

Abstract
Genes with novel cellular functions may evolve through exon shuffling which can assemble unique protein architectures. Here we show that DNA transposons provide a recurrent supply of materials to assemble protein-coding genes via exon-shuffling. We find that transposase domains have been captured, primarily via alternative splicing, to form fusion proteins at least 94 times independently over ~350 million years of tetrapod evolution. We find an excess of transposase DNA-binding domains fused to host regulatory domains, especially the Krüppel-associated Box (KRAB), and identify four independently evolved KRAB-transposase fusion proteins repressing gene expression in a sequence-specific fashion. The bat-specific KRABINER fusion protein binds its cognate transposons genome-wide and controls a network of genes and cis-regulatory elements. These results illustrate how a transcription factor and its binding sites can emerge.
One Sentence Summary:
Host-transposase domain fusion generates novel cellular genes, including deeply conserved and lineage specific transcription factors.
Gene duplications contribute to the birth and functional diversification of many genes(1), including developmental regulators (2, 3). Although gene duplicates can evolve diverging developmental functions relative to their parental gene, their domain architecture and biochemical activities tend to remain the same (2, 3), and proteins with novel biochemical functions arising via gene duplication (4, 5) appear to be rare (6). While completely new proteins can occasionally evolve ‘de novo’ from previously non-coding sequences (7, 8), the most obvious path forming proteins with new functionality is the rearrangement of domains with pre-existing function into new composite architectures via exon shuffling. Exon shuffling occurs when new combinations of exons are assembled through RNA splicing, and may have created new protein architectures in eukaryotic evolution (9). While the process of exon shuffling may account for the evolution of many new protein architectures (10–12), the source of new exons and the mechanisms by which they get assimilated have been scarcely characterized. Here we investigate the role of DNA transposons as a source of raw material for the birth of novel proteins via exon shuffling.
DNA transposons encode transposase proteins, which recognize and mobilize DNA through direct sequence-specific interaction with their cognate transposons (13). The canonical architecture of transposase proteins consists of a DNA-binding domain and a catalytic nuclease domain. Both domains may be repurposed or ‘domesticated’ for cellular function (13). Moreover, the mobility of DNA transposons may facilitate exon-shuffling by inserting these functional domains into new genomic contexts, where they may be spliced to generate host-transposase fusion (HTF) genes.
Three genes born via transposase capture have been documented; one specific to placental mammals (GTF2IRD2; (14)) and two specific to primates (SETMAR (15) and PGBD3-CSB (16)). A similar scenario has been proposed to explain the origin of the Paired DNA-binding domain of the Pax family of transcription factors (TFs). However, due to the deep ancestry of Pax genes, which coincides with the emergence of metazoans, precise steps by which these factors evolved has been obscured (13, 17). Further, the extent of transposase capture, the mechanisms facilitating it, and the functions of the resulting genes often remain unclear.
Transposase capture is a recurrent mechanism for novel gene formation in tetrapods
To identify host-transposase fusion (HTF) genes, we surveyed all tetrapod gene annotations (NCBI Refseq; Table S1) predicted to encode proteins with at least one domain of transposase origin (Pfam, Table S2) fused in-frame to a host-derived protein sequence (Conserved Domain Architecture Retrieval Tool [CDART](18)). We also required RNA-sequencing evidence supporting all annotated exon/intron junctions (19). To trace the evolutionary origin of each HTF gene, we used a homology-based approach (BLASTn) to query all vertebrate genomes available in the NCBI Refseq database for syntenic orthologs and paralogs. An HTF was considered to be orthologous in two or more lineages if it: 1) had a hit containing both the transposase domain and the host domain in the same transcript (nr/nt) or in the same orientation on the same contig (Refseq genomes) and 2) was located in a syntenic region of the genome, determined by the identity of flanking genes. This analysis yielded 106 unique HTF genes originating from 94 independent fusion events and 12 subsequent duplication events across the 596 species examined (Fig. 1; Table S3).
Fig. 1: Gene birth by transposase capture in tetrapods.

Tetrapod phylogenetic tree with boxes representing HTF fusion genes. Color indicates transposase superfamily assimilated. OWM=Old world monkeys; NWM=New world monkeys; GM=Gray mouse; H=Hystricoid; C=Castorid, M=Muroid, Miniopt=Miniopterid, Vesper=Vespertilionid, S.S=soft-shelled; B=bearded dragon; G=Green; B=Burmese python; L=Lacertid; J=Japanese; T=Tropical; A=African; M=Mountain; LCA=last common ancestor; MY=million years.
Placing fusion genes onto the species phylogeny suggest that they have evolved continuously during evolution (Fig. 1). Some fusion events (11.6%) preceded the divergence of tetrapods (>350 million year ago, Mya), while others, conserved across narrow species lineages (<5 species, 26.1%) or found in a single species (21%), arose more recently (Fig. 1; Table S3). Several species lineages experienced multiple HTFs of recent origins, as in the green anole (n=6), the Burmese python (n=3), the tropical clawed frog (n=2), and the vespertilionid bats (n=2), consistent with recent episodes of DNA transposon activity in these lineages (Fig. 1; (20–24)). Mammals generally have more HTF genes (mean = 40.8 ± 3.55) than other clades (Reptiles: mean = 29.3 ± 1.53; Amphibians: mean = 30 ± 2.65), reflecting apparent bursts of HTF evolution in mammalian (5.3% of all events), therian (3.2%) and eutherian (8.4%) ancestors. All known major eukaryotic DNA transposon superfamilies contribute to HTFs (Fig. 1), but Tc1/mariner (36.1%), hAT (23.4%), and P element/Kolobok (21.3%) transposases predominate, which mirrors the success of these superfamilies throughout tetrapod evolution (13, 25).
To validate that the transposase coding region of each HTF gene has evolved under functional constraint, we performed codon selection analysis on the transposase domain of each HTF shared by two or more species separated by >50 million years (My) of divergence. All tested HTFs (n= 81) display signatures of purifying selection on their transposase domains (dN/dS <1, p <0.05, LRT, Table S3), supporting their domestication for organismal function.
To further assess the functional capacity of HTF genes, we used publicly available data to examine the RNA expression patterns across 54 tissues for 44 HTFs present in the human genome (GTEx portal). Each of the genes were expressed (transcripts per million, TPM >1) in at least one human tissue, and could be classified into three categories: lowly expressed broadly (TPM >1 in 80% of the tissues, n=23), highly expressed broadly (TPM >10 in 80% of tissues, n=12), and tissue-restricted (TPM >1 in <20% tissues, n=9) (Fig S1). These data suggest that most human HTF genes are broadly expressed and may function in a variety of contexts. Collectively, these data demonstrate that HTF has been a recurrent mechanism for the generation of novel cellular genes in tetrapod evolution, including at least 44 HTFs in humans.
Transposase capture occurs through alternative splicing
To illuminate the mechanism by which transposase domains are captured to form new chimeric proteins, we examined the gene structure of HTFs. In all cases, the transposase-derived domains are encoded by exons distinct from the host domains, suggesting that transposase capture occurred via splicing events. To further delineate the process, we investigated in detail the birth of KRABINER, a recently evolved HTF in vespertilionid bats. KRABINER is predicted to encode a 447-amino acid protein consisting of an N-terminal Krüppel-associated box (KRAB) domain fused to a full-length Mlmar1 mariner DNA transposase (Fig. 2A). Using a combination of comparative genomics, PCR, and RT-PCR (19), we inferred that KRABINER originated in the common ancestor of the nine vespertilionids examined, but after their divergence from miniopterids, ~45 Mya, through the following steps: i) mariner insertion into the last intron of ZNF112, a gene present in all eutherian mammals; ii) alternative splicing to the upstream exons of ZNF112 using a splice acceptor site pre-existing in the ancestral Mlmar1 mariner transposon; and iii) a unique single nucleotide deletion in the transposase coding sequence which generated a single open reading frame coding the chimeric protein (Fig. S2, Fig. 2B). This sequence of events is similar to the process that resulted in SETMAR (15) and PGBD3-CSB (16) originating in the primate lineage, and suggests that DNA transposons possess features that may facilitate their capture via alternative splicing. Alternatively, acquisition of a splice site by a DNA transposon may increase its ability to generate an HTF gene.
