

Summary
Epstein-Barr virus (EBV) is an oncogenic human herpesvirus that persists as a multicopy episome in proliferating host cells. Episome maintenance is strictly dependent on EBNA1, a sequence-specific DNA binding protein with no known enzymatic activities. Here, we show that EBNA1 forms a cell cycle-dependent DNA cross-link with EBV origin of plasmid replication oriP. EBNA1 tyrosine 518 (Y518) is essential for cross-linking to oriP and functionally required for episome maintenance and generation of EBV transformed lymphoblastoid cell lines (LCLs). Mechanistically, Y518 is required for replication fork termination at oriP in vivo and for the formation of SDS-resistant complexes in vitro. EBNA1-DNA cross-linking corresponds to single-strand endonuclease activity specific to DNA structures enriched at replication-termination sites, such as 4-way junctions. Taken together, these findings reveal that EBNA1 forms tyrosine-dependent DNA-protein crosslinks and single stand cleavage at oriP required for replication termination and viral episome maintenance.
Keywords: Epstein-Barr virus, EBNA1, DNA-binding domain, RADAR, DNA-protein adducts, tyrosine resolvase, oriP
Subject categories: Epstein-Barr virus, episome maintenance, viral latency
In Brief
Cell cycle dependent recombinase-like activity of EBNA1 is required for replication termination and viral episome maintenance of oncogenic Epstein-Barr Virus.
Graphical Abstract
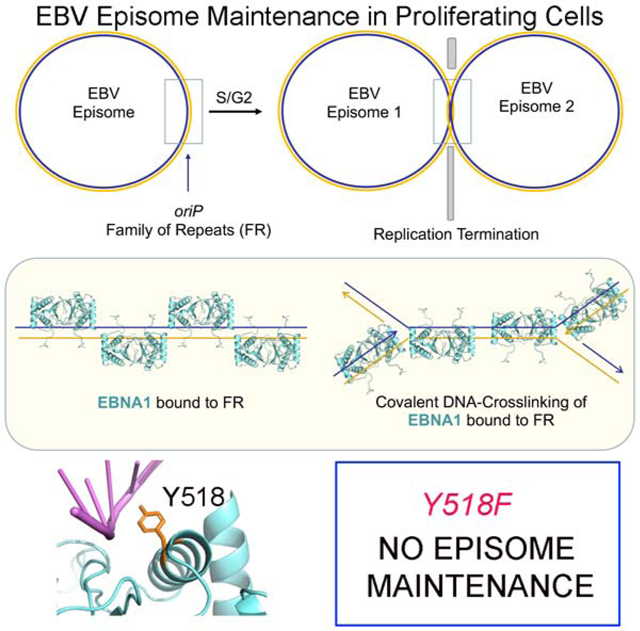
INTRODUCTION
Epstein-Barr virus (EBV) establishes long-term latent infection that increases the risk of cancer and autoimmune disease (Ascherio and Munger, 2015; Shannon-Lowe and Rickinson, 2019; Young et al., 2016). The Epstein-Barr Nuclear Antigen 1 (EBNA1) is the only viral protein required for viral DNA replication and episome maintenance during latent infection (De Leo et al., 2020; Frappier, 2015; Lindner and Sugden, 2007). EBNA1 is a high-affinity sequence specific DNA binding protein thought to lack any intrinsic enzymatic activities (Bochkarev et al., 1996; Rawlins et al., 1985). The X-ray crystal structure of the EBNA1 DNA-binding domain (DBD) has been solved (Bochkarev et al., 1996; Bochkarev et al., 1995; Deakyne et al., 2017; Malecka et al., 2019) and shares structural homology with other viral proteins required for episome maintenance of their respective viruses, including Kaposi’s Sarcoma Associated Herpesvirus (KSHV) encoded Latency Associated Nuclear Antigen (LANA), and the papillomavirus encoded E2 protein (Bochkarev et al., 1996; Bochkarev et al., 1995; Domsic et al., 2013; Edwards et al., 1998; Enemark et al., 2002; Hegde et al., 1992; Hellert et al., 2015; Hellert et al., 2013; Knight et al., 1991).
EBNA1 binds to repetitive DNA recognition elements in the viral origin of plasmid replication (oriP), which consists of a family of repeats (FR) and a dyad symmetry (DS) element. The EBNA1 DBD is essential for oriP-dependent DNA replication and episome maintenance (Frappier, 2015; Lindner and Sugden, 2007). EBNA1 binding to the DS functions as an origin of DNA replication that can recruit cellular Origin Recognition Complex (ORC) and MiniChromosome Maintenance (MCM) proteins (Dhar et al., 2001; Norseen et al., 2008; Ritzi et al., 2003). EBNA1 binding to the FR is necessary for episome maintenance and metaphase chromosome attachment, as well as enhancing replication and transcription. oriP is also a region where replication forks terminate and form recombination structures (Dhar and Schildkraut, 1991; Dheekollu et al., 2007; Dheekollu and Lieberman, 2011; Ermakova et al., 1996). Two-dimensional neutral agarose gel analysis of replication structures revealed that oriP forms a strong replication fork block and recombination-like structures. The replication fork protection factors Timeless and Tipin are recruited to oriP in a cell cycle dependent manner, and were required for formation and resolution of these structures (Dheekollu and Lieberman, 2011). Similar structures were also observed at the KSHV terminal repeats that serve as both origin of replication and episome maintenance element, similar to EBV oriP (Dheekollu et al., 2013).
OriP is thought to function as a programmed replication termination site for the circular genome of EBV. Replication termination sites in circular genomes of bacterial and yeast plasmids typically utilize a sequence specific DNA-binding factor, such as Tus, that can block replicative helicases, and site-specific recombinases, such as Xer, that can resolve catenated DNA molecules resulting from converging replication forks (Bussiere and Bastia, 1999; Neylon et al., 2005). Previous studies indicate that recombination structures form at oriP at the conclusion of DNA replication, but it is not known how EBNA1 contributes to the formation or resolution of these structures. Several cellular DNA damage repair factors associate with and function at oriP, including the MRN endonuclease complex (Dheekollu et al., 2007) and Timeless-Tipin replication fork protection complex (Dheekollu and Lieberman, 2011). Replication termination is also known to require topoisomerases and in some cases resolvases (Bastia and Zaman, 2014). Among this family of enzymes are the tyrosine recombinases that utilize a transient covalent linkage between a tyrosine residue and the phosphodiester backbone of the substrate DNA (Castillo et al., 2017; Crozat et al., 2014; Midonet and Barre, 2014; Poulter and Butler, 2015). Several tyrosine recombinases are site-specific and utilize sequence specific DNA recognition with catalytic activation in response to DNA structure formation. Topoisomerases are tyrosine-based enzymes that utilize transition state covalent interaction with the phosphate backbone leading to the transient cleavage of the DNA backbone with the function of relieving torsional strain (type I) and decatenation (type II). The structures that form at replication termination sites and collapsed replication forks require additional recombinational activities to repair the replication structure and complete synthesis of daughter DNA strands. The mechanisms required for the resolution of replication fork blocks and termination structures for the completion of EBV DNA at oriP is not yet known. Here, we provide evidence that EBNA1 forms a transient covalent DNA-adduct and single strand cleavage through a tyrosine residue and that mutation of this tyrosine impairs the episome maintenance function of EBNA1.
RESULTS
EBNA1 forms cell cycle-dependent covalent-DNA adducts.
The Rapid Approach to DNA Adduct Recovery (RADAR) was used to investigate the formation of a covalent cross-link of EBNA1 with DNA (Fig. 1A). RADAR has been shown to capture DNA-adducts such as topoisomerase II (TopoII) after treatment with etoposide (Kiianitsa and Maizels, 2013). We validated that TopoII forms a DNA-adduct in the presence of etoposide using RADAR assay (Fig. 1B, top). We next tested the ability of EBNA1 to form DNA adducts using a FLAG-tagged EBNA1 in stable cells lines carrying oriP episomes (Fig. 1B, bottom). EBNA1 DNA adducts were detected at low levels in asynchronous growing cells, but these adducts were highly enriched in cells synchronized for late S and G2/M phases of the cell cycle (Fig. 1C). EBNA1-DNA adducts were also recovered by RADAR from endogenous viral genomes in latently infected Raji Burkitt lymphoma cell line (Fig. 1D). Raji cells were fractionated by cell cycle size using centrifugal elutriation (Fig. 1C), and then assayed by RADAR followed by SDS-PAGE and Western blot (Fig. 1D). We found that EBNA1-DNA adducts were recovered mostly in elutration fractions corresponding to late S and G2/M. TopoII-DNA adducts were also recovered in S and G2 phase, while no DNA adducts were detected for DNA replication processivity factor PCNA or control protein β-Actin. Cell-cycle enriched EBNA1-DNA adducts were also observed in lymphoblastoid cell lines (LCLs) (Fig. 1E and F) indicating that this is not limited to one EBV cell line or tumor type. The specificity of RADAR for EBNA1 was further demonstrated by the lack of detectable DNA-adduct formation with single strand DNA binding protein RPA32 or viral transcription factor EBNA2 or latency membrane protein 1 (LMP1) (Fig. 1F).
FIG. 1. RADAR assay analysis of the cell-cycle dependent EBNA1-DNA adducts.

A) Schematic of RADAR assay adapted from (Kiianitsa and Maizels, 2013). B) Dot blot probed with antibody to TopoII after RADAR isolation of cells treated with 0, 25, 50, or 100 μg of etoposide (top panel). Dot blot probed with antibody to EBNA1 in 293T cells containing oriP and expressing FLAG-EBNA1 (lower panel). RADAR was performed on asynchronous (Asyn), or cell cycle synchronized cells in G1/S blocked by aphidicolin, 45’ or 90’ after release from aphidicolin block, or at G2/M border blocked by nocodazole. C) Cell cycle fractionation by centrifugal elutriation of Raji cells. Fractions were subject to RADAR and dot blot analysis for EBNA1. Elutriation fractions of 27 and 32 ml/min represent late S/G2 phase of the cell cycle. Western blot of cell cycle proteins as indicated for each elutriation fraction (right panel). D) Raji cell cycle fractions were assayed by Western blot from input (left) or after RADAR (right) for EBNA1, TopoII, PCNA, or Actin. Fraction 27 corresponds to late S/G2 phase of the cell cycle. E) LCL cell cycle elutriation profile analyzed by propidium iodide (PI) staining and FACS. F) LCL cell cycle fractions were assayed by Western blot from input (left) or after RADAR (right) for EBNA1, TopoII, PCNA, RPA32, EBNA2, and LMP1.
EBNA1 Y518 is required for EBNA1-DNA adduct formation.
Examination of the EBNA1 primary DNA sequence conservation and crystal structure revealed a highly conserved tyrosine residue (Y518) that is in close proximity (2.7Å) to the DNA phosphate backbone (Fig. 2A–E). Tyrosine 518 was mutated to phenylalanine (Y518F) and assayed for its effect on DNA-adduct formation by RADAR assay (Fig. 2F–G). FLAG-EBNA1 WT or Y518F were expressed at similar levels from oriP-dependent episomal plasmids (Fig. 2F). RADAR assay followed by FLAG-immunoprecipitation (RADAR-IP) revealed that only EBNA1 WT, but not Y518F could be recovered. This suggests that the Y518F mutation disrupts EBNA1-DNA adduct formation. To further examine the properties of the EBNA1-DNA adduct, we performed RADAR-IP followed by DNA end-labeling with T4 polynucleotide kinase (PNK) and 32 P-ATP (Fig. 2G). We found extensive radiolabeling of DNA with EBNA1 WT, but not with Y518F. Although PNK can label RNA as well as DNA, the RADAR material is resuspended in 8 mM NaOH which should hydrolyze most RNA, suggesting that the nucleic acid adduct is primarily DNA. To further characterize the EBNA1-DNA adduct, we subjected the RADAR-IP to treatment with the single strand-specific nuclease P1 (Fig. 2H). P1 nuclease treatment increased the abundance of the unmodified 55kD EBNA1 species in SDS PAGE. The modified form of EBNA1 was also observed in RADAR-IPs from native Raji cells (Fig. S1A). Several other single strand DNA nucleases, including tyrosyl DNA phosphodiesterase 1 (TDP1) and ExoVII increased the migration of the unmodified EBNA1 relative to the EBNA1-DNA adduct in SDS-PAGE (Fig. S1). These nuclease studies support the model that EBNA1 captured by RADAR-IP is modified by a single strand DNA-adduct linked through a tyrosine phosphodiester bond.
