

Abstract
We have previously demonstrated that inorganic polyphosphate (polyP) is a potent activator of the mitochondrial permeability transition pore (mPTP) in cardiac myocytes. PolyP depletion protected against Ca2+-induced mPTP opening, however it did not prevent and even exacerbated cell death during ischemia-reperfusion (I/R). The central goal of this study was to investigate potential molecular mechanisms underlying these dichotomous effects of polyP on mitochondrial function. We utilized a Langendorff-perfused heart model of I/R to monitor changes in polyP size and chain length at baseline, 20 min no-flow ischemia, and 15 min reperfusion. Freshly isolated cardiac myocytes and mitochondria from C57BL/6J (WT) and cyclophilin D knock-out (CypD KO) mice were used to measure polyP uptake, mPTP activity, mitochondrial membrane potential, respiration and ATP generation. We found that I/R induced a significant decrease in polyP chain length. We, therefore, tested, the ability of synthetic polyPs with different chain length to accumulate in mitochondria and induce mPTP. Both short and long chain polyPs accumulated in mitochondria in oligomycin-sensitive manner implicating potential involvement of mitochondrial ATP synthase in polyP transport. Notably, only short-chain polyP activated mPTP in WT myocytes, and this effect was prevented by mPTP inhibitor cyclosprorin A and absent in CypD KO myocytes. To the contrary, long-chain polyP suppressed mPTP activation, and enhanced ADP-linked respiration and ATP production. Our data indicate that 1) effect of polyP on cardiac function strongly depends on polymer chain length; and 2) short-chain polyPs (as increased in ischemia-reperfusion) induce mPTP and mitochondrial uncoupling, while long-chain polyPs contribute to energy generation and cell metabolism.
Keywords: inorganic polyphosphate, mitochondrial permeability transition pore, ischemia-reperfusion injury, bioenergetics, mitochondrial metabolism, ATP synthase
Journal Subject Terms: Metabolism, Animal Models of Human Disease
1. Introduction
Inorganic polyphosphate (polyP) is a polymer of orthophosphates linked together by phosphoanhydride bonds similar to those found in ATP (Fig. 1A) [1–5]. In eukaryotic organisms, polyP has been implicated in a wide range of physiological and pathological functions [2, 5]. In mammalian cells polyP is present in the size ranging from 5 to 800 orthophosphate residues depending on species and cell type tested [6–8]. One of the first reports of polyP presence in mammalian mitochondria dates back to over 50 years [9], however very little is still known about its roles and metabolism. The amount and turnover of polyP is much higher in tissues with high metabolic rates and demands such as brain and heart [6, 8–10]. Recent studies demonstrated that in mitochondria polyP could be involved in energy metabolism [11, 12], calcium (Ca2+) signaling [8, 13] and direct formation and/or activation of the mitochondrial Permeability Transition Pore (mPTP) [8, 14, 15]. In striking contrast to inorganic phosphate, a known inducer of mPTP [16, 17], which is typically present in the mitochondrial matrix in millimolar concentrations [2], polyP was detected in mammalian mitochondria only in micromolar concentrations [1, 6, 8], limiting its ability to buffer matrix Ca2+, Mg2+, or pH significantly and suggesting a direct regulatory role. Specifically, it has been suggested that involvement of polyP might be required for Ca2+-induced assembly of the mPTP channel-forming complex [8, 12, 15, 18, 19]. This hypothesis was built on the original discovery by Reusch and Sadoff [20] who demonstrated back in 1983 a strong correlation between bacterial transformation efficiency and the formation of poly-β-hydroxybutyrate and calcium polyphosphate (PHB/Ca2+/polyP) complexes in the plasma membranes of Escherichia coli (E. Coli) upon Ca2+-mediated induction of genetic competence. Further studies confirmed that PHB/Ca2+/polyP complexes function as voltage-activated calcium channels [21] in plasma membrane of E. Coli, and they have also been postulated to participate in co-export of Ca2+, phosphate, and deoxyribonucleic acid (DNA) transfer across the membrane during bacterial transformation [22–24]. Interestingly, a similar PHB/Ca2+/polyP complex was isolated later by Pavlov et al. [18] from rat liver mitochondria which upon reconstitution into planar lipid bilayer demonstrated the properties similar to the mPTP suggesting the role of polyP in mPTP activation. Accordingly, we previously found that enzymatic depletion of polyP from mitochondria leads to inhibition of the Ca2+-induced mPTP [8, 15]. However, polyP depletion did not prevent, and even enhanced cell death under conditions favoring reactive oxygen species (ROS)-induced mechanism of mPTP activation such as ischemia-reperfusion (I/R) [25]. Furthermore, exogenous polyP has been reported to induce death in cultured cells [26]. This suggests that involvement of polyP in mitochondrial pathology may occur through different mechanisms. The central goal of the present study was to investigate potential molecular mechanisms underlying these differential effects of polyP on mitochondrial function.