Fig. 2: Transposase capture by alternative splicing.

A) ZNF112/KRABINER locus in vespertilionid bats. B) Steps required for KRABINER birth. C) Age of fusion genes with (green) or without (gray) evidence for alternative splicing. Fusion age (bottom) determined by the midpoint of age range for each fusion as described in Table S3; top shows qualitative illustration of host transcript loss over time. D) Summary of transposon splice site usage for 9 HTFs, with canonical mammalian splice sites shown as a sequence logo. Red denotes nucleotides in the splice site that diverge from the transposon consensus sequence. SA=splice acceptor, LCA=last common ancestor. ** p<0.01 2-sample Wilcoxon Test.
We surveyed all HTF gene models for evidence of alternative splicing and found unequivocal evidence for the co-existence of both fusion and parental gene transcripts for most of the young HTFs (18 out of 33 HTFs <100 My old). As HTFs were retained within genomes over time, the ancestral parental transcripts were lost and only the fusion transcript was generally detected (Fig. 2C; Table S3). These findings suggest that most HTFs are born as alternatively spliced variants of an ancestral gene, but over time the HTF transcript often becomes the primary or sole transcript for that gene. Thus, alternative splicing may represent a mechanism for the assimilation of transposase domains by the host proteome.
The splice site enabling the capture of KRABINER’s transposase was provided by the ancestral Mlmar1 mariner transposon (Fig. S2C). To investigate whether this is a more general mechanism of novel gene evolution, we selected eight additional HTFs derived from recent transposon families to trace the origin of the splice site used for transposase capture. In all cases, we found that the splice site was directly derived from the transposon sequence. We then generated a majority-rule consensus sequence for each family to approximate the ancestral transposon (Data S1). For 6 of out 8 HTFs, the splice site sequence was strictly identical to that of the consensus sequence, while in the remaining two the splice site differed from the consensus by a single substitution (Fig. 2D). Though we cannot exclude the possibility of independent splice-site acquisition, these results suggest they preexisted in the ancestral transposon.
Fusion of transposase DNA-binding domains to KRAB is prevalent
To explore the cellular function of HTFs, we first characterized their protein domain architecture and composition (Fig. 3A, Fig. S3). Amongst transposon-derived domains, DNA-binding domains predominate (76.5%; Fig. S3), although some HTFs also include catalytic or accessory transposase domains (Fig. S3). Among host domains (i.e. not normally found in transposases), we identified 55 distinct conserved domains, most of which (76%) were involved in a single fusion event (Fig. 3A). Several of the host domains are predicted to function in transcriptional and/or chromatin regulation, such as the KRAB, SET, and SCAN domains (26–28). KRAB was the host domain most frequently fused to transposase: we inferred this domain to have been involved in 32 independent fusion events across the phylogeny, accounting for approximately one third of all HTFs (Fig. 3A). KRAB domains are abundant in tetrapod genomes and most commonly found in KRAB-Zinc Finger proteins (KRAB-ZFPs), an exceptionally diverse family of transcription factors (>200 genes in most tetrapod genomes; 487 in humans) (29). While it is possible that the frequency of KRAB-transposase fusions reflects the natural abundance of KRAB domains, the identification of two independent KRAB-transposase fusions in bird genomes, despite their paucity in KRAB-ZFPs (~8 genes per genome) (29), suggests the combination of KRAB and transposase may be evolutionarily adaptive.
Fig. 3: Biochemical activities of host-transposase fusion proteins.
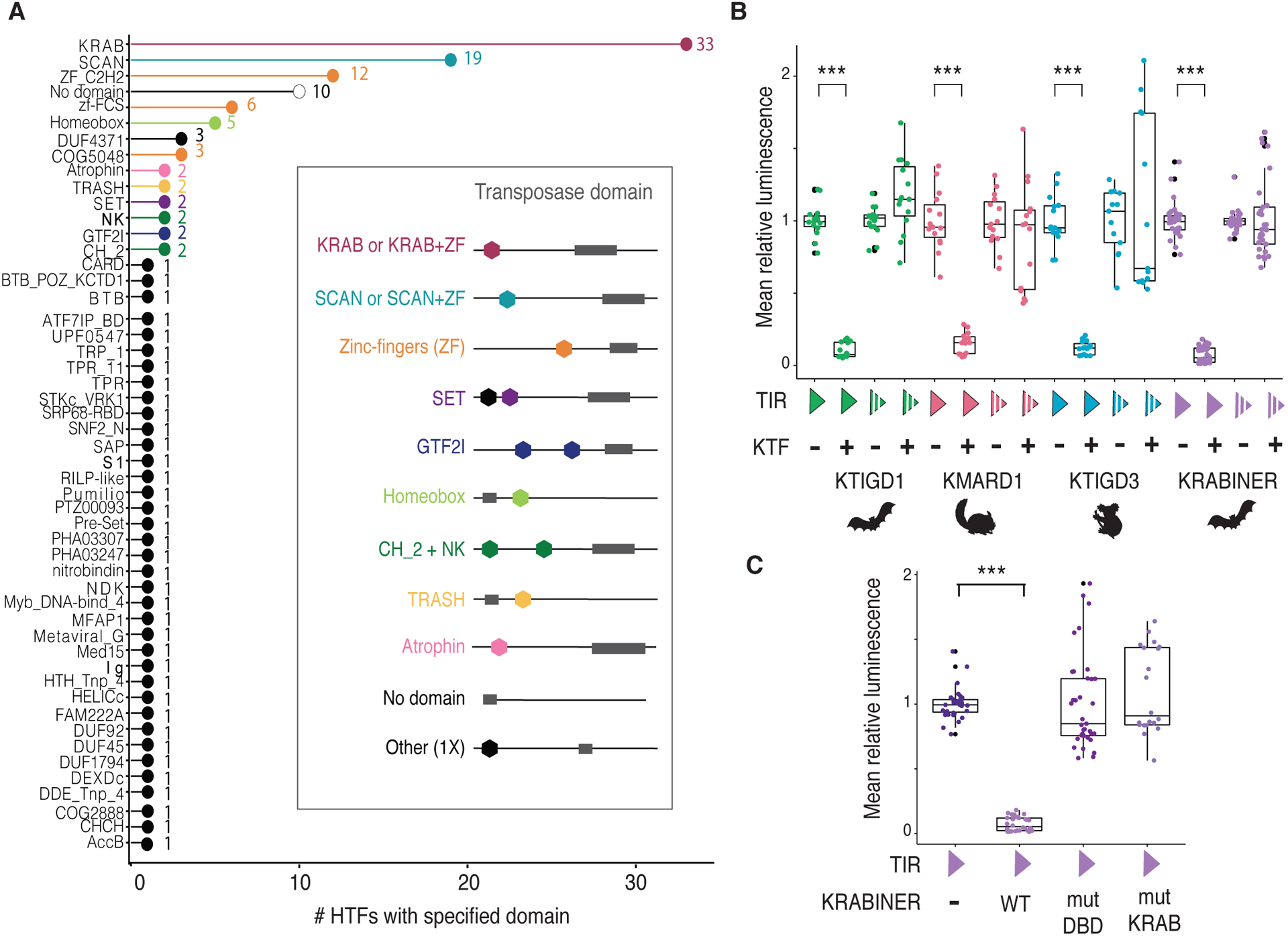
A) Diverse host domains are fused to transposases. X-axis specifies the number of HTF genes a given domain is present in; some fusions contain more than one domain. Inset shows representative domain architecture schematic for select host-transposase fusions. B) KRAB-transposase fusions repress gene expression in a sequence-specific manner. C) KRABINER requires both its KRAB and DBD domains to repress gene expression. Y axes in B-C boxplots correspond to mean luminescence relative to the KTF (−) state for each comparison (n≥15). KTF=KRAB-transposase fusion; TIR=terminal inverted repeat; filled triangle = consensus TIR, interrupted triangle = scrambled TIR; +/− = presence/absence of respectively; *** adj. p<0.001; 2-sample Wilcoxon Test, Holm-Bonferroni correction.
