

SUMMARY
Rheumatoid arthritis (RA) is an autoimmune disease characterized in seropositive individuals by the presence of anti-cyclic citrullinated protein (CCP) antibodies. RA is linked to the intestinal microbiota, yet the association of microbes with CCP serology and their contribution to RA is unclear. We describe intestinal phage communities of individuals at risk for developing RA, with or without anti-CCP antibodies, whose first-degree relatives have been diagnosed with RA. We show that at-risk individuals harbor intestinal phage compositions that diverge based on CCP serology, are dominated by Streptococcaceae, Bacteroidaceae, and Lachnospiraceae phages, and may originate from disparate ecosystems. These phages encode unique repertoires of auxiliary metabolic genes which associate with anti-CCP status, suggesting that these phages directly influence the metabolic and immunomodulatory capability of the microbiota. This work sets the stage for the use of phages as preclinical biomarkers and provides insight into a possible microbial-based causation of RA disease development.
eTOC BLURB
Mangalea et al. characterize intestinal bacteriophage communities from humans at-risk of developing rheumatoid arthritis. Bacteriophage profiles diverge based on anti-cyclic citrullinated protein autoantibody status compared to healthy controls. Bacteriophage profiling could complement existing diagnostics as a microbial biomarker for preclinical rheumatoid arthritis.
Graphical Abstract
INTRODUCTION
Rheumatoid arthritis (RA) is a systemic autoimmune disease with a global prevalence of approximately 1%. The development of RA in at-risk individuals is dependent on a combination of genetics, epidemiology, and systemic immune dysregulation (Holers et al., 2018). The heritability of RA is estimated to be 40%–60%, with increased familial risk evident among first-degree relatives (FDRs) of individuals with diagnosed RA (MacGregor et al., 2000; Deane et al., 2017). Analyses of at-risk FDRs, even those without serum RArelated autoantibodies, have identified patterns of mucosal inflammation whereby anti-cyclic citrullinated peptide (anti-CCP) antibodies, rheumatoid factors, and cytokines and chemokines are expressed locally in a subset of individuals (Chang et al., 2016; Demoruelle, 2019; Demoruelle et al., 2018). In addition, autoantibodies are present in the blood for years prior to the onset of RA and their presence as well as circulating cytokines and chemokines is predictive of future RA development (Demoruelle et al., 2017; Hughes-Austin et al., 2013; Willis et al., 2013). To probe the ‘‘mucosal origins’’ hypothesis (Holers et al., 2018) and mounting evidence implicating intestinal microbiota perturbations in RA etiopathogenesis (Brusca et al., 2014), it is necessary to characterize the ecological associations of the microbiota in at-risk individuals susceptible to RAStudies that have linked the role of the intestinal microbiota to systemic autoimmune diseases have predominantly relied on 16S ribosomal gene analyses of bacteria within the microbiome and have expanded our understanding of disease-specific alterations in the RA intestine. Individuals with established RA harbor a microbiota dominated by Prevotella copri (Scher et al., 2013; Maeda et al., 2016), enriched with Gram-positive bacteria (Zhang et al., 2015), and with decreased carriage of bifidobacteria (Vaahtovuo et al., 2008), Gram-negative Bacteroides, and Firmicutes (Toivanen et al., 2002; Zhang et al., 2015). The association of enriched Prevotellaceae, including P. copri, has also been described in individuals with preclinical RA (Alpizar-Rodriguez et al., 2019), indicating that intestinal P. copri is immunerelevant to the pathogenesis of RA (Pianta et al., 2017).
The presence of P. copri may therefore represent a biological indicator and additional risk factor for RA development and progression (Drago, 2019). However, associating a single organism to RA etiology neglects the interactions of bacteria with their surrounding environment and other bacterial community members whose populations can be influenced by predatory bacteriophages (phages).
In contrast to the recent enthusiasm for characterizing microbial links to the etiology of RA, little is known concerning the composition of phage communities in the intestine as it relates to RA disease risk. Phages of the intestinal microbiota can fluctuate in community composition in response to immune system function and disease, which suggests that they could be exploited as biomarkers for early disease detection (Duerkop, 2018). Metagenomic sequencing strategies have revealed extensive and diverse populations of phages in the human intestine (Minot et al., 2011, Manrique et al., 2016, Shkoporov et al., 2019), in which phage community dynamics correlate with distinct disease states (Duerkop et al., 2018, Clooney et al., 2019, Khan Mirzaei et al., 2020). Specific intestinal phage genomic signatures precede autoimmunity development of type 1 diabetes in a cohort of diabetes-susceptible children, with disease-associated phages correlating to the bacterial component of the microbiota (Zhao et al., 2017). Phages also adhere to mucosal surfaces, significantly impacting microbial colonization (Barr et al., 2013) and host mucosal immunity development (Quistad et al., 2017). Evidence is emerging that phages are also immunomodulatory through intrinsic anti-inflammatory properties, and are capable of direct lymphocyte regulation through the ability to translocate to multiple tissues (Gorski et al., 2017). Despite these observations and potential implications for systemic autoimmune diseases like RA, evaluation of intestinal phages in the context of RA disease risk has yet to be described.
The interplay between intestinal bacteria, their phages, and the host immune system, whose interactions have consequences not only for compositional modifications but immunomodulation, must be considered in the etiopathogenesis of RA. The microbiome, and more recently the virome, have been implicated in a range of human diseases including cancers (Minot et al., 2019, Yu et al., 2020), inflammatory bowel diseases (Norman et al., 2015, Gevers et al., 2014), and arthritis (Scher et al., 2013, Lee et al., 2019). By characterizing the phage populations in an at-risk RA cohort; further sub-grouped with regard to autoantibody status as defined by the presence of anti-CCP antibodies and compared to healthy controls, we have begun to address this question. Anti-CCP serology is a strong indicator of future RA development, but lacks sensitivity to definitively exclude disease development in seronegative individuals (Braschi et al., 2016). The cohort contains individuals that do not have inflammatory arthritis or established RA disease but are first-degree relatives to an individual with diagnosed RA, which alone increases RA risk. Studying the microbiomes of at-risk individuals could lead to the identification of novel biomarkers corresponding to existing diagnostic serology tests and therapeutic targets independent of confounding by the use of drugs in subjects with active arthritis.
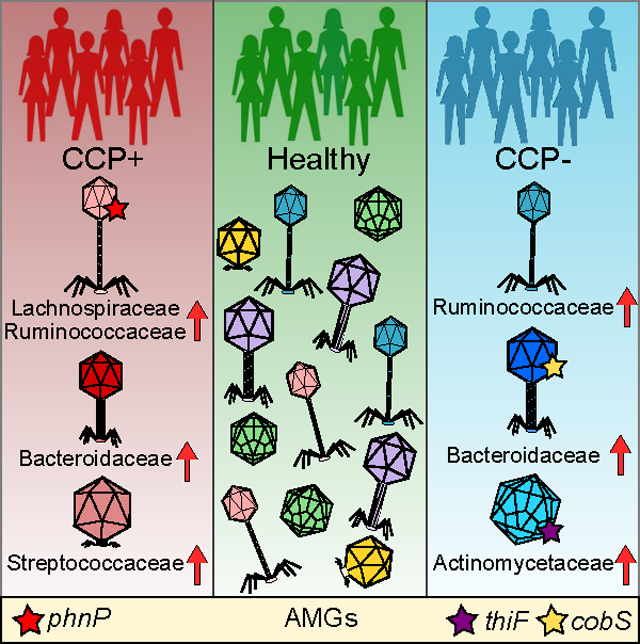
We used metagenomics to define intestinal phage populations of anti-CCP positive (CCP+) and negative (CCP-) individuals in a cohort at-risk for RA. Phage matching to bacterial hosts showed divergent intestinal phage communities dependent on anti-CCP serology status. We observed significant shifts in phages targeting Bacteroidaceae and Streptococcaceae bacteria in CCP+ at-risk FDRs as well as phages targeting Bacteroidaceae and Ruminococcaceae bacteria in CCP- at-risk FDRs. Importantly, analysis of the metabolic traits encoded in phage metagenomes revealed intra-cohort profiles reflecting distinct immunomodulatory potential. Phages with auxiliary metabolic genes (AMGs) that modify lipopolysaccharide and other outer membrane glycans of host bacteria were differentially abundant, implicating modifications to bacterial antigenicity (Van Belleghem et al., 2018) and bacterial fitness (Rodriguez-Valera et al., 2009) in RA-associated communities. Phages targeting Lachnospiraceae (Clostridium scindens) and Actinomyces (A. oris), including several AMGs, were over-abundant among CCP+ and CCP- individuals, respectively, compared to healthy controls. Our data show that there are unique and abundant intestinal phages specific to RA-susceptibility status, and this highlights their potential as biomarkers for preclinical RA and the need for further study of communitylevel bacteria-phage interactions during the development and progression of RA.
RESULTS
First-degree relatives to individuals with rheumatoid arthritis
A total of 25 human subjects were identified from the Studies of the Etiology of Rheumatoid Arthritis (SERA) (Kolfenbach et al., 2009), including 16 FDRs of individuals with RA and 9 age- and sex-matched healthy controls (HC). FDR subjects for which a detectable level of anti-CCP autoantibody was present (defined by a value ofR20 units/mL in either ELISA assay for anti-CCP3.1 IgA/IgG or anti-CCP3 IgG; Demoruelle et al., 2013) were designated the CCP+ group (n = 8). FDRs with no detected anti-CCP were designated the CCP group (n = 8) (Table 1). The mean age for the three groups in this study were 61.3 ± 11.0 for CCP+, 49.0 ± 15.7 for CCP, and 44.4 ± 13.6 for HC. The distribution of sexes for each group is reported as percent female, with 88.9% for CCP+, 62.5% for CCP, and 66.7% for HC. Among the CCP+ and HC groups, 3/9 and 2/9 of individuals reported ever smoking (a risk factor associated with RA), respectively (Table 1).
Table 1.
