

3.1. Overview
The pancreas is a glandular organ responsible for diverse homeostatic functions, including hormone production from the endocrine islet cells to regulate blood sugar levels and enzyme secretion from the exocrine acinar cells to facilitate food digestion. These pancreatic functions are essential for life; therefore, preserving pancreatic function is of utmost importance. Pancreas dysfunction can arise either from developmental disorders or adult onset disease, both of which are caused by defects in shared molecular pathways. In this chapter, we discuss what is known about the molecular mechanisms controlling pancreas development, how disruption of these mechanisms can lead to developmental defects and disease, and how essential pancreas functions can be modeled using human pluripotent stem cells. At the core of understanding of these molecular processes are animal model studies that continue to be essential for elucidating the mechanisms underlying human pancreatic functions and diseases.
3.2. Introduction
The pancreas is a multipurpose glandular organ consisting of exocrine acinar cells dedicated to secreting digestive enzymes and endocrine islet cells that produce critical hormones to regulate glucose homeostasis. Both of these functions can be disrupted by genetic mutations that lead to a wide range of developmental defects and postnatal complications that can drastically affect the life of afflicted individuals. The most severe developmental defects are often incompatible with life unless treated at birth, whereas subtler defects are often not manifested until adulthood. One of the more common diseases associated with pancreatic defects is diabetes mellitus, which is characterized by an inability to regulate blood glucose levels, leading to hyperglycemia. Over the years, significant effort and resources have been dedicated to elucidating the etiology of pancreatic diseases, including understanding the underlying congenital defects. To develop treatments for diseases of the pancreas, including diabetes, it will be important to characterize the development of the organ and determine how the highly specialized pancreatic cell types are specified and function. A complete understanding of pancreas development and function will help identify genetic risk factors and facilitate the implementation of improved therapies that could eventually cure rare and common diseases of the pancreas. Furthermore, developmental studies in animal models have greatly informed efforts to directly differentiate functional islet cell populations from human stem cells in vitro. In this chapter, we will predominantly describe what is known from rodent studies but will also highlight contributions from other model organisms and indicate when human studies have confirmed the findings from these animal models.
Most of what is known about embryonic pancreas development has been gleaned from studies in several model organisms, including mice (Mus musculus), zebrafish (Danio rerio), and frogs (Xenopus laevis). Each of these model organisms has its own unique benefits and shortcomings, but the integrated discoveries have significantly advanced our knowledge of pancreas development and its associated dysfunctions. Until recently, the most commonly used model of human pancreas development and disease has been the house mouse due to the high degree of genomic conservation between the two species, shared developmental processes, and amenability to genetic manipulations. Studies in rats (Rattus norvegicus) have also been used to characterize pancreas physiology since many physiological and metabolic responses to stress and external stimuli are more easily monitored in the rat model [1]. Although it has traditionally been more difficult to manipulate rat genomes, recent technological advances in gene editing have largely overcome these barriers, causing a recent resurgence in using rat as a model system for both physiological and developmental analyses. In addition to rodents, organisms such as chicken, zebrafish, and frogs have also provided valuable knowledge and insights into the mechanisms of pancreas development and beta cell dysfunction [2–4]. Although these models are less evolutionarily related to humans, they each have unique assets that have provided critical insights into the more fundamental questions about developmental processes. These model systems have provided information about the mechanisms of gene regulation, cell communication, and the signaling pathways that influence cellular function and contribute to congenital diseases. These studies in model organisms have also paved the way for human stem cell-derived models of pancreas development and disease, which will be discussed at the end of the chapter.
3.2.1. Overview of Pancreas Development
The pancreas is specified at embryonic days 8–9 (E8.0–9.0) in mice, which corresponds to approximately 29–31 days post conception (dpc) in humans [5]. This multifunctional organ is derived from the foregut endoderm in response to critical signals from surrounding tissues, as demonstrated in coculture experiments using ex vivo mouse tissue [6]. These early experiments were not able to identify the exact molecular pathways in play [7]; however, subsequent experiments across several model systems have provided a wealth of information regarding the molecular regulation of these early stages of pancreas development [8]. Briefly, in both mice and humans, the pancreas initially forms as two spatially distinct buds on the dorsal and ventral sides of the endodermal gut tube posterior to the lungs and anterior to the intestine (Fig. 3.1). The dorsally derived bud receives signals from the notochord and dorsal aorta, whereas the ventral bud receives signals from the septum transversum mesenchyme and cardiac mesoderm. Much of what is known about dorsal pancreas induction derives from elegant studies in chicken embryos [9]. These experiments highlighted the importance of the notochord as a signaling hub for dorsal pancreas induction. The same group of investigators went on to identify activin and FGF as the secreted notochord factors that were responsible for repressing Sonic hedgehog (Shh) signaling within the underlying foregut endoderm. Furthermore, they demonstrated that repression of Shh signaling within the prepancreatic foregut endoderm was critical for promoting pancreas induction [10]. Alternatively, the ventral pancreas develops in a noncontiguous region of the foregut endoderm in close proximity to the liver. In experiments performed predominantly in mice, it was demonstrated that BMP and FGF signaling from the adjacent septum transversum mesenchyme and cardiac mesoderm specified the ventral pancreatic bud while simultaneously repressing the liver primordium [11]. Although there are still many gaps in our understanding of these earliest events in pancreas specification and development, these studies provided a comprehensive analysis of the early tissue interactions and signaling pathways that are important for the primary steps of pancreas induction. Furthermore, many of the developmental events that have been characterized in these animal model systems have recently been shown to be conserved in human pancreas development [12, 13].
Fig. 3.1.
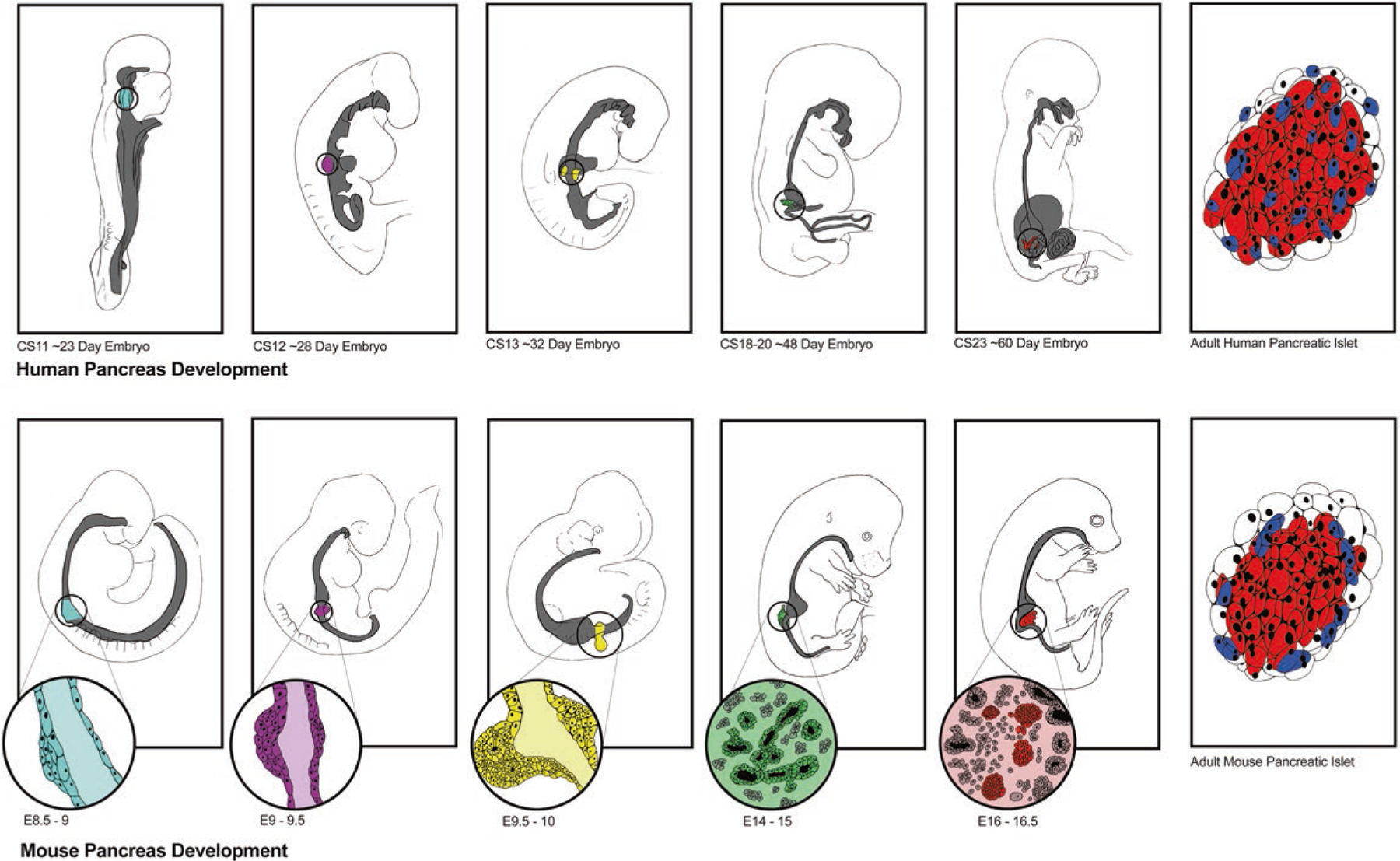
Human and mouse pancreas development. Pancreas developmental programs in humans and mice, top and bottom, respectively, are highly conserved, as shown in the panels correlating similar time points across species. Starting about 23 days after conception in humans (~CS11) and 8.5–9 days in mice (E8.5–E9) the dorsal pancreatic bud emerges from the endodermal gut tube. Several stages of development are depicted across each row demonstrating similar morphological changes occurring in both humans and mice over about 60 days in humans and 16.5 days in mice, at which point immature endocrine cells are present and producing hormones. Over the next few months in humans and weeks in mice, these endocrine cells mature as islets that contain the same cell types but in different ratios and distribution within the islet in each species
Following initial pancreas specification, the dorsal and ventral buds independently grow, branch, and initiate parallel cellular differentiation pathways in what has been referred to as the primary transition stage in mice. Between E12.5 and E15.5 (40–48 dpc in human), the two buds are brought into close proximity through morphological movements inherent to the rotating gut tube and fuse to form a single organ (Fig. 3.1). Although each anlage gives rise to the same complement of adult pancreatic cell types, they delineate distinguishable regions of the adult organ as the pancreatic head (ventrally derived, attached to the duodenum and intestines) and tail (dorsally derived, attached to the spleen and stomach). Between approximately E14.5–E16.5, FGF10 induces Notch and a cascade of unknown signaling events [12, 14, 15] to initiate the formation of exocrine lineage progenitors at the tip of the elongating branches and the emergence of endocrine progenitors in the central core of the organ. At this stage, there is a major wave of islet cell differentiation and expansion, which is referred to as the secondary transition. By birth, each of the exocrine and endocrine cell lineages have been fully specified and continues to mature and proliferate for approximately 2 weeks postnatally. Several extensive reviews describe many additional intrinsic and extrinsic signaling pathways that have been implicated at several stages of pancreatic differentiation, maturation, and expansion [14, 15].