KRAB-transposase fusions act as sequence-specific repressors of gene expression
Given the prevalence of KRAB-transposase fusions and the canonical function of KRAB domains in establishing silent chromatin when tethered to DNA (26), we next used these genes as a paradigm to test the hypothesis that transposase fusion creates novel sequence-specific transcriptional regulators. We selected four recently emerged KRAB-transposase fusions for which we had generated consensus sequences of their cognate transposon family, enabling us to identify their terminal inverted repeats (TIRs) which typically contain the transposase binding site (Fig. S4). We cloned the consensus sequence of each TIR or a scrambled version upstream of a firefly luciferase reporter and measured luciferase expression in HEK293T cells in the presence or absence of a vector expressing the cognate HTF protein. Each KRAB-transposase fusion protein repressed luciferase expression in the presence of its cognate intact TIR but not the scrambled sequence (Fig. 3B). These results indicate that KRAB-transposase proteins can repress gene expression in a sequence-specific manner.
To test whether KRAB-transposase repression is dependent on KAP1 (TRIM28), the transcriptional corepressor often recruited by the KRAB domain (26), we repeated the reporter assays in HEK293T cells knockout (KO) for KAP1 (30). The results (Fig. S5) show that repression by KMARD1 and KTIGD1 is dependent on KAP1, whereas KRABINER and KTIGD3 are only partially dependent on KAP1.
To further dissect the requirement of individual domains, we generated two mutant versions of KRABINER by altering residues predicted to (i) compromise DNA-binding activity (mutDBD) or (ii) the function of the KRAB domain (mutKRAB). To generate the DBD mutant, we exploited the similarity of KRABINER’s mariner transposase to that of Mos1 (23), a transposon from Drosophila. Electrophoretic mobility shift assays demonstrated that a single point mutation in the first helix-turn-helix motif of the Mos1 transposase was sufficient to abolish binding to its TIR (31). We mutated the homologous site in KRABINER’s DBD, as well as three additional residues that directly contact TIR DNA in the Mos1 transpososome crystal structure (32) (Fig. S4B). To generate the KRAB mutant, we introduced several point mutations altering conserved residues previously identified as critical for KRAB-mediated repression (33–36). While the mutDBD and mutKRAB proteins were expressed at comparable levels as wild-type (WT) KRABINER (Fig. S4C), both failed to repress reporter gene expression (Fig. 3C). Together the results of these reporter assays support the hypothesis that KRAB-transposase fusions yield modular proteins functioning as sequence-specific transcriptional repressors.
KRABINER regulates transcription in bat cells
To further test whether transposase capture gives birth to transcriptional regulators, we investigated the ability of KRABINER to modulate gene expression in embryonic fibroblasts of the bat Myotis velifer, where the gene is endogenously expressed (Fig. S2). We used the CRISPR-Cas9 system to engineer a KRABINER KO cell line with a pair of gRNAs designed to precisely delete the mariner transposon from the ZNF112 locus, leaving the rest of the gene intact (Fig. 4A; Fig. S6). We then used a piggyBac vector to deliver transgenes at ectopic chromosomal sites into the KO cell line to establish independent clonal lines reintroducing wild-type KRABINER (WT, n=4 cell lines), or the predicted DNA-binding mutant (mutDBD, n=3), or the predicted KRAB mutant (mutKRAB, n=3) (Fig. S6). Each transgene was cloned under the control of a tetracycline-inducible promoter and contained a C-terminal myc tag to monitor protein expression (Fig. 4A; Fig. S7). The non-induced condition showed leaky expression more closely recapitulating the level of WT KRABINER transcription (hereafter termed “rescue”, R), while transgene induction resulted in KRABINER over-expression (OE) relative to the parental cell line (Fig. S8A).
Fig. 4: KRABINER regulates transcription of genes and TREs in bat cells.

A) Strategy to generate KRABINER KO and rescue lines. TRE=tet responsive element; CMV=cytomegalovirus. B–C) Summary of transcriptional changes of genes and TREs, respectively, upon loss and restoration of KRABINER. KRABINER regulated genes (up or down) change reciprocally between KO vs WT and WT KRABINER rescue vs KO comparisons. p values calculated via a right-tailed hypergeometric test. DE 1 condition refers to differential transcription in either the KO vs WT or WT KRABINER vs KO comparison. Non-specific refers to a gene rescued by WT KRABINER and one or both mutDBD and mutKRAB variants. Unchanged refers to genes/TREs with adj. p>0.05, Wald test.
To investigate whether KRABINER modulates transcription, we profiled KRABINER KO and WT cells with Precision Run-On followed by sequencing (PRO-Seq), which provides a sensitive measurement of nascent transcription throughout the genome, including genes bodies and transcribed regulatory elements (TREs) such as promoters and enhancers (37, 38). By quantifying changes in gene body transcription, we identified 2,644 genes differentially transcribed between WT and KO cells: 1,295 were upregulated in KO (UP), 1,349 were downregulated (DOWN) (DESeq2; Wald test adj. p <0.05; (39)), suggesting KRABINER is capable of regulating genic transcription. To identify transcriptional changes which require both the DNA-binding domain and KRAB activity of KRABINER, we also assessed transcriptional changes in the transgenic rescue lines. Of the 2,644 altered genes, 121 genes (43 UP, 78 DOWN) had their transcription level consistently restored in WT transgenic lines but not in any of the mutant transgenic lines (Fig. 4B; Table S4; overlap p<0.001, right-tailed hypergeometric test). A similar pattern was observed for TREs (identified using dREG; (38)), with 3,472 differentially transcribed TREs following loss of KRABINER, of which 99 were restored exclusively in the WT lines (33 UP, 66 DOWN; Fig. 4C; Table S5; overlap p<0.001 for downregulated TREs, right-tailed hypergeometric test). A subset of these TREs are associated with restored gene body transcription (18% UP, 12% DOWN), while others are distal (>100 kb) to genes (18% UP, 33% DOWN) or associated with genes bodies that are not differentially transcribed (64% UP, 55% DOWN) (Table S4). While our reporter assays indicate that KRABINER can act as a strong repressor, our loss of function analyses in bat cells suggest that the protein exerts a range of transcriptional modulation on the bat genome.
In addition to transcriptional changes unique to the WT transgenic lines, there were several genes and TREs that were rescued by the WT transgene and either mutDBD (100 genes, 79 TREs) or mutKRAB transgenes (73 genes, 65 TREs; Fig. S9). Thus, while at some loci KRABINER’s regulatory activity appears to require both its DNA-binding and KRAB domains, its transcriptional effects on other loci only requires one of these domains. These mechanisms may also explain the ability of KRABINER to either activate or repress transcription in a locus-dependent fashion. Taken together, this data suggests that KRABINER contributes to the transcriptional regulation of a subset of genes and cis-regulatory elements in the examined embryonic cell line.
