

Abstract
Aggressive myeloid leukemias such as blast crisis chronic myeloid leukemia and acute myeloid leukemia remain highly lethal. Here we report a genome-wide in vivo CRISPR screen to identify new dependencies in this disease. Among these, RNA-binding proteins (RBPs) in general, and the double-stranded RBP Staufen2 (Stau2) in particular, emerged as critical regulators of myeloid leukemia. In a newly developed knockout mouse, loss of Stau2 led to a profound decrease in leukemia growth and improved survival in mouse models of the disease. Further, Stau2 was required for growth of primary human blast crisis chronic myeloid leukemia and acute myeloid leukemia. Finally, integrated analysis of CRISPR, eCLIP and RNA-sequencing identified Stau2 as a regulator of chromatin-binding factors, driving global alterations in histone methylation. Collectively, these data show that in vivo CRISPR screening is an effective tool for defining new regulators of myeloid leukemia progression and identify the double-stranded RBP Stau2 as a critical dependency of myeloid malignancies.
The clinical management of chronic myeloid leukemia (CML) has been revolutionized by tyrosine kinase inhibitors that target Abl kinases1. While these drugs are effective in treating patients diagnosed at the chronic phase, they are unable to effecttively control disease in the accelerated or blast crisis phase2, leading to survival rates of less than 10%3. While developmental signals such as Wnt and Smoothened, and RNA Binding Proteins (RBPs) such as Msi2 (ref. 4) and Adar1 (ref. 5), have been implicated in CML progression, concerted efforts to comprehensively map biological regulators of blast crisis CML (bcCML) at a genome-wide scale have been lacking. To address this, we focused on primary cancer-initiating and propagating leukemia stem cells (LSCs) in an in vivo CRISPR/Cas9-based screen6–8. BcCML resembles acute myeloid leukemia (AML) in the clinic and previous work has indicated that molecular dependencies of bcCML are often relevant to de novo AML4,9–11. Consistent with this, our functional genomic screen identified multiple previously known dependencies of both bcCML and AML, such as the Aurora and Plk pathways, Myc and Max transcription factors, as well as several key new regulators of cancer stem cell growth and expansion in vivo. Among these, RBPs emerged as one of the top-ranked required gene families, with ~680 RBPs scoring as a key dependency in the screen. Selection of RBPs with enriched expression in LSCs and no known role in cancer led to identification of the double-stranded RNA-binding protein Stau2.
Staufen is an RNA-binding protein, initially identified as a maternal-effect gene regulating anterior–posterior patterning in the Drosophila oocyte12,13 and has since been shown to be a critical fate determinant in dividing Drosophila neuroblasts14,15. The two mammalian homologs, Stau1 and Stau2, contain five conserved double-stranded RNA-binding domains that regulate messenger RNA stability, transport of mRNAs to different cell organelles and mRNA translation16. Stau2 in particular is highly expressed in the brain, where it plays a critical role in asymmetric division and neuronal stem cell differentiation17,18. While Staufen proteins have been well studied in the nervous system, their function in the hematopoietic system or in cancer remains unknown.
To define the role of Stau2 in myeloid leukemia, we created a Stau2 knockout mouse and found that Stau2 deletion severely impaired leukemia propagation and improved survival. An integrated computational analysis of targets bound by Stau2 identified by eCLIP and genes downregulated upon Stau2 loss via RNA sequencing (RNA-seq) identified Stau2 as a critical new direct regulator of a network of chromatin modifiers. Aligned with this, Stau2 inhibition impacted global histone methylation patterns by directly regulating the KDM family of genes and modulating the epigenetic landscape. These data demonstrate an exciting new role for Stau2 in myeloid malignancies via control of regulators of the epigenome and show that in vivo CRISPR screening can be an effective tool for defining new dependencies of myeloid leukemia progression.
Results
Genome-wide in vivo CRISPR–Cas9 screen in myeloid leukemia stem cells.
To determine the molecular effectors of myeloid leukemia growth in vivo, we carried out a genome-wide CRISPR/Cas9 screen in a BCR-ABL/NUP98-HOXA9-driven model of bcCML. This models a very aggressive bcCML-like disease or AML, where 90% of leukemia cells are undifferentiated (lineagelo/−), providing an ideal system for in vivo screens (Fig. 1a and Extended Data Fig. 1a–e). Guides depleted due to the requirement of genes they target were assessed by comparing sequencing of CRISPR guides from infected cells before and after in vitro selection and from leukemic blasts retrieved from spleen after growth in vivo.
Fig. 1 |. Genome-wide in vivo CRISPR screen identifies an essential role for RBPs in aggressive myeloid leukemia progression.
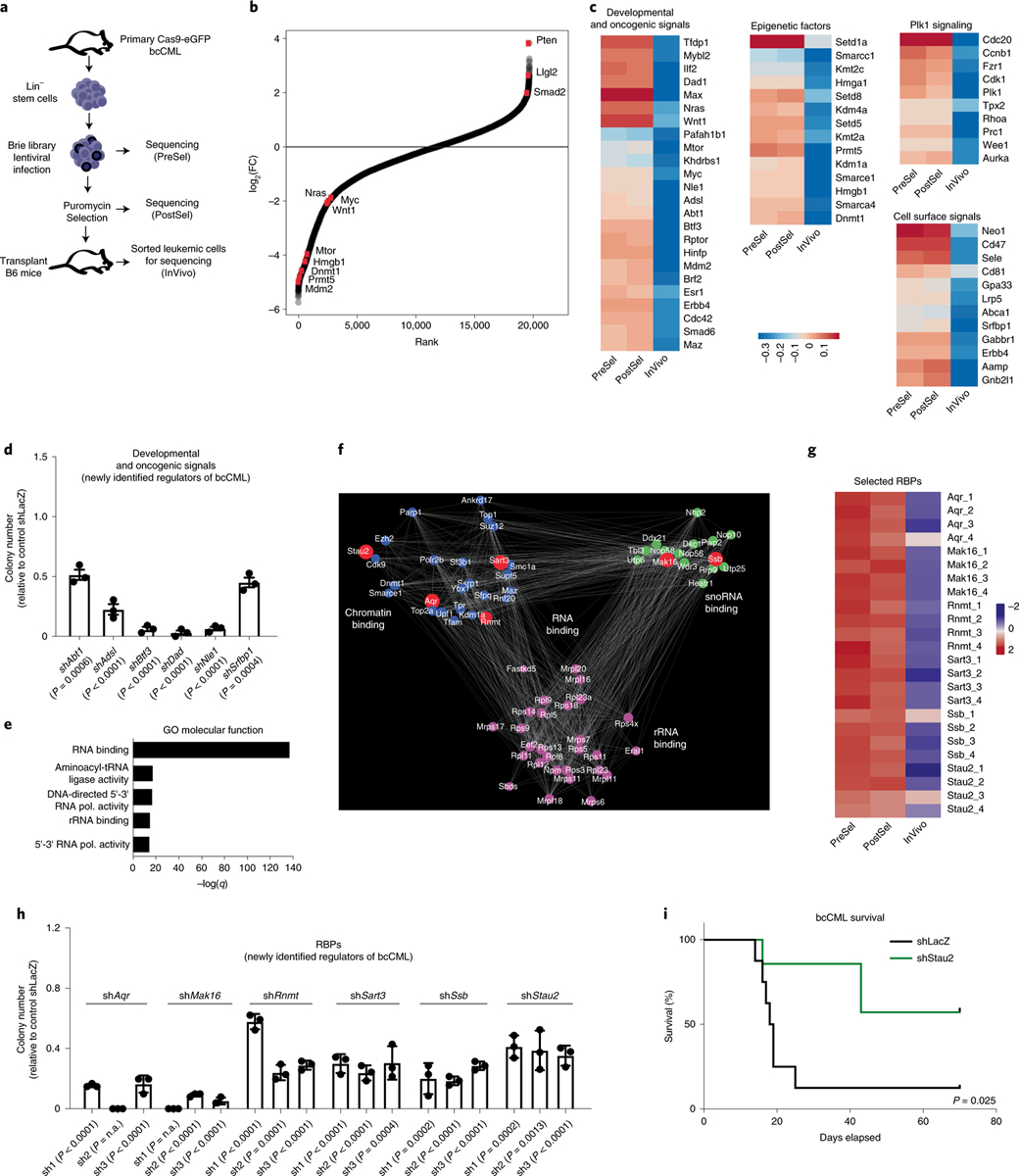
a, Schematic representation of the design strategy employed to perform in vivo CRISPR screen in LSCs. Lin−, murine lineage−; PreSel, preselection; PostSel, post-selection; InVivo, in vivo sample. b, Rank-ordered depiction plot showing the distribution of normalized CRISPR guide scores for each gene targeted by guides in the Brie library. Red dots indicate genes known to play an important role in cancer progression. c, Heat maps show impact on indicated gene representation PreSel, PostSel and in vivo. d, Impact of shRNA-mediated knockdown of indicated genes on the colony-forming ability of Lineage− bcCML cells (mean ± s.e.m.; n = 3 independent culture wells per group; two-tailed Student’s t-tests). e, GO molecular function analysis of n = 3,540 genes that were depleted by threefold or more in vivo as compared to input; Fisher’s exact test. rRNA, ribosomal RNA; tRNA, transfer RNA; pol., polymerase. f, Subnetwork of three Louvain communities of RNA-binding genes (GO: 0003723) depleted by threefold in the CRISPR screen. The communities are exclusively enriched in GO molecular function terms of chromatin binding, small nucleolar RNA binding and rRNA binding; and the genes responsible for the enrichment are colored in blue, green and magenta, respectively. The red symbols denote selected genes, which were further tested in vitro in h. Only selected genes are shown. g, Heat map shows the impact on each guide for selected six RBPs PreSel, PostSel and in vivo. h, Graph shows the impact of three independent shRNAs targeting six candidate RBPs on the colony-forming ability of Lineage− bcCML stem cells (mean ± s.d., n = 3 independent culture wells; two-tailed Student’s t-tests). n.a., not available. i, Lineage− cells from established bcCML were isolated and infected with shRNAs against control (shLacZ) or Stau2 (shStau2). Infected cells were transplanted into irradiated recipients and survival was monitored (n = 8 animals for shLacZ and n = 7 animals for shStau2; log-rank test. Data are combined from two independent experiments).
Computational analysis of the screen revealed that while few guides were depleted during the brief in vitro selection period, about 3,500 genes were depleted by threefold or more in vivo, indicating that these may be essential for leukemia growth and propagation. We also found significant overlap of these genes with those reported to be essential for survival in vitro (Extended Data Fig. 1f)19, confirming the robustness of our in vivo screen. As positive controls for cancer growth, we found that many known drivers of CML and bcCML, such as the developmental regulators cMyc20, Mdm2 (ref. 21), Wnt1 (ref. 22), Pafah1b1 (Lis1)11 and mTor23, were highly depleted. In addition, regulators of AML including the developmental and oncogenic signals Nras24 and Wnt1 (ref. 25), the stem cell self-renewal genes Cdc42 and Lis1 (refs. 11,26) and epigenetic regulators Kdm1a, Hmga1 and Smarca4 (refs. 27,28) were highly depleted and tumor suppressors such as Pten were enriched (Fig. 1b,c). Notably, we identified several genes that could be potential new regulators of leukemia, including the transcription factor Btf3 and the enzyme adenylosuccinate lyase, which are known to drive solid tumor growth29,30 but have not been explored in hematological malignancies (Fig. 1d). Finally, we identified genes that have not yet been implicated in any oncogenic context and may thus be unexplored dependencies of cancer: this included the transcription factor Abt1, the anti-apoptotic gene Dad1, the actin regulator Srfbp1 and Notchless homolog 1 (Nle1), known to regulate skeletal development31. Knocking down each of these genes using short hairpin (sh)RNAs led to a 2–30-fold reduction in colonies formed by murine LSCs as well as a 3–50-fold reduction in colonies formed by human K562 cells in vitro (Fig. 1d and Extended Data Fig. 1g–i), validating the results of the CRISPR screen and identifying this as an important and useful resource for future discovery.
To map core molecular programs involved in myeloid cancer progression with an unbiased approach, we performed an Enrichr analysis on genes depleted by threefold or more in vivo; among the programs identified were Chromatin remodeling (Biocarta), Myc and Max pathway genes (Encode) and PLK and Aurora signaling pathways (NCI Nature) (Extended Data Fig. 1j). This analysis of our in vivo screen revealed a profound enrichment of genes involved in RNA binding; the similarity to observations from in vitro screens32 suggests that RBPs may be a disproportionate dependency in myeloid leukemias (Fig. 1e). Although RBPs constitute a large varied family of proteins, interest in their function has emerged relatively recently33. In an effort to define critical nodes of interaction among RBPs, we carried out a network analysis on the 695 genes known to directly bind mRNAs under the ‘RNA-binding’ gene ontology (GO) term and defined a subnetwork that naturally separated into three Louvain communities34. The unique, significantly enriched molecular functions in these communities (LFDR < 0.1) were chromatin binding, small nucleolar RNA binding and ribosomal RNA binding (Fig. 1f), indicating that these subsets of genes may be of particular relevance to myeloid leukemia progression and would be of interest to pursue in the future.
To focus on new regulators of cancer progression, we excluded genes essential for generalized functions, such as transcription, translation, spliceosomal assembly, RNA turnover or export (such as Polr2a, Polr2b, Dicer and Nxf1). Of the 19 remaining genes, 9 had already been implicated in cancer: this included members of the RNA methyltransferase family (Mettl), the RNA-editing enzyme Adar1 and the RNA-binding Sam68 gene (Khdrbs1)5,35,36. Among the remaining ten genes with no known function in cancer, only six were enriched in human LSCs (Extended Data Fig. 2a–c)37. These were the glucose metabolism regulator Aqr, the cap methyltransferase Rnmt, mRNA splicing factor Sart3 (related to the tumor antigen Sart1), the RNA chaperone Ssb, the fate determinant Stau2 and Mak16, which has no known function (Fig. 1g). Subsequently, each of the six genes was targeted functionally and validated using three independent shRNAs per gene and one guide per gene. This resulted in impaired colony formation in vitro (Fig. 1h and Extended Data Fig. 2d,e), further validating the results of the CRISPR screen and providing new opportunities for discovery. Gene expression indicated that most were broadly expressed across the hematopoietic system (Gene Expression Commons)37; thus, we focused on Stau2 as it was enriched in immature stem/progenitor cells (Extended Data Fig. 2a,f), in line with its previously identified role in maintaining neural stem cells17,18.
Fig. 2 |. Generation and analysis of Stau2 knockout mice.
