

Summary
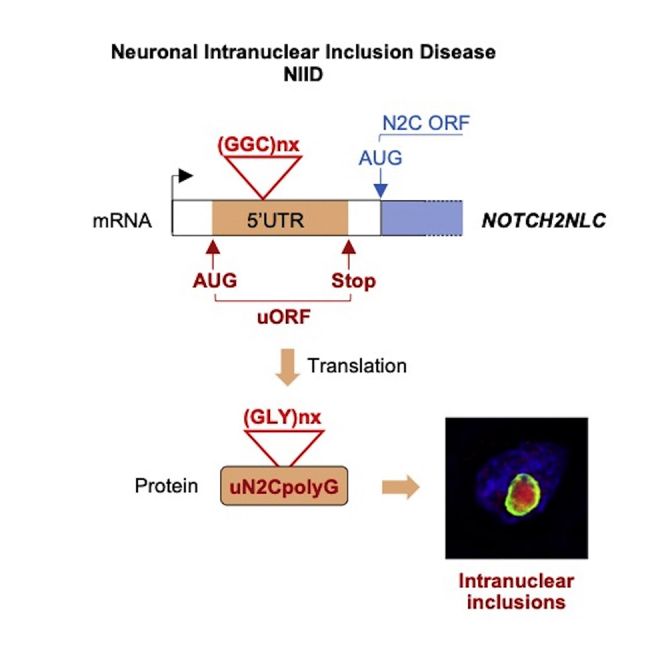
Neuronal intranuclear inclusion disease (NIID) is a neurodegenerative disease characterized by the presence of intranuclear inclusions of unknown origin. NIID is caused by an expansion of GGC repeats in the 5′ UTR of the NOTCH2NLC (N2C) gene. We found that these repeats are embedded in a small upstream open reading frame (uORF) (uN2C), resulting in their translation into a polyglycine-containing protein, uN2CpolyG. This protein accumulates in intranuclear inclusions in cell and mouse models and in tissue samples of individuals with NIID. Furthermore, expression of uN2CpolyG in mice leads to locomotor alterations, neuronal cell loss, and premature death of the animals. These results suggest that translation of expanded GGC repeats into a novel and pathogenic polyglycine-containing protein underlies the presence of intranuclear inclusions and neurodegeneration in NIID.
Keywords: trinucleotide repeat disorder, polyglycine, polyG, RAN translation, genetic diseases, neurodegeneration
Graphical abstract
Highlights
-
•
NIID is a neurodegenerative disease caused by expansion of GGC repeats in NOTCH2NLC
-
•
These GGC repeats are translated into a polyglycine (polyG) protein
-
•
The polyG protein is toxic and forms intranuclear inclusions in cells and animals
-
•
Similarities between FXTAS and NIID define a new set of disorders: polyG diseases
The neurodegenerative disease NIID is caused by an expansion of GGC repeats in NOTCH2NLC. Boivin et al. found that these repeats are translated into a toxic polyglycine (polyG) protein that forms intranuclear inclusions. An identical mechanism exists in FXTAS, unveiling a novel group of genetic pathologies, the polyG diseases.
Introduction
More than 40 inherited human diseases are caused by expansion of microsatellites, short DNA tandem repeats variable in size and sequences. These microsatellite expansions are pathogenic by three main non-exclusive mechanisms (Nelson et al., 2013; Gao and Richter, 2017; Rodriguez and Todd, 2019). First, they can promote DNA epigenetic changes that inhibit transcription, resulting in loss of function of the allele carrying the repeats. Second, RNA transcripts containing the expanded repeats can bind to specific RNA binding proteins, potentially altering their localization and function. Third, expanded repeats can be translated by canonical initiation to AUG or near-cognate start codons or by translation initiation directly within the repeat through a novel mechanism called repeat-associated non-AUG (RAN) translation (Zu et al., 2011), into proteins containing a pathogenic stretch of repeated amino acids. Progress in long-read and whole-genome sequencing has recently unveiled a dozen novel microsatellite expansions located in the “non-coding” part of the human genome as pathogenic causes, notably an intronic AG-rich repeat expansion in the RFC1 gene in cerebellar ataxia with neuropathy and bilateral vestibular areflexia syndrome (CANVAS) (Cortese et al., 2019; Rafehi et al., 2019); 6 similar intronic AT-rich repeat expansions in benign adult familial myoclonic epilepsy (BAFME) 1–6 and 5 similar 5′ UTR-embedded GC-rich repeat expansions in fragile X-associated tremor/ataxia syndrome (FXTAS), neuronal intranuclear inclusion disease (NIID), oculopharyngodistal myopathy (OPDM), and oculopharyngeal myopathy with leukoencephalopathy (OPML), neuromuscular and neurodegenerative syndromes with some overlapping symptoms and similar histopathological features (Hagerman et al., 2001; Ishiura et al., 2019; Sone et al., 2019; Deng et al., 2019, 2020; Tian et al., 2019; Xi et al., 2021; review in Ishiura and Tsuji, 2020).
Among these later disorders, NIID, also known as neuronal intranuclear hyaline inclusion disease (NIHID) and intranuclear inclusion body disease (INIBD), is a rare genetic disease characterized by the presence of intranuclear inclusions in the central and peripheral nervous systems and in multiple other organs (Lindenberg et al., 1968; Munoz-Garcia and Ludwin, 1986; Sone et al., 2016). NIID age of onset is variable, and three subgroups (infant, juvenile, and adult) have been defined (Takahashi-Fujigasaki, 2003). Clinically, NIID is also tentatively divided in three subgroups: dementia dominant, parkinsonism dominant, and muscle weakness dominant. However, NIID symptoms are highly heterogenous, and overlap between subgroups is observed frequently, with variable muscle weakness associated with various dysfunctions of the central and peripheral nervous systems, which can include progressive dementia and cognitive impairment, parkinsonism, cerebellar ataxia, sensory disturbance, autonomic dysfunction, and/or peripheral neuropathy (Takahashi-Fujigasaki, 2003; Sone et al., 2016; Takahashi-Fujigasaki et al., 2016). Moreover, individuals with NIID with atypical presentation, such as essential tremors, multiple-systems atrophy, and amyotrophic lateral sclerosis, and various acute symptoms, including stroke-like episodes, epileptic seizures, and/or encephalitic episodes, have also been reported (Sone et al., 2016; Fang et al., 2020; Li et al., 2020; Sun et al., 2020; Yuan et al., 2020). As a consequence of these diverse ages of onset and clinical presentations, NIID diagnosis is most often confirmed by the widespread presence of characteristic eosinophilic intranuclear inclusions in neurons and glial cells in the central and peripheral nervous systems and in various other tissues (Chen et al., 2020a, Liu et al., 2008, Sone et al., 2005, Sone et al., 2011, Sone et al., 2014). These intranuclear inclusions are immunoreactive for various markers of the proteasomal and autophagic degradation pathways, including ubiquitin, sumo, and p62 (Pountney et al., 2003; Mori et al., 2012; Nakamura et al., 2014; Sone et al., 2016). Importantly, an expansion of GGC repeats located in the 5′ UTR of the NOTCH2NLC (Notch 2 N-terminal like C, N2C) gene has been found recently to be associated with individuals with familial and sporadic NIID, mostly in people of Asian origin (Sone et al., 2019; Ishiura et al., 2019; Deng et al., 2019; Tian et al., 2019). The NOTCH2NLA, NOTCH2NLB, and NOTCH2NLC genes are human-specific paralogs of NOTCH2 exons 1–5, which encode Notch 2 N-terminal like (N2L) proteins that regulate Notch signaling to expand human neuronal progenitors during brain development (Fiddes et al., 2018; Suzuki et al., 2018). Alteration of NOTCH2NLC (N2C) protein function in NIID is unlikely because GGC repeats are located more than 100 nt upstream of the ATG start codon initiating the N2C open reading frame (ORF). Furthermore, NOTCH2NLC mRNA levels are unaltered in individuals with NIID (Sone et al., 2019; Ishiura et al., 2019; Tian et al., 2019). Thus, it remains to be determined how expansion of GGC repeats embedded in a predicted non-coding genomic region can lead to formation of intranuclear inclusions and cause neuronal cell death.
Here we find that the NOTCH2NLC GGC repeats are embedded in a small upstream open reading frame (uORF) located ahead of the main N2C ORF and encodes a small protein, uN2C. Of clinical importance is that GGC repeats embedded into the uN2C ORF are translated into a polyglycine stretch, resulting in expression of a uN2C polyglycine-containing protein (uN2CpolyG) in NIID. Antibodies developed against uN2CpolyG revealed its presence in the typical intranuclear inclusions in skin and brain sections of individuals with NIID. Furthermore, expression of uN2CpolyG in cell and animal models drives formation of p62-positive inclusions. Finally, expression of uN2CpolyG in mice is toxic, resulting in locomotor alterations, neuronal cell loss, and a reduced lifespan. These results are reminiscent of FXTAS, where an expansion of GGC repeats is embedded in a small upstream ORF, resulting in expression of a polyglycine-containing protein that, like uN2CpolyG, forms intranuclear inclusions and is toxic in cell and animal models (Hukema et al., 2015, Sellier et al., 2017, Todd et al., 2013). These data suggest the existence of a novel class of human genetic diseases where expanded GCC repeats are translated into pathogenic polyglycine (polyG) proteins.
