

Abstract
Herein we report the syntheses and comparative photophysical, electrochemical, in vitro, and in vivo biological efficacy of 3-(1′-hexyloxy)ethyl-3-devinylpyropheophorbide-cyanine dye (HPPH-CD) and the corresponding indium (In), gallium (Ga), and palladium (Pd) conjugates. The insertion of a heavy metal in the HPPH moiety makes a significant difference in FRET (Förster resonance energy transfer) and electrochemical properties, which correlates with singlet oxygen production [a key cytotoxic agent for photodynamic therapy (PDT)] and long-term in vivo PDT efficacy. Among the metalated analogs, the In(III) HPPH-CD showed the best cancer imaging and PDT efficacy. Interestingly, in contrast to free base HPPH-CD, which requires a significantly higher therapeutic dose (2.5 μmol/kg) than imaging dose (0.3 μmol/kg), the corresponding In(III) HPPH-CD showed excellent imaging and therapeutic potential at a remarkably low dose (0.3 μmol/kg) in BALB/c mice bearing Colon26 tumors. A comparative study of metalated and corresponding nonmetalated conjugates further confirmed that STAT-3 dimerization can be used as a biomarker for determining the level of photoreaction and tumor response.
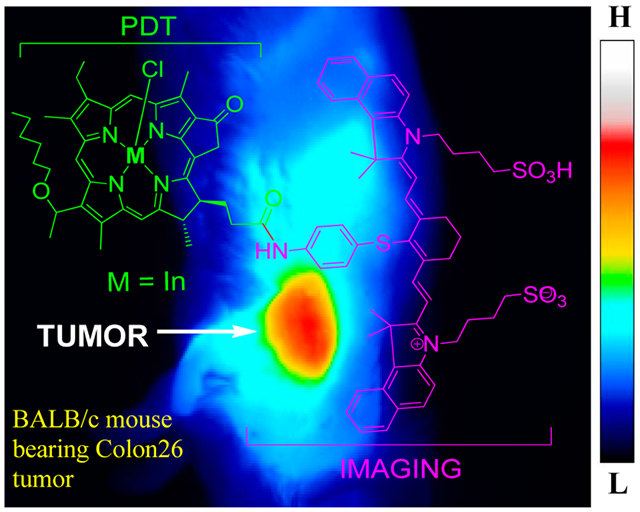
Graphical Abstract
INTRODUCTION
Since the Federal Drug Administration’s approval of the photodynamic therapy (PDT) drug Photofrin in the 1990s for various oncologic uses, there have been continued efforts in various laboratories to develop next-generation photosensitizers (PS) which can overcome the limitations associated with Photofrin.1,2 Unlike conventional treatments, PDT is unique in that it requires the combination of three factors for the treatment’s therapeutic efficacy.3 These components are (i) photosensitizing drug (PS), (ii) light of the wavelength that activates the PS, and (iii) molecular oxygen. After optimal tumor accumulation of the PS, the tumor is illuminated by guided monochromatic laser light of a specific wavelength. The result of this illumination is excitation of electrons in the PS and its promotion to higher energy states. Energy transfer via the photodynamic process between the PS excited triplet state and triplet state oxygen leads to the production of highly reactive singlet oxygen, which is the cytotoxic species created by the photoreaction. Delivery of photoactivating light is necessary to destroy tumor tissue; however, if tumor cells at the margins are missed, then a high possibility of tumor regrowth exists. Therefore, real-time visualization of the PS-containing tumor within the treatment field would enable efficient delivery of light to the entire lesion, thereby improving PDT.
In recent years, imaging modalities such as fluorescence and/or photoacoustic (PA) image guided therapy has attracted significant interest.4 Fortunately most of the porphyrin-based PSs fluoresce and this property has been extensively explored in preclinical and clinical settings to demonstrate the utility of image-guided therapy/surgery for superficial tumors and also for the resection of tumor margins.5 Some of these PSs have been shown to be useful in differentiating malignant tumors from normal tissues.6,7 In certain instances, it has been shown that due to the presence of a significant amount of blood in the surgical field, the optical properties of some PSs are not optimal for detecting fluorescence signals from the desired site(s). In general porphyrin-based PSs exhibit limited Stokes shift(s) between the longest wavelength absorption and emission and have relatively low extinction coefficients. One possible solution for developing efficient agents for imaging with PDT capability could be to conjugate a near-infrared-absorbing probe with desirable photophysical properties to tumor-avid PS, provided that the addition of the probe to the PS does not reduce its tumor-avidity.
Previous reports from our laboratory have shown that certain chlorophyll-a analogs (e.g., HPPH, which is undergoing Phase II clinical trials) can be used as a vehicle to deliver the imaging agents [e.g., cyanine dyes (CDs)] to tumors.8 The bifunctional agent showed the potential for image-guided surgery with an option of PDT and for monitoring tumor response to surgery or other treatment modalities. Due to its excellent tumor imaging ability (fluorescence) and long-term tumor curative properties via PDT, HPPH-CD is currently being developed in collaboration between Photolitec, USA and HISUN Pharma, China for fluorescence image-guided surgery of various cancer indications. However, the HPPH-CD conjugate (Figure 1) does have some drawbacks, the most significant of which being that a significantly higher dose (8-fold) of the conjugate is required for cancer therapy as compared to its imaging dose. In other words, there was a significant therapeutic dose difference between the bifunctional agent (HPPH-CD) and the PS (HPPH) alone (3.5 μmol/kg vs 0.47 μmol/kg, respectively).
Figure 1.
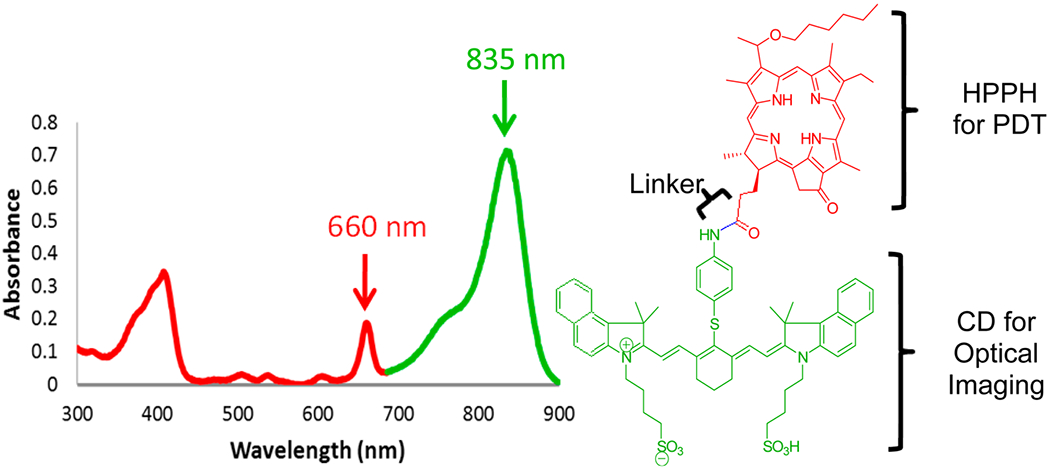
Electronic absorption spectrum of HPPH-CD conjugate in methanol. The part of the spectrum presented in red corresponds to the HPPH moiety, whereas the green band belongs to the CD part of the conjugate.
Two possible explanations for this requirement of a higher therapeutic drug dose of HPPH-CD include (i) intramolecular interactions between the two linked moieties of HPPH and CD (either stacking/aggregation or a close proximity of two chromophores via folding) and (ii) the spectral overlap of the fluorescence of HPPH and absorption of CD, which could prevent transition from the ground singlet state of HPPH to its excited triplet state in a process known as Förster resonance energy transfer (FRET).9
If the two moieties are close to each other, a portion of the reactive singlet oxygen produced by photoactivation of HPPH could destroy the CD (a singlet oxygen quencher), rather than attacking the tumor. This hypothesis was confirmed by measuring the singlet oxygen yield of the conjugate at variable time points during light exposure. Initially, the singlet oxygen yield was low but, as the experiment continued and more CD was destroyed, a significant increase in singlet oxygen production was observed.8
While several approaches could minimize the effects of the interaction between porphyrin and CD chromophores, we have pursued two methods. The first, presented here, focuses on increasing the singlet oxygen quantum yield of HPPH. The second, published elsewhere, aims at designing conjugates in which the two chromophores are separated by a variable length linker.8,10
For developing conjugates with increased singlet oxygen yields, we inserted a series of heavy metals (In, Ga, and Pd) in the HPPH moiety. It has been shown that heavy metals within the porphyrin alter its structure and favor a more planar conformation, thereby making the porphyrin more stable.11,12 Metalation has also been shown to alter the photophysical and photochemical properties of the porphyrin,13 resulting in a hypsochromic shift in the absorption spectra. This could further reduce the spectral overlap between the absorption and fluorescence of the two moieties, resulting in decreased FRET and enhanced singlet oxygen production. The presence of the heavy metal in HPPH should reduce the energy difference between the singlet state and the excited triplet state of the PS and increase its triplet state lifetime,14 in a process known as the heavy-atom effect. As a result of these changes to the triplet state, intersystem crossing should become more favorable, thereby resulting in increased singlet oxygen production due to a longer lifetime of the PS’s excited triplet state. While there are no steadfast rules as to what metals are effective in enhancing singlet oxygen generation in various tetrapyrrolic systems, it was observed that indium(III) insertion enhances singlet oxygen production and PDT efficacy.15 Since FRET is a distance-dependent process in which the donor and acceptor need to be in close proximity (1–10 nm), we have observed9 that the other approach to extend the linker region between the two chromophores also helps to prevent quenching of the CD by singlet oxygen.
We hypothesized that the insertion of heavy metals (In, Ga, Pd) to HPPH moiety of HPPH-CD conjugate will alter its photophysical properties which, in combination with reduced FRET, will enhance its PDT efficacy without diminishing the cancer-imaging ability of the CD.
RESULTS AND DISCUSSION
Chemistry.
Cyanine dye and HPPH-CD conjugate (compound 4) were synthesized following the methodologies developed in our laboratory8 and converted to the corresponding In, Ga, and Pd analogs by following the methodology illustrated in Scheme 1. In brief, the cyanine dye 2, obtained by treating 1 with p-thioaniline, was reacted with 3-[1′-hexyloxy)-ethyl]-3-devinylpyropheophorbide-a (HPPH) 3. Conjugate 4 was reacted with indium chloride, gallium chloride, and palladium chloride to provide metalated analogs 5, 6, and 7, respectively, in good yields. All new compounds were purified by silica column chromatography and silica-coated preparative plates. New conjugates were characterized by NMR, UV–vis, and mass spectrometry.
Scheme 1.
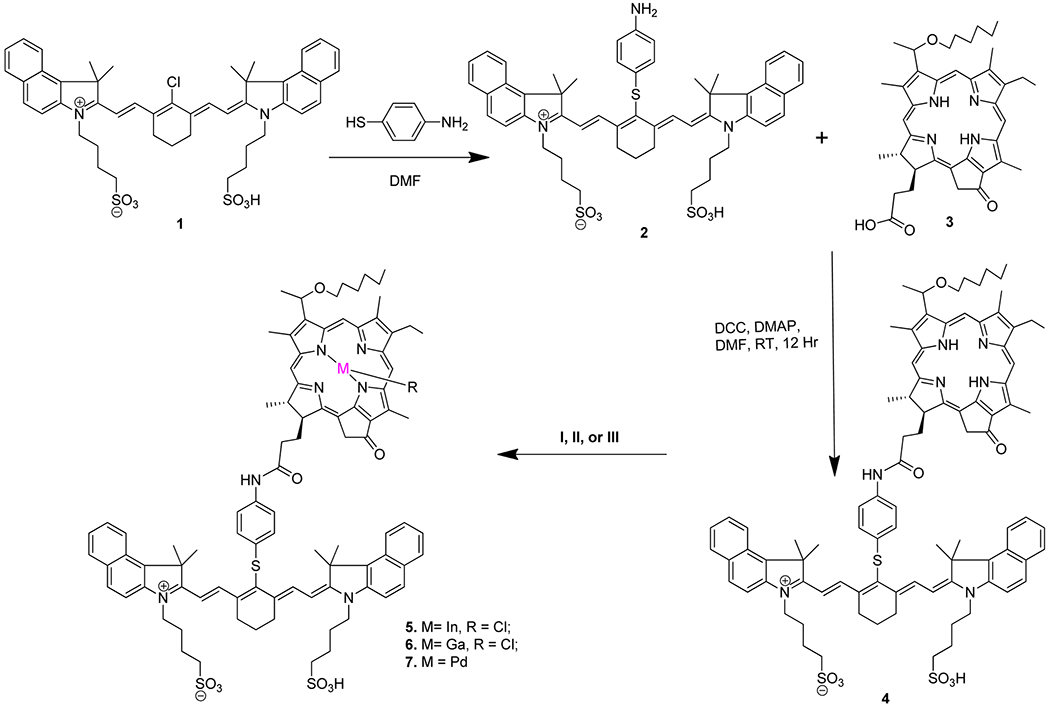
General Procedure for the Synthesis of Metalloporphyrin–Cyanine Dye Conjugates
Photophysical Characterization of HPPH-CD Conjugates (Free Base vs Metalated Analogs).
