

Highlights
-
•
Cognitively-defined subgroups among individuals with AD dementia identified.
-
•
Subgroup gray matter volume patterns were distinct, matching cognitive profiles.
-
•
Gene-set enrichment analyses revealed gray matter vs gene expression associations.
-
•
These associations were partly shared between subgroups, and partly unique.
-
•
Distinct biological drivers may be involved in heterogeneity of Alzheimer’s disease.
Abbreviations: AD, Alzheimer’s disease; ACT, Adult changes in thought cohort; ADC, Amsterdam dementia cohort; APOE, Apolipoprotein E; CSF, Cerebrospinal fluid; DARTEL, Diffeomorphic anatomical registration through exponentiated lie algebra; DSC, Sørensen–Dice coefficient; FDR, False-discovery rate; PCA, Posterior cortical atrophy; lvPPA, Logopenic variant primary progressive aphasia; GSEA, gene set enrichment analysis; MMSE, Mini-mental state examination; SPM, Statistical parametric mapping; SNP, single-nucleotide polymorphism; SD, Standard deviation
Keywords: Alzheimer’s disease, Gray matter volumes, Gene expression, Heterogeneity, Psychometrics
Abstract
The clinical presentation of Alzheimer’s disease (AD) varies widely across individuals but the neurobiological mechanisms underlying this heterogeneity are largely unknown. Here, we compared regional gray matter (GM) volumes and associated gene expression profiles between cognitively-defined subgroups of amyloid-β positive individuals clinically diagnosed with AD dementia (age: 66 ± 7, 47% male, MMSE: 21 ± 5). All participants underwent neuropsychological assessment with tests covering memory, executive-functioning, language and visuospatial-functioning domains. Subgroup classification was achieved using a psychometric framework that assesses which cognitive domain shows substantial relative impairment compared to the intra-individual average across domains, which yielded the following subgroups in our sample; AD-Memory (n = 41), AD-Executive (n = 117), AD-Language (n = 33), AD-Visuospatial (n = 171). We performed voxel-wise contrasts of GM volumes derived from 3Tesla structural MRI between subgroups and controls (n = 127, age 58 ± 9, 42% male, MMSE 29 ± 1), and observed that differences in regional GM volumes compared to controls closely matched the respective cognitive profiles. Specifically, we detected lower medial temporal lobe GM volumes in AD-Memory, lower fronto-parietal GM volumes in AD-Executive, asymmetric GM volumes in the temporal lobe (left < right) in AD-Language, and lower GM volumes in posterior areas in AD-Visuospatial. In order to examine possible biological drivers of these differences in regional GM volumes, we correlated subgroup-specific regional GM volumes to brain-wide gene expression profiles based on a stereotactic characterization of the transcriptional architecture of the human brain as provided by the Allen human brain atlas. Gene-set enrichment analyses revealed that variations in regional expression of genes involved in processes like mitochondrial respiration and metabolism of proteins were associated with patterns of regional GM volume across multiple subgroups. Other gene expression vs GM volume-associations were only detected in particular subgroups, e.g., genes involved in the cell cycle for AD-Memory, specific sets of genes related to protein metabolism in AD-Language, and genes associated with modification of gene expression in AD-Visuospatial. We conclude that cognitively-defined AD subgroups show neurobiological differences, and distinct biological pathways may be involved in the emergence of these differences.
1. Introduction
The clinical phenotype of Alzheimer’s disease (AD) dementia is typically characterized by prominent memory impairment. However, there is considerable variation in the clinical manifestation of AD that can also present with substantial deficits in cognitive domains other than memory (Lam et al., 2013). At the ends of the clinical spectrum reside the atypical variants of AD; posterior cortical atrophy (PCA) (Crutch et al., 2017) and logopenic variant primary progressive aphasia (lvPPA) (Gorno-Tempini et al., 2008), which are characterized by early and predominant impairments in a single cognitive domain (i.e., visuospatial and language impairments, respectively). While these atypical variants represent phenotypical extremes, there is substantial inter-individual cognitive variability in persons with AD dementia who do not meet clinical criteria for these specific variants and thus remain classified under the moniker of “typical AD dementia” (Ossenkoppele et al., 2019). A framework has been proposed to categorize people with typical AD dementia into cognitively-defined subgroups based on their relative performance on cognitive domains (Crane et al., 2017). A deeper understanding of the underlying mechanisms that govern differences between cognitively-defined subgroup of typical AD dementia might identify differential pathways that play a role in the pathogenesis of AD, improve the accuracy of diagnosis and prognosis, and aid in the development of personalized medicine strategies and design of clinical trials.
Cognitive phenotypes that characterize atypical AD dementia variants (i.e., lvPPA, PCA) are associated with marked clinical differences and regional variations in neurodegeneration (Lam et al., 2013, Ossenkoppele et al., 2015). Moreover, differences in regional gene expression profiles associate with regional differences in anatomical (Whitaker et al., 2016) and functional (Richiardi et al., 2015) properties of the brain, as well as with selective regional vulnerability to neurodegenerative disease (Freeze et al., 2019, Grothe et al., 2018, Sepulcre et al., 2018). Based on these observations, we aimed to address two research objectives with regard to cognitively-defined subgroups within the broad spectrum of persons with ‘typical’ AD dementia. First, to examine whether cognitively-defined subgroups associate with regional variations in gray matter (GM) volumes. And second, to explore possible biological drivers of differences in regional susceptibility to neurodegeneration, by relating subgroup-specific regional GM volumes to brain-wide gene expression profiles based on a stereotactic characterization of the transcriptional architecture of the human brain as provided by the Allen human brain atlas (Hawrylycz et al., 2015).
2. Material and methods
2.1. Participants
For this single-center study, we included participants from the Amsterdam Dementia Cohort (ADC). The ADC is located at the Alzheimer Center Amsterdam, where patient care and scientific research are performed in parallel. All patients that visit the Alzheimer Center Amsterdam undergo an elaborate one-day screening battery including extensive neuropsychological evaluation, an MRI scan and a lumbar puncture. In a subset of the ADC, an amyloid-PET scan is performed (e.g., after refusal of a lumbar puncture or in the context of a research project). After their baseline visit, patients visit the clinic annually for follow-up visits. All patients that visit the Alzheimer Center Amsterdam are asked to consent to the use of their clinical data for scientific purposes and 99% accepts (van der Flier et al., 2014), whereupon they are included into the ADC. Additional information on the set-up, content, and data collection procedures within the ADC are described elsewhere (van der Flier et al., 2018).
Written informed consent was obtained from all participants according to the Declaration of Helsinki and the local medical ethics review committee of the Amsterdam UMC approved the study.
2.1.1. AD participants under investigation
Participants that undergo categorization into AD-subgroups (see section 2.3) were selected based on the following criteria: 1) clinical diagnosis of AD dementia (McKhann et al., 2011) at time of dementia screening, 2) molecular biomarker profile indicative of AD neuropathology (i.e., annual upward drift corrected CSF amyloid-β42 < 813 pg/mL (Tijms et al., 2018); and/or a positive amyloid-β PET scan determined by visual assessment (Ossenkoppele et al., 2015a)), 3) availability of a 3Tesla MRI scan, and 4) availability of neuropsychological data to compute all cognitive domains assessed (see sections 2.2 and 2.3). Exclusion criteria for participants that undergo categorization into AD-subgroups were: 1) meeting core criteria for an atypical variant of AD dementia, i.e., PCA or lvPPA (Gorno-Tempini et al., 2008, Schott et al., 2017), 2) psychiatric or neurological disorders (other than AD dementia), and 3) known genetic mutations associated with familial AD. A total of 679 participants were included based on these criteria. There was no age restriction for inclusion into the sample and the ADC sample is characterized by a relatively low mean age (~64 years old) (van der Flier et al., 2018). Because of possible differences between younger and older individuals with AD dementia, we provide sensitivity analyses in the supplement that look at early-onset AD (<65 years at dementia screening) and late-onset AD (>65 years at dementia screening) participants separately. Of the total sample of 679, 282 (42%) were under the age of 65.
2.1.2. Control groups
We also selected a control group of subjects who were amyloid-β negative on PET/CSF and determined to be cognitively healthy in a multi-disciplinary meeting based on standardized neuropsychological assessment (Groot et al., 2018). This group provided normative GM volumes that were used to assess regional GM volumes of the AD-subgroups (see section 2.6).
Furthermore, to assess how the cognitively-defined AD-subgroups relate to established atypical variants of AD, we additionally selected “positive control” samples of individuals with lvPPA (n = 20, age 66.9 ± 5.2, 60% male, MMSE 23.1 ± 4.1) and PCA (n = 69, age 62.0 ± 6.1, 41% male, MMSE 20.2 ± 4.6) from our previous studies (Bergeron et al., 2018, Groot et al., 2020), which were diagnosed according to published clinical criteria (Gorno-Tempini et al., 2008, Schott et al., 2017). There are currently no consensus diagnostic criteria for selective amnestic or behavioral/dysexecutive variants of AD.
2.2. Cognition
Neuropsychological tests from the ADC neuropsychological test battery were categorized by an expert panel (ET, JM, AS, PC) into cognitive domains; memory, executive-functioning, language and visuospatial-functioning (Table A1). For the four cognitive domains, confirmatory factor analyses (Mplus version 7.419) (Muthén and Muthén, 1998) were implemented to generate composite cognitive domain scores from the individual test scores. Composite cognitive domain scores were then co-calibrated to normative scores that are based on metrics obtained for 4,050 people with incident AD dementia from our legacy cohort assessed in our previous publications (Crane et al., 2017, Mukherjee et al., 2020). This was achieved by implementing “anchor items” (same stimulus, and scored identically, in the ADC and the legacy cohort; A. Text) to anchor metrics and then co-calibrating the ADC composite cognitive domain scores to those from the legacy cohort using bifactor models in Mplus. Because the metrics from our legacy cohort are based on late-onset AD cases, the scores of younger ADC participants (<60 years) were co-calibrated using the parameters from the model obtained in older (>60 years) ADC participants. Detailed methods on how co-calibration of scores was achieved and brief descriptions of the legacy cohorts can be found in the supplement (A. Text) and in our previous publications (Crane et al., 2017, Mukherjee et al., 2020).
For the purposes of normalizing our scores, we z-transformed the co-calibrated cognitive domain scores using the mean and standard deviation (SD) from the corresponding score obtained in the Adult Changes in Thought (ACT) sample (which was also part of our legacy cohort). ACT was used as our reference population because it was our largest sample available. The ACT-normalized and co-calibrated cognitive scores of the ADC sample are presented in Table A2.
We additionally obtained mini-mental state examinations (MMSE) scores, which were used to assess differences in global cognition between AD-subgroups at baseline, and to assess differences in longitudinal decline in cognitive function (i.e., clinical progression). The MMSE was selected to measure global cognition and progression as it is a widely implemented test to examine clinical progression in clinical practice (Doody et al., 2001) and clinical trials (Doody et al., 2014, Salloway et al., 2014), and is administered at every follow-up visit in the ADC cohort.
2.3. Subgroup classification
Subgroup classification relies on scores across all four domains assessed (i.e., memory, executive-functioning, language and visuospatial-functioning). Classification is achieved by first averaging, for each individual, the scores across the four cognitive domains and then determining the difference for every domain score from that average. We used a difference of 0.80 units (i.e., 0.80 SD from the mean in ACT, see section 2.2) to identify domains with scores substantially lower than a person’s average score. This threshold was previously empirically determined after assessing a range of candidate thresholds (see (Crane et al., 2017) for further details). We then considered domain(s) with scores substantially lower than the person’s average score to assign people to groups (see Fig. A1 for a visual representation of categorization). This classification yields 6 groups. Consistent with our previous publications on these subgroups (Crane et al., 2017, Mukherjee et al., 2020), these were named according to which domain showed substantial relative impairment; memory (AD-Memory), executive-functioning (AD-Executive), language (AD-Language), visuospatial-functioning (AD-Visuospatial). When more than one domain was relatively impaired, subjects were classified as AD-Multiple. When none of the domains was relatively impaired a subject was classified as AD-No Domains, meaning these individuals had similar levels of impairment across all four cognitive domains. Because the AD-No Domains group does not show a particular cognitive phenotype, we used this as the reference group for comparisons against the other AD-subgroups. It is important to note that because the classification of subgroups is based in intra-individual differences in impairments across cognitive domains relative to the person’s average, the subgroups classification is based solely on cognitive profiles rather than overall level of cognitive impairment. Table A2 shows that there were differences across AD-subgroups with regards to average level of impairment across domains. Furthermore, because the normative scores were derived from the ACT cohort, classification into a subgroup is based on substantial relative impairment compared to an AD population.
