

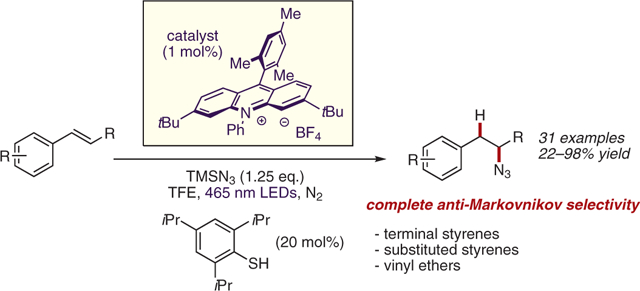
Abstract
Organic azides serve as synthetically useful surrogates for primary amines, a functional group which is ubiquitous in bioactive and medicinally relevant molecules. Historically, the formal hydroazidation of simple activated olefins and styrenes has proven difficult due to the inherent propensity of these compounds to oligomerize. Herein is disclosed a method for the anti-Markovnikov hydroazidation of activated olefins, catalyzed by an organic acridinium salt under irradiation from blue LEDs. This method is applicable to a variety of substituted and terminal styrenes and several vinyl ethers, yielding synthetically versatile hydroazidation products in moderate to excellent yield.
Keywords: photooxidation, alkenes, azides, radicals, regioselectivity
Graphical Abstract
The ubiquity of nitrogenous functional groups in industrially and medicinally important organic molecules cannot be overstated. Given their known importance, the development of reactions that can introduce nitrogen-based functionality to a molecular scaffold has been a well-traversed area of research. Organic azides serve as versatile precursors to primary amines following a simple hydrogenation or Staudinger reduction. Outside of their utility as amine surrogates, the biological stability and diverse reactivity of azides has led to the development of a variety of bioconjugation methods that hinge on so-called azide ‘click chemistry’ – typically in the form of copper-catalyzed [3+2] Huisgen type cycloadditions or Staudinger ligation processes.1–5
Organic azides are typically prepared via SN2 type substitution reactions, utilizing an inorganic azide source (often sodium azide) and a primary or secondary alkyl halide. However, this limits the scope of possible azide products to those that can be tracked to commercially available or synthetically viable alkyl halide precursors. The direct reaction of a hydrazoic acid (HN3) across olefins is known, but often limited to substrates that produce stabilized carbocations following protonation and requires the use of excess HN3.6 Compounded with the well-documented hazards associated with the use of HN3, including its explosive and toxic nature, the direct reaction of alkenes with hydrazoic acid is not a realistic solution to the preparation of organic azides in a small-scale laboratory setting.
Several transition-metal-mediated and radical hydroazidation reactions have been reported in recent years (Scheme 1). In 2005, Carreira and co-workers reported a hydroazidation reaction of simple unactivated olefins by using a cobalt Schiff base complex that was prepared in situ in the presence of a substoichiometric quantity of hydroperoxide oxidant, silane, and tosyl azide.7 While this method is mild and tolerant of a variety of functional groups, examples utilizing styrenes or other types of electron-rich olefin substrates are limited. Electrophilic nitrogen-centered azide radicals engage in anti-Markovnikov addition reactions with unactivated olefins.8–11 Azide radicals may be generated using hypervalent azido-iodine compounds in combination with a copper catalyst or photoredox catalyst, affecting the hydroazidation of simple olefins.12,13 Earlier this year, Xu and co-workers reported the generation of azide radicals using a benziodoxole organocatalyst in the presence of water and trimethylsilylazide (TMSN3).14 This reaction proceeds smoothly with a variety of mono-, diand tri-substituted olefins bearing a number of potentially sensitive functional groups. However, more electron-rich olefins, including substituted and unsubstituted styrenes, enol ethers and enamines proved unreactive under the optimized conditions.
Scheme 1.