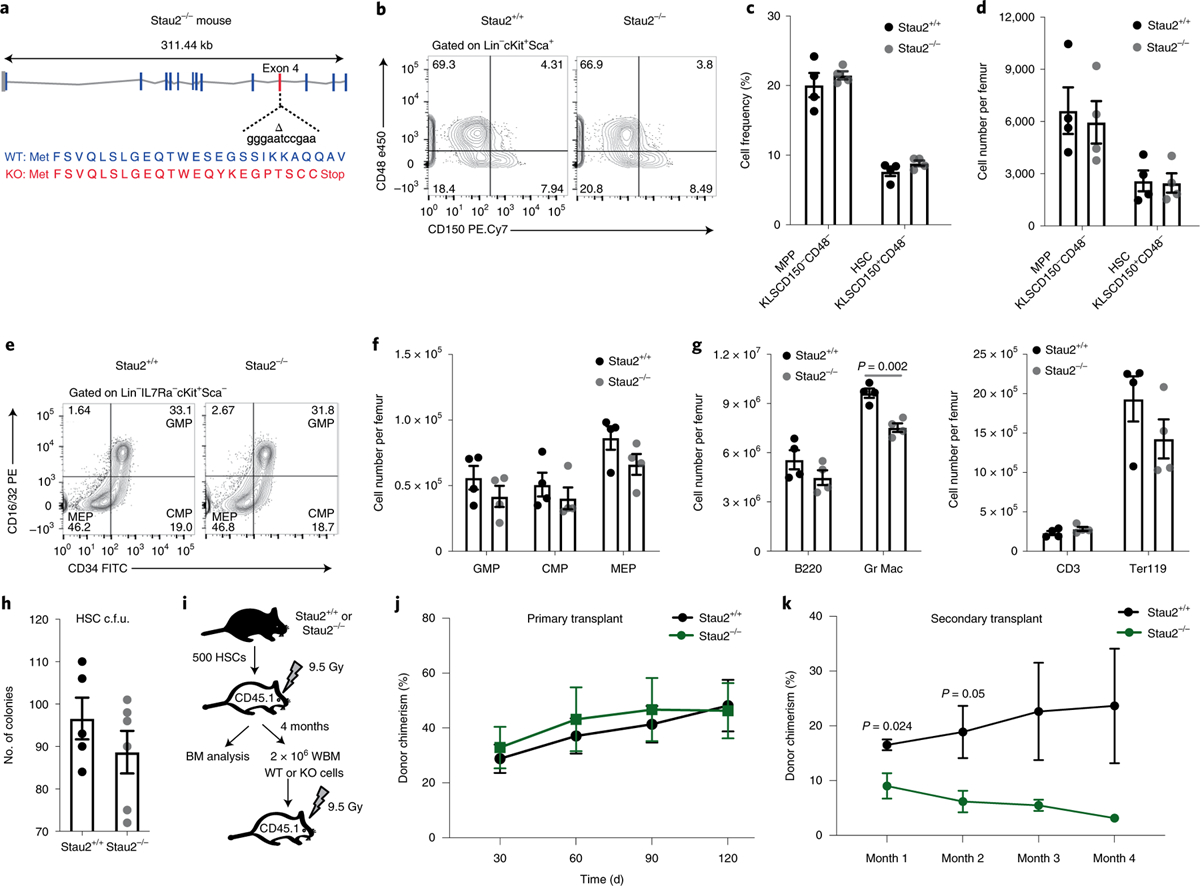
a, Schematic diagram of the site targeted by a CRISPR guide to generate Stau2 knockout (KO) mice. WT, wild type. b–d, Representative fluorescence-activated cell sorting (FACS) plots (b) show HSC frequency in Stau2+/+ and Stau2−/− mice (gated on Lin−cKit+Sca+ (KLS) cells, where Lin+ is marked by CD3e, CD4, CD8, Gr1, CD11b/Mac-1, TER119, CD45R/B220 and CD19) (c,d). Graphs show the average frequencies and numbers (mean ± s.e.m.) of HSCs (KLSCD150+CD48−) and multipotent progenitors (MPPs, KLSCD150−CD48−) (n = 4 animals for each age- and sex-matched littermates; data are combined from three independent experiments). e,f, Representative FACS plots show the frequency and graphs show the number (mean ± s.e.m.) of committed progenitors (granulocyte–macrophage progenitor (GMP), Lin−IL7Ra−Kit+Sca1−CD34+CD16/3 2+; common myeloid progenitor (CMP), Lin−IL7Ra−Kit+Sca1−CD34+CD16/32−; megakaryocyte–erythroid progenitor (MEP), Lin−IL7Ra−Kit+Sca1−CD34−CD 16/32−) in Stau2+/+ and Stau2−/− mice (mean ± s.e.m.; n = 4 animals per cohort; data are combined from three independent experiments). g, Graph shows the total number of differentiated hematopoietic cells in bone marrow of Stau2+/+ and Stau2−/− mice (mean ± s.e.m.; n = 4 animals per cohort; two-tailed Student’s t-test; data are combined from three independent experiments). h, Colony-forming ability of Stau2+/+ and Stau2−/− HSCs (mean ± s.e.m.; n = 5 replicates for Stau2+/+ and n = 6 for Stau2−/−; data are combined from two independent experiments). c.f.u., colony-forming units. i, Schematic shows the experimental strategy used to determine the impact of Stau2 loss on the in vivo repopulating capacity and self-renewal ability of HSCs. BM, bone marrow; WBM, whole bone marrow. j,k, Graphs show average donor chimerism in the peripheral blood of primary and secondary transplant mice (n = 4 animals per cohort; two-tailed Student’s t-tests).
Development and analysis of Stau2 knockout mice.
Because we noted a requirement for Stau2 in the in vivo CRISPR screen, we sought to determine whether oncogene expression could induce Stau2. While expression of BCR-ABL alone led to a small but significant 1.2-fold increase in Stau2 expression, BCR-ABL and NUP98-HOXA9 together enhanced expression by ~fourfold (Extended Data Fig. 2g,h), indicating that oncogene expression can promote Stau2 expression. Consistent with this, higher doses of the BCR-ABL kinase inhibitor Gleevec significantly reduced Stau2 expression in established bcCML stem cells (Extended Data Fig. 2i). To test whether there was a functional dependence of bcCML on Stau2, we targeted it using an shRNA-based strategy. shRNA-mediated knockdown of Stau2 in LSCs led to a dramatic improvement in survival of recipient mice: while 0% of control mice survived, ~60% of the mice transplanted with Stau2 knockdown cells survived, leading to a 4.8-fold reduced risk of death relative to control, (hazard ratio (HR) = 0.209) (Fig. 1i).
These data prompted us to test the impact of Stau2 in myeloid leukemia using a more definitive genetic approach. We thus designed and developed Stau2 knockout mice with CRISPR/Cas9 technology. Mice were engineered with an 11-bp deletion in exon 4 (first shared exon) that resulted in a frame-shift mutation leading to multiple stop codons and loss of RNA-binding domains 2–5 (Fig. 2a and Extended Data Fig. 3a). Homozygous null mice were born at ratios equal to wild-type and heterozygous littermates and showed no overt changes in morphology or behavior and bred normally. Analysis of Stau2 knockout mice showed that its loss had a minor impact on bone-marrow cellularity (1.27-fold; Extended Data Fig. 3b) and it did not affect the frequency or number of most hematopoietic stem/progenitor populations or differentiated cells (Fig. 2b–g and Extended Data Fig. 3c,d). Functionally, Stau2 deletion did not impact either hematopoietic stem cell (HSC) colony formation (Fig. 2h) or repopulation of lethally irradiated mice (Fig. 2i,j and Extended Data Fig. 3e–g). Although steady-state hematopoiesis was not detectably dependent on Stau2, Stau2 loss affected serial transplantability of bone-marrow cells (Fig. 2k and Extended Data Fig. 3h–j), possibly indicating some level of impact on long-term self-renewal and maintenance. It is possible that the impact on normal hematopoiesis is less severe because of a compensatory increase in Stau1 in normal hematopoietic cells, which was not observed in leukemia (Extended Data Fig. 3k).
Impact of Stau2 deletion on myeloid leukemia establishment and propagation.
In contrast to the small but significant impact of Stau2 loss on steady-state hematopoiesis and in the primary transplant setting, Stau2 deletion led to a twofold reduction in the establishment of bcCML both in vitro (Fig. 3a,b) and in vivo (13-fold increase in the likelihood of survival relative to wild type, HR = 0.075; Fig. 3c). Stau2 loss also led to a functional depletion in the LSC population, indicated by a threefold reduction of in vitro colony-forming ability (Fig. 3d,e) as well as a marked increase in survival of mice transplanted with Stau2−/− LSCs (72.7%) relative to controls (0%) in secondary transplant assays (Fig. 3f and Extended Data Fig. 4a; 27.4-fold higher likelihood of survival than wild type, HR = 0.036). Further, conditional inhibition of Stau2 in established leukemias led to an increase in median survival from 15 to 26 d (HR = 0.032), suggesting that the leukemias remain dependent on Stau2 for their continued propagation (Fig. 3g and Extended Data Fig. 4b,c). At a cellular level, Stau2 loss resulted in a 2.8–3.4-fold increase in the differentiated Lin+ cells (Fig. 3h,i) and a concomitant 2.5–5-fold reduction in primitive Lin−CD150−Flt3+Sca1+ LSCs38 (Fig. 3j,k and Extended Data Fig. 4d–f). Notably, while Stau2 deletion did not affect either proliferation or apoptosis of bulk cancer cells (Extended Data Fig. 4g–j) or apoptosis of LSCs (Fig. 3l,m), it led to a twofold reduction in Ki67+-proliferating LSCs (Fig. 3l,m). These data suggest that Stau2 loss delays myeloid leukemia progression at least in part via accelerated differentiation and reduced proliferation of LSCs.
Fig. 3 |. Impact of genetic loss of Stau2 on bcCML initiation and propagation.
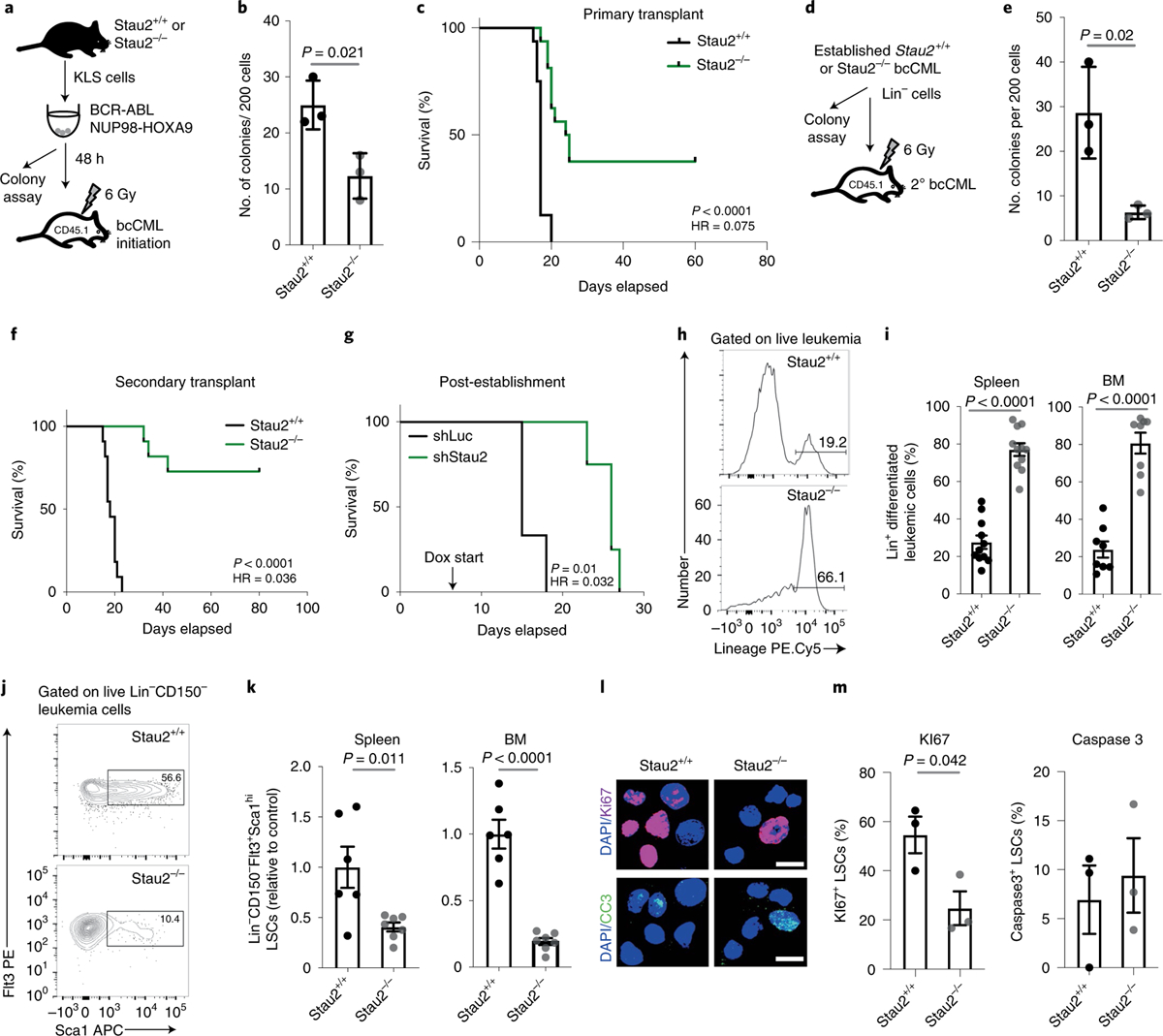
a, Schematic shows the strategy used to determine the impact of Stau2 loss on the initiation of bcCML. KLS cells from Stau2+/+ and Stau2−/− mice were retrovirally transduced with BCR-ABL and NUP98-HOXA9 and cultured in colony assays in vitro or transplanted into recipients to monitor survival. b, Graph shows the colony-forming ability of transduced BCR-ABL+(NGFR+)-NUP98-HOXA9+(CD2+)Stau2+/+ and Stau2−/− cells (mean ± s.d.; n = 3 replicates independent culture wells per cohort; two-tailed Student’s t-tests). c, Survival curve shows the impact of Stau2 loss on the initiation of bcCML in vivo in primary transplants (n = 16 animals per cohort; data are combined from four independent experiments; log-rank test). d–f, Lin− cells were isolated from Stau2+/+ and Stau2−/− established leukemia and used to determine whether Stau2 loss affects self-renewal of LSCs. Schematic of strategy used (d). Lin− Stau2+/+ and Stau2−/− cells were cultured in vitro to assess colony formation (e, mean ± s.d.; n = 3 independent culture wells per cohort; two-tailed Student’s t-tests; data are shown from one representative experiment that was repeated twice with similar results) or transplanted to monitor survival in secondary recipients (f, n = 11 animals per cohort; data are combined from three independent experiments; log-rank test). g, bcCML Lin− cells expressing doxycycline (dox) inducible control (shLuc) or Stau2 shRNAs (shStau2) were transplanted, recipients were given doxycycline water from day 6 post-transplant, and survival was monitored (n = 3 animals for shLuc and n = 4 animals for shStau2; data are shown from one representative experiment that was repeated with similar results; Extended Data Fig. 4c; log-rank test). h,i, Representative FACS plots show Lin expression in Stau2+/+ and Stau2−/− leukemia (h). The frequency of Lin+ cancer cells in spleen (i, left) and bone marrow (i, right) of recipient mice is shown (mean ± s.e.m.; n = 11 animals per cohort for spleen; data are combined from three independent experiments; n = 8 animals per cohort for bone marrow. Data are combined from two independent experiments; two-tailed Student’s t-tests). j,k, Representative FACS plots show Lin−CD150−Sca1+Flt3+ expression in Stau2+/+ and Stau2−/− leukemia (j). The frequency of these LSCs in the spleen (k, left) and the bone marrow (k, right) of recipient mice is shown (mean ± s.e.m.; n = 6 animals for Stau2+/+ and n = 7 animals for Stau2−/−; data are combined from two independent experiments; two-tailed Student’s t-tests). l, Representative images show representative Ki67 and cleaved caspase 3 staining in LSCs from Stau2+/+ and Stau2−/− cells from leukemic mice. Only nuclear staining was considered positive (scale bar, 10 μm). m, Graph shows the frequency of Ki67+ or cleaved caspase 3+ cells (mean ± s.e.m.; n = 3 animals per cohort, values are averaged from 12–40 cells per mouse; two-tailed Student’s t-tests).
Given the impact of Stau2 inhibition on mouse models of bcCML, we tested whether Stau2 was expressed in, and important for, primary human leukemia cells. Gene expression analysis showed that STAU2 expression rises with human AML progression (Fig. 4a) and is higher in AML stem cells relative to normal human HSCs (Fig. 4b), indicating that it may play a functional role in human disease. Functionally STAU2 knockdown resulted in a 9–11-fold drop in the colony-forming ability of the bcCML cell line K562 and ~sixfold drop in the AML line MV-4–11 (Fig. 4c,d and Extended Data Fig. 5a). Further, STAU2 knockdown reduced the colony-forming ability of five primary human bcCML patient samples by ~1.5–4.6-fold, of three AML samples by ~2–23-fold and of a myelodysplastic syndrome (MDS) sample by twofold (Fig. 4e and Extended Data Fig. 5b). Finally, STAU2 inhibition led to a threefold reduction in the engraftment of primary human bcCML and AML samples in NSG mice (Fig. 4f), indicating that STAU2 is essential for the growth and propagation of primary human disease in vivo. While STAU2 knockdown did not detectably impact CD34+ LSC/progenitor cell content in vivo (Extended Data Fig. 5c), there was a trend toward increased differentiation (Extended Data Fig. 5d), consistent with the impact of Stau2 loss in mouse models of the disease (Fig. 3h,i).