2.4. Neuroimaging
MRI scans were performed according to standardized acquisition protocols including a 3D T1-weighted structural MRI sequence on three different 3Tesla MRI scanners (Table A3). We adjusted for scanner type in the statistical models (see section 2.6). All the MRI scans were performed on the same visit as the neuropsychological examination (i.e., baseline dementia screening) or within a very short timeframe. The structural T1 images were segmented into gray matter, white matter and CSF volumes using the “New Segment” toolbox implemented in Statistical Parameter Mapping (SPM) 12 software (Welcome Trust Centre for Neuroimaging, UCL, London, UK). To generate a study-specific template, Diffeomorphic Anatomical Registration Through Exponentiated Lie Algebra (DARTEL) was used to align grey matter images non-linearly to a common space. Grey matter images were then spatially normalized to MNI standard space using the study specific template and individual flow fields. Modulation was applied to preserve tissue volume signal and images were smoothed using an 8 mm full-width-at-half-maximum isotropic Gaussian kernel. After each processing step, the images were visually checked for quality. The resulting normalized GM images were used as input for voxel-based morphometry analyses assessing regional variations in of GM volumes within the AD-subgroups (see section 2.6).
2.5. Regional gene expression profiles
Brain-wide gene expression profiles were obtained from microarray data from the Allen database of the human brain transcriptome, which is publicly available from the Allen brain institute (http://human.brain-map.org). This dataset contains regional gene expression data from around 61,000 microarray probes collected from ~ 3700 tissue samples, which were obtained from six control subjects who died without any evidence of neurologic disease (aged 24–57). Anatomical information for each of the probes is provided and can be used to determine the location within stereotactic standard space. Microarray data from the ~ 3700 tissue samples, with their corresponding anatomical locations in MNI space provided in the Allen database, were used to interpolate brain-wide gene expression at the voxel level using Gaussian process regression implemented in the R package gstat (see Gryglewski et al., 2018, Pebesma, 2004 for detailed methods). Briefly, the method relies on the assumption that spatially adjacent sites (i.e., voxels) are more similar than voxels that are far apart. First, this spatial dependency of each gene’s expression was captured using spatial variogram models. Subsequently, Gaussian process regression was used to obtain unbiased predictions of gene expression in unobserved voxels (between samples) based on data from all available samples (with observed gene expression data) by weighting data according to the variogram model and the distance between the observed and predicted voxel. This was performed separately for the cerebral cortex, subcortical regions and the cerebellum, due to the different degree of spatial dependency in gene expression between these (ontogenetically distant) parts of the brain. Internal cross-validation assessing correlations between predicted and observed voxel values as well as correlation of predicted gene expression and PET data of corresponding targets support the validity and utility of this method. These brain-wide gene expression maps have been made publicly available at www.meduniwien.ac.at/neuroimaging/mRNA.html.
These maps represent gene expression data across the entire cerebral cortex for each of the 18,686 protein-coding human genes. Microarray data from both hemispheres is only available in two subjects (Hawrylycz et al., 2012), and, in line with previous examinations (Grothe et al., 2018, Sepulcre et al., 2018), we restricted all our analyses assessing associations between GM volumes and gene-expression to the left hemisphere.
2.6. Statistical analysis
All statistical analyses were performed in SPM12 or R version 3.5.2. Differences in demographic and clinical characteristics between the subgroups were assessed using independent-samples t-tests (continuous variables), χ2-tests (categorical variables) and Kruskal-Wallis tests (for the non-normally distributed education variable). Linear-mixed effects models were assessed to examine differences between subgroups in global cognition (i.e., MMSE) at baseline and change over time. We ran a model using one predictor for all subgroups, with AD-No Domains as the reference and adjusted for age, sex and education. To assess the difference in rates of mortality across cognitively-defined subgroups, age-adjusted Cox proportional hazard models were performed. Again, we ran one model comparing all subgroups to the AD-No Domains group. Statistical significance for these models was set at p < 0.05.
2.6.1. Regional gray matter volumes
To assess the brain-wide spatial pattern of GM volume for each of the subgroups, SPM12 was used to perform voxel-wise contrasts between subgroups and the amyloid-β negative, cognitively normal controls, as well as between the AD-No Domains subgroup and the other AD-subgroups. These analyses were adjusted for age, sex, intracranial volume and scanner type. For the analyses assessing differences between AD-No Domains and the other groups, we aimed to consider the possibility that one group may have, on average, presented later in the disease course than another group, leading to overall greater atrophy. Therefore, we included a term for overall GM to intracranial volume ratio in the contrast models for comparisons against the AD-No Domains group. Because of the additional correction for global GM volumes, differences between AD-subgroups and AD-No Domains reflect the difference in regional GM volumes relative to the total GM volumes across the whole cortex, rather than absolute differences in volumes. All voxel-wise contrasts yield statistical parametric T-maps that represent the voxel-wise difference in GM between comparators.
The T-maps for the contrasts against cognitively normal controls were also used to assess spatial similarities between regional GM volumes among cognitively-defined AD-subgroups and the lvPPA and PCA reference groups. We determined the overlap between the most atrophied voxels from the T-maps (meant + 2*standard deviationt) with the Sørensen–Dice coefficient (DSC) as: 2*(A ∩ B)/(A + B), with 0 signifying no overlap and 1 complete overlap.
2.6.2. Associations between regional gray matter volumes and regional gene-expression profiles
First, we took the voxel-wise contrasts against the amyloid-β negative cognitively normal control group, as well as the voxel-wise gene expression data (available from www.meduniwien.ac.at/neuroimaging/mRNA.html see section 2.5; Fig. 1A) and extracted regional values for the 34 Desikan-Killiany regions of interest using the “Marsbar” software toolbox for SPM12 (Brett et al., 2002) (Fig. 1B). The 34 regional T-values (GM volume differences compared to controls) were then correlated to the 34 regional gene expression values using Spearman’s correlations (Fig. 1C) (Krienen et al., 2016). This was repeated for each of the 18,686 genes under investigation. Therefore, this results in 18,686 correlation coefficients (one for each gene), each indicating the spatial relationship between brain-wide gene expression and regional GM volumes. For explorative analyses the correlation coefficients of all 18,686 genes were rank ordered according to the strength of the association (Fig. 1D). To reduce the wealth of gene-specific correlations into more comprehensible data we used gene set enrichment analysis (GSEA). GSEA is a statistical approach developed specifically to condense data from large microarrays on single genes into more comprehensible information on functional gene sets. GSEA uses the complete spectrum of information provided by the rank ordered gene expression-GM volume correlations and determines whether known gene sets (i.e., grouped genes with related functions) are negatively or positively enriched for a specific pattern of GM volumes as a whole. In the present study, we explored 6,032 different gene sets obtained from the curated Reactome (http://reactome.org/; dataset: c2.cp.reactome.version:7.0 from the Molecular Signatures Database (MSigDB)), and gene ontology (GO; http://www.geneontology.org/; dataset: c5.all version 7.0) databases. These gene sets are defined by genes that are commonly associated with a specific biological process or GO annotation, respectively. GSEA uses the rank ordered correlation coefficients between regional gray matter volumes and gene expression as well as the known gene sets as inputs to determine positive or negative enrichment of the gene sets by assessing whether genes within a set are non-normally distributed (skewed) towards one edge of the rank-ordered correlation spectrum (Fig. 1E). We ran the “Run GSEA pre-ranked” tool on default parameters and implemented version 4.0.3 of the GSEA. software package (available from: http://software.broadinstitute.org/gsea/index.jsp). Gene sets that cluster towards the positive end show higher than expected neurotypical expression levels in brain regions with relatively low GM volumes (‘positively enriched’), whereas those clustering towards the negative end show lower expression in brain regions with lower GM volumes (‘negatively enriched’). The non-normality of the distribution is represented by the normalized enrichment score, which also accounts for different sizes of the tested gene sets, and the statistical significance (p) of this score was adjusted using the false discovery rate (FDR). The threshold for statistical significance was set to P(FDR) < 0.10 (Subramanian et al., 2005). Using this approach, information regarding the spatial correlation between GM volumes and regional gene expression is summarized into gene sets.
Fig. 1.

Gene-set enrichment analyses and grouping of gene sets. Panel A displays voxel-wise differences in gray matter volumes between AD subjects and cognitively normal controls (top row; T-maps, adjusted for covariates), and brain-wide gene expression values (MAPT as an example) obtained from www.meduniwien.ac.at/neuroimaging/mRNA.html (bottom row). Panel B displays the parcellation of the T-map and gene expression values into regions-of-interest (ROI) from the Desikan-Killiany atlas. Panel C displays Spearman’s correlations between T-values and gene expression values within ROIs. In this plot, each datapoint represents grey matter volume differences with controls within the 34 ROIs on the x-axis and gene-expression values in the corresponding ROI on the y-axis. The steps in panel A through C are repeated for all 18,686 genes, yielding a correlation spectrum that is ranked from positive to negative (Panel D). GSEA software then produces an enrichment score that indicates whether a gene set, as a whole, preferentially falls towards one end of the correlation spectrum (Panel E; with the gene set ‘synaptic plasticity’ as an example). Panel F displays results from the gene set grouping analysis in Cytoscape where significantly enriched gene sets are plotted as interrelated nodes connected by edges denoting their overlapping genes.
In order to account for partial redundancy of the enriched gene sets and to identify which biological pathways are common across the gene set produced by the GSEA approach, we organized the gene sets into a network structure using the Cytoscape plug-in ‘Enrichment Map’ (Merico et al., 2010). Grouping into clusters is achieved by assessing common genes across enriched gene sets. Results are visualized in an automated network layout where related gene sets (nodes) are connected by edges that represent the degree of overlapping genes, thus forming clusters (see Fig. 1F and figure legend). The gene sets within the resulting clusters were then manually examined to identify the common biological pathway associated with the gene sets. We also ran “Leading-edge” analyses in the GSEA 4.0.3. software package to identify the most relevant individual genes driving the enrichment signal for the different gene sets in a given cluster (“leading-edge genes”).
The whole process described in this paragraph is performed separately for each of the AD-subgroups. Therefore, each AD-subgroup yields their own set of enriched gene set clusters. These were then compared against each other by assessing whether the gene sets within a cluster are enriched in multiple subgroups or uniquely enriched in only one subgroup. A gene set cluster was considered uniquely enriched in a subgroup when the majority of the gene sets that make up that cluster were not significantly enriched in another subgroup.
3. Results
3.1. Subgroup characteristics
About one third (n = 231/679, 34%) of subjects were classified as AD-No Domains. Forty-one subjects (6%) were classified as AD-Memory, 117 (17%) as AD-Executive, 33 (5%) as AD-Language and 171 (25%) as AD-Visuospatial. In addition, 86 subjects (13%) were classified as AD-Multiple. Because of the heterogeneous composition of the AD-Multiple group, our main analyses are focused on the other subgroups. Table 1 displays the demographic and clinical characteristics for the whole sample and cognitively-defined AD-subgroups, and pairwise differences between groups are given in Table A4. Mean age of the total sample was 66.2 ± 7.4, 47% were male, MMSE was 21.2 ± 5.1 and 69% were APOEε4 positive. Using the AD-No Domains group as the reference we observed that AD-Visuospatial were younger (63.7 ± 7.2 vs 66.9 ± 7.7, p < 0.01) and AD-Language had a lower APOEε4 prevalence (51.5 vs 72.7%, p < 0.01). AD-Memory showed the highest APOEε4 prevalence (90.2%), which was significantly higher than in AD-No Domains (72.7%, p = 0.017). Furthermore, compared to AD-No Domains, AD-Executive had lower total GM to intracranial volume ratios (0.37 ± 0.04 vs 0.38 ± 0.04, p < 0.01) and AD-Memory had greater GM to intracranial volume ratios (0.41 ± 0.04 vs 0.38 ± 0.04, p < 0.01).
Table 1.