In adults, the pancreas is comprised of three major tissues: exocrine, endocrine, and ductal epithelium. The majority (>90–95%) of the pancreas is made up of exocrine tissue, which is made up of acinar cells that secrete digestive enzymes through the pancreatic ducts into the intestine. Most of the remaining pancreatic tissue (~5–10%) is composed of endocrine cells, which are organized into mini-organs known as islets of Langerhans. The islets are responsible for supplying the body with hormones to control blood sugar levels. There are four major endocrine cell types found in mouse and human adult islets: alpha, beta, delta, and PP cells, each of which produces a unique hormone: glucagon, insulin, somatostatin, and pancreatic polypeptide, respectively (Fig. 3.2). Although the same four endocrine cell populations exist in mice and humans, they are present in different relative ratios and with dissimilar spatial distribution within islets of the two species [16] (Fig. 3.2). Both mouse and human endocrine cells predominantly differentiate from an endocrine progenitor population as single hormone-expressing cells, although a subset of endocrine cells within the human embryonic pancreas are polyhormonal [17]. By birth, the polyhormonal populations are no longer present; however, it is unclear whether they have resolved into mono-hormonal cells or have undergone cell death. Both species also form small populations of ghrelin-producing epsilon cells and gastrin-producing cells in the embryonic pancreas, but these cell types disappear during postnatal stages of pancreatic maturation [18–20].
Fig. 3.2.
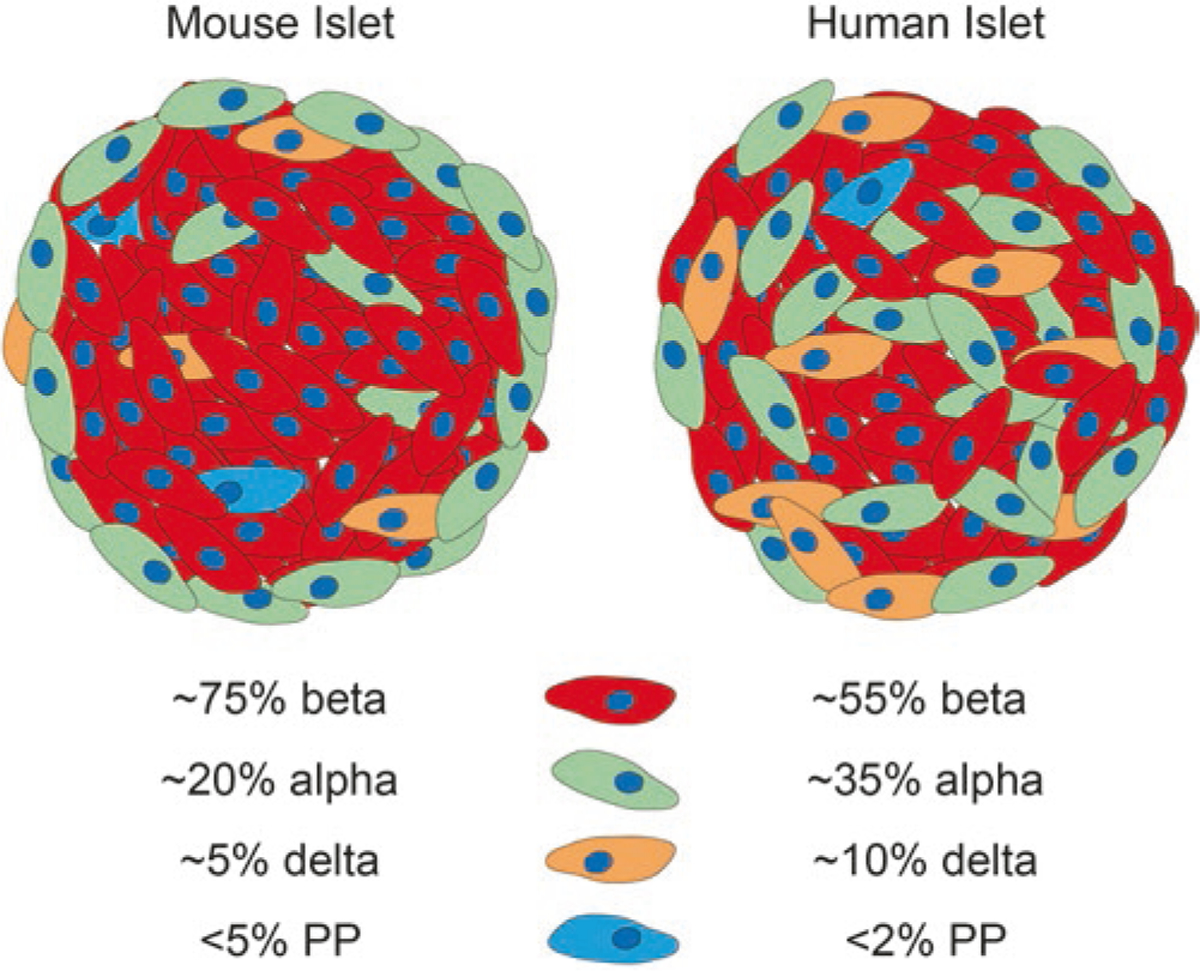
Similarities and differences between mouse and human islets. The same endocrine cell types are present in both human and mouse islets but are spatially distinct and present in different ratios in each organism. All percentages are averages and represent a range of endocrine cell composition, especially in human alpha and beta cells, which can vary greatly from islet to islet. Red represent insulin (beta cells), green represents glucagon (alpha cells), orange is somatostatin (delta cells), and blue is pancreatic polypeptide (PP cells)
3.2.2. Molecular Regulation of Pancreas Development
Like all developmental processes, pancreas induction, morphogenesis, and cellular differentiation require a carefully orchestrated series of developmental events that rely on the integration of many different signaling pathways and transcriptional regulators. Studies in animal models have identified several signaling pathways that are crucial for pancreagenesis and have laid the foundation for the successful in vitro differentiation of pancreatic progenitors and immature islet populations from human stem cells [21] (see below). Although many of these critical cell signaling pathways are conserved in human pancreas development, they have not been directly implicated in human pancreatic developmental defects and disease. This is likely because the majority of these pathways are globally required during development, and inactivation of such regulatory mechanisms would be incompatible with life.
While there are still open questions about the precise signaling mechanisms regulating pancreas development, animal models have provided substantial information about how transcription factors regulate pancreas development, including evidence demonstrating their functional conservation and importance in disease [22–26]. One of the first and most critical transcription factors identified in pancreas specification is pancreatic and duodenal homeobox 1 (PDX1, also known as IPF1 and STF1). PDX1 was first identified and characterized in Xenopus [27, 28] and has conserved expression in all model organisms studied to date, and in human. PDX1 is expressed in the pancreas, duodenum, and stomach shortly after there is morphological evidence of each respective tissue within the foregut endoderm. Within the developing pancreas, PDX1 is initially expressed throughout the pancreatic progenitor population but ultimately becomes restricted to beta and delta cells of the islet [29]. Because of its initially broad expression, global deletion of Pdx1 in mice leads to pancreas agenesis, a disease also associated with PDX1 mutations in humans (Table 3.1). Shortly following pancreas specification, PDX1 becomes coexpressed with another early transcription factor—pancreas transcription factor 1a (PTF1A) within the earliest pancreatic progenitor population [56]. Deletion of Ptf1A also leads to partial pancreas agenesis, characterized by the absence of acinar tissue and a reduced number of endocrine cells [57]. Although PDX1 and PTF1a are the first tissue-restricted transcription factors known to be expressed at the onset of pancreas specification, a rudimentary pancreatic bud still forms when either factor is deleted, suggesting that there are as yet unidentified upstream factors that initiate pancreas specification [56].
Table 3.1.
Conserved transcription factors essential for pancreas development contributing to human and mouse pancreatic defects and disease
| Transcription factor | Human disease | Mouse defect | References |
|---|---|---|---|
| PDX1 | Pancreatic agenesis | Pancreatic agenesis | De Franco et al. [30] |
| PNDM | Stoffers et al. [31] | ||
| MODY4 (het) | Jonsson et al. [32] | ||
| HNF1B | Pancreatic hypoplasia | Pancreatic agenesis (Hz) | Haumaitre et al. [33] |
| MODY5 | Haumaitre et al. [34] | ||
| GLIS3 | PNDM | Neonatal diabetes, loss of beta cells | Rubio-Cabezas et al. [35] |
| Senée et al. [36] | |||
| Kang et al. [37] | |||
| NEUROD1 | PNDM (hz) | Postnatal death diabetes | Rubio-Cabezas et al. [38] |
| MODY6 (het) | Malecki et al. [39] | ||
| T2DM | Naya et al. [40] | ||
| NKX2.2 | PNDM | Postnatal death—no beta cells | Flanagan et al. [41] |
| Sussel et al. [42] | |||
| MNX1 | PNDM | Dorsal pancreas agenesis | Flanagan et al. [41] |
| Harrison et al. [43] | |||
| Li et al. [44] | |||
| PAX6 | PNDM | No alpha cells, reduced beta cells | Solomon et al. [45] |
| St-Onge et al. [46] | |||
| PTF1A | Pancreatic agenesis | Pancreatic agenesis | Sellick et al. [47] |
| PNDM | Weedon et al. [48] | ||
| Kawaguchi et al. [49] | |||
| Krapp et al. [50] | |||
| GATA6 | Pancreatic agenesis (het) | None, but pancreatic agenesis in G6/G4 double knockout | De Franco et al. [30] |
| PNDM | Allen et al. [51] | ||
| Adult onset diabetes | Shaw-Smith et al. [52] | ||
| Xuan et al. [53] | |||
| Carrasco et al. [54] | |||
| NEUROG3 | Adult onset diabetes (hypomorphic mutation) | Perinatal death, no endocrine cells | Rubio-Cabezas et al. [35] |
| PNDM (biallelic mutations) | Gradwohl et al. [55] |
Additional early players in pancreas development are the GATA4 and GATA6 transcription factors. These factors are broadly expressed in the foregut endoderm, prior to pancreagenesis, but become more specifically restricted to the pancreatic anlagen [58]. While whole-body knockouts of these transcription factors lead to early embryonic lethality prior to pancreas formation due to extraembryonic endoderm and heart defects, deletion of both factors simultaneously within the pancreas progenitor population causes pancreas agenesis [53, 54]. Molecular analysis of these mutants revealed that deletion of Gata4 and Gata6 in the foregut endoderm causes ectopic expression of the hedgehog signaling pathway and respecification of the pancreas into intestine and stomach fates [59]. Studies are currently ongoing to determine the function of the GATA factors during pancreas development since interesting differences exist between human and mice (see human pancreas agenesis section below).