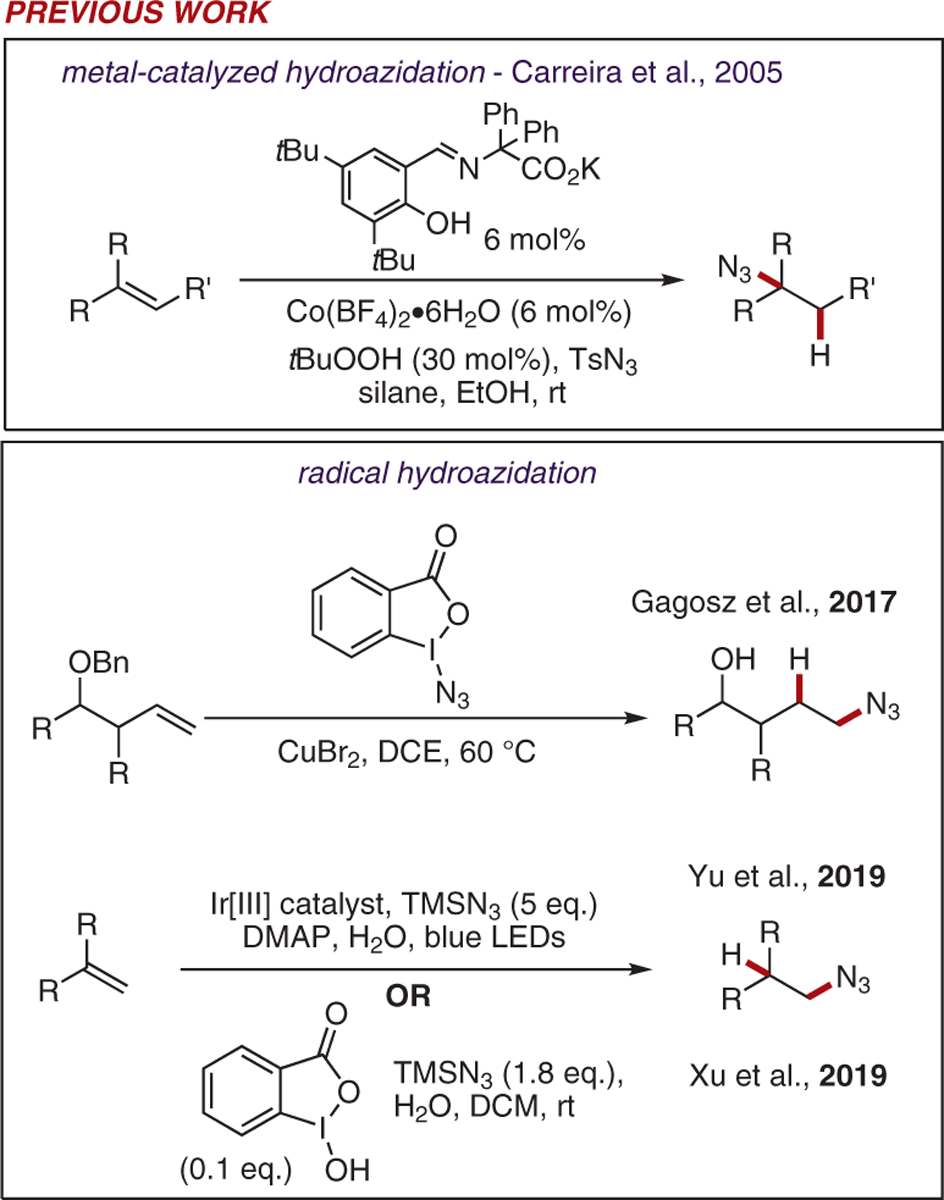
Recent work in alkene hydroazidation
To our knowledge, methods for the direct anti-Markovnikov hydroazidation of activated alkenes are nonexistent. As such, we sought to develop a hydroazidation reaction that would operate smoothly on these more electron-rich substrates. A methodology that would enable the hydroazidation of styrenyl substrates is particularly interesting given the known biological activity of phenylethylamine derivatives.15
Based on previous work from our group focused on alkene hydrofunctionalization reactions, we envisioned that photoredox catalysis may be a useful tool to develop a methodology enabling the anti-Markovnikov hydroazidation of this class of substrates.
By using a strongly oxidizing acridinium salt photoredox catalyst, alkene cation radicals may be generated in catalytic quantities via photoinduced electron transfer (PET) upon irradiation with blue light. The removal of an electron from the alkene π-system renders the resulting cation radical electrophilic and able to react with a suitably paired nucleophilic partner. Following interception of the cation radical by a nucleophile, a hydrogen atom transfer event facilitated by a hydrogen atom donor co-catalyst can trap the resulting benzylic radical, affording formal hydrofunctionalization products.16,17 Since the regioselectivity of nucleophile addition is dictated by the formation of the more stable radical species, this process proceeds with anti-Markovnikov selectivity in nearly all cases (Scheme 2).
Scheme 2.