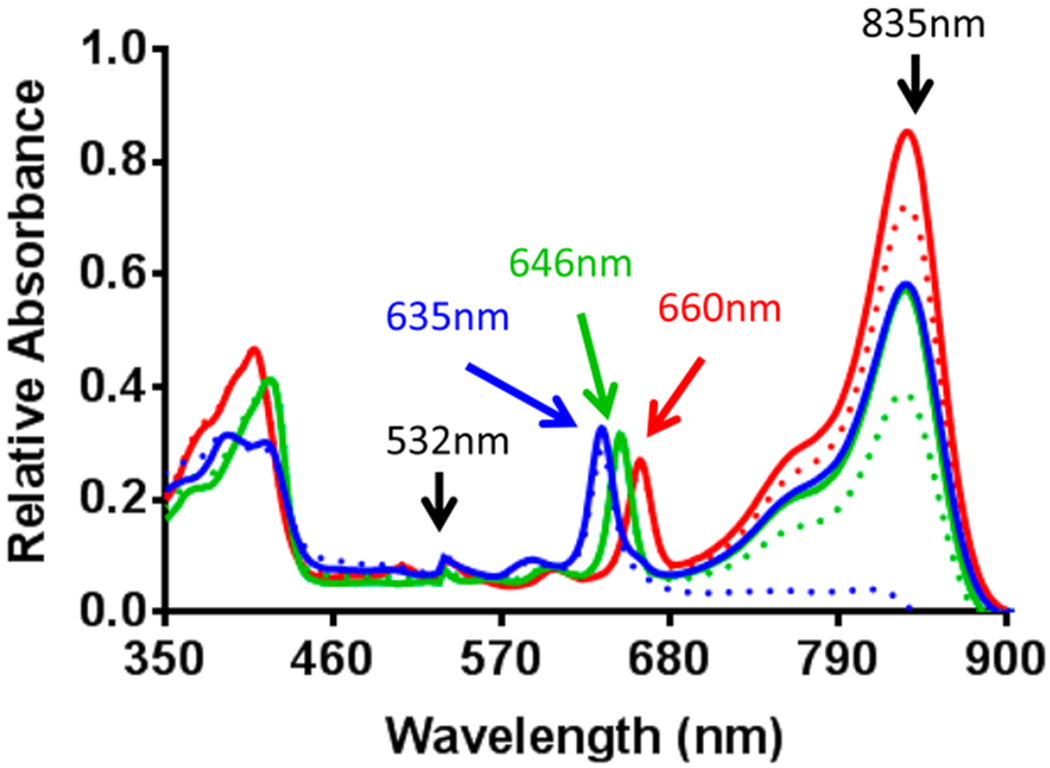
Photophysical properties for all conjugates were examined in methanol, where metalloporphyrin conjugates 5 (In), 6 (Ga), and 7 (Pd) had no difference in the absorption of the CD moiety (835 nm) compared to reference compound 4 (free-base). This was expected, as metalation of the porphyrin should not alter the optical properties of the CD moiety. However, all metalloporphyrins experienced a hypsochromic (blue) shift of ~10–25 nm in absorption of the porphyrin moiety, resulting in λmax values of 646 nm for 5, 649 nm for 6, and 635 nm for 7 as compared to the 660 nm absorption observed for 4.
The singlet oxygen quantum yield for 4 (free-base), 5 (In), and 7 (Pd) were also measured in methanol through exciting the PSs at 532 nm (125 mW). As compared to HPPH (1O2 yield: 45%), the singlet oxygen-producing efficiency of HPPH-CD conjugate 4 was low (5%). This reduction in singlet oxygen yield can be explained due to its quenching by the CD. As expected, compared to free base HPPH-CD, the singlet oxygen yields of the corresponding metalated analogs In and Pd were higher and among all the analogs, the In(III) HPPH-CD conjugate 5 producing the highest singlet oxygen efficiency (27%).
Before evaluating these compounds in detailed in vitro and in vivo studies, we determined their stability in organic and formulating solvents. Among the metalated analogs 5–7, the Ga complex showed limited stability in methanol. Leaving the solution for an extended period of time at room temperature decomposed the PS component of the conjugate, which was surprising. This limited certain in vitro experiments and characterization studies that could have been performed with the Ga compound.
Comparative in Vitro Stability of HPPH-CD Conjugates (Free Base vs Metalated Analogs).
Photobleaching (PB), or the rate of destruction of a chromophore when exposed to light of an appropriate wavelength, provides useful information in PDT studies with regard to the stability of a compound, indirect information about the singlet oxygen generation, and information with regard to the interaction between the two moieties, e.g., PS and CD joined with a linker. Generally, higher singlet oxygen yields result in faster PB rate of CD. However, other factors, such as stability of the chromophores and distance between HPPH and CD, also affect the rate of PB. To compare the PB rate of CD in various HPPH-CD conjugates (metalated and nonmetalated), absorption spectra of all the compounds were collected prior to irradiation and at regular intervals during irradiation. Photoactivation of the conjugates with 532 nm (125 mW) laser light resulted in 17% destruction of the CD moiety of free-base HPPH-CD 4 after 10 min of irradiation (Figure 2). Under the same conditions, 25% CD destruction was observed for 5 (In). On the other hand, Pd(II)HPPH complex 7 resulted in no detectable traces of CD after 10 min. Thus, a complete destruction of CD moiety was achieved. Compound 6 (Ga) was not analyzed as it was found to be unstable in organic solvents (see above). Continued irradiation of 4 (free-base) and 5 (In) for an additional 15 min resulted in approximately 49% destruction of the CD moiety of HPPH-CD 4 compared to the nonirradiated sample and approximately 66% for In(III) HPPH-CD conjugate 5 (data not shown).
Figure 2.
In vitro photobleaching of compounds 4 (free-base, red), 5 (In, green), and 7 (Pd, blue) in methanol. Absorbance at equimolar concentration (5 μM) in methanol of each compound was measured prior to (solid lines) and after 10 min (dotted lines) of irradiation with 532 nm laser light (125 mW).
In addition to different singlet oxygen yields, the difference in rates of PB between the metalated and nonmetalated analogs could be due to the oxidation state of the metals used. When metals are inserted into the core of the porphyrin systems by reacting with metal halides, the two hydrogen atoms present as –NH protons at the center core are replaced with chloride ions. For example, palladium chloride (PdCl2) has an oxidation state of +2 and, on reacting with HPPH-CD, both replaces the hydrogen of the HPPH moiety and liberates hydrogen chloride. Indium chloride (InCl3) and gallium chloride (GaCl3) have oxidation states of +3 and therefore produce the corresponding In(III) or Ga(III) complex in a similar fashion to Pd(II) by replacing the two central –NH protons. However, unlike Pd, In and Ga are still coordinated to one of their chlorine counterions. This counterion sits above the plain of the slightly tilted HPPH moiety and prevents stacking and aggregation of conjugates 5 and 7, which could also restrict the CD moiety from folding back (in space) toward the PS (HPPH), thereby reducing the rate of PB of CD.
Photochemical Reactivity of 4 and 5.
We have sought to define key aspects of the photochemical reactivity of 4 and the corresponding In(III) conjugate 5. A unique component of this system is the potential to examine the consequences of independently exciting HPPH or CD. We have evaluated the kinetics and PB products upon irradiation of the porphyrin (λmax = 655) and cyanine components (λmax = 825 nm) using LED light sources roughly tuned to each of these respective wavelengths. As shown in Figure 3, 20 μM solutions of 4 and 5 were irradiated with either LED source centered on either 690 or 780 nm (15 mW/cm2). The respective porphyrin and cyanine absorptions were then observed over time. Upon 690 nm light irradiation, the cyanine absorption (λmax = 825 nm) of the In-containing HPPH-CD conjugate 5 decayed faster (t1/2 = 37 min) than 4 (t1/2 = 55 min), which bears the free base. In both cases, the porphyrin absorbance was only minimally changed over this time period (which may derive from changes in the modest residual cyanine absorption at 655 nm), suggesting that the porphyrin ring system is much more resilient to PB. As cyanines are known to readily react with 1O2, the greater singlet oxygen quantum yield for the In-containing HPPH (ϕΔ = 0.70 for 5 and 0.45 for 4) rationalizes the increased rate of cyanine PB. Irradiation with 780 nm light near the cyanine λmax induced a somewhat slower, but still efficient, cyanine PB. Notably, in this case the kinetics of cyanine decay did not vary between 4 (t1/2 = 62 min) and 5 (t1/2 = 60 min), suggesting that the 780 nm irradiation only induces a cyanine self-sensitized photooxidation process with negligible contribution from the conjugated porphyrin. As one would expect, only minimal decrease in the porphyrin signal was observed during the 780 nm irradiation.
Figure 3.
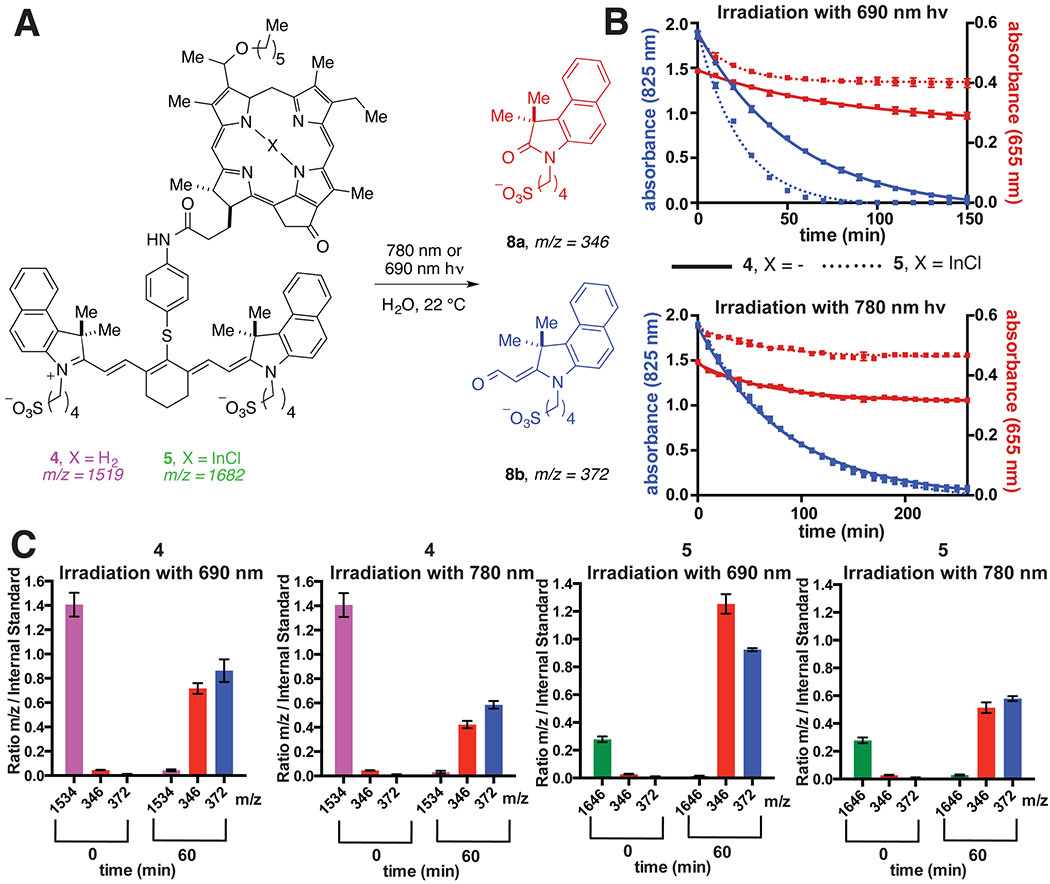
A. Conversion of 4 and 5 to 8a and 8b. B. Kinetic plots of cyanine and porphyrin absorbance (825 and 655 nm, respectively) upon exposing 20 μM solutions (3:1 MeOH:H2O) of 4 and 5 to 15 mW/cm2 light (690 or 780 nm) for the indicated times. C. Relative spectral ion counts of identical solutions of 4, 5, 8a, and 8b over the same time course. Ion counts were determined at each time point relative to an internal standard [3-(3,3-dimethyl-2-oxoindolin-1-yl)propane-1-sulfonate]. The observed ion for 5 is 1646 (M–Cl)+, which corresponds to loss of chloride.
We have also examined the identity and distribution of cyanine photodegradation products upon either porphyrin or cyanine excitation using mass spectrometry analyses. Prior studies have shown that cyanines can undergo a photooxidation process with singlet oxygen to form oxidatively cleaved products.16–20 This photooxidation occurs through a mechanism involving photosensitized generation of singlet oxygen, regioselective dioxetane formation, and final dioxetane cleavage to form carbonyl products. We demonstrated that 4 and 5 were converted to two major photooxidative cleavage products using ESI-MS: 8a and 8b. Furthermore, the relative distribution of the two photoproducts 8a and 8b appear to be similar, irrespective of excitation wavelength. Efforts to characterize products containing the porphyrin fraction were unsuccessful. Notably, this process only occurs upon irradiation, with no significant changes observed in non-irradiated samples.
In Vitro FRET Analysis between Ps and CD of HPPH-CD Conjugates.
A method to examine the level of FRET occurring between PS and CD of the conjugates is to use light to excite PS but look only at the fluorescence intensity of CD. If metalation of the HPPH-CD conjugate reduces FRET, then less CD fluorescence intensity would be expected upon photoactivation of the porphyrin. For this study, compounds were dissolved in methanol to a concentration of 5 μM, exposed to 532 nm light, and fluorescence above 800 nm recorded. Excitation at 532 nm was selected, as all compounds have strong absorption at this wavelength. Emission was monitored above 800 nm because porphyrin absorption/emission is negligible in this region. Free CD alone was also included in this study to show its inherent fluorescence when excited at 532 nm in the absence of a porphyrin. As shown in Figure 4, both metalloporphyrins had significantly lower FRET than free-base 4, suggesting that a reduction in spectral overlap is an effective method to reduce FRET for these compounds.21
Figure 4.