The pancreatic progenitor population gives rise to the three major cell lineages in the pancreas, including the islet endocrine cells. At the onset of the secondary transition, the transient expression of the transcription factor Neurogenin3 (NEUROG3) marks a critical molecular event that defines the endocrine progenitor population. NEUROG3 is expressed in both pancreas and intestinal endocrine progenitor populations. In the pancreas, NEUROG3 expression spikes just prior to the secondary transition to delineate the endocrine progenitor cell population [60]. In mice, global deletion of Neurog3 results in the complete absence of all pancreatic and intestinal endocrine populations [55, 61]. However, individuals with loss of function alleles of NEUROG3 appear to have normal islet endocrine function but suffer from severe diarrhea due to the lack of intestinal endocrine cells, suggesting that NEUROG3 may have a lesser or redundant role in human endocrine cell development [35].
Within and downstream of the NEUROG3 endocrine progenitor cell population, dozens of conserved transcription factors have been shown to regulate subsequent stages of endocrine cell type differentiation [26] (Fig. 3.3). This includes two members of the NKX family of transcription factors, NKX2.2 and NKX6.1. Although NKX2.2 and NKX6.1 are initially expressed throughout the pancreas progenitor population, they eventually become restricted to the NEUROG3 endocrine progenitor cells and continue to be expressed in subsets of endocrine lineages, where they function. NKX2.2 becomes restricted to alpha, beta, and PP cells, whereas NKX6.1 is specifically expressed in the beta cell lineage [42, 62]. Despite their widespread expression early in pancreas development, both factors predominantly function to specify the islet cell lineages: NKX2.2 functions upstream of NKX6.1 and is essential for the differentiation of several islet cell lineages, whereas NKX6.1 only affects the formation and function of the beta cell lineage. Both factors continue to be expressed in the adult islet; expression of NKX6.1 is essential for appropriate beta cell functional maturation, and NKX2.2 is necessary for the maintenance of beta cell identity [63, 64].
Fig. 3.3.
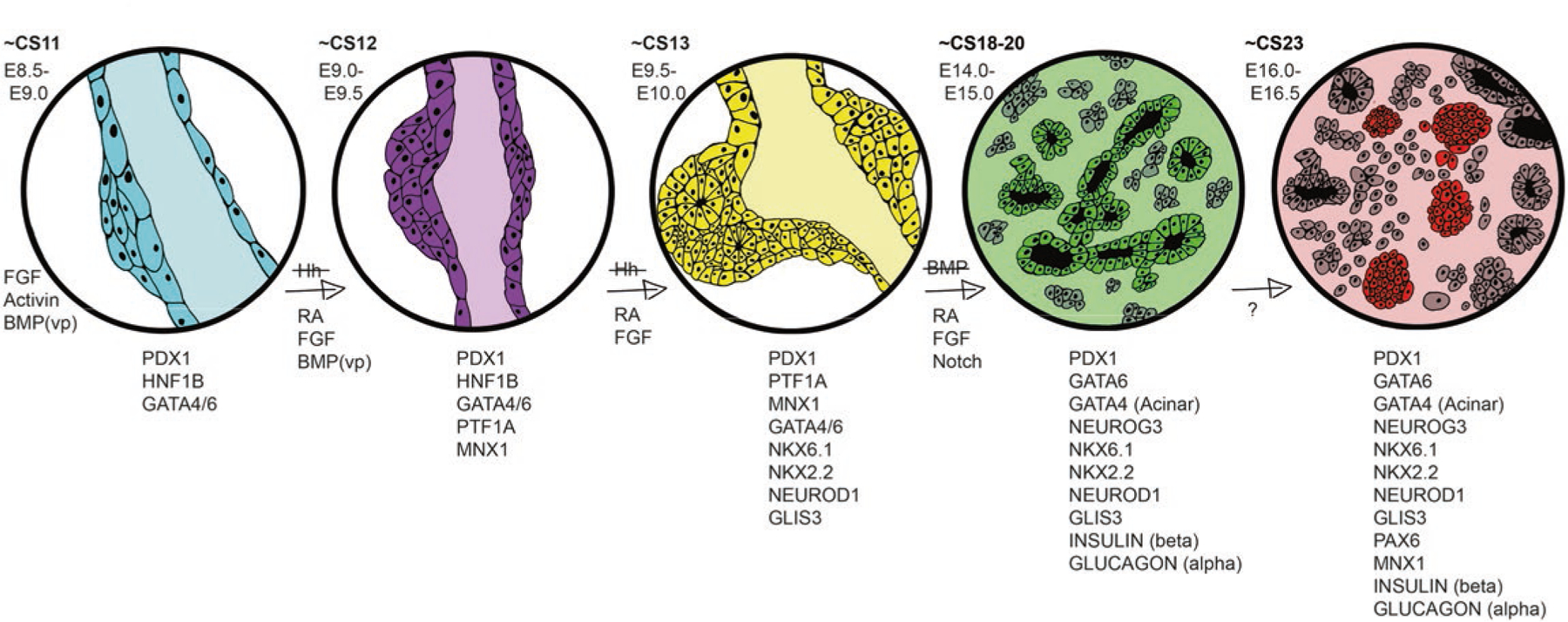
Molecular mechanisms of pancreas development in mice and humans. A selection of transcription factors and cell signaling pathways define the transition from gut endoderm to immature pancreas, which takes place in about 37 days in humans (CS labels in bold) and about 8 days in mice (E labels). While much is known about the cell signaling pathways early on, less is known about the key signaling events later in this development scheme (~E16.0–E16.5). VP represents the contribution of BMP signaling specifically to the ventral pancreatic bud, but not the dorsal bud, at this stage of development
NEUROD1 is another essential transcription factor that is broadly expressed in the pancreas at several developmental stages and in all islet lineages, but functions predominantly downstream of NKX2.2 and NEUROG3 to regulate islet cell development and survival [40, 65, 66]. Neurod1 null mice die just after birth from severe diabetes or survive and develop hyperglycemia later in life, depending on the mouse strain background [40, 67]. Finally, GLIS3 is a functionally conserved transcription factor that is coexpressed with NEUROG3 and then becomes more specific to beta and PP cells. Global deletion of Glis3 in mice results in greatly reduced beta and PP cell numbers, and Glis3 mutant mice often develop neonatal diabetes [68]. Consistently, humans carrying mutations in GLIS3 can develop permanent neonatal diabetes and/or have an increased risk of developing both type 1 and type 2 diabetes [69, 70].
Genetic studies in mice have identified a large cohort of transcriptional regulators, of which only a subset have been discussed above, that are essential for pancreas development and function [22, 23]. Importantly, the identification of these conserved factors has guided targeted sequencing of patients with defective pancreas development to identify corresponding functions in human. Furthermore, with the advent of whole genome sequencing in patients suffering from pancreas-related diseases, there is increasing evidence that many human developmental disorders are caused by the misexpression or dysregulation of specific transcription factors critical for healthy pancreas development in mice. Many of these diseases can now be traced to disrupted developmental processes or genetic mutations (see OMIM, https://www.omim.org/), while several others still remain uncharacterized [71].
3.2.3. Developmental Defects and Their Impact on Pancreatic Function and Disease
During the past ~20 years, there have been significant advancements in our understanding of the molecular pathways underlying pancreas development. Importantly, studies in human fetal pancreatic tissue have confirmed that there are conserved expression patterns for many of the aforementioned transcription factors and signaling molecules in humans [5, 12, 13, 26, 72, 73]. Furthermore, mutational analyses in mice have greatly contributed to our understanding of the developmental defects that can arise when these regulatory pathways are disrupted. As described below, genetic studies in mice have also helped identify many of the human genetic mutations that are responsible for congenital defects that lead to neonatal and adult pancreas-related diseases. It is important to note that unlike many other organs, the pancreas is not required for gestational development. However, a functioning pancreas, and specifically insulin-producing beta cells, are critical for survival after birth. Therefore, mutations that impair aspects of pancreas development can result in a range of mild-to-severe phenotypes, depending on the nature of the genetic mutation and/or where the affected gene product functions.
Pancreas agenesis
Pancreas agenesis is defined in patients who have either an incompletely formed or completely absent pancreas. The most severe cases are rare and nearly always fatal since the disrupted developmental processes often affect the formation of other tissues and are not compatible with survival. However, there are several cases of human pancreas agenesis that arise from mutations in transcription factor genes that have been characterized in mouse genetic models. For example, several cases of pancreas agenesis have been associated with mutations in PDX1 and PTF1a, two of the most critical factors for early pancreas development in mice [31, 47, 49, 57, 74]. Mice lacking either Pdx1 or Ptf1a form only the initial pancreatic rudiments and die shortly after birth due to severe hyperglycemia [32, 75, 76]. Despite PDX1 and PTF1a being two of the earliest and most critical transcription factors involved in pancreas organogenesis, mutations in these genes only account for a low percentage of pancreas agenesis patients, suggesting that mutations in other factors are responsible for the majority cases of this disease. One compelling study that examined the genomes of 27 agenesis patients showed that 56% of these cases could be attributed to haploinsufficient GATA6 mutations [77]. Studies in mice have shown that GATA6 is a critical regulator of early embryonic development—mice lacking Gata6 die between embryonic day 5.5 and embryonic day 7.5 due to early endodermal defects [78]. Surprisingly, however, pancreas specific deletion of Gata6 in mice had minor effects on pancreas development. Only when Gata6 and another family member, Gata4, were deleted from the developing pancreas simultaneously did complete agenesis occur [53, 54]. This discrepancy between the roles of GATA factors in mouse and human development is not completely understood but does suggest that there might be background genetic modifiers or nonautonomous environmental defects contributing to the pancreas agenesis phenotypes seen in humans. Although additional genetic mutations or modifiers that might contribute to the pancreas agenesis phenotype observed in patients with GATA6 mutations have not been identified, when heterozygous loss of GATA6 is modeled in a system of human pluripotent stem cell-derived pancreas differentiation (see Sect. 3.4), pancreas and beta-like cells are still able to form. However, when GATA6 mutations are combined with the inhibition of retinoic acid signaling, pancreas development is impaired. These data highlight the importance of cross-talk between signaling pathways and transcription factors that are essential for understanding the mechanisms of pancreas agenesis and other diseases [79, 80].