FIG. 2. Tyrosine 518 is required for EBNA1-DNA crosslinking.

A) Alignment of EBNA1 and conservation of Y518 from different strains of EBV and related simian herpesvirus papio. B-E) Crystal structure images for EBNA1 bound to DNA (from PDB: 1B3T and 6PW2) showing the position of Y518 relative to DNA backbone. F) FLAG-EBNA1 WT or Y518F with oriP were expressed in 293T cells and assayed by Western blot of FLAG for total cell extracts (input) or after RADAR assay and FLAG-IP (RADAR-IP). G) RADAR-IP for EBNA1 WT or Y518F were incubated with [γ-32P]-ATP and T4 polynucleotide kinase (PNK). Samples were then subject to SDS-PAGE and analyzed by autoradiography. H) EBNA1 WT or Y518F were processed by RADAR and then incubated with P1 nuclease or buffer control (Cntrl) followed by Western blot analysis. Control input material is shown in left panel.
EBNA1 Y518F can bind oriP DNA but does not form covalent DNA-adducts.
We compared the ability of EBNA1 WT and Y518F to interact with oriP DNA by chromatin immunoprecipitation (ChIP) assay (Fig. 3A). Using standard formaldehyde cross-linking for ChIP, we found that EBNA1 Y518F bound oriP similar to WT (Fig. 3A, left). To test if EBNA1 might bind covalently to oriP, we assayed whether EBNA1-DNA complexes could survive the disruptive effects of ChIP conditions of 1% SDS and high-salt without formaldehyde cross-linking (Native ChIP). We observed that EBNA1 WT bound with ~30% efficiency compared to cross-linking conditions, while Y518F binding was not detected without formaldehyde cross-linking (Fig. 3A, middle). A similar comparison with histone H3K4me3, which is known to be enriched at oriP, revealed a strong dependence on formaldehyde cross-linking for occupancy in either WT or Y518F EBNA1 expressing cells (Fig. 3A, right). These findings suggest that a significant percentage of EBNA1 WT, but not Y518F can bind oriP in a manner resistant to SDS denaturing conditions. To better understand the contribution of Y518 to EBNA1 DNA binding, we expressed and purified the EBNA1 DNA binding domain (DBD) for WT, Y518A, Y518F, Y518E, H468A, and N519A proteins (Fig. 3B–D). N519 is a neighboring amino acid that makes additional DNA contact, and H468 is in the flexible N-terminal arm and makes contact with the opposite DNA strand. DNA binding activity was assayed by electrophoretic mobility shift assay (EMSA) with a single binding site from the DS element of oriP. EMSA revealed that Y518F has similar DNA binding affinity (14.6 nM) for DS as EBNA1 WT (10.4 nM), while Y518E had no measurable binding, and Y518A and N519A had >10 fold weaker binding (>100 nM) (Fig. 3D). H468A had a 2-fold reduction in binding affinity (20 nM). These results indicate that Y518F has nearly identical DNA binding properties as EBNA1 WT in EMSA and ChIP assays, yet is incapable of forming a stable complex in native ChIP, similar to its failure to form complexes in the RADAR assay.
FIG. 3. Y518F disrupts cross-linking but not binding to oriP.

A) 293T cells expressing FLAG-EBNA1 WT or Y518F with oriP were assayed by ChIP assay under standard formaldehyde cross-linking conditions (Cross-linked) or without formaldehyde (Native) and assayed for ChIP with antibodies to IgG or Flag (EBNA1 WT or Y518F) or H3K4me2, as indicated. ChIP-DNA was assayed for oriP FR sequence by Taqman specific qPCR. student T-test, n=3., ns -not significant, * p value < 0.01 ** p value <0.001*** p value <0.0001. B) EMSA analysis of EBNA1 DBD WT, Y518A, Y518F, Y518E, H468A or N519A using DS probe with a single EBNA1 binding site. EC) Coomassie stain of SDS-PAGE for 200 ng purified EBNA1 DBD WT, Y518A, Y518F, Y518E, H468A or N519A. D) Quantification of EMSA and calculation of EC50 (nM) for each mutant.
EBNA1 Y518F is defective for plasmid maintenance.
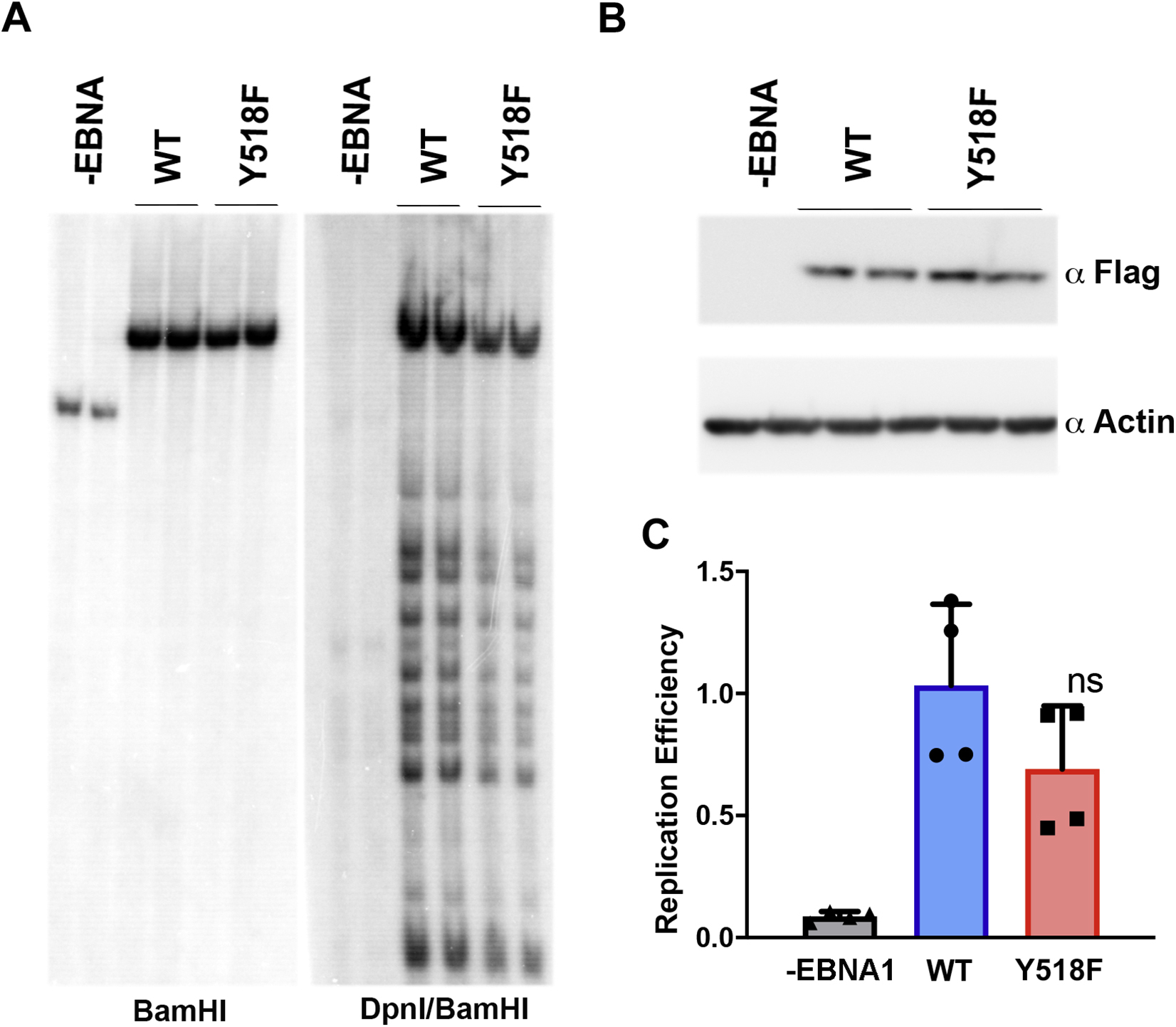
The functional impact of Y581F was assessed by DNA replication and plasmid maintenance assays in cells transfected with small oriP plasmids (~10 kb) conferring hygromycin resistance (Figs S2 and 4). Transient DNA replication was measured 3 days after transfection by DpnI resistance assay (Fig. S2A and B). Y518F trended towards a decrease in replication activity relative to WT EBNA1, but this difference did not reach statistical significance (Fig. S2C). We next assayed the episome maintenance function of oriP plasmids by comparing oriP episomes at 3 days relative to those retained after 20 days in culture (Fig. 4A and B). We found a significant defect in Y518F for episome maintenance by Southern blot analysis. As these plasmids carry the hygromycin resistance gene, we also assayed the ability to form hygromycin resistant colonies after 20 days post-transfection (Fig. 4C and S3A–C). Consistent with Southern blot analysis, Y518F was fold less efficient than EBNA1 WT in forming hygromycin resistant colonies (Fig. 4C). These findings indicate that Y518F is deficient in episome maintenance of oriP plasmids.
FIG. 4. Y518F is required for oriP-dependent episome maintenance and B-cell immortalization.

A) Southern blot analysis showing two biological replicates at day 3 and 20 post transfection of oriP plasmids expressing either WT or Y518F EBNA1. B) Western blot of FLAG-EBNA1 proteins or loading control Actin at day 3 post-transfection (lower panels). C) Quantification of Southern blot assays shown as representative in panel F. *** p value <.001, student t-test. n=6. D) Quantification of colony formation assay for oriP plasmids expressing hygromycin resistance in 293T cells with either WT or Y518F EBNA1. ** p value<.01, student t-test. n=4. E) Southern blot analysis of input EBV bacmid genomes expressing WT or Y518F EBNA1, cut with BamHI and probed with terminal repeat (TR) DNA (left panel). Cellular and EBV bacmid DNA isolated 21 days post-transfection of EBV bacmids with WT or Y518F EBNA1 probed cellular DNA fragment (Chr 17), or EBV TR or family of repeats (FR). F) Quantification of Southern blots shown in panel E. ** p<.01, student t-test, n=4. G) Quantification of colony formation assays for WT or Y518F expressing EBV bacmid genomes with GFP in 293T cells. *** P<.001, student t-test. H) Images of GFP-positive early stage LCL blast colonies from EBV bacmid virus with WT or Y518F EBNA1 at week 2 (top panels) or week 6 (lower panels). I) Quantification of early LCL clones stratified by size for WT or Y518F. * p<.05, student t-test. J) Numbers of successful LCLs generated from EBV bacmid infections wit WT or Y518F with 5 donors and two independent viral clones for each mutant.
Y518 is required for viral episome maintenance and generation of immortalized B-lymphocytes.
