

SUMMARY
Oligodendrocyte precursor cells (OPCs) are essential for developmental myelination and oligodendrocyte regeneration after CNS injury. These progenitors express calcium-permeable AMPA receptors (AMPARs) and form direct synapses with neurons throughout the CNS, but the roles of this signaling are unclear. To enable selective alteration of the properties of AMPARs in oligodendroglia, we generate mice that allow cell-specific overexpression of EGFP-GluA2 in vivo. In healthy conditions, OPC-specific GluA2 overexpression significantly increase their proliferation in an age-dependent manner but did not alter their rate of differentiation into oligodendrocytes. In contrast, after demyelinating brain injury in neonates or adults, higher GluA2 levels promote both OPC proliferation and oligodendrocyte regeneration, but do not prevent injury-induced initial cell loss. These findings indicate that AMPAR GluA2 content regulates the proliferative and regenerative behavior of adult OPCs, serving as a putative target for better myelin repair.
Graphical abstract
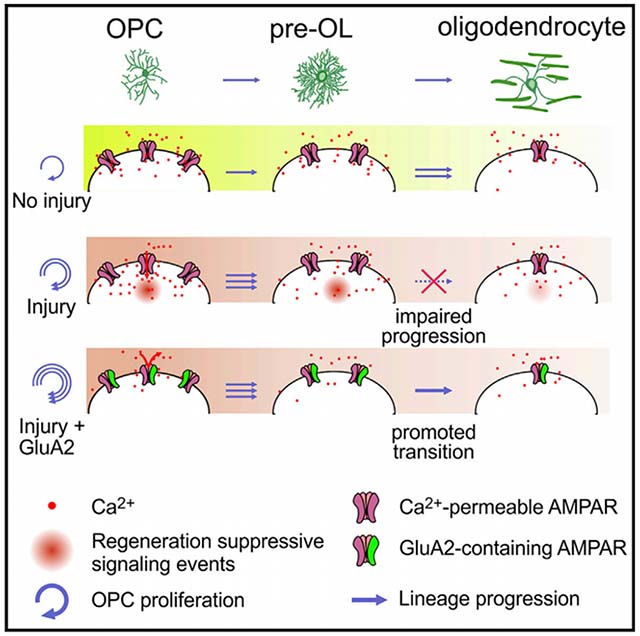
In brief
Khawaja et al. show that increasing expression of GluA2 in oligodendrocyte precursor cells (OPCs), which renders AMPA receptors calcium impermeable, did not alter oligodendrocyte generation, but promoted OPC proliferation and oligodendrocyte regeneration in demyelinating brain injury, suggesting that suppressing AMPA receptor calcium signaling in OPCs could help promote myelin repair.
INTRODUCTION
In the central nervous system (CNS), oligodendrocytes (OLs) enable rapid axonal conduction by forming myelin sheaths (Simons and Nave, 2015). Emerging evidence suggests that they also provide metabolic support to axons (Fünfschilling et al., 2012; Lee et al., 2012) and control extracellular potassium ion homeostasis (Larson et al., 2018). Consequently, OL loss and myelin damage aggravate neuronal dysfunction in CNS injury or disease (Alizadeh et al., 2015; Pan and Chan, 2017). Conversely, enhanced OL regeneration from resident oligodendrocyte precursor cells (OPCs) and improved myelin repair assist in the rapid restoration of neuronal function and limit the spread of tissue damage (Mei et al., 2016; Wang et al., 2018). However, disease- or injury-specific mechanisms of OL loss and effective approaches for promoting OL regeneration need to be identified.
OLs express α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) (McDonald et al., 1998; Patneau et al., 1994; Salter and Fern, 2005), and AMPAR-mediated glutamate excitotoxicity is considered a major mechanism of OL loss in CNS injury (Deng et al., 2003; McCarran and Goldberg, 2007; McDonald et al., 1998; Salter and Fern, 2005). The systemic administration of an AMPAR antagonist reduced OL loss in hypoxic-ischemic (H/I) injury (Follett et al., 2000; McCarran and Goldberg, 2007) and in multiple sclerosis (MS)-like conditions (Pitt et al., 2000). However, AMPARs are also expressed in OPCs at higher levels than in OLs (De Biase et al., 2010; Kukley et al., 2010; Zhang et al., 2014), and synaptic connectivity between neurons and OPCs (Bergles et al., 2000; Kukley et al., 2007; Lin et al., 2005; Ziskin et al., 2007) raises the possibility that AMPAR-mediated, axon-glia synaptic transmission is critical for neuronal activity-regulated oligodendrogenesis (Fields, 2015; Spitzer et al., 2016). Supporting this view, genetic ablation of key AMPAR subunits from all OL lineage cells impaired the survival of newly formed OLs during normal development (Kougioumtzidou et al., 2017). Moreover, local administration of an AMPAR antagonist into demyelinated white matter (WM) partially impaired remyelination (Gautier et al., 2015), pointing to the importance of AMPAR signaling in OL regeneration. Given these conflicting results of AMPAR inhibition on WM injury, OL development, and repair, a more advanced understanding is required of AMPAR signaling aspects that regulate the developmental and regenerative behavior of oligodendroglia.
Here, using a newly developed mouse line for inducible overexpression of the Gln-to-Arg (Q/R) edited GluA2, the calcium-impermeable AMPAR subunit (Hollmann et al., 1991), we identified the effects of elevated GluA2 levels in OPCs for normal OL development and postinjury regeneration. Our results highlight the regulatory role of GluA2 in OPCs for OL regeneration and myelin repair without affecting normal oligodendrogenesis.
RESULTS
A mouse line for cell-specific GluA2 overexpression
To determine how GluA2-containing AMPARs regulate OL development and regeneration, the ROSA26 locus was targeted with a transgene containing CAG promoter, loxP-flanked STOP cassette, and EGFP-fused (Q/R) edited GluA2 (EGFP-Gria2) coding sequences for a new mouse line. After these mice (termed R26-Gria2) are crossed to a Cre driver, Cre-active cells and their progeny will express EGFP-GluA2 in double transgenic (Tg) offspring, (Figure 1A), reducing AMPAR-mediated calcium entry (Figure 1B).
Figure 1. Cell-specific GluA2 overexpression disrupts AMPA-dependent calcium response in OPCs.
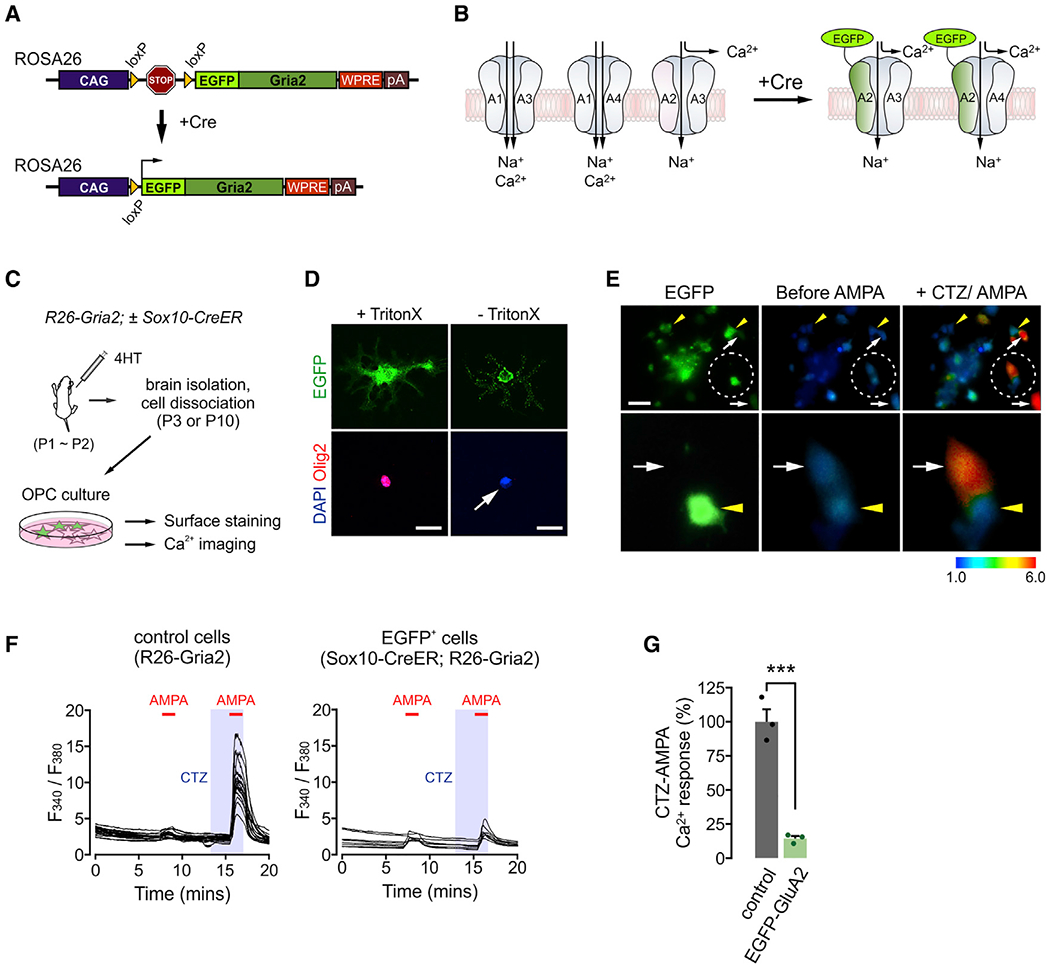
(A) Diagram of CAG-loxP-STOP-loxP-EGFP-Gria2 transgene placed on the Rosa26 locus.
(B) Expected outcomes after insertion of overexpressed EGFP-GluA2 into the cell membrane. Presence of calcium-impermeable of GluA2 in AMPAR complexes may block calcium entry through AMPARs, regardless of subunit composition. A1–A4 are GluA1–GluA4, respectively.
(C) Illustration of experiments for EGFP-GluA2 validation. After 4HT injections (0.2 mg per subcutaneous [s.c.] injection, at P1 and P2), R26-Gria2 ± Sox10-CreER mice were killed at P3 (calcium imaging) or P10 (immunopanning and surface detection of EGFP).
(D) Immunodetection of EGFP expressed on the cell surface. OPCs were isolated via immunopanning from 4HT-administered Sox10-CreER;R26-Gria2 mice and stained for EGFP and Olig2 with or without Triton X-100 for permeabilization. After secondary antibody probing, cells were permeabilized for DAPI staining. White arrow highlights no Olig2 detection. Scale bars: 25 μm.
(E) Upper panel: images of cultured EGFP+ OPCs (left) and their calcium responses before (center) and after CTZ (100 μM) + AMPA (20 μM) (right). Lower panel: enlarged views of 2 adjacent cells (EGFP+ and EGFP− OPCs) in the circled area of the upper panel. Yellow arrowheads and white arrows indicate EGFP+ and EGFP− cells, respectively. Scale bar: 50μm.
(F) Traces of the calcium responses of individual cells as ratios of fluorescence collected after excitation at 340 and 380 nm. Red bars and shaded regions represent durations of AMPA (1 min) and CTZ (2.5 min) applications, respectively. Calcium responses of OPCs collected from an R26-Gria2 (left) and EGFP+ OPCs from a Sox10-CreER; R26-Gria2 mouse (right).
(G) EGFP-GluA2 attenuates CTZ + AMPA-elicited calcium responses in OPCs. Peak responses averaged over all cells for each coverslip. n = 3 coverslips per group. Data are represented as means ± SEMs. Unpaired Student’s t test. ***p < 0.001.
See also Figures S1 and S2.
To validate the effects of EGFP-GluA2 on AMPAR-dependent calcium responses in OPCs, we bred R26-Gria2 mice with newly developed Sox10-CreER Tg mice. 4-hydroxytamoxifen (4HT) injections into Sox10-CreER; R26-Gria2 mice led to widespread EGFP expression in the brain, restricted to OL lineage (Figures 1C and S1A). After OPCs were isolated from those mice, EGFP was immunolabeled without membrane permeabilization (Figures 1C and 1D), suggesting extracellular localization of EGFP and proper targeting of EGFP-GluA2 into the cell membrane. During calcium imaging, OPCs were sequentially exposed to AMPA alone, and, after a brief wash, to cyclothiazide (CTZ), an inhibitor of AMPAR desensitization, and to CTZ + AMPA (Figure 1E). The brief pre-incubation with CTZ allowed sensitive and selective measurement of AMPAR-dependent intracellular calcium increases over non-AMPAR-mediated responses (Patneau et al., 1993). Whereas OPCs obtained from control mice or EGFP− OPCs from Sox10-CreER; R26-Gria2 mice displayed marked calcium increase following CTZ + AMPA, EGFP+ OPCs did not (Figures 1E–1G). The absence of calcium responses in EGFP+ OPCs may not be due to EGFP itself, because there were robust CTZ + AMPA-induced calcium increases in EGFP+ OPCs prepared from Sox10-CreER; R26-mEGFP mice (Figure S2). These results confirm that EGFP-GluA2 expression attenuates AMPAR-dependent calcium responses in OPCs in vitro.
Cell mosaic analysis of GluA2-overexpressing OPCs
We bred R26-Gria2 with Cspg4-CreER (Zhu et al., 2011) and R26-tdTomato (Ai14; Madisen et al., 2010), generating Cspg4-CreER; Ai14; R26-Gria2 mice (Figure 2A). When a small dose of 4HT was given, OPCs expressed either tdTomato or EGFP (i.e., EGFP-GluA2) or both throughout the brain (Figure 2B). While EGFP+ OPCs represent GluA2-overexpressing OPCs, EGFP− tdTomato+ OPCs represent control cells that are not affected by GluA2 overexpression. Thus, comparisons of EGFP+ tdTomato+ (called E-A2 cells hereafter) and EGFP− tdTomato+ cells enabled us to assess the effects of higher GluA2 levels on OPC behavior and its cell fate in the same brain. Five days after 4HT injection at postnatal day 3 (P3) (P3+5), 32.2% ± 4% of fluorescence-labeled cells expressed tdTomato alone, 16.2% ± 1% EGFP-GluA2 alone, and 51.6% ± 4% both tdTomato and EGFP in the corpus callosum (CC) (Figure 2C). A similar fractional labeling pattern was observed in other brain areas (e.g., in the cerebral cortex [CTX]: tdTomato 32.0%, EGFP 10.6%, and both 57.5%) at P8, and in the CC at a later age (P15: tdTomato 32.1%, EGFP 19.1%, and both 48.9%), even though the fluorescence-labeled cells at P15 included newly formed OLs beside OPCs. E-A2 and tdTomato+ cells from P15 Cspg4-CreER; Ai14; R26-Gria2 brains were separated via fluorescence-activated cell sorting (FACS) (Figure 2D). Subsequent RT-qPCR indicated that E-A2 cells express more Gria2 by ~3-fold (Figures 2E and 2F). These results showed that a small dose of 4HT administration to Cspg4-CreER; Ai14; R26-Gria2 mice allows mosaic identification of OPCs with different levels of GluA2.