Fig. 4 |. Stau2 loss impairs growth of primary human leukemia.
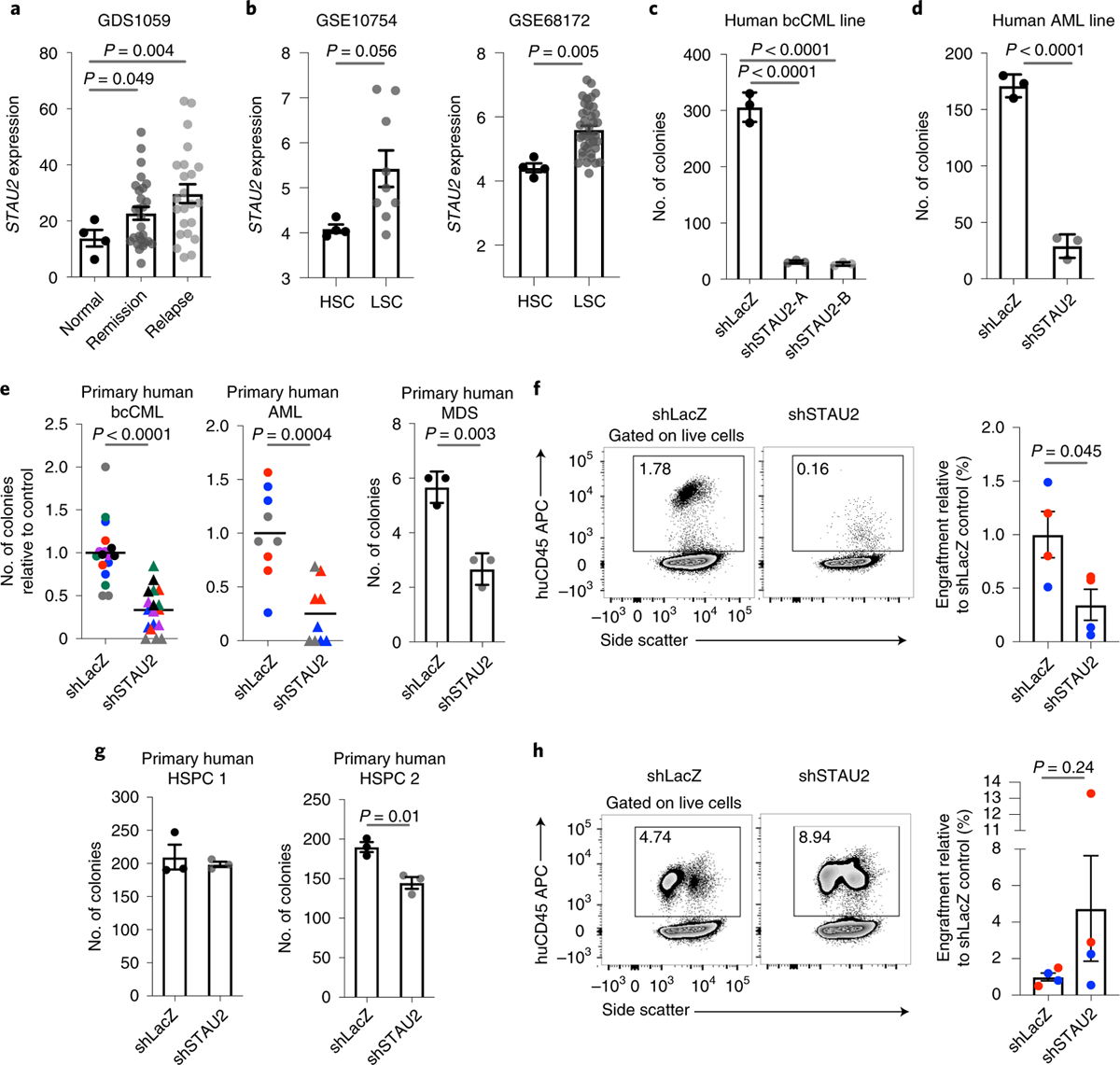
a, Gene expression analysis shows increased STAU2 expression in primary human AML remission and relapse samples compared to control (mean ± s.e.m.; n = 4 normal, n = 27 remission and n = 23 relapse; two-tailed Student’s t-tests). b, Graphs show relative STAU2 expression in primary human AML stem cells compared to normal HSCs. Data are shown from two independent datasets (mean ± s.e.m.; n = 4 HSCs and n = 9 LSCs for GSE10754 and n = 4 HSC and n = 38 LSC for GSE68172; two-tailed Student’s t-tests). c,d, Colony-forming ability of K562 (c) and MV-411 cells (d) transduced with shRNAs against control or STAU2 (mean ± s.d.; n = 3 independent culture wells per group; two-tailed Student’s t-tests) e, Primary human bcCML (left), AML (middle) or MDS (right) samples were transduced with lentiviral shRNAs targeting LacZ (control) or human STAU2. Infected cells were sorted and plated in methylcellulose. Each dot represents number of colonies, each color represents an individual patient sample, and the line represents mean (n = 17 for shLacZ and n = 18 independent culture wells for shSTAU2 from six bcCML samples; n = 9 independent culture wells per group from three AML samples; n = 3 independent culture wells per group from one MDS sample; two-tailed Student’s t-tests). f, NSG mice were transplanted with cells from primary bcCML and AML patient samples transduced with lentiviral shRNAs against control (shLacZ) or human STAU2, two mice per cohort for bcCML (blue); two mice per cohort for AML (red). Representative FACS plots show human cell chimerism in bone marrow 7–8 weeks post-transplant (left). Graph shows fold change in human leukemia cell chimerism in bone marrow of samples expressing shSTAU2 relative to shLacZ control (mean ± s.e.m.; n = 4 animals per cohort; two-tailed Student’s t-tests). g, Average number of colonies formed by normal human CD34+ bone marrow hematopoietic stem and progenitor cells (HSPCs) transduced with lentiviral shRNAs targeting either LacZ (control) or human STAU2 (mean ± s.e.m.; n = 3 independent culture wells per group; two-tailed Student’s t-tests). h, NSG mice were transplanted with primary human CD34+ HSPCs transduced with lentiviral shRNAs against control (shLacZ) or human STAU2. Representative FACS plots show human cell chimerism in bone marrow 8 weeks post-transplant (left). Graph shows fold change in bone-marrow engraftment of samples expressing shSTAU2 relative to control (shLacZ). Each dot represents a mouse, and each color represents an individual human sample (mean ± s.e.m.; n = 4 animals per cohort; two-tailed Student’s t-tests).
STAU2 knockdown had a small impact on ability of human cells to form colonies (Fig. 4g) or engraft in NSG mice (Fig. 4h and Extended Data Fig. 5e), suggesting that STAU2 inhibition could be of utility in aggressive myeloid malignancies, especially those like de novo AML which remains unresponsive to most current treatments. However, given the impact of Stau2 loss on mouse hematopoietic stem and progenitor cell function in serial transplants and the variability of responses between human samples, further work is needed to determine the therapeutic window for inhibition of STAU2 or other elements of the pathway.
Chromatin-binding factors are downstream effectors of Stau2.
To define the molecular effectors that mediate the impact of Stau2, we carried out an unbiased analysis of transcripts bound by Stau2 using enhanced crosslinking immunoprecipitation followed by high-throughput sequencing (eCLIP)39 in K562 bcCML cells (Fig. 5a and Extended Data Fig. 6a,b). This identified ~2,400 genes (~7,000 transcripts) bound by STAU2, primarily in the 3′ untranslated region (UTR) stem loop regions (Fig. 5b and Extended Data Fig. 6c–e). GO analysis of these targets showed significant enrichment in pathways related to protein localization, regulation of metabolic processes and chromatin organization (Fig. 5c), consistent with the network analysis indicating that Stau2 was closest to genes associated with chromatin binding (Fig. 1f).
Fig. 5 |. Downstream effectors of Stau2 function in myeloid leukemia.
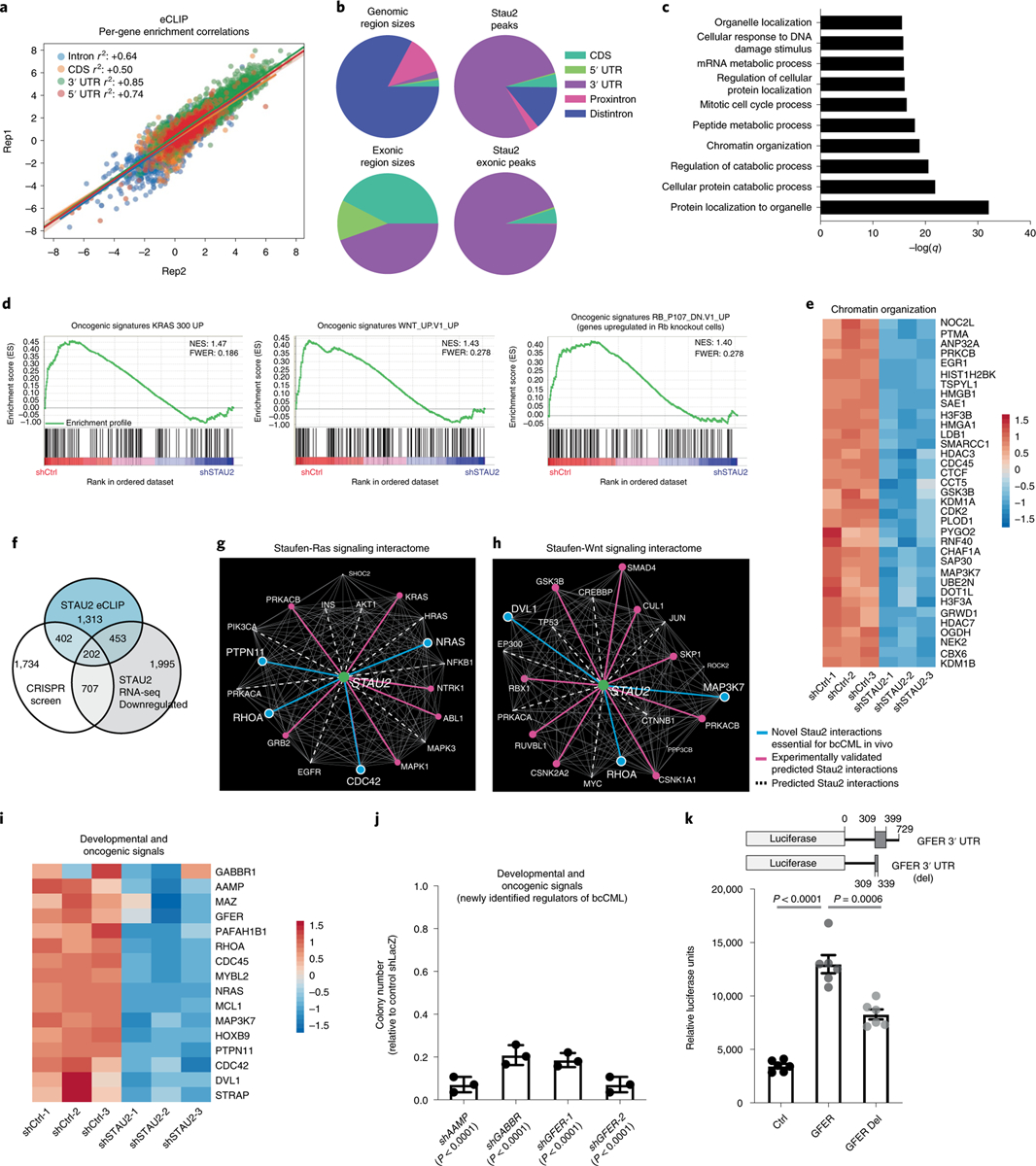
a, Graph shows per-gene enrichment correlation between two independent eCLIP experiments for STAU2 in K562 cells. Each dot represents a gene and the color represents the binding region (intron, coding sequence (CDS) or UTRs). b, Stau2 binding sites in either genomic regions or intronic regions of targets. c, GO terms most significantly enriched in genes enriched by eightfold or more (n = 2,370) by Stau2 as compared to input in eCLIP. d, Gene-set enrichment analysis shows significant downregulation of genes associated with the indicated pathways upon Stau2 knockdown. e, Heat map shows the changes in RNA expression of selected genes upon STAU2 knockdown that fall under the ‘Chromatin Organization’ GO term in c. f, Overlap between the genes that dropout by threefold or more in the genome-wide CRISPR screen in bcCML stem cells (n = 3,045), genes that Stau2 binds by eightfold or more from the CLIP-seq (n = 2,370) and genes that are depleted significantly (q < 0.05; n = 3,357) by RNA-seq upon STAU2 knockdown. g,h, Network analysis of Stau2 with genes associated with Ras (g) and Wnt (h) signaling pathways. The dotted white lines represent interactions that are closer than expected by random chance, of which the ones also identified as being regulated by STAU2 (eCLIP or RNA-seq) are highlighted in solid pink. The solid blue lines show interactions identified in the integrated dataset in f. i, Heat map shows effect of Stau2 loss on the expression of genes from the three-way overlap (in f) that play a role in development and oncogenesis. j, Impact of shRNA-mediated knockdown of developmental and oncogenic signals from the three-way overlap (from f) on the colony-forming ability of K562 cells (mean ± s.d.; n = 3 independent culture wells per group; two-tailed Student’s t-tests). k, Relative luciferase reporter activity in 293 T cells (mean ± s.e.m.) expressing control 3′ UTR reporter, GFER 3′ UTR with STAU2 binding sites (bases 309–399 downstream of the stop codon) and GFER del 3′ UTR with a partial STAU2 binding site (truncated at 339 bp downstream of the stop codon); n = 6 independent culture wells per group, data combined from two independent experiments; two-tailed Student’s t-tests.
To determine genes whose expression was also controlled by STAU2, we analyzed shControl and shSTAU2-bearing K562 cells by RNA-seq and identified ~3,500 genes as downregulated and ~2,700 genes as upregulated following STAU2 knockdown. These included genes associated with Kras and Wnt signaling and loss of Rb (q < 0.05; Extended Data Fig. 7a and Fig. 5d). Many of the upregulated genes were known tumor suppressors such as PTEN40, KLF6 (ref. 41) and VHL (Extended Data Fig. 7b). Consistent with an association of Stau2 with binding gene transcripts associated with chromatin organization (Fig. 5c), several genes within this family were significantly downregulated in cells lacking STAU2 (Fig. 5e), implying that Stau2 not only directly binds transcripts associated with epigenetic regulation, but also regulates them functionally.
To comprehensively define signals that are downstream of Stau2 and are critical for cancer progression, we analyzed the overlap of genes bound directly by STAU2 (eCLIP) and downregulated upon STAU2 knockdown (RNA-seq), with those identified as dependencies (>threefold loss) in the genome-wide in vivo CRISPR screen (Fig. 5f and Supplementary Table 1). The integrated dataset offered a unique view of Stau2’s impact on leukemogenesis: specifically, it showed that Stau2 directly controls potent oncogenic signals, such as the Ras and Wnt signaling networks (Fig. 5g,h) as well as many new determinants of bcCML (Fig. 5i). We assessed some of the genes known to have other functions but not previously implicated in myeloid leukemia, like the angiogenesis regulator angio-associated migratory protein, the liver regeneration factor GFER and γ-aminobutyric acid type b, receptor 1. Inhibition of these signals reduced colony-forming ability of K562 cells by 2–14-fold (Fig. 5j and Extended Data Fig. 7c,d).
To determine whether Stau2 binding sites in the 3′ UTRs are essential for downstream gene expression we selected GFER, which includes a binding site in the 3′ UTR, as a candidate transcript. Cells expressing luciferase linked to the GFER 3′ UTR showed a 3.7-fold increase in luciferase activity relative to control and partial deletion of the Stau2 binding site could no longer activate luciferase to the same levels (Fig. 5k and Extended Data Fig. 7e), indicating that Stau2 binding to the 3′ UTR is essential for expression. Because in this case the promoter was constitutively active in both the wild-type and mutated 3′ UTR, reduction in luciferase RNA expression levels could only have been a result of decreased RNA stability. This suggests that Stau2 can, at least in some contexts, stabilize mRNA, which may be distinct from the reported role of Stau1 in promoting mRNA decay42. These results suggest that the dataset developed here can be used to not only identify downstream effectors of Stau2 but also novel regulators of leukemia progression in general.