To investigate whether KRABINER regulates a discrete set of genes or a network of related genes, we performed gene ontology enrichment analysis (2-sided hypergeometric test, Bonferroni step-down correction) for all genes rescued by the WT transgene (n=121) as well as additional target genes whose promoters were found to be differentially transcribed in the TRE analysis (n=57) (Table S4; ClueGO (19, 40)). This analysis revealed a significant enrichment for genes involved in negative regulation of cell migration (GO: 0030336, adj. p=2.1e−6) and in gastrulation (GO: 0007369, adj. p=2.5e−5) as the most enriched terms (Fig. S10A; Table S5). Additional significant terms were linked to morphogenetic or developmental pathways, including positive regulation of Wnt signaling pathway (GO: 2000096, adj. p=5.2e−3), artery morphogenesis (GO:0048844, adj. p=2.5e−2), heart valve morphogenesis (GO: 0003179, adj. p=8.8e−3), and neural crest differentiation (GO: 0014033, adj. p=2.2e−2) (Fig. S10A; Table S5). A similar result was obtained for the downregulated genes alone, suggesting KRABINER’s direct targets may be downregulated (Fig. S10B; Table S5). These results suggest that, in the embryonic cell line examined, KRABINER regulates a set of genes enriched for developmental functions, as may be expected for a canonical transcription factor.
KRABINER binds to many genomic sites, with a preference for mariner TIRs
We next use chromatin immunoprecipitation followed by sequencing (ChIP-seq) to determine if and where KRABINER binds throughout the bat genome. We profiled binding of myc-tagged KRABINER WT protein in the KO cell line background as well as that of mutKRAB and mutDBD mutant proteins 24-hour post induction of transgene expression (n=3 each) (Fig. S11). With these samples, we called binding peaks using MACS2 (41) for each genotype relative to input and filtered out peaks identified in all three genotypes (>50% reciprocal overlap), which are likely to represent spurious or non-specific interactions (19). This analysis identified 1888 WT, 5702 mutKRAB, and 4264 mutDBD peaks respectively. The higher number of peaks obtained with mutKRAB and mutDBD likely reflects the higher expression of those transgenes relative to the WT transgene (Fig. S8). In order to identify genomic sites likely bound directly via KRABINER, we focused on the WT peaks and used the mutKRAB and mutDBD peaks as additional filters or background sets.
Of the 1888 WT KRABINER binding peaks, 56% (n=1070) were unique to the WT condition, suggesting that most of KRABINER’s binding requires both functional domains (Fig. 5A). However, there were about twice as many peaks shared between the WT and mutKRAB conditions (n=572, 28%) than the WT and mutDBD (n=291, 15%) (>50% reciprocal overlap; Fig. 5A), suggesting that most of KRABINER’s binding is dependent on its DBD. The set of peaks overlapping exclusively between WT and mutDBD conditions likely represent indirect genomic interactions, possibly mediated through protein-protein interactions via the KRAB domain (42) or other region of the protein.
Fig. 5: KRABINER binds to mariner TIRs in bat cells.

A) Heatmaps summarizing merged, library-size and input-normalized ChIP-seq coverage of each KRABINER variant centered on the summit of WT (top), WTXmutKRAB (middle), and WTXmutDBD (bottom) peak sets. B) Metaplot summarizing normalized ChIP-seq coverage of each KRABINER variant over all genomic Mlmar1 elements (top). The top enriched motif in the WT and WT & mutKRAB peak sets is identical to the predicted bipartite binding motif within the Mlmar1 mariner TIR (bottom) (HOMER). C) Enrichment of transposon families in WT only, WTXmutKRAB, and WTXmutDBD peak sets. Observed = number of overlaps between a TE family and a given peak set. Expected = # of expected overlaps between a TE family and a given peak set after shuffling TE locations 1000 times. p values determined using the binomial distribution, n=1000 shuffles. WT= purple; mutDBD=pink; mutKRAB=green. WT = wild-type, DBD = DNA binding domain, ORF = open reading frame, TIR = terminal inverted repeat.
We then looked for sequence motifs that might explain KRABINER binding to its genomic sites (HOMER; (43)). We extracted a 200-bp window centered on the peak summit for all WT peaks and asked which de-novo motifs were enriched relative to mutDBD peaks for the WT and WTxmutKRAB peak sets or to the mutKRAB peaks for the WTxmutDBD peak set. For the WT unique and WT-mutKRAB peaks, the top enriched motif (binomial test; p=1e−50 and p=1e−80) was identical, and resembled a bipartite region within the Mlmar1 mariner TIR sequence that aligns with the binding sites mapped previously for the Mos1 transposase ((44) Fig. 5B; Fig. S12). Of the additional 12 and 17 enriched motifs predicted for the WT and WT-mutKRAB peak sets respectively, all were derived from Mlmar1 elements (Fig. S12). We identified only one enriched motif in the WT-mutDBD peaks, which bore no resemblance to Mlmar1 TIRs or any known metazoan transcription factor (Data S6). These data suggested that the WT and mutKRAB, but not mutDBD, proteins bind many Mlmar1 elements dispersed throughout the genome. Consistent with this, Mlmar1 elements are enriched within WT (log2 Fold enriched [FE] = 4.43, p=2.2e−16) and WT-mutKRAB peaks (log2FE = 4.95, p=2.2e−16), but not the WT-mutDBD peaks (p=0.63) (Fig. 5C, 2-sided binomial test, 1000 bootstraps, (19)). In total, the WT KRABINER transgenic protein binds to 206 (8.5%) of all Mlmar1 elements annotated in the bat genome assembly.
To determine where in the Mlmar1 element KRABINER binds, we plotted the input-normalized ChIP-seq reads for each genotype over all Mlmar1 transposons in the genome. We found that a fraction of these transposons was bound by the WT and mutKRAB proteins, but not the mutDBD protein, consistent with the peak-based approach (Fig. 5B; Fig. S12). The ChIP-seq read coverage peaks within the TIR regions, and especially the 3’ TIR (Fig. 5B; Fig. S12), which is consistent with the binding activity of other mariner transposons (31, 32, 45). Collectively, our ChIP-seq data demonstrates that KRABINER is capable of binding numerous genomic sites, with a preference for Mlmar1 TIRs. Further, its ability to bind Mlmar1 TIRs is dependent upon its transposase DNA-binding domain.
KRABINER binding is associated with downregulation of nearby TREs
To test if KRABINER binding leads to transcriptional change, we next induced expression of the KRABINER transgenes and performed PRO-seq 24-hours post induction (OE), conditions matching our ChIP-seq experiments. We then identified TREs differentially transcribed between the OE vs rescue conditions, which are of the same exact genotype and in principle differ only in the level of KRABINER expression (Figs. S7–8). Because TREs represent discrete transcriptional units such as promoters and enhancers, we reasoned that KRABINER binding to or near these regions would more likely impact transcription than binding within a gene body. We identified 391 TREs (178 UP, 213 DOWN; Fig. 6A) differentially transcribed upon overexpression of the WT KRABINER protein but neither of the mutant proteins (mutDBD or mutKRAB). Additionally, several TREs were differentially expressed in the same direction upon OE of WT protein and either mutDBD or mutKRAB (Fig S13), consistent with the hypothesis that a subset of KRABINER’s transcriptional changes require only one of its functional domains.
Fig. 6. KRABINER regulates a network of genes and TREs in bat cells.