Results
NOTCH2NLC GGC repeats are translated into a polyG-containing protein
Because translation of expanded repeats into pathogenic stretches of repeated amino acids is an established mechanism of pathogenicity in microsatellite diseases, we investigated the potential translation of GGC repeats in NIID. First we cloned GGC repeats embedded in the human NOTCH2NLC exon 1 sequence and fused these repeats to GFP in the three possible frames (Figures 1A and S1A). These frames were called polyG, polyalanine (polyA), and polyarginine (polyR), according to the stretch of amino acids potentially encoded by the repeats. Cell transfection followed by immunoblotting against GFP or observation of GFP fluorescence indicated that NOTCH2NLC GGC repeats were translated predominantly in the glycine frame (Figures 1B and 1C). qRT-PCR quantification indicated that GGC repeats cloned in the three frames are similarly transcribed (Figure S1B). Furthermore, cell transfection of expanded GGC repeats cloned downstream of an artificial ATG start codon in the alanine or arginine frames and fused to GFP results in expression of ATG-driven polyA and polyR GFP-tagged proteins (Figures S1C and S1D). These controls indicate that the limited expression of polyA or polyR from the NOTCH2NLC 5′ UTR cannot be accounted for by lack of their expression at the RNA level or a technical bias impairing their detection and that the NOTCH2NLC GGC repeats are mainly translated into a polyG-containing protein, which was called uN2CpolyG (upstream of N2C polyG-containing protein).
Figure 1.

NOTCH2NLC GGC repeats are translated in a polyG-containing protein
(A) Schematic of NOTCH2NLC exon 1 with GGC repeats fused to GFP in the glycine, alanine, or arginine frame.
(B and C) Immunoblot against GFP (B) or direct fluorescence (C) of HEK293 cells transfected for 24 h with GGC repeats embedded in NOTCH2NLC exon 1 and fused to GFP in the three possible frames.
(D) Top panel: N-terminal sequence and corresponding LC-MS/MS spectra of GFP-immunoprecipitated and trypsin-digested protein expressed from uN2C-GFP-transfected HEK293 cells. Bottom panel: nucleotide and amino acid sequences corresponding to the NOTCH2NLC upstream ORF (uN2C) N terminus.
(E) Immunoblot against GFP or the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) of lysostaphin-digested proteins extracted from uN2CpolyG-GFP-transfected HEK293 cells.
(F) Immunoblot against GFP or the GAPDH of proteins extracted from wild-type or mutant (ΔATG) uN2CpolyG-GFP-transfected HEK293 cells.
(G) Top panel: schematic of NOTCH2NLC exons 1 and 2. Bottom panel: amino acid sequence of the NOTCH2NLC upstream ORF (uN2C).
See also Figure S1.
To better characterize this protein, GGC repeats embedded in human NOTCH2NLC exon 1 and fused to GFP in the glycine frame were expressed in HEK293 cells, GFP immunoprecipitated, trypsin digested, and analyzed by mass spectrometry. Liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis confirmed that NOTCH2NLC GGC repeats are translated into a glycine stretch (Figures 1D and S1E). As a further control, treatment with lysostaphin, a glycylglycine endopeptidase, eliminated the N-terminal glycine stretch of uN2CpolyG-GFP, confirming its polyG nature (Figure 1E). Importantly, MS also revealed that GGC repeats are translated through initiation to a canonical AUG start codon located 15 nt before the repeats (Figures 1D and S1E). Deletion of this ATG codon abolished uN2CpolyG expression (Figure 1F). These data suggest that the NOTCH2NLC 5′ UTR contains a small upstream ORF, uN2C, that spans exons 1 and 2 and ends 8 nt before the ATG start site of the main NOTCH2NLC (N2C) ORF (Figures 1G and S1F). Consequently, the uN2CpolyG protein is composed of a short 5-amino-acid N terminus, a variable central glycine stretch corresponding to the number of GGC repeats, and a 38-amino-acid C-terminal part (Figure 1G).
The uN2polyG protein is present in NIID intranuclear inclusions
To confirm that NOTCH2NLC GGC repeats are translated into a polyG-containing protein in NIID, we developed two antibodies (4D12 and 4C4) directed against the uN2C C terminus, whose amino acid sequence differs from NOTCH2 and NOTCH2NLA predicted upstream ORFs (Figures 2A and S2A). Antibody specificity was confirmed by immunoblot and immunofluorescence (Figures S2B–S2D). Importantly, immunofluorescence using these antibodies revealed the presence of uN2CpolyG in the characteristic p62- and ubiquitin-positive intranuclear inclusions in skin and brain sections of individuals with NIID, carriers of an expansion of GGC repeats in NOTCH2NLC (N2C-NIID), but no staining in control age-matched individuals (Figures 2B–2E and S2E). As a control for uN2CpolyG staining specificity, identical results were observed with 4C4 and 4D12 antibodies, which are directed against different epitopes of the uN2C protein. Moreover, neither 4D12 nor 4C4 antibodies labeled p62-positive intranuclear inclusions of FMRpolyG in brain sections of FXTAS, a neurodegenerative disease caused, like NIID, by expanded GGC repeats translated into a polyG-containing protein (Figure S2F). Finally, quantification indicated quasi-complete co-localization of uN2CpolyG with p62-positive intranuclear inclusions in NIID (Figure S2G). These results demonstrate that NOTCH2NLC GGC repeat expansions are translated into a novel polyG-containing protein that is found in the typical intranuclear inclusions in individuals with N2C NIID.
Figure 2.

The uN2polyG protein is present in NIID intranuclear inclusions
(A) Alignment of NOTCH2 and NOTCH2NLA, NOTCH2NLB, and NOTCH2NLC putative upstream ORFs. Brackets indicate the amino acid sequences against which the 4D12 and 4C4 antibodies are directed.
(B–E) Immunofluorescence against uN2CpolyG using 4D12 (B and D) or 4C4 (C and E) antibody and p62 (B, D, and E) or ubiquitin (C) on skin (B and C) and brain (D and E) sections of carriers of the NOTCH2NLC-GGC repeat expansion (N2C NIID) or age-matched control individuals. Scale bars, 10 μm. Nuclei were counterstained with DAPI.
During this analysis, we noted that various European individuals with NIID with typical p62-positive intranuclear inclusions were nevertheless negative for uN2CpolyG staining (Figure S2H; data are summarized in Table S1). Genetic analyses revealed that these individuals are negative for the NOTCH2NLC GGC expansion, which is consistent with a recent report showing that most Europeans with NIID are negative for this mutation (Chen et al., 2020b). These unexpected results suggest that NIID is a heterogenous syndrome and that other mutations causing subtypes of this disorder remain to be identified, notably in European individuals with NIID.
Expression of uN2CpolyG is pathogenic in cells
To further study the uN2CpolyG protein, we cloned the full NOTCH2NLC uORF, from its ATG start codon in exon 1 to its penultimate codon in exon 2, containing its 38 C-terminal amino acids, with a control (12×) or an expanded (100×) size of GGC repeats, and fused this uN2C ORF to GFP or a hemagglutinin (HA) tag (Figure S3A). Cell transfection of the GFP-tagged N2C uORF with 12 or 100 GGC repeats indicated that their expression occurs independent of repeat size (Figure S3B). In contrast, fusion of the N2C uORF to a smaller HA tag (∼1 kDa) resulted in detection of uN2CpolyG with 100 GGC repeats but limited expression with 12 repeats (Figures S3C and S3D). This is characteristic of short upstream ORFs that are translated into small and often unstable peptides that can be stabilized and detected when fused with a large tag such as GFP (Aspden et al., 2014). In support of this hypothesis, cell treatment with Bafilomycin A1, which inhibits the autophagy degradation pathway, increased expression of the small uN2C-HA protein with 12 GGC repeats (Figure S3D). These results suggest that translation of the N2C uORF occurs independent of the length of the GGC stretch. However, in absence of an expansion, the uN2C protein, like most uORF proteins, is small and unstable and, thus, expressed at low levels in control individuals, whereas uN2C is stabilized by the polyG expansion and accumulates in individuals with N2C NIID.
Next we investigated whether the uN2CpolyG protein could be responsible for the typical inclusions and the neuronal cell dysfunctions observed in NIID. Importantly, immunofluorescence analyses indicated that sole expression of the uN2CpolyG protein in embryonic mouse cortical neuronal cell cultures was sufficient to form cytoplasmic and intranuclear inclusions, which are p62-positive (Figure 3A). Inclusions formation is likely driven by the polyG expansion because expression of a pure polyG stretch (ATG polyG-GFP), deleted of any uN2C sequence, also forms p62-positive cellular inclusions (Figure 3A). Identical results were obtained in immortalized cell lines (Figures S3E–S3G). Further analyses indicated that accretion of uN2CpolyG into cellular aggregates is progressive and parallels its accumulation in the insoluble fraction of transfected cell lysates (Figures S3F and S3G). Because protein aggregates can be degraded by macroautophagy, we tested whether compounds known to activate this catabolic process may prevent accumulation of the uN2CpolyG protein. Interestingly, inhibition of the mammalian target of rapamycin (mTOR) kinase decreases uN2CpolyG accumulation (Figure 3B). Finally, expression of the uN2CpolyG protein is toxic and leads to neuronal cell death in embryonic mouse cortical neuronal cell cultures (Figure 3C) and in GT1-7 immortalized neuronal cells (Figure S3H). Importantly, expression of the uN2C protein with a control number of glycine repeats (GGC 12x) was not overtly toxic, whereas expression of a polyG stretch in isolation and deleted of any uN2C sequence was sufficient to cause cell death (Figures 3C and S3H). These data indicate that expression of uN2CpolyG is pathogenic in cell culture, with its polyG stretch driving aggregation and toxicity.
Figure 3.