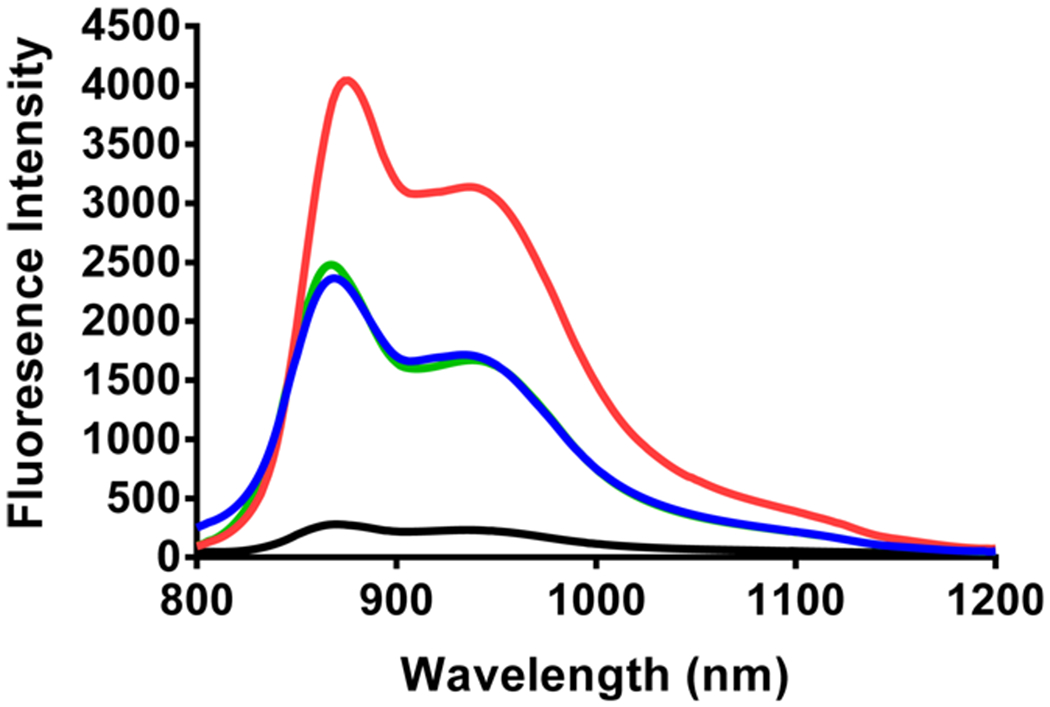
FRET analysis of PS and CD moieties as a nonmetalated analog 4 (red), indium complex 5 (green), and palladium complex 7 (blue). CD alone (2) is shown in black to show baseline CD fluorescence when excited with 532 nm light. All compounds were dissolved in methanol (5 μM), excited at 532 nm, and fluorescence emission past 800 nm collected in 2 nm steps.
FRET Dynamics.
The occurrence of FRET22–24 between PS and CD was monitored by the femtosecond transient absorption spectroscopy. Femtosecond laser excitation of 4 (HPPH–CD free base) in deaerated DMSO at 410 nm, which is the absorption band of the PS moiety, resulted in the ultrafast energy transfer from H2Ch HPPH to CD to produce the singlet excited state of CD (1CD*: * denotes the excited state). The transient absorption band was recorded at 2 ps, and observed at 586 nm taken at 2 ps (Figure 5A, black line), agreeing with the transient absorption of an unlinked CD. The absorbance at 586 nm increased by the energy transfer. The energy-transfer rate constant (kEN), which was determined from the rise of absorbance, is 1.4 × 1011 s−1 [= (7.2 ps)−1] (Figure 5B). Then, the absorption due to 1CD* decreased with the rate constant of 1.5 × 109 s−1 (Figure 5C). This value is virtually the same as that of an unlinked reference compound (1.0 × 109 s−1). When free base conjugate 4 was replaced by In complex 5, which has an identical linker (Ree = 11.3 Å), energy transfer from In-HPPH to CD also occurred to produce 1CD*, where the edge-to-edge distance (Ree) value was estimated by DFT calculation at the B3LYP/6-31G(d) level of theory. The rate constant was determined to be 1.4 × 1011 s−1 [= (7.5 ps)−1] (Figure 6A). In the case of palladium complex (7), ultrafast FRET occurred with a rate constant of 3.8 × 1011 s−1 [= (2.6 ps)−1].
Figure 5.
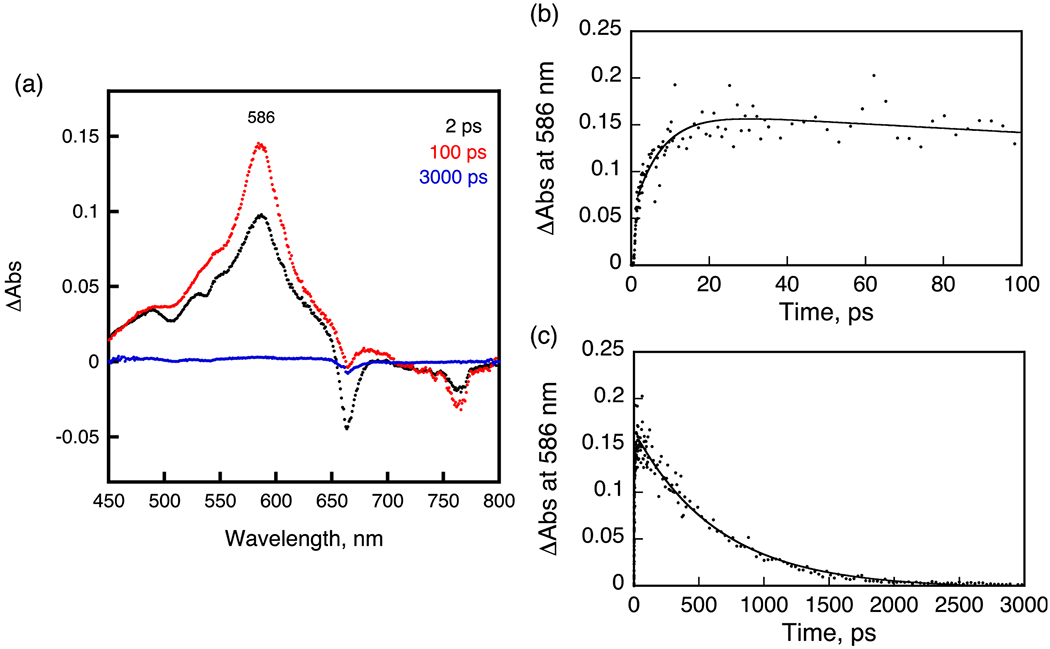
A. Transient absorption spectra of 4 in deaerated DMSO after femtosecond laser excitation at 410 nm. B,C. Time profiles at 586 nm from 0 to 100 ps and from 0 to 3000 ps.
Figure 6.
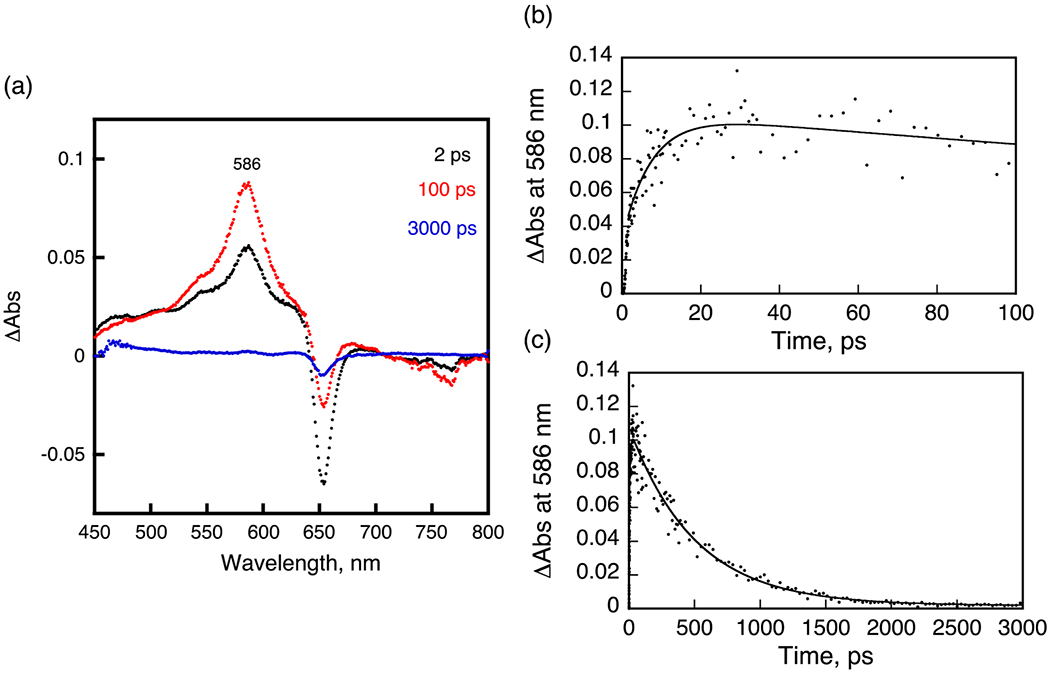
A. Transient absorption spectra of 5 in deaerated DMSO after femtosecond laser excitation at 410 nm. B, C. Time profiles at 586 nm from 0 to 100 ps and from 0 to 3000 ps.
Electrochemistry and Spectroelectrochemistry.
Redox properties of each HPPH conjugate and free CD were examined in DMSO containing 0.1 M TBAP to determine whether the specific central metal ion influences the compound’s electron-accepting and electron-donating properties. Representative cyclic voltammograms for the four conjugates are shown in Figure 7, along with the voltammogram of unlinked CD and pyropheophorbide.
Figure 7.
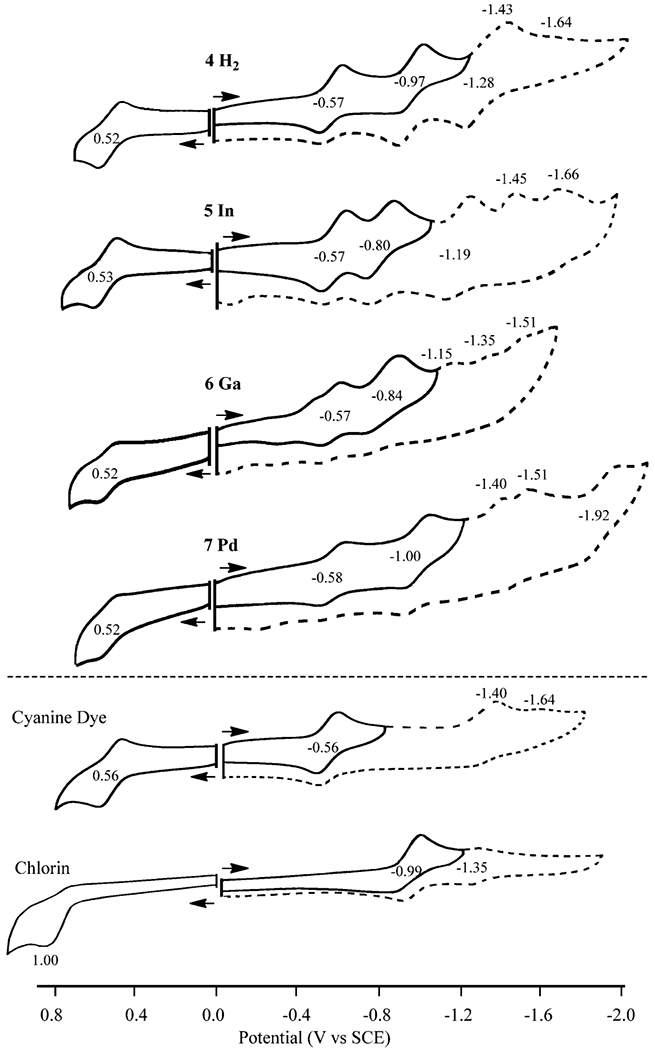
Cyclic voltammograms of investigated compounds in DMSO.
CD undergoes a reversible one-electron oxidation at E1/2 = 0.56 V and a reversible one-electron reduction at E1/2 = −0.56 V in DMSO containing 0.1 M TBAP. There is also a second irreversible reduction at Ep = −1.40 V at a scan rate of 0.1 V/s. Under the same solution conditions, pyropheophorbide undergoes an initial reversible reduction at E1/2 = −0.99 V and a second reduction with a smaller peak current at E1/2 = −1.35 V (Figure 7).
The four conjugates all undergo a reversible first oxidation and first reduction at similar half-wave potentials of 0.52 or 0.53 V and −0.57 or −0.58 V, respectively. However, significant differences are seen in the second reduction, where E1/2 values fall between −0.80 to −1.0 V vs SCE. Two or three additional reductions are also observed at more negative potentials for the investigated conjugates in DMSO. An examination of the data in Figure 7 shows that the first oxidation and first reduction of conjugates 4–7 all involve the CD portion of the molecule, while the second one-electron reduction at E1/2 = −0.80 to −1.00 V involves the pyropheophorbide part of the molecule. As expected for reduction at the π ring system of the macrocycle, the potentials for this reaction vary with the specific central metal ion.
Additional evidence for the above assignments is given by the thin-layer spectroelectrochemical data presented in Figures 7 and 8. CD in DMSO containing 0.1 M TBAP is characterized by a intense split near-IR bands at 773 and 849 nm,25 whereas conjugates 4–7 exhibit UV–vis spectra that are a simple sum of absorptions for the individual CD and pyropheophorbide units. For example, the 410 and 532 nm bands of 4 in DMSO can be attributed to the free-base pyropheophorbide part of the molecule,26 while the intense split near-IR bands of 4 at 762 and 847 nm can be attributed to the CD.
Figure 8.