In many agenesis cases, patients can survive with very small rudiments of a pancreas, making it possible to live with the disease. These cases have been primarily documented as dorsal pancreas agenesis and are classified as missing the neck, body, and tail of the pancreas, which are all derived from the dorsal anlage. In some instances, these disorders are referred to as pancreatic hypoplasia. The first instance of this disease was described in 1911, and approximately 100 additional cases have been reported since then [81]. Most of these patients were surprisingly asymptomatic until they developed a secondary complication, such as diabetes mellitus or pancreatitis. The origins of pancreatic hypoplasia are also multifactorial, but there are several mouse models that develop this phenotype and could provide insight into the human defects. For example, when a key enzyme in the retinoic acid signaling pathway, RALDH2, is deleted, mice die during gestation with apparent dorsal-specific pancreas agenesis [82]. Dorsal agenesis is also observed in mice deficient in N-cadherin, MNX1, and the activin receptor [43, 44, 83, 84]. A comprehensive list of mouse models that result in aspects of pancreatic agenesis has been previously documented [24]. There are many additional case studies in humans that describe pancreas hypoplasia, although most of them have not identified the causal genetic defect [85, 86]. With technological advances allowing cheaper, faster, and more efficient whole genome sequencing available, it is likely that we will soon have a more comprehensive list of mutations that contribute to pancreas agenesis, many of which are likely to have already been described in mice and other model organisms.
Annular pancreas
Annular pancreas is another pancreatic defect with developmental origins. It was first described as early as the nineteenth century, when a physician noticed an extension of the pancreas, which was described as a connective tissue wrapped around the duodenum of a deceased individual during an autopsy. Some of the earliest reports of this disease suggested that it was extremely rare; random examination of cadavers for the abnormality estimated its incidence at about 3 in 20,000. However, with modern technological advances, such as endoscopies, magnetic resonance imaging (MRI), and computed tomography (CT) scans that allow physicians to identify the disorder in living patients, incidence is now estimated to be as high as 1 in 1000 [87]. Although there is currently a lack of consensus on the actual number of cases, recent work from Lim et al. 2017 estimates an occurrence of only 1 in 20,000 [88]. Some of this controversy is due to a lack of consensus regarding diagnoses. While it is generally accepted that the cause of annular pancreas is due to the inappropriate development of the ventral pancreatic bud, the pathogenesis is still not completely clear. One theory is that a free end of the ventral bud fuses to the duodenum during development and grows around the duodenum as it rotates, although it has also been proposed that the ventral pancreas inappropriately develops as a bilobed organ, and one of the lobes grows around the duodenum [71, 87]. With a handful of exceptions, the majority of human cases are sporadic and have not been linked to genetic mutations [89]. Annular pancreas has also been observed in mouse models of pancreas development when Indian Hedgehog (Ihh), a ligand in the Hedgehog (Hh) signaling family, is inhibited. This leads to ectopic branching of the ventral pancreas, causing an annular pancreatic phenotype [90]. Interestingly, similar mutations in Hh pathway components have not been identified in patients with annular pancreas, suggesting that other pathways regulating Hh signaling contribute to the development of the disease. Further work is required to better understand the mechanisms underlying annular pancreas, and models in mice hold the most promise for elucidating those mechanisms.
Neonatal diabetes mellitus (NDM)
Neonatal diabetes mellitus is a rare form of diabetes that is usually diagnosed in infants at 6 months of age or younger. NDM is often confused with type 1 diabetes, the more common autoimmune form of diabetes that can also occur in young children, but patients with NDM are distinguished by their lack of autoantibodies. Young patients are diagnosed with NDM because of their inability to regulate blood glucose levels. They present at birth with severe hyperglycemia, a life-threatening condition caused by the absence of insulin production from the endocrine pancreas. NDM is rare, with estimated cases of ~1:400,000 [91]. Roughly half of the cases are transient neonatal diabetes mellitus (TNDM), and although they often resolve, many individuals become diabetic later in life, suggesting the existence of an unresolved developmental defect. The remaining cases are referred to as permanent neonatal diabetes mellitus (PNDM) and result in lifelong diabetic conditions that require continual treatment with exogenous insulin, similar to the adult onset diabetes. A subset of both PNDM and TNDM have also been shown to result from specific mutations in genes for ATP-sensitive potassium channels, which are critical for regulating beta cell function [92].
Several studies have identified single-gene mutations in patients with NDM largely based on targeted sequencing of transcription factor genes known to be important for pancreas development in mice. One study in particular examined the sequenced genomes of 37 patients with NDM and identified mutations in seven of the 29 candidate genes tested. Five of the genes (GLIS3, NEUROD1, PDX1, PTF1a, RFX6) had been previously implicated in PDM, while mutations in two additional genes (MNX1 and NKX2.2) were also identified [41]. Notably, all of these transcription factors have been studied extensively in mice, where they are known to cause severe pancreatic defects and perinatal lethality when mutated [22, 42, 93], once again highlighting the importance of animal models in providing critical information about genetic defects causing human developmental diseases.
Diffuse congenital hyperinsulinism in infancy (CHI-D)
CHI-D is the most frequent cause of severe, persistent hypoglycemia in newborns and infants. The majority (60%) of babies are diagnosed during the first month of life and the remainder diagnosed within the first year. Genetic analyses have linked hyperinsulinemia to rare mutations in genes encoding the transcription factors HNF1A and HNF4A; however, the majority of CHI-D cases are caused by mutations that specifically inactivate the critical KATP channel necessary for nutrient sensing and insulin secretion [94]. Interestingly, this defect does not fully explain the phenotype since a recent histological study of postmortem CHI-D pancreatic islets suggested that individuals with CHI-D displayed additional defects in the somatostatin-producing delta cell population [95]. Of particular interest was the discovery of aberrant expression of NKX2.2 in delta cells; both mouse and human studies have demonstrated that NKX2.2 is normally excluded from this endocrine population. In the future, experimental studies in mice to induce ectopic expression of NKX2.2 in delta cells would clarify whether NKX2.2 misexpression is a causative factor of the disease.
Maturity-onset diabetes of the young (MODY)
MODY is a monogenic form of diabetes and comprises about 2% of cases of diabetes in people under 20 years old, making this disease much more common than NDM [96]. Because MODY is broadly characterized by beta cell dysfunction, it is often misdiagnosed as type 1 diabetes. Currently, mutations in 11 different MODY genes have been described, the majority of which were identified by sequencing genes known to cause pancreatic islet defects when mutated in mouse models. Furthermore, most of the affected genes encode transcription factors that are expressed in the developing pancreas and in adult beta cells, including PDX1, NEUROD1, HNF4A, and HNF1B [31, 39, 97–99]; however, there are also a subset of nontranscription factor MODY genes (https://www.omim.org/entry/606391) that lead to diabetes as well. Since nearly all of these genes encode proteins that are critical for important developmental process during embryogenesis, MODY mutations are predominantly point mutations that cause autosomal dominant alleles. Interestingly, similar to what was observed with the GATA6 mutations in pancreas agenesis, heterozygous dominant alleles of several MODY genes, including HNF1alpha and HNF3beta in mice, do not generally cause disease [100]. This could be due to the absence of additional genetic modifiers in the inbred animal models used for these studies or nonconserved mechanisms of disease progression. To clarify whether there are underlying genetic or environmental causes for these discrepancies between human and animal models, new genomic engineering technologies such as CRISPR/Cas9 have facilitated disease modeling in human stem cell-derived beta cells (see stem cell section below). The combination of these two disease modeling approaches will significantly improve our ability to identify and characterize genes responsible for developmental diseases of the pancreas.
3.3. Juvenile and Adult Diseases of the Pancreas
While the aforementioned pancreas disorders are rare and generally monogenic in origin, there are multiple disorders of the pancreas that are much more common and result from a combination of genetic mutations and environmental influences. The most prevalent of these more common disorders is broadly classified as diabetes mellitus (DM). This polygenic disease is extremely wide-spread, affecting nearly 10% of the United States population or 30.3 million people. Remarkably, an additional 84.1 million people are estimated to have prediabetes, a condition that indicates a high likelihood to develop full diabetes in a person’s lifetime [101]. There is currently no cure for diabetes, which is part of the reason that it was the seventh leading cause of death in the US in 2015 (American Diabetes Association, http://www.diabetes.org/diabetes-basics/statistics/). Although the designation of DM represents many different diseases, nearly all of these statistics are summations of the two main types of diabetes mellitus, type 1 and type 2. In the following section, animal models of these diseases will be explored, compared, and contrasted in relation to current treatments for each disorder. Although these adult diseases are generally not considered to be “birth defects,” there are strong genetic components underlying both diseases, and many of the genetic mutations are in genes previously characterized in developmental processes.
3.3.1. Type 1 Diabetes Mellitus (T1DM)
Type 1 diabetes mellitus is a multifactorial autoimmune disease characterized by the destruction of insulin-producing beta cells in the pancreatic islet, leading to severe hyperglycemia in affected individuals. T1DM is prevalent in children; however, individuals of all ages can develop the disease. As of 2015, there have been an estimated account of 542,000 children with the disease, and the incidence appears to be increasing every decade (International Diabetes Federation, http://www.diabetesatlas.org/). In humans, approximately 20–40 different genes have been linked to T1DM, including strong linkage to particular alleles of the major histocompatibility complex (MHC) II region, which accounts for approximately 40% of the hereditary cause of T1DM. Despite the important genetic component of the disease, not all individuals with genetic risk factors develop T1DM, suggesting a role for environmental factors. The disease generally progresses through several well-defined stages. The presence of autoantibodies is the main classifier of stage 1 of disease progression and will persist throughout the life of the patient. Loss of beta cell mass also begins during stage 1. Stage 2 coincides with the advent of hyperglycemia due to further loss of beta cells, which are being destroyed by the autoimmune attack. Finally, stage 3 is defined by even further loss of beta cells past a critical threshold, as well as presentation of clinical symptoms such as polyuria, thirst, hunger, and weight loss associated with severe hyperglycemia. More serious complications of chronic hyperglycemia include retinopathies, neuropathies, nephropathies, and cardiovascular diseases. All of these serious complications warrant further study and highlight a need for even better treatments and eventually a cure for the disease.
Models of T1DM
There are several rodent models of TIDM that have greatly facilitated our understanding of disease onset and progression, despite some inherent limitations regarding how these models were generated and how their immune systems function as compared to humans. The most common murine model of T1DM is the nonobese diabetic (NOD) mouse. Created in Osaka, Japan, in the 1970s, these mice develop insulitis as early as 3–4 weeks of age leading to the destruction of beta cells, mimicking T1DM progression in humans [102]. Importantly, many of the T1DM alleles and biological pathways are shared by humans and NOD mice. Furthermore, the MHCII locus is similar in structure in NOD mice and humans [103]. While NOD mice have proven to be an extremely useful model for understanding the progression of T1D, recent access to human T1DM pancreatic tissue has revealed some important differences in immune infiltration and disease pathology between the species [104]. Furthermore, it has been relatively easy to treat NOD mice with immunotherapies and drug treatments that unfortunately have not been recapitulated in human trials [105].