Expression of uN2CpolyG is toxic in cell culture
(A) GFP fluorescence and immunofluorescence against p62 and Map2 of cortical neuronal cell cultures from mouse embryos transduced for 24 h with GFP, uN2C-GFP (12 GGC), uN2CpolyG-GFP (100 GGC), or ATG polyG-GFP (70 GGC) AAV2/PHP.eB. Scale bars, 10 μm. Nuclei were counterstained with DAPI.
(B) Immunoblot against GFP or the GAPDH of proteins extracted from uN2CpolyG-GFP-transfected HEK293 cells treated for 15 h with the indicated drugs.
(C) Cell viability of cortical neuronal cell cultures from mouse embryos transduced with GFP, uN2C-GFP, uN2CpolyG-GFP, or ATG polyG-GFP AAV2/PHP.eB. Error bars indicate SEM. Student’s t test, ∗∗∗p < 0.001.
See also Figures S3, S4, and Table S2.
To further investigate the mechanism of uN2CpolyG toxicity, we searched for potential proteins interacting with it. Cell transfection of uN2CpolyG-GFP followed by GFP immunoprecipitation and MS identified various associated proteins with clear enrichment for the Ku70 and Ku80 proteins (Figure S4A; Table S2). Ku70 and Ku80 form a heterodimeric ring that initiates nonhomologous DNA end joining (NHEJ) repair at DNA double-strand breaks (Grundy et al., 2014; Fell and Schild-Poulter, 2015). Co-immunoprecipitation confirmed that the control uN2C and uN2CpolyG proteins interact with the endogenous Ku proteins (Figure S4B). However, this interaction is independent of the glycine stretch because ku70 and Ku80 interact neither with the polyG-containing protein expressed in FXTAS (FMRpolyG) nor with a pure polyG stretch (ATG polyG-GFP) (Figure S4B). Mutation analyses of the uN2C ORF revealed that a central amino acid sequence that is absent from NOTCH2, NOTCH2NLA, and NOTCH2NLB putative upstream ORFs is essential for the interaction of uN2C with the Ku proteins (Figure S4C). Next we investigated localization of the uN2C and uN2CpolyG proteins upon induction of DNA damage. Interestingly, most of uN2CpolyG was immobilized in inclusions, with little protein recruited to sites of DNA damage (Figure S4D). In contrast, the control uN2C protein was recruited within second to sites of laser microirradiation-induced DNA damage. Mutation analyses indicated that this recruitment is dependent on the uN2C amino acid sequence required to interact with the Ku proteins (Figure S4D). These data suggest that the 5′ UTR of NOTCH2NLC contains an upstream ORF that encodes a small protein, uN2C, which binds to the Ku proteins and is recruited at DNA damage sites. In contrast, upon GGC repeat expansion, uN2C encodes a mutant polyG-containing protein, uN2CpolyG, which is immobilized in aggregates and, thus, cannot join DNA damage. These data question whether uN2C or uN2polyG may affect DNA repair. Immunoblotting and immunofluorescence against phospho-Ser139 H2AX (γH2AX), a marker of DNA double-strand breaks, indicated faster kinetics of DNA repair in irradiated cells expressing the uN2C control protein with 12 glycine as compared to control cells (Figures S4E and S4F). In contrast, the polyG expansion in uN2C leads to an inactive protein that is unable to promote DNA repair (Figures S4E and S4F). We thus tested whether this impaired activity may translate into any pathological consequences in NIID. However, immunofluorescence analyses revealed no overt mislocalization of the Ku proteins or any evident accumulation of γH2AX-positive DNA damage in uN2CpolyG-expressing cells or in skin and brain sections of individuals with adult-onset N2C NIID. These results indicate that the small uN2C protein is potentially a novel regulator of DNA damage response, whereas in NIID, a polyG expansion alters uN2C localization and DNA repair activity, but this reduced function is likely compensated by the second NOTCH2NLC allele and is not sufficient to drive overt DNA repair alterations in individuals with NIID.
Expression of uN2CpolyG is pathogenic in animals
To evaluate the pathological consequences of uN2polyG expression in animals, we injected mice with adeno-associated virus (AAV) particles expressing GFP-tagged uN2C with a control (12×) or pathogenic (100×) size of polyG. To target the central nervous system, we used an AAV serotype (PHP.eB) that crosses the blood-brain barrier upon intravenous injection. Furthermore, to focus on polyG protein toxicity and exclude any potential RNA gain-of-function mechanism, the GGC repeat sequence was modified to include alternative codons (GGA, GGT, and GGG) that also encode for glycine and, thus, avoid production of a pure GGC RNA hairpin. qRT-PCR indicated similar levels of expression between AAV GFP-, control uN2C-GFP-, and uN2CpolyG-GFP-injected mice (Figure S5A). To determine the consequences of uN2CpolyG production in mice, we conducted a series of locomotor assays. Three months after AAV injection, we noted a progressive alteration of motor performance and coordination of uN2CpolyG-expressing mice. First, AAV uN2CpolyG-injected mice were unable to sustain hindlimb extension under tail suspension compared with control AAV GFP- or uN2C-injected mice (Figure 4A). Furthermore, uN2CpolyG-expressing mice had an increased likelihood of falling from a rotarod (Figure 4B), showing increased foot faults and slips on a notched bar (Figure 4C; Videos S1, S2, and S3), and were hyperactive in the open field arena (Figure 4C) compared with control mice expressing the GFP or N2C uORF with a normal size of GGC repeats. These alterations likely originate from specific neuronal dysfunction and not from global health deterioration because animals expressing the uN2CpolyG protein were overall healthy with normal grip strength and normal weight (Figures S5B and S5C). We noted none of these locomotor changes 1.5 months after AAV injection, demonstrating that these alterations are progressive. Finally, expression of the polyG protein is deleterious because mice expressing uN2CpolyG die around 4–6 months after injection, whereas GFP- and control uN2C-injected mice exhibit normal longevity and are indistinguishable from control non-injected mice (Figure 4E). Importantly, expression of uN2CpolyG in mice leads to widespread formation of intranuclear inclusions, which are p62-, ubiquitin-, and sumo-positives (Figures 4F, 4G, and S5D–S5F), reproducing the characteristic histopathological features of NIID. Finally, we observed neuronal cell death, notably loss of Purkinje cells, in mice expressing the uN2CpolyG protein (Figures 4H and S5G), which is consistent with the progressive loss of motor balance and coordination observed in these animals (Figures 4B and 4C; Videos S1, S2, and S3). Consistent with neurodegeneration, Gfap staining indicated increased neuroinflammation in brain sections of uN2CpolyG-injected mice (Figure S5H). Overall, these data suggest that expression of the uN2CpolyG protein in animals is toxic and is sufficient to generate the typical NIID intranuclear inclusions (Figure S6).
Figure 4.

Expression of uN2CpolyG is pathogenic in animals
(A) 60-s tail suspension test shows hind limb clasping in uN2CpolyG-GFP-expressing mice compared with GFP or uN2C-GFP control animals.
(B–D) Time before falling from a rotating rod (B), numbers of paw slips and errors in the notched bar test (C), and maximal distance traveled during 30 min in an open field (D) for AAV2/PHP.eB GFP-injected (n = 6), uN2C-GFP-injected (n = 6), and uN2CpolyG-GFP-injected (n = 11) male mice tested 3 months after injection.
(E) Kaplan-Meier survival curve of AAV2/PHP.eB GFP-injected (n = 6), uN2C-GFP-injected (n = 6), and uN2CpolyG-GFP-injected (n = 8) male mice. Dates of AAV injection and locomotor tests are indicated by arrows.
(F) Immunofluorescence against uN2CpolyG and p62 on cerebellum areas of uN2C-GFP- and uN2CpolyG-GFP-expressing mice sacrificed 2 months after AAV injection. Scale bars, 10 μm.
(G) Quantification of p62- or uN2CpolyG-positive intranuclear inclusions in different brain regions of uN2CpolyG-GFP-expressing mice. Brackets indicate the percentage of co-localization between p62- and uN2polyG-positive intranuclear inclusions. N = 3 mice; at least 200 nuclei were counted per brain region and per animal.
(H) Left panel: immunofluorescence against uN2CpolyG and calbindin on the cerebellum of uN2C-GFP- and uN2CpolyG-GFP-expressing mice sacrificed 4 months after AAV injection. Scale bars, 20 μm. Right panel: quantification of Purkinje cell numbers in GFP-expressing (n = 4), uN2C-GFP-expressing (n = 4), or uN2CpolyG-GFP-expressing (n = 4) mice.
In box-and-whisker plots, box upper and lower limits represent the 25th and 75th percentiles, whiskers represent minimum and maximum values, and a horizontal line across the box represents the median. Bar graphs indicate standard error of the mean (SEM). Student’s t test, ∗∗∗p < 0.001. Nuclei were counterstained with DAPI.
See also Figures S5 and S6 and Videos S1, S2, and S3.
Mice were recorded 4 months post AAV-injections.
Mice were recorded 4 months post AAV-injections.
Mice were recorded 4 months post AAV-injections.
Discussion
NIID is a rare neurodegenerative disorder caused by an expansion of GGC repeats located in the 5′ UTR of the NOTCH2NLC gene (Sone et al., 2019; Ishiura et al., 2019; Deng et al., 2019; Tian et al., 2019). The NOTCH2NLA, NOTCH2NLB, and NOTCH2NLC genes reside in the 1q21.1 locus and result from partial duplication of the NOTCH2 N-terminal part, which encodes six epidermal growth factor (EGF)-like domains but excludes NOTCH2 peptide signal, transmembrane, and cytoplasmic domains. Important for hominid brain size evolution, NOTCH2NL proteins regulate Notch signaling and expand human cortical progenitors (Fiddes et al., 2018; Suzuki et al., 2018). Furthermore, genomic duplications or deletions encompassing the NOTCH2NLA and/ or NOTCH2NLB genes lead to neurodevelopmental syndromes, whereas a GGC expansion in the NOTCH2NLC gene causes a neurodegenerative disease. These results highlight the importance of the NOTCH2NL gene for neuronal cells in humans but question how a repeat expansion embedded in a sequence predicted to be “non-coding” can be pathogenic.
