
The human embryonic kidney-derived cell line 293T [44] and human glioma cell lines U-87, A-172, and T98-G were grown in DMEM with 10% FBS. Virus stocks were produced by transient transfection of 293T cells with plasmid pACE-GFP or pACE-CD [5] using Lipofectamine Plus (Invitrogen, Carlsbad, CA, USA). For in vitro experiments, 4 μg/ml Polybrene (Sigma, St. Louis, MO, USA) was added at the time of infection. For titer determination, after incubation of each cell line with virus for 24 h, 50 μM AZT (Sigma) was added to prevent ongoing virus replication as described previously [4], and cells were analyzed for GFP expression by using flow cytometry on a FACScan apparatus (Becton-Dickinson, Franklin Lakes, NJ, USA). Viral titer was represented as transducing units/ml.
In vitro transduction of tumor cells.
Tumor cells at 20% confluency in 6-cm dishes were infected with virus ACE-GFP at a multiplicity of infection of 0.05. At various time points after infection, the cells were trypsinized, diluted 1:5, and replated, while an aliquot was analyzed for GFP expression by using flow cytometry. In a separate experiment, ACE-GFP-infected tumor cells were mixed with uninfected cells at a ratio of 0.1, 1, or 10% of the cell population and seeded onto 6-cm dishes. Four and seven days after mixing, the total cell population for each mixture ratio was analyzed for GFP expression.
In vitro sensitivity to prodrug 5-FC.
To determine the toxicity of 5-FC (Sigma) in vitro, uninfected, ACE-GFP-transduced, or ACE-CD-transduced U-87 cells were seeded in replicate 96-well plates (2000 cells/well), cultured overnight, and exposed to 5-FC at various concentrations (0, 0.1, 0.5, 2, and 5 mM). Cell viability was determined daily by MTS assay using the CellTiter Aqueous One Solution Cell Proliferation Assay kit (Promega, Madison, WI, USA).
In vitro cytotoxic efficacy of ACE-CD/5-FC.
Uninfected and ACE-CD-transduced U-87 cells were mixed at various initial ratios as indicated (Fig. 2C) and seeded onto replicate 96-well plates (2000 cells/well). After overnight culture, the mixed cell populations were exposed to 2 mM 5-FC or to control medium without prodrug, and viable cells were determined by MTS assay.
Survival assays using intracerebral tumor models.
Intracerebral U-87 gliomas were established by stereotactic injection into the right frontal lobe in athymic mice (Harlan Sprague Dawley, Indianapolis, IN, USA) as described previously [5]. Seven days later, the mice were randomly distributed to four groups (n = 9 each): two groups of animals were stereotactically injected with 10 μl PBS vehicle control, and two groups were injected with 10 μl ACE-CD (1 × 104 transducing units). Eight days after vector transduction, one of the ACE-CD groups and one of the vehicle groups received daily intraperitoneal injections of 5-FC (500 mg/kg), while the remaining ACE-CD and vehicle groups received daily intraperitoneal injections of PBS for 15 consecutive days.
For multiple-cycle prodrug administration experiments, the ACE-CD vector was injected 5 days after tumor inoculation in two groups of mice (n = 8 each), while one additional group (n = 8) received only PBS vehicle control. Fifteen days after vector transduction, one of the ACE-CD groups and the vehicle group received daily injections of 5-FC, while the remaining ACE-CD group received daily PBS injections, for 8 consecutive days. This 8-day cycle of prodrug or vehicle administration was repeated at intervals of every 3 weeks.
Immunohistochemical analysis.
Frozen sections of brain tumor specimens were prepared as previously described [5]. Representative tumor samples sectioned through their largest diameter were incubated overnight at 4°C with 83A25 antiviral envelope antibody [45] at 1:200 dilution. Immunoreactivity was visualized by Vectastain ABC kit (Vector Laboratories, Burlingame, CA, USA), using 3-amino-9-ethyl-carbazole as the chromagen and hematoxylin for counterstaining.
PCR analyses of virus spread and stability in animals.