Among the genes in the three-way overlap, the identification of several chromatin modifiers was the most interesting (Fig. 6a); thus, we discovered six new direct interactions of Stau2 with chromatin-binding genes (Fig. 6b; KDM1A, MAZ, NOC2L, NUDT21, UBTF and TFAM) within the previously identified RNA-binding network (Fig. 1f). To determine whether these were indeed the functional effectors downstream of Stau2 in myeloid leukemia, we focused on KDM1A, given the availability of KDM1 inhibitors. Delivery of ORY-1001, a KDM1A small molecule inhibitor, resulted in a threefold reduction in colony-forming ability of wild-type LSCs, mimicking the impact of Stau2 deletion (Fig. 6c). Notably, there was no additional impact of KDM1A inhibition on the colony-forming ability of Stau2−/− cells, suggesting that Stau2’s function in bcCML is mediated, at least in part, through the regulation of the KDM1A gene (Fig. 6c). Consistent with the relatively minor impact of Stau2 deletion on steady-state hematopoiesis (Fig. 2), we saw no impact of the KDM1A inhibitor on the colony-forming ability of normal HSCs in vitro (Extended Data Fig. 7f), similarly to earlier reports28,43. Further, long-term in vivo treatment did not alter the frequency or number of HSCs, progenitors or differentiated cell populations (Extended Data Fig. 7g–j), indicating that KDM1A inhibition may be of therapeutic value in bcCML.
Fig. 6 |. Stau2 regulates chromatin-binding genes.
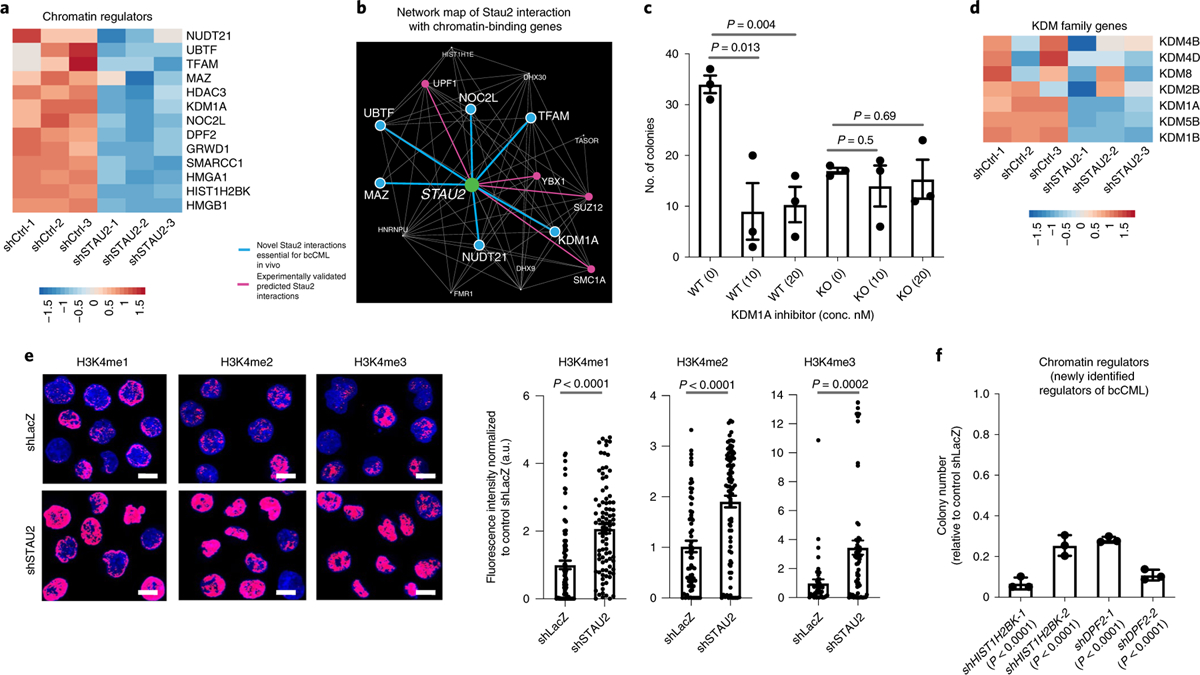
a, Heat map shows effect of Stau2 loss on the expression of genes from the three-way overlap (in Fig. 5f) that play a role in chromatin regulation. b, Nearest-neighbor interactions of STAU2 with chromatin-binding genes that also bind RNA. Pink edges denote previously known interactions (PCNet) that were also identified by either eCLIP or RNA-seq; the blue edges are newly identified interactions based on three independent lines of evidence in Fig. 5f. c, Impact of inhibiting KDM1A at the indicated concentrations on the colony-forming ability of Lin− Stau2+/+ and Stau2−/− bcCML cells (mean ± s.e.m., n = 3 independent culture wells per group. Data shown are from one representative experiment that was repeated with similar results; two-tailed Student’s t-tests). d, Heat map shows impact on indicated KDM family genes by RNA-seq upon STAU2 knockdown. e, Impact of STAU2 inhibition on H3K4me1, H3K4me2 and H3K4me3 expression in K562 cells (mean ± s.e.m., H3K4me1, n = 85 cells for shLacZ and n = 89 cells for shSTAU2; H3K4Me2, n = 66 cells for shLacZ and n = 86 cells for shSTAU2; H3K4Me3, n = 46 cells for shLacZ and n = 66 cells for shSTAU2; two-tailed Student’s t-tests. Data are combined from two independent experiments). f, Impact of shRNA-mediated knockdown of epigenetic regulators from the three-way overlap (from a) on the colony-forming ability of K562 cells (mean ± s.d., n = 3 replicates per group; two-tailed Student’s t-tests).
As gene expression analysis indicated that STAU2 loss resulted in reduced expression of not just KDM1A but a large number of the KDM family members, including other H3K4 demethylases such as KDM1B and KDM5B (Fig. 6d and Extended Data Fig. 7k), we tested the impact of STAU2 loss on global H3K4 methylation patterns. Our experiments showed that loss of STAU2 leads to a 1.6–3.4-fold accumulation in H3K4 mono-, di- and tri-methyl marks by immunofluorescence (Fig. 6e) and broad changes in H3K4Me2 and H3K4Me3 marks around the promoter regions by chromatin immunoprecipitation (ChIP)-seq (Extended Data Fig. 7l,m). These data indicate that the function of the KDM family of genes is altered in the absence of STAU2 and provide one example of how Stau2 can impact gene expression. Further, shRNA-mediated knockdown of the BRG1-interacting protein DPF2 and the histone family member HIST1H2BK also significantly reduced the colony-forming ability of K562 cells by 3.5–14-fold, identifying these as new drivers of leukemia propagation (Fig. 6f and Extended Data Fig. 7c).
Discussion
Since the discovery of CRISPR/Cas9-based genome editing, genome-wide in vitro CRISPR screens have been carried out largely in cancer cell lines to identify signals that are essential for survival19,44–46. One differentiating aspect of the genome-wide in vivo screen strategy reported here is that it was carried out in a physiologically relevant setting using LSCs, a population that drives cancer growth, progression and chemoresistance. This strategy has the advantage in that it not only comprehensively defines programs essential for cancer progression, such as developmental and oncogenic signals and chromatin remodeling, but also allows for the identification of microenvironment-responsive signals that are essential for leukemogenesis but may be missed in an in vitro setting. This includes angio-associated migratory protein and γ-aminobutyric acid type b receptor 1 surface molecules that have not previously been implicated in hematological malignancies and may be of interest in future work. These observations complement the growing evidence from previous work that adhesive signals, such as tetraspanins, integrins and integrin-pathway proteins, are key mediators of microenvironmental support and myeloid leukemia progression47–50. Because cell surface receptors can be attractive targets for antibody-mediated blocking, our work suggests that these candidates should be explored further to define their therapeutic importance in hematological malignancies.
A major discovery from the CRISPR screen was the identification of RBPs as a class of critical mediators of leukemia growth in vivo. While other CRISPR-based screens have implicated RBPs in cancer33,51, our unique network analysis of the subset of RBPs that were highly depleted led to the discovery of three distinct subcommunities of RBPs, with the chromatin-binding subfamily that included Stau2 emerging as a critical new regulator of myeloid leukemia. To conclusively define a functional role for Stau2 in aggressive myeloid leukemia progression, we generated a novel germline Stau2 knockout mouse targeting exon 4 that results in a loss of four of the five double-stranded RNA-binding domains. While a Stau2gt mouse targeting intron 7 (resulting in loss of the last two of the five annotated double-stranded RNA-binding domains) has recently been described and shows altered spatial learning behavior52, the impact of this deletion on hematopoiesis or in cancer is not known. Thus, the Stau2 knockout mouse that we generated is not only valuable in establishing the link from Stau2 to cancer, but also as a resource for identifying new functions of this RBP in other contexts.
RBPs are known to play a critical role in post-transcriptional regulation of RNA and modulate multiple pro-oncogenic functions, including epithelial-to-mesenchymal transition and invasion (Khsrp, Esrp1)53,54, self-renewal and survival (Msi2, Dcps, Rbm39)4,33,51,55, angiogenesis (Eif4e, Elavl1/HuR)56 and immune evasion (Lin28, Igf2bp3)57,58. Staufen has been shown to bind the transcripts of fate determinants such as miranda, prospero and Trim32 during development and regulate their asymmetric localization in dividing Drosophila neuroblasts and mammalian neuronal stem cells, thus promoting differential fate in daughter cells14,15,17. In the adult brain, Stau2 is also important for controlling local translation of proteins essential for synaptic plasticity and memory formation, by binding and transporting mRNAs such as Map1b in ribonucleoprotein complexes from the neuronal cell body to the synapse59,60. Whether Stau2 influences cancer development in a similar manner by binding mRNAs of fate determinants and asymmetrically localizing them is not known and will be interesting to explore.
The integrated analysis of eCLIP and RNA-seq with the in vivo CRISPR screen provides a unique tool to identify physiologically relevant downstream effectors of Stau2 function in aggressive myeloid leukemias. This approach unexpectedly identified Stau2 as an upstream regulator of broad-acting chromatin-binding factors such as the LSD/KDM family. This is particularly exciting because targeting histone methylation change is rapidly emerging as a therapeutic avenue for several cancers and this places Stau2 as an upstream regulator of such programs. This approach also identified a role for Stau2 in regulating powerful oncogenes such as Ras, Maz and RhoA and in modulating the Ras signaling network. Given that there are no known upstream regulators of NRas transcription, this indicates that Stau2 may also be a critical dependency across cancers and identifies new areas of research. These multiscale genomic data will hopefully serve as an important molecular map to identify critical dependencies of leukemia and other cancers.
Methods
Generation of experimental mice.
All animal experiments were performed according to protocols approved by the University of California San Diego Institutional Animal Care and Use Committee. Mice were bred and maintained in the animal care facilities at the University of California San Diego. For the CRISPR screen, donor cells were derived from Cas9-eGFP mice (strain, B6(C)-Gt(ROSA)26Sorem1.1(CAG−cas9*,−EGFP)Rsky/J). For all other experiments, Stau2+/+ and Stau2−/− mice described were used as donors. B6-CD45.1 (strain, B6.SJL-PtprcaPepcb/BoyJ) or C57BL6/J mice were used as transplant recipients. All mice were 6–16 weeks of age and mice of both sexes were used for all studies.
Generation of Stau2 knockout mice.
Stau2 knockout mice were generated by the Mouse Biology Program at University of California, Davis. Briefly, Cas9 (nuclease) and mRNA (from guide RNA sequence: GGGAAGCCTAAGGGTACAGACGA) was injected into fertilized zygotes of C57BL6/N mice and surviving embryos were surgically transferred into pseudopregnant recipient mice. F0 founders were bred with wild-type mice to confirm germline transmission and one founder with a single deletion (gggaatccgaa) in exon 4 of Stau2 was selected for further breeding. Sixteen loci with four mismatches were sequenced in the founder to rule out off-target effects (all other potential off-target regions had more than four mismatches and were not sequenced). F1 mice were back-crossed with C57BL6/J for two generations to further eliminate any off-target effects and the progeny were used for breeding and subsequent experiments (henceforth referred to as Stau2−/− mice).
Cell isolation and FACS analysis.
Cells were suspended in Hanks’ balanced salt solution (Gibco, Life Technologies) with 5% fetal bovine serum and 2 mM EDTA. Cells were prepared for FACS analysis and sorting as previously described47,49. The antibodies used for defining hematopoietic cell populations were as follows: CD3ε, CD4, CD8, Gr1, CD11b/Mac-1, TER119, CD45R/B220 and CD19 (all for lineage), CD117/cKit, Sca1, CD48 and CD150. Lin+ cells were marked by CD3ε, CD4, CD8, Gr1, CD11b/Mac-1, TER119, CD45R/B220 and CD19; KLS cells were defined as cKit+Lin−Sca+; adult HSCs were defined as cKit+Lin−Sca1+CD48− CD150+ (KLSCD48−CD150+, SLAM); MPPs were defined as KLSCD48−CD150−; committed progenitors were defined as GMP, Lin−IL7Ra−Kit+Sca1−CD34+CD16/3 2+; CMP, Lin−IL7Ra−Kit+Sca1−CD34+CD16/32−; and MEP, Lin−IL7Ra−Kit+Sca1−C D34−CD16/32−. All antibodies were purchased from BD Pharmigen, eBioscience or BioLegend. A detailed list of antibodies used is provided in Supplementary Table 3. Cell sorting and analysis were performed on a FACSAria III (Becton Dickinson) and the data were analyzed using FlowJo software (Tree Star).
Genome-wide in vivo CRISPR screen and bioinformatic analysis.
Lentiviral particles were made from the Brie library (Addgene 73633) using published protocols61 and used to infect 300 million freshly isolated Cas9-GFP+lineage− bcCML stem cells from spleens of premorbid mice at an multiplicity of infection of 0.25. At 60 h post-spinfection, an aliquot of cells was collected and represented a pool of infected cells with about 200 cells per guide (pre-selection). Remaining cells were selected for 48 h in puromycin to enrich for cells carrying gRNAs. Of these, some were saved for sequencing to identify genes critical for survival (post-selection at a coverage of about 200 cells per guide) and 35 million cells were transplanted in sublethally irradiated recipients (1 million cells per mouse) for a coverage of >400 cells per guide. Recipient mice were killed 7 d post-transplant, before onset of full-blown disease. Cells from the spleen of all 35 mice were pooled after sorting for equal numbers (~1.1 million cells per mouse) of leukemic cells (InVivo). Although bone marrow leukemia cells were also sorted, we could not obtain the necessary number of cells to achieve the required sequencing coverage, possibly due to damage post-irradiation. Genomic DNA was isolated from all samples using Qiagen DNeasy Blood and Tissue Kit, PCR amplified and sequenced as previously described61.
The CRISPR screen was analyzed assuming that each cellular species grows exponentially with growth rate related to its fitness. As there were only two time points (initial and final), we simply took the logarithm of the count ratio between the time points for each (single-guide RNA) species and adopted the empirical Bayes approach of B. Efron (https://www.rdocumentation.org/packages/locfdr/versions/1.1–8/topics/locfdr). Here, the main peak of distribution of log2 ratio of counts is considered to be empirically null and the significance of outliers (local false discovery rate; LFDR) is calculated with respect to this null distribution. As we do not have replicate experiments, we do not know whether the empirical null is different from the actual null. It is possible that the actual null is narrower than the empirical, meaning that the LFDR values obtained using the empirical null are conservative. If the actual null is wider than the empirical, the LFDR values are anti-conservative. Thus, only the fold change (dropout by threefold or more) and not the LFDR values were taken into consideration for all downstream validation and analysis.
In vivo transplantation assays.