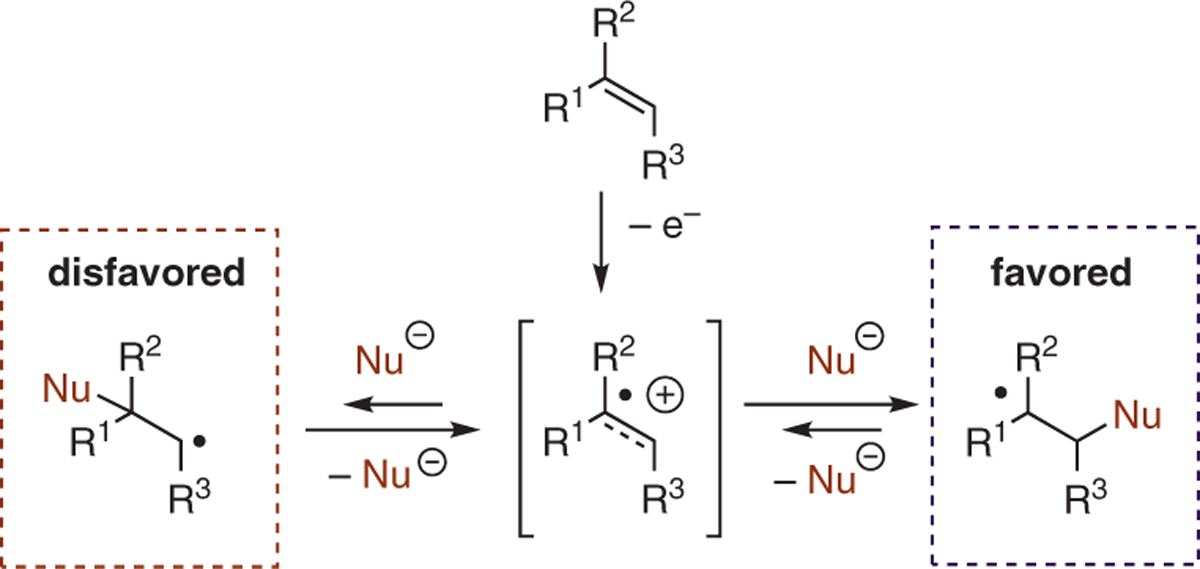
Regioselectivity in nucleophilic addition to alkene radical cations
The generation of reactive cation radical and neutral radical species in catalytic qualities renders this chemistry tolerant of a variety of synthetically useful functional handles. Our group has leveraged the reactivity of PET-generated alkene cation radicals to develop formal anti-Markovnikov hydroacetoxylation, hydroamination, and hydro-etherification reactions, among others.17–21 Based on this body of work, we envisioned that a formal hydroazidation reaction could be conceived through a similar reaction manifold.
Reaction development began with the use of indene as a substrate in the presence of 5 mol% of acridinium photooxidant tBu-Acr-BF4, 20 mol% of diphenyl disulfide, and 2.0 equivalents of sodium azide in trifluoroethanol (TFE, 0.1 M), a solvent commonly employed in our past reactions that was also thought to act as a proton source (Scheme 3). Tetrabutylammonium tetrafluoroborate (TBA BF4; 2.0 equiv) was added under the assumption that the tetrabutylammonium cation would serve as a phase-transfer agent, helping to solubilize azide ions. Following irradiation for 18 hours, the desired hydroazidation product was formed in 30% yield (based on 1H NMR analysis using HMDSO as an internal standard). Along with the desired product, a nearly equal amount of the corresponding thiol-ene product, resulting from addition of a thiyl radical formed via homolysis of diphenyl disulfide to the olefin, was observed. When acetonitrile (MeCN) was used as the solvent under otherwise identical conditions, only alkene starting material was observed following irradiation. By exchanging the hydrogen atom transfer catalyst from diphenyl disulfide to the bulkier 2,4,6-triisopropylthiophenol (TRIP-SH), the desired product was formed in 75% yield with no formation of thiol-ene byproducts. Further optimization showed that the loadings of acridinium photocatalyst and TMSN3 could be lowered to 1 mol% and 1.25 equivalents, respectively, with no adverse effect on the yield of the hydroazidation product.
Scheme 3.