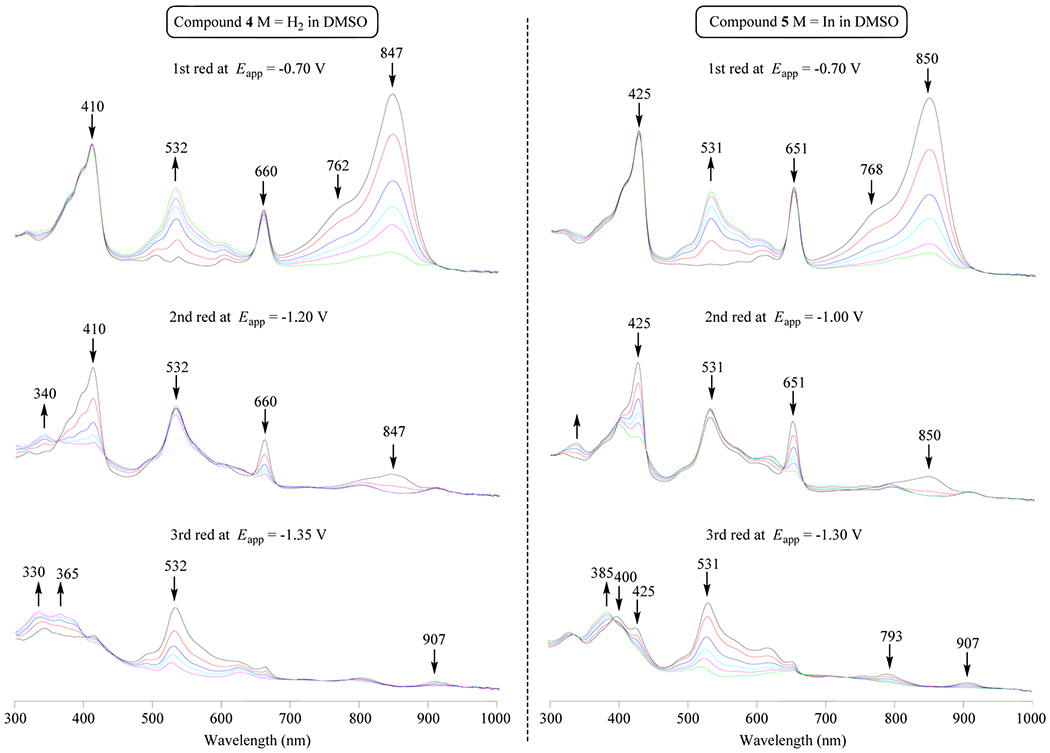
Thin-layer UV–vis spectral changes at controlled potential reduction of compounds 4 and 5 in DMSO.
Applying a controlled reducing potential of −0.70 V to a solution of 4 in DMSO containing 0.1 M TBAP leads to a decrease in intensity of these split near-IR bands as a new visible band grows in at 532 nm (Figure 8). No changes are observed in bands associated with the pyropheophorbide part of conjugate 4 (410 and 660 nm) or 5 (425 and 651 nm) at this applied potential. However, CD bands of the conjugate both undergo similar spectral changes during the first reduction of unlinked CD in the thin-layer cell, thus providing further evidence that the first reduction of the investigated conjugates is located on the CD part of the molecule.
The second reduction of 4–7 occurs at the pyropheophorbide part of the molecule, as seen by the thin-layer spectral changes depicted in Figure 8. There is a decrease in intensity of the 410 and 660 nm bands as the reduction proceeds, and the spectral charges are similar to those reported in the literature for the first reduction of purpurinimide. The electron transfer site upon oxidation of 4–7 can also be assigned to the CD rather than to the pyropheophorbide part of the macrocycle. The changes in Figure 8 are almost identical to spectral changes reported in the literature for oxidation of the cyanide dye.25
In summary, the first oxidation and first reduction of the four investigated conjugates are both centered on the CD and half-wave potentials for the first reduction are almost identical to the measured E1/2 values for first one-electron reduction of CD. Changes in the central metal ion from H2(4) to Pd(7) have minimal effect on redox potentials of CD but do effect the first reduction at the purpurinimide part of the molecule in previously examined compounds.26
The CD unit is easier to reduce than the pyropheophorbide unit in conjugates 4–7. Thus, CD could be used as an oxidant to quench the excited triplet state of the pyropheophorbide.27,28 According to the Rehm–Weller equation,29 the quenching constant would somewhat depend on the potential difference between the first oxidation and the first reduction potential (the electrochemical HOMO–LUMO gap) for molecules having similar structures. The experimentally measured HOMO–LUMO gap ranges from 1.09 to 1.10 V for the four investigated conjugates and this ΔE1/2 value is similar to the HOMO–LUMO gap of the CD (1.12 V). There is an effect of the central metal ion on the HOMO–LUMO gap of the purpurinimide conjugates (a larger HOMO–LUMO gap is observed for the Ni(II)-centered compound than for the free-base derivative), but no effect of the metal is seen for the HOMO–LUMO gap of the currently investigated conjugates.
In Vitro PDT Efficacy of HPPH-CD Conjugates (Free Base vs the Metalated Analogs).
Before the in vitro PDT efficacy of the metalloporphyrins could be determined, we first examined the toxicity of the compounds in the absence of activating light. Dark toxicity is not a desirable characteristic for PSs as they should only cause cell death in the presence of light treatment. Mouse Colon26 cells were seeded into 96-wells plates and incubated for 24 h in complete media containing variable concentrations of conjugates (0.1 to 3.0 μM). Cell viability was assessed by a MTT assay. With the exception of free base HPPH-CD 4, each of the metalated analogs 5–7 showed ~20% dark toxic cell death at the highest drug dose of 3.0 μM (not shown).
To compare the photosensitizing potential of conjugates 4–7, a modified MTT assay was performed that included a light irradiation step to activate the conjugates. Colon26 tumor cells were incubated with compounds at the concentrations ranging from 0.01 to 3.0 μM for 24 h and then exposed to light at the appropriate wavelength (4 = 665 nm, 5 = 651 nm, 6 = 654 nm, 7 = 640 nm). Light fluence delivered to the cells ranged from 0 to 3.0 J/cm2. Cell survival was determined 24 h after light treatment by MTT staining. Figure 9 shows the combined results of cells treated with 0.3 μM conjugate, tested over a light range of 0 to 3.0 J/cm2. It is evident that, under these in vitro treatment parameters, free-base HPPH-CD 4 was not significantly phototoxic, whereas metalation showed improved efficacy. Among all the compounds tested, In(III) conjugate 5 was most effective, achieving 100% reduction in cell viability at 0.3 μM drug concentration and a light dose of 3.0 J/cm2. Efficacy across all compounds trended as follows: 5 (In) ≫ 7 (Pd) > 6 (Ga) > 4 (free base). Activity by the metalated PSs and recorded by STAT3 cross-linking were in line with those determined by MTT assay (Figure 9 vs Figure 10A). The several-fold higher STAT3 cross-linking activity measured for nonconjugated HPPH relative to compound 5 (In-HPPH-CD) is in part due to the greater cellular uptake and distinct subcellular localization of the former (Figure 10B). HPPH and In-HPPH were primarily localized to mitochondria and endoplasmic reticulum (ER), whereas HPPH-CD conjugates, with or without metals, were primarily localized to lysosomes and other granular organelles, but also detectable in ER and mitochondria.
Figure 9.
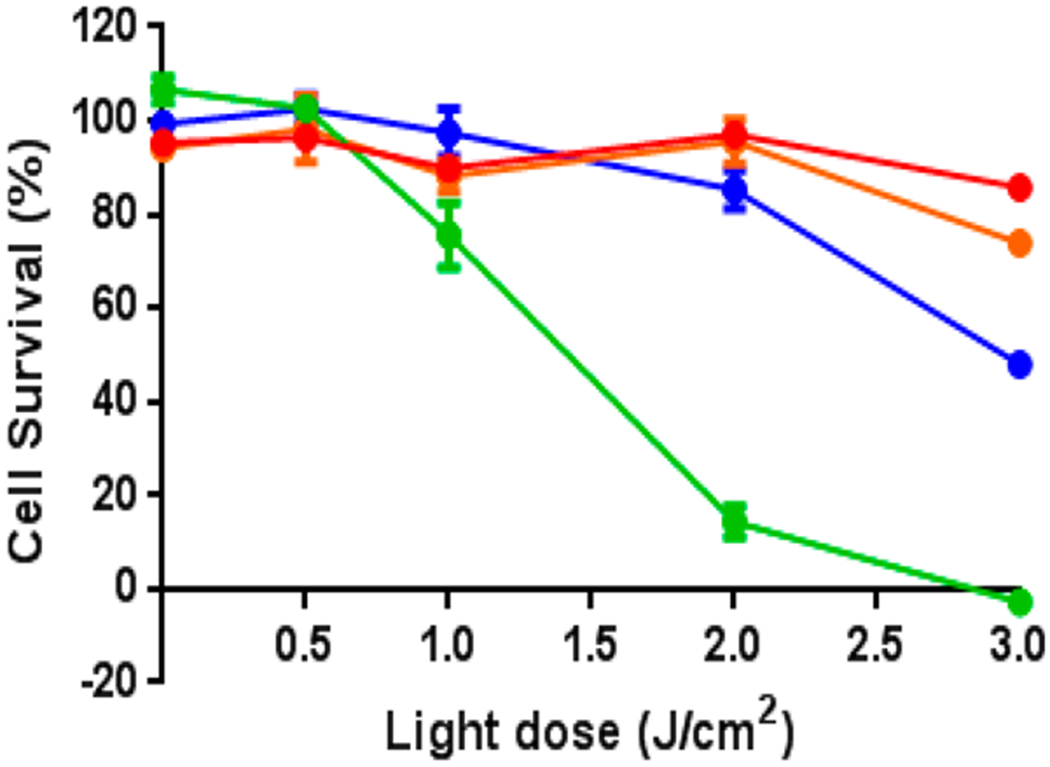
In vitro PDT activity of compounds free-base 4 (red), indium complex 5 (green), gallium complex 6 (orange), and palladium 7 (blue) via the cell viability MTT assay in colon 26 cells. Cells were incubated with each compound in 0.3 μM concentration for 24 h and then irradiated with the appropriate wavelength of light (4 = 665 nm, 5 = 651 nm, 6 = 654 nm, 7 = 640 nm) at various doses of light from 0 to 3.0 J/cm2. Values are expressed as cell percent survival of the vehicle-treated controls (set to 100%). Data represents the average of at least 3 experiments; error bars represent standard deviation.
Figure 10.
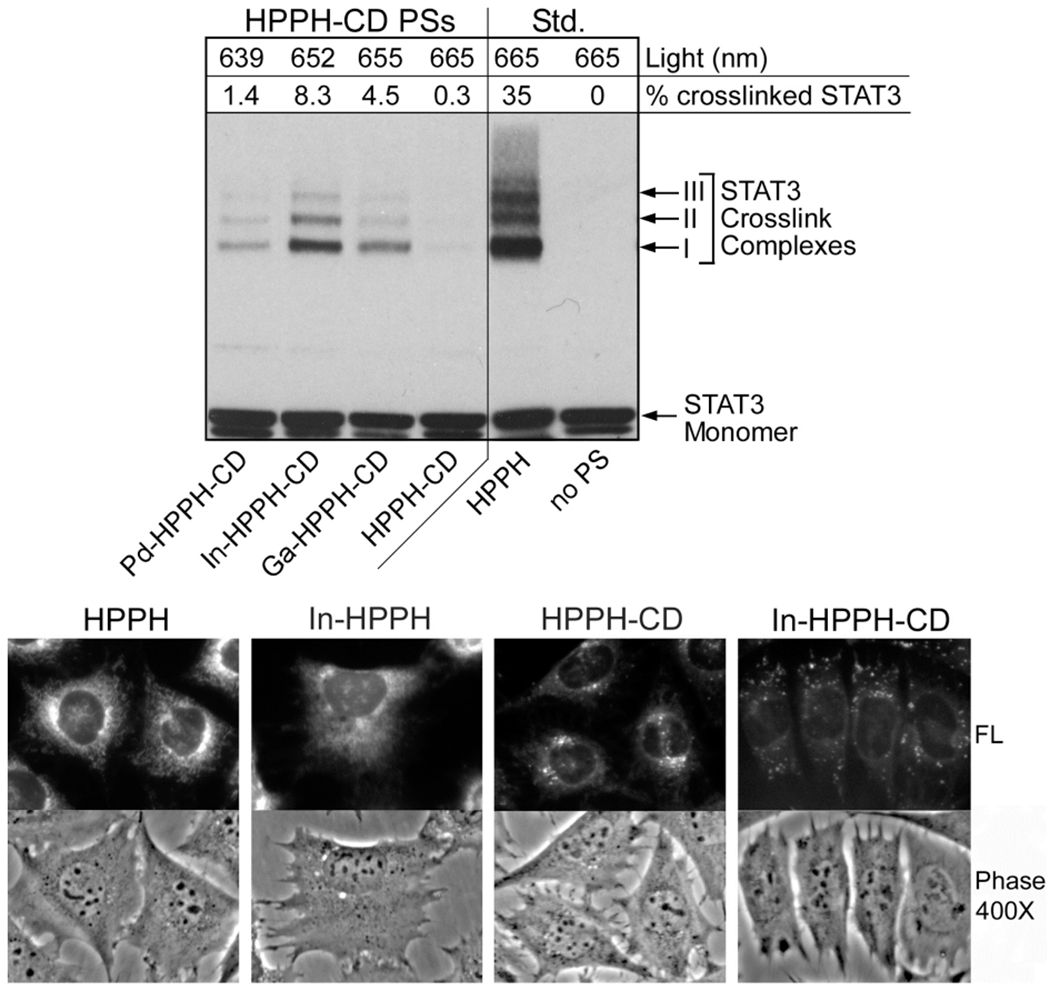
A. Identification of PS-directed photoreaction by STAT3 cross-linking. BCC1 cells were incubated for 2.5 h with culture medium containing 10% fetal bovine serum and 400 nM of the indicated compounds. The cells were washed and then exposed to light at their compound’s PS respective λmax yielding at a fluence of 3 J/cm2. Immediately after light treatment, the cells were extracted and proteins analyzed by immunoblotting for STAT3. The conversion of monomeric STAT3 to cross-linked dimer complex I was determined and expressed as a percentage of the total STAT3 (as indicated at the top of each lane). B. Subcellular localization of HPPH, In-HPPH, compounds 4 (HPPH-CD)and 5 (In-HPPH-CD). After incubation for 4.5 h, the cells were imaged under an inverted fluorescent phase microscope at 400.