For bone marrow transplants, 500 LT-HSCs (KLSCD150+CD48−) isolated from bone marrow of mice expressing CD45.2 were transplanted into lethally irradiated (9.5 Gy) congenic recipient mice (expressing CD45.1) along with 2 ×105 Sca1-depleted bone marrow rescue cells. Peripheral blood of recipient mice was collected every 4 weeks for up to 4 months after transplant. Bone marrow was analyzed at 4 weeks as previously described49.
Retroviral and lentiviral constructs and production.
Retroviral MSCV-BCR-ABL-IRES-YFP (or -tNGFR) and MSCV-NUP98-HOXA9-IRES-huCD2 (or -tNGFR) were used to generate bcCML. Lentiviral shRNA constructs were designed and cloned in the pLV-hU6-EF1a-green backbone (Biosettia) as per the manufacturer’s protocol. Individual gRNAs selected from the Brie library were cloned in Plentiguide-Puro (Addgene 52963) and inducible shRNAs were cloned in pLKO.1 Tet-On backbone (Addgene 21915) as per the manufacturer’s protocols. A detailed list of shRNA and gRNA sequences is provided in Supplementary Table 4. Virus was produced as described earlier11.
Generation and analysis of leukemic mice.
KLS cells were isolated and sorted from age- and sex-matched Stau2+/+ and Stau2−/− mice and infected with MSCV-BCR-ABL-IRES-tNGFR and MSCV-NUP98-HOXA9-IRES-huCD2 (or BCR-ABL-YFP and NUP98-HOXA9-tNGFR) to generate bcCML as previously described11. For secondary bcCML transplantations, cells recovered from terminally ill primary recipients were sorted for Lin− MSCV-BCR-ABL-IRES-tNGFR (or YFP) and MSCV-NUP98-HOXA9-IRES-huCD2 (or tNGFR) and transplanted into secondary recipients. Analysis of diseased mice was conducted as previously described11. Apoptosis assays were performed by staining cells with annexin V and 7AAD (eBioscience). Analysis of in vivo 5-bromodeoxyuridine (BrdU) incorporation was performed using the APC BrdU Flow kit (BD Pharmingen) after a single intraperitoneal injection of BrdU (2 mg at 10 mg ml−1). For inducible shRNA experiments, mice in each cohort were randomly assigned for bone marrow and spleen analyses or for survival studies.
Methylcellulose colony-formation assays.
For colony assays with mouse cells, indicated numbers of HSCs (KLSCD150+CD48−) or Lin− cells BCR-ABL/NUP98-HOXA9 leukemia were plated in complete methylcellulose medium (M3434 StemCell Technologies). HSC colonies were scored at 14 d and leukemic colonies were scored at 7 d.
Stau2 eCLIP and bioinformatic analysis.
Libraries were generated using standard eCLIP methods according to published protocols39. In summary, K562 cells (20 × 106) were UV crosslinked (400 mJ cm−2), lysed and sonicated (Bioruptor) in eCLIP lysis buffer. RNA fragments were digested (RNaseI, Ambion) and STAU2–RNA complexes were immunoprecipitated using Dynabeads bound to a STAU2-specific antibody (RN013P, MBL). A library was also generated from a size-matched input (SMInput) sample containing RNAs present in the whole-cell lysate, without RBP-specific immunoprecipitation (IP) and otherwise treated identically to the IPs. For the IPs, a series of stringent washes was followed by RNA dephosphorylation with FastAP (Thermo Scientific) and T4 PNK (NEB) then ligation of an adaptor to the 3′ ends of the RNAs with T4 RNA ligase 1 (NEB). Protein–RNA complexes were separated via SDS–PAGE, transferred to a nitrocellulose membrane and RNA was extracted from the membrane using proteinase K (NEB). Following purification, RNA was reverse transcribed with Superscript III (Invitrogen), free primers were removed with ExoSap-IT (Affymetrix) and ligated with a DNA adaptor on the 3′ end with T4 RNA ligase 1. Complementary DNA was amplified by PCR using Q5 Master Mix (NEB) and purified before Illumina sequencing.
Sequencing reads were processed using the bioinformatics pipeline described earlier39. Briefly, paired-end reads were demultiplexed using inline barcodes embedded within read 1 and trimmed to remove adaptors and low-quality sequences. Reads were mapped to human-specific RepBase (v.18.05) sequences to filter any repeat elements using STAR (2.4.0i). Surviving reads were mapped uniquely with STAR to the human genome (hg19) and PCR duplicates were removed using the randomer within read 2. Because each dataset consists of two inline barcodes, mapped reads from each were combined, sorted and the resulting merged BAM file was used to find clusters of enriched regions (peaks) with Clipper. These clusters were normalized using an identically processed size-matched input (SMInput) sample, by which the number of IP reads within clusters are compared to the number of SMInput reads in the same region. Significant peaks were determined using Fisher’s exact test with a cutoff of −log10(P value) ≥ 3 and log2(fold change) ≥ 3. A similar approach was used to determine broader regions of enrichment; instead of identifying peaks, reads were categorically binned into Gencode-defined (v.19) genic regions (CDS, 5′ UTR, 3′ UTR, 5′ UTR and 3′ UTR, intron, intergenic, noncoding_exon, noncoding_intron) and compared to the corresponding SMInput. The current processing pipeline can be found here: https://github.com/yeolab/eclip. Motif analysis was performed using HOMER (v.4.9.1), using significant peaks as input and randomly sampled regions for each region (CDS, 5′ UTR, 3′ UTR, proximal and distal intron) as background.
To determine double-stranded regions in STAU2 binding sites, computational secondary structure prediction was used to calculate base-pairing probabilities over every base in STAU2 binding sites62. Base-pairing probabilities were also calculated over equally sized regions flanking each STAU2 binding site and then compared. Significance was determined by shuffling the base-pairing probability values within the STAU2 binding sites and their corresponding flanking regions and comparing shuffled averages to actual. Although significant, it is likely that the differences are small due to limited accuracy of structural predictions without experimental structure probing data63 as well as the large size of the STAU2 binding sites, yet significant because signal is present in a large population of sites.
For metagene analysis, reproducible peaks between two replicates were found using IDR (v.2.0.2) and annotated using Gencode-defined (v.19) regions. Peaks that aligned unambiguously to genic regions (n = 7,168) were overlapped with genes that contained at least one bound peak (n = 2,305) to generate the metagene plot. For each position within a gene’s UTR or CDS region, a score of 1 was assigned if a peak was found and 0 otherwise, forming a series of positions that are bound and unbound. These positions were scaled according to the ratio of average lengths of genes expressed in K562: 5′ UTR = 13%, CDS = 100%, 3′ UTR = 49%. The number of binding events at each scaled position as a fraction of the total number of bound genes was then used to generate the metagene diagram, showing the pattern of significant binding across a generic mRNA transcript.
RNA sequencing and bioinformatic analysis.
K562 cells were infected with either control (shLacZ) or shSTAU2 lentivirus, in triplicate for each group. Infected cells were sorted and total RNA was isolated using RNeasy Micro kit (Qiagen). Libraries were generated from 200 ng of RNA using Illumina’s TruSeq Stranded mRNA Sample Prep kit following manufacturer’s protocols. Libraries were multiplexed and sequenced with 100-bp paired-end reads (PE100) to ~30 million reads per sample (Illumina HiSeq4000). Cutadapt was used to remove Truseq adaptors. The Ensembl human hg38 assembly (https://uswest.ensembl.org/info/data/ftp/index.html) was indexed and transcript quantifications were performed using kallisto with 50 bootstrap samples. All other kallisto parameters remained at default. Sleuth was used for gene-level quantification and differential expression analysis using the Wald test. Gene-set enrichment analysis was performed using Broad’s GSEA software with gene counts from sleuth analysis using the oncogenic signatures gene sets. Only those pathways that had a family-wise error rate (FWER) <0.30 were considered significant.
Network analysis.
The functional gene–protein interaction network PCNet64 was used. The human network contains 18,660 Entrez genes and 2,679,919 interactions and the mouse network was obtained by translating human gene IDs into mouse gene IDs using the National Center for Biotechnology Information gene orthology table. Network nodes corresponding to genes without murine orthologs were removed from the network; as a result, the murine PCNet had 15,714 genes and 2,348,920 interactions. The set of 3,411 genes depleted by a factor of three or more in the in vivo CRISPR screen is highly enriched in GO term RNA binding (GO:0003723, raw P value, 3 × 10−13; LFDR, 3 ×10−6).
To define a potential role of STAU2 in signaling, we selected 32 signaling pathways from the Kyoto Encyclopedia of Genes and Genomes database with relevance to cancer (Supplementary Table 2). For each signaling pathway we found genes that are significantly closer to STAU2 than expected, in a network sense and calculated mutual information ISTAU2,A between STAU2 and every other gene as described earlier65. A gene A is said to be closer to STAU2 in the network sense than gene B if ISTAU2,A > ISTAU2,B. Mutual information between two genes is not a monotonic function of the graph distance (length of shortest path) between them. The distribution of mutual information between STAU2 and a uniformly randomly chosen gene A is taken to be the null distribution. The set of values ISTAU2,A for genes A from a given pathway is a particular sample from the null distribution, albeit not random if the pathway is significantly influenced by STAU2. Thus, for each gene A in a given pathway, we calculate the expected number N(A) of genes as close or closer to STAU2 than A in a null set of genes of the same size as the pathway in question. We next define false discovery rate for each gene as the ratio N(A)/N(A), where N(A) is the actual number of genes in the pathway as close or closer to STAU2 than A. For each pathway, genes with a false discovery rate <0.1 were called significant (Supplementary Table 2). A significant gene in the context of any signaling pathway was considered a predicted putative STAU2 interactor. These are listed in the Supplementary Table 2 in decreasing order of mutual interaction with STAU2.
Normal human CD34+ cells and human leukemia samples.
Normal human CD34+ HSPCs were purchased (Stem Cell Technologies) and human leukemia samples were obtained from Singapore General Hospital, the Duke Adult Bone Marrow Transplant Clinic, the Fred Hutchinson Cancer Research Center (Fred Hutch/UW Hematopoietic Diseases Repository) and UC San Diego Moores Cancer Center from Institutional Review Board-approved protocols with written informed consent in accordance with the Declaration of Helsinki. Patient sample details are listed in Supplementary Table 5. These cells were infected with shRNAs as previously described49 and infected cells were either plated in complete methylcellulose medium (StemCell Technologies) or transplanted in sublethally irradiated (2.75 Gy) 12–20-week-old female NSG mice. Colonies were counted 7–14 d post-plating. Investigators were blinded to patient sample details at time of scoring. Mice were analyzed 6–8 weeks post-transplant.
ChIP-seq.
K562 cells infected with control or shSTAU2 were fixed with 1% formaldehyde and incubated with 125 nmol l−1 glycine for an additional 10 min to stop the crosslinking reaction. Cells were washed twice with PBS containing 1 mM PMSF and complete protease inhibitor (Roche) and lysed in SDS lysis buffer (50 mmol l−1 Tris-HCl (pH 8.2); 10 mmol l−1 EDTA; 1% SDS and 1 mmol l−1 PMSF and protease inhibitor). Lysates were sonicated using E220 Focused-ultrasonicator (Covaris), diluted fivefold in ChIP dilution buffer (12.5 mmol l−1 Tris-HCl (pH 8.2); 162.5 mmol l−1 NaCl and 1.25% Triton X-100) and incubated with anti-histone H3K4me2 or anti-histone H3K4me3 (Abcam) and protein G Dynabeads (Invitrogen) at 4 °C overnight. The immunoprecipitates were washed twice with low-salt wash buffer (20 mmol l−1 Tris-HCl (pH 8.2); 150 mmol l−1 NaCl; 2 mmol l−1 EDTA; 0.1% SDS and 1% Triton X-100), once with high-salt wash buffer (20 mmol l−1 Tris-HCl (pH 8.2); 500 mmol l−1 NaCl; 2 mmol l−1 EDTA; 0.1% SDS and 1% Triton X-100) and finally twice with TE containing 0.1% Triton X-100. The immunoprecipitated chromatin was eluted in elution buffer (1% SDS and 100 mmol l−1 NaHCO3), and crosslinking was reversed by overnight incubation at 65 °C in elution buffer with 300 mmol l−1 NaCl. The eluted chromatin was treated with RNase A (Invitrogen) and then with ProK buffer (40 mmol l−1 Tris-HCl (pH 6.5); 80 mg l−1 proteinase K and 10 mmol l−1 EDTA). The DNA was purified using ChIP DNA clean and concentrator (Zymo Research) following the manufacturer’s procedure. Library prep for sequencing was performed using KAPA Hyper Prep kit (Kapa Biosystems) following the manufacturer’s procedure. DNA fragments were sequenced on HiSeq4000 (Illumina).
The ChIP-seq fastq files were processed and aligned within the systemPipeR R package. Sequence reads were trimmed and aligned to the hg38 human assembly using bowtie2 with a -k parameter of 50 and the non-deterministic option (bowtie2). Macs2 was used for peak calling using the default settings and ChIPseeker was used for further analysis of peak locations and quantifications (Macs and ChIPseeker ref). Deeptools was used for normalization of the alignments and visualization of the read densities surrounding the promoter regions.
Statistics and reproducibility.
The number of samples and statistical analyses are described for each figure. Statistical analyses were carried out using GraphPad Prism software v.8.0 (GraphPad Software) or Enrichr where indicated. Data are mean ± s.e.m. or mean ± s.d. as indicated. Two-tailed Student’s t-tests and log-rank tests were used to determine statistical significance. No statistical method was used to predetermine sample size and no data were excluded from the analyses. Randomization and blinding were conducted as indicated. All experiments were reproducible.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The CRISPR screen, STAU2 eCLIP, STAU2 knockdown RNA-seq data and the H3K4Me ChIP data that support the findings of this study have been deposited in GenBank (accession codes GSE135300, GSE134971, GSE135012 and GSE142307, respectively). The source data associated with each figure are provided with the manuscript. All other data supporting the findings of this study are available from the corresponding author on reasonable request.
Code availability
The current processing pipeline for eCLIP can be found at https://github.com/yeolab/eclip. For ChIP-seq, data analysis was performed in R and Python with the following packages: systemPipeR (https://bioconductor.org/packages/release/bioc/html/systemPipeR.html), bowtie2 (http://bowtie-bio.sourceforge.net/bowtie2/index.shtml), MACS2 https://github.com/taoliu/MACS, ChIPseeker (https://bioconductor.org/packages/release/bioc/html/ChIPseeker.html), deepTools (https://deeptools.readthedocs.io/en/develop/) and IGV (https://software.broadinstitute.org/software/igv/). The RNA-seq data analysis was performed in R and web-based programs with the following packages: kallisto (https://pachterlabgithub.io/kallisto/), sleuth (https://github.com/pachterlab/sleuth), GSEA (https://www.gsea-msigdb.org/gsea/index.jsp) and Enrichr (https://amp.pharm.mssm.edu/Enrichr/). All computer code is available upon reasonable request. Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. Genome-wide CRISPR screen in myeloid leukemia.