Figure 2. GluA2 overexpression does not change OPC proliferation or differentiation into OLs in early life.
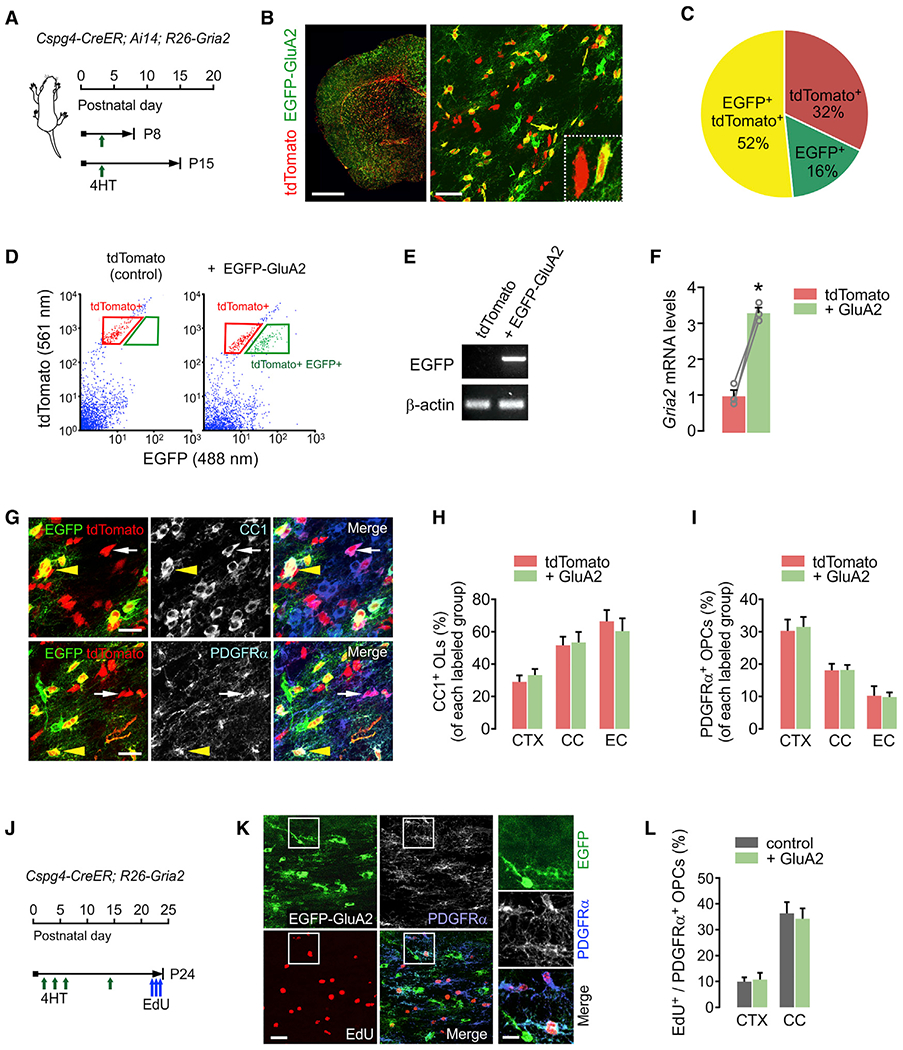
(A) Timelines for 4HT injection and mouse sampling. Cspg4-CreER; Ai14; R26-Gria2 triple Tg pups received a single 4HT injection (0.2 mg, s.c.) at P3 and were sampled at either P8 or P15.
(B) Fluorescence (left) and confocal (right) images of tdTomato or EGFP-expressing cells in the brain (left: P15; right: P8). Right panel inset: 2 cells expressing tdTomato alone (control) or both tdTomato and EGFP-GluA2 (E-A2 cell), in the external capsule (EC). Scale bars: 1 mm (left), 50 μm (right).
(C) Pie chart for cells expressing tdTomato, EGFP, or both tdTomato and EGFP in the CC at P8 (P3+5). n = 4 mice.
(D) Scatterplot of flow cytometry and gating strategy for isolation of tdTomato+ EGFP− and E-A2 cells. The tdTomato+ cells from an age-matched Cspg4-CreER; Ai14 littermate (P3+12) were used as the gating control (left).
(E) RT-PCR of EGFP with the isolated cells. The absence or presence of EGFP mRNA confirmed the effective separation of control and E-A2 cells.
(F) Relative mRNA levels of Gria2 in tdTomato+ and E-A2 (+ GluA2) cells isolated from the same brain. n = 3 mice. Paired Student’s t test. *p < 0.05.
(G) Confocal images of tdTomato+ CC1+ OLs (top panel) and tdTomato+ PDGFRα+ OPCs (bottom panel) with or without EGFP-GluA2 expression. White arrows and yellow arrowheads indicate tdTomato+ EGFP− (control) and E-A2 cells, respectively. Scale bars: 25 μm.
(H and I) Percentages of OLs (H) and OPCs (I) among tdTomato+ EGFP− cells (tdTomato) and E-A2 cells (+ GluA2) in the CTX, CC, and EC. n = 5 mice. Paired Student’s t test. (H) Control versus + GluA2 group, CTX: p = 0.227; CC: p = 0.498; EC: p = 0.102. (I) CTX: p = 0.776; CC: p = 0.957; EC: p = 0.895.
(J) Timeline for injections of 4HT and EdU, and mouse sampling. Cspg4-CreER; R26-Gria2 mice received 4HT at P2, P4, P6, and P14, and 3 injections of EdU between P22 and P23, before sampling at P24.
(K) Confocal images of EdU+ PDGFRα+ OPCs in a Cspg4-CreER; R26-Gria2 mouse (P24). Right panels: enlargement of the left panel insets. Scale bars: 25 μm (left) and 10 μm (right).
(L) Percentage of EdU+ cells among EGFP− (control) and EGFP+ OPCs (+ GluA2). Paired Student’s t test (control versus + GluA2 group, CTX: p = 0.305, n = 4 mice; CC: p = 0.613, n = 5 mice). Data are represented as means ± SEMs.
OPC-specific GluA2 overexpression does not change cell proliferation and differentiation at early ages
To determine the effects of high GluA2 levels on OPC-to-OL differentiation during early postnatal development, we examined the brains of P15 (P3+12) Cspg4-CreER; Ai14; R26-Gria2 mice. Although Cre recombination occurs only in NG2+ OPCs in these mice, new pre-OLs and OLs derived from EGFP+ (or tdTomato+) OPCs should continue to express the same fluorescent proteins because their expression is under the control of the CAG promoter. We quantified the proportions of newly developed CC1+ OLs and platelet-derived growth factor receptor α+ (PDGFRα+) OPCs among control (tdTomato+ alone) and E-A2 cells (Figure 2G). The percentages of OLs and OPCs among each group of fluorescence-labeled cells (Figures 2H and 2I) did not differ in the CTX, CC, and external capsule (EC). This indicates that elevated GluA2 levels in OPCs and their progeny did not alter the rate of oligodendrogenesis between P3 and P15.
To understand the impact of GluA2 overexpression on OPC proliferation, we also injected 5-ethynyl-2′-deoxyuridine (EdU) into the Cspg4-CreER; R26-Gria2 (no Ai14) mice (Figure 2J). We compared percentages of EdU incorporation among EGFP− OPCs (the control), and EGFP+ OPCs (Figure 2K), but observed no difference (Figure 2L). These results show that oligodendroglial GluA2 overexpression did not significantly alter OPC proliferation at young ages (before P24).
GluA2 overexpression in oligodendrocyte lineage does not change myelination in the young brain
Despite analytical advantages of mosaic cell labeling, the use of Cspg4-CreER mice and a small dose of 4HT allows for gene recombination only in a relatively small subset of OPCs, preventing the accurate assessment of the impact of GluA2 overexpression on myelination. To determine the effects of high GluA2 levels on myelination by sampling a larger fraction of oligodendroglia, we used Sox10-CreER; Ai14 (control) and Sox10-CreER; Ai14; R26-Gria2 (+ GluA2) mice (Figure 3A). 4HT-administered Sox10-CreER mice exhibited Cre activity in a greater number of OLs and OPCs than Cspg4-CreER, as assessed by the expression of tdTomato (Figure S1B) (e.g., 94.5% of OLs in Sox10-CeER; Ai14; R26-Gria2 versus 64.8% in Cspg4-CreER; Ai14; R26-Gria2) and EGFP-GluA2 in the CC (Figure S1C), and it was restricted to OL lineage (Figure S1D).
Figure 3. No overt changes in myelination after GluA2 overexpression.
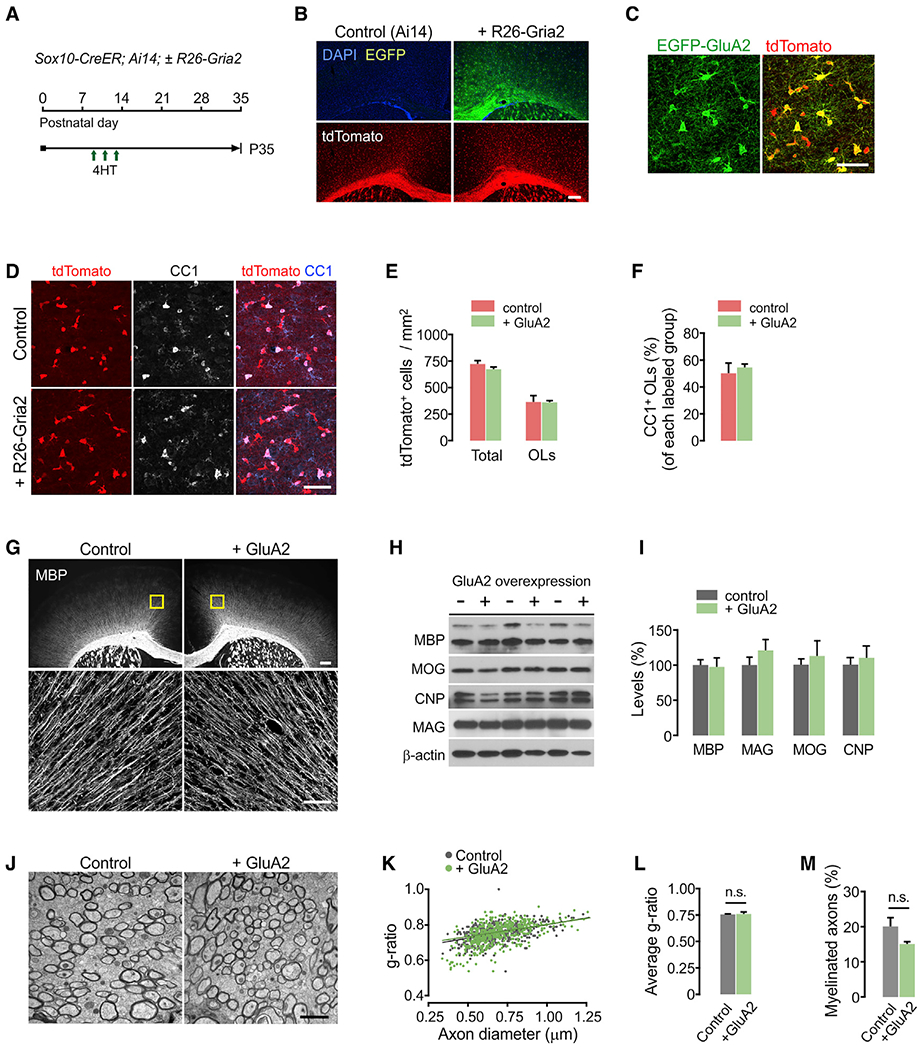
(A) Timeline for 4HT injections and mouse sampling. Sox10-CreER; Ai14 (control) and Sox10-CreER; Ai14; R26-Gria2 (+ GluA2) mice received 4HT at P9, P11, and P13 (0.2 mg per s.c. injection) and killed at P35.
(B) Fluorescence images showing widespread tdTomato expression with or without EGFP-GluA2. Scale bar: 200 μm.
(C) Confocal images of tdTomato and EGFP in the CTX of a Sox10-CreER; Ai14; R26-Gria2 mouse. Scale bar: 50 μm.
(D) Confocal images of cortical tdTomato+ CC1+ OLs. Scale bar: 50 μm.
(E) Densities of all tdTomato+ cells and tdTomato+ CC1+ OLs in the CTX (control versus + GluA2 group, total tdTomato+ cells: p = 0.246; tdTomato+CC1+ OLs: p = 0.972).
(F) Percentage of CC1+ OLs among tdTomato+ cells. n = 5 (Sox10-CreER; Ai14, control) and 4 (Sox10-CreER; Ai14; R26-Gria2, + GluA2) mice for (E and F).
(G) Fluorescence and confocal images of MBP in R26-Gria2 (control) and Sox10-CreER; R26-Gria2 (+ GluA2) mice. Upper panels: boxes correspond to confocal images in lower panels (optical thickness = 7 μm). Scale bars: 200 μm (up) and 50 μm (bottom).
(H and I) Western blot analysis of myelin proteins from cortical lysates of R26-Gria2 and Sox10-CreER; R26-Gria2 mice (H) and its quantification (I). Control versus + GluA2 group, MBP: p = 0.877; MAG: p = 0.337; MOG: p = 0.611; CNP: p = 0.636. n = 3 mice per group.
(J) Representative electron micrographs of callosal axons. Scale bar: 2 μm.
(K) Scatterplots of g-ratios. More than 150 myelinated axons per mouse were analyzed.
(L and M) Average g-ratio (L, control versus + GluA2 group, p = 0.880) and percentage of myelinated axons (M, control versus + GluA2 group. p = 0.12). For quantification of the percentage of myelinated axons, 700–900 axons were analyzed per mouse. n = 3 mice per group.
Data are represented as means ± SEMs. Unpaired Student’s t test for (E), (F), (I), (L), and (M). See also Figure S1.