The degree of cross-linking of the STAT3 protein has been shown to be an effective marker for determining the biological effectiveness of a photoreaction involving singlet oxygen production.30 To determine the relative photosensitizing activities of the compounds within cells, we treated basal cell carcinoma (BCC1) cells with 4–7, along with 3 as a standard, and quantified the light-dependent STAT3 cross-linking by Western blot. BCC1 cells were used instead of Colon26 cells because of their large size that also allowed us to assess the intracellular distribution of the compounds by fluorescence microscopy.31 The In analog 5 produced the most STAT3 cross-linking, whereas the free-base produced the least (Figure 10A). STAT3 cross-linking with Ga and Pd were intermediate. The trend in efficiency of the photoreactions for HPPH-CD conjugates as measured by the metalated PSs and recorded by magnification recording HPPH fluorescence.
STAT3 cross-linking were in line with those determined by MTT assay (Figure 9 vs Figure 10A). The several-fold higher STAT3 cross-linking activity measured for nonconjugated HPPH relative to compound 5 (In-HPPH-CD) is in part due to the greater cellular uptake and distinct subcellular localization of the former (Figure 10B). HPPH and In-HPPH were primarily localized to mitochondria and endoplasmic reticulum (ER), whereas HPPH-CD conjugates, with or without metals, were primarily localized to lysosomes and other granular organelles, but also detectable in ER and mitochondria.
In Vivo Whole Body Fluorescence Optical Imaging of All HPPH-CD Conjugates.
The in vivo fluorescence optical imaging capabilities of the CD component of each conjugate allowed us to determine the systemic distribution of the compounds, the tumor selectivity of the compounds, and the kinetics of tumor accumulation as a function of time post-injection. After imaging CD (excited at 782 nm, emission at >830 nm) at various times post-injection, it was found that all conjugates had maximal tumor accumulation at 24 h. The results summarized in Figure 11 show the accumulation at 24 h post-injection of each conjugate injected into 3 mice plus a control mouse.
Figure 11.
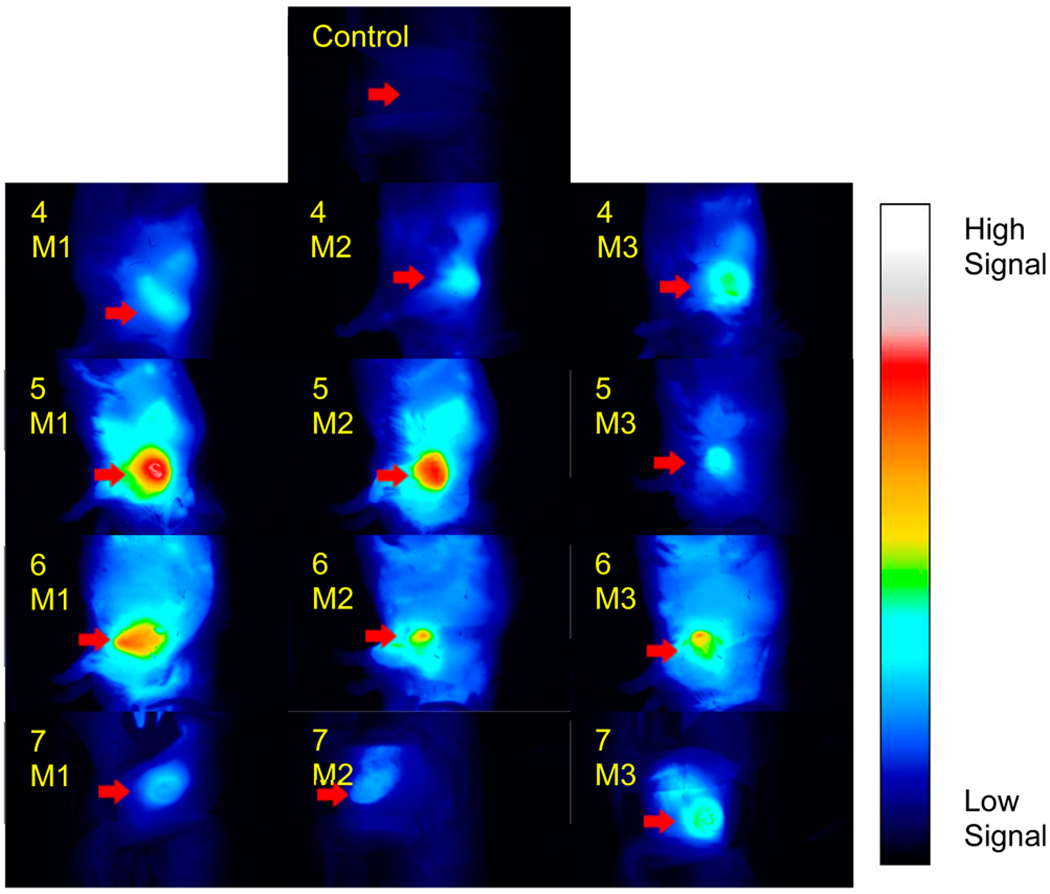
Whole body fluorescence optical imaging of free-base 4, In complex 5, Ga complex 6, and Pd complex 7 at 24 h post-injection under similar parameters. BALB/c mice (3 per group) bearing Colon26 tumors were injected intravenously with 0.3 μmol/kg of each compound, with one mouse serving as an uninjected control. Images were acquired with a Nuance camera at 782 nm excitation and >830 nm emission using 800/830 nm long-pass filters. Fluorescence intensity scaling is identical in all images. M = BALB/c mice bearing Colon26 tumors.
Although all conjugates had maximum tumor uptake at 24 h post-injection, their fluorescence intensities differed. As all images are scaled identically and initial concentrations were the same, this difference in intensity can be directly attributed to tumor accumulation. All signals from the conjugates significantly decreased after 72 hours. Overall, 5 (In) had the highest intratumoral CD fluorescence tumor followed by 6 (Ga) and 7 (Pd), while 4 (free-base), respectively, was the lowest. Of note is that the relative pattern of retention detected in Colon26 tumors was highly comparable to the photoreaction efficiency observed mediated in vitro in BCC1 cells (Figure 11 vs Figure 9 and Figure 10A).
Comparative in Vivo Photobleaching of the CD Moiety in Metalated vs Non-Metalated HPPH-CD Conjugate.
To probe for correlations (if any) between the in vitro and in vivo PB characteristics, studies were performed in mice (3 BALB/c mice/group) bearing Colon26 tumors that had received an i.v. injection of 0.3 μmol/kg of each conjugate. The mice were imaged 24 h post-injection and fluorescence intensity of CD at the tumor was recorded. Tumors were then irradiated at the appropriate wavelength (4 = 665 nm, 5 = 651 nm, 6 = 654 nm, and 7 = 640 nm) at a therapeutic light fluence of 135 J/cm2. At regular intervals during the treatment, mice were removed from the laser light, imaged, and then placed back under the laser light. CD signal from the tumor was recorded at each time point and plotted against the initial signal prior to irradiation (t = 0). Each mouse was exposed to laser irradiation for a total of 30 min. Since the tumor accumulation of the compounds was appreciably different (Figure 11), PB data are expressed as relative values. The results summarized in Figure 12 indicate that all three metalated compounds led to rapid CD PB within the first 2 min of light exposure. Among the compounds investigated, compound 7 (Pd complex) exhibited the fastest initial rate of PB, with 50% CD signal loss within 0.25 min of irradiation. Four minutes after initial irradiation, only 20% of the initial signal remained, which dropped to 15% at 30 min. Similar to compound 7, In(III) conjugate 5 lost 50% of the initial CD signal after approximately 1.0 min of irradiation. While the initial rate of CD PB was slower than that of 7, only 18% of the In conjugate’s CD signal remained after 30 min. Interestingly, the rate of CD PB in compound 6 (Ga complex) was significantly reduced, with only 52% of the CD moiety bleached after 30 min.
Figure 12.
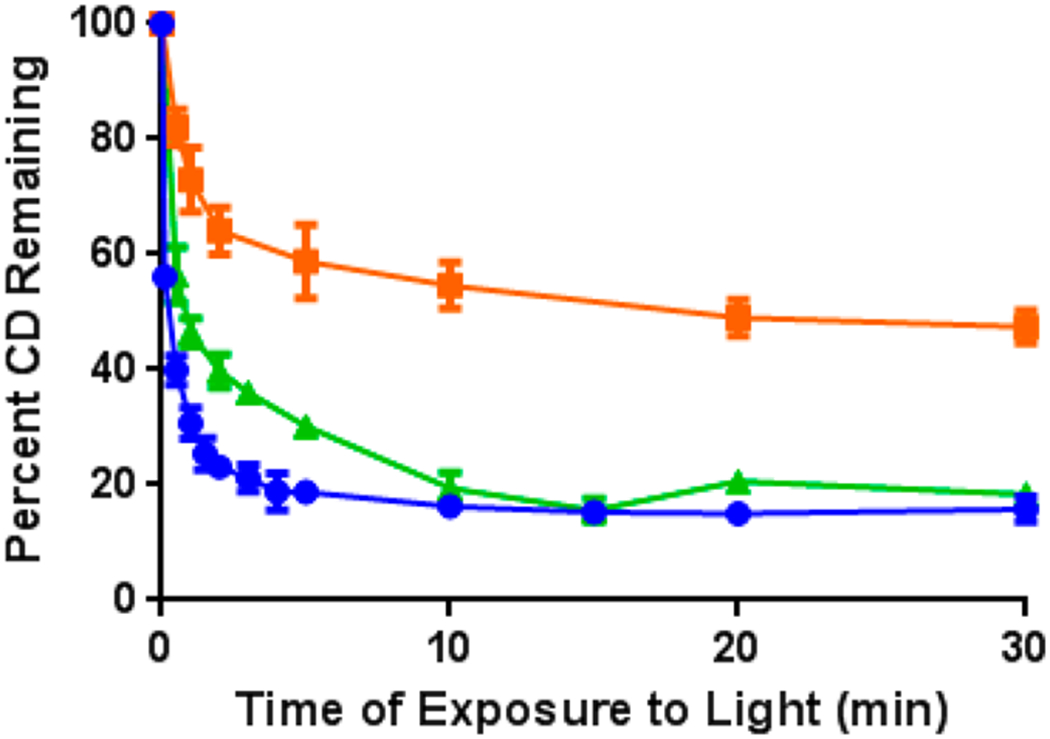
In vivo photobleaching of the CD moiety after excitation of In complex 5 (green line), Ga complex 6 (orange), and Pd complex 7 (blue) at a λmax of 651, 654, and 640 nm, respectively.Three BALB/c mice per group bearing Colon26 tumors were irradiated 24 h post-injection (0.3 μmol/kg solution) at 135 J/cm2. Mice were imaged at regular intervals over a 30 min period and the fluorescence intensity of the CD moiety plotted relative to the preirradiated signal. The results are from a single experiment. Means and standard deviations represent average data from the 3 animals.
Comparative in Vivo PDT of Metalated PS-CD Conjugates.
The first step in determining the in vivo PDT efficacy of the metalated compounds was to perform a dose–response study.24 Previous reports from our laboratory have shown that the free-base HPPH-CD conjugate gives 40–50% tumor cure (60 day) at a dose of 3.5 μmol/kg after irradiating with light (135 J/cm2).8 Moreover, we have shown that upon inserting indium (In) within free HPPH (not conjugated to CD), the corresponding compound was more effective at a lower dose than that applied for HPPH.7 Based on these findings, we began the in vivo PDT efficacy dose–response study at a lower dose of 0.3 μmol/kg (10 mice/group bearing Colon26 tumors). The in vivo long-term tumor growth assay (tumor cure) is illustrated in Figure 13. As can be seen, conjugate 5 led to 60% cure (6 out of 10 mice were tumor-free after 60 days). Under similar treatment parameters, the corresponding Ga (6) and Pd (7) conjugates showed limited efficacy.
Figure 13.