Here we found that the NOTCH2NLC GGC repeats are embedded in a small upstream ORF, resulting in their translation into a polyG-containing protein that forms intranuclear inclusions and is toxic in cell and animal models (Figure S6). Immunoblotting and direct GFP observation indicate that NOTCH2NLC GGC repeats are mainly translated in the glycine frame through canonical initiation at an AUG start codon, but these assays may not be sensitive enough to detect low levels of polyA or polyR-containing proteins translated from non-canonical translation initiation in the repeats; thus, a contribution of RAN translation in NIID cannot be formally excluded. Translation of a small upstream ORF containing an expansion of GGC repeats into a toxic polyG protein is reminiscent of another neurodegenerative disorder, FXTAS, which is caused by an expansion of 70–200 CGG repeats located in the 5′ UTR of the FMR1 gene (Hagerman et al., 2001). These repeats are embedded in a short upstream ORF that is translated through canonical initiation at an ACG near-cognate start codon into a polyG-containing protein, FMRpolyG, which, like uN2CpolyG, forms p62-positive intranuclear inclusions and causes neuronal cell death in cell and animal models (Todd et al., 2013; Hukema et al., 2015; Sellier et al., 2017). NIID and FXTAS share some common clinical features and quasi-indistinguishable histopathological characteristics with remarkably similar intranuclear inclusions (Gelpi et al., 2017; Lim et al., 2020). Thus, like neurodegenerative polyQ diseases, which are caused by CAG repeat expansions embedded in diverse ORFs that are translated into toxic polyglutamine-containing proteins, we propose that NIID and FXTAS represent a novel class of disorders, polyG diseases, where expansions of GGC repeats embedded in diverse upstream ORFs are translated into toxic polyG-containing proteins (Figure S6). Of interest, GGC repeat expansions located in the 5′ UTRs of the LRP12, GIPC1, and NUTM2E (also known as FAM22E) genes have been recently identified as the cause of the OPDM and OPML neuromuscular and neurodegenerative disorders (Ishiura et al., 2019; Deng et al., 2020; Xi et al., 2021). OPDM and OPML share some overlapping symptoms and similar histopathological features with NIID and FXTAS. Furthermore, GGC repeat expansions in NOTCH2NLC have been identified recently in individuals with oculopharyngodistal myopathy with neurological symptoms, highlighting the overlap between these diseases (Ogasawara et al., 2020; Yu et al., 2021). Whether these newly identified diseases caused by GGC repeats are pathogenic through a common mechanism, translation of ill-defined short upstream ORFs into toxic polyG-containing proteins that form intranuclear inclusions, is an exciting question for the future (Figure S6).
Another topic of discussion is the relationship between GGC repeat length and clinical features. In polyQ diseases, a clear correlation exists between the size of the CAG repeat expansion and the age of onset and disease severity. In NIID, it is noteworthy that intermediate sizes (40–80) of NOTCH2NLC GGC repeats increased the susceptibility to develop the parkinsonism-dominant form of NIID and were found recently in individuals with Parkinson’s disease (Ma et al., 2020), whereas carriers of longer GGC expansions are more at risk to develop the dementia- and muscle weakness-dominant forms of NIID (Sone et al., 2019; Ishiura et al., 2019; Deng et al., 2019; Tian et al., 2019). However, an unambiguous correlation between repeat size and clinical severity might be limited to a narrow interval for 5′ UTR-embedded GGC repeat diseases because large expansions may trigger a potential “protective” mechanism because of their potential methylation and close proximity with promoters. Indeed, expansions over 200 CGG repeats in the 5′ UTR of the X-linked FMR1 gene promote epigenetic DNA changes that silence FMR1 promoter, resulting in decreased expression of the uORF encoding the toxic FMRpolyG protein but also in decreased expression of the main ORF encoding the synaptically important FMRP protein, whose loss cannot be compensated by a second allele in males, ultimately causing the neurodevelopmental fragile X syndrome (FXS). In NIID, if large GGC repeat expansions promote similar epigenetic changes, then this would result in silencing of the NOTCH2NLC allele carrying the GGC expansion, abolishing expression of the toxic uN2CpolyG protein, whereas decreased expression of the main ORF encoding the N2C protein would be likely non-pathogenic, as compensated by the second NOTCH2NLC allele. In short, because of a potential “protective” transcriptional silencing mechanism promoted by a high number of repeats, it is possible that 5′ UTR-embedded GC-rich repeat diseases (FXTAS, NIID, OPDM, and OPML) may not systematically follow a classic autosomal dominant inheritance. This potential promoter silencing mechanism may also explain why these diseases do not show evident anticipation (increased severity and/or younger age of onset with increased number of repeats) over a certain threshold of repeats.
Regarding the potential mechanism of toxicity of these polyG proteins, how they cause cell death is unclear. We found that the control uN2C protein interacts with the Ku70 and Ku80 proteins and activates DNA repair. Expansion of the polyG stretch impairs uN2C localization and interaction with the Ku proteins, resulting in a NHEJ-inactive uN2CpolyG protein. Alterations of DNA repair mechanisms can lead to various neurodegenerative syndromes. However, potential haploinsufficiency of uN2C in NIID is unlikely because NOTCH2NLC mRNA levels are unaltered in NIID (Sone et al., 2019; Ishiura et al., 2019; Tian et al., 2019). Furthermore, individuals with NIID lack the typical sensitivity to ionizing radiation, immunodeficiency, and microcephaly observed in individuals with radiosensitivity with severe combined immunodeficiency (RS-SCID), who are carriers of loss-of-function mutations in components of the NHEJ repair mechanism (Woodbine et al., 2014). Finally, we do not observe overt mislocalization of the Ku proteins or accumulation of DNA damage in cell and animal models of NIID, nor in skin and brain sections of adult individuals with NIID. Similarly, we found neither alterations of the Ku proteins nor increased DNA damage in cells or mice expressing the FXTAS polyG-containing protein FMRpolyG. Thus, we believe that potential alteration of DNA repair is most likely not the main cause of neuronal cell death in NIID. In contrast, uN2CpolyG and FMRpolyG possess a common polyG stretch that, when expressed in isolation, is sufficient to form p62-positive inclusions and causes neuronal cell death. Formation of cellular inclusions is consistent with the known in vitro self-aggregation properties of polyG homopolypeptides, which form amyloid-like fibrils (Lorusso et al., 2011; Plumley et al., 2011), but the molecular and cellular mechanisms by which these polyG proteins move into cell nuclei and drive neuronal cell dysfunctions remain unknown.