Genomic DNA extracted from intracerebral U-87 gliomas was used as template for PCR amplification with an upstream primer hybridizing to the C-terminus of the env gene (5′-TTACCACCTTAATCTCCACCATCA-3′) and downstream primer hybridizing to the 3′ long terminal repeat (5′-TACAGGTGGGGTCTTTCATTC-3′) and visualized by ethidium bromide staining of agarose gels. Genomic DNA isolated from various extratumoral organs of ACE-CD-injected animals was also examined for virus spread by PCR using primers hybridizing to the yeast CD gene (upstream primer, 5′-ACAGGGGGAATGGCAAGC-3′; downstream primer, 5′-TCTTCAAACCAATCCTGAGG-3′). Briefly, 600 ng of each genomic DNA sample was used in a 50-μl PCR with PCR SuperMix (Invitrogen) and primers hybridizing to the yeast CD gene. Products were amplified by 30 cycles of successive incubation at 94°C for 45 s, 53°C for 45 s, and 72°C for 1 min. Eight microliters of each reaction product was resolved on a 1.1% agarose gel and visualized by ethidium bromide staining. A standard curve was generated by amplification of serially diluted ACE-CD plasmid template at specific copy numbers mixed into genomic DNA from uninfected cells. As an internal control for the PCR procedure, a 525-bp fragment of the mouse β-casein gene was also amplified, as previously described [5].
Statistical analysis.
Unpaired Student’s t tests were performed for statistical analysis of cell viability determined by MTS. Survival data were analyzed according to the method of Kaplan–Meier, using SAS software (SAS Institute, Cary, NC, USA) to calculate significance values.
Acknowledgments
We thank Christopher R. Logg, Paula Cannon, and Renata Stripecke for helpful discussion, as well as David Hinton and Florence Hofman for assistance in evaluating intracranial tumor sections. This work was funded by U.S. National Institutes of Health Grants R01-CA105171 (to N.K.) and P01-CA59318 (to N.K.) and by awards from the Connell Foundation and the Kriegel Foundation (to T.C.C.).
References
- 1.Hermiston TW, and Kuhn I (2002). Armed therapeutic viruses: strategies and challenges to arming oncolytic viruses with therapeutic genes. Cancer Gene Ther. 9: 1022–1035. [DOI] [PubMed] [Google Scholar]
- 2.Kirn D, Martuza RL, and Zwiebel J (2001). Replication-selective virotherapy for cancer: biological principles, risk management and future directions. Nat. Med 7: 781–787. [DOI] [PubMed] [Google Scholar]
- 3.Logg CR, Tai CK, Logg A, Anderson WF, and Kasahara N (2001). A uniquely stable replication-competent retrovirus vector achieves efficient gene delivery in vitro and in solid tumors. Hum. Gene Ther 12: 921–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tai CK, Logg CR, Park JM, Anderson WF, Press MF, and Kasahara N (2003). Antibody-mediated targeting of replication-competent retroviral vectors. Hum. Gene Ther 14: 789–802. [DOI] [PubMed] [Google Scholar]
- 5.Wang WJ, Tai CK, Kasahara N, and Chen TC (2003). Highly efficient and tumor-restricted gene transfer to malignant gliomas by replication-competent retroviral vectors. Hum. Gene Ther 14: 117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miller DG, Adam MA, and Miller AD (1990). Gene transfer by retrovirus vectors occurs only in cells that are actively replicating at the time of infection. Mol. Cell. Biol 10: 4239–4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rainov NG (2000). A phase III clinical evaluation of herpes simplex virus type 1 thymidine kinase and ganciclovir gene therapy as an adjuvant to surgical resection and radiation in adults with previously untreated glioblastoma multiforme. Hum. Gene Ther 11: 2389–2401. [DOI] [PubMed] [Google Scholar]
- 8.Logg CR, Logg A, Tai CK, Cannon PM, and Kasahara N (2001). Genomic stability of murine leukemia viruses containing insertions at the Env-3′ untranslated region boundary. J. Virol 75: 6989–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Solly SK, et al. (2003). Replicative retroviral vectors for cancer gene therapy. Cancer Gene Ther. 10: 30–39. [DOI] [PubMed] [Google Scholar]