A) MA plot summarizing changes in TRE transcription upon over-expression of WT KRABINER. Non-specific (black) refers to changes in TRE transcription that are shared between over-expression of WT KRABINER and one or both mutant KRABINER variants. Unchanged (gray) refers to TREs with adj. p>0.05, Wald test. B) Proposed model for KRABINER’s function as a transcription factor in bats. KRABINER directly binds to mariner TIRs within the genome and leads to direct downregulation of a subset of TREs. KRABINER also binds to other genomic regions, and indirectly regulates a number of genes and TREs. Resc. = rescue; OE = overexpression; TIR = terminal inverted repeats; Tpase = transposase.
To determine if KRABINER binding is associated with differential TRE transcription, we first examined whether WT KRABINER ChIP-seq peaks were located near (<1 kb) differentially expressed TREs. While the total number of KRABINER peaks located nearby differentially expressed TREs was small (n=6), it was a significant enrichment over the random expectation (permutation test; log2FE = 2.58, empirical p=0, 10000 bootstraps, (19)) (Fig. S14). Interestingly, all were downregulated TREs, consistent with the results of our reporter assays suggesting that tethering KRABINER to DNA induces local transcription repression. Furthermore, five out of six differentially expressed TREs were located within ~1-kb of at least one Mlmar1 element, and the TIRs of these transposons were located near the summit of the KRABINER peaks. Finally, several of the TREs connected with KRABINER binding to nearby Mlmar1 elements were also associated with changes in adjacent gene expression.
For example, a differentially expressed TRE is located in the promoter region of the bat ortholog of the DNA damage recognition and repair factor gene XPA, which is downregulated (Wald test; log2FC = −0.69, adj. p = 0.0079) upon WT KRABINER OE and located immediately adjacent to a mariner TIR bound by KRABINER (Fig. S15). A similar pattern is seen for an intergenic TRE, located between the bat homolog of the family with sequence similarity 174 member B (FAM174B) and chromodomain helicase DNA binding protein (CHD2) genes (Fig. S16). This region contains three distinct TREs, two of which are downregulated upon WT KRABINER OE (Wald test; log2FC = −1.1, adj. p=0.04 and log2FC = −1.09, adj. p=0.006 respectively), and this change is associated with KRABINER binding to the Mlmar1 TIRs immediately upstream of these TREs (Fig. S16). Other regulated TREs include one located in the promoter region of the bat homolog of the small nuclear ribonucleoprotein polypeptide A’ (SNRPA1) gene (Wald test; log2FC = −0.84, adj. p=0.02), which is located downstream of four bound Mlmar1 elements (Fig. S17), and a distal TRE (Wald test; log2FC = −1.24, adj. p=0.01) upstream of the nucleoporin 50 (NUP50) gene (Fig. S18). Importantly, each of the KRABINER-downregulated TREs are near two or more bound TIR sequences, suggesting KRABINER’s effect on TREs may be strengthened by additional binding sites.
Collectively, our PRO-seq and ChIP-seq data demonstrates that KRABINER acts as a canonical transcription factor, and that some of its transcriptional regulatory activity in bat cells is accomplished by KRABINER binding to its cognate mariner TIRs. However, only a minority of genes regulated by KRABINER appear to be direct targets, suggesting that KRABINER is integrated within a complex transcriptional network (Fig. 6B).
Discussion:
While gene birth via duplication has been extensively documented, how novel protein architectures and biological functions are born has remained poorly characterized. Here, we validate that exon-shuffling is a major evolutionary force generating genetic novelty (9) and provide evidence that DNA transposons fuel the process not only by supplying protein domains to assemble new protein architectures, but also in many cases by introducing the splice sites that enable the fusion process. While these events must be relatively rare on an evolutionary timescale, the mobility of DNA transposons likely increases the probability of generating a functional gene via exon-shuffling by introducing genetic material into new contexts. We also derived first principles of how transposase-mediated exon-shuffling occurs, providing a foundation for the identification of host-transposase fusion genes in other lineages.
Transposase-mediated exon-shuffling offers a plausible mechanism for the birth of known developmental regulatory proteins, such as Pax6, which controls eye development and patterning across animals (46). Another example is POGZ, a gene expressed predominantly in the brain and associated with autism and intellectual disability when mutated in humans (47, 48). While these are examples of HTFs with relatively deep evolutionary origins and likely serving broadly conserved functions, our functional analysis of KRABINER, a bat-specific protein with transcriptional modulatory activities, suggests that the process of gene birth via transposase capture has been a continuous source of regulatory innovation.
Many studies have implicated transposable elements in the dispersion of transcription factor binding sites and cis-regulatory elements that have rewired gene regulatory networks during evolution (49–54). However, these studies do not explain how a new regulatory network, including its associated regulatory proteins, initially evolves. The data presented here offer a plausible path by which a new trans-regulatory protein and its cis-binding sites in the genome simultaneously emerge from the same transposon family (55). Historically this model has been difficult to test because previously recognized transposase-derived transcription factors (such as Pax) and their network of regulated genes evolved hundreds of millions of years ago, which would have obscured the transposon origin of their genomic binding sites. Our study of KRABINER, a recently evolved HTF, together with those of two other young HTF genes evolved during primate evolution, SETMAR (40–58 my old;(15, 56) and PGBD3-CSB (>40 my old; (16, 57, 58)) have captured how the early steps by which a new cis-regulatory circuit, including both coding and noncoding components, can emerge from a transposon family.
Although we focused on the tetrapod lineage in this study, we propose that the principles and implications of transposase capture revealed herein extend beyond vertebrates. Transposases are ancient and possibly the most abundant and ubiquitous genes in nature (59) and a variety of host domains that regulate transcription exist in all branches of the tree of life. It is easy to envision how these sequences have provided the raw material for the assemblage of endless combination of transposase-host fusion proteins throughout evolution.
Materials and Methods
Identifying and characterizing transposase fusion genes
To identify host-transposase fusion (HTF) genes, we used a domain centric approach (Conserved Domain Architecture Tool, CDART (18)) to identify NCBI Refseq (60) tetrapod gene annotations (Table S1) that 1) contained a transposase domain (Table S2), had two or more exons (to exclude standalone transposases), and had 3) RNA-seq support for all annotated introns or were identified in de-novo RNA-seq data (Data S2–3). We further characterized each HTF by its domain structure, originating gene and transposon, and the timing of gene birth (19). We identified orthologs of each HTF gene, where present, using a combination of homology (BLASTn) and synteny (19). We further assigned each HTF an age in million years based on the estimated evolutionary divergence (Timetree; (61)) between all species possessing the HTF. Finally, we tested the transposase domains of all HTFs conserved in two or more species diverged > 50 million years for evidence of purifying selection (Phylogenetic Analysis by Maximum Likelihood, PAML; (62)), with significance determined by comparing the estimated model to a model assuming neutral evolution (Likelihood ratio test [LRT], p<0.05, chi-sq. distribution) (19).
Determining mechanism for HTF gene birth
We first stratified HTF genes into two classes: genes born via splicing or gene duplication. Of those born via splicing, we considered gene models containing both the original host transcript and the fusion transcript to have originated via alternative splicing. For a subset of HTFs for which we were able to reconstruct the consensus sequence of the originating transposon (KTIGD1, KMARD1, KTIGD3, KRABINER, KMARD4, KHATD2, TIGD-G1, TIGD-G4-Zscan29, and MARD-G1) we also inferred whether the splice site was present in the transposon and, if so, if it was also present in the consensus sequence (19).