Another rodent model of spontaneous autoimmune-induced T1DM are BB rats, which were named for the founder colonies: BBdp/Wor (inbred from Worcester, MA, USA) and BBdp (outbred from Ottawa, Canada) [106]. Male and female mice from both strains initially develop pancreatic insulitis, and the beta cells are destroyed between 50 and 90 days of life. Interestingly, persistent immune cell infiltration does not occur in these animals’ islets, which is consistent with patients with T1DM [106]. While these rats are clearly useful for modeling T1DM and the associated effects of the disease like neuropathies [107], a major drawback is that they almost always develop lymphopenia, a disorder in which individuals have very low levels of lymphocytes or white blood cells. While lymphopenia happens in nearly all BB rats, the condition is not normally associated with people who have T1DM. This additional immunological complication has affected the interpretation of many studies using BB rats and is an important reminder that animal models of disease are not exact replicas of human disease and should be used in conjunction with one another to make informed conclusions about the disease.
3.3.2. Type 2 Diabetes Mellitus (T2DM)
Unlike the types of diabetes mellitus discussed above, T2DM is frequently linked to the metabolic syndrome and the obesity epidemic rather than developmental mutations, although there is often a genetic component to this highly multifactorial disease. T2DM is the most common form of diabetes, accounting for more than 90% of cases. T2DM is characterized by metabolic dysfunction, impaired insulin secretion, and insulin resistance, alone or in combination [101]. There are approximately five stages or phases of diabetes: prediabetes and phases 1–4, each marked by a particular set of symptoms. Prediabetes and phase 1 are mainly characterized by glucose intolerance in which an afflicted individual is unable to properly clear glucose after eating, which leads to hyperglycemia. In addition, prediabetics begin to lose sensitivity to insulin, even though their beta cells are still fully functional. Phase 2 is marked by basal hyperglycemia in addition to glucose intolerance. At this phase of the disease, patients may also experience insulin resistance. In phase 3, afflicted individuals generally have fasting hyperglycemia, along with some functional beta cell loss. In phase 4, patients often require exogenous insulin due to severe loss of functional beta cells, which fail to produce sufficient amounts of insulin to regulate blood glucose levels. Complications from this late phase can also, in rare cases, lead to what some describe as phase 5 and is characterized by ketoacidosis as the body begins to look for other sources of fuel. As the disease progresses, a multitude of serious secondary health complications occur, including cardiovascular disease, neuropathies (numbness, particularly in extremities), nephropathy, retinopathy, stroke, and high blood pressure [108].
Models of T2DM
In humans, T2DM represents a heterogenous set of complex polygenic diseases; therefore, choosing the right rodent T2DM model is critical. Rodent models have been traditionally classified as spontaneous or induced, and vary greatly in the severity and phase of diabetes they represent. The most common models are monogenic or diet induced, and each has advantages and disadvantages [109]. Here, we describe a subset of these models in more detail and explain how each model is used to further understand and treat T2DM.
LepOB/OB and LeprDB/DB mice are two related monogenic models of obesity-induced T2DM that affect the function of the leptin hormone, which regulates appetite, and the receptor through which it signals, respectively. The inability to regulate feeding occurs when either of these critical components does not function normally. The LepOB/OB model was discovered as a spontaneously occurring mutation in 1949 at the Jackson Laboratories, but the genetic mutation was not identified as leptin until the mid-1990s [110]. The LepOB/OB mice generally start gaining weight around 2 weeks of age and are hyperglycemic by about 4 weeks. While these mice are characterized as having diabetes due to this hyperglycemia and impaired insulin release, they generally do not progress to stage 3 of T2DM, unless they are bred to be on the C57BLKS/J background [111]. The LeprDB/DB mice were similarly discovered as a spontaneous mutation in the 1960s, again at Jackson labs, but the mutation was not linked to the leptin receptor until years later [112]. The development of diabetes occurs in a similar fashion to the LepOB/OB model, but when placed on the C57BLKS/J, these mice will develop full-blown, late-stage T2DM, often leading to ketoacidosis and death after just a few months. Interestingly, depending on the strain background, each of these models can produce either hyper- or hypotrophic beta cells and can both be hyper- and hypoinsulinemic. Phenotypic variability that depends on strain background is analogous to the differences in disease progression and severity seen in patients with leptin-related deficiencies.
Named after the city in Japan where these mice were created, the AKITA mouse is another mouse model used to study T2DM. The origins of this model’s disease are rooted in a spontaneous mutation in the INS2 gene inhibiting normal processing of insulin in the beta cell. The inappropriate accumulation of misfolded insulin protein leads to ER stress, which results in severe insulin-dependent diabetes. This generally occurs in 3–4 weeks and is accompanied by some of the symptoms of T2DM, like polyuria and hyperglycemia resulting from loss of beta cell mass, although this mouse is not obese. Furthermore, Akita mice have also been used to study diabetes complications, like neuropathies or cardiovascular defects, and have served as a reliable model to test the utility of exogenous beta cell sources to supplement beta cell deficits [113, 114].
In addition to these common mouse models of T2DM, there are also several rat models available, each with its own unique benefits and caveats [109]. The Otsuka Long Evans Tokushima Fatty (OLETF) rat has been used as a model for late-onset T2DM since the early 1990s [115]. These rats are unable to restrict their appetite and become obese with age. With obesity, they develop the inability to regulate their blood glucose levels and become insulin resistant, leading to diabetes onset [116]. Similar to humans, the age of disease onset and severity of disease varies widely in OLEFT rats; however, this also makes it a difficult model to study. Another popular rat model of T2DM is the Zucker Diabetic Fatty (ZDF) rats. Developed in the mid-1980s, ZDF rats have dysfunctional leptin receptor, similar to the LepDB/DB mice. They are frequently used to study the transition from prediabetes to early-onset diabetes and become fully diabetic by about 12 weeks of age. While they serve as a good model to study this important transition, the Zucker rats are genetically predisposed to acquiring defects in beta cell transcription factors that also contribute to their diabetic phenotype independent of leptin signaling defects, which can complicate analyses [117]. The Goto-Kakizaki (GK) rat provides a nonobese model to study mild T2DM characterized by hyperglycemia and insulin resistance largely linked to neonatal beta cell developmental defects [118, 119]. Similar to all disease models, the correct choice of the animal model depends on the type of diabetes and questions being explored.
3.4. Human Models of Pancreas Development
While mice, rats, and other animal models have been invaluable in furthering our knowledge of pancreas development, as well as pancreatic disease onset and pathogenesis, the fact remains that they differ substantially from humans. Apart from the obvious differences, mice also exhibit subtle but fundamental differences from humans in terms of their beta cell development, beta cell mass, islet organization, and blood glucose tolerance. These differences mean that although mice and other model organisms provide a great preliminary model for human development and disease research, they are not sufficient for complete insight into the complex pathologies that affect the human pancreas.
3.4.1. Modeling Human Pancreas and Islet Development In Vitro
Studying human pancreas development is fraught with challenges, both technical and ethical. Much of our current knowledge comes from pancreatic tissues taken from aborted human fetuses at various stages of early development—up to 22 weeks [5, 120]. These tissues are generally sectioned and examined by antibody staining to reveal insights into pancreas and islet morphology, appearance and relative ratios of pancreatic cell types, and islet cell organization during fetal development. While these methods have provided some insights into human pancreas development and highlighted similarities and differences between mice and humans, they only provide a static view of developmental processes in an unperturbed state. The analysis is further complicated by the extreme sample variability that exists between different patient donors. Due to the inherent limitations associated with studying human development and the corresponding defects that lead to disease, the ability to recapitulate human development in a dish by differentiating human pluripotent stem cells (hPSC), including hESCs and hiPSCs (embryonic and induced pluripotent stem cells, respectively), represents an exciting new avenue of research. Furthermore, the advent of gene editing technologies, such as TALENs and CRISPR/Cas9, which allow researchers to induce putative human disease-causing mutations into the hPSC differentiation system, has revolutionized our ability to characterize the intrinsic contribution of particular genetic mutations to developmental defects and disease. However, to truly harness the potential of hPSCs, we need efficient protocols that allow their directed differentiation to organs and cell types of interest, including stem cell-derived pancreas and beta-like cells (SBCs).
One key approach to differentiating hPSC toward a specific cell type is to faithfully recapitulate the developmental cues in vitro that the cells would “experience” during in vivo development. As our understanding of human development is limited, directed differentiation of hPSC to SBCs is largely based on knowledge derived from animal models of development and disease that were outlined previously in this chapter. A number of protocols currently exist for the differentiation of hPSCs to pancreas and beta-like cells [121–125]. Although all of the protocols are able to accurately mimic the earliest stages of pancreas development, through to the generation of endocrine progenitor cells, the directed differentiation of the mature exocrine and islet cell types has proven more challenging. This is in part due to the relative paucity of information related to the signaling pathways that are involved in these later stages of development and differentiation.
In general, pancreas differentiation protocols can be broken down into the developmental stages defined in mice (Figs. 3.3 and 3.4). Since we know that pancreas is derived from endoderm and, in particular, foregut endoderm, the first few differentiation steps are not specific to the pancreas but are shared by all endodermally derived tissues (Fig. 3.4). First, hPSCs must transition to a definitive endoderm cell fate, which is driven by activation of the WNT and TGF beta pathways. Following 2 days of differentiation, the cells express transcription factors specific to the definitive endoderm stage of development, including SOX17. The definitive endoderm cells are then differentiated toward primitive gut tube by activation of the FGF signaling pathway. After approximately 3 days, the cells have switched on primary gut tube markers, including HNF1A, FOXA2, and HNF6. At this point, the pancreatic cell fate, which is marked by PDX1 expression, is induced by retinoic acid and the inhibition of hedgehog signaling, similar to what occurs during mouse dorsal pancreas induction. Inhibition of the BMP signaling pathway is also necessary to block the differentiation of pancreatic foregut cells toward liver lineages, similar to ventral pancreas formation in the mouse (Figs. 3.3 and 3.4, matching colors).
Fig. 3.4.