Although 75% of cortical tdTomato+ cells co-expressed EGFP (Figures 3B and 3C) in Sox10-CeER; Ai14; R26-Gria2 mice, the numbers of all tdTomato+ cells and tdTomato+ OLs did not differ from those of Sox10-CreER; Ai14 mice (Figures 3D and 3E). Consequently, percentages of CC1+ OLs among tdTomato+ cells were similar for the two mouse groups (Figure 3F). Moreover, there was no difference in immunoreactivity for cleaved caspase-3 (data not shown), indicating that higher GluA2 levels in OL lineage did not alter OL survival during early CNS maturation.
The results of myelin basic protein (MBP) immunostaining (Figure 3G) and western blot analysis for myelin proteins (Figures 3H and 3I) also indicate that the sustained increase in GluA2 expression does not cause gross myelin deficits. Consistently, electron microscopy of callosal axons (Figure 3J) revealed no difference in myelin thickness (Figures 3K and 3L) or percentage of myelinated axons (Figure 3M). Thus, elevated GluA2 expression in OL lineage cells does not appear to affect myelination in the young brain.
OPC-specific GluA2 overexpression increases cell proliferation but not oligodendrocyte development in adults
Although our results thus far suggest that the relative abundance of GluA2-containing AMPARs does not change OL development in the young brain, the lack of impact of experimentally elevated GluA2 levels raises two additional possibilities: EGFP-GluA2 transgene expression incompletely blocks AMPAR calcium currents, and/or endogenous levels of GluA2 in OPCs are already high in WM areas at the tested ages. Calcium permeability through AMPARs in WM OPCs is low at young ages (Chen et al., 2018; Ziskin et al., 2007), but increases at P50 (Ziskin et al., 2007). Accordingly, we asked whether GluA2 overexpression after P50 changes the behavior of OPC and OL development.
Multiple doses of tamoxifen injection after P50 led to the differential expression of EGFP and tdTomato in Cspg4-CreER; Ai14; R26-Gria2 mice (Figures 4A and 4B). EdU was also administered before mouse sampling at P75 (P51+24) (Figure 4A). Although densities of total tdTomato+ control and E-A2 cells did not differ significantly (Figure 4C), we observed that EdU incorporation into E-2A cells was remarkably higher in all examined areas (Figure 4D). The density of E-2A OPCs and the relative proportion of OPCs among E-A2 cells were markedly increased in the CC and EC (Figures 4E and 4F). As the last 2 days’ EdU chasing was sufficient to reveal this difference, we assumed that OPC proliferation had been increasing from an earlier time point, perhaps leading to changes in oligodendrogenesis. However, the density of newly generated OLs and their relative proportion among tdTomato+ cells were unaltered by GluA2 overexpression (Figures 4G and 4H). These results show that higher GluA2 levels in OPCs do not enhance OPC-to-OL differentiation in adult mice, although they increased OPC proliferation in the adult brain. The age-related difference in the impact of GluA2 overexpression not only supports the previous observation that the calcium permeability of AMPARs increases with age at axon-OPC synapses (Ziskin et al., 2007) but also suggests the effectiveness of EGFP-GluA2 on AMPAR-mediated signaling events.
Figure 4. GluA2 overexpression increases OPC proliferation in the adult brain.
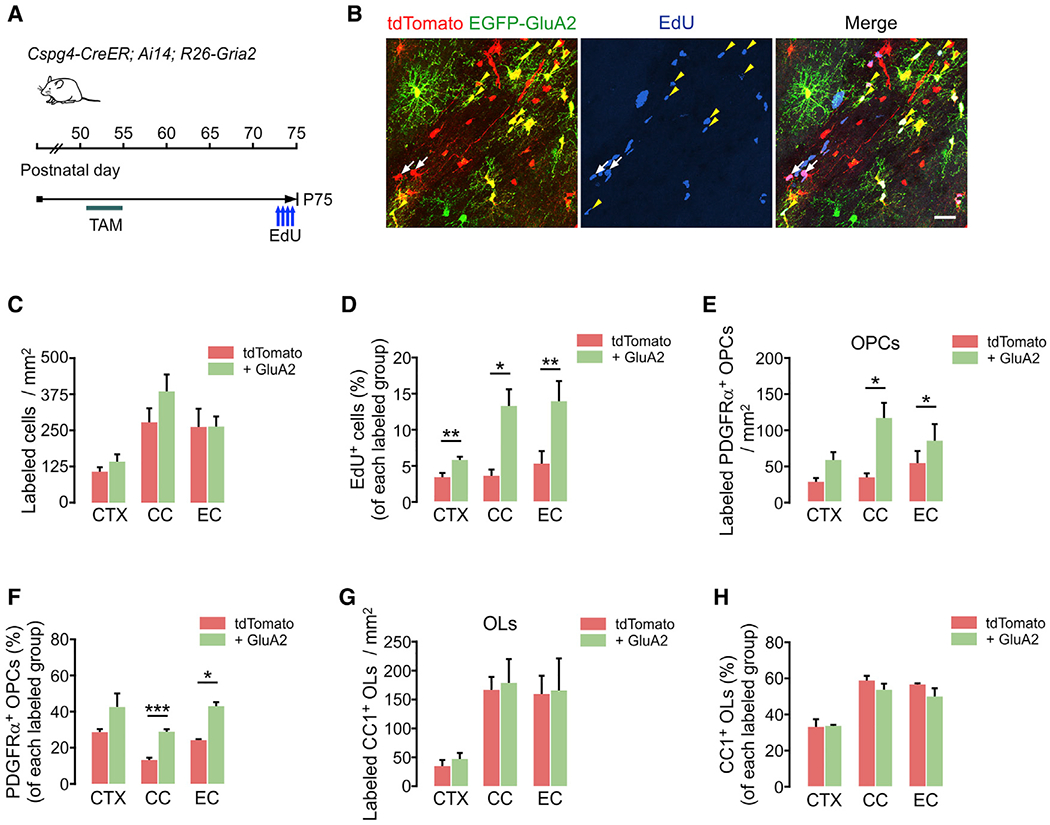
(A) Timeline for tamoxifen and EdU injections and sampling of Cspg4-CreER; Ai14; R26-Gria2 mice. Tamoxifen was injected (40 mg/kg per i.p. injection) 10 times between P51 and P55, and mice were sampled at P75. EdU was injected 4 times over 2 days before sampling.
(B) Confocal images of EdU-incorporated tdTomato+ or E-A2 cells in EC. White arrows and yellow arrowheads indicate EdU+ tdTomato+ EGFP− (control) and EdU+ E-2A (+ GluA2) cells, respectively. Scale bar: 25 μm.
(C) Densities of tdTomato+ (control) and E-2A (+ GluA2) cells (control versus + GluA2 group, CTX: p = 0.11; CC: p = 0.26; EC: p = 0.99).
(D) Percentages of EdU+ cells among tdTomato+ (control) and E-A2 cells (+ GluA2) (control versus + GluA2 group, CTX: p = 0.0084; CC: p = 0.021; EC: p = 0.0091).
(E) Densities of tdTomato+ PDGFRα+ OPCs with or without EGFP-GluA2 expression (control versus + GluA2 group, CTX: p = 0.069; CC: p = 0.048; EC: p = 0.047).
(F) Percentages of OPCs among tdTomato+ (control) and E-A2 cells (+ GluA2) (control versus + GluA2 group, CTX: p = 0.18; CC: p = 0.0009; EC: p = 0.017).
(G) Densities of tdTomato+ CC1+ OLs with or without EGFP-GluA2 expression (control versus + GluA2 group, CTX: p = 0.41; CC: p = 0.84; EC: p = 0.93).
(H) Percentages of OLs among tdTomato+ (control) and E-A2 cells (+ GluA2) (control versus + GluA2 group, CTX: p = 0.89; CC: p = 0.25; EC: p = 0.23).
Data are represented as means ± SEMs, n = 4 mice (C–H). Paired Student’s t test for (C–H). *p < 0.05, **p < 0.01, and ***p < 0.001.
Hypoxic-ischemic brain injury impairs oligodendrocyte development
In CNS injury, pathologically elevated glutamate levels in extracellular spaces lead to excessive calcium entry through AMPARs, causing excitotoxic neuronal death (Choi, 1988). Early studies suggested that OLs are also subject to a similar mechanism (Deng et al., 2003; Salter and Fern, 2005). However, it has not been determined whether the susceptibility to injury is primarily due to AMPARs expressed by oligodendroglia in vivo.
To determine whether GluA2 levels regulate OL death and/or regeneration after injury, we used a neonatal H/I brain injury model in which a P7 mouse is given a unilateral carotid artery ligation and subsequently exposed to a hypoxic condition (Shen et al., 2012) (Figure S3A). Three days later (3 days postinjury [dpi]), its brain showed marked astrogliosis (Figures 5A and S3B) and reduction in MBP immunoreactivity in the EC (Figures 5A and S3C) on the side ipsilateral to the ligated artery, which is consistent with earlier observations (Shen et al., 2012). Using Mobp-EGFP BAC Tg mice, we confirmed that the numbers of CC1+ or EGFP+ OLs were reduced by H/I injury (Figures S3D and S3E). However, PDGFRα+ OPC density was not changed by the injury at 3 dpi (Figure S3F).
Figure 5. GluA2 overexpression in OPCs promotes post-injury OL development and cell proliferation in the young brain.
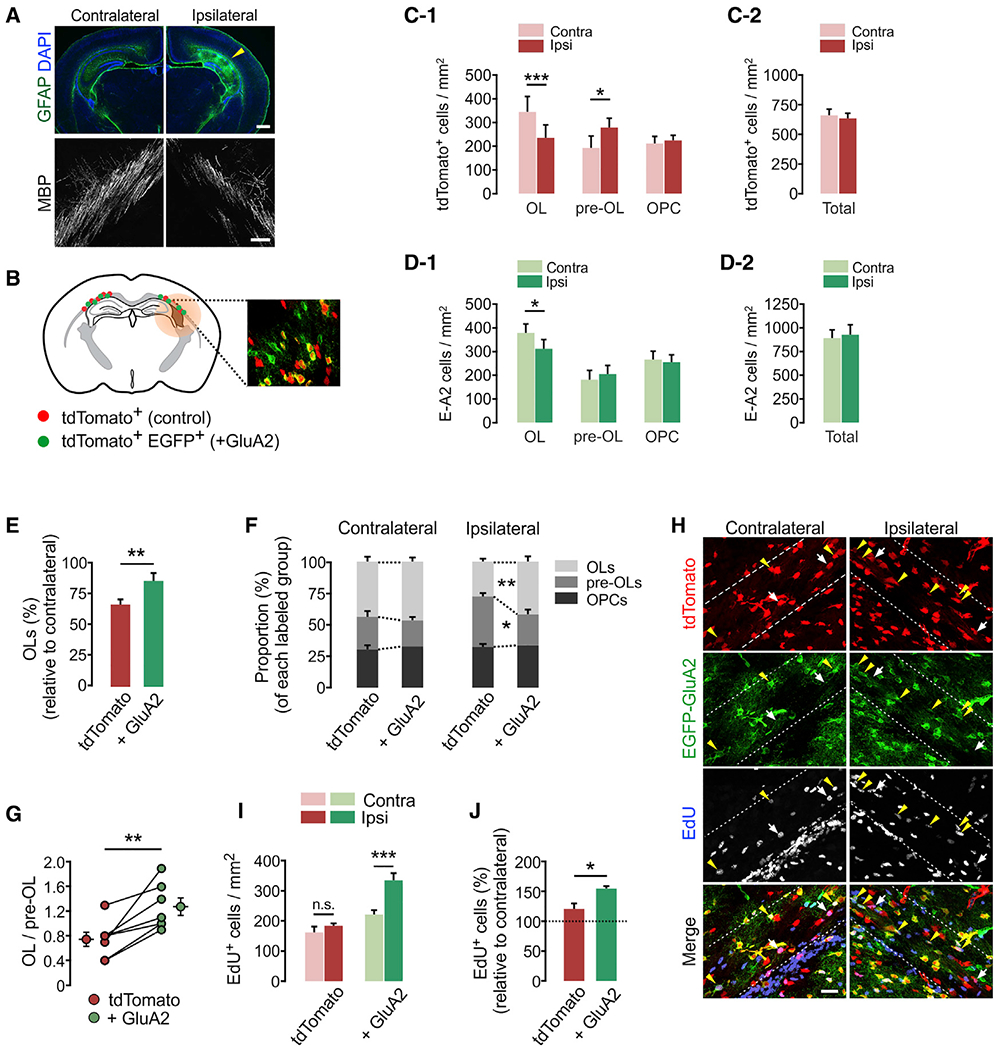
(A) Fluorescence brain images showing astrogliosis and loss of MBP on the injured side, ipsilateral to ligated artery, at 3 dpi. Scale bars: 500 μm (top) and 50 μm (bottom).
(B) Schematic diagram of mosaic analysis of tdTomato+ control (red dots) and E-A2 cells (+ GluA2, green dots) in the injured EC (shaded) in Cspg4-CreER; Ai14; R26-Gria2 mice. Inset: confocal image of tdTomato and EGFP at P10.
(C-1) Differential changes in density of tdTomato+ OL lineage cells according to maturation stage after H/I injury (contralateral versus ipsilateral side, OL: p = 0.0004, n = 11 mice; pre-OL: p = 0.0172, n = 8 mice; OPCs: p = 0.434, n = 9 mice).
(C-2) Total tdTomato+ cells in the EC at 3 dpi (P10). Contralateral versus ipsilateral side, p = 0.461; n = 11 mice.
(D-1) H/I injury-induced changes in densities of E-A2 cells according to maturation stage. Contralateral versus ipsilateral side, OLs: p = 0.031, n = 11 mice; pre-OLs: p = 0.436, n = 8 mice; OPCs: p = 0.561, n = 9 mice.
(D-2) Total E-A2 cells in the EC at 3 dpi (P10). Contralateral versus ipsilateral side, p = 0.552, n = 11 mice.
(E) Loss of tdTomato+ control and E-A2 (+ GluA2) OLs on the ipsilateral (injured) relative to the contralateral side (control versus + GluA2 group, p = 0.009, n = 11 mice).
(F) Proportion of OLs, pre-OLs and OPCs among labeled cells (control versus + GluA2 group, ipsilateral OLs: p = 0.0097; ipsilateral pre-OLs: p = 0.019, n = 8 mice).
(G) Ratios of (OL to pre-OL) on ipsilateral side of each mouse. Separate circles with error bars represent means ± SEM for each group (control versus + GluA2 group, p = 0.006, n = 7 mice).
(H) Confocal images of EdU+ cells in H/I-injured Cspg4-CreER; Ai14; R26-EGFP-Gria2 mice at P10. White arrows and yellow arrowheads indicate EdU+ tdTomato+ EGFP− (control) and EdU+ E-A2 (+ GluA2) cells, respectively. Scale bar: 25 μm.