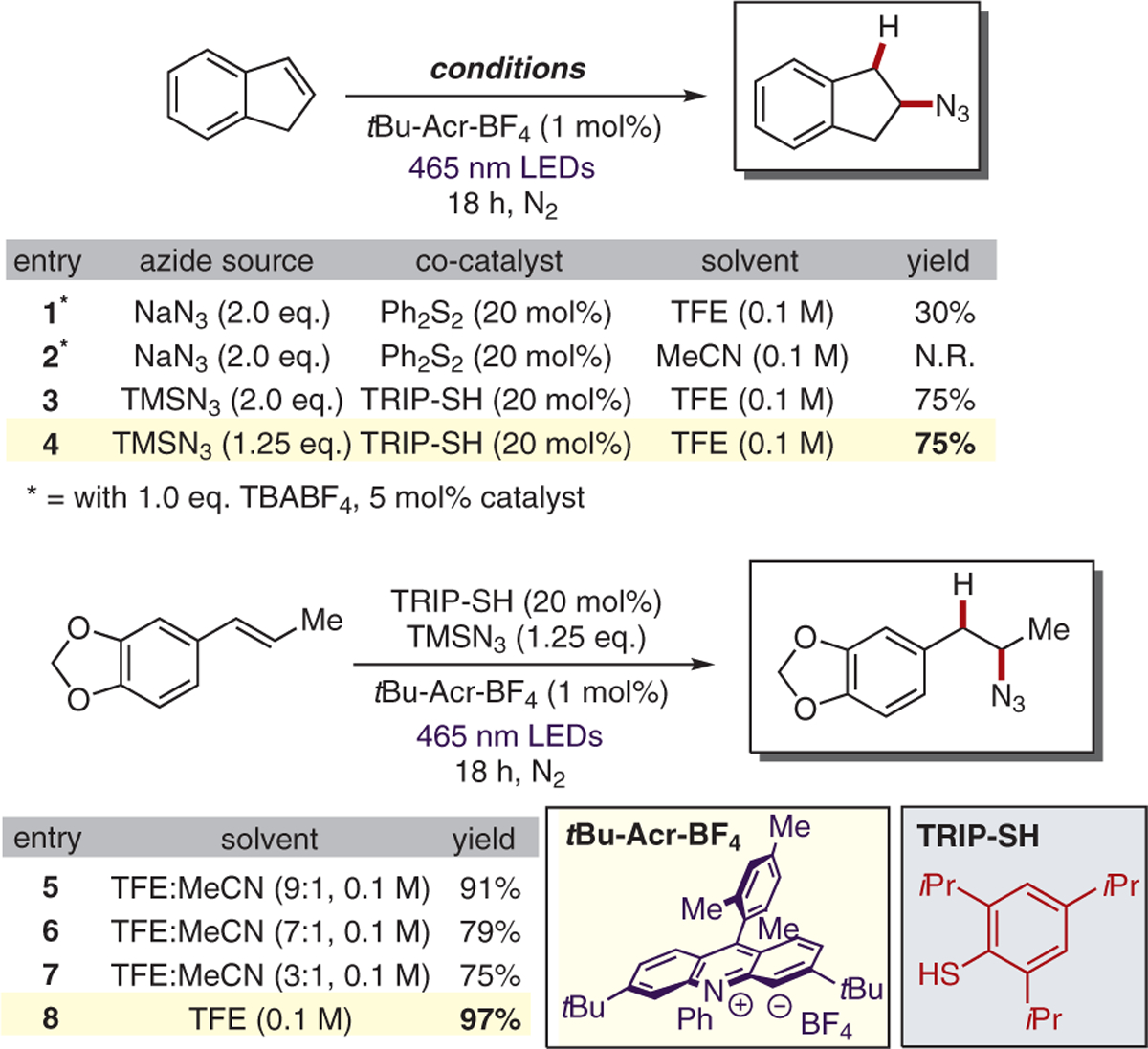
Reaction optimization
Interestingly, in a series of experiments utilizing various TFE/MeCN solvent mixtures and isosafrole as the substrate, the yield of the desired hydroazidation product decreased from 91% to 75% as the percentage of acetonitrile in the solvent mixture was increased from 10% to 25% (Scheme 3, entries 5–8).