Demographic and clinical characteristics of the sample.
| Cognitively-defined Alzheimer’s disease subgroup |
Reference
groups |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| All AD-subgroups | AD-Memory | AD-Executive | AD-Language | AD-Visuospatial | AD-Multiple | AD-No Domains | Normal controls | lvPPA | PCA | |
| N (% of all AD-subgroups) | 679 | 41 (6%) | 117 (17%) | 33 (5%) | 171 (25%) | 86 (13%) | 231 (34%) | 127 | 20 | 69 |
| Age at diagnosis | 66.3 (7.4) | 68.3 (6.6) | 68.0 (6.6) | 64.4 (6.4) | 63.7 (7.3) | 66.8 (7.4) | 66.9 (7.7) | 57.7 (8.8) | 66.9 (5.1) | 62.0 (6.1) |
| Early-onset, % | 282 (42%) | 11 (27%) | 40 (34%) | 17 (52%) | 95 (56%) | 33 (38%) | 86 (37%) | – | 15 (75%) | 19 (28%) |
| Sex, male | 318 (47%) | 17 (42%) | 60 (51%) | 18 (55%) | 79 (46%) | 48 (56%) | 96 (42%) | 53 (42%) | 10 (53%) | 28 (41%) |
| MMSE | 21.2 (5.1) | 23.7 (2.8) | 20.4 (5.5) | 17.2 (5.7) | 22.3 (4.8) | 20.0 (5.1) | 21.5 (4.8) | 28.7 (1.1) | 23.1 (4.1) | 20.2 (4.6) |
| NPI total score | 12.0 (13.8) | 12.4 (12.3) | 13.6 (15.1) | 7.9 (9.4) | 11.6 (13.7) | 8.9 (12.4) | 12.9 (14.3) | 3.9 (8.2) | 6.3 (7.3) | 8.4 (10.1) |
| GDS | 2.8 (2.6) | 2.8 (2.7) | 2.6 (2.6) | 2.4 (2.1) | 3.0 (2.3) | 2.9 (2.7) | 2.8 (2.7) | 2.4 (1.6) | 2.4 (1.7) | 3.3 (2.3) |
| Education, median (IQR)^ | 5 [4–6] | 5 [4–6] | 5 [4–6] | 5 [5–6] | 5 [4–6] | 5 [4–6] | 5 [4–6] | 6 [5–7] | 5 [4–6] | 5 [4–6] |
| APOEε4 positive (%) | 467 (69%) | 37 (90%) | 75 (64%) | 17 (52%) | 113 (66%) | 57 (66%) | 168 (73%) | 40 (32%) | 10 (53%) | 38 (55%) |
| CSF Amyloid-β in pg/ml | 504 (1 1 3) | 530 (119) | 498 (114) | 476 (108) | 499 (114) | 492 (111) | 516 (111) | 984 (195) | 546 (106) | |
| CSF Total tau in pg/ml | 734 (426) | 829 (431) | 697 (419) | 781 (438) | 735 (419) | 681 (359) | 748 (457) | 229 (94) | 741 (347) | 735 (275) |
| CSF P-tau in pg/ml | 90 (39) | 103 (37) | 87 (38) | 96 (43) | 89 (36) | 85 (37) | 91 (43) | 44 (16) | 88 (32) | 93 (34) |
| Total GM to ICV ratio | 0.38 (0.04) | 0.41 (0.04) | 0.37 (0.04) | 0.38 (0.04) | 0.39 (0.04) | 0.37 (0.04) | 0.38 (0.04) | 0.44 (0.04) | 0.38 (0.04) | 0.39 (0.03) |
Values depicted are mean (standard deviation), unless otherwise indicated. All pairwise differences between groups are displayed in Table A. 4. APOE – Apolipoprotein E, GDS – Geriatric depression scale, NPI – Neuropsychiatric inventory, GM – gray matter, P-tau – phosphorylated tau, ICV-intracranial volume.
^ - Assessed using the qualitative Dutch Verhage scale (Table A9).
3.2. Rates of clinical disease progression and mortality
Baseline MMSE scores were higher in AD-Memory (β(SE) = 1.84(0.85), p = 0.03) and AD-Visuospatial (1.19(0.51), p = 0.02), and lower in AD-Language (-4.0(0.98), p < 0.01), when compared to AD-No Domains. Furthermore, all subgroups progressed faster over time on the MMSE than AD-No Domains (-0.50(0.18), p = 0.01 for AD-Executive; -1.61(0.57), p < 0.01 for AD-Language; -0.56(0.13), p < 0.01 for AD-Visuospatial), except for AD-Memory (-0.29(0.23), p = 0.20; Fig. 2A). Using the AD-No Domains group as the reference, we observed a higher mortality rate in AD-Executive (HR[95%CI] = 1.94[1.38–2.73], p < 0.01), AD-Language (2.16[1.29–3.63], p = 0.05) and AD-Visuospatial (1.48[1.07–2.00], p = 0.02). There were no differences in mortality rates between AD-Memory and AD-No Domains (0.80[0.41–1.54], p = 0.5; Fig. 2B).
Fig. 2.

Clinical progression and mortality rates across cognitively-defined subgroups. The plot in panel A displays results from linear-mixed effects models assessing the effect of subgroup and the interaction effect between subgroup*time on MMSE scores, adjusted for age, sex and education. The model includes one predictor for all subgroups with AD-No Domains as the reference. The Kaplan-Meier curves in panel B represents the survival probability over time for the various subgroups. Hazard ratios were calculated using a Cox proportional hazard model using AD-No Domains as the reference, and was adjusted for age. Error bands in both panels are not included for visualization purposes. * - effect of subgroup on MMSE ** - interaction effect of subgroup*time on MMSE over time.
3.3. Regional gray matter volumes
Voxel-wise contrasts between cognitively-defined AD-subgroups and cognitively normal controls revealed lower GM volumes in temporoparietal areas across all subgroups (Fig. 3A, Fig. A2 and 3), as well as subgroup-specific patterns of GM volume differences. The subgroup-specific patterns are best appreciated in the voxel-wise contrasts between AD-No Domains and the other AD-subgroups (Fig. 3B). GM volume differences in AD-Memory were prominent in the medial temporal lobe – especially in bilateral hippocampus – while AD-Language showed temporal predominant GM volume differences with lateralization to the disadvantage of the left hemisphere. AD-Executive displayed a widespread pattern with greater GM volume differences in widespread cortical areas compared to AD-No Domains. For AD-Visuospatial, the pattern of GM volume differences with AD-No Domains markedly occupied posterior brain areas.
Fig. 3.

Regional gray matter volumes differences across cognitively-defined Alzheimer’s disease-subgroups. Results from voxel-based morphometry analyses, displayed as T-maps, representing differences in regional gray matter volumes, adjusted for age, sex, scanner, intracranial volume. Higher T-values signify lower GM volumes (indicating more atrophy). Panel A displays results from voxel-wise contrasts between AD-subgroups and cognitively normal controls. The T-values from the comparisons against controls in Panel A were used as input for the GM volume vs gene expression analyses. Panel B displays results from voxel-wise contrasts between the domain-specific subgroups and the AD-No Domains group. These analyses were additionally corrected for overall GM to intracranial volume ratios, resulting in images that are indicative of differences in spatial patterns rather than overall level of GM volumes. Voxel-wise contrasts showing only significant voxels are displayed in the supplement (Fig. A3). The coronal slice was taken at y = -8 within the MNI template.
3.4. Regional gray matter volumes in AD-subgroups vs atypical variants of Alzheimer’s disease
Given their clinical resemblances, we aimed to compare the spatial patterns of GM volumes differences between AD-Visuospatial and PCA, and between AD-Language and lvPPA. A striking similarity can be appreciated from Fig. 4A, which displays the voxel-wise contrasts vs cognitively normal controls (see Fig. A4 for a more detailed depiction of GM volume differences compared to controls in PCA and lvPPA). Spearman correlation analyses between GM volumes within the 34 Desikan-Killiany ROIs for AD-Language vs lvPPA (rho = 0.92, p < 0.01), and AD-Visuospatial vs PCA (rho = 0.65, p < 0.01) confirmed a high spatial correspondence (Fig. 4B). Furthermore, the Sørensen–Dice coefficient (DSC) for overlap between the voxels that showed the greatest difference with controls (Fig. 4C) for AD-Language and lvPPA was 0.73, which far exceeded the DSC for overlap between lvPPA and all other groups (AD-Memory: 0.19, AD-Executive: 0.23, and AD-Visuospatial: 0.19) and also far exceeded the DSC of overlap between AD-Language and the other subgroups (AD-Memory: 0.23, AD-Executive: 0.22, and AD-Visuospatial: 0.19). The DSC for overlap between AD-Visuospatial and PCA (0.33) exceeded the DSC for overlap between PCA and all other groups (AD-Memory: 0.04, AD-Executive: 0.17 and AD-Language: 0.04; Fig. 4D). This DSC between AD-Visuospatial and PCA (0.33) was also higher than the DSC for overlap between AD-Visuospatial and AD-Memory (0.27) or AD-Language (0.19), though lower than DSC for overlap between AD-Visuospatial and AD-Executive (0.50; Fig. 4D).
Fig. 4.

Similarity between AD-Language and lvPPA, and between AD-Visuospatial and PCA. Panel A displays the voxel-wise contrasts for AD-Language, lvPPA, AD-Visuospatial and PCA vs cognitively normal controls, adjusted for age, sex, scanner and intracranial volume. Panel B displays Spearman correlations between T values within 34 ROIs from the voxel-based contrasts against cognitively normal controls for AD-Language and lvPPA (top), and AD-Visuospatial and PCA (bottom). Panel C displays binarized maps of voxels with the greatest GM volume difference with controls for each T-map according to the threshold: (Meant + 2*SDt), with the mean denoting the average within each T-map and the SD the standard deviation within that image. Panel D displays the Sørensen–Dice coefficient (DSC) for overlap between voxels with the greatest GM volume difference, which is calculated as: 2*(A ∩ B)/(A + B). 0 indicates no overlap and 1 indicates complete overlap.
3.5. Gene expression profiles associated with gray matter volumes across multiple subgroups
To assess biological drivers that might explain differences in subgroup-specific regional GM volumes, we performed exploratory GSEA using gene-expression data for the 18,686 genes provided in the Allen human brain atlas. Table A5 lists all enriched gene sets for the different subgroups that were identified through this approach. Fig. 5 displays the gene sets grouped into a network structure based on common biological functions across gene sets. Some gene sets could not be clustered based on shared biological functions with other gene sets. These are displayed as idle nodes and the corresponding gene sets are listed in Table A5. Fig. 7 is an overview of the clusters displayed in Fig. 5 across AD-subgroups in order to facilitate easier comparison of identified gene set clusters between subgroups.
Fig. 5.


GSEA results grouped into clusters of gene sets. Displayed are the results from gene set enrichment analyses and the clusters represent groupings of gene sets, variations in gene expression levels of which are spatially associated with regional GM volumes in AD-Memory, AD-Executive, AD-Language, AD-Visuospatial and AD-No Domains. Red nodes represent gene sets that are positively enriched (lower volumes -> higher gene expression) while blue nodes are negatively enriched. The size of the nodes represent the size of the gene set, larger nodes signify gene sets with more genes included. The thickness of the edges corresponds to the number of genes that overlap between two gene sets (i.e., nodes). Grouping of gene sets into clusters was achieved by assessing shared biological functions across gene sets (by Cytoscape software) and clusters were named (by the authors) according to that common biological function. Idle nodes represent gene sets that did not overlap with other gene sets and could therefore not be clustered into groups. Leading-edge analyses (to detect the most relevant genes driving the groupings) are presented in Fig. 6. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. 7.

Overview of shared and unique gene set clusters associated with regional patterns of gray matter volumes across AD-subgroups. The venn-diagram displays which gene set clusters were negatively (left) and positively (right) enriched in each of the AD-subgroups. These are taken from the named clusters in Fig. 5. The circles and ovals were shaped in order to show which clusters are unique or shared between AD-subgroups but the shape and size of the shaded areas have no inherent meaning.
Clusters of genes associated with synaptic function and plasticity (e.g., dopamine release cycle, and long-term synaptic and neuronal plasticity) were positively enriched in all subgroups. A cluster comprised of gene sets associated with the immune system (e.g., interleukin-7 and regulation of alpha/beta t-cell activation) was positively enriched in AD-Executive and AD-Language but negatively enriched in AD-Memory, AD-Visuospatial and AD-No Domains. In AD-Memory and AD-Visuospatial, we observed a large negatively enriched cluster with gene sets implicated in mitochondrial respiration (e.g., ATP synthesis, respiratory electron transport), a cluster that was also present in AD-No Domains. Another large negatively enriched cluster present in multiple subgroups (AD-Memory, AD-Executive and AD-Visuospatial) comprised gene sets associated with protein metabolism (e.g., methylation, amino acetylation), which was also present in AD-No Domains. A smaller negatively enriched cluster associated with autophagy (e.g., mitophagy, mitochondrial depolarization) was present in AD-Memory and AD-No Domains (Fig. 5, Fig. 7; Table A5).