These results unveil a novel class of genetic disorders, polyG diseases, in which expanded GGC repeats embedded in upstream ORFs are translated into toxic polyG-containing proteins. Pharmaceutical compounds modulating autophagy could be of therapeutic interest to prevent toxic accumulation of these proteins.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-uN2CpolyG (SVEMAMNPV), Mouse Monoclonal Clone 4C4 | This paper | N/A |
| Anti-uN2CpolyG (AARPPRMH), Mouse Monoclonal Clone 4D12 | This paper | N/A |
| Anti-FLAG (DYKDDDDK) tag, Rabbit Polyclonal, | Thermo Fisher Scientific | Cat# PA1-984B; RRID: AB_347227 |
| Anti-HA (YPYDVPDYA) tag, Mouse Monoclonal Clone 16B12 | Abcam | Cat# ab130275; RRID: AB_11156884 |
| Anti-GFP, Rabbit Polyclonal, | Abcam | Cat# ab290; RRID: AB_303395 |
| Anti-GAPDH, Mouse Monoclonal Clone GA1R | Abcam | Cat# ab125247; RRID: AB_11129118 |
| Anti-Calbindin, Mouse Monoclonal Clone CB-955 | Abcam | Cat# ab82812; RRID: AB_1658451 |
| Anti-Ubiquitin, Rabbit Monoclonal Recombinant EPR8830 | Abcam | Cat# ab134953; RRID: AB_2801561 |
| Anti-Sumo 2 and 3, Rabbit Polyclonal | Abcam | Cat# ab3742; RRID: AB_304041 |
| Anti-p62 (SQSTM1), Rabbit Monoclonal Recombinant EPR4844 | Abcam | Cat# ab109012; RRID: AB_2810880 |
| Anti-MAP2, Mouse Monoclonal Clone MT-01 | Abcam | Cat# ab7756; RRID: AB_306050 |
| Anti-Histone H2A.X phospho S139, Rabbit Monoclonal Recombinant EP854(2)Y | Abcam | Cat# ab81299; RRID: AB_1640564 |
| Anti-Histone H2A.X, Rabbit Monoclonal Recombinant EPR22820-23 | Abcam | Cat# ab229914 |
| Anti-Ku80 (XRCC5), Mouse Monoclonal Clone 5C5 | Abcam | Cat# ab119935; RRID: AB_10899161 |
| Anti-Ku-70 (XRCC6), Mouse Monoclonal Clone N3H10 | Santa Cruz Biotechnology | Cat# sc-56129; RRID: AB_794205 |
| Anti-GFAP, Rabbit Polyclonal, | Proteintech | Cat# 16825-1-AP; RRID: AB_2109646 |
| Bacterial and virus strains | ||
| AAV2/PHP.eB CAG GFP | This paper | N/A |
| AAV2/PHP.eB CAG uN2C-GFP | This paper | N/A |
| AAV2/PHP.eB CAG uN2CpolyG-GFP | This paper | N/A |
| One Shot Stbl3 Chemically Competent E. coli | Invitrogen | Cat# C737303 |
| One Shot TOP10 Chemically Competent E. coli | Invitrogen | Cat# C404003 |
| Biological samples | ||
| Human skin and brain tissues | Described in Table S1 | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Lysostaphin Enzyme Protein Recombinant | ProSpec | Cat# ENZ-269 |
| Histosol Plus | LifeScience Products | Cat# HS-100-5GL |
| TO-PRO-3 Iodide (642/661) - 1 mM Solution in DMSO | Fisher Scientific | Cat# T-3605 |
| Lipofectamine 2000 Transfection reagent | Fisher Scientific | Cat# 11668019 |
| Complete Mini EDTA-free Protease Inhibitor Cocktail | Roche | Cat# 11836170001 |
| Bafilomycin A1 | Sigma | Cat# B1793 |
| MG132 | Sigma | Cat# M8699 |
| Doxycycline hydrochloride | Sigma | Cat# D3447 |
| Critical commercial assays | ||
| Anti-GFP Magnetic beads | Abcam | Cat# ab193983 |
| Anti-HA Magnetic Beads | Pierce | Cat# 88837 |
| EC Prime Western Blotting Detection Reagent | Sigma | Cat# GERPN2236 |
| DAB Substrate Kit | Abcam | Cat# ab64238 |
| TRI Reagent (Guanidine Thiocyanate & Phenol) | Merck | Cat# T9424-25ML |
| Transcriptor High Fidelity cDNA Synthesis Kit | Sigma | Cat# 5081955001 |
| LightCycler 480 SYBR Green I Master | Roche | Cat# 04707516001 |
| Experimental models: Cell lines | ||
| Human: U2OS cells | ATCC | Cat# 300364/p489_U-2_OS; RRID: CVCL_0042 |
| Human: HEK293 cells | ATCC | Cat# 300192/p777_HEK293; RRID: CVCL_0045 |
| Mouse: GT1-7 cells | ATCC | Cat# SCC116; RRID: CVCL_0281 |
| Human: U2OS T-REx TO-uN2C-GFP | This paper | N/A |
| Human: U2OS T-REx TO-uN2CpolyG-GFP | This paper | N/A |
| Experimental models: Organisms/strains | ||
| Wild type Mouse: C57BL/6J Mus musculus | The Jackson, Laboratory | JAX:000664, RRID: IMSR_JAX:000664 |
| Oligonucleotides | ||
| Primer RT-qPCR GFP Forward ACGTAAACGGCCACAAGTTC | This paper | N/A |
| Primer RT-qPCR GFP Reverse AAGTCGTGCTGCTTCATGTG | This paper | N/A |
| Primer RT-qPCR RPLP0 Forward GAAGTCACTGTGCCAGCCCA | This paper | N/A |
| Primer RT-qPCR RPLP0 Reverse GAAGGTGTAATCCGTCTCCA | This paper | N/A |
| Recombinant DNA | ||
| Plasmid: pcDNA3-TetOn uN2C-GFP (12 GLY) | This paper | N/A |
| Plasmid: pcDNA3-TetOn uN2CpolyG-GFP (100 GLY) | This paper | N/A |
| Plasmid: pAAV2-CAG uN2C-GFP (12 GLY) | This paper | N/A |
| Plasmid: pAAV2-CAG uN2CpolyG-GFP (100 GLY) | This paper | N/A |
| Plasmid: pcDNA3 NOTCH2NLC Exon1-GFP Gly frame | This paper | N/A |
| Plasmid: pcDNA3 NOTCH2NLC Exon1-GFP Ala frame | This paper | N/A |
| Plasmid: pcDNA3 NOTCH2NLC Exon1-GFP Arg frame | This paper | N/A |
| Plasmid: pcDNA3 ATG GLY 70x-GFP | This paper | N/A |
| Plasmid: pcDNA3 ATG ALA 100x-GFP | This paper | N/A |
| Plasmid: pcDNA3 ATG ARG 100x-GFP | This paper | N/A |
| Software and algorithms | ||
| Fiji (ImageJ) | NIH | RRID: SCR_002285 |
| Prism | GraphPad | RRID: SCR_005375 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Nicolas Charlet-Berguerand (ncharlet@igbmc.fr).
Materials availability
All unique reagents (e.g., uN2CpolyG constructs, 4C4 and 4D12 antibodies, etc.) generated in this study are available from the Lead Contact under MTA subject to restrictions from commercial source.
Data and code availability
Proteomics datasets related to Figure S4 in the paper are available in Table S2. Complete proteomics source data are available from the corresponding author upon reasonable request. No further unique datasets or codes were generated in this study.
Experimental model and subject details
Human samples
Patient available information is described in Table S1. Human skin and brain samples were sampled with the informed consent of individuals and families and approved by the Institutional Review Board of the Peking University First Hospital and Suzuka National Hospital.
Mice
All animal work was performed with approval from the IGBMC/ICS Animal Care Committee and of the French agency for research on animal (DGRI) authorization number APAFIS#11459-2017092208308760. C57BL/6 wild-type male mice were retro-orbitally AAV-injected at 2 months and then housed for 6 to 8 months in a temperature-controlled room (19–22°C) with a 12:12-hours light/dark cycle and free access to food and water. Mice were sacrificed by cervical dislocation in order to dissect the brain and spinal cord which were subsequently frozen for molecular biology or PFA-fixed and embedded in paraffin for histology.
Cell cultures and models
Mouse primary cortical neurons were prepared from E18 C57BL/6 wild-type mice embryos. Cortical regions were dissected and digested in 1X HBSS (ThermoFisher) with 0.25% trypsin (ThermoFisher) at 37°C for 10 minutes. Trypsin digestion was stopped by addition of DMEM (ThermoFisher), 10% horse serum (Life Technologies) and 1x GlutaMAX (ThermoFisher). Then, cortical neurons were dissociated, centrifuged, resuspended and plated on 0.1 mg/ml poly L-lysine (Sigma) pre-coated 24-well plates in Neurobasal Medium (ThermoFisher) supplemented with 1x B27, 0.5 mM L-glutamine and 100 IU/ml penicillin/streptomycin (ThermoFisher) at 37°C with 5% CO2. Neuronal GT1-7 cells were grown in DMEM 4.5 g/l glucose with 10% fetal calf serum and 100 IU/ml penicillin/streptomycin at 37°C in 5% CO2. U2OS and HEK293 cells were grown in DMEM 1 g/l glucose with 10% FCS and gentamycin at 37°C in 5% CO2. U2OS T-REx cells (ThermoFisher) stably expressing uN2C-GFP or uN2CpolyG-GFP were Lipofectamine-transfected with Pci1-linearized pcDNA3-TetOn expressing uN2C with 12 or 100 optimized GGC fused to the GFP and sectioned for neomycin resistance for two weeks.
Method details
Constructs
Human NOTCH2NLC exon 1 sequence containing 166 nts upstream of the GGC repeats was cloned into pcDNA3.1 fused to a the GFP deleted of its ATG and in all three frames. Mutations of the repeats size, ATG start codon or within the uN2C ORF were achieved by inverse PCR or by oligonucleotide ligations. NOTCH2NLC upstream ORF fused to the GFP with either 12 or 100 optimized GGN repeats was cloned into the pcDNA3-TetOn and pAAV2-CAG vector. To insure stability of repeat expansions, all GGC repeat-containing plasmids were transformed into STBL3 bacterial strain (Invitrogen). All constructs were confirmed by sanger sequencing.
Cell transfection and treatments
For AAV transduction of mouse primary cortical neurons, 4 to 5 days differentiated mouse primary cortical neurons were incubated with 5x10EXP10 vg/ml of AAV2/PHP.eB expressing the GFP, uN2C-GFP or uN2CpolyG-GFP, half of the media was changed every 48 hours and cells were let differentiated for 4 to 6 more days. For transient transfection, cells were plated in DMEM and 0.1% fetal bovine serum and transfected for 24 hours using Lipofectamine 2000 (Fisher Scientific). After 1 to 6 days post transient transfection, induction with 1 μg/mL doxycycline (Sigma) or AAV transduction, cells were analyzed by immunofluorescence or western blotting. For cell viability, mouse cortical neurons primary cultures were incubated 10 min with 1 μg/ml of propidium iodide and analyzed by microscopy. For GT1-7 cell viability, cells were detached by trypsin and resuspended in PBS with 20 nM TO-PRO-3 iodide (Fisher scientific) and FACS analyzed. Cells were treated with 100 nM of Bafilomycin A1, 1 μM MG132 or the indicated concentration of drug (Sigma) during 15 hours before analysis.
Mass spectrometry analysis
HEK293 cells were transfected with uN2C-GFP plasmid using Lipofectamine 2000 (Fisher Scientific) for 24 hours. Proteins were purified by GFP-immunoprecipitation, trypsin digested and the peptides were extracted twice with acetonitrile/water/formic acid-45/45/10-v/v/v followed by a final extraction with acetonitrile /formic acid (FA)-95/05-v/v. Extracted peptides were then analyzed using an Ultimate 3000 nano-RSLC (Thermo Scientific) coupled in line with an Orbitrap ELITE (Thermo Scientific). Peptides were separated on a C18 nano-column with a linear gradient of acetonitrile and analyzed with in a Top 20 collision-induced dissociation data-dependent mass spectrometry with an inclusion list. Data were processed by database searching using SequestHT (Thermo Fisher Scientific) with Proteome Discoverer 1.4 software (Thermo Fisher Scientific) or against a homemade database of all potential three frames translated proteins or peptides from the human NOTCH2NLC 5’UTR sequences. Precursor and fragment mass tolerance were set at 7 ppm and 0.5 Da respectively. Oxidation (M) and Nterminal Acetylation were set as variable modification, and Carbamidomethylation (C) as fixed modification. Peptides were filtered with the Fixed value node of Proteome Discoverer 1.4.