The functional role of Y518 is likely to be more apparent in the context of the larger (~170 kb) viral genome and the process of EBV-infection induced B-cell immortalization. To this end, we introduced the Y518F mutation into the EBV bacmid genome by recombineering (Fig. 4E). Two independently generated WT and Y518F bacmid clones were assayed for episome maintenance in 293T cells. We found that cells carrying Y518F were significantly reduced for total EBV DNA copy number at day 21 relative to day 3 (Fig. 4E and F), and this correlated with a significant defect in forming stable GFP-positive, hygromycin resistant colonies (Fig. 4G). To test the effect of Y518F on EBV immortalization of primary B-cells, we generated infectious virus from a single clone of transfected 293T cells with WT and Y518F bacmids. Virus was normalized by GFP units for infection of Raji cells and by viral DNA content by PCR. We then assayed identical GFP units of virus for their abilities to immortalize primary B-cells from five different donors into lymphoblastoid cells lines (LCLs) (Fig. 4H and I). While many lymphoblast transformants could be detected for EBV WT, very few were observed with Y518F (Fig. 4J). Out of 20 individual infections with 5 different donor and two independently generated viruses, only one Y518F infection produced a slow growing LCL. While no mutations were identified in the EBNA1 DBD in this rare clone, we observed a ~2 fold increase in EBNA1 and EBNA2 protein expression levels relative to WT EBV bacmid transformed cells (Fig. S3D).
To better understand the molecular basis for Y518 function in DNA replication and episome maintenance, we tested additional EBNA1 mutations in DNA replication and episome maintenance (Fig. 5). Examination of the EBNA1 crystal structure suggested that histidine 468 may contribute to Y518 DNA adduct formation since it is located in close proximity to the DNA backbone on the opposite strand to Y518 (Fig. 5A and B). We found that Y518A was incapable of supporting transient DNA replication (Fig. 5C–E), consistent its defect in DNA binding (Fig. 3B). H468A showed modest defects in DNA replication and episome maintenance by itself (Fig. 5C–E), but exacerbated the defects of Y518F in both DNA replication (Fig. 5E) and especially episome maintenance (Fig. 5F). These findings suggest that H468 contributes to the functional activity of EBNA1 in the context of Y518F.
Fig. 5. Functional analysis of EBNA1 Y518A and H468A mutations.

A-B) X-ray crystal structure of EBNA1 DNA showing Y518 and H468 proximity to DNA. C) Western blot of FLAG-EBNA1 WT, Y518F, Y518A, and H468A transfected 293T cells probed with FLAG and β-Actin. D) DNA replication assay for EBNA1 WT and mutants shown in panel C. Recovered DNA cut with BamHI (top) or BamHI/DpnI (lower) probed by Southern blot. E) Quantification of replication assay shown in panel D. F) Replication assay for EBNA1 WT, H468A, and H468A/Y518F. Recovered DNA cut with BamHI (left) or BamHI/DpnI (right). Quantification of DNA replication assay shown as bar graph. Error bars are sdm. G) Plasmid maintenance assay for EBNA1 H468A and H468A/Y518F shown at 3 day (top) and 21 days (lower) panels. Quantification for plasmid maintenance shown as bar graph. *** p<.001, student t-test n=4.
Loss of DNA replication fork pausing and recombination structures in Y518F.
To explore the mechanistic basis of Y518 in episome maintenance, we performed 2D neutral agarose gel analysis to assay DNA replication and recombination structures formed in living cells (Fig. 6A–C). Previous studies revealed that episome maintenance correlated with cell cycle-dependent replication fork pausing and formation of recombination structures formation at oriP (Dheekollu et al., 2011). Cells carrying oriP plasmids with either EBNA1 WT or Y518F were cell cycle synchronized and then subjected to two-dimensional neutral agarose gel analysis. As expected, we observed strong replication fork pausing (red arrow) and recombination structures typical of oriP in episomes maintained with EBNA1 WT (Fig. 6C, upper panel). In contrast, the major replication fork pausing structures were not observed at any time points for Y518F EBNA1 containing episomes (Fig 6C, lower panel). Similar loss of replication termination and recombination structures with Y518F was observed in 2D alkaline-agarose gels (Fig. S4A–B). These findings suggest that Y518F is required for efficient replication fork pausing at oriP, and that these activities correlate with EBNA1-DNA crosslinking and episome maintenance function.
FIG. 6. Biochemical analysis of recombination structures and EBNA1-DNA adduct formation.

A) Schematic of 2D agarose gel analysis of replication and recombination structures. B) Sites of restriction enzyme DraI used to generate OriP probe for Southern blot. C) Southern blot analysis of 2D gels at different stages of the cell cycle for EBNA1 WT or Y518F. Cell cycle times post aphidicolin release are indicated below. 2.30 h is late S/G2. Red arrow indicates termination and recombination-like structures in WT, but not detected in Y518F. D) DNA probe for 2xFR was assayed for binding with EBNA1 DBD WT, Y518A, Y518F, Y518E, H468A, or N519A. Reactions were analyzed by native EMSA (top) or by 0.1% SDS-EMSA (lower). Red arrows indicate positions of EBNA1-DNA protein complexes resistant to SDS-PAGE.
EBNA1 Y518 is required for SDS-resistant DNA binding in vitro.
We next assayed whether EBNA1 could bind and form SDS-resistant complexes with various recombination-like DNA structures in EMSA (Fig. 6D). We assayed the ability of highly purified EBNA1 DBD WT, Y518A, Y518F, Y518E, H468A, and N519A to bind to an oligonucleotide DNA probe containing two tandem EBNA1 sites derived from the family of repeats region (2xFR), which corresponds to site where we observe replication termination. As expected, WT, Y518F, and H468A bound to similarly to the 2xFR probe in native EMSA. Remarkably, we found that only WT EBNA1, but none of the other mutants could form an SDS-resistant complex with the 2xFR probe (Fig. 6D, lower panel). These findings indicate that highly purified bacterial DBD WT has capacity to form an SDS-resistant complex with 2xFR DNA.
We next compared the ability of full-length FLAG-tagged EBNA1 proteins purified from 293T cells to also form the SDS resistant complex in EMSA (Fig. S6). We purified FLAG-EBNA1 WT, Y518F, Y518A, Y518E, and H468A and assayed these proteins by Western blot and Coomassie showing similar levels of purity (Fig. S4C). We then assayed these proteins for binding to the 2xFR probe with or without addition of 0.1% SDS in the binding reaction. We found that FLAG-EBNA1 WT, Y581F, and H468A bound similarly to 2xFR in native conditions, but only WT and H468A showed partial resistance to 0.1% SDS (Fig. S4D). We also assayed WT and Y518F for binding to different DNA structures, including a Y-fork (Y), a nicked double strand DNA (N), or a 4-way Holiday junction (4WJ) all containing EBNA1 binding sites (Fig. S5). These were then assayed for binding with or without addition of 0.1% SDS. EMSA revealed that DBD and FL-EBNA1 could bind all such structures, and that the complexes formed with the 4WJ showed partial resistance to SDS (Fig. S5C). The same samples were further analyzed by electrophoresis in SDS-EMSA at high temperatures (Fig. S5D). Under these stringent conditions, only full length EBNA1 WT, but not the DBD, was able to form SDS-resistant complexes with the 4WJ, and to a lesser extent with the Y or N probes. Y518F did not form SDS resistant complexes with Y or N probes, although showed some residual binding with the 4WJ in the context of the full-length EBNA1.
EBNA1 Y518 is required for DNA endonuclease activity.
To investigate whether EBNA1 cross-linking to the DNA phosphate backbone corresponded to endonuclease activity, we isolated the FL-EBNA1 complexes bound to 2xFR or 4WJ probes (Fig. 7A) and assayed the radiolabeled single strand DNA molecules by denaturing urea-PAGE (Fig. 7B, C and S6). We found that FL-EBNA1 WT generated strong nicks at specific sites in the DNA substrate, while these same cleavage sites were not observed with FL-EBNA1 Y518F or Y518A (Fig. 7B, C and S6, arrows). The pattern of DNA breakage is consistent with cleavage at each of the Y518 contact sites in the 4WJ probe. These findings suggest that full length EBNA1 derived from mammalian cells has site-specific cleavage activity dependent on Y518.
FIG. 7. Site-specific endonuclease activity EBNA1.

A-B) Endonuclease cleavage assay for 2xFR or 4WJ probes with either no protein, FLAG-EBNA1 WT or Y518F. Reactions were subjected to electrophoresis on native (A) or denaturing 7M urea (B) polyacrylamide gel electrophoresis. Single strand (SS) probe alone. Arrows indicate major cleavage products. C) Same as in panel B, except a longer gel is used to resolve DNA incubated with either EBNA1 WT or Y518A. D-E) Model depicting EBV episomes during replication termination and separation and (E) EBNA1 Y518-dependent endonuclease activity at 4-way recombination junctions formed at FR element of oriP during replication termination.
DISCUSSION
Oncogenic herpesviruses persist in tumor cells as multicopy episomes. Unraveling the mechanisms controlling this viral persistence is fundamental to our understanding of viral tumorigenesis. The episome maintenance systems of oncogenic herpesviruses typically require a viral-encoded sequence-specific DNA-binding protein (e.g. EBNA1 and LANA) that binds to multiple repeat binding sites in the genetic maintenance element of the viral genome (e.g. oriP and TR). These protein-DNA structures are known to recruit cellular replication and segregation machinery and tether viral genomes to host chromosomes as passengers during mitotic division to maintain stable episome copy number in proliferating cells (De Leo et al., 2020). Here, we provide evidence that the EBV EBNA1-oriP episome maintenance system involves a tyrosine-dependent covalent adduct at oriP that is important for resolution of replication termination and essential for viral episome persistence. We used RADAR assay to show that the EBV EBNA1-oriP complex is resistant to chaotropic and denaturing agents consistent with formation of a covalent protein-DNA adduct. EBNA1-DNA adducts formed in a cell cycle-dependent fashion that were enriched in late S/G2 phase. We show that a highly conserved tyrosine 518 in close proximity to DNA phosphodiester backbone is essential for EBNA1 crosslinking to DNA. The Y518F mutation did not affect EBNA1 DNA binding under standard reaction conditions in vitro or by ChIP assay in vivo, and had only minor effects in transient DNA replication assays. In contrast, Y518F severely compromised episome maintenance function for plasmids and viral episomes. Viral genomes with Y518F were crippled in their ability to immortalize primary B-cells into LCLs. Mechanistically, Y518F failed to generate the recombination structures associated with replication fork termination that were readily observed with WT EBNA1 in 2D agarose gel analyses. Finally, we show that SDS-resistant EBNA1-DNA complexes formed with full-length EBNA1 bound to recombinational DNA structures, such as the 4WJ, and has single strand cleavage activity in vitro. Taken together, we propose that EBNA1 possesses a previously unknown DNA cross-linking activity that proceeds through a covalent protein-tyrosyl intermediate. This activity is critical for site-specific replication termination and episome maintenance of viral genomes (Fig. 7C and D).
Orthologs of EBNA1
EBNA1 is a member of a family of viral episome maintenance protein that include KSHV, MHV68 LANA, and HPV E2 (De Leo et al., 2020). More distant structural relatives can be found among betaherpesviruses, such as the HHV6 encoded IE2 protein, but it is not yet known if this protein functions in episome maintenance of its respective viruses (Nishimura et al., 2017). While these proteins share structural and functional features, they also have viral-specific idiosyncrasies. The EBNA1 Y518 has been shown to be part of a core domain essential for high-affinity sequence-specific DNA recognition (Cruickshank et al., 2000). Similar tyrosine residues can be identified in LANA and E2. LANA contacts DNA through several tyrosine residues, and mutation of Y1014 abolishes the episome maintenance function of LANA (Domsic et al., 2013). LANA binding at the TR also results in recombinational structures during G2 phase of the cell cycle, but the specific-structures and DNA organization is different than that of EBNA1 binding at oriP (Dheekollu et al., 2013). E2 appears to also use conserved tyrosine residues to contact DNA, but these may be restricted to subclasses of papillomaviruses (Bussiere et al., 1998; Hegde and Androphy, 1998; McBride, 2013). Thus, while orthologs of EBNA1 may share some of these features, it is likely that they represent specialized activities best adapted to the replication strategies of their virus families.