Early spectroscopic investigations of the reaction between alkene cation radicals and various nucleophiles have demonstrated that hydrogen bonding and solvent polarity have profound effects on the rates of nucleophilic addition to these reactive species.22,23 More specifically, previous work from the Schepp group has demonstrated that the addition of azide anion to alkene radical cations is strongly affected by hydrogen-bond attenuation of redox potentials. The kinetics of the reaction between styrene cation radicals and azide anion has been previously studied via flash photolysis transient absorption spectroscopy. In non-hydrogen-bonding solvents, such as acetonitrile, the azide ion (N3–) is oxidized by the alkene cation radical to yield azide radical (N3•) and the corresponding neutral alkene. The azide radical then quickly equilibrates with a second equivalent of azide ion to generate an inactive, non-nucleophilic N6•– radical anion dimer with an estimated equilibrium constant of ca. 200 M–1 (in MeCN), which is identifiable by an absorption centered at 700 nm.24 However, when trifluoroethanol is used as the solvent, no absorption attributed to the formation of N6•– is identifiable. Following flash photolysis of TFE solutions of styrene derivatives in the presence of azide ion, an absorption maxima between 350 and 380 nm is observed, indicating the formation of a benzylic radical resulting from nucleophilic addition of azide ion to the alkene cation radical.25 Furthermore, the peak potential observed via cyclic voltammetry for oxidation of azide ion is shifted +0.5 V in TFE versus MeCN, indicating that hydrogen bonding is capable of dramatically attenuating the redox potential of this anionic species (E°peak (MeCN) = 0.275 V vs. Fc, E°peak (TFE) = 0.802 V vs. Fc). When reacted in TFE solution, the alkene cation radical is no longer able to oxidize the azide ion, and productive nucleophilic addition takes place. This previous work is in good accordance with our observations during reaction optimization.
With optimized conditions in hand, the scope of this transformation with respect to alkene partners was explored (Scheme 4). β-Substituted styrene derivatives were found to be excellent substrates for this transformation, with β-methylstyrene affording the desired hydroazidation product 9 in 98% yield (based on NMR analysis, using HMDSO as an internal standard). Simple alkyl-, aryl-, and chlorostyrene derivatives were all smoothly converted into the corresponding secondary azide products 11–14 in good yield. A variety of phenolic substrates were well-tolerated under the optimized reaction conditions, including those bearing benzoyl (22), benzyl (24), and silyl (25) protecting groups. Aromatic ester product 27 was isolated in 70% yield with no transesterification observed. A more oxidizable naphthalene-derived substrate also afforded the hydroazidation product 26 in good yield. Substrates containing potentially labile benzylic C–H bonds were converted into the desired products 10, 17, and 28 in excellent yield and no functionalization was detected at these reactive C–H sites. Notably, a substrate containing a terminal alkene was converted into the anti-Markovnikov hydroazidation product 30 in 71% isolated yield with no functionalization of the unactivated alkene detected, highlighting the complementarity of this method to other known radical hydroazidation reactions. Heterocyclic quinoline and thiophene substrates were functionalized to give the corresponding azide products 31 and 32, in 55% and 65% yield, respectively. Terminal styrene derivatives underwent the desired transformation in poor to moderate yields. Notably, diphenyl disulfide was identified as a more efficient hydrogen atom transfer catalyst than TRIP-SH for these substrates. Due to the lack of substituents in the β-position, these styrenes are prone to oligomerization via radical mechanisms. To combat this, a less sterically hindered HAT catalyst must be employed to accelerate the rate of HAT versus oligomerization. Oxidizable vinyl ethers were also competent reactants, with products 6 and 7 isolated in 58% and 79% yield. Product 7 is formed following elimination of the tertiary alcohol in hydroazidation product of substrate 8 during chromatography.
Scheme 4.