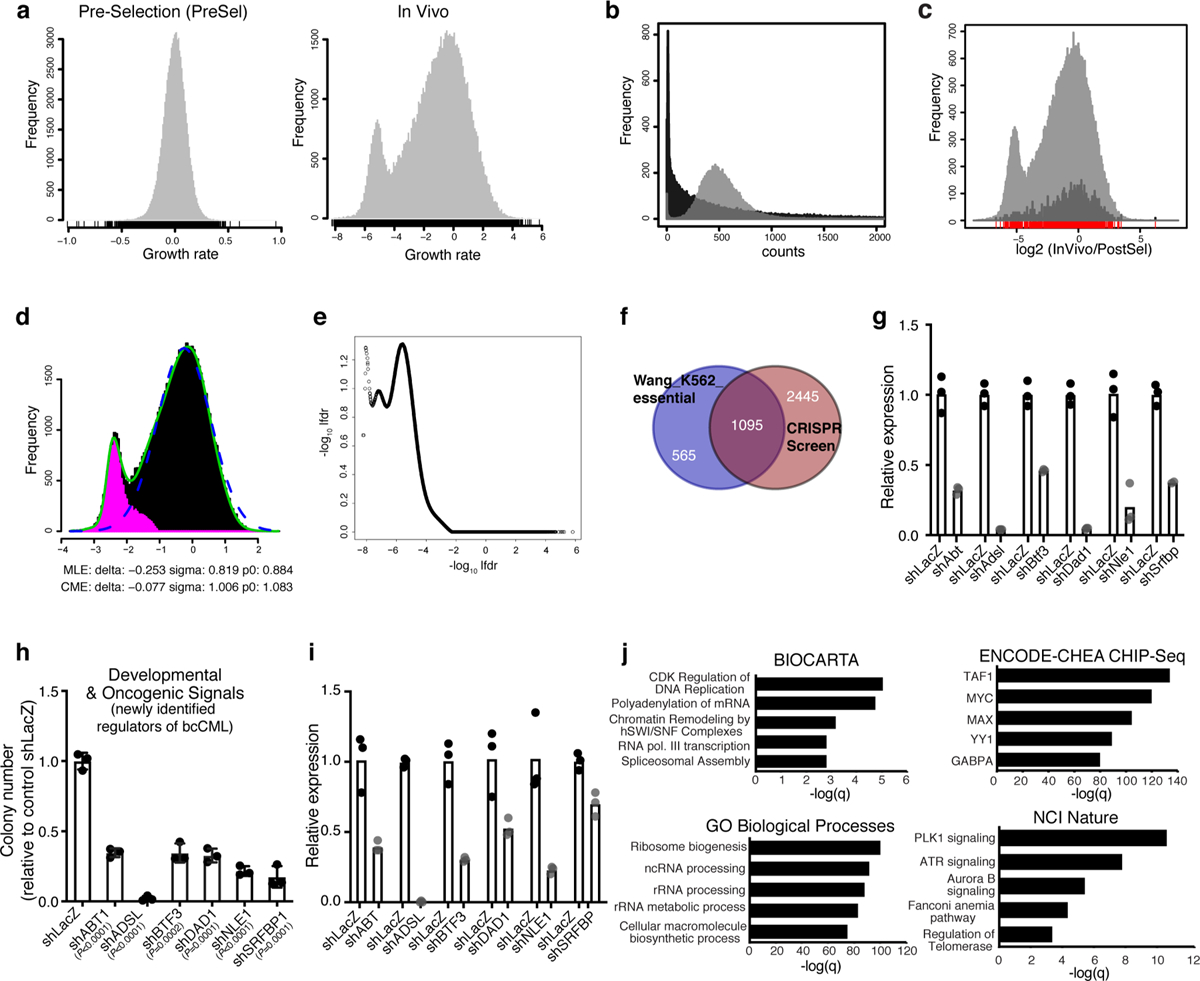
a, Histograms show the growth rates for samples Pre-selection (PreSel) and InVivo with respect to Post-selection (PostSel, control) for all sgRNAs in the Brie library. b, Histograms of sgRNA counts after in vitro selection (PostSel, grey histogram) and after in vivo selection (InVivo, black histogram). c, Graphical representation of sgRNA count ratio [log2 (InVivo/PostSel)] of all sgRNA species, including the controls (grey) or control sgRNA species alone (amplified by a factor of 10 for visibility, black). d, Plot showing method to identify genes with true selection in vivo. The x-coordinate is the z-statistic of the log2 count ratio at the gene level, between InVivo and PostSel. The green line is the actual distribution with the secondary peak; the dashed blue line is the empirical null distribution, and the pink histogram signifies estimated non-null genes (genes with true selection). e, Graphical representation of the CRISPR screen on a plot showing −log10lfdr versus log2(InVivo/PostSel). f, Venn diagram shows overlap between reported cell essential genes for K562 bcCML cells19 (n = 1660) and genes that are depleted by 3-fold or more in the in vivo CRISPR screen (n = 3540); p < 0.0001 (hypergeometric probability formula). g, Relative RNA expression of the indicated genes in NIH-3T3 cells expressing the indicated shRNAs against novel leukemia regulators relative to control shLacZ (n = 3 technical replicates per group). h, Impact on colony forming ability of K562 cells transduced with lentiviral shRNAs against the indicated novel leukemia regulators relative to control (shLacZ) (n = 3 independent culture wells per group; two-tailed Student’s t-tests). i, Relative RNA expression of the indicated genes in K562 cells expressing the indicated shRNAs against novel leukemia regulators relative to control shLacZ (n = 3 technical replicates). j, Enriched molecular programs identified from the Enrichr analysis of genes that were depleted by 3-fold or more in vivo as compared to input (n = 3540; Fisher’s exact test).
Extended Data Fig. 2 |. Genome-wide CRISPR screen identifies Staufen2 as a regulator of myeloid leukemia.
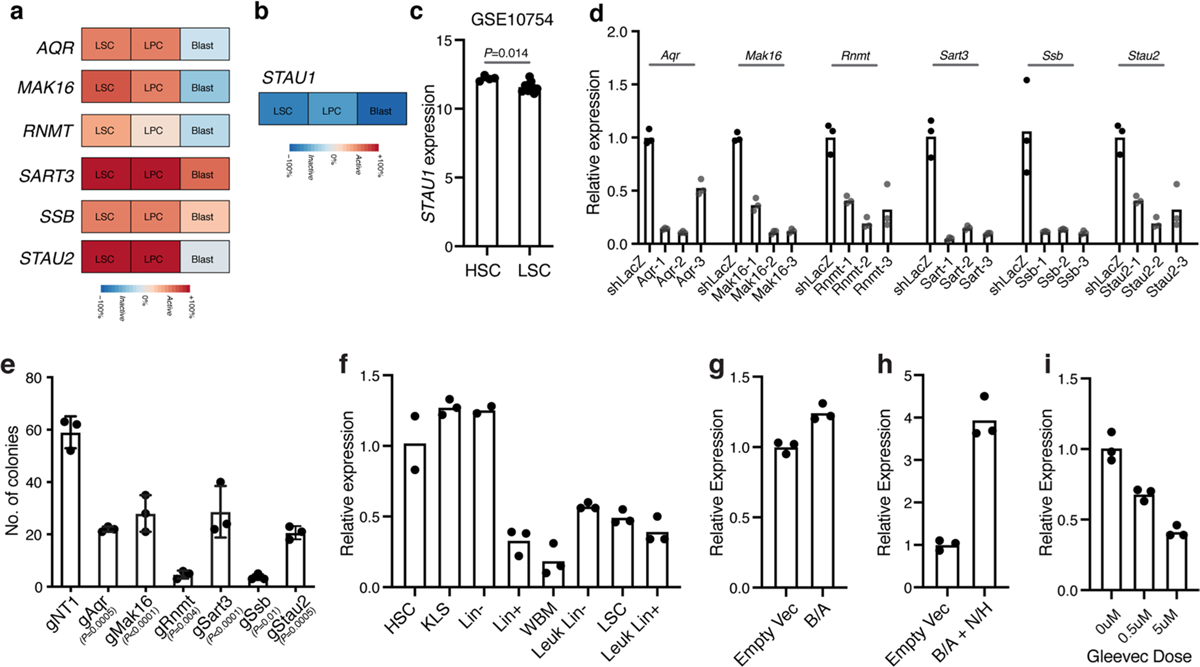
a, Relative expression of the indicated genes in human leukemia stem cells as compared to more differentiated cancer populations from Gene Expression Commons. b, Relative expression of STAU1 in human leukemia stem cells as compared to more differentiated cancer populations from the Riken database. c, STAU1 expression in human HSCs and human leukemia stem cells (mean±S.E.M.; n = 4 independent HSC and n = 9 independent LSC samples; two-tailed Student’s t-test). d, Relative RNA expression of the indicated genes in NIH-3T3 cells expressing shRNAs against RNA-binding proteins relative to control shLacZ (n = 3 technical replicates per group). e, Number of colonies formed by Cas9+ bcCML lin- cells transduced with the CRISPR guides against the indicated genes relative to a control non-targeting gRNA (mean±S.D.; n = 3 independent culture wells per group; two-tailed Student’s t-test). f, Relative expression of Stau2 transcript in the indicated populations isolated from the normal bone marrow of wild-type mice and sorted Lin+ and Lin− and leukemia stem cell (LSC) populations from established wild-type bcCML (n = 3 technical replicates for KLS, Lin+, Leukemic Lin+, Lin−, LSC, and n = 2 technical replicates for HSC and Lin−). g-h, Relative Stau2 expression in KLS cells transduced with BCR-ABL (g) or BCR-ABL and NUP98−HOXA9 (h) 72–96 h post-infection (n = 3 technical replicates per group). i, Impact of indicated doses of Gleevec for 48 h on Stau2 expression in established Lin− bcCML cells (n = 3 technical replicates per group).
Extended Data Fig. 3 |. Impact of genetic loss of Stau2 on normal HSC function.
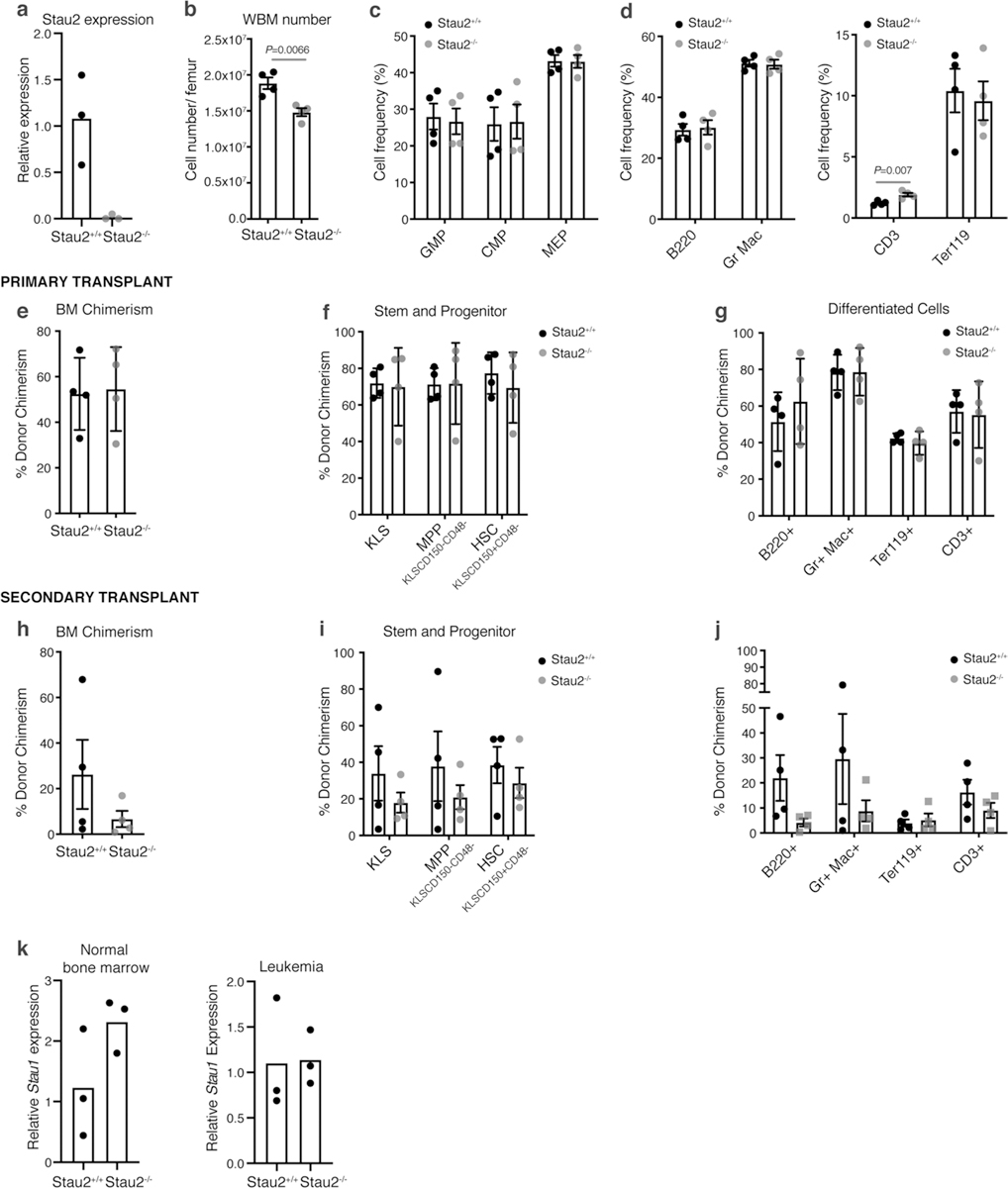
a, Relative Stau2 expression in whole bone marrow cells isolated from Stau2+/+ and Stau2−/− mice (n = 3 technical replicates per group). b, Total number of cells (mean±S.E.M.) in the bone marrow of Stau2+/+ and Stau2−/− mice (n = 4 animals per cohort; data combined from 3 independent experiments; two-tailed Student’s t-test). c-d, Frequency of committed progenitors (c) and differentiated cells (d) in the bone marrow of Stau2+/+ and Stau2−/− mice (mean±S.E.M.; n = 4 animals per cohort; data combined from 3 independent experiments; two-tailed Student’s t-test). e, Frequency of donor-derived Stau2+/+ or Stau2−/− cells in the bone marrow of recipient mice transplanted with 500 HSCs of each genotype, 4 months post-transplant (mean±S.D.; n = 4 animals per cohort; data combined from 3 independent experiments). f-g, Frequency of indicated donor-derived hematopoietic stem and progenitor and differentiated cell populations in the bone marrow of recipient mice transplanted with Stau2+/+ or Stau2−/− HSCs 4 months post-transplant (mean±S.D.; n = 4 animals per cohort; data combined from 3 independent experiments). h, Frequency of donor-derived Stau2+/+ or Stau2−/− cells in the bone marrow of secondary transplant recipients, 4 months post-transplant (mean±S.E.M; n = 4 animals per cohort; data combined from 3 independent experiments). i-j, Frequency of indicated donor-derived hematopoietic stem and progenitor and differentiated cell populations in the bone marrow of secondary transplant recipients, 4 months post-transplant (mean±S.D.; n = 4 animals per cohort; data combined from 3 independent experiments). k, Relative Stau1 expression in normal hematopoietic cells from Stau2+/+ and Stau2−/− mice (left panel) or in Lin- leukemia cells (right panel).
Extended Data Fig. 4 |. Role of Stau2 in bcCML progression.