- 10.Bachrach E, Duch M, Pelegrin M, Dreja H, Pedersen FS, and Piechaczyk M (2003). In vivo infection of mice by replication-competent MLV-based retroviral vectors. Methods Mol. Med 76: 343–352. [DOI] [PubMed] [Google Scholar]
- 11.Kievit E, et al. (1999). Superiority of yeast over bacterial cytosine deaminase for enzyme/prodrug gene therapy in colon cancer xenografts. Cancer Res. 59: 1417–1421. [PubMed] [Google Scholar]
- 12.Nakamura H, Mullen JT, Chandrasekhar S, Pawlik TM, Yoon SS, and Tanabe KK (2001). Multimodality therapy with a replication-conditional herpes simplex virus 1 mutant that expresses yeast cytosine deaminase for intratumoral conversion of 5-fluorocytosine to 5-fluorouracil. Cancer Res. 61: 5447–5452. [PubMed] [Google Scholar]
- 13.Logg CR, Logg A, Matusik RJ, Bochner BH, and Kasahara N (2002). Tissue-specific transcriptional targeting of a replication-competent retroviral vector. J. Virol 76: 12783–12791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huber BE, Austin EA, Richards CA, Davis ST, and Good SS (1994). Metabolism of 5-fluorocytosine to 5-fluorouracil in human colorectal tumor cells transduced with the cytosine deaminase gene: significant antitumor effects when only a small percentage of tumor cells express cytosine deaminase. Proc. Natl. Acad. Sci. USA 91: 8302–8306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tamargo RJ, and Brem H (1992). Drug delivery to the central nervous system: a review. Neurosurg. Q 2: 259–279. [Google Scholar]
- 16.Menei P, Boisdron-Celle M, Croue A, Guy G, and Benoit JP (1996). Effect of stereotactic implantation of biodegradable 5-fluorouracil-loaded microspheres in healthy and C6 glioma-bearing rats. Neurosurgery 39: 117–123 discussion 123–114. [DOI] [PubMed] [Google Scholar]
- 17.Menei P, et al. (2004). Stereotaxic implantation of 5-fluorouracil-releasing microspheres in malignant glioma. Cancer 100: 405–410. [DOI] [PubMed] [Google Scholar]
- 18.Fournier E, et al. (2003). Therapeutic effectiveness of novel 5-fluorouracil-loaded poly(methylidene malonate 2.1.2)-based microspheres on F98 glioma-bearing rats. Cancer 97: 2822–2829. [DOI] [PubMed] [Google Scholar]
- 19.Benoit JP, Faisant N, Venier-Julienne MC, and Menei P (2000). Development of microspheres for neurological disorders: from basics to clinical applications. J. Controlled Release 65: 285–296. [DOI] [PubMed] [Google Scholar]
- 20.Westphal M, et al. (2003). A phase 3 trial of local chemotherapy with biodegradable carmustine (BCNU) wafers (Gliadel wafers) in patients with primary malignant glioma. Neuro-Oncology 5: 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brem H, et al. (1995). Placebo-controlled trial of safety and efficacy of intraoperative controlled delivery by biodegradable polymers of chemotherapy for recurrent gliomas. The Polymer-Brain Tumor Treatment Group. Lancet 345: 1008–1012. [DOI] [PubMed] [Google Scholar]
- 22.Tamiya T, et al. (1995). Transgene inheritance and retroviral infection contribute to the efficiency of gene expression in solid tumors inoculated with retroviral vector producer cells. Gene Ther. 2: 531–538. [PubMed] [Google Scholar]
- 23.Vermes A, Guchelaar HJ, and Dankert J (2000). Flucytosine: a review of its pharmacology, clinical indications, pharmacokinetics, toxicity and drug interactions. J. Antimicrob. Chemother 46: 171–179. [DOI] [PubMed] [Google Scholar]
- 24.Mesnil M, Piccoli C, Tiraby G, Willecke K, and Yamasaki H (1996). Bystander killing of cancer cells by herpes simplex virus thymidine kinase gene is mediated by connexins. Proc. Natl. Acad. Sci. USA 93: 1831–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Touraine RL, Ishii-Morita H, Ramsey WJ, and Blaese RM (1998). The bystander effect in the HSVtk/ganciclovir system and its relationship to gap junctional communication. Gene Ther. 5: 1705–1711. [DOI] [PubMed] [Google Scholar]
- 26.McMasters RA, Saylors RL, Jones KE, Hendrix ME, Moyer MP, and Drake RR (1998). Lack of bystander killing in herpes simplex virus thymidine kinase-transduced colon cell lines due to deficient connexin43 gap junction formation. Hum. Gene Ther 9: 2253–2261. [DOI] [PubMed] [Google Scholar]