Tyrosine recombinases
Circular genomes from yeast and bacteria utilize site-specific recombinases to complete replication and enable faithful segregation (Crozat et al., 2014). One of the functions of these recombinases is to reduce the risk of homologous recombination between multi-plasmid systems and suppress the formation of dimers during DNA replication. The yeast 2 micron plasmid encodes a tyrosine recombinase FLP that is required to resolve post-replication genome dimers (Jayaram et al., 2004; McQuaid et al., 2019; Sau et al., 2019). Like FLP, we found that EBNA1 can bind with high-affinity to 4-way junctions and introduce single strand nicks. Some recombinases like XerD require accessory factors like the FtsK translocase to induce an active conformation required for recombinase activity (Bigot et al., 2005). EBNA1 may also require accessory factors, such as replication helicases, that induce DNA conformations suitable for endonuclease activation. EBNA1 may also share some functional features with the HUH site-specific endonuclease superfamily members, such as the AAV Rep protein (Chandler et al., 2013). The AAV Rep 78 protein uses a conserved tyrosine to catalyze a DNA cleavage reaction required for viral integration and resolution of replication termination. While EBNA1 lacks many of the features of these tyrosine recombinases, we suggest that it functions to facilitate replication termination within the repetitive elements of the FR region of oriP and suppress non-homologous recombination that would otherwise lead to genome rearrangements and chromosomal integrations. In this respect, EBNA1 may be considered an anti-integrase.
Essential role in plasmid maintenance
EBNA1 is essential for maintaining the episomal form of the viral genome during latent infection in proliferating cells. EBNA1 is not absolutely essential for immortalization of B-lymphocytes, as lymphoblastoid cells can be generated, albeit at much lower efficiencies, from viral genomes that have integrated into the host genome (Humme et al., 2003). EBNA1 may be dispensable for DNA replication of the viral genome, as cellular replication machinery can initiate at alternative origins of DNA replication within the viral genome (Norio and Schildkraut, 2004; Ott et al., 2011). While oriP is an efficient origin, it is not absolutely required for the DNA replication of the viral genome (Ott et al., 2011). However, EBNA1 binding to oriP is absolutely required for episome maintenance. The primary mechanism of EBNA1-mediated episome maintenance is by tethering the viral genome to the host chromosome. The viral genome is bound through the carboxy-terminal DBD and the host chromosome is bound through the amino-terminal RGG/AT-hook domains (reviewed in (De Leo et al., 2020)). Our findings suggest that EBNA1 performs an additional activity in episome maintenance that is required for efficient resolution and segregation of newly replicated circular viral genomes.
The EBNA1-DNA cross-linking activity may also contribute to the protection of repetitive DNA elements in oriP, especially at the FR where lagging strand synthesis may be particularly vulnerable to secondary DNA structure formation and repeat loss (Fricker and Peters, 2014). EBNA1 cross-linking activity may be particularly important for maintenance of large viral genomes and thus important for viral genome survival during latent infection in proliferating B-cells and EBV-associated tumors. Cellular mechanisms may also provide redundant and alternative mechanisms for segregating viral episomes. We also note that the endonuclease activity could only be reconstituted with the full-length EBNA1 isolated from mammalian cells. This suggest that either additional domains outside of the DBD, post-translational modifications, or associated cellular proteins are required for endonuclease activity. This is also consistent with the cell-cycle dependence of the EBNA1-DNA complex that could be isolated by RADAR assay in latently infected cells. Host cofactors have been identified for many bacteriophage tyrosine recombinases, so there is precedence that EBNA1 may require host factors to generate appropriate DNA substrates and/or complete DNA-crosslinking at oriP (Rajeev et al., 2009).
Implications for drugging EBNA1
EBNA1 is the only viral protein that is consistently expressed in EBV-associated tumors, suggesting that it provides essential activities for tumorigenesis and therefore an attractive target for small molecule inhibition. Our evidence that EBNA1 has tyrosine-dependent single-strand endonuclease activity provides new opportunities for inhibiting EBNA1 function in viral oncogenesis. Inhibitors of topoisomerases, such as etoposide, work by inhibiting the religation of the protein-DNA phosphodiester bond, resulting in the trapping of the protein-DNA adduct (Bax et al., 2019). Inhibitors of PARP1/2 can also trap PARP bound with high affinity to DNA, but this trapping is not through a covalent protein-DNA linkage (Shen et al., 2015). Our findings leave open the possibility that small molecules could be developed to trap EBNA1 bound to DNA and potentially inhibit DNA replication termination and episome maintenance. Such inhibitors may be more potent than recently developed small molecule inhibitors that block EBNA1 binding to DNA (Messick et al., 2019).
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Paul M. Lieberman, Ph.D.
The Wistar Institute, 3601 Spruce Street, Philadelphia, PA 19104
Email: Lieberman@wistar.org
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
This study did not generate any unique datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
The experimental models in this study were human derived cell lines available from ATCC or from the corresponding authors laboratory. All newly generated human lymphoblastoid cell lines were generated from donor derived B-lymphocytes immortalized with recombinant EBV with EBNA1 WT or Y518F. Both male and female donors were included as the source of these LCLs.
METHOD DETAILS
Cell culture and plasmid.
EBV-positive Burkitt lymphoma cell lines RAJI (gift of Diane Hayward, Johns Hopkins University) and EBV bacmid immortalized LCLs (generated at the Wistar Institute) were grown in RPMI medium (Gibco BRL) containing 15% fetal bovine serum, and antibiotics penicillin and streptomycin (50U/ml). 293T (ATCC) cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 100 μg/ml streptomycin, and 100U/ml penicillin. Cells were cultured in an incubator set at 37 °C and 5% CO2. Mammalian expression vector for Flag-EBNA1 contained B95–8 EBNA1 lacking the GA repeats (aa 101–324) under the control of CMV-3XFLAG promoter in a plasmid derived from pREP10 (Clontech) containing, oriP, GFP, and hygromycin resistance (Dheekollu et al., 2017). WT EBNA1 DBD expression construct was made by PCR amplification of EBNA1 DBD (aa 459–607) using B95–8 as a template and cloned into a modified pET DUET vector containing the hexahistidine SUMO protein tag at the N-terminus.
Site-directed mutagenesis.
Primers were designed to generate the mutation (Y518F) in WT EBNA1 DBD and Flag EBNA1 in pRep10 plasmid (ThermoFisher). A two-stage PCR protocol for site-directed mutagenesis was adapted from Stratagene (Wang and Malcolm, 1999). Following DpnI digestion and heat inactivation, PCR products were transformed into DH5α cells. Purified plasmids from colonies were sequenced to confirm the mutation.
Cell Cycle Synchronization.
Cell cycle fractionation using centrifugal elutriation was performed with a modified Beckman JE 5.0 using counter flow rates for RAJI cells as described previously (Ritzi et al., 2003). 293T cells transfected with either WT or Y518F plasmids were synchronized at G1/S phase of the cell cycle with 1ug/ml Aphidicolin overnight, released into S phase and collected as indicated (Zhou et al., 2005).
Cell cycle profile analysis.
To determine the cell cycle profiles of cells, cultures were either treated or left untreated and fixed in ice-cold 70% ethanol for at least 30 min. After fixation, the cells were mixed with staining solution (0.5 mg/ml propidium iodide, 100 mg/ml RNase A) for 30 min in the dark. Samples were analyzed using an EPICS XL (Beckman-Coulter, Inc., Miami, FL), and 50,000 events were recorded. For all flow cytometry experiments, the WINMDI software program (Scripps Institute) was used to analyze the data.
Western blots.
The PVDF membranes were blotted with the following antibodies: anti-β-actin-peroxidase (AC-15) (Sigma-Aldrich, cat no. A3854), anti-EBNA1 mouse monoclonal antibody (Acris, cat no. BM3127), and anti-Flag M2-peroxidase (horseradish peroxidase [HRP]) (Sigma-Aldrich, cat no. A8592), anti-Topoisomerase II rabbit polyclonal antibody (Abcam, cat no. ab52934), anti PCNA rabbit monoclonal antibody (Abcam, cat no. ab92552) and imaging on a Amersham Imager 680.
RADAR assay.
Cell lysis and DNA protein covalent crosslink (DPCC) isolation was performed according to Maizels et al (Kiianitsa and Maizels, 2013, 2014). Cells were washed once in ice cold phosphate buffered saline (PBS) and then resuspended in lysis buffer MB (6 M guanidinium ithiocyanate, 10 mM Tris–HCl (pH 6.5), 20 mM EDTA, 4% Triton X100, 1% Sarkosyl and 1% dithiothreitol) at a ratio of 1 ml MB for every 2 × 106 cells. A total of 20 × 106 cells was used for each cell cycle sample. An aliquot of lysate (10–20%) was saved for analysis of the unfractionated extract, and nucleic acids and DPCC were precipitated from the remainder by addition of 0.5 volume of 100% ethanol followed by centrifugation. The precipitate was washed twice in 75% ethanol and immediately resuspended in freshly prepared 8 mM NaOH. To quantify DNA, a small aliquot was digested with 50 μg/ml proteinase K (Invitrogen) for 3 hours at 50°C and quantified using nanodrop 2000C (Thermo-scientific) according to manufacturer instructions to determine DNA concentration.
Detection of DNA-protein crosslinks detection (DPC).
Specific DPCs were detected using a vacuum Dot-blot manifold (Bio-Rad) or SDS PAGE followed by immunodetection. In brief, equal amounts of DNA were diluted in Tris-buffered saline (TBS; 10 mM Tris pH 7.5, 150 mM NaCl) and applied to either a nitrocellulose (Bio-Rad, Hercules CA) membrane using a vacuum Dot-blot manifold (Bio-Rad) or analyzed on an SDS PAGE and transferred onto a polyvinylidene difluoride (PVDF, Millipore) membrane.
Immunoprecipitation of EBNA1-DNA crosslinks.
Equal amounts of DPCs based on DNA concentration were diluted 10-fold in IP buffer (50 mM Tris pH 7.4, 150 mM NaCl, 10 mM NEM, 0.5% Nonidet P40, 1% Triton X-100, 1 mM EDTA) and 1 mM phenylmethane sulfonyl fluoride (PMSF), Protease inhibitors (Sigma) and Phosphatase inhibitors (Roche) and precleared with Sepharose beads (Millipore) for 1 h. After incubation for 4 h to overnight with Anti-Flag M2 affinity beads (Sigma-Aldrich), complexes were washed twice with IP buffer containing 300 mM NaCl, and twice with IP buffer containing no detergents. Samples were then eluted with 3 x Flag peptide (250 ng/ml) in Elution buffer (25 mM Na-HEPES 100 mM NaCl).
Labeling of EBNA1-DNA crosslinks (EDC).
Kinase assays were performed in 20 μl reaction volume, containing 5 μl EDC ( ~0.5 ng), 2 μl of 10x reaction buffer (700 mM Tris pH 7.6, 100 mM MgCl2, 50 mM dithiothreitol), 5 μl [γ−32P]-ATP, 5 μl H2O, and 3 μl T4 PNK incubated at 37° C for 1 h. The reaction was stopped by adding EDTA to a final concentration of 30 mM. The samples were then run on 10% SDS PAGE and were visualized using a Typhoon 9410 PhosphorImager (Cytiva).
Nuclease digestion of EBNA1-DNA crosslinks.