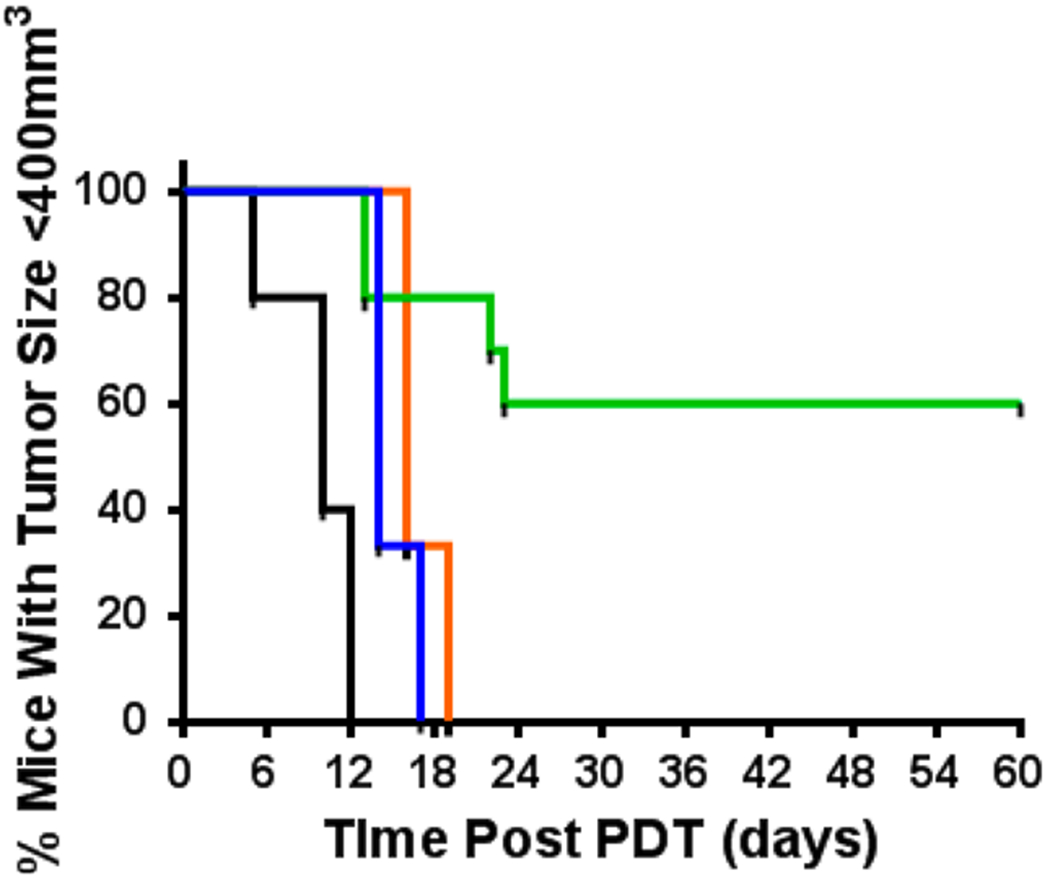
In vivo PDT evaluation of metalated analogs: vehicle alone (black, n = 5), indium complex 5 (green, n = 10, 0.3 μmol/kg), gallium complex 6 (orange, n = 3, 1.0 μmol/kg), and palladium complex 7 (blue, n = 3, 0.3 μmol/kg). BALB/c mice with Colon26 tumors were treated with light at an appropriate wavelength (vehicle control = 651 nm, 5 = 651 nm, 6 = 654 nm, 7 = 640 nm) at 135 J/cm2/ 75 mW/C m2 (light fluence and fluence rate) at 24 h post-injection.
At a slightly higher dose of 0.5 μmol/kg, Pd complex 7 also showed limited PDT efficacy (Figure 13). However, raising the dose to 1.0 μmol/kg was found to be toxic, as mice displayed hunched backs, inability to groom, and >10% weight loss. One possible explanation for this toxicity can be gleaned from the in vitro observation illustrated in Figure 2, where the Pd complex showed photodestruction of the PS upon irradiation, which could lead to release of toxic Pd metal into the system. In the case of Ga compound 6, all mice treated at 0.5 and 1.0 μmol/kg (Figure 12) experienced tumor regrowth. No visual damage to healthy tissue (scabbing, edema, and/or inflammation) was observed at the site of light treatment.
CONCLUSIONS
In this study, we demonstrated the impact of the presence of heavy metals, e.g., In, Ga, or Pd, on the photophysical and electrochemical properties as well as tumor uptake and PDT efficacy of a bifunctional agent in which HPPH was conjugated with a near-infrared CD. All metalated analogs showed advantages over nonmetalated analogs, with significantly reduced FRET and improved in vitro PDT efficacy. In(III) HPPH-CD showed enhanced in vivo tumor uptake and PDT efficacy. In contrast to free-base HPPH-CD conjugate, the corresponding In(III) analog as a single drug dose can be used for both fluorescence tumor-imaging and phototherapy. The in vivo results demonstrate that compound 5 [In(III)HPPH-CD] maintains excellent imaging capabilities, but has drastically improved therapeutic response compared to the corresponding nonmetalated conjugate. However, additional studies, e.g., pharmacokinetic/pharmacodynamic properties and safety profile (especially heavy metal toxicity) of the indium conjugate 5, necessary for clinical translation are currently underway.
EXPERIMENTAL PROCEDURES
All reactions were carried out in dried glassware under an atmosphere of nitrogen and in the presence of magnetic stirring. Thin-layer chromatography (TLC) was performed on pre-coated silica gel (layer thickness: 0.25 mm) or aluminum oxide plates. Column chromatography was performed over silica gel 60 (70–230 mesh) or neutral alumina (grade III). In some cases, preparative TLC was used for the purification of compounds. Solvents were dried following standard methodology. 1H NMR spectra were recorded at room temperature in CDCl3 using a Varian VNMRS-400 spectrometer. All chemicals shifts are reported in parts per million (δ). 1H NMR (400 MHz) spectra were referenced to residual CHCl3 (7.26 ppm) or TMS (0.00 ppm). Mass spectrometry analyses were performed at the Mass Spectrometry Facility, Michigan State University, East Lansing, MI. UV–vis spectra were recorded on an FT UV–visible spectrophotometer using dichloromethane/THF as solvent.
Conjugate 5.
HPPH-CD conjugate 4 (100 mg), indium(III) chloride (100 mg, Sigma-Aldrich, St. Louis, MO), and sodium bicarbonate (600 mg) were put in the solvent mixture of toluene (60 mL) and ethanol (20 mL). The reaction mixture was refluxed for 1 h. After evaporation of the solvents, the residue was purified by vacuum silica column chromatography using MeOH/CH2Cl2 (1:4) as the elute solvent. After evaporation of the solvent 86 mg of compound was obtained (~80% yield). UV–vis in MeOH (ε, M−1 cm−1): 835 nm (ε = 1.59 × 105), 646 nm (5.65 × 104), 602 nm (1.24 × 104), 563 nm (8.76 × 103), 416 nm (7.51 × 104). NMR (CDCl3), δ (ppm) for compound 5: 9.87 (ss looks like a doublet, 1H, meso-H in HPPH part), 9.70 (s, 1H, meso-H in HPPH part), 8.39 (s, 1H, meso-H in HPPH part), 7.98 (m, 4H, aromatic-H of cyanine dye), 7.84 (br s, 4H, aromatic-H of cyanine dye), 7.50 (br s, 4H, aromatic-H of cyanine dye), 7.37 (br s, 4H, =CH– of cyanine dye), 7.07 (m, 4H, 4H of the linker phenyl group), 5.78 (m, 1H, H-31), 5.17 (m, 1H, H-17), 5.03 (m, 1H, H-18), 4.60 (m, 2H, H-132), 4.40 (m, 2H, N+-CH2), 4.01 (br, 10H, 2H for N–CH2, 2H for H-171, 4H for – CH2SO3, 2H for – OCH2(CH2)4CH3), 3.78 (s, 3H, 7-CH3), 3.59 (s, 3H, 12-CH3), 3.68–3.45 (m, 6H, 4H for SO3-CH2CH2-(CH2)2, 2H for 8-CH2CH3), 3.34, (m, 2H, H-172), 3.31 (s, 3H, 2-CH3), 3.12 (m, 4H, SO3-(CH2)2CH2-CH2), 2.04 (m, 11H, 3H for 3-CH3, 2H for –OCH2CH2(CH2)3CH3), 1.73 (s, 12H, 4X-CH3 of cyanine dye), 1.33 (m, 3H, 18-CH3), 1.26 (m, 3H, 8-CH2CH3), 1.15 (m, 6H, -O(CH2)2(CH2)3CH3), 0.71 (m, 3H, -O(CH2)5CH3). C-13 NMR: 173.92 (C), 169.29 (C), 159.64 (C), 150.37 (C), 145.93 (C), 145.35 (C), 145.16 (C), 143.99 (C), 139.36 (C), 138.79 (C), 136.52 (C), 136.28 (C), 135.00 (C), 133.68 (C), 131.81 (C), 130.83 (C), 130.54 (C), 129.9 (C), 129.56 (C), 129.16 (C), 127.84 (C), 127.59 (C), 127.2 (C), 127.0 (C), 126.6 (C), 125.1 (C), 124.5 (C), 123.5 (C), 122.7 (C), 121.6 (C), 120.6 (C), 115.8 (C), 114.3 (C), 112.9 (C), 112.5 (CH), 110.8 (CH), 110.0 (CH), 109.0 (CH), 107.3 (CH), 106.9 (CH), 105.2 (CH), 105.0 (CH), 103.8 (CH), 102.7 (CH), 100.4 (CH), 94.9 (CH), 94.2 (CH), 93.2 (CH), 92.1 (CH), 87.8 (CH), 80.4 (CH), 76.2 (CH), 75.3 (CH), 74.7 (CH), 72.8 (CH), 72.2 (CH), 71.2 (CH), 71.0 (CH), 69.9 (CH), 69.3 (CH), 61.8 (CH2), 59.6 (CH2), 56.5 (CH2), 50.6 (CH2), 45.7 (CH2), 44.0 (CH2), 39.6 (CH2), 39.2 (CH2), 32.0 (CH2), 31.4 (CH2), 30.7 (CH2), 29.9 (CH2), 29.3 (CH2), 27.0 (CH2), 26.2 (CH2), 25.1 (CH2), 24.5 (CH2), 24.1 (CH2), 23.9 (CH2), 23.5 (CH2), 22.9 (CH3), 22.2 (CH3), 22.0 (CH3), 21.6 (CH3), 20.6 (CH3), 19.3 (CH3), 18.7 (CH3), 16.6 (CH3), 13.3 (CH3), 12.1 (CH3), 10.4 (CH3). MS for 5: Calculated for C91H102N7O9S3InCl: 1682.6, Found: 1682.5. HRMS for 5: Calculated for C91H102N7O9S3In: 1647.5937, Found: 1647.5998.
Conjugate 6.
HPPH-CD conjugate 4 (100 mg), gallium chloride (400 mg, Alfa Aesar, Ward Hill, Massachusetts), and sodium bicarbonate (400 mg) were put in the solvent mixture of toluene (65 mL) and ethanol (25 mL). The reaction mixture was refluxed for 30 min. After evaporation of the solvents, the residue was purified by vacuum silica column chromatography using MeOH/CH2Cl2 (1:4) as the elute solvent. After evaporation of the solvent 48 mg of compound was obtained (~45% yield). UV–vis in MeOH (ε, M−1 cm−1): 839 nm (1.59 × 105), 649 nm (5.51 × 104), 606 nm (1.25 × 104), 563 nm (8.77 × 103), 419 nm (ε = 7.52 × 104). NMR (CDCl3), δ (ppm) for compound 6: 9.70 (s, 1H, meso-H in HPPH part), 9.22 (s, 1H, meso-H in HPPH part), 8.64 (s, 1H, meso-H in HPPH part), 8.12 (m, 4H, aromatic-H of cyanine dye), 8.02 (br s, 4H, aromatic-H of cyanine dye), 7.73 (br s, 4H, aromatic-H of cyanine dye), 7.46 (br s, 4H, ═CH– of cyanine dye), 7.18 (m, 4H, 4H of the linker phenyl group), 5.79 (m, 1H, H-31), 5.19 (m, 1H, H-17), 5.01 (m, 1H, H-18), 4.63 (m, 2H, H-132), 4.45 (m, 2H, N+-CH2), 4.07 (br, 10H, 2H for N–CH2, 2H for H-171, 4H for – CH2SO3, 2H for –OCH2(CH2)4CH3), 3.79 (s, 3H, 7-CH3), 3.55 (s, 3H, 12-CH3), 3.69–3.44 (m, 6H, 4H for SO3-CH2CH2-(CH2)2, 2H for 8-CH2CH3), 3.32 (m, 2H, H-172), 3.36 (s, 3H, 2-CH3), 3.12 (m, 4H, SO3-(CH2)2CH2-CH2), 2.02 (m, 11H, 3H for 3-CH3, 2H for –OCH2CH2(CH2)3CH3), 1.75 (s, 12H, 4X-CH3 of cyanine dye), 1.31 (m, 3H, 18-CH3), 1.24 (m, 3H, 8-CH2CH3), 1.17 (m, 6H, -O-(CH2)2(CH2)3CH3), 0.73 (m, 3H, -O(CH2)5CH3). C-13 NMR: 173.42 (C), 159.52 (C), 150.06 (C), 145.53 (C), 143.69 (C), 141.96 (C), 139.06 (C), 138.67 (C), 138.29 (C), 136.16 (C), 134.62 (C), 133.27 (C), 131.34 (C), 130.18 (C), 129.60 (C), 129.21 (C), 128.64 (C), 127.67 (C), 127.28 (C), 126.51 (C), 126.32 (C), 124.58 (C), 124.20 (C), 123.23 (C), 122.26 (C), 121.49 (C), 120.91 (C), 120.14 (C), 119.75 (C), 114.93 (C), 114.35 (C), 113.77 (C), 111.45 (C), 110.30 (C), 109.72 (CH), 107.59 (CH), 107.01 (CH), 106.43 (CH), (CH), 101.22 (CH), 100.45 (CH), 99.29 (CH), 97.94 (CH), 96.00 (CH), 94.46 (CH), 92.34 (CH), 91.76 (CH), 76.32 (CH), 74.00 (CH), 72.65 (CH), 72.07 (CH), 70.72 (CH), 68.98 (CH), 68.21 (CH), 67.24 (CH), 62.42 (CH), 61.64 (CH), 60.87 (CH), 60.10 (CH), 59.71 (CH), 56.43 (CH2), 51.99 (CH2), 51.80 (CH2), 50.64 (CH2), 45.24 (CH2), 43.69 (CH2), 42.92 (CH2), 39.25 (CH2), 38.86 (CH2), 31.94 (CH2), 30.95 (CH2), 30.18 (CH2), 29.40 (CH2), 28.83 (CH2), (CH2), 25.93 (CH2), 25.54 (CH2), 24.96 (CH2), 24.00 (CH2), 23.61 (CH2), 23.03 (CH3), 22.45 (CH3), 21.68 (CH3), 21.30 (CH3), 20.33 (CH3), 18.98 (CH3), 18.59 (CH3), 16.27 (CH3), 12.99 (CH3), 11.45 (CH3), 9.90 (CH3). MS for 6: Calculated for C91H102N7O9S3GaCl: 1636.6, Found: 1636.1. HR-MS for 6: Calculated for C91H102N7O9S3Ga: 1601.6158, Found: 1601.6147.