(I) Densities of EdU+ cells. Contralateral versus ipsilateral side, control: p = 0.181; + GluA2: p = 0.0005, n = 5 mice.
(J) Percentages of EdU+ cells in the ipsilateral EC relative to the contralateral EC (control versus + GluA2 group, p = 0.041, n = 5 mice).
Data are represented as means ± SEMs. Paired Student’s t test for (E), (G), (I), (J). *p < 0.05, **p < 0.01, and ***p < 0.001. See also Figures S3 and S4.
To better define oligodendroglial responses to H/I injury according to maturation stage, we induced H/I injury in 4HT-administered Cspg4-CreER; Ai14; R26-Gria2 mice (Figure S4A) and analyzed tdTomato+ EGFP− OL lineage cells (Figure S4B). In our analysis, tdTomato-labeled pre-OLs were indirectly quantified by subtracting the number of PDGFRα+ OPCs and CC1+ OLs from tdTomato+ cells that were all Olig2+, except for tdTomato+ pericytes, easily distinguishable by their morphology (Figure S4B). As expected, tdTomato+ OLs were reduced, but the density of tdTomato+ OPCs was unchanged (Figure 5C-1). In contrast, pre-OLs (tdTomato+ CC1− PDGFRα−) significantly increased (Figure 5C-1), leaving the total of tdTomato+ cells unaltered (Figure 5C-2). Thus, increased pre-OL and decreased OL fractions may be the characteristic responses of oligodendroglia according to their maturation states 3 days after H/I injury (Figure S4C). The proportion of EdU+ among tdTomato+ cells was also increased by injury, but OPC density was unchanged (Figure S4D), suggesting that the proliferation and differentiation of OPCs into pre-OLs were enhanced in the lesions. The postinjury accumulation of pre-OLs and reduced OLs may be attributed to impaired lineage progression from pre-OL to OLs, as suggested before (Buser et al., 2012; Segovia et al., 2008).
GluA2 overexpression promotes oligodendrocyte lineage progression and enhances OPC proliferation after neonatal brain injury
Next, we explored how high GluA2 levels regulate OPC responses to H/I injury (Figures 5B–5D). As with tdTomato+ cells (Figures 5C-1 and 5C-2), H/I injury reduced E-A2 OLs (Figure 5D-1) without changing the number of E-A2 cells (Figure 5D-2). However, the loss of E-A2 OLs relative to their contralateral counterparts was far less than that of tdTomato+ OLs (14.1% versus 32.9%) (Figure 5E). Moreover, unlike tdTomato+ pre-OLs (Figure 5C-1), E-A2 pre-OLs were not increased after H/I (Figure 5D-1). The fraction of OLs among labeled cells was markedly increased from 27.8% to 41.7% by GluA2 overexpression, whereas that of pre-OLs was reduced from 39.9% to 24.7% on the injured side (Figure 5F, right). In contrast, GluA2-overexpression did not affect the fraction of OLs and pre-OLs on the contralateral side (Figure 5F, left). In each injured area, E-A2 cells exhibited higher OL to pre-OL (OL:pre-OL) ratios than did tdTomato+ control cells (Figure 5G).
We also examined the effects of OPC-specific GluA2 overexpression on cell proliferation after H/I injury (Figures 5H and S4A). In general, injury increased EdU incorporation into OPCs in both groups (Figure S4D). However, the accumulation of EdU+ cells (Figure 5I) and the percentage increase in EdU+ E-A2 cells on the injured side relative to the contralateral side were much greater than in control cells (55% increase among E-A2 cells versus 21% increase among tdTomato+ cells) (Figure 5J).
These results indicate that higher levels of GluA2 in OPCs, and possibly in immature OLs, promote oligodendroglial lineage progression arrested by WM lesions in the young brain. GluA2-overexpressing OPCs may further support OL regeneration by enhancing cell proliferation and differentiating into pre-OLs.
GluA2 overexpression from mature oligodendrocytes does not prevent oligodendrocyte loss in H/I injury
The observed benefits from GluA2 overexpression (Figures 5E–5G) can be attributed to increased OL survival via OL protection from excitotoxicity in injury. To test this possibility, we crossed R26-Gria with Plp1-CreER mice (Doerflinger et al., 2003) and induced GluA2 overexpression primarily in OLs (Figure 6A). Due to the sparsity of Plp1-CreER-affected cells at the young age (P7), differential labeling OLs with tdTomato or EGFP-GluA2 with a small dose of 4HT did not generate a sample size that was sufficient for analysis. To increase sampling power, we used Plp1-CreER; R26-EYFP mice as controls and compared them with Plp1-CreER; R26-Gria2 mice. In the EC of these mice at P10 (P5+5), >90% of EYFP+ (in Plp1-CreER; R26-EYFP) and EGFP+ cells (in Plp1-CreER; R26-Gria2 mice) were CC1+ OLs (data not shown). Following the H/I induction, both groups exhibited a significant loss of CC1+ Olig2+ OLs on the injured side (Figure 6B), but we did not observe any difference in degree of cell loss, which was also assessed by EYFP or EGFP (Figures 6C–6E). These results suggest that the beneficial effects of GluA2 overexpression in oligodendroglia on neonatal H/I injury cannot be attributed to OL protection from AMPAR-mediated excitotoxicity.
Figure 6. Beneficial effects of GluA2 overexpression become evident through post-injury OL development.
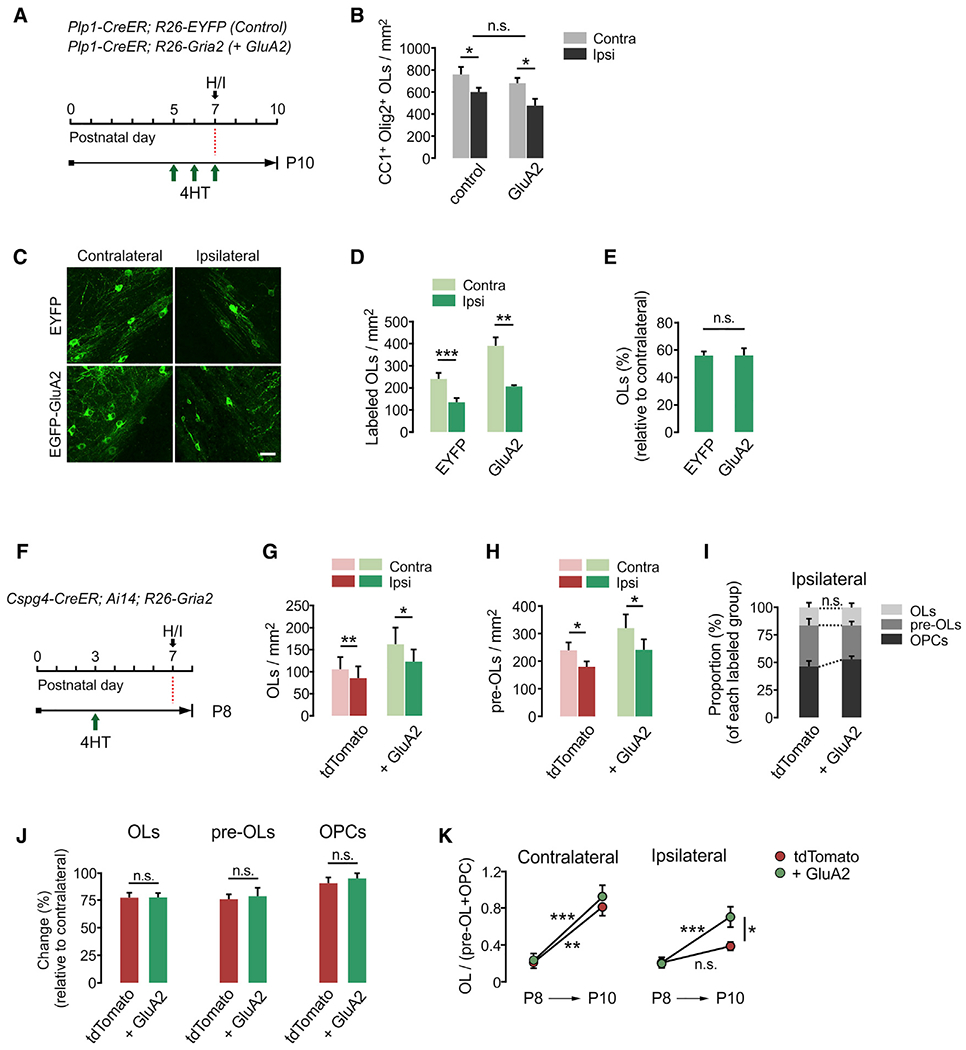
(A) Timeline for 4HT injection, H/I injury, and mouse sampling. Plp1-CreER; R26-EYFP (control) and Plp1-CreER; R26-Gria2 (GluA2) mice received daily 4HT injections (0.2 mg per s.c. injection) between P5 and P7, before injury induction.
(B) Densities of CC1+ Olig2+ OLs in EC at 3 dpi. Contralateral versus ipsilateral side, control: p = 0.019; GluA2: p = 0.016. Paired Student’s t test. n = 4 (Plp1-CreER; R26-EYFP) and 5 (Plp1-CreER; R26-Gria2) mice.
(C) Confocal images of OLs expressing EYFP or EGFP-GluA2 at 3 dpi. Scale bar: 25 μm.
(D) Densities of EYFP (or EGFP-GluA2)-labeled CC1+ OLs in EC. Contralateral versus ipsilateral side, EYFP: p = 0.004; GluA2: p = 0.005. Paired Student’s t test.
(E) Percentage losses of EYFP (or EGFP-GluA2)-labeled OLs in the ipsilateral EC relative to the contralateral (EYFP versus GluA2, p = 0.987). Unpaired Student’s t test. n = 5 (Plp1-CreER; R26-EYFP) and 6 (Plp1-CreER; R26-Gria2) mice for (D) and (E).
(F) Timeline for 4HT injection, H/I injury, and sampling of Cspg4-CreER; Ai14; R26-Gria2 mice. Mice were sampled at 1 dpi (P8) to examine early cell loss after injury.
(G and H) Densities of fluorescent protein-labeled CC1+ OLs (G) (contralateral versus ipsilateral side, tdTomato: p = 0.006; + GluA2: p = 0.026) and CC1− PDGFRα− pre-OLs (H) (contralateral versus ipsilateral side, tdTomato: p = 0.025; + GluA2: p = 0.0348) in EC at P8.
(I) Proportion of OLs, pre-OLs, and OPCs among tdTomato+ EGFP− and E-A2 cells on the ipsilateral side at P8 (tdTomato versus + GluA2, OLs: p = 0.969; pre-OLs: p = 0.115; OPCs: p = 0.099).
(J) Changes in percentages of fluorescent protein-labeled OLs, pre-OLs, and OPCs in the ipsilateral EC relative to contralateral at P8 (tdTomato versus + GluA2, OLs: p = 0.975; pre-OLs: p = 0.932; OPCs: p = 0.139).
(K) Ratios of (OL/Pre-OL + OPC) at P8 and P10. tdTomato versus + GluA2 on the contralateral side at P8: p > 0.05; at P10: p > 0.05; P8 versus P10 on the contralateral side, tdTomato: p < 0.01; + GluA2: p < 0.001; tdTomato versus + GluA2 on the ipsilateral side at P8: p > 0.05; P10:p< 0.05; P8 versus P10 on the ipsilateral side, tdTomato: p > 0.05; + GluA2: p < 0.001. Two-way ANOVA with post hoc comparison by Bonferroni test. n = 5 (P8) and 9 (P10) mice.
Data are represented as means ± SEMs. *p < 0.05, **p < 0.01, and ***p < 0.001. Paired Student’s t test for (G)–(J). n = 5 mice for (G) and (H).
GluA2 overexpression manifests its beneficial effects in H/I injury through new oligodendrocyte generation
GluA2 overexpression may protect OPCs or immature OLs from excitotoxic cell death in injury. Some studies suggested that immature OLs (O4+ OPCs or pre-OLs) are more vulnerable than OLs to H/I injury because they express calcium-permeable AMPARs at higher levels (Deng et al., 2003; Liu et al., 2013; Volpe, 2001). To determine whether high GluA2 levels enhance the survival of OPCs or pre-OLs, we examined the loss of OL lineage cells 1 day after H/I (i.e., P8) (Figure 6F). Like P10, there was a significant reduction in tdTomato+ CC1+ OLs in the injured EC (Figure 6G), but the degree of reduction did not differ between the 2 groups (22.8% for control versus 22.6% for E-A2 OLs) (Figures 6G, 6I, and 6J, left). Notably, unlike P10, pre-OLs were reduced at 1 dpi (Figure 6H), and the degree of reduction was similar (24.1% for control versus 23.3% for E-A2 pre-OLs) (Figures 6I and 6J, center). OPC density on the ipsilateral side remained unaltered in both groups (Figure 6J, right). Therefore, the effects of injury on the different stages of oligodendroglia were not changed by GluA2 overexpression at this early time of injury (Figure 6J), suggesting that oligodendroglial vulnerability to injury is not directly correlated with GluA2 levels.
Further analysis of time-dependent changes in the proportion of each OL lineage stage revealed that the ratio of mature OLs to immature oligodendroglia (OL:pre-OLs + OPCs) increased from P8 to P10 on the uninjured contralateral side, regardless of GluA2 overexpression (Figure 6K, left). In contrast, on the injured side, the ratio (OL:pre-OLs + OPCs) did not increase for tdTomato+ control cells, indicating that injury impaired OL development. However, GluA2 overexpression markedly enhanced this ratio (Figure 6K, right). Therefore, the increased OL:pre-OL ratio at P10 should be ascribed to enhanced OL differentiation. These findings suggest that GluA2 overexpression manifests its beneficial effects during the regenerative process, rather than preventing oligodendroglial loss at an early time point of the neonatal H/I injury.