Antibody production
To generate monoclonal antibodies directed against uN2polyG, two months old female BALB/c mice were injected intraperitoneally with KLH conjugated peptides (4D12: CAARPPRMH, 4C4: CSVEMAMNPV) with 200 ug of poly(I/C) as adjuvant. Three injections were performed at 2 weeks intervals and four days prior to hybridoma fusion, mice with positively reacting sera were re-injected. Spleen cells were fused with Sp2/0.Agl4 myeloma cells. Supernatants of hybridoma cultures were tested at day 10 by ELISA for cross-reaction with peptides. Positive supernatants were then tested by Immunofluorescence and western blot on transfected HEK293 cells. Specific cultures were cloned twice on soft agar. Specific hybridomas were established and ascites fluid was prepared by injection of 2x106 hybridoma cells into Freund adjuvant-primed BALB/c mice. All animal experimental procedures were performed according to the French and European authority guidelines.
AAV production and retro-orbital injection
Recombinant AAV2/PHP.eB were generated by tri-transfection of HEK293 cells with pAAV2-GFP, -uN2C-GFP or -uN2CpolyG-GFP with pUCmini-iCAP-PHP.eB and pAD-DELTA-F16. Recombinant vectors were purified by double cesium chloride ultracentrifugation gradients from cell lysates, followed by dialysis and concentration against sterile PBS. Particles were quantified by real time PCR and vector titers were expressed as viral genomes per ml (vg/ml). 2 months old C57BL/6 male mice were injected retro-orbitally with 100 μL of sterile PBS with 1.5x10EXP13 vg/kg of AAV2/PHP.eB.
Animal phenotyping
Rotarod test (Bioseb, Chaville, France) was performed with three testing trials during which the rotation speed accelerated from 4 to 40 rpm in 5 min. Trials were separated by 10-15 min interval. The average latency was used as index of motor coordination performance. Grip test: this test measures the maximal muscle strength (g) using an isometric dynamometer connected to a grid (Bioseb). Mice were allowed to grip the grid with all its paws then they were pulled backward until they released it. Each mouse was submitted to 3 consecutive trials. The maximal strength developed by the mouse before releasing the grid was recorded and the average value of the three trials adjusted to body weight. Notched bar test: mice were tested under 100-lux lighting on a 2 cm-wide and 50 cm-long natural wooden piece notched bar comprising 12 platforms of 2 cm spaced by 13 gaps of 2 cm and bearing a 6 cm2 terminal platform. Animals had to cross the notched bar twice for training and 3 times for the test. Every instance of a back paw going through the gap was considered an error, and the global error percentage was calculated. Open field test: mice were tested in automated open fields (Panlab, Barcelona, Spain), each virtually divided into central and peripheral regions. The open fields were placed in a room homogeneously illuminated at 120 Lux. Each mouse was placed in the periphery of the open field and allowed to explore freely the apparatus for 30 min, with the experimenter out of the animal’s sight. The distance traveled, the number of rears, and time spent in the central and peripheral regions were recorded over the test session. The number of entries and the percent time spent in center area are used as index of emotionality/anxiety
X-ray and UVA-laser irradiation
U2OS cells expressing uN2C with 12 or 100 glycine GFP-tagged were X-ray irradiated for 10 minutes at 10 Gy in a CellRad (Precision). UV-laser micro-irradiation of U2OS cells expressing GFP-tagged U2C with 12 or 100 glycine seeded onto glass-bottom dishes was performed with a spinning disk Yokogawa X1 equipped with a Nikon microscope. For scanning irradiation, we use UV laser 266 nm pulse width < 1 ns power 15 mW and one repetition for each irradiation area. For GFP signal, we use time-lapse spinning disk acquisition with 491 nm 100 mW diode laser, the complete system is customized by Gataga System (Massy-France) and controlled with Metamorph Software.
Immunofluoresence and immunchemistry
For immunofluorescence, mouse or human brain sections were deparaffinized for 10 min in Histosol Plus (LifeScienceProducts) and dehydrated as follows: ethanol 100% (10 min), ethanol 90% (5 min), ethanol 70% (5 min), and rinsed in water. Antigen retrieval was performed in pressure cooker in 10 mM Tris PH9, 1 mM EDTA followed by blocking 1 h with PBS, 0,5% Triton X-100 and 5% Horse Serum. Glass coverslips containing plated cells or brain sections treated as described above were fixed for 15 min in PBS with 4% paraformaldehyde, washed with PBS and incubated in PBS plus 0.5% Triton X-100 during 10 min. The cells were washed three times with PBS and the coverslips were incubated during one hour with primary antibody against Calbindin (Abcam ab82812, 1/200), γH2AX (Abcam ab81299, 1/300), ubiquitin (Abcam ab134953, 1/100), Sumo2/3 (Abcam ab3742, 1/100), p62/Sqstm1 (Abcam ab109012, 1/1000), GFAP (Proteintech 16825-1-AP, 1/250), MAP2 (Abcam ab7756, 1/200) and uN2C 4C4 or 4D12 (mouse monoclonal homemade, 1/100). After washing with PBS, the coverslips were incubated with goat anti mouse secondary antibody conjugated with Alexa 488 or CY3 (Interchim SA) for one hour, washed twice with PBS and incubated for 3 min in PBS/DAPI (1/10 000 dilution). Coverslips were rinsed twice before mounting in Pro-Long media (Molecular Probes) and were examined using a Leica microscope. For immunochemistry, mouse brain sections were deparaffinized 10 min in Histosol Plus (LifeScienceProducts) and dehydrated as follows: ethanol 100% (10 min), ethanol 90% (5 min), ethanol 70% (5 min), and rinsed in water. Antigen retrieval was performed in pressure cooker in 10 mM Tris PH9, 1 mM EDTA followed with 10 μg/ml protein kinase treatment for 20 min at 37°c. Endogenous peroxidase activity was blocked, and blocking 1 h with PBS, 0,5% Triton X-100 and 5% Horse Serum and immunostaining was performed overnight at 4°C using antibody against Calbindin (Abcam ab82812, 1/50), Gfap (Proteintech, 16825-1-AP, 1/100), Sumo2/3 (Abcam ab3742, 1/100) and uN2C 4C4 or 4D12 (mouse monoclonal homemade, 1/100). Antigen–antibody complexes were visualized by incubation with DAB substrate (Abcam) and slides were counterstained with hematoxylin and eosin.
Lysostaphin treatment
HEK293 cells transfected with a uN2CpolyGFP construct were scrapped in PBS 1X and centrifuged during 10 min at 3000 rpm at 4°C. The cell pellet was resuspended in 400 μl of RIPA and 16 μl of cell extract was incubated with 1 μg of lysostaphin (Prospec, ENZ-269) during 5 to 15 minutes at 37°C. Laemmli buffer was add to the mix and proteins were analyze by western blot.
Co-immunoprecipitation assay
24 h after transfection of HEK293 cells with 1 μg of uN2C-GFP constructs in Lipofectamine 2000 (Invitrogen), cells were lysed in RIPA buffer (50 mM Tris-HCl pH 7.5, 0.15 M NaCl, 0.5% Triton X-100) supplemented with protease inhibitor cocktail (Roche) and clarified by centrifugation at 14000 rpm for 10 min. Immunoprecipitations were performed at 4°C for 1 h using pre-washed Anti-GFP (Abcam ab193983) or anti-HA (Pierce 88837) Magnetic Beads in RIPA buffer, washed three time, then bound proteins were eluted by 3 min denaturation step at 95°C with Laemmli buffer followed by mass spectrometry or western blot.
Western blotting
Proteins were denatured 3 min at 95°C, separated on 4%–12% bis-Tris Gel (NuPAGE), transferred on nitrocellulose membranes (Whatman Protan), blocked with 5% non-fat dry milk in Tris Buffer Saline buffer (TBS), incubated with anti-GFP (Abcam ab290), HA (Abcam ab130275), FLAG (Thermofisher PA1-984B), γH2AX (Abcam ab81299), H2AX (Abcam ab229914), Ku70 (SantaCruz sc-56129), Ku80 (Abcam ab 119935), GAPDH (Abcam ab125247) in TBS plus 5% non-fat dry milk, washed 3 times and incubated with anti-rabbit or mouse Peroxidase antibody (1:10,000, Cell Signaling) 1 hour in TBS, followed by washing and ECL Prime chemiluminescence revelation kit (Sigma).
Quantitative real time RT-PCR
Total RNAs from mouse tissues or cells were isolated by TriReagent (Merck). cDNAs were generated using the Transcriptor High Fidelity cDNA synthesis kit (Sigma) for quantification of mRNAs. qPCR were realized using the LightCycler 480 SYBR Green I Master (Roche) in a Lightcycler 480 (Roche) with 15 min at 94°C followed by 50 cycles of 15 s at 94°C, 20 s at 58°C and 20 s at 72°C. RPLPO mRNA was used as standard and data were analyzed using the Lightcycler 480 analysis software (2ΔCt method).