Summary of Subject Characteristics for the Samples Included in the Study
| VARIABLE | HC | CCP+ | CCP− |
|---|---|---|---|
| Count | 9 | 8 | 8 |
| Age (mean) | 44.4 | 61.3 | 49 |
| Age (SD) | 13.6 | 11 | 15.7 |
| Sex (% female) | 66.7 | 88.9 | 62.5 |
| Serum CCP+ (%) | 0 | 100 | 0 |
| Ever smokers (%) | 22.2 | 33.3 | 0 |
Generation and curation of de novo-assembled VLP contigs
Individual fecal samples from subjects were obtained at the time of autoantibody and clinical evaluations and were used for total genomic DNA isolation for shotgun metagenomic sequencing using an untargeted amplification-independent approach (Duerkop et al., 2018; Kang et al., 2017). In total, 3.56 million (Mio) contigs were assembled and pooled from the 25 individual metagenomes, with 80,762 contigs longer than 5 kb (Figure 1A).We used a three-pronged approach of independent phage discovery methods (Figures 1A and S1); (i) P/M ratio, a mapping strategy comparing ratios of reads mapped to VLP contigs (Duerkop et al., 2018), (ii) viral protein family (VPF) homology (Paez-Espino et al., 2017), and (iii) VIBRANT (Virus Identification By iteRative ANnoTation) (Kieft et al., 2020), identifying 2117, 4785, and 4758 putative phage contigs respectively (see STAR Methods). To consolidate this list, we identified contigs that were shared between all three phage discovery methods, resulting in a curated list of 660 contigs (Figures 1A and 1B). To assess host bacterial contamination among these contigs, we employed CheckV to assess the quality of viral genomes (Nayfach et al., 2020a). CheckV analysis revealed a reduced level of host bacterial contamination and an increase of pure viral genomes in the final list of 660 curated contigs (Figure 1C). We estimated completeness of our curated contigs using CheckV and determined a greater distribution of “high quality” contigs relative to contig length, in comparison to the three independent methods (Figure S2) (Roux et al., 2019). Further, using the VIBRANT platform for integrated provirus prediction, we describe communities of predominantly lytic viral genomes belonging to the Siphoviridae (Figure S3). By using a combination of approaches for viral contig discovery and assessing the overlap among these methods, we have extracted a set of 660 predicted phages which are of overall high quality, both in terms of viral contig completeness and lack of bacterial contamination than those from each of the individual methods (Figures 1C and S2), which to date have been used primarily in isolation to identify and characterize viral metagenomes.
Figure 1.

Generation and curation of de novo assembled VLP contigs. (A) De novo assembled contigs resulted in a total of 3,557,500 contigs for the entire sample set. Three independent methods were used to identify putative phages from the list of 80,762 contigs resulting in 2,117 contigs from the P/M ratio method, 4,785 contigs from the Viral Protein Families method, and 4,758 contigs using VIBRANT. (B) A Venn diagram shows the overlap of redundant contigs identified among the three methods. 660 unique contigs were identified independently by all phage identification methods. (C) CheckV contamination analysis of the three separate methods as well as the final set of curated contigs.
Clustering of metagenomic viral contigs reveals distinct viral ecological composition.
Next we compared our set of curated contigs to over 2.3 Mio viral whole genome and metagenome sequences from the IMG/VR database (Roux et al., 2020). Of the 660 contigs, 346 (52.4%) clustered into 255 clusters that contained 7,736 additional metagenomic viral contigs (mVCs) from IMG/VR. The remaining 314 contigs (47.6%) did not cluster with any other sequence and were classified as singletons, which distributed evenly among all three groups (Figure S4A). Of the curated contigs that did cluster, cluster sizes ranged from 2 to 646 members with 78.4% of the groups containing more than 2 members, 36.5% containing more than 10 members, and 65.9% between 2 – 10 members (Figure S4B). Among these 255 clusters, 14 included reference prophages and lytic phages, and 318 (48.2%) of our original contigs clustered with classified mVCs, thus assigning multiple levels of taxonomy (Figures 2A, 2B, and Table S1).
Figure 2.

Clustering with metagenomic viral contigs reveals viral ecological composition. (A) Host assignments for the set of curated phages based on Markov clustering to the IMG/VR database metagenomic viral clusters or direct match to bacterial CRISPR spacers, based on cohort abundance. (B) Cladogram of the complete host phylogeny at the genus level for all spacers identified from total RA virome via the VPF method. The pie chart at the center represents all 958 CRISPR spacers from the family level quantified in panel A. Total host hits were quantified at the genus level and are represented in relative size by colored circles, indicating host assignments that were discerned via clustering (dark grey) and those that were identified via direct CRISPR spacer matching (light grey). Total CRISPR spacers per contig with family level host taxonomy assignments were tabulated per cohort group (C) and differentiated as narrow or broad phage host ranges (D) based on target uniformity to bacterial family. See also Tables S1, S2, S3.
Although host assignments were made using sequence-based clustering, host specificity was further determined by aligning Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR) spacer sequences to our 660 curated contigs. CRISPR spacer host assignments at the family level were present in 207 of 660 contigs (31.4%). All CRISPR spacer queries considered for these analyses, ranging in length from 18 to 70 bp, were matches of 93.1–100% identity across the full length of the query and allowing for 0–2 mismatches and up to 1 gap throughout (Paez-Espino et al., 2016) (Table S2). Among predicted phages, total assigned CRISPR spacers were evenly distributed, yet CCP+ sample containing phages predicted to target Lachnospiraceae, Ruminococcaceae, Streptococcaceae, and Veillonellaceae bacterial families were disproportionately abundant (Figures 2A and 2B). In total, 21 bacterial families were identified as hosts via CRISPR spacer matching, supplementing the phage-host interactions discerned from sequence-based clustering (Figure 2A). Among all samples in this study, phages were predicted to target Lachnospiraceae (261 CRISPR spacers), Ruminococcaceae (126), Clostridiaceae (98) and Bacteroidaceae (96) bacteria with highest frequency of total CRISPR spacers (Figure 2A, Table S3)). Phage-host interactions were also measured in terms of host range specificity, showing that while the majority of the phages were predicted to have narrow host ranges, several spacers were linked to multiple hosts across family level and higher taxa (Figure 2C), consistent with prior observations of diverse viromes (Paez-Espino et al., 2016). Broad host range phages were found across all cohorts, with a slight yet insignificant skew among CCP+ sample contigs (Figure 2D).
We further explored the association of sample cohorts to phage hosts using read mapping to determine differential host abundance profiles (Figure 3). Reads from all samples were mapped to assembled phage contigs whose host assignments were deduced using CRISPR-spacer matching and Markov clustering to quantify sequence abundances by measuring cohort-based read recruitment (Moreno-Gallego et al., 2019, Duerkop et al., 2018, Roux et al., 2017, Liang et al., 2020). In comparing the differential read recruitment to phages predicted to infect separate bacterial families, we observed differences based on reads originating from either the CCP+ or CCP- groups in relation to the HC cohort (Figure 3). Phage contigs targeting Bacteroidaceae recruited significantly more reads from CCP+ viromes than either HC or CCP- individuals (Figure 3A). In contrast, phages predicted to target Clostridiaceae bacteria were evenly abundant across all three groups (Figure 3B). Lachnospiraceae phages were evenly distributed among the groups with a slight elevation in CCP+ individuals that was not statistically significant (Figure 3C). Ruminococcaceae phages were significantly skewed when comparing HC to CCP- individuals (Figure 3D) and a major shift in phage read recruitment abundance was evident for Streptococcaceae phages, as a greater percentage of CCP+ reads were mapped to these phages in relation to either HC or CCP- virome reads (Figure 3E). This skew among CCP+ individuals is supported by prior works showing elevated Streptococcal phage abundances in intestinal viromes of humans with inflammatory bowel disease (Norman et al., 2015) and a murine model of colitis (Duerkop et al., 2018). No significant differences were observed for read recruitment to Veillonellaceae-targeting phages (Figure 3F). Thus, differences in the host specificities were evident between CCP+, CCP-, and HC groups with respect to read mapping abundance profiles for Bacteroidaceae, Ruminococcaceae, and Streptococcaceae phages.
Figure 3.

Phage-host assignments for curated VLP contigs reveal cohort-based differential read recruitment among several bacterial families. Relative abundances were calculated for all VLP reads mapped to phages predicted to target Bacteroidaceae (A), Clostridiaceae (B), Lachnospiraceae (C), Ruminococcaceae (D), Streptococcaceae (E), and Veillonellaceae (F) bacterial families. Scaffold abundances were averaged across all samples and statistics were determined by nonparametric Wilcoxon tests (* p < 0.05, ** p < 0.01, **** p < 0.0001).
CRISPR spacer host metadata reveal CCP+ phages represent greater variability in microbial host ecology.
To further explore the phage ecology from our subject cohort, we analyzed the host and mVC metadata from the Joint Genome Institute’s (JGI) Genomes OnLine Database (GOLD) (Mukherjee et al., 2019). Using the GOLD Biosample Ecosystem Classification system, we analyzed the ecosystem distributions for all CRISPR spacers identified in our curated contig list and discovered that the majority of host assigned contigs fell within four distinct ecosystem classification levels; from broad to specific environments: host-associated, human-associated, digestive system, and large intestine (Figure 4). For each of the four ecosystem categories, the most abundant classifications were used to compare across the study cohorts. At the highest order GOLD Ecosystem distribution, the host-associated (i.e., human, mammal, plant, arthropod, fungi) origin classification per contig was comparable for the HC and CCP- groups but not for the CCP+ group (Figure 4A). A similar pattern was evident at the lower order metadata distributions, with phage contigs derived from CCP+ individuals being more divergent from the other cohorts for contigs of human-associated origin (Figure 4B), digestive system origin (Figure 4C), and large intestine origin for the Ecosystem Subtype (Figure 4D).
Figure 4.

CRISPR spacer host metadata reveal CCP+ phages represent greater variability in microbial host ecology. (A) JGI/GOLD Ecosystem Distribution showing the percent host-associated spacers calculated for each contig based on cohort distribution. (B) Ecosystem Category distribution showing the percent human-associated spacers. (C) Ecosystem Type distribution showing the percent of contigs that contain spacers originating from the digestive system. (D) Ecosystem Subtype showing the percent of contigs that contain spacers originating from the large intestine microenvironment. Statistical significance was determined using pairwise Wilcoxon rank sum tests for comparisons between the three groups, using the Benjamini-Hochberg correction for multiple testing comparisons (* p = 0.023, **** p < 2 × 10−16). See also Figure S4 and Table S6.
Multiple CRISPR spacer ecosystem distributions revealed homogeneity among phages derived from HC and CCP- samples, and indicated greater disparity in communities across CCP+ samples, suggesting that CCP+ individuals harbor disparate phage communities that are more likely to originate from non-host associated sources. The putative origins of these phages are related to environmental metadata of CRISPR spacers in the JGI GOLD database describing the origin of bacterial DNA samples across ecologically diverse biomes worldwide (Nayfach et al., 2020b); and increased heterogeneity in the CCP+ phages suggests a condition-dependent host intestinal environment with increased diversity. At the highest Ecosystem classification level, with only three unique classification terms, these non-host associated sources that are more prevalent in the CCP+ group, correspond to a higher degree of spacers matching organisms originating from environmental and/or engineered habitats as archived in GOLD (Figure S5). Examples of engineered habitats include wastewater and food production, while the environmental ecosystem broadly encompasses microbes originally identified as aquatic, terrestrial, or airborne, indicating multiple possible combinations for organism habitats. Our analysis of GOLD metadata for all phages with predicted host isolates within our study reveals the possibility of divergent habitat origins for CCP+ derived contigs.