GluA2 overexpression in OPCs promotes oligodendrocyte regeneration in demyelinated lesions in the adult brain
Next, to determine whether high GluA2 levels also promote OL regeneration in the adult brain, lysolecithin (LPC) was injected into the CC of tamoxifen-administered Cspg4-CreER; Ai14; R26-Gria2 mice at P65. Also, to assess OPC proliferation, 5-bromo-2’-deoxyuridine (BrdU) (7 day-chasing) and EdU (2 day-chasing) were injected, each of which targeted different time windows (Figure 7A). Comparing tdTomato+ control and E-A2 cells at 10 dpi (P75) (Figures 7B and 7C), we observed that 81.2% of tdTomato+ BrdU+ and 78.4% of tdTomato+ EdU+ cells were also EGFP+, indicating that E-A2 cells were far more proliferative than controls (Figures 7D and 7E). Despite the different durations of thymidine analog chasing, the similar increases of BrdU and EdU incorporation into E-A2 cells relative to the tdTomato+ cells indicate that the increased OPC proliferation had been sustained during the remyelination period. Consistent with these observations, many more E-A2 cells accumulated in the injured area (Figure 7F). More surprisingly, most of these had become either OLs or pre-OLs, and thus, there were more E-A2 OLs and pre-OLs than tdTomato+ control counterparts (Figures 7G and 7H). However, the number of E-A2 OPCs were far fewer than tdTomato+ OPCs in the lesions (Figure 7I). These results suggest that upon demyelinated injury, GluA2 overexpression in adult OPCs enhances both cell proliferation and OL lineage progression. Nonetheless, the rate of OL maturation (or regeneration) was greater than the rate of OPC proliferation among E-A2 cells.
Figure 7. GluA2 overexpression in adult OPCs promotes cell proliferation and OL regeneration after acute demyelination.
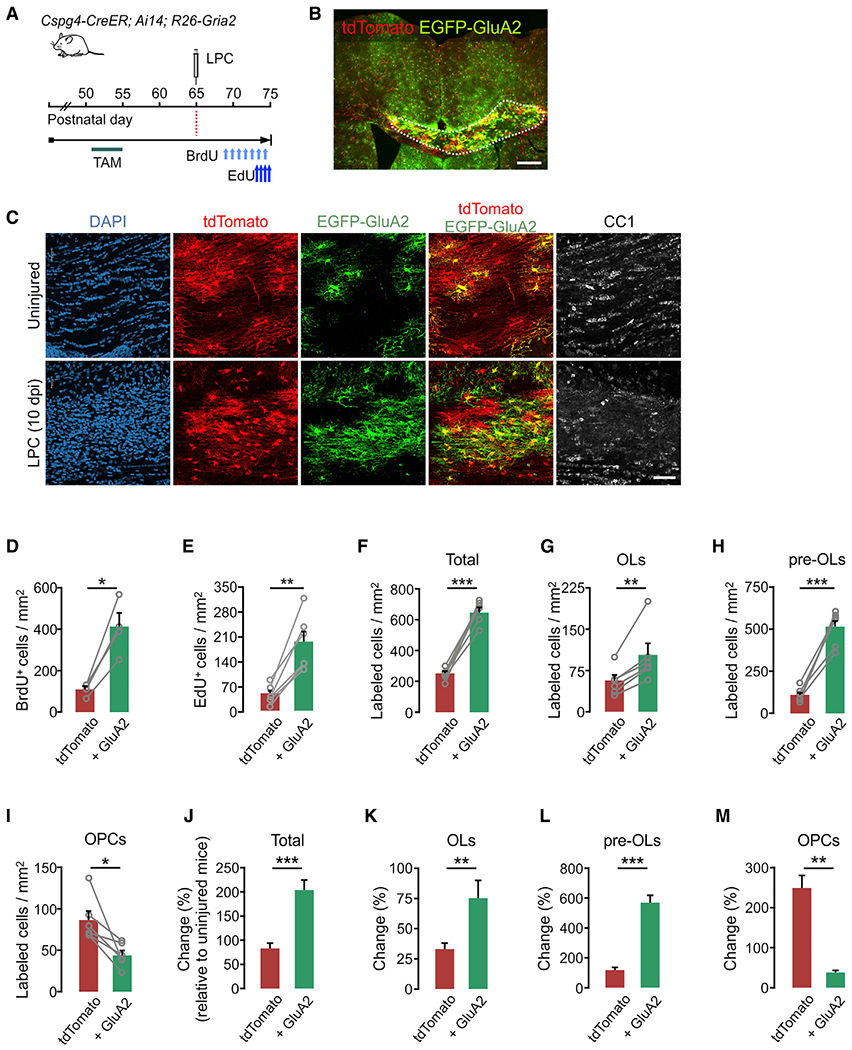
(A) Timeline for tamoxifen injections, LPC, BrdU, and EdU, and sampling of Cspg4-CreER; Ai-14; R26-Gria2 mice. Tamoxifen (40 mg/kg per intraperitoneal [i.p.] injection) was administered 10 times between P51 and P55, and mice were sampled at P75. Demyelination was induced by 1% LPC injection into the CC at P65. BrdU (between P68 and P74) and EdU (between P73 and P74) were also administered daily.
(B) Fluorescence image of tdTomato+ and EGFP+ cells in the LPC-administered CC and surrounding regions. Scale bar: 500 μm.
(C) Confocal images of tdTomato, EGFP-GluA2, and CC1+ cells in the CC of uninjured (top) or the LPC-injected (bottom) mice. Scale bar: 50 μm.
(D) Densities of BrdU-incorporated tdTomato+ and E-A2 cells (tdTomato+ control versus E-A2, p = 0.011).
(E) Densities of EdU-incorporated tdTomato+ and E-A2 cells (tdTomato+ control versus E-A2, p = 0.0025).
(F–I) Densities of tdTomato+ and E-A2 cells analyzed according to cell stage.
(F) Total tdTomato+ and E-A2 cells (tdTomato+ control versus E-A2, p = 0.0006).
(G) CC1+ OLs (tdTomato+ control versus E-A2, p = 0.0088).
(H) CC1− PDGFRα− pre-OLs (tdTomato+ control versus E-A2, p = 0.0004).
(I) PDGFRα+ OPCs (tdTomato+ control versus E-A2, p = 0.02).
Paired Student’s t test (for D–I). n = 4 (D) and n = 6 (E—I) mice.
(J–M) Percentage of changes in densities of tdTomato+ and E-A2 cells in LPC-injected (10 dpi) mice compared to uninjured mice (P51 + 24) at P75.
(J) Total labeled cells (tdTomato+ control versus E-A2, p = 0.0005).
(K) CC1+ OLs (tdTomato+ control versus E-A2, p = 0.0066).
(L) CC1− PDGFRα− pre-OLs (tdTomato+ control versus E-A2, p = 0.0004).
(M) PDGFRα+ OPCs (tdTomato+ control versus E-A2, p = 0.0012). Unpaired Student’s t test. n = 4 (uninjured) and n = 6 (LPC-injected) mice.
Data are represented as means ± SEMs. *p < 0.05, **p < 0.01, and ***p < 0.001.
To understand how E-A2 cells behave in the injured adult brain relative to the uninjured conditions, we also compared the oligodendroglial regeneration patterns (from P65 to P75) with normal adult oligodendrogenesis (from P51 to P75). This analysis further confirmed that, in an injury, GluA2 expression not only enhances cell proliferation but also markedly facilitates cell maturation (Figures 7J–7M).
DISCUSSION
CNS glia often express ionotropic glutamate receptors, but the functions of these receptors are not completely understood. Notably, these receptor-mediated events do not elicit prominent action potentials in glial cells due to the low density of voltage-gated sodium channels, highly negative resting potential, and high resting membrane potassium conductance (De Biase et al., 2010; McKhann et al., 1997; Berret et al., 2017). However, it is possible that calcium influx through AMPARs triggers intracellular signaling cascades and induces diverse subcellular events in these cells. Blocking AMPAR-mediated calcium entry into cerebellar Bergmann glia by GluA2 overexpression disrupts cell morphology and impairs the Bergmann glia’s ability to clear extracellular glutamate (Iino et al., 2001), which likely affects motor functions (Saab et al., 2012). It has also been suggested that AMPAR calcium regulates the strength of axo-OPC synapses (Ge et al., 2006). Most interestingly, recent studies have demonstrated that myelin growth requires an increase in intracellular calcium in newly forming myelin sheaths (Baraban et al., 2018; Krasnow et al., 2018), which is also dependent on neuronal activity. Therefore, it is important to identify the mechanisms of this calcium increase in oligodendroglia and to understand how AMPAR-mediated calcium regulates OPC behavior and/or myelin growth. This study assessed the specific role of GluA2-containing AMPARs, which are calcium impermeable, in normal oligodendrogenesis and postinjury OL regeneration in both developing and adult brains.
In two recent studies, AMPAR functions were modified in OL-lineage cells during early oligodendrogenesis; either most AMPAR currents were removed from the entire OL lineage (Kougioumtzidou et al., 2017) or AMPAR-mediated calcium permeability was increased in OPCs (Chen et al., 2018). However, they did not directly explore the roles of calcium influx through AMPARs during both normal and postinjury OL development via selective inhibition of calcium permeability. Our genetic approach for enhancing GluA2 expression was OL-lineage and maturation stage specific, and calcium imaging confirmed that it effectively inhibits AMPAR calcium responses in OPCs in vitro. Moreover, tagging GluA2 with extracellular EGFP enabled unambiguous tracing of GluA2-over-expressing cells and comparison with neighboring, unaffected cells. Thus, R26-EGFP-Gria2, in combination with various cell-type-specific Cre mice, provide a powerful tool for cell-specific manipulation of AMPARs that is impossible to achieve using pharmacological manipulations.
The lack of significant impact of GluA2 overexpression on early oligodendrogenesis or OPC proliferation in the young brain suggests that AMPAR calcium is not the primary regulatory mechanism for OPC proliferation and OL development at early ages. Interestingly, the deletion of GluA2 alone from OL lineage, which presumably increases AMPAR calcium permeability, resulted in neither oligodendrogenesis impairment nor myelin defects (Kougioumtzidou et al., 2017). Our results are in line with those of Kougioumtzidou et al. (2017), in that levels of GluA2 alone, whether elevated or depleted, did not affect normal oligodendrogenesis. On the contrary, when dividing OPCs were transduced with GluA2 mutants that increased AMPAR calcium permeability, they exhibited a modest increase in cell proliferation and reduced differentiation into OLs (Chen et al., 2018). Why some results of these three studies differ is unclear, although differences in the methods used to control GluA2 levels or modify its properties (e.g., germline-based transgenesis versus acute viral transfection) may be a factor. We used a tamoxifen-inducible genetic control, and thus, our results are less likely to be confounded by compensatory mechanisms that otherwise mitigate mutant phenotypes, especially when a gene is ablated at the embryonic stage. Also, our study sampled many OPCs, whether dividing or not, and OLs from multiple brain regions and ages. If, as our data indicate, OL numbers are not changed by higher GluA2 levels or disrupting AMPAR calcium influx, but are affected by the complete abolition of AMPAR currents, as reported by Kougioumtzidou et al. (2017), the combined results would suggest that different aspects of AMPAR signaling other than calcium influx, such as membrane depolarization, play a more significant role in early OL development and/or myelination.
However, in the mature brain, higher GluA2 levels significantly increased OPC proliferation. The age-dependent differential phenotypes of OPC proliferation probably were due to high endogenous levels of synaptic GluA2 in WM of the early postnatal brain (Ziskin et al., 2007). This suggests that AMPAR calcium regulates OPC behavior more significantly in adults than at young ages. To fully understand the underlying biology of AMPARs in OPCs, this possibility needs to be further addressed in detail.
What role does AMPAR calcium play in OL lineage cells after an injury? Our results challenge the view that ischemic CNS injury directly causes excitotoxic cell death of OLs via AMPAR-mediated mechanisms (Deng et al., 2003; Follett et al., 2000; Salter and Fern, 2005), unlike the case for neurons (Choi, 1988; Larm et al., 1997). As pharmacological approaches used in previous studies lacked target cell-type specificity (Follett et al., 2000; McDonald et al., 1998; Salter and Fern, 2005), it is possible that the observed OL protection with AMPAR blockers was due to non-cell autonomous mechanisms.
By analyzing the spatiotemporal dynamics of OL lineage cells, we showed that the reduction of OLs following neonatal H/I injury results from a combination of initial OL loss and subsequent impairment of OL regeneration. Our results show that GluA2 overexpression benefits oligodendroglial repair mostly by promoting cell lineage progression from pre-OL to OL (from P7 to P10), as well as by enhancing OPC proliferation. If the overexpression of Q/R edited GluA2 impedes calcium influx without altering the amplitude of synaptic AMPAR currents, as shown at hippocampal synapses (Shi et al., 2001), these findings suggest that calcium entry through OPC AMPARs suppresses post-injury OL regeneration. Extracellular environments of WM lesions inhibit oligodendroglial regeneration (Gruchot et al., 2019), probably via AMPAR-mediated calcium.
We do not exclude the possibility that our GluA2 overexpression enhanced overall synaptic AMPAR transmission, in addition to altering relative calcium permeability. Given the report that the loss of oligodendroglial AMPARs negatively affects OL accumulation during normal CNS maturation (Kougioumtzidou et al., 2017), this idea seems plausible. However, it does not account for the failure of GluA2 overexpression to increase normal oligodendrogenesis at all ages.
Why do the outcomes of GluA2 overexpression for OL generation become pronounced only in the context of WM injury? Although we have not addressed this question, other studies suggest that injury-induced changes in extracellular environments (e.g., inflammation) alter GluA2 subcellular localization. Ischemic injury selectively reduces surface expression of GluA2 in OPCs, without altering GluA2 expression levels (Deng et al., 2003). Whole-cell recordings of OPCs revealed increases in inward current-voltage (I-V) rectification of AMPAR excitatory postsynaptic current (EPSC) in H/I-injured mice, indicating enhanced calcium permeability of AMPARs (Shen et al., 2012). Another possibility is that injury-induced calcium entry is largely mediated by extrasynaptic AMPARs (Beattie et al., 2010; Ferrario et al., 2013), while physiological AMPAR calcium signaling exerts its effects only through receptors localized at synaptic membranes.
In contrast to studies showing the protective effects of AMPAR antagonists on OLs in ischemic injury (Chen et al., 2007; Deng et al., 2003), Gautier et al. (2015) showed that the local injection of AMPAR antagonists into demyelinated WM impaired remyelination, pointing to a positive role for these receptors in OL regeneration. However, as noted above, such approaches affect both neuronal and glial AMPARs, and influence not only calcium influx but also receptor-mediated depolarization. In this context, our findings suggest a cellular mechanism that may reconcile the two opposing results concerning oligodendroglial AMPARs signaling in the injured brain (harmful for OL survival versus beneficial for oligodendrogenesis). In injury, while calcium influx through AMPARs on OPCs inhibits OL lineage progression, other aspects of AMPAR signaling may support remyelination, pointing to a critical role for OPC GluA2 in cell regeneration. Given the emerging recognition of the heterogeneous nature of OPCs (Foerster et al., 2019; Spitzer et al., 2019), it is critical to determine whether there are OPC subpopulations expressing different GluA2 levels or AMRAR calcium permeability, and if so, whether they have a different potential to differentiate into OLs after demyelinating injury.