3.6. Gene expression profiles uniquely associated with subgroup-specific patterns of regional gray matter volume
For AD-Memory, two negatively enriched clusters were not shared with AD-No Domains or any of the other subgroups, indicating that these are uniquely implicated in AD-Memory. These clusters were associated with the cell cycle (e.g., DNA replication, regulation of apoptosis), and membrane proteins (e.g., MHC protein and cell lumen). Another negatively enriched cluster associated with RNA metabolism (e.g., mRNA splicing, precatalytic spliceosome) was present only in AD-Memory, while this cluster was positively enriched in AD-Executive. For AD-Language, two unique clusters were identified, comprising negatively enriched gene sets associated with taste receptor activity and positively enriched gene sets associated with metabolism of proteins (e.g., genes associated with axon guidance and angiogenesis, and cytosolic ribosome), respectively. While other subgroups also showed enrichment for gene sets implicated in protein metabolism, these did not overlap with the AD-Language specific gene sets and were also negatively rather than positively enriched. For AD-Visuospatial, a large negatively enriched cluster associated with modification of gene expression (e.g., epigenetic regulation and depurination), a smaller negatively enriched cluster associated with metabolism of carbohydrates (e.g., gluconeogenesis, lysosomal lumen), and a small positively enriched cluster of gene sets associated with keratinization were unique to this subgroup. There were no clusters for AD-No Domains that were unique to this group (Fig. 5, Fig. 7; Table A5).
4. Discussion
We categorized 679 amyloid-β positive individuals clinically diagnosed with AD dementia into subgroups based on the distribution of impairments across cognitive domains. We found that all subgroups except AD-Memory showed faster clinical disease progression and had a higher mortality rate compared to AD-No Domains (i.e., no substantial relative cognitive impairments). In accordance with findings in atypical variants of AD (Crutch et al., 2017; Gorno-Tempini et al., 2011) this illustrates that AD-subgroup membership (i.e., displaying a specific cognitive phenotype) has clinical implications. Furthermore, all cognitively-defined subgroups displayed distinct patterns of regional GM volumes, suggesting that cognitive heterogeneity is associated with differences in regional susceptibility to neurodegeneration, even within the spectrum of typical AD dementia. More specifically, we observed lower medial-temporal GM volumes in AD-Memory, lower medial-frontal/parietal GM volumes in AD-Executive, left < right temporal GM volumes in AD-Language and lower GM volumes in posterior parts of the brain in AD-Visuospatial. The regional GM volume patterns of AD-Language and AD-Visuospatial were highly analogous to those observed in lvPPA and PCA groups, which might suggest that atypical variants and AD-subgroups are part of a clinical-radiological spectrum. To explore potential biological drivers that might underlie the observed clinical and neurobiological heterogeneity among subgroups, we associated regional GM volume patterns in AD-subgroups with brain-wide gene expression profiles. We found biological pathways that were associated with GM volume patterns across multiple subgroups, including gene sets involved in metabolism of proteins, mitochondrial respiration, the immune system, and synaptic function and plasticity. There were also biological pathways that were unique to specific cognitively-defined subgroups, including pathways involved in cell cycle for the AD-Memory group, certain sets of protein metabolism in AD-Language, and modification of gene expression in AD-Visuospatial. These findings point to potential biological drivers behind the emergence of clinical and neurobiological heterogeneity in AD.
4.1. Associations between genetics and clinical phenotype in Alzheimer’s disease
The mechanisms underlying clinical-biological heterogeneity in AD are still largely unknown. However, previous examinations have revealed that there are specific genetic risk factors that influence the clinical manifestation of AD. For instance, it is has repeatedly been shown that APOEε4 carriers have more extensive medial temporal atrophy and memory deficits (e.g., van der Flier et al., 2011). In line with these findings, the prevalence of APOEε4 in our AD-Memory group was a striking 90%, which is substantially higher than what is typically reported in AD cohort studies (66% (Mattsson et al., 2018)). The reason for this specific association between APOEε4 and AD-Memory is not known but the consistency of this finding across examinations (Crane et al., 2017, Mukherjee et al., 2020, van der Flier et al., 2011) highlights APOEε4 positivity as a determinant to develop a memory-predominant clinical presentation of AD. In addition to the usual suspect in AD research, the APOE gene, previously published genetic association studies using the same subgroup classification scheme also observed that genetic loci previously associated with AD-subgroup-specific associations showed varying odds-ratios (Crane et al., 2017) and novel loci were specific for different subgroups (Mukherjee et al., 2020). While these examinations of specific loci and individual genes may one day be instrumental in determining potential therapeutic targets and personalized medicine strategies, these approaches are reliant on large sample sizes and often require additional fine mapping because top hits are often found outside of coding regions and are very rarely the causal single-nucleotide polymorphism (SNP).
4.2. Brain-wide gene expression profiles associated to gray matter volume patterns across subgroups
In order to identify possible biological drivers behind the emergence of heterogeneity in AD, we implemented a brain imaging vs gene expression co-localization approach that has recently shown promise in identifying potential biological pathways implicated in regional susceptibility to pathologic alterations in AD and other neurodegenerative diseases (Freeze et al., 2019, Grothe et al., 2018, Sepulcre et al., 2018). We observed that gene sets that were enriched in regions that show lower GM volumes across multiple subgroups could be grouped into genes associated with; mitochondrial respiration, metabolism of proteins, immune system and synaptic plasticity. Mitochondrial dysfunction is thought to be (potentially causally (Swerdlow et al., 2014)) implicated in the pathogenesis of AD (Flannery and Trushina, 2019), possibly through an interaction with APOE (Yin et al., 2020). Our findings regarding the association between lower GM volumes and lower expression of genes associated with mitochondrial respiration could be due to increased susceptibility in these regions to mitochondrial dysfunction, subsequent oxidative stress, and eventual cell-death and atrophy. With regard to gene sets associated with protein metabolism, ribosomal protein synthesis is lower in brain tissue that is affected by AD (Langstrom et al., 1989), which is in line with our results. This process has usually been regarded as a downstream consequence of pathology rather than an upstream process (Langstrom et al., 1989) and points to a possible avenue for investigations into the pathogenesis of AD. Interestingly, leading-edge analyses of the most relevant genes driving the enrichment signal for the gene sets in this cluster pointed to genes from the mitochondrial ribosomal protein (MRP*) family (see Fig. 6), which has previously been proposed as a target to combat mitochondrial dysfunction (Sylvester et al., 2004). Other gene sets that were enriched in multiple groups consisted of genes associated with immune function. We found that lower regional GM volumes in AD-Memory, AD-Executive and AD-No Domains were associated with lower expression of genes associated with interleukin-7, a cytokine involved in T-cell development. These findings are in line with a previous study showing that genes involved in the immune response are negatively enriched in regions vulnerable to AD pathology (Freer et al., 2016) and supports a role of inflammation in AD pathogenesis (Gjoneska et al., 2015). However, in contrast to these findings, we also observed that expression of genes associated with regulation of T-cell activation and differentiation were positively enriched in areas where GM volumes were lower in AD-Language and AD-Executive.
Fig. 6.

Leading edge gene analysis across subgroups. These panels display which genes (x-axis) overlapped between gene sets (y-axis) and drive the clustering results for each of the subgroups, panel A = AD-Memory, B = AD-Executive, C = AD-Language, D = AD-Visuospatial and E = AD-NoDomains. Some gene families which showed a lot of overlap are displayed on the borders of the leading-edge results. Axis-labels are removed for visualization purposes.
We also observed that genes associated with synaptic plasticity were enriched in regions with lower GM volumes across all AD-subgroups, which replicates a previous examination in an independent sample of AD subjects (Grothe et al., 2018). In AD, the spatial pattern of neurodegeneration closely matches regional distributions of tau-pathology (Braak and Braak, 1991, Whitwell et al., 2012), and the most plastic brain regions, such as the medial temporal lobe (Gonçalves et al., 2016), also appear to be the most vulnerable to initial deposition of tau pathology (Braak and Braak, 1991, Walhovd et al., 2016). The correspondence of regions with both heightened synaptic plasticity and lower GM volumes therefore becomes apparent. Initially, plastic brain regions, such as the medial temporal lobe, would be assumed to be the most resistant to pathology and neurodegeneration, but in the long-term this may become maladaptive as the brain ages and pathology sets in (Hillary and Grafman, 2017). While in the present study, GM volume difference with controls were most pronounced in AD-Memory, the other subgroups were still characterized by an AD-characteristic pattern with GM volume differences with controls including the medial temporal lobe, which might explain why this cluster was observed across multiple subgroups.
4.3. Gene-expression profiles associated to patterns of gray matter volumes in one subgroup
We also observed clusters of gene sets that were uniquely associated with regional GM volumes in a single subgroup. For AD-Memory, we found a large cluster of negatively enriched gene sets associated with the cell-cycle. Associations between the cell-cycle and AD have been demonstrated before and involves the dysfunction of neuronal cell-cycle re-entry, leading to the two-hit hypothesis. The first hit involves dysfunctional cycle re-entry, which would normally result in apoptosis and no development of AD pathology. However, chronic oxidative stress can cause a second hit that prevents normal apoptosis and allows the build-up of AD pathology (Moh et al., 2011). More research is necessary to determine why this mechanism would be more pronounced in AD-Memory than in other subgroups. For AD-Language, we found that gene sets associated with metabolism of proteins were positively enriched in the regions with lowest GM volumes (indicating more atrophy) in this subgroup, while in the other subgroups gene sets associated with metabolism of proteins were negatively enriched in the most atrophied regions. However, while the gene sets were related to overlapping biological functions (protein metabolism), the specific gene sets implicated in AD-Language were different form the ones implicated in the other subgroups. While, given the current state of research, it is hard to establish a causative role between higher ribosomal protein expression and AD-Language specific GM volumes (i.e., lower left temporal GM volumes than right), this finding does indicate that potential therapeutic approaches aimed at modulating expression of these proteins might not be beneficial to all AD patients and may even be detrimental in some cases (Caccamo et al., 2015). In AD-Visuospatial, we found a rather large, unique cluster of negatively enriched gene sets associated with gene expression modification. Gene sets within this cluster were mainly associated with epigenetic modifications (e.g., methylation, acetylation) and enrichment within this gene set cluster was mainly driven by the histone cluster protein (HIST*) gene family (Fig. 6), which is associated with packaging and ordering DNA into nucleosomes (Esposito and Sherr, 2019). Previous studies have shown that gene expression modification is implicated in AD through what is called an epigenetic blockade, referring to a large-scale decline in gene expression that is affected by post-translational histone modification (Gräff et al., 2012). Studies in mouse models have shown that this epigenetic blockade might be reversible (Gräff et al., 2012), opening up potential targets for therapeutic interventions. This epigenetic modification has previously been linked to an AD phenotype in animal models (Kosik et al., 2012), but there is no previous evidence that this points to a specific relation with visuospatial impairments and GM volume reduction in posterior brain regions. However, a recent genome-wide association study examining genetic loci associated with regional gray matter volumes (van der Lee et al., 2019) reported a distinct locus (rs12411216) specifically associated with occipital volumes. This locus is located in an intron of MIR92B and THBS3, showing a signal peak covering >20 genes with promotor histone marks overlapping the variant. This is an intriguing similarity to our findings and warrants further investigation.
Taken together, our results regarding variations in gene expression associated with distinct GM volume patterns across subgroups revealed several distinct clusters of biologically coherent gene sets, some of which showed unique associations with subgroup-specific GM volume patterns. Of note, several of the identified biological pathways have been previously implicated in AD through diverse lines of genetic and molecular research. We further add to these findings by showing that expression levels of genes associated with these pathways are spatially linked to region-specific brain GM volume patterns, and we provide several potential molecular targets for future investigations. By outlining that certain biological pathways are uniquely implicated in specific cognitively-defined subgroups, we also show that not all therapeutic targets might be equally beneficial for everybody with AD and highlight the need for examinations into personalized medicine strategies.