Quantitative read mapping reveals differentially abundant contigs despite sample cohesiveness.
We next asked whether certain phage community members are present in different abundances among the members of the cohort at-risk for rheumatoid arthritis compared to healthy controls. To assess differences between phages among the sample groups, we used a viral read recruitment strategy whereby VLP reads from all samples were mapped to the 660 curated contigs (Moreno-Gallego et al., 2019, Duerkop et al., 2018). Using read count matrices for all contigs as input in the DEseq2 statistical package for differential analysis of comparative count data (Love et al., 2014), we analyzed three pairwise comparisons for over- or under-abundant viral contigs (Figure 5). Initial comparisons of normalized and log-transformed count matrices were performed to evaluate differences across all samples. Principal component analyses reveal minimal variance explained by the first two principal components for CCP+ vs HC samples (Figure 5A), CCP- vs HC samples (Figure 5B), and CCP+ vs CCP- samples (Figure 5C), indicating that total sample community signatures cannot be readily differentiated based on at-risk or healthy control cohorts. We further explored the sample similarities by comparing Euclidian sample-to-sample distances of normalized log-transformed count matrices. Hierarchical clustering of sample-to-sample distances did not reveal any discernable clustering for CCP+ vs HC samples (Figure 5D), and only minimal similarities between two CCP- samples when compared to the HC (Figure 5E) and CCP+ (Figure 5F) groups, suggesting general sample cohesiveness between cohorts.
Figure 5.

Quantitative read mapping exposes differentially abundant contigs despite sample cohesiveness. (A, B, C) Analyses of the first and second principal components of sample-to-sample DESeq2 analyses revealed minimal variance explained across all comparisons. (D, E, F) Euclidian distances for sample-sample read-based coverages were used for hierarchical clustering across all pairwise comparisons reveal minimal clustering based on sample type. (G, H, I) Volcano plots reveal 9%, 10%, and 8% of contigs included in our analysis are differentially abundant respective to CCP+ vs. HC, CCP- vs. HC, and CCP+ vs. CCP- group-based comparisons of specific contig community members. See also Table S5.
Considering the rationale that samples with complex communities are better explored at the level of each unique member (Gevers et al., 2014), we next analyzed specific members of the intestinal phage community. Visualization of the principal components incorporating the viral identification metrics used in the VIBRANT neural network for our 660 curated contigs shows minimal differentiation among phage scaffolds based on scaffold quality (Figure S6A) or predicted phage state (i.e., lytic or lysogenic) (Figure S6B), although fragmentation of smaller sized contigs is evident for both analyses. Further, grouping of contigs at the sample type level does not differentiate any specific cluster (Figure S6C), which is consistent with the minimal variance observed at the sample level (Figures 5A, 5B, and 5C). Finally, we assessed the differential abundance of read recruitment counts for the set of 660 contigs and estimated fold changes based on the negative binomial generalized linear model provided by DESeq2 (Love et al., 2014). Using thresholds of log2-fold change greater than 1 or less than −1 (equivalent to fold change of ± 2) and Benjamini-Hochberg adjusted p-values < 0.001, we identified a total of 178 differentially abundant contigs (27% of the 660 phages) across three pair-wise abundance comparisons. For CCP+ vs HC samples a total of 59 contigs (30 over- and 29 under-abundant) (Figure 5G), for CCP- vs HC a total of 66 contigs (27 over- and 39 under-abundant) (Figure 5H), and for CCP+ vs CCP- a total of 53 contigs (27 over- and 21 under-abundant) (Figure 5I) passed our thresholds for significance. These data indicate that these cohort groups represent minimal sample-sample variation, but may provide clues related to detection of biomarkers via specific community members. The top phage contigs associated with either CCP+ or CCP- individuals were Clostridium scindens (Lachnospiraceae) and Actinomyces oris (Actinomycetaceae), respectively, over-abundant at log2 fold changes of 25.9 and 23.5 compared to the healthy control samples. A comparison of the bacterial relative abundances via 16S amplicon sequencing confirmed an expansion of Lachnospiraceae bacteria among samples originating from CCP+ individuals (Figure S7A). The bacterial composition across all cohorts was relatively even in terms of richness (Figures S7B and S7C), evenness (Figure S7D), and species diversity (Figure S7E). Conversely, phage host abundances in the CCP- cohort relative to healthy controls were not correlated to a family-level differentiation in bacterial taxa relative abundance.
Phage auxiliary metabolic gene abundances highlight cohort-associated disparities in metabolic potential.
To determine the functional potential and metabolic capabilities of intestinal phages, we quantified AMGs assigned to specific metabolic pathways in the Kyoto Encyclopedia of Genes and Genomes (KEGG) database across at-risk and healthy cohorts. Since their identification as viral drivers of host metabolism (Breitbart et al., 2007), phage-encoded AMGs have been recognized to redirect host functional capacities thereby directly influencing bacterial ecology (Thompson et al., 2011, Breitbart et al., 2018). We compared our set of phage contigs against a previously curated list of 2,835 AMGs with KEGG annotations (Kieft et al., 2020). Among our 660 phage contigs, 161 (24%) were found to encode at least 1 AMG, with 252 AMGs in total across all samples (Table S4). HC phages accounted for 131 metabolic signatures, while CCP+ and CCP- had less AMGs with 77 and 44, respectively (Figure S8A). Among the most represented metabolic categories across all phages, amino acid metabolism and the metabolism of cofactors and vitamins contained 121 and 88 AMGs, respectively, with energy metabolism being the next largest category with 22 AMG hits (Figure S8B). These general pathway results indicate that phages in the intestine presumably affect host metabolism through the consumption of metabolic resources needed for their own biogenesis, as described in phage-host infection studies of pathogens (Chevallereau et al., 2016, De Smet et al., 2016, Chatterjee et al., 2020) and marine virocells (Howard-Varona et al., 2020).
Hierarchical clustering grouped all AMGs into 5 distinct metabolic clusters relative to HC and at-risk CCP cohorts (Figure 6A). Among these groups, the gene coding for phnP (K06167), a phosphonate phosphodiesterase, stands apart from the others, both in terms of clustering and also for relative pathway abundance (Figure 6A). For group-associated differences in AMG abundances, there are notable absences among both CCP+ and CCP- groups, including several clustered transferases such as the mannose-phosphate transferases (algA, xanB, rfbA, wbpW, pslB), manno-heptose transferases (gmhC, hldE, waaE, rfaE), and the galE epimerase and glmS transaminase (Figure 6A). Considering the impact of such transferases on bacterial cell wall polysaccharides and biofilm formation (Valvano et al., 2002, Nakao et al., 2006), these results point to a baseline of phage-driven bacterial surface modifications from HC-derived phages. Conversely, AMGs involved in lipopolysaccharide (LPS) biosynthesis such as the waaL O-antigen ligase and the gmhB phosphatase are only present in CCP+ phages or at greater abundance in CCP+ phages, respectively, indicating a possible role in immune evasion. Within the CCP- cohort, one of the most abundant AMGs, KEGG ortholog entry K23144 encoding for a polyketide sugar transferase important in peptidoglycan biosynthesis is completely absent from the HC cohort and present at lower levels for CCP+ samples. Phage-encoded bacterial surface modifying enzymes such as the sugar transferases and LPS/peptidoglycan biosynthetic genes are differentially represented across the cohorts in this study, which has implications for bacterial fitness in the intestinal ecosystems and their interactions with the immune system.
Figure 6.

Phage auxiliary metabolic gene abundances highlight cohort-associated disparities in potential metabolic function. (A) Total counts per KEGG Pathway were used to normalize relative abundance of AMGs per sample, which were clustered using the ComplexHeatmap package in R. Areas in black indicate no AMG hits were present for the entire cohort for the 660 contig samples. See also Table S4. (B) Differentially abundant contig for the CCP+ to HC pairwise comparison, visualizing only the contigs which had CRISPR spacer-predicted hosts. Color-coded stars belong to a list of AMGs and indicate association with the contig they are adjacent to. (C) Differentially abundant contigs for the CCP- vs HC comparison.
We next incorporated the AMG characterization of genomes within our curated set of phages to those that were significantly over- or under-abundant in previous differential abundance analyses (Figures 5G, 5H, and 5I). Among the 20 differentially abundant contigs from the CCP+ vs HC pairwise comparison that contained CRISPR spacer-predicted hosts, 8 of these encoded at least one AMG (Figure 6B). The 9 under-abundant phages in this comparison encode 5 AMGs, including manno-heptose transferases (gmhC, hldE, waaE, rfaE), mannose-1-phostphate transferases (algA, xanB, rfbA, wbpW, pslB) and ahbD AdoMet-dependent heme synthase all together on 1 contig, and cysH and iscS genes on 2 other contigs (Figure 6B). Among the 11 significantly over-abundant contigs, 3 of these encode the phnP phosphodiesterase; 3 phages predicted to infect Flavonifractor sp. (Ruminococcaceae) and one predicted to infect Clostridiales bacteria. The remaining AMG found in CCP+-associated over-abundant phages encodes for a cobalamin biosynthesis protein cobS, which is considered a core component of marine phage genomes (Ignacio-Espinoza et al., 2012) and also ubiquitous in phage genomes that infect E. coli (Breitbart, 2012). Our identification of a CCP+ over-abundant phage contig targeting Bacteroides fragilis and carrying the cobS AMG (Figure 6B) reinforces the universal nature of this AMG that is conserved across hosts and environments (Kieft et al., 2020). We also identified 16 unique phage contigs with definitive CRISPR spacer-predicted hosts that were differentially abundant and associated with the CCP- cohort (Figure 6C). Within these contigs, 9 are significantly under-abundant compared to healthy controls, with 3 of these encoding AMGs. CCP- associated phages were identified as carrying cobS, DNMT3A, thiF, and iscS metabolic genes (Figure 6C).
DISCUSSION
RA is a complex disease with an unknown etiology that puts a burden on quality of life resulting in a strong societal impact (Markenson, 1991, Hunter et al., 2017). In addition to multiple epidemiological factors being associated with RA development, including genetic and familial risk, environmental factors and biological sex (Deane et al., 2017), the microbiota remains an important and understudied aspect that likely influences RA autoimmunity (Scher et al., 2011). Given the widespread occurrence and diversity of phages in the human intestinal microbiota and their impact on intestinal microbial ecology during health and disease (Duerkop, 2018, Minot et al., 2011, Mirzaei et al., 2017), we analyzed this neglected component of the microbiota as it relates to RA etiopathogenesis.