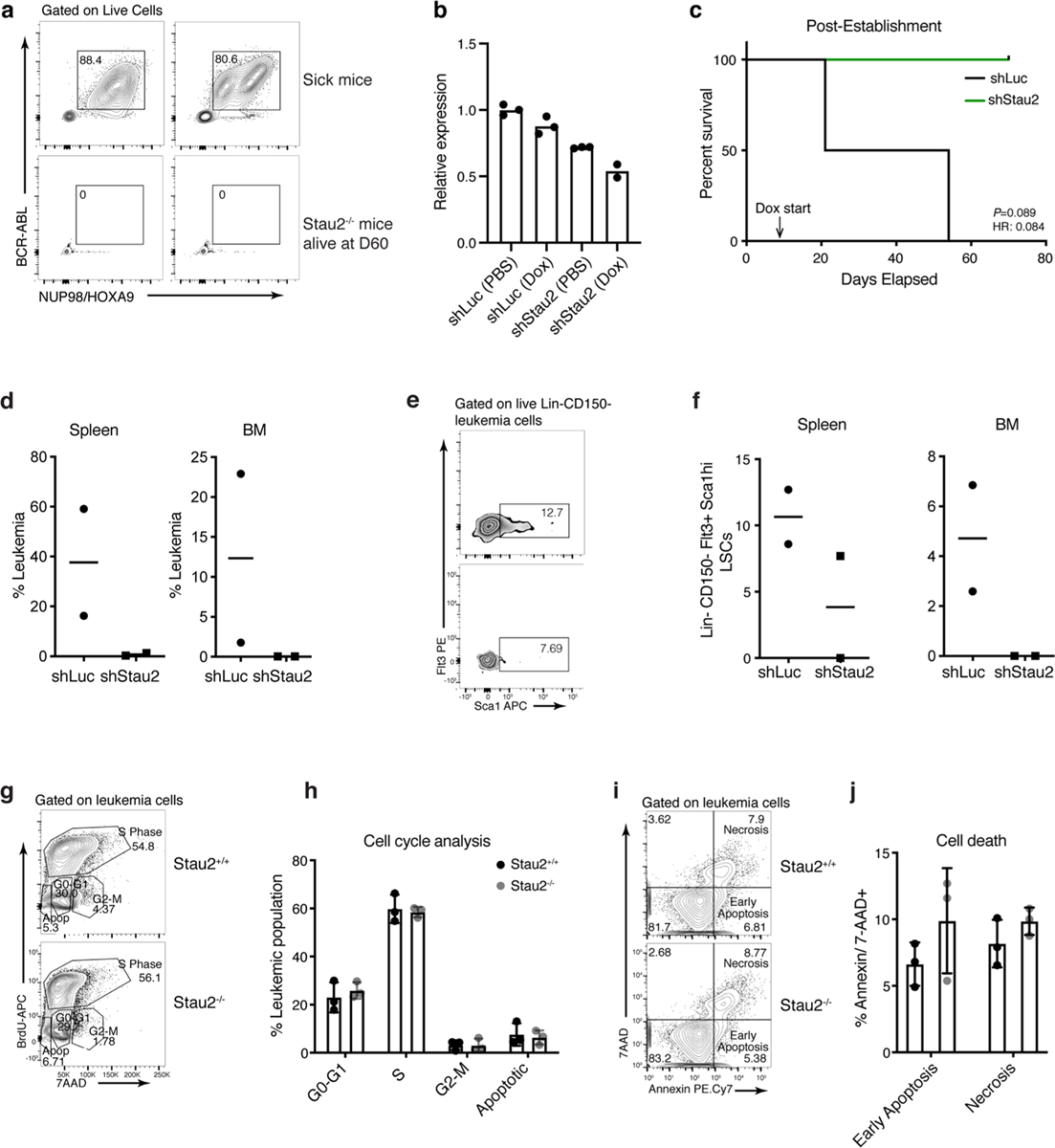
a, Representative FACS plots show leukemia burden in control sick mice at disease end-point (top panel) and in Stau2−/− mice that did not develop disease 60d post-transplant (bottom panel; n = 2 animals per cohort). b, Relative Stau2 expression in Lin- bcCML cells transduced with inducible shLuc or shStau2. Cells were treated with PBS (control) or Doxycycline (to induce the shRNA expression) for 48 h in vitro and analyzed by qRT-PCR (n = 3 technical replicates per group except shStau2 Dox where n = 2). c, bcCML Lin- cells expressing doxycycline inducible control (shLuc) or Stau2 shRNAs (shStau2) were transplanted and recipients were given doxycycline water from day 6 post-transplant and survival monitored (n = 2 animals per cohort; data shown from one representative experiment, which was repeated with similar results, see Fig. 3g). d, Frequency of leukemia cells in the bone marrow and spleen of mice transplanted with shLuc and shStau2. shRNA expression was induced with doxycycline 6 days post-transplant and the bone marrow and spleen were analyzed 14d post-transplant (n = 2 animals per cohort, line represents median). e-f, Representative FACS plots (e) and graph (f) show the LSC frequency in mice transplanted with Lin-bcCML cells transduced with inducible shRNAs against control (shLuc) or Stau2 (n = 2 animals per cohort, line represents median). g-h, Lin- Stau2+/+ and Stau2−/− cells were transplanted into recipients. Thirteen days after transplant, mice were injected with bromodeoxyuridine (BrdU) and cells analyzed for incorporation 18 h later. Representative FACS plots show BrdU and 7AAD staining of BCR-ABL+Nup98−HOXA9+ Stau2+/+ and Stau2−/− leukemic cells (g). Average frequency of cells in distinct phases of the cell cycle (mean±S.D.; n = 3 animals per cohort) is shown (h). i-j, Representative FACS plots (i) and graph (j) show analysis of early and late apoptosis in BCR-ABL+NUP98-HOXA9+ leukemia cells 13 days post-transplant (mean±S.E.M; n = 3 animals per cohort).
Extended Data Fig. 5 |. Impact of STAu2 knockdown on human myeloid leukemia and normal human HSPCs.
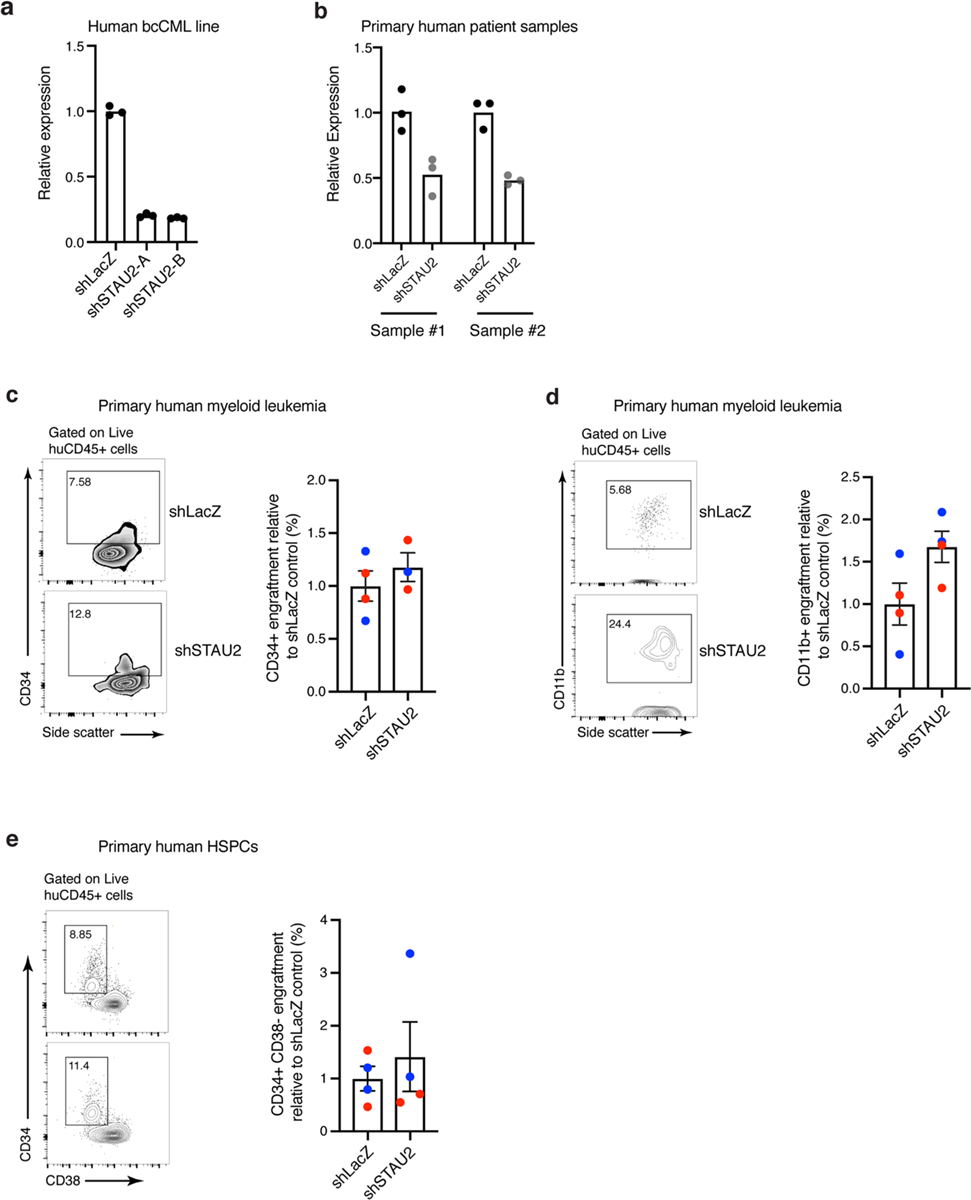
a, Graphs show relative STAU2 expression in K562 cells transduced with either LacZ (control) or STAU2 shRNAs (A and B; n = 3 technical replicates per group). b, Relative STAU2 expression in two representative primary human myeloid leukemia samples transduced with shLacZ or shSTAU2. Expression was analyzed 72 h post transduction (n = 3 technical replicates per group). c, Representative FACS plot (left) and graph (right) showing the frequency of CD34+ cells in engrafted human leukemia samples in the bone marrow of NSG mice 7–8 weeks post-transplant. Each dot represents a mouse and each color represents a primary human sample (bcCML in blue and AML in red; mean±S.E.M.; n = 4 animals per cohort). d, Representative FACS plot (left) and graph (right) showing the frequency of CD11b+ differentiated cells in the engrafted human leukemia samples in the bone marrow of NSG mice 7–8 weeks post-transplant. Each dot represents a mouse and each color represents a primary human sample (bcCML in blue and AML in red; mean±S.E.M.; n = 4 animals per cohort). e, Representative FACS plot (left) and graph (right) showing the relative frequency of CD34+ CD38− cells in the engrafted normal primary human CD34+ HSPC samples in the bone marrow of NSG mice 8 weeks post-transplant. Each dot represents a mouse and each color represents an individual human sample (mean±S.E.M.; n = 4 animals per cohort).
Extended Data Fig. 6 |. STAu2 targets genes identified by eCLIP Analysis.
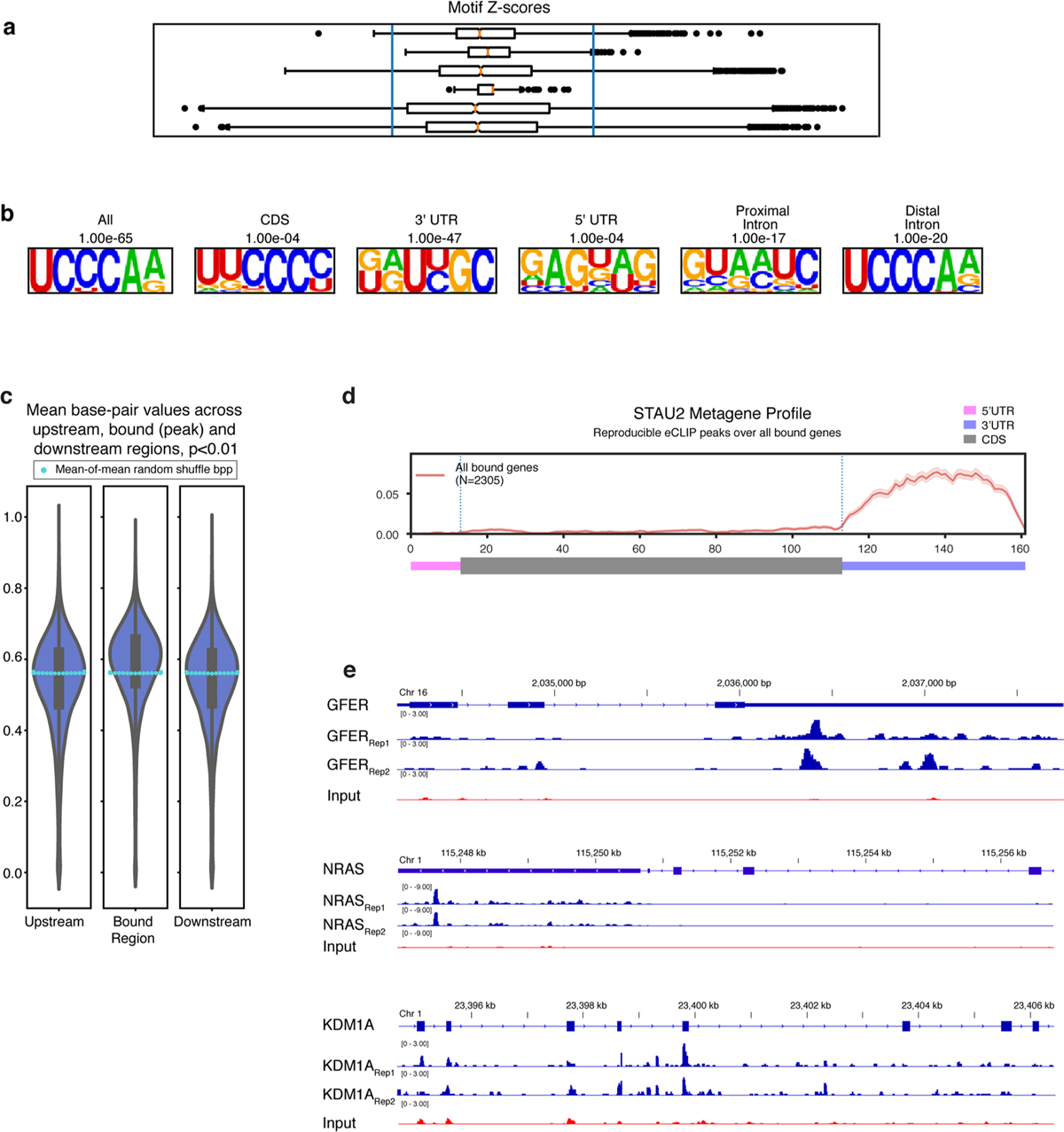
a, Z-scores of individual k-mers from Stau2 eCLIP (in the CDS, the UTRs and introns). b, Motifs identified by HOMER analysis of Stau2 eCLIP peaks located within different annotated regions. c, Violin plot of the average base pairing probabilities across each reproducible STAU2 peak and their flanking regions. The swarmplot of light-blue dots represent median values of 100 iterations of random shuffled base pair probability means to show significance (compared to the white dots showing actual median bpp). d, Metagene diagram showing the pattern of significant binding across a generic mRNA transcript. e, eCLIP traces showing STAU2 binding peaks in the indicated 3’UTR regions of GFER and NRAS and in the CDS of KDM1A.
Extended Data Fig. 7 |. Impact of Stau2 and Kdm1a inhibition on normal and malignant hematopoiesis.