Quantification and statistical analysis
To eliminate bias, image or animal analyzes were either completely automated or blinded. All statistical analyses were performed using Excel (Microsoft) and Prism (GraphPad). Experiments are represented as either mean value ± Standard Error of Mean (SEM) or box-and-whisker plots with box upper and lower limits representing the 25th and 75th quartiles, respectively, the whiskers depicting the lowest and highest data points and the horizontal line through the box represent the median. The statistical tests used are two-tailed paired Student’s t test or ANOVA. Significance was set as ∗p < 0.05; ∗∗p < 0.005 and ∗∗∗p < 0.001. Sample-sizes were determined based on past experiments and to minimize the number of mice used. No statistical method was used to determine whether data meet assumptions of the statistical approach. Detailed statistical information, including the statistical test, measures, number “n” of animals, cells and/ or experiments are indicated in the figures and their respective legends.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81571219, 82071409, and U20A20356 to Z.W.); the Double Thousand Talents Program of Jiangxi Province (to D.H.); the Peking University Medicine Fund of Fostering Young Scholars’ Scientific & Technological Innovation (to J.D.); the Japan Society for the Promotion of Science (KAKENHI JP19H03577) and the MHLW FC Program (JPMH19189624) (to J.S.); ERC-2012-StG 310659, ANR-18-CE16-0019, and FRM EQU202103012936 (to N.C.B.); and ANR-10-LABX-0030-INRT and ANR-10-IDEX-0002-02 (to I.G.B.M.C.).
Author contributions
Experiments were performed by M.B., J.D., J.S., V.P., B.M., F. Ruffenach, F. Riet, E.G., P.K., H.J., and M.O.-A. Control and NIID cases originated from K.M., W.A.C., A.A.D., D.H., H.M., Y.I., J.S., and Z.W. Data were collected and analyzed by M.B., J.D., J.S., Z.W., W.A.C., A.A.D., E.G., L.N., and N.C.-B. The study was designed, coordinated, and written by N.C.-B. with input from all authors.
Declaration of interests
The authors declare no competing interests.
Published: April 21, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.neuron.2021.03.038.
Supporting citations
The following references appear in the Supplemental information: Cupidi et al. (2019), McFadden et al. (2005).
Supplemental information
References
- Aspden J.L., Eyre-Walker Y.C., Phillips R.J., Amin U., Mumtaz M.A., Brocard M., Couso J.P. Extensive translation of small Open Reading Frames revealed by Poly-Ribo-Seq. Elife. 2014;3:e03528. doi: 10.7554/eLife.03528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Lu L., Wang B., Cui G., Wang X., Wang Y., Raza H.K., Min Y., Li K., Cui Y. Re-defining the clinicopathological spectrum of neuronal intranuclear inclusion disease. Ann. Clin. Transl. Neurol. 2020;7:1930–1941. doi: 10.1002/acn3.51189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Yan Yau W., Jaunmuktane Z., Tucci A., Sivakumar P., Gagliano Taliun S.A., Turner C., Efthymiou S., Ibáñez K., Sullivan R., Genomics England Research Consortium Hardy J, Ryten M, Vandrovcova J, Houlden H. Neuronal intranuclear inclusion disease is genetically heterogeneous. Ann. Clin. Transl. Neurol. 2020;7:1716–1725. doi: 10.1002/acn3.51151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortese A., Simone R., Sullivan R., Vandrovcova J., Tariq H., Yau W.Y., Humphrey J., Jaunmuktane Z., Sivakumar P., Polke J. Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nat. Genet. 2019;51:649–658. doi: 10.1038/s41588-019-0372-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cupidi C., Dijkstra A.A., Melhem S., Vernooij M.W., Severijnen L.A., Hukema R.K., Rozemuller A.J.M., Neumann M., van Swieten J.C., Seelaar H. Refining the Spectrum of Neuronal Intranuclear Inclusion Disease: A Case Report. J. Neuropathol. Exp. Neurol. 2019;78:665–670. doi: 10.1093/jnen/nlz043. [DOI] [PubMed] [Google Scholar]
- Deng J., Gu M., Miao Y., Yao S., Zhu M., Fang P., Yu X., Li P., Su Y., Huang J. Long-read sequencing identified repeat expansions in the 5’UTR of the NOTCH2NLC gene from Chinese patients with neuronal intranuclear inclusion disease. J. Med. Genet. 2019;56:758–764. doi: 10.1136/jmedgenet-2019-106268. [DOI] [PubMed] [Google Scholar]
- Deng J., Yu J., Li P., Luan X., Cao L., Zhao J., Yu M., Zhang W., Lv H., Xie Z. Expansion of GGC Repeat in GIPC1 Is Associated with Oculopharyngodistal Myopathy. Am. J. Hum. Genet. 2020;106:793–804. doi: 10.1016/j.ajhg.2020.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang P., Yu Y., Yao S., Chen S., Zhu M., Chen Y., Zou K., Wang L., Wang H., Xin L. Repeat expansion scanning of the NOTCH2NLC gene in patients with multiple system atrophy. Ann. Clin. Transl. Neurol. 2020;7:517–526. doi: 10.1002/acn3.51021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fell V.L., Schild-Poulter C. The Ku heterodimer: function in DNA repair and beyond. Mutat. Res. Rev. Mutat. Res. 2015;763:15–29. doi: 10.1016/j.mrrev.2014.06.002. [DOI] [PubMed] [Google Scholar]
- Fiddes I.T., Lodewijk G.A., Mooring M., Bosworth C.M., Ewing A.D., Mantalas G.L., Novak A.M., van den Bout A., Bishara A., Rosenkrantz J.L. Human-Specific NOTCH2NL Genes Affect Notch Signaling and Cortical Neurogenesis. Cell. 2018;173:1356–1369.e22. doi: 10.1016/j.cell.2018.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F.B., Richter J.D. Microsatellite Expansion Diseases: Repeat Toxicity Found in Translation. Neuron. 2017;93:249–251. doi: 10.1016/j.neuron.2017.01.001. [DOI] [PubMed] [Google Scholar]
- Gelpi E., Botta-Orfila T., Bodi L., Marti S., Kovacs G., Grau-Rivera O., Lozano M., Sánchez-Valle R., Muñoz E., Valldeoriola F. Neuronal intranuclear (hyaline) inclusion disease and fragile X-associated tremor/ataxia syndrome: a morphological and molecular dilemma. Brain. 2017;140:e51. doi: 10.1093/brain/awx156. [DOI] [PubMed] [Google Scholar]
- Grundy G.J., Moulding H.A., Caldecott K.W., Rulten S.L. One ring to bring them all--the role of Ku in mammalian non-homologous end joining. DNA Repair (Amst.) 2014;17:30–38. doi: 10.1016/j.dnarep.2014.02.019. [DOI] [PubMed] [Google Scholar]
- Hagerman R.J., Leehey M., Heinrichs W., Tassone F., Wilson R., Hills J., Grigsby J., Gage B., Hagerman P.J. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology. 2001;57:127–130. doi: 10.1212/wnl.57.1.127. [DOI] [PubMed] [Google Scholar]
- Hukema R.K., Buijsen R.A., Schonewille M., Raske C., Severijnen L.A., Nieuwenhuizen-Bakker I., Verhagen R.F., van Dessel L., Maas A., Charlet-Berguerand N. Reversibility of neuropathology and motor deficits in an inducible mouse model for FXTAS. Hum. Mol. Genet. 2015;24:4948–4957. doi: 10.1093/hmg/ddv216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiura H., Tsuji S. Advances in repeat expansion diseases and a new concept of repeat motif-phenotype correlation. Curr. Opin. Genet. Dev. 2020;65:176–185. doi: 10.1016/j.gde.2020.05.029. [DOI] [PubMed] [Google Scholar]
- Ishiura H., Shibata S., Yoshimura J., Suzuki Y., Qu W., Doi K., Almansour M.A., Kikuchi J.K., Taira M., Mitsui J. Noncoding CGG repeat expansions in neuronal intranuclear inclusion disease, oculopharyngodistal myopathy and an overlapping disease. Nat. Genet. 2019;51:1222–1232. doi: 10.1038/s41588-019-0458-z. [DOI] [PubMed] [Google Scholar]
- Li M., Li K., Li X., Tian Y., Shen L., Wu G., Zhang Z., Chen W. Multiple reversible encephalitic attacks: a rare manifestation of neuronal intranuclear inclusion disease. BMC Neurol. 2020;20:125. doi: 10.1186/s12883-020-01712-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S.Y., Ishiura H., Ramli N., Shibata S., Almansour M.A., Tan A.H., Houlden H., Lang A.E., Tsuji S. Adult-onset neuronal intranuclear inclusion disease mimicking Fragile X-associated tremor-ataxia syndrome in ethnic Chinese patients. Parkinsonism Relat. Disord. 2020;74:25–27. doi: 10.1016/j.parkreldis.2020.03.025. [DOI] [PubMed] [Google Scholar]
- Lindenberg R., Rubinstein L.J., Herman M.M., Haydon G.B. A light and electron microscopy study of an unusual widespread nuclear inclusion body disease. A possible residuum of an old herpesvirus infection. Acta Neuropathol. 1968;10:54–73. doi: 10.1007/BF00690510. [DOI] [PubMed] [Google Scholar]
- Liu Y., Mimuro M., Yoshida M., Hashizume Y., Niwa H., Miyao S., Ujihira N., Akatsu H. Inclusion-positive cell types in adult-onset intranuclear inclusion body disease: implications for clinical diagnosis. Acta Neuropathol. 2008;116:615–623. doi: 10.1007/s00401-008-0442-7. [DOI] [PubMed] [Google Scholar]