Using three separate database-independent approaches, we describe a collection of 660 phage contigs, their potential metabolic capability, and their differential abundance. Through a combination of CRISPR spacer matching and Markov clustering with viral metagenomic sequences from diverse environments, we predicted host assignments for 285 or 43.2% of these phages, which is a high level of taxonomic assignments relative to recent reports of approximately 10 – 30% host assignment identification (Moreno-Gallego et al., 2019, Bin Jang et al., 2019, Duerkop et al., 2018). By analyzing a core set of de novo assembled phage contigs paired with taxonomy, we identified differential phage communities associated with the at-risk RA individuals compared to healthy controls, while adding phage-host assignments to previously unidentified intestinal phages (Sutton et al., 2019, Roux et al., 2015).
Phage-host assignments were dominated by Lachnospiraceae-targeting phages, some of which were over-abundant in CCP+ individuals. This expansion of phages also correlated with increased abundances of Lachnospiraceae bacteria in the CCP+ cohort compared to either CCP- or the healthy cohort, suggesting a link to this family of Firmicutes and CCP autoantibody production in the human intestine. Increased abundance of Lachnospiraceae has been observed in two previous studies of intestinal microbiotas of mice following collagen-induced arthritis (Liu et al., 2016, Jubair et al., 2018). We report increased Lachnospiraceae phage-host interactions in CCP+ individuals at-risk for developing RA. Given that the FDR individuals included in this study do not show clinical signs of established RA, our identification of a preclinical cohort with increased Lachnospiraceae phage-host interactions could serve as a biological indicator of disease. The contribution of phages targeting Lachnospiraceae and thus influencing important bacterial metabolites warrants further investigation considering a potential link to self-antigen tolerance and autoimmune disease (Vacca et al., 2020). We also describe an expansion of Bacteroidaceae-targeting phages associated with the CCP+ and CCP- groups, yet no change in Bacteroidaceae bacteria (Figure S7A), which have been shown to be expanded following induced arthritis in mice (Liu et al., 2016). In addition to these phages serving as potential biomarkers of disease in humans at risk for RA, our data indicate that Bacteroidaceae and Lachnospiraceae-targeting phages designate a distinction between CCP serology status that may serve as an additional indicator of disease progression and/or future disease severity (Braschi et al., 2016). Notably, bacteria in the Lachnospiraceae and Ruminococcaceae families have been linked to the pre-diabetic intestinal microbiota and diabetic pathogenesis, while Bacteroidaceae are associated with disease protection in a murine model of diabetes (Krych et al., 2015). The identification of cohort-specific phage-host interactions sheds light on potential preclinical biomarkers connecting specific intestinal microbial communities to possible regulation of microbiota-mediated mucosal inflammation (Holers et al., 2018, Chriswell et al., 2019).
We observed over-abundant phages targeting Clostridium scindens, Flavonifractor sp., Actinomyces oris, among others, when comparing CCP+ to HC. A member of the Lachnospiraceae, C. scindens is an intestinal commensal bacterium involved in maintaining homeostatic large intestinal bile acid composition and providing host protection from opportunistic Clostridioides difficile blooms (Studer et al., 2016, Buffie et al., 2015). Phages targeting C. scindens in CCP+ at-risk individuals may influence bile acid dysmetabolism, which is linked to inflammatory bowel diseases (Duboc et al., 2013, Atarashi et al., 2013). Comparing CCP- to HC groups revealed several phages targeting Bacteroidaceae and Bacteroides species, bacteria involved in multiple reactions of bile acid metabolism promoting host metabolic health (Gerard, 2013, Yao et al., 2018). Recently, phage BV01 was shown to reduce Bacteroides bile acid metabolism (Campbell et al., 2020), further implicating phages in the mammalian intestinal metabolic cycle. Our findings suggest individuals at risk for RA harbor divergent communities of phages with potential to alter intestinal metabolism through either reduction of key bacterial species and thus reducing endogenous metabolic function, or through phage-derived introduction of specific AMGs.
Changes to the intestinal metabolome can lead to compositional microbiota transitions that impact host nutrient uptake and immune homeostasis (Lozupone et al., 2012). Considering that manipulations of microbial metabolic pathways in the intestine can influence inflammation and dysbiosis (Zhu et al., 2018), our identification of phage communities with differential abundances of AMGs points to divergent metabolic landscapes associated with at-risk RA cohorts. We were surprised to identify three phages that were over-abundant in the CCP+ cohort, two with Flavonifractor sp. predicted hosts and one Clostridiales-targeting phage, encoding phnP. The phosphonate phosphodiesterase accounts for 10% of the total AMGs represented in our phage genomes and is differentially abundant among the CCP+ cohort samples. The PhnP phosphodiesterase, part of a 14-gene operon originally described in Escherichia coli, plays a crucial intermediary role in the carbon-phosphorous lyase pathway by degrading a dead-end cyclic phosphonate byproduct (He et al., 2011). The presence of phnP across phages derived from at-risk and healthy cohorts (Figure 6A), suggests phage-driven organophosphonate degradation. Phosphonate degradation is important for phosphorus assimilation in enteric bacteria (Lee et al., 1992), although phosphonate metabolism has not been described for Flavonifractor species and a phnP homolog is not available for this genus in the KEGG database (K06167). In a recent study characterizing microbiota KEGG orthologs as predictors of methotrexate responsiveness for RA treatment, a gene in the phosphonate transport system, phnC (K02041), exhibited high median random forest importance as a predictor of drug response in new-onset RA subjects (Artacho et al., 2020). The contribution of the phosphonate metabolic pathway in bacteria and phages will require further exploration in the context of RA pathogenesis and treatment. It is possible that these phage-encoded metabolic products are supplementing phosphorous uptake among Ruminococcaceae and Lachnospiraceae bacteria that predominate in CCP+ individuals prior to RA clinical symptoms. Our analysis is limited in that we did not measure a longitudinal progression of microbial metabolic pathways in these human samples, yet these metabolic associations warrant further investigations into causality and the potentially cascading effects on interbacterial interactions (Hsu et al., 2019).
Our results point to divergent communities of phages with multiple bacterial host targets that group according to anti-CCP serology in individuals predisposed to developing RA. At-risk individuals endure a prolonged asymptomatic period before pathological early RA develops in those who are at a higher disease susceptibility in the preclinical RA state (Holers et al., 2018). Current approaches for RA diagnosis rely mostly on anti-CCP serology which has up to 93% specificity but as low as 67% sensitivity (for the CCP3.1 assay used here) (Demoruelle et al., 2013), indicating that a negative result does not preclude development of clinical RA. Phage community composition analyses may complement existing diagnoses for RA, considering that intestinal phages can play important roles in immune tolerance, mucosal immunity, and microbial homeostasis (Chatterjee et al., 2018). Given that phage community alterations have been shown to precede autoimmunity development in children at risk for developing type 1 diabetes (Zhao et al., 2017), phage community structure should be considered as a biomarker for diseases such as RA that are influenced by non-genetic microbial factors (Duerkop, 2018). To that end, we have characterized the intestinal viromes of RA at-risk individuals corresponding to anti-CCP serology. We measured species-specific phage-host interactions and identified over-abundances of C. scindens and A. oris targeting phages in CCP+ and CCP- individuals, respectively. Divergent metabolic profiles evident by differential abundance of AMG-encoding phages in both conditions warrant further interrogation during models of RA-like disease. Future work should investigate the potential of phages in a murine arthritis model to determine the influence of RA-associated phages with immunomodulation and inflammatory disease progression. This preclinical RA study implicates specific intestinal phages that could open new avenues to assess the basis for phage involvement in other microbiota-associated diseases.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Breck A. Duerkop (breck.duerkop@cuanschutz.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The VLP and whole metagenome DNA sequencing reads as well as the final curated phage contigs generated in this study are available at the European Nucleotide Archive under the Study titled “Intestinal VLP reads and predicted phage contigs for at-risk RA individuals” (accession numbers PRJEB42612 and ERP126498). The VLP and whole metagenome raw unmapped read sets are available for each of the 25 individual samples included in this study and are available under the Study Primary Accession PRJEB42612. The 660 curated contigs are compiled in a multifasta file deposited as Sample SAMEA7856466 under the same Study PRJEB42612.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Study Subjects and Fecal Samples
Fecal samples were obtained from individuals recruited for the SERA (Studies of the Etiology of Rheumatoid Arthritis) initiative, aimed at understanding the mechanisms that prelude the preclinical development of RA. SERA is a multicenter prospective cohort study that has identified first-degree relative (FDR) probands defined as a parent, full sibling, or offspring of individuals with diagnosed clinical RA (Kolfenbach et al., 2009). FDR probands were evaluated in extensive clinical research visits, longitudinal follow-ups, and autoantibody testing to determine CCP status (Kolfenbach et al., 2009). FDR probands were split into cohorts VLPs were further treated with 50 mg/mL proteinase K and 0.5% SDS for 30 min at 56°C before addition of 100 μL phage lysis buffer (4.5 M guanidiniumisothiocyanate, 44 mM sodium citrate pH 7.0, 0.88% sarkosyl, 0.72% 2-mercaptoethanol) (Shkoporov et al., 2018) and incubated for 10 min at 65°C. VLP DNA was precipitated and extracted with an equal volume of phenol/chloroform/isoamyl alcohol 25:24:1, spun at 7800g for 5 min, and further extracted with an equal volume of chloroform. VLP DNA was precipitated with 0.3M NaOAc (pH 5.2) and an equal volume of isopropanol, washed with ice-cold 70% ethanol, and resuspended in sterile water.
Metagenomic DNA Sequencing
Samples were physically separated into whole metagenome (M), including all genomic DNA present in the sample, and virus-like particle (VLP) fractions, which were subjected to phage-specific precipitation (Figure S1A). VLP sequencing reads were used for de novo contig assembly of VLP metagenomes. Illumina sequencing resulted in an average of 123.8 ± 32.2, 135.2 ± 40.4, and 104.7 ± 45.9 million (M) paired end reads per sample for CCP+, CCP- and HC whole metagenomes, respectively, and an average of 67.3 ± 29.5, 73.2 ± 33.7, and 89.6 ± 47.8 M paired reads per sample for CCP+, CCP- and HC VLP fractions, respectively (Figure S1B). VLP contigs longer than 5 kb were distributed evenly across the three sample groups, totaling 2908.6 ±1461.3, 3209.0 ± 2573.8, and 3535.7 ±2826.4 contigs per sample for CCP+, CCP- and HC respectively (Figure S1C). VLP and whole metagenomic DNA was sequenced using the Illumina NovaSEQ 6000 platform with paired-end 150-cycle sequencing chemistry. DNA libraries were generated using the Ovation Ultralow System v2 (Nugen, part no. 0334) library preparation kit including 12 cycles of amplification. TruSeq adapters (Illumina) were used for multiplexing. Libraries were quantified using a Qubit and quality was measured using a Tapestation. All library preparation, quantification, quality assessment and control, were performed by the University of Colorado Anschutz Medical Campus Genomics and Microarray Core.