Nuclease assays were performed in 20 μl reaction volume, containing 10 μl EDC (~ 1ng), 2 μl of 10x reaction buffer specific for each nuclease. P1 nuclease buffer (200 mM acetic acid, 10 mM CaCl2, pH 6.0), 7 μl H2O, and 1 μl (10 Units) P1 nuclease. TDP1 10x buffer (500 mM Tris-HCl (pH 7.5), 800 mM KCl, 20 mM EDTA, 10 mM dithiothreitol (DTT), 400 ug/ml bovine serum albumin (BSA), and 0.1% Tween 20), TDP2 10x buffer (500 mM Tris-HCl ( pH 7.5), 800 mM KCl, 50 mM MgCl2, 1.0 mM EDTA, 10 mM dithiothreitol (DTT), 400 ug/ml BSA, and 0.1% Tween 20). ExoVII 10x buffer (500 mM Tris-HCl (pH 8.0), 500 mM sodium phosphate, 100 mM 2-mercaptoethanol 80 mM EDTA). The reactions were incubated at 37° C for 30 min and then inactivated by the addition of 0.8 μl of 0.5 M EDTA and Laemli buffer. The samples were then run on SDS PAGE and electroblotted to a polyvinylidene difluoride (PVDF, Millipore) membrane and visualized as above.
Chromatin Immunoprecipitation (ChIP).
ChIP assays were performed as previously described (Chau and Lieberman, 2004). Briefly 293T cells were plated and transfected as in the plasmid replication assay with FLAG-EBNA1 WT or the Y518F point mutant. Seven days post transfection, cells were synchronized with aphidicolin and S phase cells were harvested for either native or crosslinked ChIP. TaqMan qPCR performed using primers and probe detecting EBV B95.8 genome coordinates 8401–8521 adjacent to FR was designed by Themo-Fisher. Antibodies used were as follows: mouse anti-IgG (Santa Cruz Biotechnology), Trimethyl-histone H3 (Lys4) (Millipore, cat no. 07–473), anti-flag M2 mouse monoclonal (Sigma Aldrich, cat no. F1804).
EBNA1 DBD purification.
EBNA1 WT or mutant (Y518F, Y518A, Y518E, H468A, N519A) DNA-binding domain (459–607) were expressed as a 6xHis SUMO fusion protein in E. coli (BL21(DE3)) competent cells (NEB) using an autoinduction procedure (Studier, 2005). Briefly, 500 mL of 2X Luria Broth supplemented with 1 mM MgSO4, 1000x trace metals, 50×5052 and 20xNPS were inoculated with 10 ml overnight culture and grown at 25º C for 24–30 hours. Cell pellets were resuspended in 20 mM Tris HCl pH 8.5, 1 M NaCl, 20 mM imidazole, 0.01% Tween 20, 5 mM β-mercaptoethanol, 1 mM PMSF, and 1 mg/ml of lysozyme. Cells were lysed, sonicated, and centrifugation for 40 mins at 16000 rpm in a SS-34 fixed angle Sorvall rotor, lysate was poured over a gravity Nickel-NTA resin column. The resin was washed with 20 mM Tris HCl pH 8.5, 1 M NaCl, 30 mM imidazole, and 5 mM β-mercaptoethanol. Protein was eluted in the presence of wash buffer with 400 mM Imidazole. Eluted protein was concentrated using an Amicon 10 MWCO filter and further purified using a Superose 6 10/300 gel filtration column (Cytiva), which also removed the imidazole. Fractions containing His-SUMO-EBNA1 protein were digested with SUMO protease overnight at 4 °C. Purified tagless protein was obtained by running the cleaved protein over a HisTrap FF prepacked column (1 ml or 5 ml, Cytiva) and collecting the flow through. Protein was concentrated and quantified using Nanodrop spectrophometer (ThermoFisher) at A280 nm with an extinction coefficient of 9.6 (E 1% = 1 mg/ml). Equalized protein concentrations were verified by SDS-PAGE.
Native Electrophoretic Mobility Shift Assays (EMSA).
Purified untagged EBNA1 WT DNA-binding domain (459–607) or its point mutant derivatives were incubated at the indicated concentrations (6.25 nM to 200 nM) with DNA probes (160 femtomoles). Oligonucleotide DNA probes were end-labeled with 32P-γ-ATP and T4 polynucleotide kinase (PNK), and annealed in annealing buffer (50mM Tris HCl (pH 7.5); 400mM NaCl) as described (Rass and West, 2006). Oligonucleotides were purchased from IDT Inc. in standard desalted form. A list of oligonucleotides in provided in Key Resources Table. Binding reactions were incubated for 10 mins at room temperature in 20 μl binding buffer containing 10 mM HEPES pH 7.5, 5 mM MgCl2, 2.5 mM DTT, 0.25% Tween, 200 mM NaCl, and 10% glycerol. DNA-protein complexes were resolved by electrophoresis at 250 Volts in 5% non-denaturing polyacrylamide gels (Bio-rad, 30% Acrylamide/Bis Solution 29:1, 1610156) in 0.5X Tris borate-EDTA buffer. The gels were subjected to autoradiography on X-ray films followed by exposure to phosphorimager screens, scanned in Typhoon, and signals quantified using ImageQuant software.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-EBNA1 | Acris | Cat# BM3127 |
| Anti-Actin | Sigma | Cat# A3854 |
| Anti-Flag HRP | Sigma | Cat# A8592 |
| Anti-TOPO II | Abcam | Cat# ab52934 |
| Anti-PCNA | Abcam | Cat# ab92552 |
| Anti-Cyclin A | Invitrogen | Cat# PA5-99162 |
| Anti-Cyclin E | SCBT Inc | Cat# sc-377100 |
| Ati-CyclinB1 | Invitrogen | Cat# MA5-13128 |
| Anti-pH3S10 | Abcam | Cat# ab5176 |
| Anti-Ebna2 | Millipore | Cat# MABE8 |
| Anti-Lmp1 | Millipore | Cat# MABF2248 |
| Anti-Rpa32 | Abcam | Cat# ab7642 |
| Anti-EAD | Millipore | Cat# MAB8186 |
| Anti-IgG | SCBT Inc | Cat# sc-2025 |
| Anti-flag M2 | Sigma | Cat# F1804 |
| Anti-Trimethyl-histone H3 (Lys4) | Millipore | Cat# 07-473 |
| Anti-Rabbit IgG HRP | BioRad | Cat# 1706515 |
| Anti-Mouse IgG HRP | BioRad | Cat# 1706516 |
| Anti-Zta | In House | N/A |
| Bacterial and Virus Strains | ||
| Max Efficiency DH5a | Invitrogen | Cat# 128258-012 |
| E. coli strain GS1783 | Oesterdider/Cornell | N/A |
| E. coli strain DH10b | Oesterdider/Cornell | N/A |
| EBV B95-8 Bacmid | H.J.Delecluse/DKFZ | doi:10.1073/pnas.95.14.8245 |
| Biological Samples | ||
| ID#599 PBMC | Wistar | N/A |
| ID#180 PBMC | Wistar | N/A |
| ID#367 PBMC | Wistar | N/A |
| ID#296 PBMC | Wistar | N/A |
| ID# 323 PBMC | Wistar | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Dulbecco’s Phosphate buffer saline(PBS) | Corning | 21-031-CM |
| Dulbecco’s Modified Eagle Medium (DMEM) | GIBCO | 2491-015 |
| RPMI Medium 1640 | GIBCO | 11875-085 |
| Fetal Bovine Serum | Hyclone | Cat# SH30396-03 |
| Trypsin | Corning | 25-053-CI |
| Penicillin/streptomycin | Corning | 30-002-CI |
| Primocin | InvivoGen | Cat# ant-pm05 |
| Lipofectamine 2000 | Invitrogen | 11668-019 |
| Hygromycin | Corning | 30-240-CR |
| Protease inhibitors | Sigma | P8340 |
| Phosphatase inhibitors | Roche | 04 906 837 001 |
| Aphidicolin | Sigma | A0781 |
| DTT | Sigma | D9779 |
| Gaunidine thiocyanate | Sigma | G5277 |
| Flag Magnetic beads | Sigma | M8823 |
| EBV-Ebna1 Agarose beads | SBCT | Cat# sc-81581AC |
| Protein A Sepharose | GE Healthcare | Cat# 17513801 |
| Protein G Seoharose | GE Healthcare | Cat#17061801 |
| His Trap FF | GE Healthcare | 17524802 |
| 3XFlag peptide | Sigma | F4799 |
| Tdp1 Human | Prospec | Cat# enz-685-b |
| Tdp2 Human | Prospec | Cat# enz-698-b |
| Proteinase K | Roche | 03115852001 |
| Critical Commercial Assays | ||
| B-cell enrichment Kit II | Stem Cell Tech | Cat#19054 |
| Bacmid purification kit | Clonetech | Cat# 740579 |
| Random Prime DNA labeling Kit | Tankara | Cat# 6045 |
| Custom TaqMan Gene Expression assays | Thermo-Fisher | ID APU667U |
| TaqMan Master Mix | Applied Biosystems | 4324018 |
| PureLink Plasmid Maxiprep Kit | Invitrogen | Cat#20220831 |
| Experimental Models: Cell Lines | ||
| Human: Raji | ATCC | Cat# CRL-7936 |
| Human: 293T | ATCC | Cat# CRL-11268 |
| Human: BCBL | Yan Yuan/UPENN | N/A |
| Marmoset B-cell line (B95-8) | ATCC | Cat# CRL-1612 |
| B95-8 LCL | This paper | N/A |
| Bac Wt LCL | This paper | N/A |
| BacY518F LCL | This paper | N/A |
| Oligonucleotides | ||
| 5’ tct gga tag cat atg cta tcc aga 3’ | DS (1 x binding site; self complementray used in Fig 3) | IDT |
| 5’ attaggatagcctatgctacccagatatagatta ggatagcatatgctacccagatatagagagcgtcacctaagcc 3’ 5’ggcttaggtgacgctctatatctgggtagcata tgctatcctaatctatatctgggtagcataggctatcctaat 3’ |
2X FR | IDT |
| 5’gcagctgacgcgtaattcgatagcatatgcttcccgttggcgctagc 3’ 5’ gctagcgccaacgggaagcatatgcttaggctaattccggactggtcg 3’ 5’ cgaccagtccggaattagccta 3 5’ atcgaattacgcgtcagctgc 3’ |
Y Arc | IDT |
| 5’attaggatagcctatgc 3’ 5’tacccagatatagattaggatagcatatgctacccagatatag 3’ FR3’top 5’ ctatatctgggtagcatatg 3’ 5’ctatcctaatctatatctgggtagcataggctatcctaat 3’ 3’bottom |
Nick DS | IDT |
| 5’ attaggatagcctatgctacccagatatagatta ggatagcatatgctacccagatatagag 3’ 5’ ctctatatctgggtagcatatgctatcctaatctaa attaggatagcatatactaccctatagc 3’ 5’ gctatagggtagtatatgctatcctaatttaaatat gggtagcctaatgcatccaactg 3’ 5’ cagttggatgcattaggctacccata tttatatctgggtagcataggctatcctaat 3’ |
4 WHJ | IDT |
| Recombinant DNA | ||
| pEP-KanS | Oesterdider/Cornell | N/A |
| pET-Duet Sumo EBNA1 DBD Wt | Juliana et al 2018 | N/A |
| pET-Duet Sumo EBNA1 DBD Y518F | This Paper | N/A |
| pET-Duet Sumo EBNA1 DBD Y518A | This Paper | N/A |
| pET-Duet Sumo EBNA1 DBD Y518E | This Paper | N/A |
| pET-Duet Sumo EBNA1 DBD H468A | This Paper | N/A |
| pET-Duet Sumo EBNA1 DBD N519A | This Paper | N/A |
| EBV Rep10 Flag EBNA 1 | Dheekollu et al 2017 | N/A |
| EBV Rep10 Flag Y518F | This Paper | N/A |
| EBV Rep10 Flag Y518A | This Paper | N/A |
| EBV Rep10 Flag H468A | This Paper | N/A |
| EBV Rep10 Flag H468A/Y518F | This Paper | N/A |
| EBV Bac95-8 | Delecluse HJ. | N/A |
| EBV Bac95-8 Y518F | This Paper | N/A |
| Software and Algorithms | ||
| NIS Elements AR | Nikon | Version 5. |
| ImageQuant TL | GE Healthcare | Version 8.2 |
| PRISM | Graph Pad Software | Version 6 |
| Pymol | Schrodinger | https://www.pymol.org/ |
| WINMDI software | Scripps Institute | N/A |
| Other | ||
| Enzymes | ||
| P1 Nuclease | NEB | Cat#M0660S |
| Exonuclease VII | NEB | Cat# M0379S |
| RecJF | NEB | Cat# M0264S |
| Mung bean nuclease | NEB | Cat# M0250S |
| T4PNK | NEB | Cat# M0201S |
| DpnI | NEB | R0176L |
| BamHI | NEB | R3136L |
| Dra1 | NEB | R0129L |
| DNase I | Roche | Cat# 04-716728-001 |
| RNaseA | Invitrogen | Cat# 12091-021 |
| Benzonase | Millipore | Cat# 71206-3 |
SDS-EMSA.