4.4. Strengths and limitations
Strengths of the study include a relatively large sample size from a single center with consistent assessments of AD biomarker positivity and well phenotyped individuals with dementia who had 3Tesla MRI available as well as information from multiple sources (i.e., clinical, imaging and genetics). Furthermore, in contrast to previous examinations that focused on categorization of individuals with AD based on structural properties (Ossenkoppele et al., 2019, Risacher et al., 2017, ten Kate et al., 2018, Zhang et al., 2016), structural properties in combination with cognition (Sun et al., 2019), neuropathological features (Murray et al., 2011, Whitwell et al., 2012), or clustering analyses (Scheltens et al., 2017, Stopford et al., 2008) and factor scoring (Sevush et al., 2003) of cognitive data, we used a classification scheme that relies on the intra-individual distribution of impairments across cognitive domains. This relatively simple method relies on patient-specific profiles of impairments across cognitive domains and can be performed on an individual basis, which is in contrast to approaches such as clustering analyses that rely on large sample sizes and sufficiently distributed data. Our study also has several limitations. First, the relative group sizes of the cognitively-defined subgroups presented in this work may not be representative of group sizes in other cohorts. The ADC is a tertiary memory clinic which specializes in, and is therefore enriched for, early onset and atypical AD clinical presentations (van der Flier et al., 2014). Indeed, 41.5% of our sample consisted of participants with early-onset AD (EOAD), while EOAD comprises only around 5–6% of all AD cases (Mendez, 2017). Since the classification of subgroups was based on normative scores from an late-onset AD (LOAD) population (the ACT cohort) and LOAD is generally characterized by less non-amnestic impairment compared to EOAD (Mendez, 2017), EOAD subjects might have had an increased chance of being classified into the single-domain subgroups other than AD-Memory. This might have contributed to our observation that there are more AD-Visuospatial EOAD subjects compared to LOAD (Table A6, 7 and 8). While this might limit the direct generalizability of our findings to other cohorts, and replication in more clinically representative cohorts is needed, the variety in clinical profiles in the ADC cohort allowed us to obtain sufficiently large subgroup sizes to obtain robust estimates of the differential regional susceptibilities to reductions in GM volumes and their related gene expression profiles. Furthermore, we have outlined results from stratified groups of early-onset (<65 years) and late-onset (>65 years) subjects in the supplement (Tables A6, 7 and 8; Fig. A5 and 6 and 8), and show that, although GM volume differences compared to controls are generally more pronounced in EOAD, the spatial pattern across subgroups (which determines the GM vs gene-expression associations) is very similar to what we see in LOAD (Fig. A5 and 6). Another limitation of the present study is that the Allen human brain database contains limited bi-hemispheric data (Hawrylycz et al., 2012), which prevented us from assessing possible biological drivers to lateralization of GM volumes. This may particularly be an issue for the AD-Language subgroup, as the GM volume pattern in this subgroup showed a marked hemispheric asymmetry. Our cross-sectional study design also comes with inherent limitations, which includes our inability to conceptually assess atrophy (i.e., gray matter volume loss over time) but rather GM volume differences at a specific timepoint (i.e., time of AD dementia diagnosis). Longitudinal assessments are needed in order to establish whether the spatial patterns of GM volume differences observed in the present study correspond to areas displaying faster rates of atrophy.
4.5. Clinical-radiological spectrum of Alzheimer’s disease
Elucidating associations between clinical and neurobiological heterogeneity in AD is crucial in understanding pathogenesis and meaningful stratification into distinct subgroups based on cognitive data might prove useful in future diagnostic and prognostic work-ups, and may even aid in developing future personalized medicine strategies. Although none of the participants in our AD-subgroups fulfilled diagnostic criteria for recognized atypical variants of AD, the subgroups were both clinically and radiologically distinct and the AD-Language and AD-Visuospatial groups were very reminiscent to lvPPA and PCA. Our results regarding distinct associations between regional GM volumes and gene expression also suggest that specific biological pathways may differentially affect the emergence of differences in AD-related neurodegeneration between subgroups.
While atypical presentations are increasingly recognized and included in diagnostic criteria, the vast majority of AD patients do not meet the rather strict clinical criteria for an atypical variant and are, therefore, by default regarded as typical AD. We would propose that future diagnostic criteria should also account for the considerable heterogeneity among these individuals and. in line with observations in previous work (Snowden et al., 2007, Stopford et al., 2008), we propose that phenotypic presentations of AD are more accurately arrayed along a clinical-radiological spectrum (Fig. 8). The hypothetical model depicted in Fig. 8 is a work in progress and more research is needed to map the clinical and neurobiological heterogeneity in AD in all its complexity into one model. For instance, it is necessary to properly identify and define a distinctive, selective amnestic variant of AD, if it exists. Findings concerning the clinical (slower progression (Mez et al., 2013b, Mez et al., 2013a)), neurodegenerative (medial temporal atrophy (Lam et al., 2013)), and genetic characteristics (APOEε4 prevalence (Crane et al., 2017, Mukherjee et al., 2020); partly unique GM volume-related gene expression profile) all indicate that there is a distinction between memory-predominant (amnestic) AD and the typical clinical presentation with heterogeneous impairments across domains that is most often observed in AD (such as seen in our AD-No Domains group). The degree to which this amnestic variant overlaps with the AD-Memory subgroup defined by our approach is currently unclear. Largely the same arguments hold true for the relationship between the AD-Executive subgroup of our study and the dysexecutive variant of AD (Dickerson and Wolk, 2011, Ossenkoppele et al., 2015), for which provisional research criteria were recently proposed (Townley et al., 2020). Furthermore, the framework we have developed to categorize subgroups emphasizes patterns of cognitive functioning at the time of AD diagnosis, and ignores behavioral and personality changes. We suspect there may be a behavioral variant where behavioral aspects are more prominent than expected for the overall level of clinical impairment. While previous research has tried to delineate such a behavioral variant of AD (Dubois et al., 2007, Ossenkoppele et al., 2015), this is difficult to study as behavior is rarely assessed as comprehensively as cognition in the research evaluation of people with newly diagnosed AD dementia. Furthermore, it has yet to be determined whether a possible behavioral variant can be distinguished from a dysexecutive variant (Ossenkoppele et al., 2015, Townley et al., 2020). We denote these uncertainties in our model, and highlight this as a potential area for future investigation.
Fig. 8.
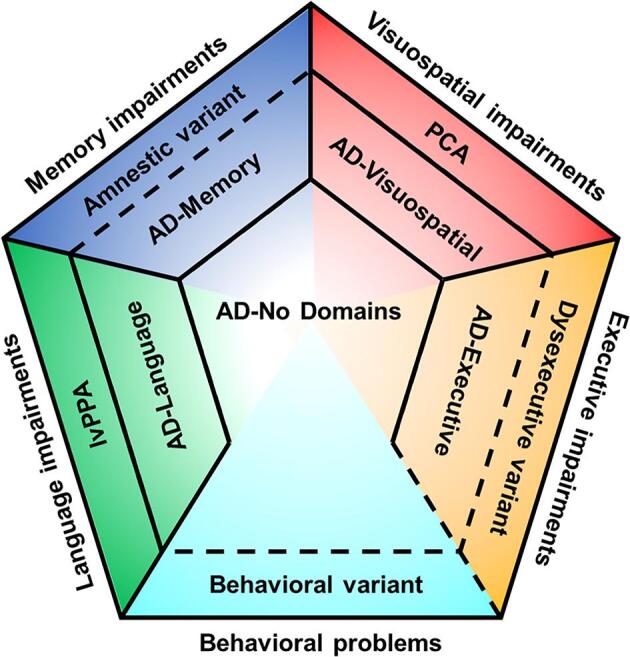
Hypothetical model of the Alzheimer’s disease clinical-neurobiological spectrum. Solid lines represent differences in categories that are either outlined in this manuscript or provided by established clinical criteria (i.e., for lvPPA and PCA). Dashed lines represent suspected differences in categories that are not yet established and are under investigation or need to be assessed in new lines of research. Note that relative sizes of the partitions are not representative for prevalence of the categories.
Future research will need to continue outlining the clinical and neurobiological disparities within the spectrum of AD (e.g., by mapping tau (Braak and Braak, 1991, Whitwell et al., 2008) and amyloid-β (Lehmann et al., 2013) pathology) and to examine factors that are involved in their emergence (e.g., pre-morbid learning disabilities (Miller et al., 2018, Miller et al., 2013) and structural properties of the pre-morbid brain (Batouli et al., 2014)). These efforts will advance the ongoing quest to answer fundamental questions about the mechanisms that are involved in the etiology of AD and the emergence of clinical and radiological heterogeneity among individuals with AD dementia.
5. Conclusions
We demonstrate that classifying individuals within the spectrum of typical AD based on cognitive profiles yields subgroups that show different rates of clinical progression, mortality rates and which show differential patterns of regional GM volumes. We also show that the cognitively-defined subgroups show similarities to established atypical variants of AD, suggesting that cognitive subgroups may be an intermediate category between individuals without a specific cognitive phenotype and the atypical variants of AD. Our gene-set enrichment analyses revealed that GM volume patterns of AD-subgroups are differentially associated to gene-expression profiles, which suggest that specific biological drivers might underlie clinical and neurobiological heterogeneity in AD. These findings may inform future investigations into possible targets for disease-modifying treatments against AD and one day aid in the development of personalized medicine strategies.
Funding
This work was supported by R01 AG 029672 (Paul K Crane, PI). Wiesje van der Flier is recipient of JPND-funded E-DADS (ZonMW project #733051106). Michel J Grothe is supported by the “Miguel Servet” program [CP19/00031] of the Spanish Instituto de Salud Carlos III (ISCIII-FEDER). Frederik Barkhof is supported by the NIHR biomedical research center at UCLH. Jesse Mez is supported by P30AG13846 and K23AG046377. Research of Alzheimer center Amsterdam is part of the neurodegeneration research program of Amsterdam Neuroscience. Alzheimer Center Amsterdam is supported by Stichting Alzheimer Nederland and Stichting VUmc fonds. Wiesje van der Flier holds the Pasman chair. The clinical database structure was developed with funding from Stichting Dioraphte. The sponsors had no role in the writing of the report; and in the decision to submit the article for publication.
CRediT authorship contribution statement
Colin Groot: Conceptualization, Formal analysis, Investigation, Writing - original draft, Visualization. Michel J. Grothe: Conceptualization, Methodology, Software, Formal analysis, Writing - review & editing. Shubhabrata Mukherjee: Methodology, Software, Formal analysis, Data curation, Writing - review & editing. Irina Jelistratova: Methodology, Software, Formal analysis, Writing - review & editing. Iris Jansen: Writing - review & editing. Anna Catharina van Loenhoud: Writing - review & editing. Shannon L. Risacher: Writing - review & editing. Andrew J. Saykin: Writing - review & editing. Christine L. Mac Donald: Writing - review & editing. Jesse Mez: Writing - review & editing. Emily H. Trittschuh: Writing - review & editing. Gregor Gryglewski: Methodology, Software, Formal analysis, Investigation, Resources, Data curation, Writing - review & editing. Rupert Lanzenberger: Methodology, Software, Formal analysis, Investigation, Resources, Data curation, Writing - review & editing. Yolande A.L. Pijnenburg: Writing - review & editing. Frederik Barkhof: Writing - review & editing. Philip Scheltens: Resources, Data curation, Writing - review & editing. Wiesje M. van der Flier: Resources, Data curation, Writing - original draft, Writing - review & editing, Supervision. Paul K. Crane: Conceptualization, Methodology, Resources, Writing - original draft, Writing - review & editing, Supervision, Funding acquisition. Rik Ossenkoppele: Conceptualization, Methodology, Resources, Writing - original draft, Writing - review & editing, Supervision, Funding acquisition.