Tracing the birth and evolution of KRABINER
We first determined the timing of the Mlmar1 mariner insertion into the ZNF112 locus using both homology-based approaches (BLAST) against publicly available bat genomes and PCR amplification followed by sequencing of the mariner insertion in an additional seven bat species (19). We then performed RT-PCR on cDNA from cell lines derived from three bat species (Myotis velifer, M. lucifugus, and Eptesicus fuscus) using primers designed to either amplify the key splice junction (exons 4–5 and 5–6) or to amplify the full length (exons 1 and 6) KRABINER (Fig. S2, Table S6).
Luciferase Assays
To assess the ability of four KRAB-transposase fusion (KTF) genes (KRABINER, KMARD1, KTIGD1, and KTIGD3), we performed luciferase reporter assays. For KRABINER we also included DNA-binding domain or KRAB mutant variants, which were generated based on preexisting studies of a closely related transposon (31, 32) or studies that identified residues critical for KRAB domain function (33–36) respectively. For each HTF we transfected either WT or KAP1 KO HEK293T cells (30) with three plasmids: a KRAB-transposase fusion expression vector or empty vector, a firefly luciferase expression vector with the cognate consensus or scrambled TIR sequence located immediately upstream of the promoter, and a renilla luciferase expression vector as an internal control. We lysed cells 48 hours post transfection and split the lysate into 5 wells of a 96-well plate (n=5 technical replicates). We then measured firefly and renilla luminescence via plate reader, and normalized each value as follows to obtain a final relative luminescence per replicate : (KTF-lumfirefly/KTF-lumrenilla)/mean(empty-lumfirefly/empty-lumrenilla) (19). We repeated each experiment a minimum of three times and considered differences in mean relative luminescence significant at adj. p<0.05 (pairwise Wilcoxon Test with Bonferroni multiple testing correction).
Generating and validating KRABINER KO and rescue cell lines
We generated a KRABINER KO cell line using the CRISPR-Cas9 system as previously described (63). We used a pair of gRNAs (gRNA 1 (GL430169:87458-87477) - CATTTAGTTTCAGCCTCTCATGG, gRNA 2 (GL430169:89264-89286) - TAATACGTAAGCTGCTGTGTGGG) that flank the Mlmar1 mariner insertion at the ZNF112 locus to precisely delete the mariner element (Fig. S6A), leaving the parental gene intact (19). Following clonal expansion, we identified a single cell line with a homozygous deletion that we verified via Sanger sequencing. We further verified absence of KRABINER transcription by RT-PCR (Fig. S6B; Table S6).
To rescue KRABINER expression, we used the piggyBac system to introduce transgenes encoding inducible forms of either wild-type, DNA binding mutant (mutDBD), or KRAB mutant (mutKRAB) variants of KRABINER into our KO cell line (19). Transduced cells were selected via puromycin treatment (1.5 μg/mL) for one week, and then clonally expanded (n=3 minimum). We verified insertion of the transgene using PCR followed by sequencing (Fig. S7B, Table S6) and expression of the transgene at the RNA level via RTPCR (Fig. S7C, Table S6) and Precision-Run-on-Sequencing (PROSeq; Fig. S8A) and at the protein level via Western-blot and immunofluorescence assays (Fig. S7) (19).
PRO-seq analysis
We performed PRO-seq on KRABINER WT (n=2), KO (n=2), and rescue cell lines (min. n=3). Rescue cell lines were treated with 1μg/mL doxycycline (induced, OE) or not (non-induced, rescue) 24 hours prior to PRO-seq. PRO-seq libraries (20 million cells per genotype per treatment, >90% viability) were prepared as previously described (19, 37, 64, 65). Libraries were sequenced on an Illumina NextSeq 500 platform with 37×37bp chemistry (Table S7), and raw reads for all samples are accessible at SRP256595. We processed the resulting PRO-seq data using a pipeline available at (19,70). To quantify gene body transcription, we defined gene body regions as the transcription start site (TSS) plus 500bp to transcription end site (TES) and TSS minus 500bp to TES for + and − strand Myoluc2 ENSEMBL gene annotations respectively. We called transcribed regulatory elements (TREs) for each sample using (dREG (38)) and merged (bedtools merge; (66)) to generate a comprehensive TRE set. We quantified read counts in both gene bodies and TREs at single nucleotide resolution using a custom script and bedtools map (66). TRE annotations and bigWig coverage files for each sample are available at GSE148789.
We performed differential transcription analysis for the gene bodies and TREs separately using DESeq2 (39). We performed three comparisons: KRABINER KO vs WT, WT/mutDBD/or mutKRAB Rescue vs KO, and WT/mutDBD/or mutKRAB OE vs Rescue. We considered a gene or TRE to be regulated by KRABINER if it exhibited significant (adj. p<0.05; Wald test) reciprocal changes in the KO vs WT and the WT R vs KO comparison or if it was differentially transcribed in the OE vs R comparison (adj. p<0.05; Wald test). Raw expression counts for genes and TREs as well as DESeq2 outputs for all comparisons are available at GSE148789. We also performed Gene Ontology Enrichment for KRABINER regulated genes (Table S4 (19)).
ChIP-seq analysis
We performed ChIP-seq on KRABINER rescue cell lines (min. n=3, 20 million cells each, >90% viability) 24 hours post treatment with 1μg/mL doxycycline to induce transgene expression (19). We prepared libraries for each IP and input (3.33% total sonicated chromatin for each sample, pooled across genotype) using the NEBNext® Ultra™ II DNA Library Prep Kit for Illumina® (New England Biolabs, Ipswich, MA) according to manufacturer instructions with 4 cycles of PCR amplification. Libraries were sequenced on the HiSeq 4000 platform in PE150 mode by Novogene Corporation Inc (Sacramento CA). Reads for all samples are available at SRP256596.
We then quality processed reads, mapped them to the Myoluc2.0 assembly, and called KRABINER binding peaks for each variant (WT, mutDBD, and mutKRAB) relative to matched input samples using MACS2 (19, 41). We removed peaks called in all three genotypes, which are likely to be spurious, and further subset the WT peaks into three categories: those unique to the WT transgene and those shared between either the WT and mutDBD or WT and mutKRAB transgenes (19). To annotate the peaks, we identified sequence motifs enriched in each of the WT peak categories relative to a background set (all mutDBD peaks for WT unique and shared WT-mutKRAB or all mutKRAB peaks for the shared WT-mutDBD peaks) (HOMER, Data S4–6 (19, 43)). We then determined which TEs were enriched for KRABINER binding, as previously described (67), and considered a TE family to be enriched if it overlapped more than expected by chance (binomial p value<.05 (1000 shuffles). We further determined if KRABINER binding peaks were enriched in or near differentially transcribed genes/TREs using a pseudo-random shuffling method to calculate empirical p values (significant at p<0.05, 10000 shuffles) (19). All raw and normalized bigWig files (DeepTools,(19, 68)) and peak files are available at GSE148789.
Supplementary Material
Acknowledgements:
We acknowledge D. Ray, W. Wright, N. Elde, J. Lis, H. Rowe, J. Wysocka, and T. Macfarlan for providing reagents. We also thank members of the Feschotte lab for helpful discussion.
Funding:
This work was supported by R35GM122550, from the National Institutes of Health to CF. JJ was supported by NHGRI fellowship F31HG010820. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Competing Interests: The authors declare no competing interests.
Data and Materials Availability: All processed sequencing data is deposited in the Gene Expression Omnibus (GSE148789) and raw data is deposited in the SRA archive (PRO-Seq: SRP256595; ChIP-Seq: SRP256596). Supplemental sequence and motif data (S1-S6) is available at (69), and code for processing the PRO-Seq data is available at (70). All cell lines and reagents are available upon request.