In SDS-EMSA experiments, subsequent to DNA-protein complex formation, SDS was added to 0.1% final concentration and incubated at room temperature for 10 min. The reaction mixtures were then electrophoresed on 5% denaturing polyacrylamide gels containing 0.1% SDS in 0.5X Tris borate-EDTA-0.1% SDS buffer at 300 Volts in cold room.
Plasmid replication assays.
Plasmid DNA replication assays have been described previously (Deng et al., 2003; Dheekollu et al., 2017). Briefly, 293T (~1 × 106 cells) were plated in 10 cm dishes. 24 h later cells were transfected with Lipofectamine 2000 (12 μl, Invitrogen) and 4 μg oriP plasmids expressing either FLAG-B95–8 EBNA, Y518F, or control plasmid containing oriP alone. Cells were split after 48 h, and then harvested at 72 h post transfection for both episomal DNA and protein. Episomal DNA was extracted by Hirt Lysis (Hirt, 1967). The DNA pellets were dissolved in 150 μl of 10 mM Tris HCl, 1 mM EDTA buffer (pH 7.6) and 15 μl was subjected to restriction digestion with BamHI alone and 135 μl was subjected to BamHI and DpnI digestion overnight at 37° C. Pure DNA was extracted with phenol: chloroform (1:1), precipitated, and electrophoresed on a 0.9% agarose gel and transferred to a nylon membrane (PerkinElmer) for Southern blotting. Blots were visualized and quantified using a Typhoon 9410 PhosphorImager (Cytiva).
Plasmid maintenance assays.
Plasmid maintenance assays have been described previously (Dheekollu et al., 2017). Briefly, 293T cells were seeded in 6 well plates at concentrations of 0.75–1.5 × 105 cells/ml. Twenty-four hours later, cells were transfected with Lipofectamine 2000 (10 μl) and 2 μg oriP plasmids expressing CMV promoter-FLAG-EBNA1, with either WT or Y518F EBNA1, as indicated. Two or three days post transfection an equal portion of the cells were split into 15 cm plates to continue passaging and the remaining cells were harvested for Hirt Lysis and Western blot analysis. Isolated DNA was subjected to BamHI digestion and resolved on 0.9% agarose gels. Southern blotting was performed as described above. After the first passage, 100 μg/ml hygromycin was added for 3–5 days. After initial selection and first passage, cells were continuously split upon reaching confluency for an additional 20–30 days without selection.
Colony formation assay.
As described previously (Dheekollu et al., 2017), 293T cells were co-transfected with 10 μg of oriP plasmids (REP10) expressing WT or Y518F EBNA1. The cells were split after 24 h and selected with 0.5 μg/ml of hygromycin. After 2 weeks of selection, the resulting hygromycin-resistant colonies were stained with toluidine blue (0.5 mg of toluidine blue in 2% sodium carbonate). Colonies that were at least 2 mm in size were scored as positive. Colonies were counted using a colony counting macro written with NIH Image.
Flag-EBNA1 purification.
293T cells were transfected with either FLAG-EBNA1 WT or Y518F containing oriP plasmid and cells were collected after 7 days post-transfection and washed once in 1X PBS. Cells (~108) were lysed in 50 ml of Lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 05% Nonidet P40, 1 mM EDTA), 1 mM PMSF containing protease inhibitors (Sigma, cat no. P8340) and Phosphatase inhibitors (Roche, cat no. 4906837001). Lysate were spun at 16000 for 10 min and immunoprecipitated with 100 μl of anti-Flag resin (Sigma, cat no. M8823). Complexes were washed with either lysis buffer or Cleavage Buffer (50 mM Tris pH 8.0, 5 mM MgCl2, 40 mM NaCl, 1 mM DTT, 100 mg/ml BSA), 1 mM PMSF containing protease inhibitors (Sigma, cat no. P8340) and Phosphatase inhibitors (Roche, cat no. 4906837001), and eluted with Flag peptide.
Recombinant EBV genomes.
Recombinant EBV B95–8 genome carries the prokaryotic F factor origin of replication, the green fluorescent protein (GFP) gene, the chloramphenicol (CAM) resistance gene, and the hygromycin (HYG) resistance gene as described previously (Delecluse et al., 2008; Delecluse and Hammerschmidt, 2000). The B95–8/Y518F EBNA1 genome was obtained by homologous recombination exchanging the DNA binding domain contain Y518F mutation in EBNA1 gene using two-step recombination method (Tischer et al., 2006). Positive selected clones were purified and analyzed with the BamHI restriction enzyme to confirm correct recombination, which was further confirmed by Sanger sequencing.
Bacmid (Bac) genome maintenance assays.
Bac maintenance assay was similar to plasmid maintenance assay with the following modifications: Briefly, 293T cells were seeded in 6 well plates at concentrations of 0.75–1.5 × 105 cells/ml. Twenty-four hours later cells were transfected with Lipofectamine 2000 (10 μl) and 1 mg Bac B95–8 with WT or Y518F EBNA1 genes, as indicated. Two to three days post transfection an equal portion of the cells were split into 15 cm plates to continue passaging and the remaining cells were harvested Bac DNA analysis. Isolated DNA was subjected to BamHI digestion and resolved on 0.9% agarose gels. Southern blotting was performed as described above with TR DNA probe. After the first passage, 100 μg/ml hygromycin was added for 21 days and harvested the plates for Bac DNA Southern analysis with either TR, FR or human ch17 probes.
Virus producer cell lines.
EBV producer cell lines and virus were generated as previously described (Shannon-Lowe et al., 2005). Briefly, rB95–8 and B95–8/Y518FEBNA1 Bac DNA was purified from 4 ml of bacterial culture according to Bacmid purification kit (NucleoBond 740579, Clonetech) and the DNA was transfected into HEK293 cells (1 mg DNA per 4×105 cells) using lipofectamine 2000 (Invitrogen). Cells were seeded onto 150 mm culture plates in RPMI supplemented with 10% FBS and hygromycin (100 mg/ml). Eight GFP-positive colonies were expanded four weeks post transfection and tested for their ability to produce virus upon induction of the lytic cycle (Western blot, qPCR, and PFGE analysis) and selected for viral production.
Recombinant virus production.
Recombinant EBV was produced as described previously (Shannon-Lowe et al., 2005). Briefly, 293 producer cells for rB95–8 or B95–8/Y518FEBNA1 were induced by transfection of a BZLF1 expression plasmid with co-transfection of BALF4 expression plasmid. The supernatants were collected 4 days post-transfection and filtered through a 0.45 mm filter to remove the cell debris. To concentrate virus, the supernatant was layered onto a 10% sucrose cushion and spin at 26,000 rpm for 1 h and virus pellet was resuspended in RPMI medium. Viral gene equivalents were determined by qPCR and by infection of 105 Raji B cells with concentrated virus stock using quantitation methods described (Tsai et al., 2013).
Primary B cell infections and colony formation assays.
B cells were purified from PBMCs (Peripheral blood mononuclear cells) using B-cell enrichment Kit II (Stem Cell Technologies, cat no.19054;). 2 ×106 B-cells were infected with 10 infectious particle units of recombinant virus at room temperature. After 2 weeks post infection, B-cell colonies were identified and counted by light microscope using a colony counting macro written for NIH Image.
Analysis of replication DNA structures.
DNA for the two-dimensional (2D) gels was extracted by the modified Hirt’s method as previously described (Follonier and Lopes, 2014). Two-dimensional gel electrophoresis. was performed essentially as described previously (Willis et al., 2014). For 2D gels, DNA was cut with DraI restriction enzyme to generate an ~ 5.6-kb fragment containing OriP. Neutral/alkaline (N/A) electrophoresis, as described by (Huberman et al., 1987), was used to detect replication pausing on nascent strands. The first-dimension conditions were identical to the N/N 2D gel method, while the second dimension gel was with 0.9% agarose in water. The solidified gels were placed in electrophoresis apparatus containing alkaline electrophoresis buffer, 50 mM NaOH, 2 mM EDTA, and incubated at room temperature for 1 hr. Electrophoresis was performed at 0.7 V/cm for 20 hr. DNA in both methods was then detected by hybridization with α−32P-labeled probe specific for the oriP region as above.
Endonuclease cleavage assay.
Cleavage assays were carried out in 20 μl cleavage buffer (50 mM Tris-HCl (pH 8.0), 5 mM MgCl2, 40 mM NaCl, 1 mM DTT, 100 mg/ml BSA) containing ~1 nM of the 5′−32P-end-labeled DNA substrates with either ~500 ng of WT or Y518F Flag EBNA1 proteins. After incubation at 37°C for 30 min, cleaved DNA products were deproteinized by addition of 5 μl of stop buffer (100 mM Tris-HCl pH 7.5, 50 mM EDTA, 2.5% SDS and 10 mg/ml proteinase K) and incubation for 30 min at 37°C. The radiolabeled products were then resuspended in formamide-dye loading buffer for electrophoresis on 8 M Urea-6% PAGE denaturing gels and analyzed as above.
QUANTIFICATION AND STATISTICAL ANALYSIS
All biological experiments were repeated at least 3 times (n=3) and data was analyzed for error variation by standard deviation from the mean (sdm) and student two-tailed t-test for p-values using Exel and PRISM GraphPad software.
Supplementary Material
A) RADAR-IP from RAJI showing slower migrating species associated with native EBNA1. B) RADAR-IP with FLAG-EBNA1 was purified and incubated with 100 nM TDP1 or TDP2 and assayed by Western blot with FLAG-EBNA1 antibody. C) Quantification of unmodified (free) EBNA1 after TDP1 or TDP2, as represented in panel B. * p<.05, Student t-test, n=3. D) Quantification of unmodified EBNA1 after P1 nuclease of RADAR-IP, as represented in Fig. 2D. E) Western blot of RADAR-IP with FLAG-EBNA1 and nucleases ExoVII, RecJF, Mung Bean nuclease, Benzonase, DNase, RNase, as indicated above each lane. Arrows indicate slower mobility, modified EBNA1. F) Quantification of unmodified (Free) EBNA1 for nucleases shown as representative in panel E. *p<.05, Student t-test, n=3.
{kind=link}
A) Transient DNA replication assay for oriP plasmids lacking EBNA1 (−) or expressing WT or Y518F EBNA1 in 293T cells. Plasmids were isolated by Hirt lysis at 72 hrs post-transfection and cut with BamHI (left) or BamHI/DpnI (right). B) Quantification of transient DNA replication assays representative shown in panel A. Replication efficiency is the ratio of DpnI/BamHI to BamHI DNA. C) Western blot of cells transfected with oriP plasmid -EBNA1, WT, or Y518F probed for EBNA1 and β-Actin.