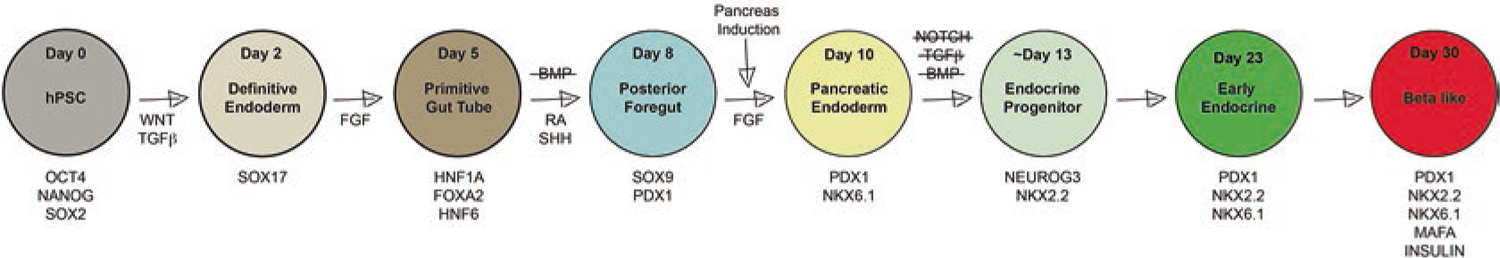
Directed differentiation of hPSCs to beta-like cells in a dish. Based largely on knowledge gleaned from animal models of pancreas development, the stepwise addition of chemicals and small molecules to human pluripotent stem cells leads to the activation and inhibition of many of the cell signaling pathways required for differentiation into insulin-producing beta-like cells. A selection of the critical transcription factors expressed at each stage is noted underneath each time point, noted by day of differentiation and largely corresponds to the same transcription factors found at similar stages of mouse development (Fig. 3.3)
Once pancreatic cell fates have been specified (corresponding to approximately E9.5 of mouse pancreas development,) the PDX1 positive pancreatic foregut cells are then differentiated into pancreatic endoderm cells that continue to express PDX1 and have also activated NKX6.1 via FGF signaling. These cells undergo endocrine differentiation and are subsequently exposed to a cocktail of empirically defined signals that drive them toward pancreatic beta cells while blocking differentiation to other pancreatic cell types. Similar to the differentiation of mouse pancreatic endocrine cells, the pancreatic endoderm cells will transiently express the transcription factor NEUROG3 and then NKX2.2 to delineate the endocrine progenitor population around day 13 of the differentiation protocol. After approximately 10 more days of endocrine differentiation, the early endocrine cell population emerges, classified by the expression of key beta cell markers. While these cells do begin to produce the insulin hormone, they are not yet functional as they do not regulate insulin secretion in response to glucose stimulation. A further 7 days of maturation in a minimal media is required to allow the cells to become functional beta-like cells. While 95% of cells reach the NKX2.2 stage, only 20–40% go on to become insulin positive SBCs, indicating that additional research is needed both in mice and in the hPSC system to optimize the pancreas differentiation protocol.
3.4.2. Disease Modeling with Stem Cell-Derived Pancreatic Cells
Although the hPSC pancreatic differentiation protocol still needs to be refined, it has already proven to be extremely useful for validating conserved gene functions in human pancreas development. For example, in a tour de force study by the Huangfu group, TALEN and CRISPR/Cas-mediated gene editing was used to analyze the role of eight essential pancreatic transcription factors (PDX1, RFX6, PTF1A, GLIS3, MNX1, NEUROG3, HES1, and ARX) in the hPSC-directed differentiation of pancreas [80]. These studies not only verified conserved gene requirements between mice and humans but also revealed a number of previously unsuspected developmental mechanisms with implications for type 2 diabetes. Similar studies were also instrumental in addressing potential discrepancies between mouse and human gene functions. Using gene-edited hESCs and patient-specific IPSCs, two groups were able to partially resolve the discrepancy between mouse and human phenotypes caused by mutations in GATA6 by implicating the contribution of genetic modifiers and nonautonomous cell defects to the pancreas agenesis phenotype seen in human patients [79, 80]. Similarly, McGrath and colleagues used hESC-derived pancreatic cells to confirm that NEUROG3 had similar essential roles in mouse and human pancreas endocrine cell development but determined that patients with mutations in NEUROG3 retained sufficient functional protein to avoid disease [126]. These are just a small number of studies that demonstrate the utility of stem cell-derived pancreas tissue to model development and disease. With increasing technological advances and a better understanding of pancreas development from animal models, these in vitro differentiation models hold great potential for making even greater strides in elucidating outstanding questions about human pancreas development and disease.
Acknowledgement
Illustrations in this chapter were drawn by Jennifer Colquhoun.
References
- 1.Dolgin E. The knockout rat pack. Nat Med. 2010;16:254–7. [DOI] [PubMed] [Google Scholar]
- 2.Kim SK, Hebrok M, Melton DA. Pancreas development in the chick embryo. Cold Spring Harb Symp Quant Biol. 1997;62:377–83. [PubMed] [Google Scholar]
- 3.Matsuda H. Zebrafish as a model for studying functional pancreatic β cells development and regeneration. Dev Growth Differ. 2018;60(6):393–9. [DOI] [PubMed] [Google Scholar]
- 4.Chalmers AD, Slack JM. Development of the gut in Xenopus laevis. Dev Dyn. 1998;212(4):509–21. [DOI] [PubMed] [Google Scholar]
- 5.Jennings RE, Berry AA, Kirkwood-Wilson R, Roberts NA, Hearn T, Salisbury RJ, et al. Development of the human pancreas from foregut to endocrine commitment. Diabetes. 2013;62(10):3514–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wessells NK, Cohen JH. Early pancreas organogenesis: morphogenesis, tissue interactions, and mass effects. Dev Biol. 1967;15(3):237–70. [DOI] [PubMed] [Google Scholar]
- 7.Slack JM. Developmental biology of the pancreas. Development. 1995;121(6):1569–80. [DOI] [PubMed] [Google Scholar]
- 8.Zorn AM, Wells JM. Vertebrate endoderm development and organ formation. Annu Rev Cell Dev Biol. 2009;25(1):221–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim SK, Hebrok M, Melton DA. Notochord to endoderm signaling is required for pancreas development. Development. 1997;124(21):4243–52. [DOI] [PubMed] [Google Scholar]
- 10.Hebrok M, Kim SK, Melton DA. Notochord repression of endodermal Sonic hedgehog permits pancreas development. Genes Dev. 1998;12(11):1705–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wandzioch E, Zaret KS. Dynamic signaling network for the specification of embryonic pancreas and liver progenitors. Science. 2009;324(5935):1707–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pan FC, Brissova M. Pancreas development in humans. Curr Opin Endocrinol Diabetes Obes. 2014;21(2):77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jennings RE, Berry AA, Strutt JP, Gerrard DT, Hanley NA. Human pancreas development. Development. 2015;142(18):3126–37. [DOI] [PubMed] [Google Scholar]
- 14.Gittes GK. Developmental biology of the pancreas: a comprehensive review. Dev Biol. 2009;326(1):4–35. [DOI] [PubMed] [Google Scholar]
- 15.Serup P. Signaling pathways regulating murine pancreatic development. Semin Cell Dev Biol. 2012;23(6):663–72. [DOI] [PubMed] [Google Scholar]
- 16.Hart NJ, Powers AC. Use of human islets to understand islet biology and diabetes: progress, challenges and suggestions. Diabetologia. 2019;62(2):212–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Riopel M, Li J, Fellows GF, Goodyer CG, Wang R. Ultrastructural and immunohistochemical analysis of the 8–20 week human fetal pancreas. Islets. 2014;6(4):e982949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arnes L, Hill JT, Gross S, Magnuson MA, Sussel L. Ghrelin expression in the mouse pancreas defines a unique multipotent progenitor population. PLoS One. 2012;7(12):e52026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prado CL, Pugh-Bernard AE, Elghazi L, Sosa-Pineda B, Sussel L. Ghrelin cells replace insulin-producing beta cells in two mouse models of pancreas development. Proc Natl Acad Sci U S A. 2004;101(9):2924–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suissa Y, Magenheim J, Stolovich-Rain M, Hija A, Collombat P, Mansouri A, et al. Gastrin: a distinct fate of neurogenin3 positive progenitor cells in the embryonic pancreas. PLoS One. 2013;8(8):e70397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krentz NAJ, Lee MYY, Xu EE, Sproul SLJ, Maslova A, Sasaki S, et al. Single-cell transcriptome profiling of mouse and hESC-derived pancreatic progenitors. Stem Cell Reports. 2018;11(6):1551–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jørgensen MC, Ahnfelt-Rønne J, Hald J, Madsen OD, Serup P, Hecksher-Sørensen J. An illustrated review of early pancreas development in the mouse. Endocr Rev. 2007;28(6):685–705. [DOI] [PubMed] [Google Scholar]
- 23.Pan FC, Wright C. Pancreas organogenesis: from bud to plexus to gland. Dev Dyn. 2011;240(3):530–65. [DOI] [PubMed] [Google Scholar]
- 24.Mastracci TL, Sussel L. The endocrine pancreas: insights into development, differentiation, and diabetes. Wiley Interdiscip Rev Dev Biol. 2012;1(5):609–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cano DA, Soria B, Martín F, Rojas A. Transcriptional control of mammalian pancreas organogenesis. Cell Mol Life Sci. 2013;71(13):2383–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hanley N. Closing in on pancreatic beta cells. Nat Biotechnol. 2014;32(11):1100–2. [DOI] [PubMed] [Google Scholar]
- 27.Peshavaria M, Gamer L, Henderson E, Teitelman G, Wright CV, Stein R. XIHbox 8, an endoderm-specific Xenopus homeodomain protein, is closely related to a mammalian insulin gene transcription factor. Mol Endocrinol. 1994;8(6):806–16. [DOI] [PubMed] [Google Scholar]
- 28.Gamer LW, Wright CV. Autonomous endodermal determination in Xenopus: regulation of expression of the pancreatic gene XlHbox 8. Dev Biol. 1995;171(1):240–51. [DOI] [PubMed] [Google Scholar]
- 29.Potter LA, Choi E, Hipkens SB, Wright CVE, Magnuson MA. A recombinase-mediated cassette exchange-derived cyan fluorescent protein reporter allele for Pdx1. Genesis. 2012;50(4):384–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.De Franco E, Shaw-Smith C, Flanagan SE, et al. Biallelic PDX1 (insulin promoter factor 1) mutations causing neonatal diabetes without exocrine pancreatic insufficiency. Diabet Med. 2013;30(5):e197–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stoffers DA, Ferrer J, Clarke WL, Habener JF. Early-onset type-II diabetes mellitus (MODY4) linked to IPF1. Nat Genet. 1997;17(2):138–9. [DOI] [PubMed] [Google Scholar]
- 32.Jonsson J, Carlsson L, Edlund T, Edlund H. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature. 1994;371(6498):606–9. [DOI] [PubMed] [Google Scholar]
- 33.Haumaitre C, Fabre M, Cormier S, Baumann C, Delezoide AL, Cereghini S. Severe pancreas hypoplasia and multicystic renal dysplasia in two human fetuses carrying novel HNF1beta/MODY5 mutations. Hum Mol Genet. 2006;15(15):2363–75. [DOI] [PubMed] [Google Scholar]