In summary, this study identified the cell-autonomous, subunit-specific roles of GluA2 levels in OPC proliferation and oligodendroglial development, demonstrating that the effects of higher GluA2 levels are both age and injury dependent.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents, except for Rosa26-EGFP-Gria2 mice, should be directed to and will be fulfilled by the Lead Contact, Shin H. Kang.
Materials availability
In this study, Rosa26-EGFP-Gria2 (R26-Gria2) and Sox10-CreER BAC Tg mice were generated at the Johns Hopkins University and Temple University, respectively. Before these mice are deposited to The Jackson Laboratory, they will be made available upon request. The relevant information and requests for R26-Gria2 mice should be directed to Dwight E. Bergles, and for Sox10-CreER BAC Tg mice to the Lead contact.
Data and code availability
This study did not generate any unique datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Generation of mouse lines
ROSA26-Gria2 mice were generated by the electroporation of mouse ES cells with an engineered construct containing ROSA26-CAG-loxP-stop-loxP-EGFP-Gria2-WPRE (R26-Gria2) and homologous recombination. This transgene construct is almost identical to that of ROSA26-CAG-(l-s-l)-tdTomato (Ai14) (Madisen et al., 2010) except for sequences encoding tdTomato that had been replaced with sequences for EGFP-GluA2. The EGFP-Gria2 cDNA was obtained from Dr. Richard Huganir (Johns Hopkins University). Sox10-CreER BAC mice were generated by a BAC modification method (Yang et al., 1997), and in the modified BAC, the coding sequences for CreERT2 and the rabbit beta-globin polyadenylation signal were inserted into the endogenous translation start site (ATG) of the mouse Sox10.
Other mice
Cspg4-CreER (JAX stock# 008538), Plp1-CreER (JAX stock# 005975), ROSA26-tdTomato (Ai14; JAX stock# 007914), ROSA26-EYFP (JAX stock# 006148) and ROSA26-mEGFP (mT/mG; JAX stock# 007576) mice were purchased from the Jackson Laboratory. Mobp-EGFP BAC mice were generated by GENSAT and described previously (Kang et al., 2013). All mice were maintained with a 12-hour light/ 12-hour dark cycle. The genetic background of Cspg4-CreER; Ai14; R26-Gria2 Tg mice used for neonatal hypoxic-ischemic brain injury was C57BL/6. Other mice for other experiments had mixed genomic backgrounds of B6SJL, C57BL/6 and 129. Both sexes of mice were used in an unbiased manner. The ages of mice for each experiment are described in Results, timelines in figures, or figure legends. All experiments were carried out in compliance with the animal protocols approved by the Institutional Animal Care and Committee (IACUC) at Temple University School of Medicine.
Primary OPC culture
OPC primary culture was prepared according to a method described previously (Chen et al., 2007). P3 pups of either sex were decapitated. The cortices were dissected from the brain, chopped into small pieces with a blade, and digested with papain and DNase I for 15 min at 37°C. Cells were further mechanically dissociated with gentle pipetting and resuspended in Dulbecco’s Modified Eagle Medium (DMEM; Corning, Cat# 10-013-CM) supplemented with 10% fetal bovine serum (FBS; Corning, Cat# 35-010-CV) and 1% penicillin/streptomycin (Thermo Fisher Scientific, Cat# BP2959-50). These cells were plated on T75 flasks coated with poly-D-lysine (PDL; Sigma-Aldrich, Cat# P6407), and maintained for 7–10 days. The T75 flasks were shaken (200 rpm) for 1 hour at 37°C to remove microglial cells. After changing media, the flasks were shaken (200 rpm) overnight (18–20 hours) at 37°C to detach OPCs. OPCs were obtained from the media by centrifugation, seeded onto PDL-coated coverslips (12 mm) (Deckgläser), and kept in the basal defined medium (BDM; DMEM containing 2 mM glutamine, 1% penicillin-streptomycin, 1 mM sodium pyruvate, 5 μg/ml insulin, 5 μg/ml N-Acetyl-L-cysteine, 1% Trace Elements B (Cellgro, 99-175-CI), 10 ng/ml d-Biotin, 0.1 mg/ml BSA, 0.1 mg/ml transferrin, 16 μg/ml putrescine, 60 ng/ml progesterone, 40 ng/ml sodium selenite) complemented with 20 ng/ml PDGF-AA and 20 ng/ml bFGF for an additional 2–3 days.
Primary OPCs isolated by immunopanning
OPCs were isolated from P10 mice of either sex with the immunopanning method described previously (Emery and Dugas, 2013). In brief, one day before cells were collected, a 100-mm dish was coated with goat anti-rat IgG (Jackson Immunoresearch, Cat# 112-005-167) at 4°C overnight, and then incubated with rat anti-CD140 (PDGFRα) antibodies (BD Biosciences, Cat# 558774) for 3 hours at room temperature (RT). The panning dish was rinsed with Dulbecco’s phosphate-buffered saline (DPBS) (Corning, Cat# 21-030-CV) three times. Mouse cortices were digested with papain and DNase I, and cells were further mechanically dissociated. After re-suspension, cells were placed and incubated on the panning dish for 45 min at RT. Non-adherent cells were discarded, and the remaining cells (OPCs) were trypsinized, seeded onto PDL-coated coverslips, and kept in a BDM containing 20 ng/ml PDGF-AA and 20 ng/ml bFGF for 5 days.
METHOD DETAILS
Administration of 4-hydroxytamoxifen or tamoxifen
The injectable solution of (Z)-4-Hydroxytamoxifen (4HT, Sigma-Aldrich, Cat# H7904) was prepared as described before (Kang et al., 2010). Briefly, 4HT was dissolved in 100% ethanol (20 mg/ml) with bath sonication, and aliquots of 1 mg (in 50 μl) were prepared and kept at −80°C until use. For each application, aliquots were emulsified after adding 250 μl of sunflower seed oil (Sigma-Aldrich, Cat# 88921), and the ethanol was evaporated for 30 min using a vacuum centrifuge. The total 4HT dose and number of injections for each set of experiments are stated in either results or figure legends. For example, a single dose of 4HT (0.2 mg s.c. per injection) was injected to Cspg4-CreER; Ai14; R26-EGFP-Gria2 mouse pups at P3 before the H/I injury was applied.
For P51 or older Cspg4-CreER; Ai14; R26-EGFP-Gria2 mice, tamoxifen (Sigma-Aldrich, Cat# T5648) was administered. After tamoxifen was dissolved (20 mg/ml) in a mixture of sunflower seed oil-ethanol (10:1), ethanol was evaporated for 30 min using a vacuum concentrator. 40 mg/kg (b.w.) of tamoxifen was intraperitoneally (i.p.) administered twice a day with at least 6 hours interval between injections. A total of 10 doses was injected into the mice between P51 and P55.
Detection of surface-expressed EGFP-GluA2
The OPCs on coverslips were fixed with 4% paraformaldehyde (PFA; in 0.1M phosphate buffer, pH 7.4) for 20 min and immunostained in the absence of Triton X-100 throughout entire staining procedures until DAPI addition. OPCs were blocked with 10% normal donkey serum for 1 hour at RT and incubated with goat anti-GFP antibodies (Rockland, Cat# 600-101-215; 1:500) and rabbit anti-Olig2 antibodies (EMD Millipore, Cat# AB9610; 1:500) at 4°C overnight. After washing with PBS (each washing for 5 min, three times), OPCs were incubated with secondary antibodies for 2 hours at RT. After secondary antibody incubation, OPCs were exposed to 0.1% Triton X-100 solution containing DAPI for 20 min. As the experimental control, OPCs on a separate coverslip were permeabilized with 0.1% Triton X-100 from the blocking step, and subjected to the immunostaining for the detection of intracellular EGFP and Olig2.
Calcium imaging
To measure calcium responses to AMPA, cultured OPCs were loaded with Fura-2 acetoxymethyl ester (Fura-2 AM; Thermo Fisher Scientific, Cat# F1201; 2 μM) in cation-safe solution (107 mM NaCl, 7.2 mM KCl, 1.2 mM MgCl2, 11.5 mM glucose, 20 mM HEPES-NaOH, 1 mM CaCl2, pH 7.2) for 30 min at RT. Cells were washed and incubated for an additional 30 min for complete de-esterification of the dye. A Leica DMI 6000B fluorescence microscope controlled by SlideBook Software (Intelligent Imaging Innovations) was used to monitor intracellular calcium levels, assessed by fluorescence emission at 505 nm, while alternating excitation wavelengths between 340 and 380 nm. AMPA-dependent calcium responses in OPCs were induced by the addition of cyclothiazide (CTZ; Hello Bio, Cat# HB0221; 100 μM) and AMPA (Tocris, Cat# 0254; 20 μM). The peak values of CTZ-sensitized, AMPA-dependent calcium responses minus peak values of AMPA (with no CTZ) responses of all recorded cells were averaged for each coverslip, which was counted as one replicate.
Fluorescence-activated cell sorting (FACS)
The entire cerebra were isolated from P15 pups and chopped with a blade into small pieces. The brain cells were dissociated using the Neural Dissociation Kit (Miltenyi Biotec, Cat# 130-092-628) according to the manufacturer’s instruction. After enzymatic digestion, the cells were mechanically dissociated with gentle pipetting and suspended in Hank’s Balanced Salt Solution (HBSS; GIBCO, Cat# 14175-075). The cell suspension was passed through a 40-μm cell strainer (Corning, Cat# 352340). Cells were re-suspended in 0.5% FBS in HBSS. Cells were isolated with BD influx (BD Biosciences) at the Flow Core Facility of Temple University School of Medicine.
Real-time PCR
Total RNAs were extracted from the FACS-isolated cells with RNAeasy Plus Micro Kit (QIAGEN, Cat# 74034). cDNAs were generated using SuperScript III First-Strand Synthesis System (Thermo Fisher Scientific, Cat# 18080051). Quantitative PCR was performed using a SYBR Green PCR kit (QIAGEN, Cat# 204143) and StepOnePlus™ Real-Time PCR system (Applied Biosystems).
Induction of hypoxic-ischemic brain injury to mouse neonates
The mouse neonates underwent a surgical procedure for permanent unilateral common carotid artery (CCA) ligation and subsequent exposure to hypoxia (Levine, 1960; Rice et al., 1981) with minor modifications (Shen et al., 2012). After being anesthetized with isoflurane (4% for induction and 2.5% for maintenance), P7 pups were fixed on a surgical platform. After a midline incision of the skin was made in the upper anterior thorax region, the right CCA was identified by key anatomical landmarks. Fascia, the vagus nerve, and sympathetic ganglia were carefully moved aside, and the CCA was isolated using a small hook, and cauterized with a fine-tipped cauterizer (Fine Scientific Tools, Cat# 18000-00). After confirming lack of blood flow into the external and internal carotid artery, muscles and superficial fascia were gently moved back into the proper anatomical position, and the incision was closed using 3M Vetbond Tissue Adhesive (3M Science). After recovery from anesthesia, the pups were returned to the dam and kept with it for an additional 30 min. They were then moved to a sealed hypoxia chamber (BioSpherix) infused with nitrogen to maintain the oxygen level at 6.0% for 30 min at 35°C and returned to the dam.
Injection of lysolecithin
After being anesthetized with a mixture of Ketamine (200 mg/kg) and Xylazine (15mg/kg) (Sigma-Aldrich, Cat# K4138), mice were fixed on a stereotaxic instrument (Stoelting). One μl of 1% L-α-Lysophosphatidylcholine (LPC or lysolecithin in physiological saline; Sigma-Aldrich, Cat# L4129) was injected into right CC (AP: +1.0 mm; ML: −0.8 mm; DV: 2.1 mm from bregma) with Hamilton syringe. The Hamilton syringe was equipped with a 33-gauge needle (45° beveled tip) attached to a motorized stereotaxic injector (Stoelting), and LPC was injected at a rate of 0.05 μl/min. After the injection, the needle was held in place for an additional 10 min and gently pulled out. The scalp was sutured, and the mice were allowed to recover from anesthesia.
Administration of EdU and BrdU
Ethynyl-2′-deoxyuridine (EdU; Lumiprobe, Cat# 40540; 10 mg/kg per injection) or 5-bromo-2′-deoxyuridine (BrdU; Thermo Fisher, Cat# BP-2508-5; 50 mg/kg per injection) was injected intraperitoneally to analyze cell proliferation of OPCs. The number of doses and frequency of injections varied depending on the sampling age, and the details are described in figure legends. For example, EdU was injected daily at P8 and P9 (a total of two doses) for the sampling at P10, whereas it was injected three times between P22 and P23 for the sampling at P24. Seven doses of BrdU were injected daily to the mice that had received LPC between P68 and P74, and a total of 4 doses of EdU were injected between P73 and P74.
Immunofluorescence
Mice were anesthetized with sodium pentobarbital (70 mg/kg, i.p.) and subjected to trans-cardiac perfusion with PBS for 3 min and 4% PFA. Brains were removed from the crania and incubated in 4% PFA at 4°C for an additional 5 hours for post-fixation. The fixed brains were moved to and stored in 30% sucrose (in PBS) at 4°C for at least 36 hours for cryoprotection until sectioning. Brain samples were frozen in Tissue-Tek optimum cutting temperature (O.C.T.) compound (Sakura, Cat# 4586) with dry ice, and sectioned 35 μm thick using a cryostat (Leica). Brain sections were collected into 6-well plates in a stereological manner, and 4 or 5 free-floating sections were used for each set of immunostaining. The sections were first permeabilized with 0.3% Triton X-100, and then blocked with blocking solution (5% normal donkey serum, 0.3% Triton X-100) for 1 hour at RT. The sections were then incubated with primary antibodies in the same blocking solution at 4°C overnight. The primary antibodies used for immunofluorescence were rabbit anti-GFP (Proteintech, Cat#50430-2-AP; 1:500), goat anti-GFP (Rockland, Cat# 600-101-215; 1:500), mouse anti-APC (clone CC1, EMD Millipore, Cat#OP80; 1:60), rat anti-CD140a (PDGFRα, BD Biosciences, Cat# 558774; 1:500), rabbit anti-PDGFRα (a gift from Dr. William Stallcup, Sanford Burnham Prebys Medical Discovery Institute; 1:500), mouse anti-MBP (Covance, Cat#SMI-99P; 1:500), rabbit anti-MBP (Cell Signaling Technology, Cat# 78896; 1:500), guinea pig anti-NG2 (generated by Bergles’ Lab, Johns Hopkins University; 1:4,000), rabbit anti-Olig2 (EMD Millipore, Cat# AB9610; 1:500), rabbit anti-NeuN (Cell Signaling Technology, Cat# 24307; 1:500), rabbit anti-GFAP (Agilent, Cat# Z033429-2; 1:500), rat anti-BrdU (Abcam, Cat# ab6326; 1:500), mouse anti-GluT1 (Abcam, Cat# ab40084; 1:500) and goat anti-Sox9 (R & D Systems, Cat# AF3075, 1:500). After brief washing with PBS (for 5 min, three times), the sections were incubated with secondary antibodies for 2 hours at RT. The secondary antibodies used in this study were Alexa Fluor 488-, Cy3-, or Cy5-conjugated donkey IgG against rat, rabbit, mouse, goat, chicken, or guinea pig (Jackson Immuno Research; 1:500). Nuclei were co-stained with DAPI. After brief washing (for 5 min, three times), the sections were mounted on slide glasses with ProLong antifade reagents (Thermo Fisher Scientific, P36970).