16S rRNA Amplicon Sequencing and Analysis
16S rRNA gene analysis was performed using fecal samples that were processed for isolation of whole metagenomic DNA using a ZymoBIOMICS DNA kit (Zymo Research) and stored at 80 °C. Amplicons of the 16S rRNA gene V4 region were amplified using Earth Microbiome Project primers 515F and 806R (Caporaso et al., 2011) with custom barcodes. Samples were sequenced on the Illumina MiSeq platform with paired end 250 bp reads using bTEFAP technology (Dowd et al., 2008) by MR DNA (Molecular Research LP, Shallowater, TX), and processed using mothur v.1.44.0 (Schloss et al., 2009). Sequenced reads, which averaged 607,915 ± 112,641.7 per sample, were demultiplexed, assembled as contigs, and processed to remove chimeras and erroneous sequences per the Kozich protocol (Kozich et al., 2013) and mothur MiSeq SOP (https://mothur.org/wiki/miseq_sop/ accessed 07/16/2020). Sequences were aligned to the Greengenes core reference alignment for taxonomy using the 2013 release (gg_13_8_99) (DeSantis et al., 2006). Sequences were differentiated into amplicon sequence variants (ASVs) using the make.shared command, resulting in a total of 8,108,071 sequences. Subsampling was performed using 186,745 sequences, which corresponded to the smallest sample in our dataset. Diversity measurements were performed using mothur calculators to estimate community richness (Chao1 estimator), community evenness (Shannon evenness), and community diversity (inverse Simpson index).
Decontamination and Read Processing
Metagenomic reads were decontaminated and trimmed as previously described (Duerkop et al., 2018) using BBMap short read aligner v38.56 (Bushnell, 2019). Briefly, raw reads were mapped to the internal Illumina phage genome control phiX174 (J02482.1), human reference genome (hg38), and potential laboratory contaminants including mouse genome (mm10), Enterococcus faecalis V583 genome (NC_004668.1), E. faecalis OG1RF genome (NC_017316.1), and E. faecalis phage VPE25 (LT546030.1) using the bbsplit algorithm with default settings. Unmapped reads were trimmed of adapter sequences, with low quality reads and reads of insufficient length removed using the bbduk algorithm with the following parameters: ktrim = lr, k = 20, mink = 4, minlength = 20, qtrim = f, as previously described (Duerkop et al., 2018).
Metagenomic Assembly
Decontaminated and trimmed R1 and R2 reads were interleaved using the fq2fa --merge command from the IDBA-UD package (Peng et al., 2012). Whole metagenome and VLP assemblies were performed using the MEGAHIT assembler v1.2.7 (Li et al., 2016) using the default setting plus the following flags: --presets meta-large (--k-min 27 --k-max 127 --k-step 10) for large and complex metagenomes.
Quantitative Read Mapping and Construction of the Curated VLP Contig Database VLP reads were assembled into 25 individual sample sets, corresponding to the 25 individual fecal samples included in our study. All contigs resulting from MEGAHIT assembly were filtered to a minimum length of 5kb, resulting in a pool of 80,762 total contigs from all samples. Three separate independently published methods were employed to identify putative phages from the pooled set of contigs over 5kb in length. First, the P/M read mapping approach was used whereby each sample’s VLP and whole metagenome reads were mapped to their corresponding assembled contigs, using BBMap as previously described (Duerkop et al., 2018). After pooling, the top 100 largest ratios of VLP reads to whole metagenome reads for all 25 read-mapping sets for each sample were identified and pooled. Redundancy was removed using cd-hit-est at an identity threshold of 95% resulting in 2117 unique contig sequences. Next, we identified an independent set of phage contigs by aligning all open reading frames of the 80,762 VLP contigs against a set of 25,281 curated viral protein families (VPFs) (Paez-Espino et al., 2017). Separate filters were applied for VPF hits calculated in relation to total genes, microbial genes, and percent non-viral Pfams. 2,902 contigs were identified that contained 5 or more VPF hits and with non-viral Pfam hits below 20%. 263 contigs were identified with 5 or more VPF hits with less than 50% non-viral Pfam hits on a contig, and 644 contigs were identified with 2 – 4 VPF hits and 0 microbial gene hits. Finally, 976 contigs were identified with only 1 VPF hit per contig, and were included regardless of microbial gene content. In total, after dereplication, the viral contigs arising from all above filters resulted in 4,785 unique viral contigs using the VPF method. For the third and final approach we employed VIBRANT (Virus Identification By iteRative ANnoTation) v1.2.1, a sequence-independent algorithm that uses neural networks of viral protein signatures to identify lytic and lysogenic phages (Kieft et al., 2020). VIBRANT identified 4,758 unique phage contigs. After combining these three independent approaches used to identify unique sets of phages, all sets were combined and the overlapping 660 contigs were used for analysis as the curated contig set. To assess contig completion and contamination levels, CheckV v0.6.0 was used with standard operating parameters.
Differential Abundance Analyses
To calculate differential abundance in pairwise analyses, we first generated read mapping count matrices by mapping all VLP reads to the curated contig set of 660 contigs. The bbmap algorithm from the BBMap suite of tools was used with the following parameters: ambiguous = random, qtrim = lr, minid = 0.97. Total raw read counts were aggregated per contig and assembled into 25 count matrices for all samples, which were then used as input for DESeq2 v1.20.0 (Love et al., 2014) running in R version 3.6.3 for comprehensive differential abundance analysis. Raw un-normalized read count coverage values were used to compare fold changes across three pairwise comparisons: CCP+ vs. HC, CCP- vs. HC, and CCP+ vs. CCP- groups. The standard workflow for differential analysis within DESeq2 was used, producing logarithmic fold-change values incorporating Wald tests for p-value calculations and the Benjamini-Hochberg multiple testing correction for the adjusted p-value. In total, 178 phage contigs from our set of 660 were found to be differentially abundant using thresholds of log2 Fold Change < −1 or > 1 and adjusted p-value < 0.001 (Table S5).
VLP Clustering, Phage Host Matching, and AMG Identification
Clustering of all viral contigs within the RA virome described in this study was performed using two lists of contigs, the total 4,785 viral sequences identified by all filters of the VPF method, as well as the final curated set of 660 contigs. First, all 4,785 sequences were screened against the most recent iteration of the public viral database IMG/VR v3.0 (Roux et al., 2020) using blastn with 95% sequence similarity over 85% of each 1kb region of the contig, which resulted in 19,892 viral sequences. Then, a total of 24,926 sequences were screened against each other using blastn with the same parameters and omitting duplicate hits. Markov clustering of these 9.4 million connections resulted in a total of 1,193 clusters encompassing 22,306 total sequences. Overall, 2,420 of the 4,785 total RA virome sequences were clustered into 1,184 clusters. Of these clusters, 41 contained a reference viral isolate, 1,037 contained another metagenomic viral contig from IMG/VR, and 106 were identified as originating from RA metagenomic sequencing projects. Lastly, clustering was also calculated for the 660 curated viral sequences, which resulted in 266 individual clusters containing 336 (roughly 48% of curated set) unique sequences.
Phage host assignments were determined via bacterial CRISPR spacer matching as previously described (Duerkop et al., 2018), requiring at least 93% sequence identity match over the entire spacer length and allowing for up to 2 mismatches. CRISPR-Cas serves as a snapshot of previous phage infections in the form of acquired spacer sequences that represent invading viral genomes (Barrangou et al., 2007), and these sequences can be used for accurate identification of phage-host interactions in intestinal microbiomes (Stern et al., 2012, Duerkop et al., 2018). Of our 660 curated contig list, 207 (31.4%) had CRISPR spacers matching reference isolates therefore leading to host predictions for a third of our final contigs. VIBRANT v1.2.1 was used to identify auxiliary metabolic genes (AMGs) according to KEGG metabolic pathway annotations. VIBRANT annotates using VOG, Pfam, and KEGG databases; therefore, if the best HMM hit is to the KEGG database and also if the annotation is in a metabolic pathway, the hit gets called as an AMG. A majority of the AMGs identified in our analysis make up a group of 14 genes conserved across many environments (Kieft et al., 2020), indicating their functional importance in core metabolism.
GOLD Ecosystem Metadata Analysis
The JGI GOLD database contains metadata from over 100,000 biosamples and over 350,000 sequencing projects involving genomic and metagenomic sequencing data from biological isolates worldwide. Moreover, recent work has contributed an additional 52,515 metagenome-assembled genomes from diverse ecologies and geographic distributions (Nayfach et al., 2020b), further enhancing microbial host ecosystem analysis. For phages that were previously identified as having CRISPR spacer host assignments, total spacer alignments as identified by blastn ranging from 1 to 825 per contig, were tallied and used to calculate the uniformity of spacer origins per contig (Table S6). In total, 438 contigs were identified with CRISPR spacers matched to queries from the JGI GOLD database annotated with metadata at the following levels: GOLD Ecosystem, GOLD Ecosystem Category, GOLD Ecosystem Subtype, and GOLD Ecosystem Type. CRISPR spacer alignments to the GOLD database ranged from 18 to 116 bp, at 100% identity, with up to 1 mismatch and 0 gaps for all queries (Table S6). Percentages for each metadata level were calculated relative to the majority designations corresponding to each category; “Host-associated” (Ecosystem), “Human” (Ecosystem Category), “Large intestine” (Ecosystem Subtype), and “Digestive system” (Ecosystem Type). Data points per contig were plotted in R v3.6.3 using tidyverse v1.3.0 and ggplot2 v3.3.3. Statistical significance was calculated in R using the pairwise Wilcoxon test with the Benjamini-Hochberg adjustment method. An additional analysis was performed to calculate the percentages of metadata uniformity relative to the other 2 Ecosystem categories “Environmental” and “Engineered”, (Supplemental Figure 5) which were performed in similar conditions to the “Host-associated” analysis (Figure 4).