- 34.Haumaitre C, Barbacci E, Jenny M, Ott MO, Gradwohl G, Cereghini S. Lack of TCF2/vHNF1 in mice leads to pancreas agenesis. Proc Natl Acad Sci U S A. 2005;102(5):1490–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rubio-Cabezas O, Jensen JN, Hodgson MI, Codner E, Ellard S, Serup P, et al. Permanent neonatal diabetes and enteric anendocrinosis associated with biallelic mutations in NEUROG3. Diabetes. 2011;60(4):1349–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Senée V, Chelala C, Duchatelet S, et al. Mutations in GLIS3 are responsible for a rare syndrome with neonatal diabetes mellitus and congenital hypothyroidism. Nat Genet. 2006;38(6):682–7. [DOI] [PubMed] [Google Scholar]
- 37.Kang HS, Kim YS, ZeRuth G, et al. Transcription factor Glis3, a novel critical player in the regulation of pancreatic beta-cell development and insulin gene expression. Mol Cell Biol. 2009;29(24):6366–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rubio-Cabezas O, Minton JA, Kantor I, Williams D, Ellard S, Hattersley AT. Homozygous mutations in NEUROD1 are responsible for a novel syndrome of permanent neonatal diabetes and neurological abnormalities. Diabetes. 2010;59(9):2326–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Malecki MT, Jhala US, Antonellis A, Fields L, Doria A, Orban T, et al. Mutations in NEUROD1 are associated with the development of type 2 diabetes mellitus. Nat Genet. 1999;23(3):323–8. [DOI] [PubMed] [Google Scholar]
- 40.Naya FJ, Huang HP, Qiu Y, Mutoh H, DeMayo FJ, Leiter AB, et al. Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/neuroD-deficient mice. Genes Dev. 1997;11(18):2323–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Flanagan SE, De Franco E, Lango Allen H, Zerah M, Abdul-Rasoul MM, Edge JA, et al. Analysis of transcription factors key for mouse pancreatic development establishes NKX2–2 and MNX1 mutations as causes of neonatal diabetes in man. Cell Metab. 2014;19(1):146–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sussel L, Kalamaras J, Hartigan-O’Connor DJ, Meneses JJ, Pedersen RA, Rubenstein JL, et al. Mice lacking the homeodomain transcription factor Nkx2.2 have diabetes due to arrested differentiation of pancreatic beta cells. Development. 1998;125(12):2213–21. [DOI] [PubMed] [Google Scholar]
- 43.Harrison KA, Thaler J, Pfaff SL, Gu H, Kehrl JH. Pancreas dorsal lobe agenesis and abnormal islets of Langerhans in Hlxb9-deficient mice. Nat Genet. 1999;23(1):71–5. [DOI] [PubMed] [Google Scholar]
- 44.Li H, Arber S, Jessell TM, Edlund H. Selective agenesis of the dorsal pancreas in mice lacking homeobox gene Hlxb9. Nat Genet. 1999;23(1):67–70. [DOI] [PubMed] [Google Scholar]
- 45.Solomon BD, Pineda-Alvarez DE, Balog JZ, et al. Compound heterozygosity for mutations in PAX6 in a patient with complex brain anomaly, neonatal diabetes mellitus, and microophthalmia. Am J Med Genet A. 2009;149A(11):2543–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.St-Onge L, Sosa-Pineda B, Chowdhury K, Mansouri A, Gruss P. Pax6 is required for differentiation of glucagon-producing alpha-cells in mouse pancreas. Nature. 1997;387(6631):406–9. [DOI] [PubMed] [Google Scholar]
- 47.Sellick GS, Barker KT, Stolte-Dijkstra I, Fleischmann C, Coleman RJ, Garrett C, et al. Mutations in PTF1A cause pancreatic and cerebellar agenesis. Nat Genet. 2004;36(12):1301–5. [DOI] [PubMed] [Google Scholar]
- 48.Weedon MN, Cebola I, Patch AM, et al. Recessive mutations in a distal PTF1A enhancer cause isolated pancreatic agenesis. Nat Genet. 2014;46(1):61–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kawaguchi Y, Cooper B, Gannon M, Ray M, MacDonald RJ, Wright CV. The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat Genet. 2002;32(1):128–34. [DOI] [PubMed] [Google Scholar]
- 50.Krapp A, Knofler M, Frutiger S, Hughes GJ, Hagenbuchle O, Wellauer PK. The p48 DNA-binding subunit of transcription factor PTF1 is a new exocrine pancreas-specific basic helix-loop-helix protein. EMBO J. 1996;15(16):4317–29. [PMC free article] [PubMed] [Google Scholar]
- 51.Allen HL, Flanagan SE, Shaw-Smith C, et al. GATA6 haploinsufficiency causes pancreatic agenesis in humans. Nat Genet. 2011;44(1):20–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shaw-Smith C, De Franco E, Lango Allen H, et al. GATA4 mutations are a cause of neonatal and childhood-onset diabetes. Diabetes. 2014;63(8): 2888–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xuan S, Borok MJ, Decker KJ, Battle MA, Duncan SA, Hale MA, et al. Pancreas-specific deletion of mouse Gata4 and Gata6 causes pancreatic agenesis. J Clin Invest. 2012;122(10):3516–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Carrasco M, Delgado I, Soria B, Martín F, Rojas A. GATA4 and GATA6 control mouse pancreas organogenesis. J Clin Invest. 2012;122(10): 3504–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gradwohl G, Dierich A, LeMeur M, Guillemot F. Neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci U S A. 2000;97(4):1607–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Burlison JS, Long Q, Fujitani Y, et al. Pdx-1 and Ptf1a concurrently determine fate specification of pancreatic multipotent progenitor cells. Dev Biol. 2008;316(1):74–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krapp A, Knöfler M, Ledermann B, Bürki K, Berney C, Zoerkler N, et al. The bHLH protein PTF1-p48 is essential for the formation of the exocrine and the correct spatial organization of the endocrine pancreas. Genes Dev. 1998;12(23):3752–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Decker K, Goldman DC, Grasch CL, Sussel L. Gata6 is an important regulator of mouse pancreas development. Dev Biol. 2006;298(2):415–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xuan S, Sussel L. GATA4 and GATA6 regulate pancreatic endoderm identity through inhibition of hedgehog signaling. Development. 2016; 143(5):780–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Villasenor A, Chong DC, Cleaver O. Biphasic Ngn3 expression in the developing pancreas. Dev Dyn. 2008;237(11):3270–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee CS, Perreault N, Brestelli JE, Kaestner KH. Neurogenin 3 is essential for the proper specification of gastric enteroendocrine cells and the maintenance of gastric epithelial cell identity. Genes Dev. 2002;16(12):1488–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sander M, Sussel L, Conners J, Scheel D, Kalamaras J, Cruz Dela F, et al. Homeobox gene Nkx6.1 lies downstream of Nkx2.2 in the major pathway of beta-cell formation in the pancreas. Development. 2000;127(24):5533–40. [DOI] [PubMed] [Google Scholar]
- 63.Schaffer AE, Taylor BL, Benthuysen JR, Liu J, Thorel F, Yuan W, et al. Nkx6.1 controls a gene regulatory network required for establishing and maintaining pancreatic Beta cell identity. PLoS Genet. 2013;9(1):e1003274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gutiérrez GD, Bender AS, Cirulli V, Mastracci TL, Kelly SM, Tsirigos A, et al. Pancreatic β cell identity requires continual repression of non–β cell programs. J Clin Invest. 2017;127(1):244–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Anderson KR, Torres CA, Solomon K, Becker TC, Newgard CB, Wright CV, et al. Cooperative transcriptional regulation of the essential pancreatic islet gene NeuroD1 (beta2) by Nkx2.2 and neurogenin 3. J Biol Chem. 2009;284(45):31236–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chao CS, Loomis ZL, Lee JE, Sussel L. Genetic identification of a novel NeuroD1 function in the early differentiation of islet alpha, PP and epsilon cells. Dev Biol. 2007;312(2):523–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huang H-P, Chu K, Nemoz-Gaillard E, Elberg D, Tsai M-J. Neogenesis of beta-cells in adult BETA2/NeuroD-deficient mice. Mol Endocrinol. 2002;16(3):541–51. [DOI] [PubMed] [Google Scholar]
- 68.Kang HS, Takeda Y, Jeon K, Jetten AM. The spatiotemporal pattern of Glis3 expression indicates a regulatory function in bipotent and endocrine progenitors during early pancreatic development and in beta, PP and ductal cells. PLoS One. 2016;11(6):e0157138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. 2009;41(6):703–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dupuis J, Langenberg C, Prokopenko I, Saxena R, Soranzo N, Jackson AU, et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet. 2010;42(2):105–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cano DA, Hebrok M, Zenker M. Pancreatic development and disease. Gastroenterology. 2007;132(2):745–62. [DOI] [PubMed] [Google Scholar]
- 72.Piper K, Brickwood S, Turnpenny LW, Cameron IT, Ball SG, Wilson DI, et al. Beta cell differentiation during early human pancreas development. J Endocrinol. 2004;181(1):11–23. [DOI] [PubMed] [Google Scholar]
- 73.Jennings RE, Berry AA, Gerrard DT, Wearne SJ, Strutt J, Withey S, et al. Laser capture and deep sequencing reveals the transcriptomic programmes regulating the onset of pancreas and liver differentiation in human embryos. Stem Cell Reports. 2017;9(5):1387–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schwitzgebel VM, Mamin A, Brun T, Ritz-Laser B, Zaiko M, Maret A, et al. Agenesis of human pancreas due to decreased half-life of insulin promoter factor 1. J Clin Endocrinol Metab. 2003;88(9):4398–406. [DOI] [PubMed] [Google Scholar]
- 75.Guz Y, Montminy MR, Stein R, Leonard J, Gamer LW, Wright CV, et al. Expression of murine STF-1, a putative insulin gene transcription factor, in beta cells of pancreas, duodenal epithelium and pancreatic exocrine and endocrine progenitors during ontogeny. Development. 1995;121(1):11–8. [DOI] [PubMed] [Google Scholar]
- 76.Offield MF, Jetton TL, Labosky PA, Ray M, Stein RW, Magnuson MA, et al. PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development. 1996;122(3):983–95. [DOI] [PubMed] [Google Scholar]