Conjugate 7.
HPPH-CD conjugate 4 (100 mg), l-ascorbic acid 6-palmitate (220 mg), and palladium(II) acetate (160 mg, Sigma-Aldrich, St. Louis, MO) were put into the solvent mixture of methanol (80 mL) and chloroform (80 mL). Under argon the reaction mixture was stirred overnight at room temperature. After the standard workup and evaporating the solvents, the residue was purified by vacuum silica column chromatography using MeOH/CH2Cl2 (1:5) as the elute solvent. After evaporation of the solvent, 90 mg of compound was obtained (~85% yield). UV–vis in MeOH (ε, M−1 cm−1): 839 nm (1.32 × 105), 635 nm (6.51 × 104), 589 nm (1.19 × 104), 536 nm (9.09 × 103), 415 nm (5.95 × 104), 389.9 nm (6.22 × 104). NMR (CHCl3), δ (ppm) for compound 7: 9.75 (s, 1H, meso-H in HPPH part), 9.61 (s, 1H, meso-H in HPPH part), 8.45 (s, 1H, meso-H in HPPH part), 7.72 (m, 4H, aromatic-H of cyanine dye), 7.53 (br s, 4H, aromatic-H of cyanine dye), 7.12 (br s, 4H, aromatic-H of cyanine dye), 6.93 (br s, 4H, ═CH– of cyanine dye), 6.80 (m, 4H, 4H of the linker phenyl group), 5.64 (m, 1H, H-31), 5.32 (m, 1H, H-17), 5.02 (m, 1H, H-18), 4.64 (m, 2H, H-132), 4.47(m, 2H, N+-CH2), 4.09 (br, 10H, 2H for N–CH2, 2H for H-171, 4H for –CH2SO3, 2H for –OCH2(CH2)4CH3), 3.76 (s, 3H, 7-CH3), 3.57 (s, 3H, 12-CH3), 3.67–3.46 (m, 6H, 4H for SO3-CH2CH2-(CH2)2, 2H for 8-CH2CH3), 3.35, (m, 2H, H-172), 3.31 (s, 3H, 2-CH3), 3.16 (m, 4H, SO3-(CH2)2CH2-CH2), 2.05 (m, 11H, 3H for 3-CH3, 2H for –OCH2CH2(CH2)3CH3), 1.73 (s, 12H, 4X-CH3 of cyanine dye), 1.34 (m, 3H, 18-CH3), 1.21 (m, 3H, 8-CH2CH3), 1.15 (m, 6H, -O(CH2)2(CH2)3CH3), 0.71 (m, 3H, -O(CH2)5CH3). C-13 NMR: 173.15 (C), 160.02 (C), 154.81 (C), 150.76 (C), 138.02 (C), 132.80 (C), 130.29 (C), 128.94 (C), 128.56 (C), 127.20 (C), 126.82 (C), 126.24 (C), 123.92 (C), 123.34 (C), 122.76 (C), 121.22 (C), 116.0 (C), 113.69 (C), 110.02 (C), 109.44 (C), 106.93 (C), 106.55 (C), 104.81 (C), 104.42 (C), 103.84 (C), 100.76 (C), 99.98 (C), 99.79 (C), 98.82 (C), 97.28 (C), 96.31 (C), 95.16 (C), 94.38 (C), 93.42 (C), 92.26 (CH), 91.49 (CH), 89.56 (CH), 87.82 (CH), 87.43 (CH), 86.86 (CH), 86.47 (CH), 85.89 (CH), 84.54 (CH), 83.19 (CH), 81.45 (CH), 76.04 (CH), 73.34 (CH), 71.60(CH), 69.48 (CH), 68.51 (CH), 67.74 (CH), 66.00 (CH), 65.23 (CH), 63.69 (CH), 62.34 (CH), 61.76 (CH), 56.35 (CH), 55.97(CH), 54.03 (CH), 53.46 (CH), 49.60 (CH2), 44.96 (CH2), 40.91 (CH2), 39.75 (CH2), 38.40 (CH2), 36.85 (CH2), 31.45 (CH2), 30.48 (CH2), 29.32 (CH2), 28.36 (CH2), 27.01 (CH2), 25.85 (CH2), 25.46 (CH2), 24.30 (CH2), 23.34 (CH2), 22.34 (CH2), 21.99, (CH2) 21.60 (CH2), 21.02 (CH2), 20.44 (CH2), 19.86 (CH3), 19.28 (CH3), 18.13 (CH3), 15.62 (CH3), 14.85 (CH3), 12.53 (CH3), 10.60 (CH3), 10.40 (CH3), 10.02 (CH3), 8.08 (CH3), 7.12 (CH3). MS for 7: Calculated for C91H102N7O9S3Pd: 1638.6, Found: 1638.5. HR-MS for 7: Calculated for C91H102N7O9S3Pd: 1638.5930, Found: 1638.5972.
General Plate Irradiation Procedure.
A stock solution of either 4 or 5 (1 mM in DMSO) was diluted into 75% (v/v) methanol/water to afford a final concentration of 20 μM. Samples were plated in 96-well plates (Corning UV-transparent acrylic copolymer, 300 μL/well) were irradiated using a 690 or 780 nm LED (L690–66–60 or L780–66–60, Marubeni America Co.) at an intensity of 15 mW/cm2 as measured using a power meter. Plates were covered with a plastic lid during the course of irradiation to minimize solvent evaporation. Wells were irradiated and analyzed at 10 min intervals by single-point absorption (655 and 825 nm). Experiments were run in duplicate with error bars derived from the standard deviation (<5% in all cases).
Procedure for LC/MS Relative Ion Analysis of Conjugate 4 and 5 Photolysis Mixture.
A stock solution of 4 or 5 (1 mM in DMSO) was diluted into 75% (v/v) methanol/water to a final concentration of 20 μM. Sodium 3-(3,3-dimethyl-2-oxoindolin-1-yl)propane-1-sulfonate (5 μM) was used as an internal standard. The samples were irradiated in 1.5 mL HPLC vials as above.. Samples were analyzed by a flow injection analysis method using an Agilent LCMS-1200 Single Quadrupole instrument (normal resolution) at t = 0 (prior to irradiation) and t = 60 min. The relative ion counts in Figure 3 were calculated by integrating the extracted ion chromatogram (EIC) of the m/z of 4 or 5, oxindole 8a, and aldehyde 8b, and then dividing the area of this extracted peak by the area of the internal standard. Experiments were run in triplicate and plotted with error bars derived from the standard deviation.
Electrochemical Studies.
Dimethyl sulfoxide (DMSO) was purchased from Sigma-Aldrich and used as received. Tetra-n-butylammonium perchlorate (TBAP) was purchased from Sigma Chemical or Fluka Chemika Co., recrystallized from ethyl alcohol, and dried under vacuum at 40 °C for at least 1 week prior to use.
Instrumentation.
UV–visible spectra were recorded in a Hewlett-Packard Model 8453 diode array spectrophotometer. Cyclic voltammetry was carried out at 298 K using an EG&G Princeton Applied Research 173 potentiostat/galvanostat. A three-electrode system was used for cyclic voltammetric measurements and consisted of a glassy carbon electrode electrode, a platinum counter electrode, and a homemade saturated calomel reference electrode (SCE). The SCE was separated from the bulk of the solution by a fritted glass bridge of low porosity which contained the solvent/supporting electrolyte mixture. High purity N2 was used to deoxygenate the solution and kept over the solution during each electrochemical experiment.
Thin-layer UV–visible spectroelectrochemical experiments were performed with a home-built thin-layer cell which has a light transparent platinum net working electrode. Potentials were applied and monitored with an EG&G PAR Model 173 potentiostat. Time-resolved UV–visible spectra were recorded with a Hewlett-Packard Model 8453 diode array spectrophotometer. High purity N2 from Trigas was used to deoxygenate the solution and kept over the solution during each electrochemical and spectroelectrochemical experiment.
Drug Formulation.
1% Tween 80.
All compounds for this study were prepared in 1% Tween 80/D5W formulation. Amount of compound and volume of solution were calculated for desired concentration. Final solution was made to be a 1% Tween 80/D5W solution. The desired compound and necessary volume of Tween 80 were added to a mortar and mulled to a paste. The paste was allowed to sit overnight and the next day the calculated amount of D5W was added to the paste and mixed. Solution was filtered through a 0.2 μm syringe filter and concentration was measured spectrophotometrically. Drug solutions were stored at 4 °C before and after use.
Cell Culture and PDT Treatment Parameters.
Cell Maintenance.
Murine Colon26 carcinoma cells were cultured and maintained in sterile RPMI-1640, containing 1× l-glutamine, supplemented with 10% fetal calf serum (FCS) (Atlanta Biologicals, triple 0.1 μm filtered, Lawrenceville, GA), and 1% Penicillin/Steptomycin/l-glutamine (P/S/l-G 10,000 IU/mL penicillin, 10,000 μg/mL streptomycin, 29.2 mg/mL l-glutamine) maintained in a humidified incubator at 37 °C in atmosphere of 5% CO2. Human BCC1 cells were maintained in DMEM containing 10% FCS.
Flow Cytometry.
Colon26 cells were harvested with trypsin, filtered through 30 μm mesh, and seeded at low density (400,000 cells) in 5 mL sterile polystyrene tubes. Cells were then incubated in triplicate with 0.8 μM with conjugates for 4 h at 37 °C. After incubation time, tubes were placed on ice and immediately analyzed by flow cytometry (Ex. 785 Em. 830 LP).
In Vitro Photodynamic Therapy.
Colon26 cells are plated into 96 well plates (3600 cells per well), allowed to adhere to the plate, and treated with a range of compound concentrations for 24 h. After the 24 h drug incubation, cells are irradiated with light from various light sources based on wavelength of irradiation. The light source consists of dye lasers (375; Spectra-Physics, Mt. View, CA) pumped by an argon-ion laser (either 171 or 2080; Spectra-Physics). The dye lasers are tuned to the appropriate wavelength for the compound being tested (4 = 665 nm, 5 = 651 nm, 6 = 654 nm, 7 = 640 nm). Total light doses range 0.5–3 J/cm2 at a fluence rate of 3.2 mW/cm2. After 48 h post PDT treatment, MTT (3-(4,5-dimethythiazol-2-yl)-2,5-diphenyl tetrazolium bromide) is added to each well. The solubilized formazan product (by the addition of DMSO) is measured spectrophotometrically using a BIOTEK EL-800 plate reader. Data is processed using GEN5 software. A medium blank value is subtracted from all samples and optical density (O.D.) values of treated cells are divided by mean O.D. value of untreated cells for each light dose. Results are expressed as the percent cell survival ± SD and are plotted as % control vs concentration of compound or % control vs light dose (at one concentration) using Graph Pad Prism 5.
To quantify the photoreaction mediated by the compounds, BCC1 cells were plated into 24-well cluster plates and cultured until the cells reach confluence. The cells were then incubated with culture medium containing 400 nM compounds for 2.5 h. The cells were washed free of extracellular compounds and treated with the laser light at the excitation wavelength specific for each PS to a fluence of 3 J/cm2 over 9 min at 37 °C. Immediately after light treatment the cells were lysed in the well with RIPA buffer. Extracts were cleared by centrifugation and the proteins in the supernatant analyzed for STAT3 by immunoblotting on 6% denaturating SDS gels as described.30 The percent conversion of STAT3 monomer into the homodimeric complex was quantified based on the enhanced chemiluminescent signal derived from the immunodetection complex as described previously (ref). Subcellular localization of internalized compounds was determined in BCC1 cells that had been cultured for additional 4 h in normal culture medium. The phase image and HPPH-fluorescence was recorded on a Zeiss Axiovert 200 inverted microscope with a Q-Imaging camera.30 The filters for HPPH fluorescence were Ex 410/40 nm and Em 675/750 nm.
Animal Husbandry.
All animals used in experiments were housed and cared for under the strict guidelines of the Institutional Animal Care and Use Committee (IACUC) in the Department of Laboratory and Animal Resources (DLAR) core facility at Roswell Park Cancer Institute (Buffalo, NY).
Animal and Tumor Systems.
All animals used in this study were BALB/cAnNCr mice obtained from Fredrick National Laboratory (Fredrick, MD). Eight- to twelve-week-old animals were inoculated subcutaneously with 1 × 106 Colon26 cells on the right shoulder where the fur was removed.
Fluorescence Optical Imaging.
In vivo imaging was performed using a 12 bit Nuance camera (CRI, Worburn, MA) imaging system. When tumors reach measurable dimensions of approximately 4 mm × 4 mm, three mice per drug were injected i.v. with compound such that final concentration of each dye would equal 0.3 μmol compound per kg body weight of the mice. At regular intervals mice were anesthetized by i.p. injection of ketamine/xylene mixture (100/10 mg/kg) and images were acquired for 5 s with a continuous wave laser for an excitation source at 782 nm (B & W Tek Inc., Newark, DE). Fluorescent light past 830 nm was captured using 800 and 830 nm long-pass filters over various time points.
Post-Acquisition Image Processing.
Nuance Camera images were processed using ImageJ 1.44p (NIH) software. Grayscale images were enhanced using Lookup Tables setting Royal and brightness was adjusted using Image Adjust setting of Brightness/Contrast. All images shown were set to the same scales.
In Vivo Photodynamic Therapy.