Data Visualizations
Various R packages were used, including DESEq2, ggplot2, ComplexHeatmap, pheatmap, corrplot, RColorBrewer, and EnhancedVolcano. Graphpad Prism v8.4.3 was used for all supplemental calculations. Lastly, SankeyMATIC (https://github.com/nowthis/sankeymatic) and meta-chart (https://www.meta-chart.com/venn) were used to create the Sankey and Venn diagrams depicted in Figure 1, respectively.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses for each experiment were performed as described in each figure legend and detailed in the Results and corresponding Method Details sections. Sample groups were quantified as follows: CCP+ (n=8), HC (n=9), CCP- (n=8), for a total of 25 individual stool samples. Statistical significance was calculated with either Prism version 8.4.3 or R version 3.6.3
Supplementary Material
Table S7. Viral morphotypes identified for all 660 phages. Related to Figure 1.
Table S6. CRISPR spacer metadata matching to JGI GOLD database. Related to GOLD Ecosystem Metadata Analysis in the Star Methods.
Table S5. Differentially abundant contigs from three pairwise comparisions: CCP+ vs HC (tab 1), CCP- vs HC (tab 2), CCP+ vs CCP- (tab 3). Related to Differential Abundance Analysis section in Star Methods.
Table S4. Auxiliary metabolic genes (AMGs) identified among the 660 phage contigs. Related to Figure 6 and Figure S8.
Table S3. Total CRISPR spacer hits and clustering identification of host bacterial families. Related to Figure 2.
Table S2. CRISPR spacer matching for curated contigs. Related to Figure 2.
Table S1. Curated list of final 660 contigs, including clustering information, and CRISPR host assignments. Related to Figure 2 and Figure 3.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological Samples | ||
| 25 individual fecal samples | SERA study | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| SM+ buffer (50mM Tris, 5mM NaCl, 8mM MgSO4, 5mM CaCl2, pH = 7, prepare beforehand and filter sterilize) | Duerkop et al., 2016 | N/A |
| DNase buffer (10 mM CaCl2, 50 mM MgCl2) | Shkoporov et al., 2018 | N/A |
| DNase I | Roche | 11284932001 |
| RNase | Roche | 10109134001 |
| proteinase K | Sigma-Aldrich | 1.24568 |
| phage lysis buffer (4.5 M guanidiniumisothiocyanate, 44 mM sodium citrate pH 7.0, 0.88% sarkosyl, 0.72% 2-mercaptoethanol) | Shkoporov et al., 2018 | N/A |
| phenol/chloroform/isoamyl alcohol 25:24:1 | Sigma-Aldrich | P3803 |
| PEG8000 | Fisher BioReagents | BP233 |
| Critical Commercial Assays | ||
| Zymo BashingBead Lysis tube | ZymoResearch | S6012–50 |
| ZymoBIOMICS DNA kit | ZymoResearch | D4303 |
| Ovation Ultralow System v2 | Nugen | 0334 |
| TruSeq Nano DNA Library Prep Kit | Illumina | 20015965 |
| Deposited Data | ||
| Raw virome sequencing data | This paper | PRJEB42612 |
| Raw metagenome sequencing data | This paper | PRJEB42612 |
| 660 curated phage contigs | This paper | PRJEB42612 |
| Oligonucleotides | ||
| Earth Microbiome Project primers 515F and 806R | Caporaso et al., 2011 | N/A |
| Software and Algorithms | ||
| GraphPad Prism v8.4.3 | GraphPad Software | N/A |
| RStudio version 1.2.5001 | RStudio, Inc. | N/A |
| R version 3.6.3 | R Foundation for Statistical Computing | N/A |
| MegaHit assembler v1.2.7 | Li et al., 2016; https://github.com/voutcn/megahit | RRID:SCR_018551 |
| IDBA-UD | Peng et al., 2012; http://i.cs.hku.hk/~alse/hkubrg/projects/idba_ud/ | RRID:SCR_011912 |
| SankeyMATIC | https://github.com/nowthis/sankeymatic | N/A |
| meta-chart | https://www.meta-chart.com/venn | N/A |
| BBtools (BBmap) v38.56 | Bushnell, 2019; http://sourceforge.net/projects/bbmap | RRID:SCR_016965 |
| VIBRANT v1.2.1 | Kieft et al., 2020 | N/A |
| IMG/VR v3.0 | Roux et al., 2020 | N/A |
| tidyverse v1.3.0 | https://cran.r-project.org/package=tidyverse | RRID:SCR_019186 |
| DESeq2 v1.24.0 | Love et al., 2014; https://bioconductor.org/packages/release/bioc/html/DESeq2.html | RRID:SCR_015687 |
| ggplot2 v3.3.3 | https://cran.r-project.org/web/packages/ggplot2/index.html | RRID:SCR_014601 |
| ComplexHeatmap | https://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html | RRID:SCR_017270 |
| Pheatmap v1.0.12 | https://www.rdocumentation.org/packages/pheatmap/versions/0.2/topics/pheatmap | RRID:SCR_016418 |
| EnhancedVolcano v1.7.16 | https://bioconductor.org/packages/EnhancedVolcano/ | RRID:SCR_018931 |
| corrplot v0.84 | https://github.com/taiyun/corrplot | N/A |
| matrixStats v0.57.0 | https://github.com/HenrikBengtsson/matrixStats | N/A |
| Mothur v1.44.0 | Schloss et al., 2009; http://www.mothur.org/ | RRID:SCR_011947 |
HIGHLIGHTS.
Unique intestinal phage compositions correlate to at-risk RA anti-CCP serology
Lachnospiraceae phage-host interactions dominate in CCP+ individuals at-risk for RA
Phages from CCP+ individuals may originate from disparate ecological niches
Phage auxiliary metabolic genes (AMGs) contribute to cohort-associated differences
ACKNOWLEDGEMENTS
The authors thank all participants in the SERA studies at the University of Colorado and elsewhere. We thank Rodolphe Barrangou for comments and suggestions to improve this manuscript. We thank Monica Ransom and Katrina Diener from the University of Colorado Anschutz Medical Campus Genomics and Microarray Core for performing Illumina library preparation and sequencing. The following funding sources supported this work: NIH U01AI101981 (V.M.H., K.A.K.), NIH U01AI130830 (B.A.D.), startup funds from the University of Colorado School of Medicine (B.A.D.), NIH Research Training in Rheumatology T32AR007534 (M.R.M.), Rheumatology Research Foundation Future Scientist Award (M.E.C.).
Footnotes
DECLARATION OF INTERESTS
D.P.E is a co-founder/employee of Ancilia Biosciences. A.S. is the founder/employee of Ancilia Biosciences. B.A.D. is a co-founder and shareholder of Ancilia Biosciences.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Alpizar-Rodriguez D, Lesker TR, Gronow A, Gilbert B, Raemy E, Lamacchia C, Gabay C, Finckh A. & Strowig T. (2019). Prevotella copri in individuals at risk for rheumatoid arthritis. Ann Rheum Dis 78, 590–593. [DOI] [PubMed] [Google Scholar]
- Artacho A, Isaac S, Nayak R, Flor-Duro A, Alexander M, Koo I, Manasson J, Smith PB, Rosenthal P, Homsi Y. et al. (2020). The pre-treatment gut microbiome is associated with lack of response to methotrexate in new onset rheumatoid arthritis. Arthritis Rheumatol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, Fukuda S, Saito T, Narushima S, Hase K. et al. (2013). Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 500, 232–236. [DOI] [PubMed] [Google Scholar]
- Ayyappan P, Harms RZ, Seifert JA, Bemis EA, Feser ML, Deane KD, Demoruelle MK, Mikuls TR, Holers VM & Sarvetnick NE (2020). Heightened levels of antimicrobial response factors in patients with rheumatoid arthritis. Front Immunol 11, 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr JJ, Auro R, Furlan M, Whiteson KL, Erb ML, Pogliano J, Stotland A, Wolkowicz R, Cutting AS, Doran KS et al. (2013). Bacteriophage adhering to mucus provide a non-host-derived immunity. Proc Natl Acad Sci U S A 110, 10771–10776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA & Horvath P. (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712. [DOI] [PubMed] [Google Scholar]
- Bin Jang H, Bolduc B, Zablocki O, Kuhn JH, Roux S, Adriaenssens EM, Brister JR, Kropinski AM, Krupovic M, Lavigne R. et al. (2019). Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat Biotechnol 37, 632–639. [DOI] [PubMed] [Google Scholar]
- Braschi E, Shojania K. & Allan GM (2016). Anti-CCP: a truly helpful rheumatoid arthritis test? Can Fam Physician 62, 234. [PMC free article] [PubMed] [Google Scholar]
- Breitbart M. (2012). Marine viruses: truth or dare. Ann Rev Mar Sci 4, 425–448. [DOI] [PubMed] [Google Scholar]
- Breitbart M, Bonnain C, Malki K. & Sawaya NA (2018). Phage puppet masters of the marine microbial realm. Nat Microbiol 3, 754–766. [DOI] [PubMed] [Google Scholar]
- Breitbart M, Thompson LR, Suttle CA & Sullivan MB (2007). Exploring the vast diversity of marine viruses. Oceanography 20, 135–139. [Google Scholar]
- Brusca SB, Abramson SB & Scher JU (2014). Microbiome and mucosal inflammation as extra-articular triggers for rheumatoid arthritis and autoimmunity. Curr Opin Rheumatol 26, 101–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffie CG, Bucci V, Stein RR, Mckenney PT, Ling L, Gobourne A, No D, Liu H, Kinnebrew M, Viale A. et al. (2015). Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 517, 205–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushnell B. 2019. BBMap short read aligner, and other bioinformatic tools. 38.56 ed. https://sourceforge.net/projects/bbmap/. [Google Scholar]
- Campbell DE, Ly LK, Ridlon JM, Hsiao A, Whitaker RJ & Degnan PH (2020). Infection with Bacteroides phage BV01 alters the host transcriptome and bile acid metabolism in a common human gut microbe. Cell Rep 32, 108142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N. & Knight R. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A 108 Suppl 1, 4516–4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HH, Liu GY, Dwivedi N, Sun B, Okamoto Y, Kinslow JD, Deane KD, Demoruelle MK, Norris JM, Thompson PR et al. (2016). A molecular signature of preclinical rheumatoid arthritis triggered by dysregulated PTPN22. JCI Insight 1, e90045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee A. & Duerkop BA (2018). Beyond bacteria: bacteriophage-eukaryotic host interactions reveal emerging paradigms of health and disease. Front Microbiol 9, 1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee A, Willett JLE, Nguyen UT, Monogue B, Palmer KL, Dunny GM & Duerkop BA (2020). Parallel Genomics Uncover Novel Enterococcal-Bacteriophage Interactions. mBio 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevallereau A, Blasdel BG, De Smet J, Monot M, Zimmermann M, Kogadeeva M, Sauer U, Jorth P, Whiteley M, Debarbieux L. et al. (2016). Next-generation “-omics” approaches reveal a massive alteration of host RNA metabolism during bacteriophage infection of Pseudomonas aeruginosa. PLoS Genet 12, e1006134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chriswell ME & Kuhn KA (2019). Microbiota-mediated mucosal inflammation in arthritis. Best Pract Res Clin Rheumatol 33, 101492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clooney AG, Sutton TDS, Shkoporov AN, Holohan RK, Daly KM, O’regan O, Ryan FJ, Draper LA, Plevy SE, Ross RP et al. (2019). Whole-virome analysis sheds light on viral dark matter in inflammatory bowel disease. Cell Host Microbe 26, 764–778 e765. [DOI] [PubMed] [Google Scholar]