For anti-APC (CC1) immunostaining, brain sections were pretreated with 5 mM citrate buffer (5 mM citric acid, 0.05% Tween 20, pH 6.0) at 95°C for 1.5 min (for sections from P8 or P10 mice) or for 5 min (for sections from P35 mice) before permeabilization. For the detection of EGFP-GluA2 at P35 mice, sections were pretreated with 0.3% H2O2 for 30 min before permeabilization, and anti-EGFP signals were amplified using Tyramide Signal Amplification kit (TSA; PerkinElmer, Cat# NEL753001KT) according to the manufacturer’s instruction.
For BrdU immunostaining, sections were pretreated with 2 N HCl for 30 min at 37°C, followed by incubating with 0.1 M borate buffer (pH 8.5) twice (5 min each time) before the initial washing with 0.01 M PBS. In case of EGFP and BrdU co-immunostaining, EGFP immunopositive signals were amplified with the TSA kit before the sections are incubated with 2 N HCl for BrdU immunostaining.
Detection of EdU
Brains sections were treated with Alexa Fluor-647 Click-iT reaction cocktail solution (Thermo Fisher Scientific, Cat# C10340) according to manufacturer’s instructions before blocking. For assessing the co-localization of EGFP and EdU, GFP immunostaining was performed prior to the incubation of the brain sections with EdU detection reagents.
Image acquisition and analysis
Widefield fluorescence images were collected with an epifluorescence microscope Axio-Imager M2 (Zeiss). Confocal images were obtained with a laser scanning microscope TCS SP8 (Leica), and processed with LAS X software (Leica).
Western blot analysis
Mice cortices were isolated, snap frozen in dry ice, and kept at −80°C until use. The cortices were homogenized in RIPA buffer (Cell Signaling Technology, Cat# 9806) supplemented with protease inhibitor cocktail (Thermo Fisher Scientific, Cat# 1861281) and EDTA (Thermo Fisher Scientific, Cat# 1861274). 5 μg of protein lysate per each sample was mixed with an equal volume of 2X Laemmli Sample Buffer (Bio-Rad, Cat# 1610737), and resolved with a 12% Bis-Tris gel (Bio-Rad, Cat# 3450118). Proteins were transferred to a PVDF membrane (EMD Millipore, Cat# IPVH00010) by the electrophoretic transfer method. The blot membranes were blocked in 5% skim milk (in TBST) for 1 hour, and incubated with primary antibody for 1 hour at RT. The primary antibodies used in this study were rabbit anti-MBP (Cell Signaling Technology, Cat# 78896; 1:2,000), rabbit anti-CNPase (Phosphosolutions, Cat#325-CNP; 1:2,000), mouse anti-MAG (Santa Cruz Biotechnology, Cat# sc-166849; 1:4,000), mouse anti-MOG (Santa Cruz Biotechnology, Cat# sc-166172; 1:2,000), and mouse anti-β-actin (Santa Cruz Biotechnology, Cat# sc-47778; 1:2,000). After washing the blot membrane with TBST for 10 min (three times), the membranes were incubated with HRP-conjugated anti-rabbit or anti-mouse IgG (Jackson Immuno Research; 1:20,000) in 5% skim milk for 1 hour at RT. After washing with TBST for 10 min (four times), the membranes were subjected to chemiluminescence reaction using an ECL kit (Supersignal West Dura, Thermo-Fisher Scientific, Cat# 34075), and signal detection using X-ray film. Intensities of protein bands on X-ray films were quantified with ImageJ. For sequential probing with different antibodies, membranes were stripped with a stripping solution (2% SDS, 0.06 M Tris-HCl, pH 6.8,0.8% 2-mercaptoethanol) at 55°C for 10 min, and washed with TBST for 5 min (five times) before re-blocking.
Electron microscopy
Mice were deeply anesthetized with pentobarbital (70 mg/kg, i.p.) and perfused with PBS followed by 2.5% PFA, 2% glutaraldehyde (in 0.1 M phosphate buffer, pH 7.4). After brain isolation, samples were post-fixed in the same fixative for 18 hours at 4°C, transferred to, and kept in 0.1 M phosphate buffer at 4°C. Samples were treated with 2% OsO4 for 1 hour, and 2% uranyl acetate for 30 min. Samples were sequentially dehydrated using 50%, 70%, 90%, 100% ethanol, and propylene oxide. Samples were embedded in Epon 812 resin (Ted Pella), and ultrathin sections were obtained using Ultracut UCT (Leica). Sections were stained with 2% uranyl acetate and lead citrate, and imaged with an H-7600 microscope (Hitachi, Tokyo, Japan). ImageJ was used to measure the calibers of axons and myelin sheaths.
QUANTIFICATION AND STATISTICAL ANALYSIS
In cell mosaic analysis with Cspg4-CreER; Ai14; R26-Gria2 mice, tdTomato+ control and tdTomato+ EGFP-GluA2-expressing cells (E-A2 cells) were used to rule out biases in assessing cellular activity affected by tdTomato expression. Cell counting with fluorescence and confocal images was performed with the help of Zen lite (Zeiss) and LAS-X (Leica), respectively. The numbers of counted cells varied according to the genetic method used for cell labeling (e.g., Cre driver and tamoxifen dose), brain region, age, and cell maturation stage. The ranges of counted cells were: (1) 50 – 150 cells per field for early oligodendrocyte development, (2) 10 – 50 cells per field in confocal images for oligodendroglial maturation in the adult CC, (3) 50 – 250 cells for neonatal H/I injury, and (4) 10 – 50 cells per field in confocal images for oligodendroglial regeneration in LPC-injected lesions. Prism 5 (Graphpad) was used for graph drawing and statistical analyses.
With the assumption that the quantitative variables of this study are normally distributed, two tailed Student’s t test, one-way or two-way ANOVA was used to test the statistical significance. A paired t test compared the ipsilateral and contralateral sides in corresponding areas, as well as tdTomato+ EGFP− and tdTomato+ EGFP+ cells (E-A2 cells) of each mouse for responses to H/I injury. An unpaired t test was used to compare cellular responses in biologically independent groups (e.g., when control and GluA2 overexpressing cells were quantified from different mice). A two-way ANOVA with Bonferroni post hoc analysis was used to assess new oligodendrocyte generation from P8 to P10 for control and GluA2-overexpressing cells. Bars and error bars of graphs represent means and standard errors of the mean (SEM), respectively. Unless otherwise stated, the sample size (n number) represents either number of coverslips (Figure 1) or number of mice used for each experiment. All of the statistical details of experiments (method, n number, and p value) can be found in figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| mouse anti-β-actin | Santa Cruz Biotechnology | Cat# sc-47778; RRID: AB_2714189 |
| rat anti-BrdU | Abcam | Cat# ab6326; RRID: AB_305426 |
| mouse anti-CC1 (APC) | EMD Millipore | Cat# OP80; RRID: AB_2057371 |
| rabbit anti-CNPase | PhosphoSolutions | Cat# 325-CNP; RRID: AB_2492062 |
| rabbit anti-GFAP | Agilent | Cat# Z033429-2; RRID: AB_10013382 |
| goat anti-GFP | Rockland | Cat# 600-101-215; RRID: AB_218182 |
| rabbit anti-GFP | Proteintech | Cat# 50430-2-AP; RRID: AB_11042881 |
| mouse anti-GluT1 | Abcam | Cat# ab40084; RRID: AB_2190927 |
| mouse anti-Mag | Santa Cruz Biotechnology | Cat# sc-166849; RRID: AB_2250078 |
| rabbit anti-MBP | Cell Signaling Technology | Cat# 78896; RRID: AB_2799920 |
| mouse anti-MBP | Covance | Cat# SMI-99P; RRID: AB_10120130 |
| mouse anti-MOG | Santa Cruz Biotechnology | Cat# sc-166172; RRID: AB_2145540 |
| rabbit anti-NeuN | Cell Signaling Technology | Cat# 24307; RRID: AB_2651140 |
| guinea pig anti-NG2 | Laboratory of Dwight Bergles | N/A |
| rabbit anti-Olig2 | EMD Millipore | Cat# AB9610; RRID: AB_57066 |
| rat anti-PDGFRα (CD140a) | BD Biosciences | Cat# 558774; RRID: AB_397117 |
| rabbit anti-PDGFRα | Gift from Dr. William Stallcup (SBP institute) | N/A |
| goat anti-Sox9 | R & D Systems | Cat# AF3075; RRID: AB_2194160 |
| Chemicals | ||
| (S)-AMPA | Tocris | Cat# 0254 |
| 5-Bromo-2′-deoxyuridine (BrdU) | Thermo Fisher Scientific | Cat# BP-2508-5 |
| Cyclothiazide | Hello Bio | Cat# HB0221 |
| 5-Ethynyl-2′-deoxyuridine (EdU) | Lumiprobe | Cat# 40540 |
| Fura-2 acetoxymethyl ester | Thermo Fisher Scientific | Cat# F1201 |
| 4-Hydroxytamoxifen (4HT) | Sigma-Aldrich | Cat# H7904 |
| L-α-Lysolecithin (LPC) | Sigma-Aldrich | Cat# L4129 |
| Tamoxifen | Sigma-Aldrich | Cat# T5648 |
| Critical commercial assays | ||
| Click-iT EdU Proliferation Kit | Thermo Fisher Scientific | Cat# C10340 |
| Neural Tissue Dissociation Kit (P) | Miltenyi Biotec | Cat# 130-092-628 |
| Pierce BCA protein assay | Thermo Fisher Scientific | Cat# 23227 |
| QuantiTect SYBR Green PCR Kit | QIAGEN | Cat# 204143 |
| Rneasy Plus Micro Kit | QIAGEN | Cat# 74034 |
| SuperSignal West Dura | Thermo Fisher Scientific | Cat# 34075 |
| SuperScript III First-Strand Synthesis System | Thermo Fisher Scientific | Cat# 18080051 |
| TSA (+) Cyanine 3 and Fluorescein System | PerkinElmer | Cat# NEL753001KT |
| Experimental model: organisms/strains | ||
| Cspg4-CreER | The Jackson Laboratory | Cat# JAX: 008538; RRID: IMSR_JAX:008538 |
| Mobp-EGFP | MMRRC / GENSAT. Kang et al., 2013 | RRID: MMRRC_030438-UCD |
| Plp1-CreER | The Jackson Laboratory | Cat# JAX: 005975; RRID: IMSR_JAX:005975 |
| ROSA26-tdTomato (Ai14) | The Jackson Laboratory | Cat# JAX: 007914; RRID: IMSR_JAX:007914 |
| ROSA26-EYFP | The Jackson Laboratory | Cat# JAX: 006148; RRID: IMSR_JAX:006148 |
| ROSA26-mEGFP | The Jackson Laboratory | Cat# JAX: 007576; RRID: IMSR_JAX:007576 |
| ROSA26-EGFP-Gria2 | This paper | N/A |
| Sox10-CreER | This paper | N/A |
| Oligonucleotides | ||
| GluA2 (Forward) | This paper: 5′ AACGAATGAAGGTGGCAAAG 3′ | N/A |
| WPRE (Reverse) | This paper: 5′ GAATTGTCAGTGCCCAACAG 3′ | N/A |
| R26U1 (Forward) | This paper: 5′ CTCTGCTGCCTCCTGGCTTCT 3′ | N/A |
| R26U2 (Reverse) | This paper: 5′ CGAGGCGGATCACAAGCAATA 3′ | N/A |
| Sox10-CreER (Forward) | This paper: 5′ CACCTAGGGTCTGGCATGTG 3′ | N/A |
| Sox10-CreER (Reverse) | This paper: 5′ ATCGCGAACATCTTCAGG 3′ | N/A |
| β-actin mRNA (Forward) | This paper; 5′ TGACAGGATGCAGAAGGAGA 3′ | N/A |
| β-actin mRNA (Reverse) | This paper; 5′ CGTTCAGGAGGAGCAATG 3′ | N/A |
| Gria2 mRNA (Forward) | This paper; 5′ TCGACAATTTGGAGGTAGCC 3′ | N/A |
| Gria2 mRNA (Reverse) | This paper; 5′ ACATGCAGTGTCCCACAGAA 3′ | N/A |
| Software and algorithms | ||
| Adobe Photoshop | Adobe | RRID: SCR_014199; https://www.adobe.com/products/photoshop.html |
| AxioVision | Zeiss Microscope | RRID:SCR_002677; https://www.micro-shop.zeiss.com/en/us/system/software-axiovision+software-products/1007/ |
| CorelDRAW | Corel Corporation | RRID:SCR_014235; https://www.coreldraw.com/en/product/coreldraw/?hp=hero-pc |
| ImageJ | NIH | RRID:SCR_003073; https://imagej.nih.gov/ij/ |
| Leica Application Suite X (LAS X) | Leica | RRID:SCR_013673; https://www.leica-microsystems.com/products/microscope-software/p/leica-las-x-ls/ |
| Prism 5 | GraphPad | RRID: SCR_002798; https://www.graphpad.com/scientific-software/prism/ |
| SlideBook | Intelligent Imaging Innovations | RRID: SCR_014300; https://www.intelligent-imaging.com/slidebook |
| StepOne software 2.1 | Thermo Fisher Scientific | RRID:SCR_014281; http://downloads.thermofisher.com/Instrument_Software/qPCR/Step-1/SOP23_Release%20Notes_4482516.pdf |
| Zen 3.0 | Zeiss Microscope | RRID: SCR_013672; https://www.zeiss.com/microscopy/int/products/microscope-software/zen.html |
| Other | ||
| BD Influx Cell Sorter | BD Biosciences | N/A |
| Hamilton syringe 701 | Hamilton company | Cat# 7635-01 |
| Hamilton small hub RN needle | Hamilton company | Cat# 7803-05 |
| O2 controller for hypoxia chamber | BioSpherix | Cat# ProOx P110 |
| Small vessel cauterizer | Fine Science Tools | Cat# 18000-00 |
| Stereotaxic injector (motorized) | Stoelting | Cat# 53311 |
Highlights.