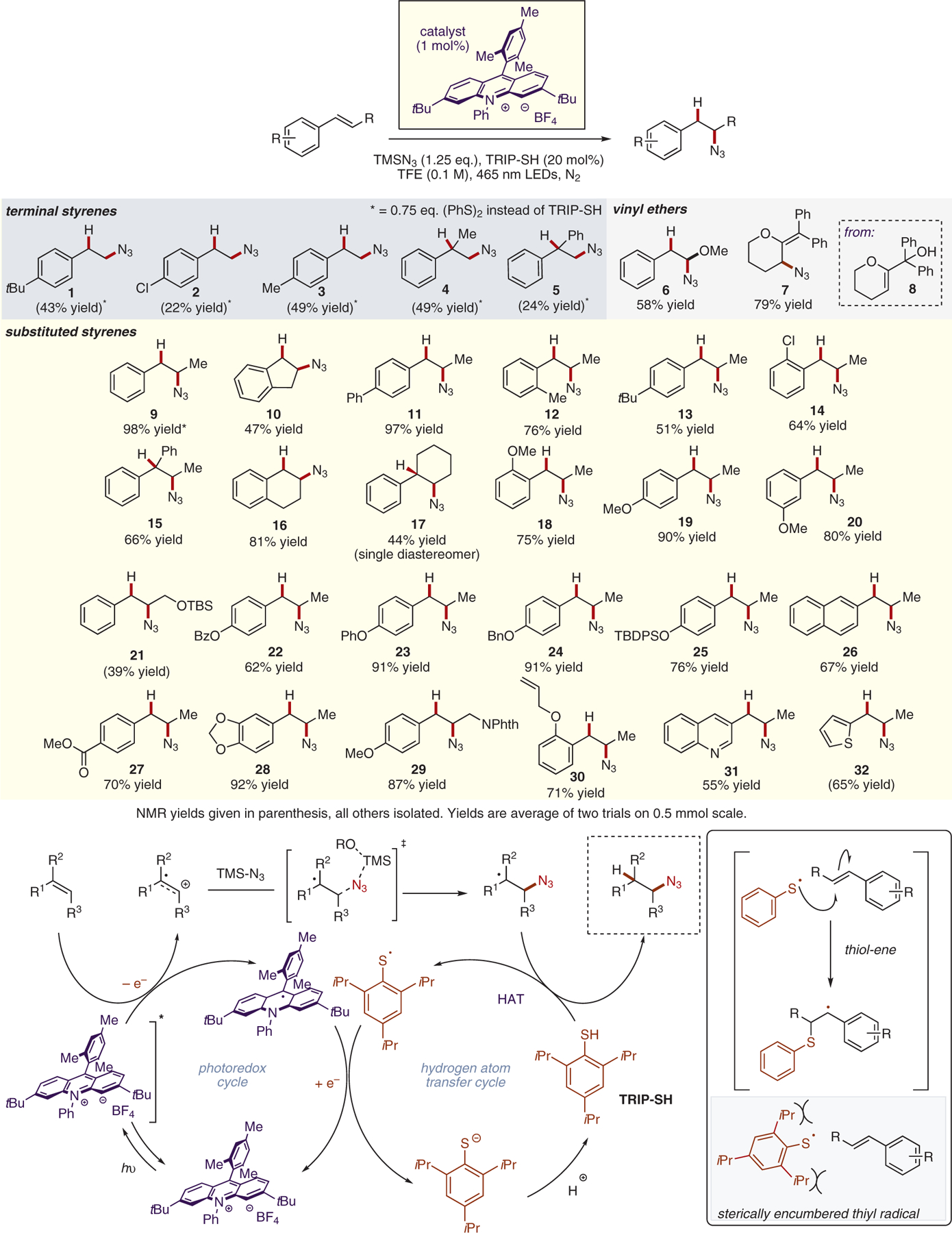
Substrate scope and proposed mechanism for photoredox alkene hydroazidation
Based on previous mechanistic investigations, the following mechanism is proposed. Following excitation by 465 nm light, the excited state of the acridinium catalyst engages in photoinduced electron transfer with the alkene substrate, yielding the corresponding alkene cation radical and the reduced form of the catalyst. Nucleophilic addition of azide ion generates a neutral radical species. Based on our observations, it is not clear whether free azide ion is generated prior to addition to the alkene cation radical. It is possible that a termolecular transition state involving a solvent molecule-assisted desilylation/nucleophilic addition is operative or that an azide silicate species could be the nucleophile. However, it is likely that some free azide ion is in solution due to hydrolysis of TMSN3 by advantageous water in the reaction mixture, as strict exclusion of water was not maintained. The benzylic radical engages in hydrogen atom transfer with TRIP-SH, generating the desired anti-Markovnikov hydroazidation product and a thiyl radical. This thiyl radical then oxidizes the reduced form of the catalyst, regenerating the ground state tBu-Acr-BF4 as well as a thio-late anion. This anion is then protonated by solvent to yield the starting co-catalyst and close the catalytic cycle.
In conclusion, we have developed an organic photoredox anti-Markovnikov hydroazidation reaction of electron-rich olefin substrates. By utilizing electrophilic cation radical intermediates, previously problematic hydroazidation reactions involving activated olefins now proceed efficiently and in high yields with low loadings of both photocatalyst and TMSN3. Furthermore, the transformation proceeds in the absence of any transition metals using TMS-N3 as the only stoichiometric reagent.26 This method fills the gap in the literature with regard to alkene hydroazidation chemistry.