References and Notes:
- 1.Ohno S, Evolution by Gene Duplication. (Springer-Verlag, Berlin, Heidelberg, 1970). [Google Scholar]
- 2.Ruddle FH et al. , Evolution of Hox Genes. Annual review of genetics 28, 423–442 (1994). [DOI] [PubMed] [Google Scholar]
- 3.Bouchard M, Schleiffer A, Eisenhaber F, Busslinger M, PaxGenes: Evolution and Function. (John Wiley & Sons, Ltd, Chichester, UK, 2008), vol. 20, pp. 5736. [Google Scholar]
- 4.Deng C, Cheng CHC, Ye H, He X, Chen L, Evolution of an antifreeze protein by neofunctionalization under escape from adaptive conflict. Proc Natl Acad Sci USA 107, 21593 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lynch VJ, Inventing an arsenal: adaptive evolution and neofunctionalization of snake venom phospholipase A2 genes. BMC Evolutionary Biology 7, 2–2 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Innan H, Population genetic models of duplicated genes. Genetica 137, 19 (2009). [DOI] [PubMed] [Google Scholar]
- 7.Van Oss SB, Carvunis A-R, De novo gene birth. PLoS Genetics 15, e1008160 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luis Villanueva-Cañas J et al. , New Genes and Functional Innovation in Mammals. Genome Biol Evol 9, 1886–1900 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gilbert W, Why genes in pieces? Nature 271, 501–501 (1978). [DOI] [PubMed] [Google Scholar]
- 10.Long M, Rosenberg C, Gilbert W, Intron phase correlations and the evolution of the intron/exon structure of genes. Proc Natl Acad Sci USA 92, 12495 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patthy L, Modular Assembly of Genes and the Evolution of New Functions. Genetica 118, 217–231 (2003). [PubMed] [Google Scholar]
- 12.Long M, VanKuren NW, Chen S, Vibranovski MD, New Gene Evolution: Little Did We Know. Annual Review of Genetics 47, 307–333 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feschotte C, Pritham E, DNA Transposons and the Evolution of Eukaryotic Genomes. Annual review of genetics 41, 331–368 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tipney HJ et al. , Isolation and characterisation of GTF2IRD2, a novel fusion gene and member of the TFII-I family of transcription factors, deleted in Williams–Beuren syndrome. European Journal of Human Genetics 12, 551–560 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Cordaux R, Udit S, Batzer MA, Feschotte C, Birth of a chimeric primate gene by capture of the transposase gene from a mobile element. Proceedings of the National Academy of Sciences of the United States of America 103, 8101–8106 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Newman JC, Bailey AD, Fan H-Y, Pavelitz T, Weiner AM, An Abundant Evolutionarily Conserved CSB-PiggyBac Fusion Protein Expressed in Cockayne Syndrome. PLoS Genetics 4, e1000031 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Breitling R, Gerber J-K, Origin of the paired domain. Development Genes and Evolution 210, 655–650 (2000). [DOI] [PubMed] [Google Scholar]
- 18.Geer LY, Domrachev M, Lipman DJ, Bryant SH, CDART: Protein Homology by Domain Architecture. Genome Research 12, 1619–1623 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. See Supplemental Materials.
- 20.Alföldi J et al. , The genome of the green anole lizard and a comparative analysis with birds and mammals. Nature 477, 587–591 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Castoe TA et al. , The Burmese python genome reveals the molecular basis for extreme adaptation in snakes. Proc Natl Acad Sci USA 110, 20645 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mitros T et al. , A chromosome-scale genome assembly and dense genetic map for Xenopus tropicalis. Developmental Biology 452, 8–20 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Ray DA et al. , Multiple waves of recent DNA transposon activity in the bat, Myotis lucifugus. Genome Research 18, 717–728 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pritham EJ, Feschotte C, Massive amplification of rolling-circle transposons in the lineage of the bat Myotis lucifugus. Proc Natl Acad Sci USA 104, 1895 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sotero-Caio CG, Platt II RN, Suh A, Ray DA, Evolution and Diversity of Transposable Elements in Vertebrate Genomes. Genome Biol Evol 9, 161–177 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bruno M, Mahgoub M, Macfarlan TS, The Arms Race Between KRAB–Zinc Finger Proteins and Endogenous Retroelements and Its Impact on Mammals. Annual Review of Genetics 53, 393–416 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Herz H-M, Garruss A, Shilatifard A, SET for life: biochemical activities and biological functions of SET domain-containing proteins. Trends in Biochemical Sciences 38, 621–639 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Edelstein LC, Collins T, The SCAN domain family of zinc finger transcription factors. Gene 359, 1–17 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Imbeault M, Helleboid P-Y, Trono D, KRAB zinc-finger proteins contribute to the evolution of gene regulatory networks. Nature 543, 550–554 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Tie CH et al. , KAP1 regulates endogenous retroviruses in adult human cells and contributes to innate immune control. EMBO Rep, e45000 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang L, Dawson A, Finnegan DJ, DNA-binding activity and subunit interaction of the mariner transposase. Nucleic Acids Research 29, 3566–3575 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Richardson JM, Colloms SD, Finnegan DJ, Walkinshaw MD, Molecular Architecture of the Mos1 Paired-End Complex: The Structural Basis of DNA Transposition in a Eukaryote. Cell 138, 1096–1108 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Margolin JF et al. , Krüppel-associated boxes are potent transcriptional repression domains. Proceedings of the National Academy of Sciences of the United States of America 91, 4509–4513 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Witzgall R, O’Leary E, Leaf A, Onaldi D, Bonventre JV, The Krüppel-associated box-A (KRAB-A) domain of zinc finger proteins mediates transcriptional repression. Proceedings of the National Academy of Sciences of the United States of America 91, 4514–4518 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Friedman JR et al. , KAP-1, a novel corepressor for the highly conserved KRAB repression domain. Genes and Development 10, 2067–2078 (1996). [DOI] [PubMed] [Google Scholar]
- 36.Murphy KE et al. , The Transcriptional Repressive Activity of KRAB Zinc Finger Proteins Does Not Correlate with Their Ability to Recruit TRIM28. PLoS ONE 11, e0163555–0164513 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kwak H, Fuda NJ, Core LJ, Lis JT, Precise Maps of RNA Polymerase Reveal How Promoters Direct Initiation and Pausing. Science 339, 950 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Z, Chu T, Choate LA, Danko CG, Identification of regulatory elements from nascent transcription using dREG. Genome Research 29, 293–303 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bindea G et al. , ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25, 1091–1093 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y et al. , Model-based Analysis of ChIP-Seq (MACS). Genome Biology 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Helleboid P-Y et al. , The interactome of KRAB zinc finger proteins reveals the evolutionary history of their functional diversification. EMBO J 38, e101220 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Heinz S et al. , Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Molecular cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Richardson JM et al. , Mechanism of Mos1 transposition: insights from structural analysis. EMBO J 25, 1324–1334 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang G, Nagel DH, Feschotte C, Hancock CN, Wessler SR, Tuned for Transposition: Molecular Determinants Underlying the Hyperactivity of a Stowaway MITE. Science 325, 1391 (2009). [DOI] [PubMed] [Google Scholar]
- 46.Kozmik Z, Pax genes in eye development and evolution. Current opinion in genetics & development 15, 430–438 (2005). [DOI] [PubMed] [Google Scholar]