Development of a mouse line for inducible, cell-specific GluA2 expression
Oligodendrocyte generation is not altered by high GluA2 levels in the healthy CNS
GluA2 overexpression increases OPC proliferation in the adult CNS
High OPC GluA2 levels promote oligodendrocyte regeneration after white matter injury
ACKNOWLEDGMENTS
We thank Jason Yang, Yumi Jeong, and Ilan Crawley for their excellent technical assistance. A.A. and H.-K.J. were supported by the National Multiple Sclerosis Society postdoctoral fellowship (FG 1927-A-1) and the Shriners Hospitals for Children postdoctoral fellowship (84298-PHI), respectively. Funding was provided from the NIH (R01GM117907 to J.S., R21NS092009 to D.E.B., and R01NS089586 to S.H.K.); Deutsche Forschungsgemeinschaft (FOR2289-P8 to A.A.); the Chica and Heinz Schaller Foundation (A.A.); the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (D.E.B.); the National Multiple Sclerosis Society (PP1988 to S.H.K.); the Ellison Medical Foundation (AG-NS-1101-13 to S.H.K.); and Shriners Hospitals for Children (85500-PHI-14 to S.H.K.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109147.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Alizadeh A, Dyck SM, and Karimi-Abdolrezaee S (2015). Myelin damage and repair in pathologic CNS: challenges and prospects. Front. Mol. Neurosci 8, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baraban M, Koudelka S, and Lyons DA (2018). Ca2+ activity signatures of myelin sheath formation and growth in vivo. Nat. Neurosci 21, 19–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beattie MS, Ferguson AR, and Bresnahan JC (2010). AMPA-receptor trafficking and injury-induced cell death. Eur. J. Neurosci 32, 290–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergles DE, Roberts JD, Somogyi P, and Jahr CE (2000). Glutamatergic synapses on oligodendrocyte precursor cells in the hippocampus. Nature 405, 187–191. [DOI] [PubMed] [Google Scholar]
- Berret E, Barron T, Xu J, Debner E, Kim EJ, and Kim JH (2017). Oligodendroglial excitability mediated by glutamatergic inputs and Nav1.2 activation. Nat. Commun 8, 557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buser JR, Maire J, Riddle A, Gong X, Nguyen T, Nelson K, Luo NL, Ren J, Struve J, Sherman LS, et al. (2012). Arrested preoligodendrocyte maturation contributes to myelination failure in premature infants. Ann. Neurol 71, 93–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Kintner DB, Jones M, Matsuda T, Baba A, Kiedrowski L, and Sun D (2007). AMPA-mediated excitotoxicity in oligodendrocytes: role for Na(+)-K(+)-Cl(−) co-transport and reversal of Na(+)/Ca(2+) exchanger. J. Neurochem 102, 1783–1795. [DOI] [PubMed] [Google Scholar]
- Chen TJ, Kula B, Nagy B, Barzan R, Gall A, Ehrlich I, and Kukley M (2018). In Vivo Regulation of Oligodendrocyte Precursor Cell Proliferation and Differentiation by the AMPA-Receptor Subunit GluA2. Cell Rep. 25, 852–861.e7. [DOI] [PubMed] [Google Scholar]
- Choi DW (1988). Glutamate neurotoxicity and diseases of the nervous system. Neuron 1, 623–634. [DOI] [PubMed] [Google Scholar]
- De Biase LM, Nishiyama A, and Bergles DE (2010). Excitability and synaptic communication within the oligodendrocyte lineage. J. Neurosci 30, 3600–3611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Rosenberg PA, Volpe JJ, and Jensen FE (2003). Calcium-permeable AMPA/kainate receptors mediate toxicity and preconditioning by oxygen-glucose deprivation in oligodendrocyte precursors. Proc. Natl. Acad. Sci. USA 100, 6801–6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerflinger NH, Macklin WB, and Popko B (2003). Inducible site-specific recombination in myelinating cells. Genesis 35, 63–72. [DOI] [PubMed] [Google Scholar]
- Emery B, and Dugas JC (2013). Purification of oligodendrocyte lineage cells from mouse cortices by immunopanning. Cold Spring Harb. Protoc 2013, 854–868. [DOI] [PubMed] [Google Scholar]
- Ferrario CR, Ndukwe BO, Ren J, Satin LS, and Goforth PB (2013). Stretch injury selectively enhances extrasynaptic, GluN2B-containing NMDA receptor function in cortical neurons. J. Neurophysiol 110, 131–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields RD (2015). A new mechanism of nervous system plasticity: activity-dependent myelination. Nat. Rev. Neurosci 16, 756–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foerster S, Hill MFE, and Franklin RJM (2019). Diversity in the oligodendrocyte lineage: plasticity or heterogeneity? Glia 67, 1797–1805. [DOI] [PubMed] [Google Scholar]
- Follett PL, Rosenberg PA, Volpe JJ, and Jensen FE (2000). NBQX attenuates excitotoxic injury in developing white matter. J. Neurosci 20, 9235–9241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fünfschilling U, Supplie LM, Mahad D, Boretius S, Saab AS, Edgar J, Brinkmann BG, Kassmann CM, Tzvetanova ID, Möbius W, et al. (2012). Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 485, 517–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier HO, Evans KA, Volbracht K, James R, Sitnikov S, Lundgaard I, James F, Lao-Peregrin C, Reynolds R, Franklin RJ, and Káradóttir RT (2015). Neuronal activity regulates remyelination via glutamate signalling to oligodendrocyte progenitors. Nat. Commun 6, 8518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge WP, Yang XJ, Zhang Z, Wang HK, Shen W, Deng QD, and Duan S (2006). Long-term potentiation of neuron-glia synapses mediated by Ca2+-permeable AMPA receptors. Science 312, 1533–1537. [DOI] [PubMed] [Google Scholar]
- Gruchot J, Weyers V, GÖttle P, FÖrster M, Hartung HP, Küry P, and Kremer D (2019). The Molecular Basis for Remyelination Failure in Multiple Sclerosis. Cells 8, 825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollmann M, Hartley M, and Heinemann S (1991). Ca2+ permeability of KA-AMPA–gated glutamate receptor channels depends on subunit composition. Science 252, 851–853. [DOI] [PubMed] [Google Scholar]
- Iino M, Goto K, Kakegawa W, Okado H, Sudo M, Ishiuchi S, Miwa A, Takayasu Y, Saito I, Tsuzuki K, and Ozawa S (2001). Glia-synapse interaction through Ca2+-permeable AMPA receptors in Bergmann glia. Science 292, 926–929. [DOI] [PubMed] [Google Scholar]
- Kang SH, Fukaya M, Yang JK, Rothstein JD, and Bergles DE (2010). NG2+ CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life and following neurodegeneration. Neuron 68, 668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SH, Li Y, Fukaya M, Lorenzini I, Cleveland DW, Ostrow LW, Rothstein JD, and Bergles DE (2013). Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat. Neurosci 16, 571–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kougioumtzidou E, Shimizu T, Hamilton NB, Tohyama K, Sprengel R, Monyer H, Attwell D, and Richardson WD (2017). Signalling through AMPA receptors on oligodendrocyte precursors promotes myelination by enhancing oligodendrocyte survival. eLife 6, e28080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasnow AM, Ford MC, Valdivia LE, Wilson SW, and Attwell D (2018). Regulation of developing myelin sheath elongation by oligodendrocyte calcium transients in vivo. Nat. Neurosci 21, 24–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukley M, Capetillo-Zarate E, and Dietrich D (2007). Vesicular glutamate release from axons in white matter. Nat. Neurosci 10, 311–320. [DOI] [PubMed] [Google Scholar]
- Kukley M, Nishiyama A, and Dietrich D (2010). The fate of synaptic input to NG2 glial cells: neurons specifically downregulate transmitter release onto differentiating oligodendroglial cells. J. Neurosci 30, 8320–8331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larm JA, Cheung NS, and Beart PM (1997). Apoptosis induced via AMPA-selective glutamate receptors in cultured murine cortical neurons. J. Neurochem 69, 617–622. [DOI] [PubMed] [Google Scholar]
- Larson VA, Mironova Y, Vanderpool KG, Waisman A, Rash JE, Agarwal A, and Bergles DE (2018). Oligodendrocytes control potassium accumulation in white matter and seizure susceptibility. eLife 7, e34829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Morrison BM, Li Y, Lengacher S, Farah MH, Hoffman PN, Liu Y, Tsingalia A, Jin L, Zhang PW, et al. (2012). Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487, 443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine S (1960). Anoxic-ischemic encephalopathy in rats. Am. J. Pathol 36, 1–17. [PMC free article] [PubMed] [Google Scholar]
- Lin SC, Huck JH, Roberts JD, Macklin WB, Somogyi P, and Bergles DE (2005). Climbing fiber innervation of NG2-expressing glia in the mammalian cerebellum. Neuron 46, 773–785. [DOI] [PubMed] [Google Scholar]
- Liu XB, Shen Y, Plane JM, and Deng W (2013). Vulnerability of premyelinating oligodendrocytes to white-matter damage in neonatal brain injury. Neurosci. Bull 29, 229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarran WJ, and Goldberg MP (2007). White matter axon vulnerability to AMPA/kainate receptor-mediated ischemic injury is developmentally regulated. J. Neurosci 27, 4220–4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JW, Althomsons SP, Hyrc KL, Choi DW, and Goldberg MP (1998). Oligodendrocytes from forebrain are highly vulnerable to AMPA/kainate receptor-mediated excitotoxicity. Nat. Med 4, 291–297. [DOI] [PubMed] [Google Scholar]
- McKhann GM 2nd, D’Ambrosio R, and Janigro D (1997). Heterogeneity of astrocyte resting membrane potentials and intercellular coupling revealed by whole-cell and gramicidin-perforated patch recordings from cultured neocortical and hippocampal slice astrocytes. J. Neurosci 17, 6850–6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei F, Lehmann-Horn K, Shen YA, Rankin KA, Stebbins KJ, Lorrain DS, Pekarek K,A Sagan S, Xiao L, Teuscher C, et al. (2016). Accelerated remyelination during inflammatory demyelination prevents axonal loss and improves functional recovery. eLife 5, e18246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan S, and Chan JR (2017). Regulation and dysregulation of axon infrastructure by myelinating glia. J. Cell Biol 216, 3903–3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patneau DK, Vyklicky L Jr., and Mayer ML (1993). Hippocampal neurons exhibit cyclothiazide-sensitive rapidly desensitizing responses to kainate. J. Neurosci 13, 3496–3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patneau DK, Wright PW, Winters C, Mayer ML, and Gallo V (1994). Glial cells of the oligodendrocyte lineage express both kainate- and AMPA-preferring subtypes of glutamate receptor. Neuron 12, 357–371. [DOI] [PubMed] [Google Scholar]
- Pitt D, Werner P, and Raine CS (2000). Glutamate excitotoxicity in a model of multiple sclerosis. Nat. Med 6, 67–70. [DOI] [PubMed] [Google Scholar]
- Rice JE 3rd, Vannucci RC, and Brierley JB (1981). The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann. Neurol 9, 131–141. [DOI] [PubMed] [Google Scholar]
- Saab AS, Neumeyer A, Jahn HM, Cupido A, šimek AA, Boele HJ, Scheller A, Le Meur K, Götz M, Monyer H, et al. (2012). Bergmann glial AMPA receptors are required for fine motor coordination. Science 337, 749–753. [DOI] [PubMed] [Google Scholar]
- Salter MG, and Fern R (2005). NMDA receptors are expressed in developing oligodendrocyte processes and mediate injury. Nature 438,1167–1171. [DOI] [PubMed] [Google Scholar]
- Segovia KN, McClure M, Moravec M, Luo NL, Wan Y, Gong X, Riddle A, Craig A, Struve J, Sherman LS, and Back SA (2008). Arrested oligodendrocyte lineage maturation in chronic perinatal white matter injury. Ann. Neurol 63, 520–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Liu XB, Pleasure DE, and Deng W (2012). Axon-glia synapses are highly vulnerable to white matter injury in the developing brain. J. Neurosci. Res 90, 105–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi S, Hayashi Y, Esteban JA, and Malinow R (2001). Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell 105, 331–343. [DOI] [PubMed] [Google Scholar]
- Simons M, and Nave KA (2015). Oligodendrocytes: Myelination and Axonal Support. Cold Spring Harb. Perspect. Biol 8, a020479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer S, Volbracht K, Lundgaard I, and Káradóttir RT (2016). Glutamate signalling: a multifaceted modulator of oligodendrocyte lineage cells in health and disease. Neuropharmacology 110(Pt B), 574–585. [DOI] [PubMed] [Google Scholar]
- Spitzer SO, Sitnikov S, Kamen Y, Evans KA, Kronenberg-Versteeg D, Dietmann S, de Faria O Jr., Agathou S, and Káradóttir RT (2019). Oligodendrocyte Progenitor Cells Become Regionally Diverse and Heterogeneous with Age. Neuron 101, 459–471.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe JJ (2001). Neurobiology of periventricular leukomalacia in the premature infant. Pediatr. Res 50, 553–562. [DOI] [PubMed] [Google Scholar]
- Wang F, Yang YJ, Yang N, Chen XJ, Huang NX, Zhang J, Wu Y, Liu Z, Gao X, Li T, et al. (2018). Enhancing Oligodendrocyte Myelination Rescues Synaptic Loss and Improves Functional Recovery after Chronic Hypoxia. Neuron 99, 689–701.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XW, Model P, and Heintz N (1997). Homologous recombination based modification in Escherichia coli and germline transmission in transgenic mice of a bacterial artificial chromosome. Nat. Biotechnol 15, 859–865. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Hill RA, Dietrich D, Komitova M, Suzuki R, and Nishiyama A (2011). Age-dependent fate and lineage restriction of single NG2 cells. Development 138, 745–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziskin JL, Nishiyama A, Rubio M, Fukaya M, and Bergles DE (2007). Vesicular release of glutamate from unmyelinated axons in white matter. Nat. Neurosci 10, 321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.