Supplementary Material
Funding Information
This project was supported by Award No. R01 GM098340 from the National Institute of General Medical Sciences. M.E.S.H. is grateful for an NSF Graduate Fellowship. J.L.R.C. was supported by a National Science Foundation REU SUROC Award to UNC (NSF-REU 1757413).
Footnotes
Supporting Information
Supporting information for this article is available online at https://doi.org/10.1055/s-0039-1690691.
References and Notes
- 1.Liang L; Astruc D Coord. Chem. Rev 2011, 255, 2933. [Google Scholar]
- 2.Bräse S; Gil C; Knepper K; Zimmermann V Angew. Chem. Int. Ed 2005, 44, 5188. [DOI] [PubMed] [Google Scholar]
- 3.Meldal M; Tornøe CW Chem. Rev 2008, 108, 2952. [DOI] [PubMed] [Google Scholar]
- 4.Aldhoun M; Massi A; Dondoni AJ Org. Chem 2008, 73, 9565. [DOI] [PubMed] [Google Scholar]
- 5.Schilling CI; Jung N; Biskup M; Schepers U; Bräse S Chem. Soc. Rev 2011, 40, 4840. [DOI] [PubMed] [Google Scholar]
- 6.Breton GW; Daus KA; Kropp PJ J. Org. Chem 1992, 57, 6646. [Google Scholar]
- 7.Waser J; Nambu H; Carreira EM J. Am. Chem. Soc 2005, 127, 8294. [DOI] [PubMed] [Google Scholar]
- 8.Tsuchimochi I; Kitamura Y; Aoyama H; Akai S; Nakai K; Yoshimitsu T Chem. Commun 2018, 54, 9893. [DOI] [PubMed] [Google Scholar]
- 9.Hassner A; Boerwinkle FJ Am. Chem. Soc 1968, 90, 216. [Google Scholar]
- 10.Biermann U; Linker U; Metzger JO Eur. J. Lipid Sci. Technol 2013, 115, 94. [Google Scholar]
- 11.Zhang P; Sun W; Li G; Hong L; Wang R Chem. Commun 2015, 51, 12293. [DOI] [PubMed] [Google Scholar]
- 12.Wang J; Yu W Chem. Eur. J 2019, 25, 3510. [DOI] [PubMed] [Google Scholar]
- 13.Lonca GH; Ong DY; Tran TMH; Tejo C; Chiba S; Gagosz F Angew. Chem. Int. Ed 2017, 56, 11440. [DOI] [PubMed] [Google Scholar]
- 14.Li H; Shen S-J; Zhu C-L; Xu HJ Am. Chem. Soc 2019, 141, 9415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shulgin A; Shulgin A PIHKAL: A Chemical Love Story; Transform Press: Berkeley, 1991. [Google Scholar]
- 16.Romero NA; Nicewicz DA J. Am. Chem. Soc 2014, 136, 17024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Margrey KA; Nicewicz DA Acc. Chem. Res 2016, 49, 1997. [DOI] [PubMed] [Google Scholar]
- 18.Nicewicz D; Hamilton D Synlett 2014, 25, 1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nguyen TM; Nicewicz DA J. Am. Chem. Soc 2013, 135, 9588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perkowski AJ; Nicewicz DA J. Am. Chem. Soc 2013, 135, 10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu F; Wang L; Chen J; Nicewicz DA; Huang Y Angew. Chem. Int. Ed 2018, 57, 2174. [DOI] [PubMed] [Google Scholar]
- 22.Brede O; David F; Steenken SJ Chem. Soc., Perkin Trans. 2 1995, 23. [Google Scholar]
- 23.Johnston LJ; Schepp NP Pure Appl. Chem 1995, 67, 71. [Google Scholar]
- 24.Workentin MS; Schepp NP; Johnston LJ; Wayner DDM J. Am. Chem. Soc 1994, 116, 1141. [Google Scholar]
- 25.Johnston LJ; Schepp NP J. Am. Chem. Soc 1993, 115, 6564. [Google Scholar]