- 27.Gagandeep S, et al. (1996). Prodrug-activated gene therapy: involvement of an immunological component in the bbystander effectQ. Cancer Gene Ther. 3: 83–88. [PubMed] [Google Scholar]
- 28.Vile RG, Nelson JA, Castleden S, Chong H, and Hart IR (1994). Systemic gene therapy of murine melanoma using tissue specific expression of the HSVtk gene involves an immune component. Cancer Res. 54: 6228–6234. [PubMed] [Google Scholar]
- 29.Sanai N, et al. (2004). Unique astrocyte ribbon in adult human brain contains neural stem cells but lacks chain migration. Nature 427: 740–744. [DOI] [PubMed] [Google Scholar]
- 30.Bilello JA, Pitts OM, and Hoffman PM (1986). Characterization of a progressive neurodegenerative disease induced by a temperature-sensitive Moloney murine leukemia virus infection. J. Virol 59: 234–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zachary JF, Knupp CJ, and Wong PK (1986). Noninflammatory spongiform polioencephalomyelopathy caused by a neurotropic temperature-sensitive mutant of Moloney murine leukemia virus TB. Am. J. Pathol 124: 457–468. [PMC free article] [PubMed] [Google Scholar]
- 32.Wong PK, et al. (1992). Murine leukemia virus induced central nervous system diseases. Leukemia 6(Suppl. 3): 161S–165S. [PubMed] [Google Scholar]
- 33.Oldstone MB, Jensen F, Elder J, Dixon FJ, and Lampert PW (1983). Pathogenesis of the slow disease of the central nervous system associated with wild mouse virus. III. Role of input virus and MCF recombinants in disease. Virology 128: 154–165. [DOI] [PubMed] [Google Scholar]
- 34.Traister RS, and Lynch WP (2002). Reexamination of amphotropic murine leukemia virus neurovirulence: neural stem cell-mediated microglial infection fails to induce acute neurodegeneration. Virology 293: 262–272. [DOI] [PubMed] [Google Scholar]
- 35.Peterson KE, Robertson SJ, Portis JL, and Chesebro B (2001). Differences in cytokine and chemokine responses during neurological disease induced by polytropic murine retroviruses map to separate regions of the viral envelope gene. J. Virol 75: 2848–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Donahue RE, et al. (1992). Helper virus induced T cell lymphoma in nonhuman primates after retroviral mediated gene transfer. J. Exp. Med 176: 1125–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hacein-Bey-Abina S, et al. (2003). LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 302: 415–419. [DOI] [PubMed] [Google Scholar]
- 38.Marshall E (2002). Gene therapy a suspect in leukemia-like disease. Science 298: 34–35. [DOI] [PubMed] [Google Scholar]
- 39.Li Z, et al. (2002). Murine leukemia induced by retroviral gene marking. Science 296: 497. [DOI] [PubMed] [Google Scholar]
- 40.Cornetta K, et al. (1991). No retroviremia or pathology in long-term follow-up of monkeys exposed to a murine amphotropic retrovirus. Hum. Gene Ther 2: 215–219. [DOI] [PubMed] [Google Scholar]
- 41.Cornetta K, et al. (1990). Amphotropic murine leukemia retrovirus is not an acute pathogen for primates. Hum. Gene Ther 1: 15–30. [DOI] [PubMed] [Google Scholar]
- 42.Mitsuya H, and Broder S (1987). Strategies for antiviral therapy in AIDS. Nature 325: 773–778. [DOI] [PubMed] [Google Scholar]
- 43.Takeuchi Y, Cosset FL, Lachmann PJ, Okada H, Weiss RA, and Collins MK (1994). Type C retrovirus inactivation by human complement is determined by both the viral genome and the producer cell. J. Virol 68: 8001–8007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.DuBridge RB, Tang P, Hsia HC, Leong PM, Miller JH, and Calos MP (1987). Analysis of mutation in human cells by using an Epstein-Barr virus shuttle system. Mol. Cell. Biol 7: 379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Evans LH, Morrison RP, Malik FG, Portis J, and Britt WJ (1990). A neutralizable epitope common to the envelope glycoproteins of ecotropic, polytropic, xenotropic, and amphotropic murine leukemia viruses. J. Virol 64: 6176–6183. [DOI] [PMC free article] [PubMed] [Google Scholar]