Figure 1. Inorganic polyphosphate molecular weight and chain length is decreasing during ischemia-reperfusion.
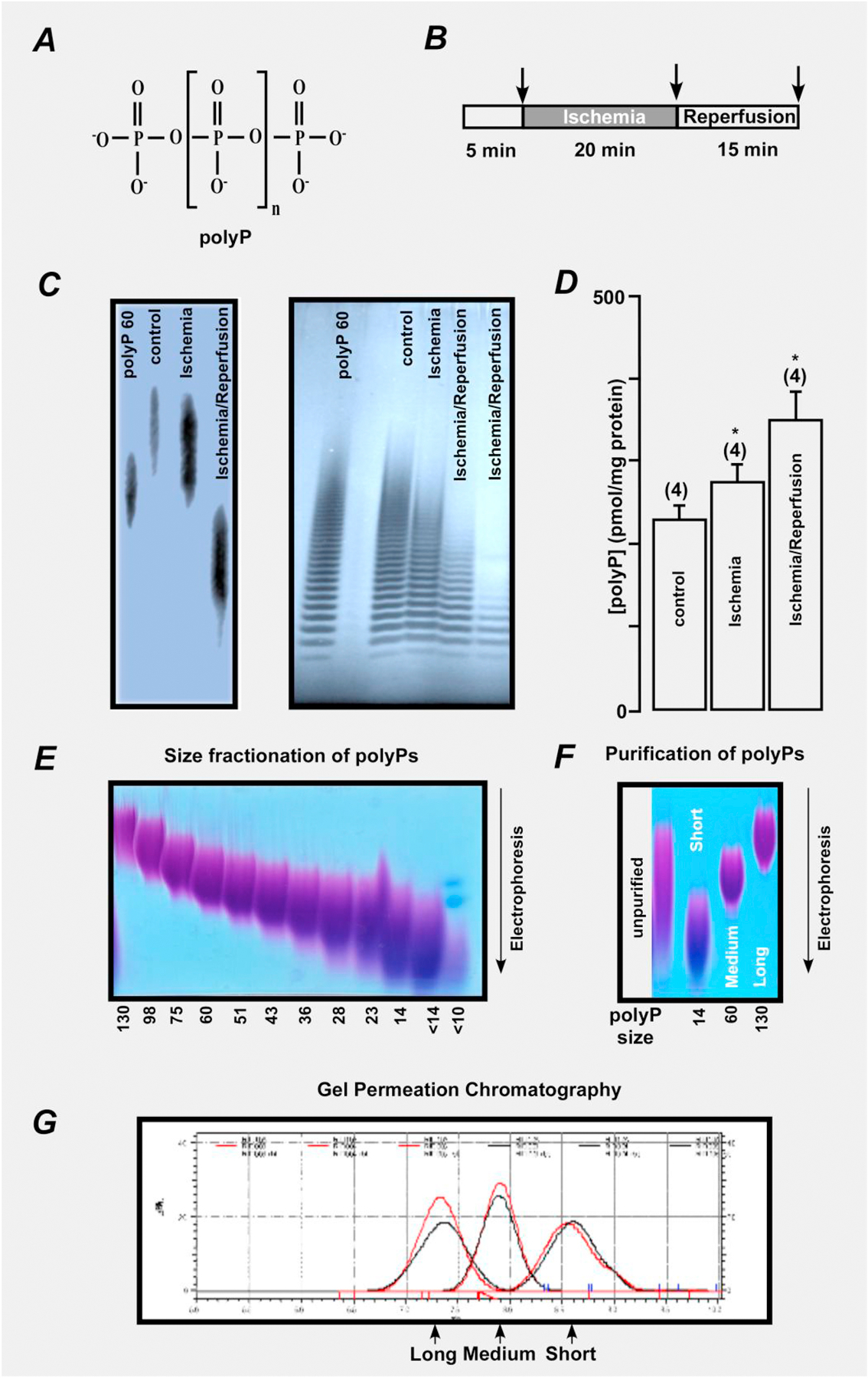
(A) Structure of inorganic polyphosphate (polyP). The n represents the number of phosphate residues in the polyP chain. It could vary from ten to hundreds of units. (B) Schematic representation of the protocol for ischemia and reperfusion. (C) Changes in polyP molecular weight (left) and polyP chain length (right) in mitochondrial lysates from mouse hearts perfused with Krebs-Henseleit buffer (control) and hearts exposed to 20 min ischemia or 20 min ischemia followed by 15 min of reperfusion. (D) Changes in polyP concentration in mitochondrial lysates from mouse hearts perfused with Krebs-Henseleit buffer (control) and hearts exposed to 20 min ischemia or 20 min ischemia followed by 15 min of reperfusion. (E) Sizes of fractionated polyPs ranging from long (130 phosphate residues) to short (14 phosphate residues and less) are separated by electrophoresis. (F) Purification of polyP is verified by electrophoresis. Sizes of unpurified polyP (left) and polyP with short (14 phosphate residues), medium (60 phosphate residues), and long (130 phosphate residues) chain length are shown. G) Size of polyP was confirmed by gel permeation chromatography.