- De Smet J, Zimmermann M, Kogadeeva M, Ceyssens PJ, Vermaelen W, Blasdel B, Bin Jang H, Sauer U. & Lavigne R. (2016). High coverage metabolomics analysis reveals phage-specific alterations to Pseudomonas aeruginosa physiology during infection. ISME J 10, 1823–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane KD, Demoruelle MK, Kelmenson LB, Kuhn KA, Norris JM & Holers VM (2017). Genetic and environmental risk factors for rheumatoid arthritis. Best Pract Res Clin Rheumatol 31, 3–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demoruelle MK (2019). Mucosa biology and the development of rheumatoid arthritis: potential for prevention by targeting mucosal processes. Clin Ther 41, 1270–1278. [DOI] [PubMed] [Google Scholar]
- Demoruelle MK, Bowers E, Lahey LJ, Sokolove J, Purmalek M, Seto NL, Weisman MH, Norris JM, Kaplan MJ, Holers VM et al. (2018). Antibody responses to citrullinated and noncitrullinated antigens in the sputum of subjects with rheumatoid arthritis and subjects at risk for development of rheumatoid arthritis. Arthritis Rheumatol 70, 516–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demoruelle MK, Harrall KK, Ho L, Purmalek MM, Seto NL, Rothfuss HM, Weisman MH, Solomon JJ, Fischer A, Okamoto Y. et al. (2017). Anti-citrullinated protein antibodies are associated with neutrophil extracellular traps in the sputum in relatives of rheumatoid arthritis patients. Arthritis Rheumatol 69, 1165–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demoruelle MK, Parish MC, Derber LA, Kolfenbach JR, Hughes-Austin JM, Weisman MH, Gilliland W, Edison JD, Buckner JH, Mikuls TR et al. (2013). Performance of anti-cyclic citrullinated peptide assays differs in subjects at increased risk of rheumatoid arthritis and subjects with established disease. Arthritis Rheum 65, 2243–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P. & Andersen GL (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72, 5069–5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowd SE, Sun Y, Wolcott RD, Domingo A. & Carroll JA (2008). Bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP) for microbiome studies: bacterial diversity in the ileum of newly weaned Salmonella-infected pigs. Foodborne Pathog Dis 5, 459–472. [DOI] [PubMed] [Google Scholar]
- Drago L. (2019). Prevotella copri and microbiota in rheumatoid arthritis: fully convincing evidence? J Clin Med 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duboc H, Rajca S, Rainteau D, Benarous D, Maubert MA, Quervain E, Thomas G, Barbu V, Humbert L, Despras G. et al. (2013). Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut 62, 531–539. [DOI] [PubMed] [Google Scholar]
- Duerkop BA (2018). Bacteriophages shift the focus of the mammalian microbiota. PLoS Pathog 14, e1007310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duerkop BA, Huo W, Bhardwaj P, Palmer KL & Hooper LV (2016). Molecular basis for lytic bacteriophage resistance in Enterococci. mBio 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duerkop BA, Kleiner M, Paez-Espino D, Zhu W, Bushnell B, Hassell B, Winter SE, Kyrpides NC & Hooper LV (2018). Murine colitis reveals a disease-associated bacteriophage community. Nat Microbiol 3, 1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerard P. (2013). Metabolism of cholesterol and bile acids by the gut microbiota. Pathogens 3, 14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gevers D, Kugathasan S, Denson LA, Vazquez-Baeza Y, Van Treuren W, Ren B, Schwager E, Knights D, Song SJ, Yassour M. et al. (2014). The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 15, 382–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski A, Dabrowska K, Miedzybrodzki R, Weber-Dabrowska B, Lusiak-Szelachowska M, Jonczyk-Matysiak E. & Borysowski J. (2017). Phages and immunomodulation. Future Microbiol 12, 905–914. [DOI] [PubMed] [Google Scholar]
- He SM, Wathier M, Podzelinska K, Wong M, Mcsorley FR, Asfaw A, Hove-Jensen B, Jia Z. & Zechel DL (2011). Structure and mechanism of PhnP, a phosphodiesterase of the carbon-phosphorus lyase pathway. Biochemistry 50, 8603–8615. [DOI] [PubMed] [Google Scholar]
- Holers VM, Demoruelle MK, Kuhn KA, Buckner JH, Robinson WH, Okamoto Y, Norris JM & Deane KD (2018). Rheumatoid arthritis and the mucosal origins hypothesis: protection turns to destruction. Nat Rev Rheumatol 14, 542–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard-Varona C, Lindback MM, Bastien GE, Solonenko N, Zayed AA, Jang H, Andreopoulos B, Brewer HM, Glavina Del Rio T, Adkins JN et al. (2020). Phage-specific metabolic reprogramming of virocells. ISME J 14, 881–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu BB, Gibson TE, Yeliseyev V, Liu Q, Lyon L, Bry L, Silver PA & Gerber GK (2019). Dynamic modulation of the gut microbiota and metabolome by bacteriophages in a mouse model. Cell Host Microbe 25, 803–814 e805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes-Austin JM, Deane KD, Derber LA, Kolfenbach JR, Zerbe GO, Sokolove J, Lahey LJ, Weisman MH, Buckner JH, Mikuls TR et al. (2013). Multiple cytokines and chemokines are associated with rheumatoid arthritis-related autoimmunity in first-degree relatives without rheumatoid arthritis: Studies of the Aetiology of Rheumatoid Arthritis (SERA). Ann Rheum Dis 72, 901–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter TM, Boytsov NN, Zhang X, Schroeder K, Michaud K. & Araujo AB (2017). Prevalence of rheumatoid arthritis in the United States adult population in healthcare claims databases, 2004–2014. Rheumatol Int 37, 1551–1557. [DOI] [PubMed] [Google Scholar]
- Ignacio-Espinoza JC & Sullivan MB (2012). Phylogenomics of T4 cyanophages: lateral gene transfer in the ‘core’ and origins of host genes. Environ Microbiol 14, 2113–2126. [DOI] [PubMed] [Google Scholar]
- Jubair WK, Hendrickson JD, Severs EL, Schulz HM, Adhikari S, Ir D, Pagan JD, Anthony RM, Robertson CE, Frank DN et al. (2018). Modulation of inflammatory arthritis in mice by gut microbiota through mucosal inflammation andautoantibody generation. Arthritis Rheumatol 70, 1220–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang DW, Adams JB, Gregory AC, Borody T, Chittick L, Fasano A, Khoruts A, Geis E, Maldonado J, Mcdonough-Means S. et al. (2017). Microbiota Transfer Therapy alters gut ecosystem and improves gastrointestinal and autism symptoms: an open-label study. Microbiome 5, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan Mirzaei M., Khan Ma.A., Ghosh P, Taranu ZE, Taguer M, Ru J, Chowdhury R, Kabir MM, Deng L, Mondal D. et al. (2020). Bacteriophages isolated from stunted children can regulate gut bacterial communities in an age-specific manner. Cell Host Microbe 27, 199–212 e195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieft K, Zhou Z. & Anantharaman K. (2020). VIBRANT: automated recovery, annotation and curation of microbial viruses, and evaluation of viral community function from genomic sequences. Microbiome 8, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiner M, Hooper LV & Duerkop BA (2015). Evaluation of methods to purify virus-like particles for metagenomic sequencing of intestinal viromes. BMC Genomics 16, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolfenbach JR, Deane KD, Derber LA, O’donnell C, Weisman MH, Buckner JH, Gersuk VH, Wei S, Mikuls TR, O’dell J. et al. (2009). A prospective approach to investigating the natural history of preclinical rheumatoid arthritis (RA) using first-degree relatives of probands with RA. Arthritis Rheum 61, 1735–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozich JJ, Westcott SL, Baxter NT, Highlander SK & Schloss PD (2013). Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol 79, 5112–5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krych L, Nielsen DS, Hansen AK & Hansen CH (2015). Gut microbial markers are associated with diabetes onset, regulatory imbalance, and IFN-gamma level in NOD mice. Gut Microbes 6, 101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Mannaa M, Kim Y, Kim J, Kim GT & Seo YS (2019). Comparative analysis of fecal microbiota composition between rheumatoid arthritis and osteoarthritis patients. Genes (Basel) 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KS, Metcalf WW & Wanner BL (1992). Evidence for two phosphonate degradative pathways in Enterobacter aerogenes. J Bacteriol 174, 2501–2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Luo R, Liu CM, Leung CM, Ting HF, Sadakane K, Yamashita H. & Lam TW (2016). MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 102, 3–11. [DOI] [PubMed] [Google Scholar]
- Liang G, Zhao C, Zhang H, Mattei L, Sherrill-Mix S, Bittinger K, Kessler LR, Wu GD, Baldassano RN, Derusso P. et al. (2020). The stepwise assembly of the neonatal virome is modulated by breastfeeding. Nature 581, 470–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Zeng B, Zhang J, Li W, Mou F, Wang H, Zou Q, Zhong B, Wu L, Wei H. et al. (2016). Role of the gut microbiome in modulating arthritis progression in mice. Sci Rep 6, 30594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W. & Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK & Knight R. (2012). Diversity, stability and resilience of the human gut microbiota. Nature 489, 220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macgregor AJ, Snieder H, Rigby AS, Koskenvuo M, Kaprio J, Aho K. & Silman AJ (2000). Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheum 43, 30–37. [DOI] [PubMed] [Google Scholar]
- Maeda Y, Kurakawa T, Umemoto E, Motooka D, Ito Y, Gotoh K, Hirota K, Matsushita M, Furuta Y, Narazaki M. et al. (2016). Dysbiosis contributes to arthritis development via activation of autoreactive T cells in the intestine. Arthritis Rheumatol 68, 2646–2661. [DOI] [PubMed] [Google Scholar]
- Manrique P, Bolduc B, Walk ST, Van Der Oost J, De Vos WM & Young MJ (2016). Healthy human gut phageome. Proc Natl Acad Sci U S A 113, 10400–10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markenson JA (1991). Worldwide trends in the socioeconomic impact and long-term prognosis of rheumatoid arthritis. Semin Arthritis Rheum 21, 4–12. [DOI] [PubMed] [Google Scholar]