Declaration of Competing Interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: Philip Scheltens has received consultancy/speaker fees (paid to the institution) from Biogen, People Bio, Roche (Diagnostics), Novartis Cardiology. He is PI of studies with Vivoryon, EIP Pharma, IONIS, CogRx, AC Immune and Toyama. Research programs of Wiesje van der Flier have been funded by ZonMW, NWO, EU-FP7, EU-JPND, Alzheimer Nederland, CardioVascular Onderzoek Nederland, Health ~ Holland, Topsector Life Sciences & Health, stichting Dioraphte, Gieskes-Strijbis fonds, stichting Equilibrio, Pasman stichting, Biogen MA Inc, Boehringer Ingelheim, Life-MI, AVID, Roche BV, Janssen Stellar, Combinostics. Wiesje van der Flier has performed contract research for Biogen MA Inc and Boehringer Ingelheim. Wiesje van der Flier has been an invited speaker at Boehringer Ingelheim and Biogen MA Inc. All funding is paid to her institution. Frederik Barkhof has been consulting for Biogen, Merck, Bayer, Novartis, Roche and IXICO. Rupert Lanzenberger received travel grants and/or conference speaker honoraria within the last three years from Bruker BioSpin MR, Heel, and support from Siemens Healthcare regarding clinical research using PET/MR. He is a shareholder of BM Health GmbH since 2019. The other authors report no disclosures.
Acknowledgements
The authors would like to thank Murray Reed (funded by Austrian Academy of Sciences, DOC 928), Matej Murgaš (funded by FWF Austrian Science Fund DOC 33-B27) and Gregory Miles James.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nicl.2021.102660.
Contributor Information
Colin Groot, Email: colin.groot@gmail.com.
Michel J. Grothe, Email: neurosev@gmail.com, michel.grothe@dzne.de.
Shubhabrata Mukherjee, Email: smukherj@uw.edu.
Irina Jelistratova, Email: irina.jelistratova@dzne.de.
Iris Jansen, Email: i.e.jansen@vu.nl.
Anna Catharina van Loenhoud, Email: a.vanloenhoud@amsterdamumc.nl.
Shannon L. Risacher, Email: srisache@iupui.edu.
Andrew J. Saykin, Email: asaykin@iupui.edu.
Christine L. Mac Donald, Email: cmacd@neurosurgery.washington.edu.
Jesse Mez, Email: jessemez@bu.edu.
Emily H. Trittschuh, Email: etritt@gmail.com.
Gregor Gryglewski, Email: gregor.gryglewski@meduniwien.ac.at.
Rupert Lanzenberger, Email: rupert.lanzenberger@meduniwien.ac.at.
Yolande A.L. Pijnenburg, Email: yal.pijnenburg@amsterdamumc.nl.
Frederik Barkhof, Email: f.barkhof@amsterdamumc.nl.
Philip Scheltens, Email: p.scheltens@amsterdamumc.nl.
Wiesje M. van der Flier, Email: wm.vdflier@amsterdamumc.nl.
Paul K. Crane, Email: pcrane@uw.edu.
Rik Ossenkoppele, Email: r.ossenkoppele@amsterdamumc.nl.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- Batouli S.A.H., Sachdev P.S., Wen W., Wright M.J., Ames D., Trollor J.N. Heritability of brain volumes in older adults: the Older Australian Twins Study. Neurobiol. Aging. 2014;35(4):937.e5–937.e18. doi: 10.1016/j.neurobiolaging.2013.10.079. [DOI] [PubMed] [Google Scholar]
- Bergeron D., Gorno‐Tempini M.L., Rabinovici G.D., Santos‐Santos M.A., Seeley W., Miller B.L., Pijnenburg Y., Keulen M.A., Groot C., van Berckel B.N.M., van der Flier W.M., Scheltens P., Rohrer J.D., Warren J.D., Schott J.M., Fox N.C., Sanchez‐Valle R., Grau‐Rivera O., Gelpi E., Seelaar H., Papma J.M., van Swieten J.C., Hodges J.R., Leyton C.E., Piguet O., Rogalski E.J., Mesulam M.M., Koric L., Nora K., Pariente J., Dickerson B., Mackenzie I.R., Hsiung G.-Y., Belliard S., Irwin D.J., Wolk D.A., Grossman M., Jones M., Harris J., Mann D., Snowden J.S., Chrem‐Mendez P., Calandri I.L., Amengual A.A., Miguet‐Alfonsi C., Magnin E., Magnani G., Santangelo R., Deramecourt V., Pasquier F., Mattsson N., Nilsson C., Hansson O., Keith J., Masellis M., Black S.E., Matías‐Guiu J.A., Cabrera‐Martin M.-N., Paquet C., Dumurgier J., Teichmann M., Sarazin M., Bottlaender M., Dubois B., Rowe C.C., Villemagne V.L., Vandenberghe R., Granadillo E., Teng E., Mendez M., Meyer P.T., Frings L., Lleó A., Blesa R., Fortea J., Seo S.W., Diehl‐Schmid J., Grimmer T., Frederiksen K.S., Sánchez‐Juan P., Chételat G., Jansen W., Bouchard R.W., Laforce R.J., Visser P.J., Ossenkoppele R. Prevalence of amyloid-β pathology in distinct variants of primary progressive aphasia. Ann. Neurol. 2018;84(5):729–740. doi: 10.1002/ana.v84.510.1002/ana.25333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H., Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82(4):239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Brett M., Anton J.-L., Valabregue R., Poline J.-B. Region of interest analysis using an SPM toolbox. NeuroImage. 2002 [Google Scholar]
- Caccamo A., Branca C., Talboom J.S., Shaw D.M., Turner D., Ma L., Messina A., Huang Z., Wu J., Oddo S. Reducing ribosomal protein S6 kinase 1 expression improves spatial memory and synaptic plasticity in a mouse model of Alzheimer’s disease. J. Neurosci. 2015;35(41):14042–14056. doi: 10.1523/JNEUROSCI.2781-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane P.K., Trittschuh E., Mukherjee S., Saykin A.J., Sanders R.E., Larson E.B., McCurry S.M., McCormick W., Bowen J.D., Grabowski T., Moore M., Bauman J., Gross A.L., Keene C.D., Bird T.D., Gibbons L.E., Mez J. Incidence of cognitively defined late-onset Alzheimer’s dementia subgroups from a prospective cohort study. Alzheimer’s Dement. 2017;13(12):1307–1316. doi: 10.1016/j.jalz.2017.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crutch S.J., Schott J.M., Rabinovici G.D., Murray M., Snowden J.S., van der Flier W.M., Dickerson B.C., Vandenberghe R., Ahmed S., Bak T.H., Boeve B.F., Butler C., Cappa S.F., Ceccaldi M., de Souza L.C., Dubois B., Felician O., Galasko D., Graff-Radford J., Graff-Radford N.R., Hof P.R., Krolak-Salmon P., Lehmann M., Magnin E., Mendez M.F., Nestor P.J., Onyike C.U., Pelak V.S., Pijnenburg Y., Primativo S., Rossor M.N., Ryan N.S., Scheltens P., Shakespeare T.J., Suárez González A., Tang-Wai D.F., Yong K.X.X., Carrillo M., Fox N.C. Alzheimer’s Association ISTAART Atypical Alzheimer’s Disease and Associated Syndromes Professional Interest Area. Consensus classification of posterior cortical atrophy. Alzheimers Dement. 2017 Aug;13(8):870-884. doi: 10.1016/j.jalz.2017.01.014. Epub 2017 Mar 2. PMID: 28259709; PMCID: PMC5788455. [DOI] [PMC free article] [PubMed]
- Dickerson B.C., Wolk D.A. Dysexecutive versus amnesic phenotypes of very mild Alzheimer’s disease are associated with distinct clinical, genetic and cortical thinning characteristics. J. Neurol. Neurosurg. Psychiatry. 2011;82(1):45–51. doi: 10.1136/jnnp.2009.199505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doody R.S., Massman P., Dunn J.K. A method for estimating progression rates in Alzheimer disease. Arch. Neurol. 2001;58:449–454. doi: 10.1001/archneur.58.3.449. [DOI] [PubMed] [Google Scholar]
- Doody R.S., Thomas R.G., Farlow M., Iwatsubo T., Vellas B., Joffe S., Kieburtz K., Raman R., Sun X., Aisen P.S., Siemers E., Liu-Seifert H., Mohs R. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2014;370(4):311–321. doi: 10.1056/NEJMoa1312889. [DOI] [PubMed] [Google Scholar]
- Dubois B., Feldman H.H., Jacova C., DeKosky S.T., Barberger-Gateau P., Cummings J., Delacourte A., Galasko D., Gauthier S., Jicha G., Meguro K., O'Brien J., Pasquier F., Robert P., Rossor M., Salloway S., Stern Y., Visser P.J., Scheltens P. Research criteria for the diagnosis of Alzheimer’s disease: revising the NINCDS-ADRDA criteria. Lancet Neurol. 2007;6(8):734–746. doi: 10.1016/S1474-4422(07)70178-3. [DOI] [PubMed] [Google Scholar]
- Esposito M., Sherr G.L. Epigenetic modifications in Alzheimer’s neuropathology and therapeutics. Front. Neurosci. 2019;13 doi: 10.3389/fnins.2019.00476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannery, P.J., Trushina, E., 2019. Mitochondrial dynamics and transport in Alzheimer’s disease. Mol. Cell. Neurosci. https://doi.org/10.1016/j.mcn.2019.06.009. [DOI] [PMC free article] [PubMed]
- Freer R., Sormanni P., Vecchi G., Ciryam P., Dobson C.M., Vendruscolo M. A protein homeostasis signature in healthy brains recapitulates tissue vulnerability to Alzheimer’s disease. Sci. Adv. 2016;2(8):e1600947. doi: 10.1126/sciadv.1600947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeze B., Pandya S., Zeighami Y., Raj A. Regional transcriptional architecture of Parkinson’s disease pathogenesis and network spread. Brain. 2019;142:3072–3085. doi: 10.1093/brain/awz223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gjoneska E., Pfenning A.R., Mathys H., Quon G., Kundaje A., Tsai L.-H., Kellis M. Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer’s disease. Nature. 2015;518(7539):365–369. doi: 10.1038/nature14252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves J.T., Schafer S.T., Gage F.H. Adult neurogenesis in the hippocampus: from stem cells to behavior. Cell. 2016;167(4):897–914. doi: 10.1016/j.cell.2016.10.021. [DOI] [PubMed] [Google Scholar]
- Gorno-Tempini M.L., Brambati S.M., Ginex V., Ogar J., Dronkers N.F., Marcone A., Perani D., Garibotto V., Cappa S.F., Miller B.L. The logopenic/phonological variant of primary progressive aphasia. Neurology. 2008;71(16):1227–1234. doi: 10.1212/01.wnl.0000320506.79811.da. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorno-Tempini M.L., Hillis A.E., Weintraub S., Kertesz A., Mendez M., Cappa S.F., Ogar J.M., Rohrer J.D., Black S., Boeve B.F., Manes F., Dronkers N.F., Vandenberghe R., Rascovsky K., Patterson K., Miller B.L., Knopman D.S., Hodges J.R., Mesulam M.M., Grossman M. Classification of primary progressive aphasia and its variants. Neurology. 2011;76(11):1006–1014. doi: 10.1212/WNL.0b013e31821103e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gräff J., Rei D., Guan J.-S., Wang W.-Y., Seo J., Hennig K.M., Nieland T.J.F., Fass D.M., Kao P.F., Kahn M., Su S.C., Samiei A., Joseph N., Haggarty S.J., Delalle I., Tsai L.-H. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature. 2012;483(7388):222–226. doi: 10.1038/nature10849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groot C., van Loenhoud A.C., Barkhof F., van Berckel B.N.M., Koene T., Teunissen C.C., Scheltens P., van der Flier W.M., Ossenkoppele R. Differential effects of cognitive reserve and brain reserve on cognition in Alzheimer disease. Neurology. 2018;90(2):e149–e156. doi: 10.1212/WNL.0000000000004802. [DOI] [PubMed] [Google Scholar]