{kind=link}
A) oriP plasmids expressing EBNA1 WT or Y518F were transfected into 293T cells and assayed for episome maintenance at 7, 14, 17, 21, or 24 day post-transfection. Western blot of FLAG-EBNA1 or Actin (left panel). Southern blot of recovered oriP plasmid (right panel). C) Quantification of Southern blot by densitometry. D) EBV protein expression was assayed by Western blot for EBNA1 WT or Y518F LCLs, or control sample from MutuI treated with TPA and sodium butyrate (NaB) probed. Proteins were assayed for EBNA1, EBNA2, LMP1, EA-D, Zta, and β-Actin.
{kind=link}
A) Analysis of oriP DNA structures by 2D Alkaline-Agarose Gel Electrophoresis. Schematic of DNA replication structures including replication pause sites and their predicted positions in 2D alkaline-agarose gel. B) EBNA1 WT (top row) or Y518F (lower row) was isolated at various stages of S/G2 phase as indicated by 2.15, 2.30, 2.45, or 3.0 hrs post-release from S-phase block with aphidicolin. C) Western blot analysis of FLAG-tagged EBNA1 WT, Y518F, Y518A, Y518E, and H468A purified from 293T cells probed with α-FLAG (left panel), α-EBNA1, or stained with Coomassie blue (right panel). D) EMSA of FLAG-EBNA1 proteins shown in panel A with the 2xFR probe with (+) or without (−) 0.1% SDS added into the binding reaction.
{kind=link}
Cleavage assay for 2xFR or 4WJ probes with either no protein, FLAG-EBNA1 WT, or Y518F. Reactions were subjected to electrophoresis on denaturing 7M urea polyacrylamide electrophoresis. Lanes marked SS denote single strand probe alone. Arrows indicate major cleavage products.
{kind=link}
A) Probes used for EMSA and endonuclease cleavage assays as shown in Figs 8 and 9. B) Analysis of radiolabeled probes by PAGE. C) DNA oligonucleotide probes for replication fork Y-structure (Y), nicked double stranded DNA (N), or 4-way Holliday junction (4WJ). D) Native EMSA with either bacterial DBD or FLAG-EBNA1 derived from 293T cells (FL) for WT or Y518F. All samples were tested with (+) or without (−) addition of 0.1% SDS in binding reaction. E) Same samples shown in panel B were assayed by SDS-EMSA at room temperature followed by autoradiography. SDS-resistant EBNA1-DNA adducts are indicated by red arrows and arrowheads.
{kind=link}
Highlights.
EBNA1 is required for episome maintenance of oncogenic Epstein-Barr Virus
EBNA1 forms cell cycle-dependent covalent DNA-crosslinks with oriP
EBNA1 Y518F disrupts DNA cross-linking and EBV episome maintenance
EBNA1 binding induces site-specific endonuclease activity at oriP
ACKNOWLEDGEMENTS
We thank members of the Lieberman lab for their helpful comments, and the Imaging and Genomics Shared Resources Facilities for their technical support. This work was funded with grants from NIH RO1 CA093606, RO1 DE017336, P30 CA010815, and the Wistar Training Grant T32 CA09171 (JSD).
Abbreviations List:
- EBV
Epstein-Barr Virus
- EBNA1
Epstein-Barr Nuclear Antigen 1
- DBD
DNA binding domain
Footnotes
Declaration of Interests
P.M.L. is a founder and consultant for Vironika, LLC.
J.S.D. is currently employed at GlaxoSmithKline.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Ascherio A, and Munger KL (2015). EBV and Autoimmunity. Curr Top Microbiol Immunol 390, 365–385. [DOI] [PubMed] [Google Scholar]
- Bastia D, and Zaman S. (2014). Mechanism and physiological significance of programmed replication termination. Semin Cell Dev Biol 30, 165–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bax BD, Murshudov G, Maxwell A, and Germe T. (2019). DNA Topoisomerase Inhibitors: Trapping a DNA-Cleaving Machine in Motion. J Mol Biol 431, 3427–3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigot S, Saleh OA, Lesterlin C, Pages C, El Karoui M, Dennis C, Grigoriev M, Allemand JF, Barre FX, and Cornet F. (2005). KOPS: DNA motifs that control E. coli chromosome segregation by orienting the FtsK translocase. EMBO J 24, 3770–3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochkarev A, Barwell JA, Pfuetzner RA, Bochkareva E, Frappier L, and Edwards AM (1996). Crystal structure of the DNA-binding domain of the Epstein-Barr virus origin-binding protein, EBNA1, bound to DNA. Cell 84, 791–800. [DOI] [PubMed] [Google Scholar]
- Bochkarev A, Barwell JA, Pfuetzner RA, Furey W Jr., Edwards AM, and Frappier L. (1995). Crystal structure of the DNA-binding domain of the Epstein-Barr virus origin-binding protein EBNA 1. Cell 83, 39–46. [DOI] [PubMed] [Google Scholar]
- Bussiere DE, and Bastia D. (1999). Termination of DNA replication of bacterial and plasmid chromosomes. Mol Microbiol 31, 1611–1618. [DOI] [PubMed] [Google Scholar]
- Bussiere DE, Kong X, Egan DA, Walter K, Holzman TF, Lindh F, Robins T, and Giranda VL (1998). Structure of the E2 DNA-binding domain from human papillomavirus serotype 31 at 2.4 A. Acta Crystallogr D Biol Crystallogr 54, 1367–1376. [DOI] [PubMed] [Google Scholar]
- Castillo F, Benmohamed A, and Szatmari G. (2017). Xer Site Specific Recombination: Double and Single Recombinase Systems. Front Microbiol 8, 453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler M, de la Cruz F, Dyda F, Hickman AB, Moncalian G, and Ton-Hoang B. (2013). Breaking and joining single-stranded DNA: the HUH endonuclease superfamily. Nat Rev Microbiol 11, 525–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chau CM, and Lieberman PM (2004). Dynamic chromatin boundaries delineate a latency control region of Epstein-Barr virus. J Virol 78, 12308–12319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crozat E, Fournes F, Cornet F, Hallet B, and Rousseau P. (2014). Resolution of Multimeric Forms of Circular Plasmids and Chromosomes. Microbiol Spectr 2. [DOI] [PubMed] [Google Scholar]
- Cruickshank J, Shire K, Davidson AR, Edwards AM, and Frappier L. (2000). Two domains of the epstein-barr virus origin DNA-binding protein, EBNA1, orchestrate sequence-specific DNA binding. J Biol Chem 275, 22273–22277. [DOI] [PubMed] [Google Scholar]
- De Leo A, Calderon A, and Lieberman PM (2020). Control of Viral Latency by Episome Maintenance Proteins. Trends Microbiol 28, 150–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deakyne JS, Malecka KA, Messick TE, and Lieberman PM (2017). Structural and Functional Basis for an EBNA1 Hexameric Ring in Epstein-Barr Virus Episome Maintenance. J Virol 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delecluse HJ, Feederle R, Behrends U, and Mautner J. (2008). Contribution of viral recombinants to the study of the immune response against the Epstein-Barr virus. Semin Cancer Biol 18, 409–415. [DOI] [PubMed] [Google Scholar]
- Delecluse HJ, and Hammerschmidt W. (2000). The genetic approach to the Epstein-Barr virus: from basic virology to gene therapy. Mol Pathol 53, 270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Z, Atanasiu C, Burg JS, Broccoli D, and Lieberman PM (2003). Telomere repeat binding factors TRF1, TRF2, and hRAP1 modulate replication of Epstein-Barr virus OriP. J Virol 77, 11992–12001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar SK, Yoshida K, Machida Y, Khaira P, Chaudhuri B, Wohlschlegel JA, Leffak M, Yates J, and Dutta A. (2001). Replication from oriP of Epstein-Barr virus requires human ORC and is inhibited by geminin. Cell 106, 287–296. [DOI] [PubMed] [Google Scholar]
- Dhar V, and Schildkraut CL (1991). Role of EBNA-1 in arresting replication forks at the Epstein-Barr virus oriP family of tandem repeats. Mol Cell Biol 11, 6268–6278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dheekollu J, Chen HS, Kaye KM, and Lieberman PM (2013). Timeless-dependent DNA replication-coupled recombination promotes Kaposi’s Sarcoma-associated herpesvirus episome maintenance and terminal repeat stability. J Virol 87, 3699–3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dheekollu J, Deng Z, Wiedmer A, Weitzman MD, and Lieberman PM (2007). A role for MRE11, NBS1, and recombination junctions in replication and stable maintenance of EBV episomes. PLoS One 2, e1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dheekollu J, and Lieberman PM (2011). The replisome pausing factor Timeless is required for episomal maintenance of latent Epstein-Barr virus. J Virol 85, 5853–5863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dheekollu J, Malecka K, Wiedmer A, Delecluse HJ, Chiang AK, Altieri DC, Messick TE, and Lieberman PM (2017). Carcinoma-risk variant of EBNA1 deregulates Epstein-Barr Virus episomal latency. Oncotarget 8, 7248–7264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dheekollu J, Wiedmer A, Hayden J, Speicher D, Gotter AL, Yen T, and Lieberman PM (2011). Timeless links replication termination to mitotic kinase activation. PLoS One 6, e19596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domsic JF, Chen HS, Lu F, Marmorstein R, and Lieberman PM (2013). Molecular basis for oligomeric-DNA binding and episome maintenance by KSHV LANA. PLoS Pathog 9, e1003672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards AM, Bochkarev A, and Frappier L. (1998). Origin DNA-binding proteins. Curr Opin Struct Biol 8, 49–53. [DOI] [PubMed] [Google Scholar]
- Enemark EJ, Stenlund A, and Joshua-Tor L. (2002). Crystal structures of two intermediates in the assembly of the papillomavirus replication initiation complex. EMBO J 21, 1487–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermakova OV, Frappier L, and Schildkraut CL (1996). Role of the EBNA-1 protein in pausing of replication forks in the Epstein-Barr virus genome. J Biol Chem 271, 33009–33017. [DOI] [PubMed] [Google Scholar]
- Follonier C, and Lopes M. (2014). Combined bidimensional electrophoresis and electron microscopy to study specific plasmid DNA replication intermediates in human cells. Methods Mol Biol 1094, 209–219. [DOI] [PubMed] [Google Scholar]
- Frappier L. (2015). Ebna1. Curr Top Microbiol Immunol 391, 3–34. [DOI] [PubMed] [Google Scholar]
- Fricker AD, and Peters JE (2014). Vulnerabilities on the lagging-strand template: opportunities for mobile elements. Annu Rev Genet 48, 167–186. [DOI] [PubMed] [Google Scholar]
- Hegde RS, and Androphy EJ (1998). Crystal structure of the E2 DNA-binding domain from human papillomavirus type 16: implications for its DNA binding-site selection mechanism. J Mol Biol 284, 1479–1489. [DOI] [PubMed] [Google Scholar]
- Hegde RS, Grossman SR, Laimins LA, and Sigler PB (1992). Crystal structure at 1.7 A of the bovine papillomavirus-1 E2 DNA-binding domain bound to its DNA target. Nature 359, 505–512. [DOI] [PubMed] [Google Scholar]
- Hellert J, Weidner-Glunde M, Krausze J, Lunsdorf H, Ritter C, Schulz TF, and Luhrs T. (2015). The 3D structure of Kaposi sarcoma herpesvirus LANA C-terminal domain bound to DNA. Proc Natl Acad Sci U S A 112, 6694–6699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellert J, Weidner-Glunde M, Krausze J, Richter U, Adler H, Fedorov R, Pietrek M, Ruckert J, Ritter C, Schulz TF, et al. (2013). A structural basis for BRD2/4-mediated host chromatin interaction and oligomer assembly of Kaposi sarcoma-associated herpesvirus and murine gammaherpesvirus LANA proteins. PLoS Pathog 9, e1003640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirt B. (1967). Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol 26, 365–369. [DOI] [PubMed] [Google Scholar]