- Minot S, Sinha R, Chen J, Li H, Keilbaugh SA, Wu GD, Lewis JD & Bushman FD (2011). The human gut virome: inter-individual variation and dynamic response to diet. Genome Res 21, 1616–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minot SS & Willis AD (2019). Clustering co-abundant genes identifies components of the gut microbiome that are reproducibly associated with colorectal cancer and inflammatory bowel disease. Microbiome 7, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzaei MK & Maurice CF (2017). Menage a trois in the human gut: interactions between host, bacteria and phages. Nat Rev Microbiol 15, 397–408. [DOI] [PubMed] [Google Scholar]
- Moreno-Gallego JL, Chou SP, Di Rienzi SC, Goodrich JK, Spector TD, Bell JT, Youngblut ND, Hewson I, Reyes A. & Ley RE (2019). Virome diversity correlates with intestinal microbiome diversity in adult monozygotic twins. Cell Host Microbe 25, 261–272 e265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, Stamatis D, Bertsch J, Ovchinnikova G, Katta HY, Mojica A, Chen IA, Kyrpides NC & Reddy T. (2019). Genomes OnLine database (GOLD) v.7: updates and new features. Nucleic Acids Res 47, D649–D659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakao R, Senpuku H. & Watanabe H. (2006). Porphyromonas gingivalis galE is involved in lipopolysaccharide O-antigen synthesis and biofilm formation. Infect Immun 74, 6145–6153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayfach S, Camargo AP, Eloe-Fadrosh E, Roux S. & Kyrpides N. (2020a). CheckV: assessing the quality of metagenome-assmebled viral genomes. bioRxiv preprint. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayfach S, Roux S, Seshadri R, Udwary D, Varghese N, Schulz F, Wu D, Paez-Espino D, Chen IM, Huntemann M. et al. (2020b). A genomic catalog of Earth’s microbiomes. Nat Biotechnol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman JM, Handley SA, Baldridge MT, Droit L, Liu CY, Keller BC, Kambal A, Monaco CL, Zhao G, Fleshner P. et al. (2015). Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 160, 447–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paez-Espino D, Eloe-Fadrosh EA, Pavlopoulos GA, Thomas AD, Huntemann M, Mikhailova N, Rubin E, Ivanova NN & Kyrpides NC (2016). Uncovering Earth’s virome. Nature 536, 425–430. [DOI] [PubMed] [Google Scholar]
- Paez-Espino D, Pavlopoulos GA, Ivanova NN & Kyrpides NC (2017). Nontargeted virus sequence discovery pipeline and virus clustering for metagenomic data. Nat Protoc 12, 1673–1682. [DOI] [PubMed] [Google Scholar]
- Peng Y, Leung HC, Yiu SM & Chin FY (2012). IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28, 1420–1428. [DOI] [PubMed] [Google Scholar]
- Pianta A, Arvikar S, Strle K, Drouin EE, Wang Q, Costello CE & Steere AC (2017). Evidence of the immune relevance of Prevotella copri, a gut microbe, in patients with rheumatoid arthritis. Arthritis Rheumatol 69, 964–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quistad SD, Grasis JA, Barr JJ & Rohwer FL (2017). Viruses and the origin of microbiome selection and immunity. ISME J 11, 835–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Valera F, Martin-Cuadrado AB, Rodriguez-Brito B, Pasic L, Thingstad TF, Rohwer F. & Mira A. (2009). Explaining microbial population genomics through phage predation. Nat Rev Microbiol 7, 828–836. [DOI] [PubMed] [Google Scholar]
- Roux S, Adriaenssens EM, Dutilh BE, Koonin EV, Kropinski AM, Krupovic M, Kuhn JH, Lavigne R, Brister JR, Varsani A. et al. (2019). Minimum Information about an Uncultivated Virus Genome (MIUViG). Nat Biotechnol 37, 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux S, Emerson JB, Eloe-Fadrosh EA & Sullivan MB (2017). Benchmarking viromics: an in silico evaluation of metagenome-enabled estimates of viral community composition and diversity. PeerJ 5, e3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux S, Hallam SJ, Woyke T. & Sullivan MB (2015). Viral dark matter and virus-host interactions resolved from publicly available microbial genomes. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux S, Paez-Espino D, Chen IA, Palaniappan K, Ratner A, Chu K, Reddy TBK, Nayfach S, Schulz F, Call L. et al. (2020). IMG/VR v3: an integrated ecological and evolutionary framework for interrogating genomes of uncultivated viruses. Nucleic Acids Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scher JU & Abramson SB (2011). The microbiome and rheumatoid arthritis. Nat Rev Rheumatol 7, 569–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scher JU, Sczesnak A, Longman RS, Segata N, Ubeda C, Bielski C, Rostron T, Cerundolo V, Pamer EG, Abramson SB et al. (2013). Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife 2, e01202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75, 7537–7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shkoporov AN & Hill C. (2019). Bacteriophages of the human gut: the “known unknown” of the microbiome. Cell Host Microbe 25, 195–209. [DOI] [PubMed] [Google Scholar]
- Shkoporov AN, Ryan FJ, Draper LA, Forde A, Stockdale SR, Daly KM, Mcdonnell SA, Nolan JA, Sutton TDS, Dalmasso M. et al. (2018). Reproducible protocols for metagenomic analysis of human faecal phageomes. Microbiome 6, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern A, Mick E, Tirosh I, Sagy O. & Sorek R. (2012). CRISPR targeting reveals a reservoir of common phages associated with the human gut microbiome. Genome Res 22, 1985–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studer N, Desharnais L, Beutler M, Brugiroux S, Terrazos MA, Menin L, Schurch CM, Mccoy KD, Kuehne SA, Minton NP et al. (2016). Functional intestinal bile acid 7alpha-dehydroxylation by Clostridium scindens associated with protection from Clostridium difficile infection in a gnotobiotic mouse model. Front Cell Infect Microbiol 6, 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton TDS & Hill C. (2019). Gut bacteriophage: current understanding and challenges. Front Endocrinol (Lausanne) 10, 784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson LR, Zeng Q, Kelly L, Huang KH, Singer AU, Stubbe J. & Chisholm SW (2011). Phage auxiliary metabolic genes and the redirection of cyanobacterial host carbon metabolism. Proc Natl Acad Sci U S A 108, E757–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toivanen P, Vartiainen S, Jalava J, Luukkainen R, Mottonen T, Eerola E. & Manninen R. (2002). Intestinal anaerobic bacteria in early rheumatoid arthritis (RA). Arthritis Res Ther 4. [Google Scholar]
- Vaahtovuo J, Munukka E, Korkeamaki M, Luukkainen R. & Toivanen P. (2008). Fecal microbiota in early rheumatoid arthritis. J Rheumatol 35, 1500–1505. [PubMed] [Google Scholar]
- Vacca M, Celano G, Calabrese FM, Portincasa P, Gobbetti M. & De Angelis M. (2020). The Controversial Role of Human Gut Lachnospiraceae. Microorganisms 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valvano MA, Messner P. & Kosma P. (2002). Novel pathways for biosynthesis of nucleotide-activated glycero-manno-heptose precursors of bacterial glycoproteins and cell surface polysaccharides. Microbiology (Reading) 148, 1979–1989. [DOI] [PubMed] [Google Scholar]
- Van Belleghem JD, Dabrowska K, Vaneechoutte M, Barr JJ & Bollyky PL (2018). Interactions between bacteriophage, bacteria, and the mammalian immune system. Viruses 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis VC, Demoruelle MK, Derber LA, Chartier-Logan CJ, Parish MC, Pedraza IF, Weisman MH, Norris JM, Holers VM & Deane KD (2013). Sputum autoantibodies in patients with established rheumatoid arthritis and subjects at risk of future clinically apparent disease. Arthritis Rheum 65, 2545–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao L, Seaton SC, Ndousse-Fetter S, Adhikari AA, Dibenedetto N, Mina AI, Banks AS, Bry L. & Devlin AS (2018). A selective gut bacterial bile salt hydrolase alters host metabolism. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu AI, Zhao L, Eaton KA, Ho S, Chen J, Poe S, Becker J, Gonzalez A, Mckinstry D, Hasso M. et al. (2020). Gut microbiota modulate CD8 T cell responses to influence colitis-associated tumorigenesis. Cell Rep 31, 107471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhang D, Jia H, Feng Q, Wang D, Liang D, Wu X, Li J, Tang L, Li Y. et al. (2015). The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat Med 21, 895–905. [DOI] [PubMed] [Google Scholar]
- Zhao G, Vatanen T, Droit L, Park A, Kostic AD, Poon TW, Vlamakis H, Siljander H, Harkonen T, Hamalainen AM et al. (2017). Intestinal virome changes precede autoimmunity in type I diabetes-susceptible children. Proc Natl Acad Sci U S A 114, E6166–E6175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Winter MG, Byndloss MX, Spiga L, Duerkop BA, Hughes ER, Buttner L, De Lima Romao E, Behrendt CL, Lopez CA et al. (2018). Precision editing of the gut microbiota ameliorates colitis. Nature 553, 208–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S7. Viral morphotypes identified for all 660 phages. Related to Figure 1.
Table S6. CRISPR spacer metadata matching to JGI GOLD database. Related to GOLD Ecosystem Metadata Analysis in the Star Methods.
Table S5. Differentially abundant contigs from three pairwise comparisions: CCP+ vs HC (tab 1), CCP- vs HC (tab 2), CCP+ vs CCP- (tab 3). Related to Differential Abundance Analysis section in Star Methods.
Table S4. Auxiliary metabolic genes (AMGs) identified among the 660 phage contigs. Related to Figure 6 and Figure S8.
Table S3. Total CRISPR spacer hits and clustering identification of host bacterial families. Related to Figure 2.
Table S2. CRISPR spacer matching for curated contigs. Related to Figure 2.
Table S1. Curated list of final 660 contigs, including clustering information, and CRISPR host assignments. Related to Figure 2 and Figure 3.
Data Availability Statement
The VLP and whole metagenome DNA sequencing reads as well as the final curated phage contigs generated in this study are available at the European Nucleotide Archive under the Study titled “Intestinal VLP reads and predicted phage contigs for at-risk RA individuals” (accession numbers PRJEB42612 and ERP126498). The VLP and whole metagenome raw unmapped read sets are available for each of the 25 individual samples included in this study and are available under the Study Primary Accession PRJEB42612. The 660 curated contigs are compiled in a multifasta file deposited as Sample SAMEA7856466 under the same Study PRJEB42612.