- Groot C., Yeo B.T.T., Vogel J.W., Zhang X., Sun N., Mormino E.C., Pijnenburg Y.A.L., Miller B.L., Rosen H.J., La Joie R., Barkhof F., Scheltens P., van der Flier W.M., Rabinovici G.D., Ossenkoppele R. Latent atrophy factors related to phenotypical variants of posterior cortical atrophy. Neurology. 2020;95(12):e1672–e1685. doi: 10.1212/WNL.0000000000010362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grothe M.J., Sepulcre J., Gonzalez-Escamilla G., Jelistratova I., Schöll M., Hansson O., Teipel S.J. Molecular properties underlying regional vulnerability to Alzheimer’s disease pathology. Brain. 2018;141:2755–2771. doi: 10.1093/brain/awy189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gryglewski G., Seiger R., James G.M., Godbersen G.M., Komorowski A., Unterholzner J., Michenthaler P., Hahn A., Wadsak W., Mitterhauser M., Kasper S., Lanzenberger R. Spatial analysis and high resolution mapping of the human whole-brain transcriptome for integrative analysis in neuroimaging. Neuroimage. 2018;176:259–267. doi: 10.1016/j.neuroimage.2018.04.068. [DOI] [PubMed] [Google Scholar]
- Hawrylycz M., Miller J.A., Menon V., Feng D., Dolbeare T., Guillozet-Bongaarts A.L., Jegga A.G., Aronow B.J., Lee C.-K., Bernard A., Glasser M.F., Dierker D.L., Menche J., Szafer A., Collman F., Grange P., Berman K.A., Mihalas S., Yao Z., Stewart L., Barabási A.-L., Schulkin J., Phillips J., Ng L., Dang C., Haynor D.R., Jones A., Van Essen D.C., Koch C., Lein E.d. Canonical genetic signatures of the adult human brain. Nat. Neurosci. 2015;18(12):1832–1844. doi: 10.1038/nn.4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawrylycz M.J., Lein E.S., Guillozet-Bongaarts A.L., Shen E.H., Ng L., Miller J.A., van de Lagemaat L.N., Smith K.A., Ebbert A., Riley Z.L., Abajian C., Beckmann C.F., Bernard A., Bertagnolli D., Boe A.F., Cartagena P.M., Chakravarty M.M., Chapin M., Chong J., Dalley R.A., Daly B.D., Dang C., Datta S., Dee N., Dolbeare T.A., Faber V., Feng D., Fowler D.R., Goldy J., Gregor B.W., Haradon Z., Haynor D.R., Hohmann J.G., Horvath S., Howard R.E., Jeromin A., Jochim J.M., Kinnunen M., Lau C., Lazarz E.T., Lee C., Lemon T.A., Li L., Li Y., Morris J.A., Overly C.C., Parker P.D., Parry S.E., Reding M., Royall J.J., Schulkin J., Sequeira P.A., Slaughterbeck C.R., Smith S.C., Sodt A.J., Sunkin S.M., Swanson B.E., Vawter M.P., Williams D., Wohnoutka P., Zielke H.R., Geschwind D.H., Hof P.R., Smith S.M., Koch C., Grant S.G.N., Jones A.R. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature. 2012;489(7416):391–399. doi: 10.1038/nature11405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillary, F.G., Grafman, J.H., 2017. Injured Brains and Adaptive Networks: The Benefits and Costs of Hyperconnectivity. Trends Cogn. Sci. https://doi.org/10.1016/j.tics.2017.03.003. [DOI] [PMC free article] [PubMed]
- Kosik K.S., Rapp P.R., Raz N., Small S.A., Sweatt J.D., Tsai L.-H. Mechanisms of age-related cognitive change and targets for intervention: epigenetics. J. Gerontol. - Ser. A Biol. Sci Med. Sci. 2012;67(7):741–746. doi: 10.1093/gerona/gls110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krienen F.M., Yeo B.T., Ge T., Buckner R.L., Sherwood C.C. Transcriptional profiles of supragranular-enriched genes associate with corticocortical network architecture in the human brain. Proc Natl Acad Sci U S A. 2016;113(4):E469–78. doi: 10.1073/pnas.1510903113. Epub 2016 Jan 6. PMID: 26739559; PMCID: PMC4739529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam B., Masellis M., Freedman M., Stuss D.T., Black S.E. Clinical, imaging, and pathological heterogeneity of the Alzheimer’s disease syndrome. Alzheimer’s Res. Ther. 2013;5(1):1. doi: 10.1186/alzrt155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langstrom N.S., Anderson J.P., Lindroos H.G., Winbland B., Wallace W.C. Alzheimer’s disease-associated reduction of polysomal mRNA translation. Brain Res. Mol. Brain Res. 1989;5(4):259–269. doi: 10.1016/0169-328X(89)90060-0. [DOI] [PubMed] [Google Scholar]
- Lehmann M., Ghosh P.M., Madison C., Laforce R., Corbetta-Rastelli C., Weiner M.W., Greicius M.D., Seeley W.W., Gorno-Tempini M.L., Rosen H.J., Miller B.L., Jagust W.J., Rabinovici G.D. Diverging patterns of amyloid deposition and hypometabolism in clinical variants of probable Alzheimer’s disease. Brain. 2013;136(3):844–858. doi: 10.1093/brain/aws327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattsson N., Groot C., Jansen W.J., Landau S.M., Villemagne V.L., Engelborghs S., Mintun M.M., Lleo A., Molinuevo J.L., Jagust W.J., Frisoni G.B., Ivanoiu A., Chételat G., Resende de Oliveira C., Rodrigue K.M., Kornhuber J., Wallin A., Klimkowicz-Mrowiec A., Kandimalla R., Popp J., Aalten P.P., Aarsland D., Alcolea D., Almdahl I.S., Baldeiras I., van Buchem M.A., Cavedo E., Chen K., Cohen A.D., Förster S., Fortea J., Frederiksen K.S., Freund-Levi Y., Gill K.D., Gkatzima O., Grimmer T., Hampel H., Herukka S.-K., Johannsen P., van Laere K., de Leon M.J., Maier W., Marcusson J., Meulenbroek O., Møllergård H.M., Morris J.C., Mroczko B., Nordlund A., Prabhakar S., Peters O., Rami L., Rodríguez-Rodríguez E., Roe C.M., Rüther E., Santana I., Schröder J., Seo S.W., Soininen H., Spiru L., Stomrud E., Struyfs H., Teunissen C.E., Verhey F.R.J., Vos S.J.B., van Waalwijk van Doorn L.J.C., Waldemar G., Wallin Å.K., Wiltfang J., Vandenberghe R., Brooks D.J., Fladby T., Rowe C.C., Drzezga A., Verbeek M.M., Sarazin M., Wolk D.A., Fleisher A.S., Klunk W.E., Na D.L., Sánchez-Juan P., Lee D.Y., Nordberg A., Tsolaki M., Camus V., Rinne J.O., Fagan A.M., Zetterberg H., Blennow K., Rabinovici G.D., Hansson O., van Berckel B.N.M., van der Flier W.M., Scheltens P., Visser P.J., Ossenkoppele R. Prevalence of the apolipoprotein E ε4 allele in amyloid β positive subjects across the spectrum of Alzheimer’s disease. Alzheimer’s Dement. 2018;14(7):913–924. doi: 10.1016/j.jalz.2018.02.009. [DOI] [PubMed] [Google Scholar]
- McKhann G.M., Knopman D.S., Chertkow H., Hyman B.T., Jack C.R., Kawas C.H., Klunk W.E., Koroshetz W.J., Manly J.J., Mayeux R., Mohs R.C., Morris J.C., Rossor M.N., Scheltens P., Carrillo M.C., Thies B., Weintraub S., Phelps C.H. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011;7(3):263–269. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez M.F. Early-onset Alzheimer disease. Neurol. Clin. 2017;35(2):263–281. doi: 10.1016/j.ncl.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merico D., Isserlin R., Stueker O., Emili A., Bader G.D. Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS One. 2010;5(11) doi: 10.1371/journal.pone.0013984. PMID: 21085593; PMCID: PMC2981572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mez J., Cosentino S., Brickman A.M., Huey E.D., Manly J.J., Mayeux R., Brucki S. Faster cognitive and functional decline in dysexecutive versus amnestic Alzheimer’s subgroups: a longitudinal analysis of the National Alzheimer’s Coordinating Center (NACC) database. PLoS One. 2013;8(6):e65246. doi: 10.1371/journal.pone.0065246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mez J., Cosentino S., Brickman A.M., Huey E.D., Mayeux R. Different demographic, genetic, and longitudinal traits in language versus memory Alzheimer’s subgroups. J. Alzheimer’s Dis. 2013;37(1):137–146. doi: 10.3233/JAD-130320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller Z.A., Mandelli M.L., Rankin K.P., Henry M.L., Babiak M.C., Frazier D.T., Lobach I.V., Bettcher B.M., Wu T.Q., Rabinovici G.D., Graff-Radford N.R., Miller B.L., Gorno-Tempini M.L. Handedness and language learning disability differentially distribute in progressive aphasia variants. Brain. 2013;136:3461–3473. doi: 10.1093/brain/awt242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller Z.A., Rosenberg L., Santos-Santos M.A., Stephens M., Allen I.E., Hubbard H.I., Cantwell A., Mandelli M.L., Grinberg L.T., Seeley W.W., Miller B.L., Rabinovici G.D., Gorno-Tempini M.L. Prevalence of mathematical and visuospatial learning disabilities in patients with posterior cortical atrophy. JAMA Neurol. 2018;75(6):728. doi: 10.1001/jamaneurol.2018.0395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moh C., Kubiak J.Z., Bajic V.P., Zhu X., Smith M.A., Lee H.G. Cell cycle deregulation in the neurons of Alzheimer’s disease. Results Probl. Cell Differ. 2011;53:565–576. doi: 10.1007/978-3-642-19065-0_23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S., Mez J., Trittschuh E.H., Saykin A.J., Gibbons L.E., Fardo D.W., Wessels M., Bauman J., Moore M., Choi S.-E., Gross A.L., Rich J., Louden D.K.N., Sanders R.E., Grabowski T.J., Bird T.D., McCurry S.M., Snitz B.E., Kamboh M.I., Lopez O.L., De Jager P.L., Bennett D.A., Keene C.D., Larson E.B., Crane P.K. Genetic data and cognitively defined late-onset Alzheimer’s disease subgroups. Mol. Psychiatry. 2020;25(11):2942–2951. doi: 10.1038/s41380-018-0298-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray M.E., Graff-Radford N.R., Ross O.A., Petersen R.C., Duara R., Dickson D.W. Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: a retrospective study. Lancet Neurol. 2011;10(9):785–796. doi: 10.1016/S1474-4422(11)70156-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthén, L.K., Muthén, B.O., 1998. Statistical Analysis With Latent Variables User’s Guide.