PDT was performed as outlined in Spernyak et al.14 Briefly, when tumors reach a measurable volume of ~62.5 mm3, mice were injected (t = 0) intravenously with compound in a dose–response manner such that final concentration of each dye ranged from 0.3 to 1.0 μmol compound per kg body weight of the mice. At peak drug accumulation time in tumor, mice were restrained in Plexiglass holders which were designed to expose one side of the mouse to open air. Tumors were irradiated with laser light corresponding to longest excitation peak at a fluence of 135 J/cm2 and fluence rate of 75 mW/cm2 for 30 min and then removed from the laser source and returned to normal housing. Mice were monitored daily for signs of toxicity/duress and, if noted, were sacrificed. The end point for mice implanted with tumors was regrowth after treatment to tumor volume of 400 mm3 or 60 days post PDT without regrowth. At the time of end point, mice were euthanized in accordance with animal protocols.
In Vivo Photobleaching.
When tumors reach a measurable volume of ~62.5 mm3, mice were injected (t = 0) intravenously with compound such that the final concentration of each dye would be 0.3 μmol compound per kg body weight of the mice. At peak drug accumulation time in tumor, mice were restrained in Plexiglass holders which were designed to expose one side of the mouse to open air. Mice were imaged using Nuance Camera for t = 0 with 5 s exposure at excitation 782 nm emission 800/830 nm long-pass filters. Tumors were irradiated with laser light corresponding to longest excitation peak (4 = 665 nm, 5 = 651 nm, 6 = 654 nm, 7 = 640 nm) at a fluence of 135 J/cm2 and fluence rate of 75 mW/cm2 for 30 min. At regular intervals over the 30 min treatment time frame, mice were removed from laser and reimaged with Nuance Camera. After treatment, mice were euthanized in accordance with animal protocols.
UV–visible Spectroscopy.
UV–vis absorption spectra of compounds were obtained using a Shimadzu UV-3600 spectrophotometer by diluting drug solutions in methanol to a final concentration of 5 μM. For photobleaching studies, 5 μM solutions in methanol were irradiated with 532 nm laser light at 125 mW over time and absorbance was measured using a Shimadzu UV-3600.
Fluorescence Spectroscopy.
Fluorescence spectra were obtained using a Fluorolog-3 spectrofluorometer (532 nm excitation, 800 nm and longer emission 2 nm slits).
Transient Absorption Measurements.
Femtosecond transient absorption spectroscopy experiments were conducted using an ultrafast source: Integra–C (Quantronix Corp.), an optical parametric amplifier: TOPAS (Light Conversion Ltd.) and a commercially available optical detection system: Helios provided by Ultrafast Systems LLC. The source for the pump and probe pulses were derived from the fundamental output of Integra-C (780 nm, 2 mJ/pulse, and fwhm = 130 fs) at a repetition rate of 1 kHz. 75% of the fundamental output of the laser was introduced into TOPAS which has optical frequency mixers resulting in tunable range from 285 to 1660 nm, while the rest of the output was used for white light generation. Prior to generating the probe continuum, a variable neutral density filter was inserted in the path in order to generate stable continuum, and then the laser pulse was fed to a delay line that provides an experimental time window of 3.2 ns with a maximum step resolution of 7 fs. In our experiments, a wavelength at 355 nm of TOPAS output, which is fourth harmonic of signal or idler pulses, was chosen as the pump beam. As this TOPAS output consists of not only desirable wavelength but also unnecessary wavelengths, the latter was deviated using a wedge prism with wedge angle of 18°. The desirable beam was irradiated at the sample cell with a spot size of 1 mm diameter where it was merged with the white probe pulse in a close angle (<10°). The probe beam after passing through the 2 mm sample cell was focused on a fiber optic cable that was connected to a CCD spectrograph for recording the time-resolved spectra (410–800 nm). Typically, 2500 excitation pulses were averaged for 5 s to obtain the transient spectrum at a set delay time. Kinetic traces at appropriate wavelengths were assembled from the time-resolved spectral data. All measurements were conducted at room temperature, 295 K.
Nanosecond time-resolved transient absorption measurements were carried out using the laser system provided by UNISOKU Co., Ltd. Measurements of nanosecond transient absorption spectrum were performed according to the following procedure. A deaerated solution containing a sample in a quartz cell (1 cm × 1 cm) was excited by a Nd:YAG laser (Continuum SLII-10, 4–6 ns fwhm, λex = 355 nm, 5 mJ/pulese). The photodynamics were monitored by continuous exposure to a xenon lamp (150 W) as a probe light and a photomultiplier tube (Hamamatsu 2949) as a detector. The solution was oxygenated by nitrogen purging for 15 min prior to measurements.
Near-IR Emission Measurements.
Singlet oxygen generation was detected by its phosphorescence emission at 1270 nm. An O2-saturated benzene-d6 solution containing samples in a quartz cell (optical path length 10 mm) was excited by an LED laser at 532 nm using an Otsuka Electronics Co. Ltd. Vis-NIR emission spectrophotometer.
Statistical Analysis.
All data were analyzed using Graph Pad Prism 5 software (GraphPad Software, San Diego, CA). To test for differences between two unpaired groups with normal distribution, a two-tailed Student’s t test (unless otherwise stated) was used with Confidence Intervals of 95%.
ACKNOWLEDGMENTS
The Financial support from the NIH (PO1 CA55791, RKP & HB), an RPCI support grant (P30 CA16056) and the Robert A. Welch Foundation (K.M.K., Grant E-680) is appreciated. Nayan Patel is thankful to the Department of Molecular Pharmacology and Cancer Therapeutics for providing fellowship from the NIH funded graduate student research training grant. This work was also supported by Grants-in-Aid (nos. 26620154 and 26288037 to K.O.) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT); ALCA and SENTAN projects from JST, Japan (to S.F.). We thank Mary Jo Bowman for STAT3 analyses.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Jori G (1996) Tumour photosensitizers: approaches to enhance the selectivity and efficiency of photodynamic therapy. J. Photochem. Photobiol., B 36 (2), 87–93. [DOI] [PubMed] [Google Scholar]
- (2).Dougherty TJ (1996) A brief history of clinical photodynamic therapy development at Roswell Park Cancer Institute. J. Clin. Laser Med. Surg. 14 (5), 219–21. [DOI] [PubMed] [Google Scholar]
- (3).Agostinis P, Berg K, Cengel KA, Foster TH, Girotti AW, Gollnick SO, Hahn SM, Hamblin MR, Juzeniene A, Kessel D, Korbelik M, Moan J, Mroz P, Nowis D, Piette J, Wilson BC, and Golab J (2011) Ca-Cancer J. Clin. 61 (4), 250–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Mallidi S, Huang H-C, Liu JY, Mensah L, Mai Z, and Hasan T (2013) Photoacoustic guided photodynamic therapy of glioblastoma. Cancer Res. 73, 3923. [Google Scholar]
- (5).Eljamel S (2010) Photodynamic applications in brain tumors: a comprehensive review of the literature. Photodiagn. Photodyn. Ther. 7 (2), 76–85. [DOI] [PubMed] [Google Scholar]
- (6).Eljamel S, Petersen M, Valentine R, Buist R, Goodman C, and Moseley H (2013) Comparison of intraoperative fluorescence and MRI image guided neuronavigation in malignant brain tumours, a prospective controlled study. Photodiagn. Photodyn. Ther. 10 (4), 356–61. [DOI] [PubMed] [Google Scholar]
- (7).Stummer W, Pichlmeier U, Meinel T, Wiestler OD, Zanella F, Reulen H-J, and for the ALA-Glioma Study Group (2006) Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: a randomised controlled multicentre phase III trial. Lancet Oncol. 7 (5), 392–401. [DOI] [PubMed] [Google Scholar]
- (8).Chen Y, Gryshuk A, Achilefu S, Ohulchansky T, Potter W, Zhong T, Morgan J, Chance B, Prasad PN, Henderson BW, Oseroff A, and Pandey R K. (2005) A novel approach to a bifunctional photosensitizer for tumor imaging and phototherapy. Bioconjugate Chem. 16 (5), 1264–74. [DOI] [PubMed] [Google Scholar]
- (9).James NS and Pandey RK. Manuscript in preparation. [Google Scholar]
- (10).Lovell JF, Chen J, Jarvi MT, Cao WG, Allen AD, Liu Y, Tidwell TT, Wilson BC, and Zheng G (2009) FRET quenching of photosensitizer singlet oxygen generation. J. Phys. Chem. B 113 (10), 3203–11. [DOI] [PubMed] [Google Scholar]
- (11).Ethirajan M, Patel N, and Pandey R (2010) Porphyrin-based multifunctional agents for tumor-imaging and photodynamic therapy (PDT), in Handbook of Porphyrin Science (Kadish K, Smith K, and Guilard R, Eds.) World Scientific Publishing, Hackensack, New Jersey, USA. [Google Scholar]
- (12).Maduray K, and Odhav B (2013) The in vitro photodynamic effect of laser activated gallium, indium and iron phthalocyanine chlorides on human lung adenocarcinoma cells. J. Photochem. Photobiol., B 128, 58–63. [DOI] [PubMed] [Google Scholar]
- (13).Mitra S, and Foster TH (2000) Photochemical oxygen consumption sensitized by a porphyrin phosphorescent probe in two model systems. Biophys. J. 78 (5), 2597–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Sharman WM, Allen CM, and van Lier JE (2000) Role of activated oxygen species in photodynamic therapy, In Methods in Enzymology (Packer HSL, Ed.) pp 376–400, Academic Press. [DOI] [PubMed] [Google Scholar]
- (15).Saenz C, Ethirajan M, lacobucci G, Pandey A, Missert JR, Dobhal MP, and Pandey RK (2011) Indium as a central metal enhances the photosensitizing efficacy of benzoporphyrin derivatives. J. Porphyrins Phthalocyanines 15, 1310–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Byers GW, Gross S, and Henrichs PM (1976) Direct and sensitized photooxidation of cyanine dyes. Photochem. Photobiol. 23 (1), 37–43. [DOI] [PubMed] [Google Scholar]
- (17).Nani RN, Kelley JA, Ivanic J, and Schnermann MJ (2015) Reactive species involved in the regioselective photooxidation of heptamethine cyanines. Chem. Sci. 6, 6556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Gorka AP, Nani RR, and Schnermann MJ (2015) Cyanine polyene reactivity: scope and biomedical applications. Org. Biomol. Chem. 13 (28), 7584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Gorka AP, Nani RR, Zhu J, Mackem S, and Schnermann MJ (2014) A near-IR Uncaging Strategy Based on Cyanine Photochemistry. J. Am. Chem. Soc. 136 (40), 14153–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Nani RR, Gorka AP, Nagaya T, Kobayashi H, and Schnermann MJ (2015) Near-IR Light-Mediated Cleavage of Antibody-Drug Conjugates Using Cyanine Photocages. Angew. Chem., Int. Ed. 54, 13635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Sekar RB, and Periasamy A (2003) Fluorescence resonance energy transfer (FRET) microscopy imaging of live cell protein localizations. J. Cell Biol. 160 (5), 629–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Guo Z, Park S, Yoon J, and Shin I (2014) Recent progress in the development of near-infrared fluorescent probes for bioimaging applications. Chem. Soc. Rev. 43, 16–29. [DOI] [PubMed] [Google Scholar]
- (23).Gorka AP, Nani RR, and Schnermann MJ (2015) Cyanine polyene reactivity: scope and biomedical applications. Org. Biomol. Chem. 13, 7584–7598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Fan J, Hu M, Zhan P, and Peng X (2013) Energy transfer cassettes based on organic fluorophores construction and applications in ratiometric sensing. Chem. Soc. Rev. 42, 29–43. [DOI] [PubMed] [Google Scholar]
- (25).Ethirajan M, Chen P, Ohulchanskyy TY, Goswami LN, Gupta A, Srivatsan A, Dobhal MP, Missert JR, Prasad PN, Kadish KM, and Pandey RK (2013) Regioselective synthesis, photophysical and electrochemical studies of 20-substituted cyanine dye-purpurinimide conjugates: Incorporation if Ni(II) into the conjugate enhances its tumor uptake and fluorescence imaging ability. Chem. - Eur. J. 19, 6670–6684. [DOI] [PubMed] [Google Scholar]
- (26).Goswami LN, Ethirajan M, Dobhal MP, Zhang M, Missert JR, Shibata M, Kadish KM, and Pandey R K. (2009) J. Org. Chem. Remarkable features of the McMurry reaction conditions in dimerization of formyl- and 2-formylvinylpurpurinimides. Electrochemistry of monomeric Ni(II) purpurinimide and the corresponding dyads. J. Org. Chem. 74, 568–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Salice P, Arnbjerg J, Pedersen BW, et al. (2010) Photophysics of squaraine dyes: Tole of charge-transfer in singlet oxygen production and removal. J. Phys. Chem. A 114, 2518–2525. [DOI] [PubMed] [Google Scholar]
- (28).Finikova OS, Chen P, Ou Z, and Kadish KM (2008) Dynamic quenching of porphyrin triplet states by two-photon absorbing dyes: Towards two-photon-enhanced oxygen nanosensors. J. Photochem. Photobiol., A 198, 75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Rehm D, and Weller A (1970) Kinetics of fluorescence quenching by electron and hydrogen-atom transfer. Isr. J. Chem. 8, 259–271. [Google Scholar]
- (30).Henderson BW, Daroqui C, Tracy E, Vaughan LA, Loewen GM, Cooper MT, and Baumann H (2007) Cross-linking of signal transducer and activator of transcription 3 a molecular marker for the photodynamic reactions in cells and tumors. Clin. Cancer Res. 13, 3156–3163. [DOI] [PubMed] [Google Scholar]
- (31).Tracy EC, Bowman MJ, Pandey RK, Henderson BW, and Baumann H (2012) Cell-type selective phototoxicity achieved with chlorophyll-a derived photosensitizers in a co-culture system of primary human tumor and normal lung cells. Photochem. Photobiol. 87, 1405–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]