- 26.General Procedure (CAUTION: use care when handling TMSN3): A flame-dried 2-dram borosilicate vial (purchased from Fisher Scientific, catalogue # 03-339-22D), equipped with a stir bar, was charged with 3,6-di-tert-butyl-9-mesityl-10-phenylac ridin-10-ium tetrafluoroborate (0.01 equiv, 0.01 mmol) and 2,5,6-triisopropylthiophenol (0.10 mmol, 0.20 equiv).For solid/non-volatile substrates, the substrate (0.50 mmol) was then added. 2,2,2-Trifluoroethanol (5.0 mL) was added and the vials were capped tightly with a Teflon-lined phenolic resin septum cap (purchased through VWR international, Microliter Product # 15–0060K). The reaction mixture was then sparged by bubbling with nitrogen or argon for 5 minutes. Trimethylsilylazide (0.625 mmol, 1.25 equiv) was added by using a micro-liter syringe. Prior to irradiation, vials were sealed with Teflon tape and electrical tape to ensure maximal oxygen exclusion. The reaction vial was then placed into the reactor and irradiated for 18 hours unless otherwise noted.Following irradiation, the reaction mixture was concentrated under reduced pressure and the desired products were isolated by flash column chromatography (see the Supporting Information substrate/product details for solvent information). Unless otherwise noted, all reaction yields are reported as the average of two separate trials (including chromatography).Example products4-(2-Azidopropyl)-1,1′-biphenyl (11): Following irradiation, the crude reaction mixture was concentrated under reduced pressure and dry-loaded onto silica gel. The desired product was isolated as a pale-yellow oil following column chromatography (100% hexane to 1% EtOAc/hexane). Yield: 97% (n = 2).1H NMR (600 MHz, CDCl3): δ = 7.63 (dd, J = 26.1, 7.7 Hz, 4 H), 7.49 (t, J = 7.6 Hz, 2 H), 7.39 (t, J = 7.4 Hz, 1 H), 7.32 (d, J = 7.8 Hz, 2 H), 3.77 (h, J = 6.6 Hz, 1 H), 2.92 (dd, J = 13.7, 7.3 Hz, 1 H), 2.82 (dd, J = 13.7, 6.4 Hz, 1 H), 1.35 (d, J = 6.5 Hz, 3 H). 13C NMR (151 MHz, CDCl3): δ = 140.90, 139.73, 136.91, 129.79, 128.84, 127.29, 127.10, 59.04, 42.24, 19.22. HRMS (APCI, positive mode): m/z [M + H, – N2] calcd: 210.1277; found: 210.1278.1-(2-Azidopropyl)-2-chlorobenzene (14): Following irradiation, the crude reaction mixture was concentrated under reduced pressure and dry-loaded onto silica gel. The desired product was isolated as a pale-yellow oil following column chromatography (100% hexane to 3% EtOAc/hexane). Yield: 64% (n = 2).1H NMR (600 MHz, CDCl3): δ = 7.40 (dd, J = 7.2, 1.9 Hz, 1 H), 7.28 (dd, J = 7.1, 2.3 Hz, 1 H), 7.25–7.19 (m, 2 H), 3.85 (h, J = 6.7 Hz, 1 H), 3.00–2.84 (m, 2 H), 1.33 (d, J = 6.5 Hz, 3 H). 13C NMR (151 MHz, CDCl3): δ = 135.70, 134.32, 131.85, 129.74, 128.42, 126.95, 57.57, 40.40, 19.43. MS (EI): m/z [M]+ calcd: 195.056; found: 195.05.1-(2-Azidopropyl)-4-phenoxybenzene (23)Following irradiation, the crude reaction mixture was concentrated under reduced pressure and dry-loaded onto silica gel. The desired product was isolated as a clear oil following purification by column chromatography (100% hexane to 1% EtOAc/hexane). Yield: 91% (n = 2).1H NMR (600 MHz, CDCl3): δ = 7.38–7.30 (m, 2 H), 7.16 (d, J = 8.5 Hz, 2 H), 7.13–7.06 (m, 1 H), 7.01 (dd, J = 8.7, 1.1 Hz, 2 H), 6.96 (d, J = 8.5 Hz, 2 H), 3.77–3.47 (m, 1 H), 2.80 (dd, J = 13.8, 7.4 Hz, 1 H), 2.72 (dd, J = 13.8, 6.3 Hz, 1 H), 1.28 (d, J = 6.5 Hz, 3 H). 13C NMR (151 MHz, CDCl3): δ = 157.31, 156.03, 132.62, 130.57, 129.72, 123.18, 118.96, 118.78, 59.14, 41.87, 19.16. HRMS (APCI, positive mode): m/z [M + H, – N2] calcd: 226.1226; found: 226.1227.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.