- 77.Allen HL, Flanagan SE, Shaw-Smith C, De Franco E, Akerman I, Caswell R, et al. GATA6 haploinsufficiency causes pancreatic agenesis in humans. Nat Genet. 2011;44(1):20–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Morrisey EE, Tang Z, Sigrist K, Lu MM, Jiang F, Ip HS, et al. GATA6 regulates HNF4 and is required for differentiation of visceral endoderm in the mouse embryo. Genes Dev. 1998;12(22):3579–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tiyaboonchai A, Cardenas-Diaz FL, Ying L, Maguire JA, Sim X, Jobaliya C, et al. GATA6 plays an important role in the induction of human definitive endoderm, development of the pancreas, and functionality of pancreatic β cells. Stem Cell Reports. 2017;8(3):589–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shi Z-D, Lee K, Yang D, Amin S, Verma N, Li QV, et al. Genome editing in hPSCs reveals GATA6 haploinsufficiency and a genetic interaction with GATA4 in human pancreatic development. Cell Stem Cell. 2017;20(5):675–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Robert AP, Iqbal S, John M. Complete agenesis of the dorsal pancreas: a rare clinical entity. Int J Appl Basic Med Res. 2016;6(4):290–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Molotkov A, Molotkova N, Duester G. Retinoic acid generated by Raldh2 in mesoderm is required for mouse dorsal endodermal pancreas development. Dev Dyn. 2005;232(4):950–7. [DOI] [PubMed] [Google Scholar]
- 83.Esni F, Johansson BR, Radice GL, Semb H. Dorsal pancreas agenesis in N-cadherin-deficient mice. Dev Biol. 2001;238(1):202–12. [DOI] [PubMed] [Google Scholar]
- 84.Kim SK, Hebrok M, Li E, et al. Activin receptor patterning of foregut organogenesis. Genes Dev. 2000;14(15):1866–71. [PMC free article] [PubMed] [Google Scholar]
- 85.Jain A, Singh M, Dey S, Kaura A, Diwakar G. A rare case of complete agenesis of dorsal pancreas. Euroasian J Hepatogastroenterol. 2017;7(2):183–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Erotokritou A, Gerharz CD, Sagir A. Agenesis of dorsal pancreas associated with pancreatic neuroendocrine tumor: a case report and review of the literature. J Med Case Rep. 2018;12(1):185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Etienne D, John A, Menias CO, Ward R, Tubbs RS, Loukas M. Annular pancreas: a review of its molecular embryology, genetic basis and clinical considerations. Ann Anat. 2012;194(5):422–8. [DOI] [PubMed] [Google Scholar]
- 88.Lim J, Porter J, Varia H, Pettit S. Annular pancreas causing duodenal obstruction in an adult. BMJ Case Rep. 2017;2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lainakis N, Antypas S, Panagidis A, Alexandrou I, Kambouri K, Kyriazis C, et al. Annular pancreas in two consecutive siblings: an extremely rare case. Eur J Pediatr Surg. 2005;15(5):364–8. [DOI] [PubMed] [Google Scholar]
- 90.Hebrok M, Kim SK, St Jacques B, McMahon AP, Melton DA. Regulation of pancreas development by hedgehog signaling. Development. 2000;127(22):4905–13. [DOI] [PubMed] [Google Scholar]
- 91.Kanakatti Shankar R, Pihoker C, Dolan LM, Standiford D, Badaru A, Dabelea D, et al. Permanent neonatal diabetes mellitus: prevalence and genetic diagnosis in the SEARCH for Diabetes in Youth Study. Pediatr Diabetes. 2013;14(3):174–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Flanagan SE, Patch A-M, Mackay DJG, Edghill EL, Gloyn AL, Robinson D, et al. Mutations in ATP-sensitive K+ channel genes cause transient neonatal diabetes and permanent diabetes in childhood or adulthood. Diabetes. 2007;56(7):1930–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Li H, Edlund H. Persistent expression of Hlxb9 in the pancreatic epithelium impairs pancreatic development. Dev Biol. 2001;240(1):247–53. [DOI] [PubMed] [Google Scholar]
- 94.Demirbilek H, Hussain K. Congenital hyperinsulinism: diagnosis and treatment update. J Clin Res Pediatr Endocrinol. 2017;9(Suppl 2):69–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Salisbury RJ, Han B, Jennings RE, Berry AA, Stevens A, Mohamed Z, et al. Altered phenotype of β-cells and other pancreatic cell lineages in patients with diffuse congenital hyperinsulinism in infancy caused by mutations in the ATP-sensitive K-channel. Diabetes. 2015;64(9):3182–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pihoker C, Gilliam LK, Ellard S, Dabelea D, Davis C, Dolan LM, et al. Prevalence, characteristics and clinical diagnosis of maturity onset diabetes of the young due to mutations in HNF1A, HNF4A, and glucokinase: results from the SEARCH for Diabetes in Youth. J Clin Endocrinol Metab. 2013;98(10):4055–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Stoffel M, Le Beau MM, Espinosa R, Bohlander SF, Le Paslier D, Cohen D, et al. A yeast artificial chromosome-based map of the region of chromosome 20 containing the diabetes-susceptibility gene, MODY1, and a myeloid leukemia related gene. Proc Natl Acad Sci U S A. 1996;93(9):3937–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Vaxillaire M, Boccio V, Philippi A, Vigouroux C, Terwilliger J, Passa P, et al. A gene for maturity onset diabetes of the young (MODY) maps to chromosome 12q. Nat Genet. 1995;9(4):418–23. [DOI] [PubMed] [Google Scholar]
- 99.Horikawa Y, Iwasaki N, Hara M, Furuta H, Hinokio Y, Cockburn BN, et al. Mutation in hepatocyte nuclear factor-1 beta gene (TCF2) associated with MODY. Nat Genet. 1997;17(4):384–5. [DOI] [PubMed] [Google Scholar]
- 100.Shih DQ, Heimesaat M, Kuwajima S, Stein R, Wright CVE, Stoffel M. Profound defects in pancreatic beta-cell function in mice with combined heterozygous mutations in Pdx-1, Hnf1alpha, and Hnf-3beta. Proc Natl Acad Sci U S A. 2002;99(6):3818–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Centers for Disease Control and Prevention (CDC). National Diabetes Statistics Report, 2017; 2017. p. 1–20. [Google Scholar]
- 102.Makino S, Kunimoto K, Muraoka Y, Mizushima Y, Katagiri K, Tochino Y. Breeding of a nonobese, diabetic strain of mice. Jikken Dobutsu. 1980;29(1):1–13. [DOI] [PubMed] [Google Scholar]
- 103.Wicker LS, Clark J, Fraser HI, Garner VES, Gonzalez-Munoz A, Healy B, et al. Type 1 diabetes genes and pathways shared by humans and NOD mice. J Autoimmun. 2005;25(Suppl):29–33. [DOI] [PubMed] [Google Scholar]
- 104.Katsarou A, Gudbjörnsdottir S, Rawshani A, Dabelea D, Bonifacio E, Anderson BJ, et al. Type 1 diabetes mellitus. Nat Rev Dis Primers. 2017;3:17016. [DOI] [PubMed] [Google Scholar]
- 105.Roep BO. Are insights gained from NOD mice sufficient to guide clinical translation? Another inconvenient truth. Ann N Y Acad Sci. 2007;1103(1):1–10. [DOI] [PubMed] [Google Scholar]
- 106.Mordes JP, Bortell R, Blankenhorn EP, Rossini AA, Greiner DL. Rat models of type 1 diabetes: genetics, environment, and autoimmunity. ILAR J. 2004;45(3):278–91. [DOI] [PubMed] [Google Scholar]
- 107.Zhang W, Kamiya H, Ekberg K, Wahren J, Sima AAF. C-peptide improves neuropathy in type 1 diabetic BB/Wor-rats. Diabetes Metab Res Rev. 2006;23(1):63–70. [DOI] [PubMed] [Google Scholar]
- 108.DeFronzo RA, Ferrannini E, Groop L, Henry RR, Herman WH, Holst JJ, et al. Type 2 diabetes mellitus. Nat Rev Dis Primers. 2015;1:15019. [DOI] [PubMed] [Google Scholar]
- 109.Chen D, Wang M-W. Development and application of rodent models for type 2 diabetes. Diabetes Obes Metab. 2005;7(4):307–17. [DOI] [PubMed] [Google Scholar]
- 110.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372(6505):425–32. [DOI] [PubMed] [Google Scholar]
- 111.Lindström P. The physiology of obese-hyperglycemic mice [ob/ob mice]. ScientificWorldJournal. 2007; 7:666–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chen H, Charlat O, Tartaglia LA, Woolf EA, Weng X, Ellis SJ, et al. Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell. 1996;84(3):491–5. [DOI] [PubMed] [Google Scholar]
- 113.Drel VR, Pacher P, Stavniichuk R, Xu W, Zhang J, Kuchmerovska TM, et al. Poly(ADP-ribose) polymerase inhibition counteracts renal hypertrophy and multiple manifestations of peripheral neuropathy in diabetic Akita mice. Int J Mol Med. 2011;28(4):629–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chen H, Zheng C, Zhang X, Li J, Li J, Zheng L, et al. Apelin alleviates diabetes-associated endoplasmic reticulum stress in the pancreas of Akita mice. Peptides. 2011;32(8):1634–9. [DOI] [PubMed] [Google Scholar]
- 115.Kawano K, Hirashima T, Mori S, Natori T. OLETF (Otsuka Long-Evans Tokushima Fatty) rat: a new NIDDM rat strain. Diabetes Res Clin Pract. 1994;24(Suppl):S317–20. [DOI] [PubMed] [Google Scholar]
- 116.Bi S, Moran TH. Obesity in the Otsuka Long Evans Tokushima Fatty rat: mechanisms and discoveries. Front Nutr. 2016;3:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tokuyama Y, Sturis J, DePaoli AM, Diabetes JT. Evolution of β-cell dysfunction in the male Zucker diabetic fatty rat. Diabetes. 1995;44(12):1447–57. [DOI] [PubMed] [Google Scholar]
- 118.Gauguier D, Froguel P, Parent V, Bernard C, Bihoreau MT, Portha B, et al. Chromosomal mapping of genetic loci associated with non-insulin dependent diabetes in the GK rat. Nat Genet. 1996;12(1):38–43. [DOI] [PubMed] [Google Scholar]
- 119.Kuwabara WMT, Panveloski-Costa AC, Yokota CNF, Pereira JNB, Filho JM, Torres RP, et al. Comparison of Goto-Kakizaki rats and high fat diet-induced obese rats: are they reliable models to study Type 2 Diabetes mellitus? PLoS One. 2017;12(12):e0189622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Meier JJ, Butler AE, Saisho Y, Monchamp T, Galasso R, Bhushan A, et al. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes. 2008;57(6):1584–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pagliuca FW, Millman JR, Gürtler M, Segel M, Van Dervort A, Ryu JH, et al. Generation of functional human pancreatic β cells. Cells. 2014;159(2): 428–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rezania A, Bruin JE, Arora P, Rubin A, Batushansky I, Asadi A, et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat Biotechnol. 2014;32(11):1121–33. [DOI] [PubMed] [Google Scholar]
- 123.Nostro MC, Sarangi F, Yang C, Holland A, Elefanty AG, Stanley EG, et al. Efficient generation of NKX6–1+ pancreatic progenitors from multiple human pluripotent stem cell lines. Stem Cell Reports. 2015;4(4):591–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Russ HA, Parent AV, Ringler JJ, Hennings TG, Nair GG, Shveygert M, et al. Controlled induction of human pancreatic progenitors produces functional beta-like cells in vitro. EMBO J. 2015;34(13):1759–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhu S, Russ HA, Wang X, Zhang M, Ma T, Xu T, et al. Human pancreatic beta-like cells converted from fibroblasts. Nat Commun. 2016;7:10080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.McGrath PS, Watson CL, Ingram C, Helmrath MA, Wells JM. The basic helix-loop-helix transcription factor NEUROG3 is required for development of the human endocrine pancreas. Diabetes. 2015;64(7):2497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]