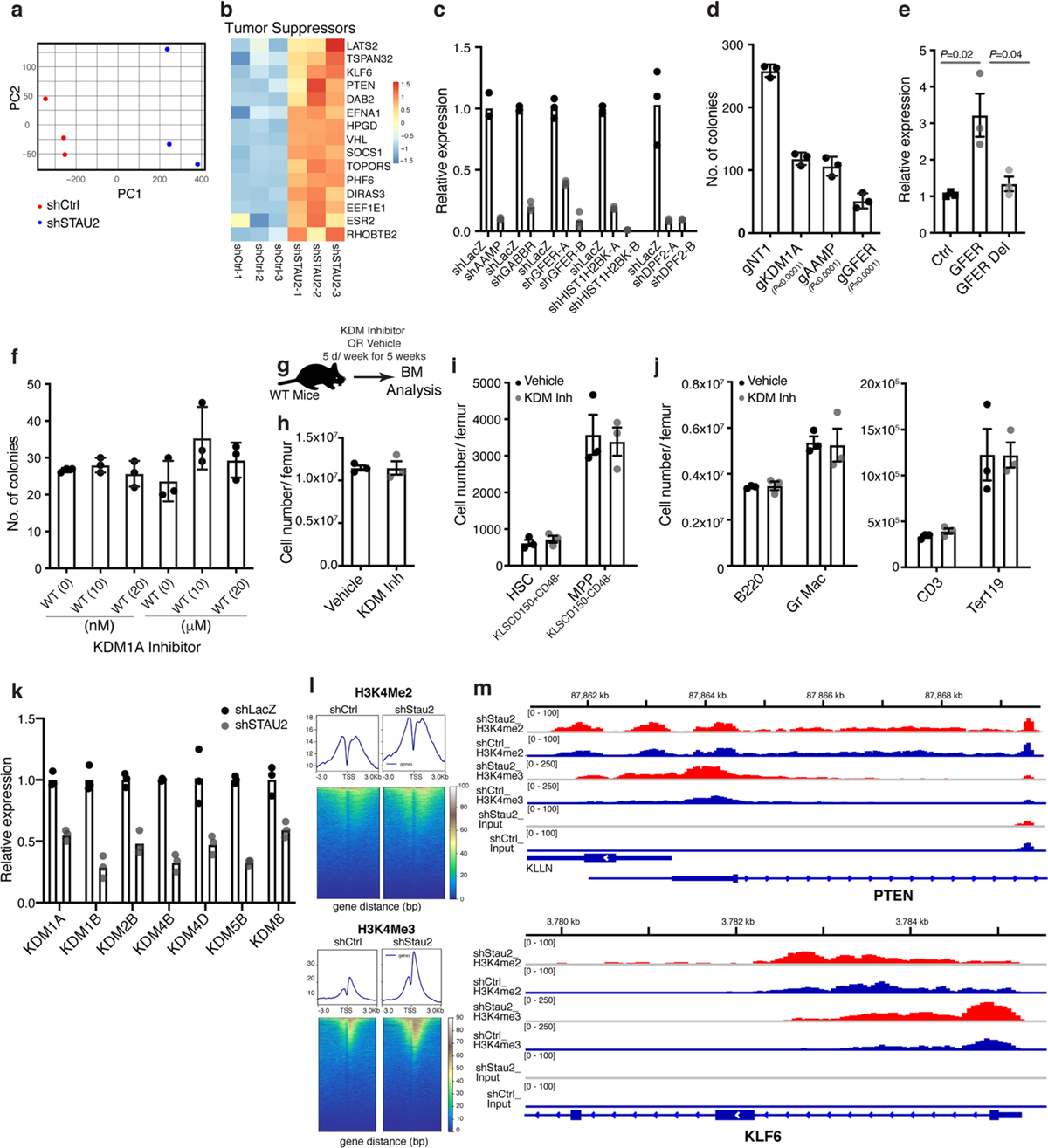
a, PCA-plot showing the distribution of the three control (Ctrl, shLacZ) and three STAU2 knockdown (shSTAU2) samples. b, Heatmap shows the changes in RNA expression of selected tumor suppressor genes upon STAU2 knockdown. c, Relative RNA expression of indicated genes in K562 cells expressing indicated shRNAs, normalized to control shLacZ (n = 3 technical replicates per group). d, Number of colonies formed by K562 cells transduced with the CRISPR-Cas9 guides against the indicated genes relative to a control non-targeting gRNA (mean±S.D.; n = 3 independent culture wells per group; two-tailed Student’s t-test). e, Relative luciferase transcript levels in 293 T cells expressing control 3′UTR reporter, GFER 3’UTR with STAU2 binding sites (bases 309–399 downstream of the stop codon) and GFER del 3’UTR with a partial STAU2 binding site (truncated at 339 bp downstream of the stop codon) (mean±S.E.M.; n = 3 independent samples per group, each in triplicate; two-tailed Student’s t-tests). f, Impact of inhibiting KDM1A at the indicated concentrations on the colony forming ability of normal hematopoietic stem cells (Li n−cKit+Sca1+CD150+CD48−) (mean±S.D.; n = 3 independent culture wells per group). g, Schematic shows KDM1A inhibitor treatment strategy to determine impact of KDM1A inhibition on normal hematopoiesis. h-j, Graphs show the average numbers of bone marrow cells (h), stem cells (HSC: KLSCD150+CD48−) and multipotent progenitors (MPP: KLSCD150−CD48−) (i), and differentiated hematopoietic cells (j) in the bone marrow of mice treated with vehicle or KDM1A inhibitor (mean±S.D.; n = 3 animals per cohort). k, Relative RNA expression of indicated chromatin regulators in K562 cells expressing control (shLacZ) or STAU2 shRNA (n = 3 technical replicates per group). l, Heatmaps showing H3K4Me2 and H3K4Me3 marks in K562 cells expressing control (shLacZ) or STAU2 shRNA (shSTAU2). m, ChIP-Seq traces showing H3K4Me2 and H3K4Me3 binding in the promoter regions of PTEN and KLF6 genes.
Supplementary Material
Acknowledgements
We are grateful to S. Levi for technical support and M. Kritzik for help with manuscript preparation. We thank P. Adams and P.M. Vertino for scientific advice, W. Pear (University of Pennsylvania) and A.M. Pendergast (Duke University) for the BCR-ABL construct and D.G. Gilliland for the NUP98-HOXA9 construct. J.B. is a recipient of a Scholar Award from the American Society of Hematology and a postdoctoral fellowship from the National Cancer Center. M.H. was supported by the National Institutes of Health (NIH) Training Grant T32HL086344 and K.S. received support from NIH Training Grants T32HL086344 and T32CA009523. E.L.V.N. was a Merck Fellow of the Damon Runyon Cancer Research Foundation (DRG-2172–13) and is supported by the National Human Genome Research Institute (HG009530). J.S.-B. was the Fraternal Order of Eagles Fellow of the Damon Runyon Cancer Research Foundation. This work was supported by NIH grants R35 CA210043 awarded to A.R; U54HG007005 and U41HG009889 awarded to G.W.Y.; and DK099335, DP1 CA174422 and R35 CA197699 awarded to T.R.
Footnotes
Competing interests
G.W.Y. is co-founder, member of the Board of Directors, on the SAB, equity holder and paid consultant for Locana and Eclipse BioInnovations. G.W.Y. is also a visiting professor at the National University of Singapore and receives travel reimbursement. E.L.V.N. is co-founder, member of the Board of Directors, on the SAB, equity holder and paid consultant for Eclipse BioInnovations. G.W.Y.’s and E.L.V.N.’s interests have been reviewed and approved by the University of California San Diego in accordance with its conflict of interest policies.
Additional information
Extended data is available for this paper at https://doi.org/10.1038/s43018-020-0054-2.
Supplementary information is available for this paper at https://doi.org/10.1038/s43018-020-0054-2.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Druker BJ et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl-positive cells. Nat. Med 2, 561–566 (1996). [DOI] [PubMed] [Google Scholar]
- 2.Saussele S & Silver RT Management of chronic myeloid leukemia in blast crisis. Ann. Hematol 94(Suppl. 2), S159–S165 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Kantarjian HOB et al. Improved survival in chronic myeloid leukemia since the introduction of imatinib therapy: a single-institution historical experience. Blood 119, 1981–1987 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ito T et al. Regulation of myeloid leukaemia by the cell-fate determinant Musashi. Nature 466, 765–768 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiang Q et al. ADAR1 promotes malignant progenitor reprogramming in chronic myeloid leukemia. Proc. Natl Acad. Sci. USA 110, 1041–1046 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cong L et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jinek M et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mali P et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kharas MG et al. Musashi-2 regulates normal hematopoiesis and promotes aggressive myeloid leukemia. Nat. Med 16, 903–908 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park SM et al. Musashi2 sustains the mixed-lineage leukemia-driven stem cell regulatory program. J. Clin. Invest 125, 1286–1298 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zimdahl B et al. Lis1 regulates asymmetric division in hematopoietic stem cells and in leukemia. Nat. Genet 46, 245–252 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schupbach T & Wieschaus E Maternal-effect mutations altering the anterior–posterior pattern of the Drosophila embryo. Roux’s Arch. Dev. Biol 195, 302–317 (1986). [DOI] [PubMed] [Google Scholar]
- 13.St Johnston D, Beuchle D & Nusslein-Volhard C Staufen, a gene required to localize maternal RNAs in the Drosophila egg. Cell 66, 51–63 (1991). [DOI] [PubMed] [Google Scholar]
- 14.Li P, Yang X, Wasser M, Cai Y & Chia W Inscuteable and Staufen mediate asymmetric localization and segregation of prospero RNA during Drosophila neuroblast cell divisions. Cell 90, 437–447 (1997). [DOI] [PubMed] [Google Scholar]
- 15.Slack C, Overton PM, Tuxworth RI & Chia W Asymmetric localisation of Miranda and its cargo proteins during neuroblast division requires the anaphase-promoting complex/cyclosome. Development 134, 3781–3787 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Heraud-Farlow JE & Kiebler MA The multifunctional Staufen proteins: conserved roles from neurogenesis to synaptic plasticity. Trends Neurosci 37, 470–479 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kusek G et al. Asymmetric segregation of the double-stranded RNA binding protein Staufen2 during mammalian neural stem cell divisions promotes lineage progression. Cell Stem Cell 11, 505–516 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vessey JP et al. An asymmetrically localized Staufen2-dependent RNA complex regulates maintenance of mammalian neural stem cells. Cell Stem Cell 11, 517–528 (2012). [DOI] [PubMed] [Google Scholar]
- 19.Wang T, Wei JJ, Sabatini DM & Lander ES Genetic screens in human cells using the CRISPR-Cas9 system. Science 343, 80–84 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reavie L et al. Regulation of c-Myc ubiquitination controls chronic myelogenous leukemia initiation and progression. Cancer Cell 23, 362–375 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trotta R et al. BCR/ABL activates mdm2 mRNA translation via the La antigen. Cancer Cell 3, 145–160 (2003). [DOI] [PubMed] [Google Scholar]
- 22.Zhao C et al. Loss of β-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell 12, 528–541 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Airiau K, Mahon FX, Josselin M, Jeanneteau M & Belloc F PI3K/mTOR pathway inhibitors sensitize chronic myeloid leukemia stem cells to nilotinib and restore the response of progenitors to nilotinib in the presence of stem cell factor. Cell Death Dis 4, e827 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bacher U, Haferlach T, Schoch C, Kern W & Schnittger S Implications of NRAS mutations in AML: a study of 2502 patients. Blood 107, 3847–3853 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Wang Y et al. Th e Wnt/β-catenin pathway is required for the development of leukemia stem cells in AML. Science 327, 1650–1653 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mizukawa B et al. The cell polarity determinant CDC42 controls division symmetry to block leukemia cell differentiation. Blood 130, 1336–1346 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi J et al. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Devel 27, 2648–2662 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harris WJ et al. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell 21, 473–487 (2012). [DOI] [PubMed] [Google Scholar]
- 29.Park H et al. Adenylosuccinate lyase enhances aggressiveness of endometrial cancer by increasing killer cell lectin-like receptor C3 expression by fumarate. Lab. Investig 98, 449–461 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Zhang DZ et al. Basic transcription factor 3 is required for proliferation and epithelial-mesenchymal transition via regulation of FOXM1 and JAK2/STAT3 signaling in gastric cancer. Oncol. Res 25, 1453–1462 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beck-Cormier S et al. Notchless is required for axial skeleton formation in mice. PLoS ONE 9, e98507 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fei T et al. Genome-wide CRISPR screen identifies HNRNPL as a prostate cancer dependency regulating RNA splicing. Proc. Natl Acad. Sci. USA 114, E5207–E5215 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang E et al. Targeting an RNA-binding protein network in acute myeloid leukemia. Cancer Cell 35, 369–384 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blondel VD, Guillaume J-L, Lambiotte R & Lefebvre E Fast unfolding of communities in large networks. J. Stat. Mech 10, 10008–10020 (2008). [Google Scholar]
- 35.Deng X, Su R, Feng X, Wei M & Chen J Role of N(6)-methyladenosine modification in cancer. Curr. Opin. Genet. Dev 48, 1–7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bielli P, Busa R, Paronetto MP & Sette C The RNA-binding protein Sam68 is a multifunctional player in human cancer. Endocr. Relat. Cancer 18, R91–R102 (2011). [DOI] [PubMed] [Google Scholar]
- 37.Seita J et al. Gene Expression Commons: an open platform for absolute gene expression profiling. PLoS ONE 7, e40321 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Neering SJ et al. Leukemia stem cells in a genetically defined murine model of blast-crisis CML. Blood 110, 2578–2585 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van Nostrand EL et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat. Methods 13, 508–514 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peng C et al. PTEN is a tumor suppressor in CML stem cells and BCR-ABL-induced leukemias in mice. Blood 115, 626–635 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Humbert M et al. Deregulated expression of Kruppel-like factors in acute myeloid leukemia. Leukemia Res 35, 909–913 (2011). [DOI] [PubMed] [Google Scholar]
- 42.Kim YK, Furic L, Desgroseillers L & Maquat LE Mammalian Staufen1 recruits Upf1 to specific mRNA 3′ UTRs so as to elicit mRNA decay. Cell 120, 195–208 (2005). [DOI] [PubMed] [Google Scholar]
- 43.Maes T et al. ORY-1001, a potent and selective covalent KDM1A inhibitor, for the treatment of acute leukemia. Cancer Cell 33, 495–511 e412 (2018). [DOI] [PubMed] [Google Scholar]
- 44.Lytle NK et al. A multiscale map of the stem cell state in pancreatic adenocarcinoma. Cell 177, 572–586 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang T et al. Gene essentiality profiling reveals gene networks and synthetic lethal interactions with oncogenic Ras. Cell 168, 890–903 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yau EH et al. Genome-wide CRISPR screen for essential cell growth mediators in mutant KRAS colorectal cancers. Cancer Res 77, 6330–6339 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bajaj J et al. CD98-mediated adhesive signaling enables the establishment and propagation of acute myelogenous leukemia. Cancer Cell 30, 792–805 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jin L, Hope KJ, Zhai Q, Smadja-Joffe F & Dick JE Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat. Med 12, 1167–1174 (2006). [DOI] [PubMed] [Google Scholar]
- 49.Kwon HY et al. Tetraspanin 3 is required for the development and propagation of acute myelogenous leukemia. Cell Stem Cell 17, 152–164 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miller PG et al. In vivo RNAi screening identifies a leukemia-specific dependence on integrin β 3 signaling. Cancer Cell 24, 45–58 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yamauchi T et al. Genome-wide CRISPR-Cas9 screen identifies leukemia-specific dependence on a pre-mRNA metabolic pathway regulated by DCPS. Cancer Cell 33, 386–400 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Popper B et al. Staufen2 deficiency leads to impaired response to novelty in mice. Neurobiol. Learn. Mem 150, 107–115 (2018). [DOI] [PubMed] [Google Scholar]
- 53.Mager LF et al. The ESRP1-GPR137 axis contributes to intestinal pathogenesis. eLife 6, e28366 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Puppo M et al. miRNA-mediated KHSRP silencing rewires distinct post-transcriptional programs during TGF-β-induced epithelial-to-mesenchymal transition. Cell Rep 16, 967–978 (2016). [DOI] [PubMed] [Google Scholar]
- 55.Fox RG et al. Image-based detection and targeting of therapy resistance in pancreatic adenocarcinoma. Nature 534, 407–411 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chang SH et al. ELAVL1 regulates alternative splicing of eIF4E transporter to promote postnatal angiogenesis. Proc. Natl Acad. Sci. USA 111, 18309–18314 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jiang S & Baltimore D RNA-binding protein Lin28 in cancer and immunity. Cancer Lett 375, 108–113 (2016). [DOI] [PubMed] [Google Scholar]
- 58.Schmiedel D et al. The RNA binding protein IMP3 facilitates tumor immune escape by downregulating the stress-induced ligands ULPB2 and MICB. eLife 5, e13426 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dubnau J et al. The Staufen/pumilio pathway is involved in Drosophila long-term memory. Curr. Biol 13, 286–296 (2003). [DOI] [PubMed] [Google Scholar]
- 60.Lebeau G et al. Staufen 2 regulates mGluR long-term depression and Map1b mRNA distribution in hippocampal neurons. Learn. Mem 18, 314–326 (2011). [DOI] [PubMed] [Google Scholar]
- 61.Doench JG et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol 34, 184–191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McCaskill JS The equilibrium partition function and base pair binding probabilities for RNA secondary structure. Biopolymers 29, 1105–1119 (1990). [DOI] [PubMed] [Google Scholar]
- 63.Deigan KE, Li TW, Mathews DH & Weeks KM Accurate SHAPE-directed RNA structure determination. Proc. Natl Acad. Sci. USA 106, 97–102 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huang JK et al. Systematic evaluation of molecular networks for discovery of disease genes. Cell Sys 6, 484–495 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wallace ZS, Rosenthal SB, Fisch KM, Ideker T & Sasik R On entropy and information in gene interaction networks. Bioinformatics 35, 815–822 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The CRISPR screen, STAU2 eCLIP, STAU2 knockdown RNA-seq data and the H3K4Me ChIP data that support the findings of this study have been deposited in GenBank (accession codes GSE135300, GSE134971, GSE135012 and GSE142307, respectively). The source data associated with each figure are provided with the manuscript. All other data supporting the findings of this study are available from the corresponding author on reasonable request.