- 47.Stessman HAF et al. , Disruption of POGZ Is Associated with Intellectual Disability and Autism Spectrum Disorders. The American Journal of Human Genetics 98, 541–552 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.White J et al. , POGZ truncating alleles cause syndromic intellectual disability. Genome Medicine 8, 3 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chuong EB, Elde NC, Feschotte C, Regulatory activities of transposable elements: from conflicts to benefits. Nature reviews. Genetics 18, 71–86 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fuentes DR, Swigut T, Wysocka J, Systematic perturbation of retroviral LTRs reveals widespread long-range effects on human gene regulation. eLife 7, e35989 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pontis J et al. , Hominoid-Specific Transposable Elements and KZFPs Facilitate Human Embryonic Genome Activation and Control Transcription in Naive Human ESCs. Cell Stem Cell 24, 724–735.e725 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sundaram V, Wysocka J, Transposable elements as a potent source of diverse cis-regulatory sequences in mammalian genomes. Philosophical Transactions of the Royal Society B: Biological Sciences 375, 20190347 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lynch VJ et al. , Ancient Transposable Elements Transformed the Uterine Regulatory Landscape and Transcriptome during the Evolution of Mammalian Pregnancy. Cell Reports 10, 551–561 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Trizzino M, Kapusta A, Brown CD, Transposable elements generate regulatory novelty in a tissue-specific fashion. BMC Genomics 19, 261–468 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Feschotte C, Transposable elements and the evolution of regulatory networks. Nature reviews. Genetics 9, 397–405 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tellier M, Chalmers R, Human SETMAR is a DNA sequence-specific histone-methylase with a broad effect on the transcriptome. Nucleic Acids Research 47, 122–133 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bailey AD et al. , The conserved Cockayne syndrome B-piggyBac fusion protein (CSB-PGBD3) affects DNA repair and induces both interferon-like and innate antiviral responses in CSB-null cells. DNA Repair 11, 488–501 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gray LT, Fong KK, Pavelitz T, Weiner AM, Tethering of the Conserved piggyBac Transposase Fusion Protein CSB-PGBD3 to Chromosomal AP-1 Proteins Regulates Expression of Nearby Genes in Humans. PLoS Genetics 8, e1002972–e1002972 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aziz RK, Breitbart M, Edwards RA, Transposases are the most abundant, most ubiquitous genes in nature. Nucleic Acids Research 38, 4207–4217 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.O’Leary NA et al. , Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Research 44, D733–D745 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kumar S, Stecher G, Suleski M, Hedges SB, TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Molecular Biology and Evolution 34, 1812–1819 (2017). [DOI] [PubMed] [Google Scholar]
- 62.Yang Z, PAML 4: Phylogenetic Analysis by Maximum Likelihood. Molecular Biology and Evolution 24, 1586–1591 (2007). [DOI] [PubMed] [Google Scholar]
- 63.Ran FA et al. , Genome engineering using the CRISPR-Cas9 system. Nat. Protocols 8, 2281–2308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mahat DB et al. , Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq). Nature protocols 11, 1455 EP - (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Judd J et al. , A rapid, sensitive, scalable method for Precision Run-On sequencing (PRO-seq). bioRxiv, 2020.2005.2018.102277 (2020). [Google Scholar]
- 66.Quinlan AR, Hall IM, BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kapusta A et al. , Transposable Elements Are Major Contributors to the Origin, Diversification, and Regulation of Vertebrate Long Noncoding RNAs. PLoS Genetics 9, e1003470 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ramírez F et al. , deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Research 44, W160–W165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cosby R et al. Cosby_et_al_2020_Supplemental_Data. 10.5281/zenodo.4060329. [DOI]
- 70.JAJ256. PROseq_alignment.sh: PRO-seq alignment pipeline used for “Recurrent evolution of vertebrate transcription factors by transposase capture.” 10.5281/zenodo.4019173 [DOI] [PMC free article] [PubMed]
- 71.Gomes NMV et al. , Comparative biology of mammalian telomeres: hypotheses on ancestral states and the roles of telomeres in longevity determination. Aging Cell 10, 761–768 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pietrokovski S, Henikoff S, A helix-turn-helix DNA-binding motif predicted for transposases of DNA transposons. Molecular and General Genetics MGG 254, 689–695 (1997). [DOI] [PubMed] [Google Scholar]
- 73.Aravind L, Anantharaman V, Balaji S, Babu MM, Iyer LM, The many faces of the helix-turn-helix domain: Transcription regulation and beyond. FEMS Microbiology Reviews 29, 231–262 (2005). [DOI] [PubMed] [Google Scholar]
- 74.Yuan Y-W, Wessler SR, The catalytic domain of all eukaryotic cut-and-paste transposase superfamilies. Proceedings of the National Academy of Sciences 108, 7884–7889 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bao W, Kapitonov VV, Jurka J, Ginger DNA transposons in eukaryotes and their evolutionary relationships with long terminal repeat retrotransposons. Mobile DNA 1, 3–3 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Aravind L, The BED finger, a novel DNA-binding domain in chromatin-boundary-element-binding proteins and transposases. Trends in Biochemical Sciences 25, 421–423 (2000). [DOI] [PubMed] [Google Scholar]
- 77.Hayward A, Ghazal A, Andersson G, Andersson L, Jern P, ZBED Evolution: Repeated Utilization of DNA Transposons as Regulators of Diverse Host Functions. PLoS ONE 8, e59940 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Craig et al. , Eds., Mobile DNA III, (American Society of Microbiology, Washington, UNITED STATES, 2015), pp. 775–802. [Google Scholar]
- 79.Roussigne M et al. , The THAP domain: a novel protein motif with similarity to the DNA-binding domain of P element transposase. Trends in Biochemical Sciences 28, 66–69 (2003). [DOI] [PubMed] [Google Scholar]
- 80.Kapitonov V, Jurka J, Kolobok, a novel superfamily of eukaryotic DNA transposons. Repbase Rep 7, 111–122 (2010). [Google Scholar]
- 81.Babu MM, Iyer LM, Balaji S, Aravind L, The natural history of the WRKY-GCM1 zinc fingers and the relationship between transcription factors and transposons. Nucleic Acids Research 34, 6505–6520 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dupeyron M, Singh KS, Bass C, Hayward A, Evolution of Mutator transposable elements across eukaryotic diversity. Mobile DNA 10, 12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kojima KK, Jurka J, Crypton transposons: identification of new diverse families and ancient domestication events. Mobile DNA 2, 12 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lu S et al. , CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Research 48, D265–D268 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bao W, Kojima KK, Kohany O, Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6, 11–11 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wickham H, ggplot2: Elegant Graphics for Data Analysis. (Springer-Verlag, New York, 2016). [Google Scholar]
- 87.Madeira F et al. , The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Research 47, W636–W641 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Seim I et al. , Genome analysis reveals insights into physiology and longevity of the Brandt’s bat Myotis brandtii. Nature Communications 4, 2212 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang G et al. , Comparative Analysis of Bat Genomes Provides Insight into the Evolution of Flight and Immunity. Science 339, 456 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Eckalbar WL et al. , Transcriptomic and epigenomic characterization of the developing bat wing. Nature Genetics 48, 528–536 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gu B et al. , Transcription-coupled changes in nuclear mobility of mammalian cis-regulatory elements. Science 359, 1050 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Oliveros JC et al. , Breaking-Cas-interactive design of guide RNAs for CRISPR-Cas experiments for ENSEMBL genomes. Nucleic Acids Research 44, W267–W271 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Haeussler M et al. , Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome biology 17, 148 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bae S, Park J, Kim J-S, Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30, 1473–1475 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chen S, Zhou Y, Chen Y, Gu J, fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Langmead B, Salzberg SL, Fast gapped-read alignment with Bowtie 2. Nature Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Smith T, Heger A, Sudbery I, UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Research 27, 491–499 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Conway JR, Lex A, Gehlenborg N, UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics 33, 2938–2940 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Li H, Durbin R, Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England) 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lawrence M et al. , Software for Computing and Annotating Genomic Ranges. PLoS computational biology 9, e1003118 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.