- Lorusso M., Pepe A., Ibris N., Bochicchio B. Molecular and supramolecular studies on polyglycine and poly-l-proline. Soft Matter. 2011;7:6327. [Google Scholar]
- Ma D., Tan Y.J., Ng A.S.L., Ong H.L., Sim W., Lim W.K., Teo J.X., Ng E.Y.L., Lim E.C., Lim E.W. Association of NOTCH2NLC Repeat Expansions With Parkinson Disease. JAMA Neurol. 2020;77:1–5. doi: 10.1001/jamaneurol.2020.3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFadden K., Hamilton R.L., Insalaco S.J., Lavine L., Al-Mateen M., Wang G., Wiley C.A. Neuronal intranuclear inclusion disease without polyglutamine inclusions in a child. J. Neuropathol. Exp. Neurol. 2005;64:545–552. doi: 10.1093/jnen/64.6.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori F., Tanji K., Odagiri S., Hattori M., Hoshikawa Y., Kono C., Yasui K., Yokoi S., Hasegawa Y., Kamitani T. Ubiquitin-related proteins in neuronal and glial intranuclear inclusions in intranuclear inclusion body disease. Pathol. Int. 2012;62:407–411. doi: 10.1111/j.1440-1827.2012.02812.x. [DOI] [PubMed] [Google Scholar]
- Munoz-Garcia D., Ludwin S.K. Adult-onset neuronal intranuclear hyaline inclusion disease. Neurology. 1986;36:785–790. doi: 10.1212/wnl.36.6.785. [DOI] [PubMed] [Google Scholar]
- Nakamura M., Murray M.E., Lin W.L., Kusaka H., Dickson D.W. Optineurin immunoreactivity in neuronal and glial intranuclear inclusions in adult-onset neuronal intranuclear inclusion disease. Am. J. Neurodegener. Dis. 2014;3:93–102. [PMC free article] [PubMed] [Google Scholar]
- Nelson D.L., Orr H.T., Warren S.T. The unstable repeats--three evolving faces of neurological disease. Neuron. 2013;77:825–843. doi: 10.1016/j.neuron.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogasawara M., Iida A., Kumutpongpanich T., Ozaki A., Oya Y., Konishi H., Nakamura A., Abe R., Takai H., Hanajima R. CGG expansion in NOTCH2NLC is associated with oculopharyngodistal myopathy with neurological manifestations. Acta Neuropathol. Commun. 2020;8:204. doi: 10.1186/s40478-020-01084-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plumley J.A., Tsai M.I., Dannenberg J.J. Aggregation of capped hexaglycine strands into hydrogen-bonding motifs representative of pleated and rippled β-sheets, collagen, and polyglycine I and II crystal structures. A density functional theory study. J. Phys. Chem. B. 2011;115:1562–1570. doi: 10.1021/jp111501d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pountney D.L., Huang Y., Burns R.J., Haan E., Thompson P.D., Blumbergs P.C., Gai W.P. SUMO-1 marks the nuclear inclusions in familial neuronal intranuclear inclusion disease. Exp. Neurol. 2003;184:436–446. doi: 10.1016/j.expneurol.2003.07.004. [DOI] [PubMed] [Google Scholar]
- Rafehi H., Szmulewicz D.J., Bennett M.F., Sobreira N.L.M., Pope K., Smith K.R., Gillies G., Diakumis P., Dolzhenko E., Eberle M.A. Bioinformatics-Based Identification of Expanded Repeats: A Non-reference Intronic Pentamer Expansion in RFC1 Causes CANVAS. Am. J. Hum. Genet. 2019;105:151–165. doi: 10.1016/j.ajhg.2019.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez C.M., Todd P.K. New pathologic mechanisms in nucleotide repeat expansion disorders. Neurobiol. Dis. 2019;130:104515. doi: 10.1016/j.nbd.2019.104515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellier C., Buijsen R.A.M., He F., Natla S., Jung L., Tropel P., Gaucherot A., Jacobs H., Meziane H., Vincent A. Translation of Expanded CGG Repeats into FMRpolyG Is Pathogenic and May Contribute to Fragile X Tremor Ataxia Syndrome. Neuron. 2017;93:331–347. doi: 10.1016/j.neuron.2016.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sone J., Hishikawa N., Koike H., Hattori N., Hirayama M., Nagamatsu M., Yamamoto M., Tanaka F., Yoshida M., Hashizume Y. Neuronal intranuclear hyaline inclusion disease showing motor-sensory and autonomic neuropathy. Neurology. 2005;65:1538–1543. doi: 10.1212/01.wnl.0000184490.22527.90. [DOI] [PubMed] [Google Scholar]
- Sone J., Tanaka F., Koike H., Inukai A., Katsuno M., Yoshida M., Watanabe H., Sobue G. Skin biopsy is useful for the antemortem diagnosis of neuronal intranuclear inclusion disease. Neurology. 2011;76:1372–1376. doi: 10.1212/WNL.0b013e3182166e13. [DOI] [PubMed] [Google Scholar]
- Sone J., Kitagawa N., Sugawara E., Iguchi M., Nakamura R., Koike H., Iwasaki Y., Yoshida M., Takahashi T., Chiba S. Neuronal intranuclear inclusion disease cases with leukoencephalopathy diagnosed via skin biopsy. J. Neurol. Neurosurg. Psychiatry. 2014;85:354–356. doi: 10.1136/jnnp-2013-306084. [DOI] [PubMed] [Google Scholar]
- Sone J., Mori K., Inagaki T., Katsumata R., Takagi S., Yokoi S., Araki K., Kato T., Nakamura T., Koike H. Clinicopathological features of adult-onset neuronal intranuclear inclusion disease. Brain. 2016;139:3170–3186. doi: 10.1093/brain/aww249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sone J., Mitsuhashi S., Fujita A., Mizuguchi T., Hamanaka K., Mori K., Koike H., Hashiguchi A., Takashima H., Sugiyama H. Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nat. Genet. 2019;51:1215–1221. doi: 10.1038/s41588-019-0459-y. [DOI] [PubMed] [Google Scholar]
- Sun Q.Y., Xu Q., Tian Y., Hu Z.M., Qin L.X., Yang J.X., Huang W., Xue J., Li J.C., Zeng S. Expansion of GGC repeat in the human-specific NOTCH2NLC gene is associated with essential tremor. Brain. 2020;143:222–233. doi: 10.1093/brain/awz372. [DOI] [PubMed] [Google Scholar]
- Suzuki I.K., Gacquer D., Van Heurck R., Kumar D., Wojno M., Bilheu A., Herpoel A., Lambert N., Cheron J., Polleux F. Human-Specific NOTCH2NL Genes Expand Cortical Neurogenesis through Delta/Notch Regulation. Cell. 2018;173:1370–1384.e16. doi: 10.1016/j.cell.2018.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi-Fujigasaki J. Neuronal intranuclear hyaline inclusion disease. Neuropathology. 2003;23:351–359. doi: 10.1046/j.1440-1789.2003.00524.x. [DOI] [PubMed] [Google Scholar]
- Takahashi-Fujigasaki J., Nakano Y., Uchino A., Murayama S. Adult-onset neuronal intranuclear hyaline inclusion disease is not rare in older adults. Geriatr. Gerontol. Int. 2016;16(Suppl 1):51–56. doi: 10.1111/ggi.12725. [DOI] [PubMed] [Google Scholar]
- Tian Y., Wang J.L., Huang W., Zeng S., Jiao B., Liu Z., Chen Z., Li Y., Wang Y., Min H.X. Expansion of Human-Specific GGC Repeat in Neuronal Intranuclear Inclusion Disease-Related Disorders. Am. J. Hum. Genet. 2019;105:166–176. doi: 10.1016/j.ajhg.2019.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd P.K., Oh S.Y., Krans A., He F., Sellier C., Frazer M., Renoux A.J., Chen K.C., Scaglione K.M., Basrur V. CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron. 2013;78:440–455. doi: 10.1016/j.neuron.2013.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodbine L., Gennery A.R., Jeggo P.A. The clinical impact of deficiency in DNA non-homologous end-joining. DNA Repair (Amst.) 2014;16:84–96. doi: 10.1016/j.dnarep.2014.02.011. [DOI] [PubMed] [Google Scholar]
- Xi J., Wang X., Yue D., Dou T., Wu Q., Lu J., Liu Y., Yu W., Qiao K., Lin J. 5′ UTR CGG repeat expansion in GIPC1 is associated with oculopharyngodistal myopathy. Brain. 2021;144:601–614. doi: 10.1093/brain/awaa426. [DOI] [PubMed] [Google Scholar]
- Yu J., Deng J., Guo X., Shan J., Luan X., Cao L., Zhao J., Yu M., Zhang W., Lv H. The GGC repeat expansion in NOTCH2NLC is associated with oculopharyngodistal myopathy type 3. Brain. 2021 doi: 10.1093/brain/awab077. Published online March 9, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y., Liu Z., Hou X., Li W., Ni J., Huang L., Hu Y., Liu P., Hou X., Xue J. Identification of GGC repeat expansion in the NOTCH2NLC gene in amyotrophic lateral sclerosis. Neurology. 2020;95:e3394–e3405. doi: 10.1212/WNL.0000000000010945. [DOI] [PubMed] [Google Scholar]
- Zu T., Gibbens B., Doty N.S., Gomes-Pereira M., Huguet A., Stone M.D., Margolis J., Peterson M., Markowski T.W., Ingram M.A. Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. USA. 2011;108:260–265. doi: 10.1073/pnas.1013343108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Mice were recorded 4 months post AAV-injections.
Mice were recorded 4 months post AAV-injections.
Mice were recorded 4 months post AAV-injections.
Data Availability Statement
Proteomics datasets related to Figure S4 in the paper are available in Table S2. Complete proteomics source data are available from the corresponding author upon reasonable request. No further unique datasets or codes were generated in this study.