- Ossenkoppele, R., Lyoo, C.H., Sudre, C.H., van Westen, D., Cho, H., Ryu, Y.H., Choi, J.Y., Smith, R., Strandberg, O., Palmqvist, S., Westman, E., Tsai, R., Kramer, J., Boxer, A.L., Gorno-Tempini, M.L., La Joie, R., Miller, B.L., Rabinovici, G.D., Hansson, O., 2019. Distinct tau PET patterns in atrophy-defined subtypes of Alzheimer’s disease. Alzheimers. Dement. https://doi.org/10.1016/j.jalz.2019.08.201. [DOI] [PMC free article] [PubMed]
- Ossenkoppele R., Mattsson N., Teunissen C.E., Barkhof F., Pijnenburg Y., Scheltens P., van der Flier W.M., Rabinovici G.D. Cerebrospinal fluid biomarkers and cerebral atrophy in distinct clinical variants of probable Alzheimer’s disease. Neurobiol. Aging. 2015;36(8):2340–2347. doi: 10.1016/j.neurobiolaging.2015.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossenkoppele R., Pijnenburg Y.A.L., Perry D.C., Cohn-Sheehy B.I., Scheltens N.M.E., Vogel J.W., Kramer J.H., van der Vlies A.E., Joie R.L., Rosen H.J., van der Flier W.M., Grinberg L.T., Rozemuller A.J., Huang E.J., van Berckel B.N.M., Miller B.L., Barkhof F., Jagust W.J., Scheltens P., Seeley W.W., Rabinovici G.D. The behavioural/dysexecutive variant of Alzheimer’s disease: clinical, neuroimaging and pathological features. Brain. 2015;138(9):2732–2749. doi: 10.1093/brain/awv191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pebesma E.J. Multivariable geostatistics in S: the gstat package. Comput. Geosci. 2004;30(7):683–691. doi: 10.1016/j.cageo.2004.03.012. [DOI] [Google Scholar]
- Richiardi J., Altmann A., Milazzo A.-C., Chang C., Chakravarty M.M., Banaschewski T., Barker G.J., Bokde A.L.W., Bromberg U., Buchel C., Conrod P., Fauth-Buhler M., Flor H., Frouin V., Gallinat J., Garavan H., Gowland P., Heinz A., Lemaitre H., Mann K.F., Martinot J.-L., Nees F., Paus T., Pausova Z., Rietschel M., Robbins T.W., Smolka M.N., Spanagel R., Strohle A., Schumann G., Hawrylycz M., Poline J.-B., Greicius M.D., Albrecht L., Andrew C., Arroyo M., Artiges E., Aydin S., Bach C., Banaschewski T., Barbot A., Barker G., Boddaert N., Bokde A., Bricaud Z., Bromberg U., Bruehl R., Buchel C., Cachia A., Cattrell A., Conrod P., Constant P., Dalley J., Decideur B., Desrivieres S., Fadai T., Flor H., Frouin V., Gallinat J., Garavan H., Briand F.G., Gowland P., Heinrichs B., Heinz A., Heym N., Hubner T., Ireland J., Ittermann B., Jia T., Lathrop M., Lanzerath D., Lawrence C., Lemaitre H., Ludemann K., Macare C., Mallik C., Mangin J.-F., Mann K., Martinot J.-L., Mennigen E., Mesquita de Carvahlo F., Mignon X., Miranda R., Muller K., Nees F., Nymberg C., Paillere M.-L., Paus T., Pausova Z., Poline J.-B., Poustka L., Rapp M., Robert G., Reuter J., Rietschel M., Ripke S., Robbins T., Rodehacke S., Rogers J., Romanowski A., Ruggeri B., Schmal C., Schmidt D., Schneider S., Schumann M., Schubert F., Schwartz Y., Smolka M., Sommer W., Spanagel R., Speiser C., Spranger T., Stedman A., Steiner S., Stephens D., Strache N., Strohle A., Struve M., Subramaniam N., Topper L., Whelan R., Williams S., Yacubian J., Zilbovicius M., Wong C.P., Lubbe S., Martinez-Medina L., Fernandes A., Tahmasebi A. Correlated gene expression supports synchronous activity in brain networks. Science. 2015;348(6240):1241–1244. doi: 10.1126/science:1255905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risacher S.L., Anderson W.H., Charil A., Castelluccio P.F., Shcherbinin S., Saykin A.J., Schwarz A.J. Alzheimer disease brain atrophy subtypes are associated with cognition and rate of decline. Neurology. 2017;89(21):2176–2186. doi: 10.1212/WNL.0000000000004670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salloway S., Sperling R., Fox N.C., Blennow K., Klunk W., Raskind M., Sabbagh M., Honig L.S., Porsteinsson A.P., Ferris S., Reichert M., Ketter N., Nejadnik B., Guenzler V., Miloslavsky M., Wang D., Lu Y., Lull J., Tudor I.C., Liu E., Grundman M., Yuen E., Black R., Brashear H.R. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2014;370(4):322–333. doi: 10.1056/NEJMoa1304839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheltens N.M.E., Tijms B.M., Koene T., Barkhof F., Teunissen C.E., Wolfsgruber S., Wagner M., Kornhuber J., Peters O., Cohn-Sheehy B.I., Rabinovici G.D., Miller B.L., Kramer J.H., Scheltens P., van der Flier W.M. Cognitive subtypes of probable Alzheimer’s disease robustly identified in four cohorts. Alzheimer’s Dement. 2017;13:1226–1236. doi: 10.1016/j.jalz.2017.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schott J.M., Lehmann M., Primativo S., Rossor M.N., Ryan N.S., Shakespeare T.J., Suárez González A., Yong K.X.X., Fox N.C., Rabinovici G.D., Lehmann M., Murray M., Snowden J.S., Snowden J.S., van der Flier W.M., Pijnenburg Y., Scheltens P., van der Flier W.M., Pijnenburg Y., Scheltens P., Dickerson B.C., Vandenberghe R., Ahmed S., Butler C., Bak T.H., Boeve B.F., Graff-Radford J., Cappa S.F., Ceccaldi M., de Souza L.C., Dubois B., Felician O., Felician O., Galasko D., Graff-Radford N.R., Hof P.R., Hof P.R., Krolak-Salmon P., Magnin E., Mendez M.F., Nestor P.J., Onyike C.U., Pelak V.S., Pelak V.S., Suárez González A., Tang-Wai D.F., Carrillo M. Consensus classification of posterior cortical atrophy. Alzheimer’s Dement. 2017;13:870–884. doi: 10.1016/j.jalz.2017.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepulcre J., Grothe M.J., d’Oleire Uquillas F., Ortiz-Terán L., Diez I., Yang H.-S., Jacobs H.I.L., Hanseeuw B.J., Li Q., El-Fakhri G., Sperling R.A., Johnson K.A. Neurogenetic contributions to amyloid beta and tau spreading in the human cortex. Nat. Med. 2018;24(12):1910–1918. doi: 10.1038/s41591-018-0206-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevush S., Peruyera G., Bertran A., Cisneros W. A three-factor model of cognition in Alzheimer disease. Cogn. Behav. Neurol. 2003;16(2):110–117. doi: 10.1097/00146965-200306000-00004. [DOI] [PubMed] [Google Scholar]
- Snowden J.S., Stopford C.L., Julien C.L., Thompson J.C., Davidson Y., Gibbons L., Pritchard A., Lendon C.L., Richardson A.M., Varma A., Neary D., Mann D.M.A. Cognitive phenotypes in Alzheimer’s disease and genetic risk. Cortex. 2007;43(7):835–845. doi: 10.1016/s0010-9452(08)70683-x. [DOI] [PubMed] [Google Scholar]
- Stopford C.L., Snowden J.S., Thompson J.C., Neary D. Variability in cognitive presentation of Alzheimer’s disease. Cortex. 2008;44(2):185–195. doi: 10.1016/j.cortex.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun N., Mormino E.C., Chen J., Sabuncu M.R., Yeo B.T.T. Multi-modal latent factor exploration of atrophy, cognitive and tau heterogeneity in Alzheimer’s disease. Neuroimage. 2019;201:116043. doi: 10.1016/j.neuroimage.2019.116043. [DOI] [PubMed] [Google Scholar]
- Swerdlow, R.H., Burns, J.M., Khan, S.M., 2014. The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim. Biophys. Acta - Mol. Basis Dis. https://doi.org/10.1016/j.bbadis.2013.09.010. [DOI] [PMC free article] [PubMed]
- Sylvester, J.E., Fischel-Ghodsian, N., Mougey, E.B., O’Brien, T.W., 2004. Mitochondrial ribosomal proteins: candidate genes for mitochondrial disease. Genet. Med. https://doi.org/10.1097/01.GIM.0000117333.21213.17. [DOI] [PubMed]
- Ten Kate, M., Dicks, E., Visser, P.J., Van Der Flier, W.M., Teunissen, C.E., Barkhof, F., Scheltens, P., Tijms, B.M., 2018. Atrophy subtypes in prodromal Alzheimer’s disease are associated with cognitive decline. Brain 141, 3443–3456. https://doi.org/10.1093/brain/awy264. [DOI] [PMC free article] [PubMed]
- Tijms B.M., Willemse E.A.J., Zwan M.D., Mulder S.D., Visser P.J., van Berckel B.N.M., van der Flier W.M., Scheltens P., Teunissen C.E. Unbiased approach to counteract upward drift in cerebrospinal fluid amyloid-β 1–42 analysis results. Clin. Chem. 2018;64:576–585. doi: 10.1373/clinchem.2017.281055. [DOI] [PubMed] [Google Scholar]
- Townley, R.A., Graff-Radford, J., Mantyh, W.G., Botha, H., Polsinelli, A.J., Przybelski, S.A., Machulda, M.M., Makhlouf, A.T., Senjem, M.L., Murray, M.E., Reichard, R.R., Savica, R., Boeve, B.F., Drubach, D.A., Josephs, K.A., Knopman, D.S., Lowe, V.J., Jack, C.R., Petersen, R.C., Jones, D.T., 2020. Progressive dysexecutive syndrome due to Alzheimer’s disease: a description of 55 cases and comparison to other phenotypes. Brain Commun. 2. [DOI] [PMC free article] [PubMed]
- van der Flier W.M., Pijnenburg Y.A.L., Fox N.C., Scheltens P. Early-onset versus late-onset Alzheimer’s disease: the case of the missing APOE ε4 allele. Lancet Neurol. 2011 doi: 10.1016/S1474-4422(10)70306-9. [DOI] [PubMed] [Google Scholar]
- van der Flier W.M., Pijnenburg Y.A.L., Prins N., Lemstra A.W., Bouwman F.H., Teunissen C.E., van Berckel B.N.M., Stam C.J., Barkhof F., Visser P.J., van Egmond E., Scheltens P. Optimizing patient care and research: the Amsterdam dementia cohort. J. Alzheimer’s Dis. 2014;41(1):313–327. doi: 10.3233/JAD-132306. [DOI] [PubMed] [Google Scholar]
- van der Flier W.M., Scheltens P., Perry G., Avila J., Tabaton M., Zhu X. Amsterdam dementia cohort: performing research to optimize care. J. Alzheimers. Dis. 2018;62(3):1091–1111. doi: 10.3233/JAD-170850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Lee S.J., Knol M.J., Chauhan G., Satizabal C.L., Smith A.V., Hofer E., Bis J.C., Hibar D.P., Hilal S., van den Akker E.B., Arfanakis K., Bernard M., Yanek L.R., Amin N., Crivello F., Cheung J.W., Harris T.B., Saba Y., Lopez O.L., Li S., van der Grond J., Yu L., Paus T., Roshchupkin G.V., Amouyel P., Jahanshad N., Taylor K.D., Yang Q., Mathias R.A., Boehringer S., Mazoyer B., Rice K., Cheng C.Y., Maillard P., van Heemst D., Wong T.Y., Niessen W.J., Beiser A.S., Beekman M., Zhao W., Nyquist P.A., Chen C., Launer L.J., Psaty B.M., Ikram M.K., Vernooij M.W., Schmidt H., Pausova Z., Becker D.M., De Jager P.L., Thompson P.M., van Duijn C.M., Bennett D.A., Slagboom P.E., Schmidt R., Longstreth W.T., Ikram M.A., Seshadri S., Debette S., Gudnason V., Adams H.H.H., DeCarli C. A genome-wide association study identifies genetic loci associated with specific lobar brain volumes. Commun. Biol. 2019;2(1) doi: 10.1038/s42003-019-0537-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walhovd K.B., Westerhausen R., de Lange A.M.G., Bråthen A.C.S., Grydeland H., Engvig A., Fjell A.M. Premises of plasticity - and the loneliness of the medial temporal lobe. Neuroimage. 2016;131:48–54. doi: 10.1016/j.neuroimage.2015.10.060. [DOI] [PubMed] [Google Scholar]
- Whitaker K.J., Vértes P.E., Romero-Garcia R., Váša F., Moutoussis M., Prabhu G., Weiskopf N., Callaghan M.F., Wagstyl K., Rittman T., Tait R., Ooi C., Suckling J., Inkster B., Fonagy P., Dolan R.J., Jones P.B., Goodyer I.M., Bullmore E.T. Adolescence is associated with genomically patterned consolidation of the hubs of the human brain connectome. Proc. Natl. Acad. Sci. U. S. A. 2016;113(32):9105–9110. doi: 10.1073/pnas.1601745113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell J.L., Dickson D.W., Murray M.E., Weigand S.D., Tosakulwong N., Senjem M.L., Knopman D.S., Boeve B.F., Parisi J.E., Petersen R.C., Jack C.R., Josephs K.A. Neuroimaging correlates of pathologically defined subtypes of Alzheimer’s disease: a case-control study. Lancet Neurol. 2012;11(10):868–877. doi: 10.1016/S1474-4422(12)70200-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell J.L., Josephs K.A., Murray M.E., Kantarci K., Przybelski S.A., Weigand S.D., Vemuri P., Senjem M.L., Parisi J.E., Knopman D.S., Boeve B.F., Petersen R.C., Dickson D.W., Jack C.R. MRI correlates of neurofibrillary tangle pathology at autopsy: a voxel-based morphometry study. Neurology. 2008;71(10):743–749. doi: 10.1212/01.wnl.0000324924.91351.7d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J., Reiman E.M., Beach T.G., Serrano G.E., Sabbagh M.N., Nielsen M., Caselli R.J., Shi J. Effect of ApoE isoforms on mitochondria in Alzheimer disease. Neurology. 2020;94(23):e2404–e2411. doi: 10.1212/WNL.0000000000009582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Mormino E.C., Sun N., Sperling R.A., Sabuncu M.R., Yeo B.T.T. Bayesian model reveals latent atrophy factors with dissociable cognitive trajectories in Alzheimer’s disease. Proc. Natl. Acad. Sci. 2016;113(42):E6535–E6544. doi: 10.1073/pnas.1611073113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.