- Huberman JA, Spotila LD, Nawotka KA, el-Assouli SM, and Davis LR (1987). The in vivo replication origin of the yeast 2 microns plasmid. Cell 51, 473–481. [DOI] [PubMed] [Google Scholar]
- Humme S, Reisbach G, Feederle R, Delecluse HJ, Bousset K, Hammerschmidt W, and Schepers A. (2003). The EBV nuclear antigen 1 (EBNA1) enhances B cell immortalization several thousandfold. Proc Natl Acad Sci U S A 100, 10989–10994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaram M, Mehta S, Uzri D, Voziyanov Y, and Velmurugan S. (2004). Site-specific recombination and partitioning systems in the stable high copy propagation of the 2-micron yeast plasmid. Prog Nucleic Acid Res Mol Biol 77, 127–172. [DOI] [PubMed] [Google Scholar]
- Kiianitsa K, and Maizels N. (2013). A rapid and sensitive assay for DNA-protein covalent complexes in living cells. Nucleic Acids Res 41, e104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiianitsa K, and Maizels N. (2014). Ultrasensitive isolation, identification and quantification of DNA-protein adducts by ELISA-based RADAR assay. Nucleic Acids Res 42, e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight JD, Li R, and Botchan M. (1991). The activation domain of the bovine papillomavirus E2 protein mediates association of DNA-bound dimers to form DNA loops. Proc Natl Acad Sci U S A 88, 3204–3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner SE, and Sugden B. (2007). The plasmid replicon of Epstein-Barr virus: mechanistic insights into efficient, licensed, extrachromosomal replication in human cells. Plasmid 58, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malecka KA, Dheekollu J, Deakyne JS, Wiedmer A, Ramirez UD, Lieberman PM, and Messick TE (2019). Structural Basis for Cooperative Binding of EBNA1 to the Epstein-Barr Virus Dyad Symmetry Minimal Origin of Replication. J Virol 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride AA (2013). The papillomavirus E2 proteins. Virology 445, 57–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuaid ME, Mereshchuk A, and Dobson MJ (2019). Insights into the DNA sequence elements required for partitioning and copy number control of the yeast 2-micron plasmid. Curr Genet 65, 887–892. [DOI] [PubMed] [Google Scholar]
- Messick TE, Smith GR, Soldan SS, McDonnell ME, Deakyne JS, Malecka KA, Tolvinski L, van den Heuvel APJ, Gu BW, Cassel JA, et al. (2019). Structure-based design of small-molecule inhibitors of EBNA1 DNA binding blocks Epstein-Barr virus latent infection and tumor growth. Sci Transl Med 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midonet C, and Barre FX (2014). Xer Site-Specific Recombination: Promoting Vertical and Horizontal Transmission of Genetic Information. Microbiol Spectr 2. [DOI] [PubMed] [Google Scholar]
- Neylon C, Kralicek AV, Hill TM, and Dixon NE (2005). Replication termination in Escherichia coli: structure and antihelicase activity of the Tus-Ter complex. Microbiol Mol Biol Rev 69, 501–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura M, Wang J, Wakata A, Sakamoto K, and Mori Y. (2017). Crystal Structure of the DNA-Binding Domain of Human Herpesvirus 6A Immediate Early Protein 2. J Virol 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norio P, and Schildkraut CL (2004). Plasticity of DNA replication initiation in Epstein-Barr virus episomes. PLoS Biol 2, e152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norseen J, Thomae A, Sridharan V, Aiyar A, Schepers A, and Lieberman PM (2008). RNA-dependent recruitment of the origin recognition complex. EMBO J 27, 3024–3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott E, Norio P, Ritzi M, Schildkraut C, and Schepers A. (2011). The dyad symmetry element of Epstein-Barr virus is a dominant but dispensable replication origin. PLoS One 6, e18609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulter RTM, and Butler MI (2015). Tyrosine Recombinase Retrotransposons and Transposons. Microbiol Spectr 3, MDNA3–0036-2014. [DOI] [PubMed] [Google Scholar]
- Rajeev L, Malanowska K, and Gardner JF (2009). Challenging a paradigm: the role of DNA homology in tyrosine recombinase reactions. Microbiol Mol Biol Rev 73, 300–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rass U, and West SC (2006). Synthetic junctions as tools to identify and characterize Holliday junction resolvases. Methods Enzymol 408, 485–501. [DOI] [PubMed] [Google Scholar]
- Rawlins DR, Milman G, Hayward SD, and Hayward GS (1985). Sequence-specific DNA binding of the Epstein-Barr virus nuclear antigen (EBNA-1) to clustered sites in the plasmid maintenance region. Cell 42, 859–868. [DOI] [PubMed] [Google Scholar]
- Ritzi M, Tillack K, Gerhardt J, Ott E, Humme S, Kremmer E, Hammerschmidt W, and Schepers A. (2003). Complex protein-DNA dynamics at the latent origin of DNA replication of Epstein-Barr virus. J Cell Sci 116, 3971–3984. [DOI] [PubMed] [Google Scholar]
- Sau S, Ghosh SK, Liu YT, Ma CH, and Jayaram M. (2019). Hitchhiking on chromosomes: A persistence strategy shared by diverse selfish DNA elements. Plasmid 102, 19–28. [DOI] [PubMed] [Google Scholar]
- Shannon-Lowe C, Baldwin G, Feederle R, Bell A, Rickinson A, and Delecluse HJ (2005). Epstein-Barr virus-induced B-cell transformation: quantitating events from virus binding to cell outgrowth. J Gen Virol 86, 3009–3019. [DOI] [PubMed] [Google Scholar]
- Shannon-Lowe C, and Rickinson A. (2019). The Global Landscape of EBV-Associated Tumors. Front Oncol 9, 713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Aoyagi-Scharber M, and Wang B. (2015). Trapping Poly(ADP-Ribose) Polymerase. J Pharmacol Exp Ther 353, 446–457. [DOI] [PubMed] [Google Scholar]
- Studier FW (2005). Protein production by auto-induction in high density shaking cultures. Protein Expr Purif 41, 207–234. [DOI] [PubMed] [Google Scholar]
- Tischer BK, von Einem J, Kaufer B, and Osterrieder N. (2006). Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40, 191–197. [DOI] [PubMed] [Google Scholar]
- Tsai MH, Raykova A, Klinke O, Bernhardt K, Gartner K, Leung CS, Geletneky K, Sertel S, Munz C, Feederle R, et al. (2013). Spontaneous lytic replication and epitheliotropism define an Epstein-Barr virus strain found in carcinomas. Cell Rep 5, 458–470. [DOI] [PubMed] [Google Scholar]
- Wang W, and Malcolm BA (1999). Two-stage PCR protocol allowing introduction of multiple mutations, deletions and insertions using QuikChange Site-Directed Mutagenesis. Biotechniques 26, 680–682. [DOI] [PubMed] [Google Scholar]
- Willis NA, Chandramouly G, Huang B, Kwok A, Follonier C, Deng C, and Scully R. (2014). BRCA1 controls homologous recombination at Tus/Ter-stalled mammalian replication forks. Nature 510, 556–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young LS, Yap LF, and Murray PG (2016). Epstein-Barr virus: more than 50 years old and still providing surprises. Nat Rev Cancer 16, 789–802. [DOI] [PubMed] [Google Scholar]
- Zhou J, Chau CM, Deng Z, Shiekhattar R, Spindler MP, Schepers A, and Lieberman PM (2005). Cell cycle regulation of chromatin at an origin of DNA replication. EMBO J 24, 1406–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A) RADAR-IP from RAJI showing slower migrating species associated with native EBNA1. B) RADAR-IP with FLAG-EBNA1 was purified and incubated with 100 nM TDP1 or TDP2 and assayed by Western blot with FLAG-EBNA1 antibody. C) Quantification of unmodified (free) EBNA1 after TDP1 or TDP2, as represented in panel B. * p<.05, Student t-test, n=3. D) Quantification of unmodified EBNA1 after P1 nuclease of RADAR-IP, as represented in Fig. 2D. E) Western blot of RADAR-IP with FLAG-EBNA1 and nucleases ExoVII, RecJF, Mung Bean nuclease, Benzonase, DNase, RNase, as indicated above each lane. Arrows indicate slower mobility, modified EBNA1. F) Quantification of unmodified (Free) EBNA1 for nucleases shown as representative in panel E. *p<.05, Student t-test, n=3.
A) Transient DNA replication assay for oriP plasmids lacking EBNA1 (−) or expressing WT or Y518F EBNA1 in 293T cells. Plasmids were isolated by Hirt lysis at 72 hrs post-transfection and cut with BamHI (left) or BamHI/DpnI (right). B) Quantification of transient DNA replication assays representative shown in panel A. Replication efficiency is the ratio of DpnI/BamHI to BamHI DNA. C) Western blot of cells transfected with oriP plasmid -EBNA1, WT, or Y518F probed for EBNA1 and β-Actin.
A) oriP plasmids expressing EBNA1 WT or Y518F were transfected into 293T cells and assayed for episome maintenance at 7, 14, 17, 21, or 24 day post-transfection. Western blot of FLAG-EBNA1 or Actin (left panel). Southern blot of recovered oriP plasmid (right panel). C) Quantification of Southern blot by densitometry. D) EBV protein expression was assayed by Western blot for EBNA1 WT or Y518F LCLs, or control sample from MutuI treated with TPA and sodium butyrate (NaB) probed. Proteins were assayed for EBNA1, EBNA2, LMP1, EA-D, Zta, and β-Actin.
A) Analysis of oriP DNA structures by 2D Alkaline-Agarose Gel Electrophoresis. Schematic of DNA replication structures including replication pause sites and their predicted positions in 2D alkaline-agarose gel. B) EBNA1 WT (top row) or Y518F (lower row) was isolated at various stages of S/G2 phase as indicated by 2.15, 2.30, 2.45, or 3.0 hrs post-release from S-phase block with aphidicolin. C) Western blot analysis of FLAG-tagged EBNA1 WT, Y518F, Y518A, Y518E, and H468A purified from 293T cells probed with α-FLAG (left panel), α-EBNA1, or stained with Coomassie blue (right panel). D) EMSA of FLAG-EBNA1 proteins shown in panel A with the 2xFR probe with (+) or without (−) 0.1% SDS added into the binding reaction.
Cleavage assay for 2xFR or 4WJ probes with either no protein, FLAG-EBNA1 WT, or Y518F. Reactions were subjected to electrophoresis on denaturing 7M urea polyacrylamide electrophoresis. Lanes marked SS denote single strand probe alone. Arrows indicate major cleavage products.
A) Probes used for EMSA and endonuclease cleavage assays as shown in Figs 8 and 9. B) Analysis of radiolabeled probes by PAGE. C) DNA oligonucleotide probes for replication fork Y-structure (Y), nicked double stranded DNA (N), or 4-way Holliday junction (4WJ). D) Native EMSA with either bacterial DBD or FLAG-EBNA1 derived from 293T cells (FL) for WT or Y518F. All samples were tested with (+) or without (−) addition of 0.1% SDS in binding reaction. E) Same samples shown in panel B were assayed by SDS-EMSA at room temperature followed by autoradiography. SDS-resistant EBNA1-DNA adducts are indicated by red arrows and arrowheads.
Data Availability Statement
This study did not generate any unique datasets or code.