We hypothesize that the variable effects of polyP are linked to differences in polyP chain length under different physiological and pathological conditions. We, therefore, examined the size distribution of polyP in normal and pathological conditions and investigated effects of the synthetic polyPs with different chain length (short, medium, and long) on their ability to participate in energy metabolism and on Ca2+-induced mPTP. We found that polyP effects were strongly size dependent. Short chain polyP (14 phosphate residues) led to the mPTP activation, induction of proton leak, mitochondrial uncoupling and metabolic failure. Long chain polyP (130 phosphate residues) demonstrated protection against mPTP, enhanced mitochondrial coupling and improved energy generation. Medium chain polyPs (60 phosphate residues) had an intermediate effect on ATP generation, and did not affect mPTP activity. These data further support the idea that involvement of polyP in mitochondrial function should be considered in connection with specific metabolic and functional state of the organelle.
2. Materials and methods
2.1. Animal models
All protocols were in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH Publication NO. 85–23, revised 1996) and approved by the University of California Davis Institutional Animal Care and Use Committee and by the University of Würzburg Institutional Animal Care and Use Committee. Experiments were performed in isolated hearts, cardiac myocytes or mitochondria from 10 week old wild type C57BL/6J controls (WT) and age-matched cyclophilin D (CypD) knock-out (KO) mice (mice with genetically deleted Ppif gene (Ppif−/−) [27] as we described before [28].
2.2. Cell Isolation
Mouse ventricular myocytes were isolated using a standard enzymatic technique using a Langendorff perfusion system [28]. Briefly, 5 min after mice were heparinized (1000 USP units) they were anesthetized by inhalation of isofluorane. When reflexes were absent, hearts were excised, placed into the Langendorff system and perfused for 5 min with a nominally Ca2+-free washing solution of composition (mM, unless specified): 140 NaCl, 4 KCl, 1 MgCl2, 5 HEPES, 10 Glucose, 1% (v/v) heparin; pH 7.4 adjusted with NaOH, followed by an enzyme solution containing 0.12 mg/mL of Liberase TM (Research Grade, Roche) in the above described solution. Upon digestion, hearts were minced and gently agitated to obtain the cells. All solutions were saturated with oxygen. Once myocytes were isolated, [Ca] was gradually raised to 1 mM using heparin-free Tyrode solution containing (in mM): 140 NaCl, 4 KCl, 1 MgCl2, 5 HEPES, 10 Glucose, 1 CaCl2, pH 7.4 adjusted with NaOH. Freshly isolated ventricular myocytes were plated on laminin-coated glass coverslips and used within 8 hours of isolation.
2.3. Permeabilized ventricular myocytes
The sarcolemma was permeabilized with digitonin (10 μM for 60 s) as described previously [8, 28]. Digitonin was added to intracellular solution containing (in mM): 135 KCl, 10 NaCl, 20 HEPES, 5 pyruvate, 2 glutamate, 2 malate, 0.5 KH2PO4, 0.5 MgCl2, 15 2,3-butanedione monoxime, 5 EGTA, and 1.86 CaCl2 to yield a free [Ca2+]i of 100 nM with pH 7.2. After permeabilization, the bath solution was changed to the same intracellular solution but without digitonin. Free Ca2+ concentrations were calculated using the MaxChelator program (http://www.stanford.edu/~cpatton/maxc.html).
2.4. Ischemia-reperfusion protocol and mitochondrial isolation from mouse hearts
Hearts were excised from mice and placed in ice-cold Krebs-Henseleit buffer (KHB) containing in mM: 120 NaCl, 4.7 KCl, 1.2 KH2PO4, 25 NaHCO3, 1.2 MgSO4, 11 D-glucose, 1 CaC2, 1% (v/v) heparin; pH 7.4 which was oxygenized by 95% O2/5% CO2. The aorta was cannulated and hearts were perfused in a retrograded fashion on a Langendorff apparatus with Krebs-Henseleit buffer for 5 min and exposed to global no flow ischemia for 20 min followed by restoration of perfusion flow for 15 min (reperfusion). Mitochondria from 2–4 mouse hearts were isolated using the protocol of Rehncrona et al. [29] with some modifications [8, 30]. The minced heart tissue was subjected to protease treatment: it was incubated with 5 mg proteinase (bacterial; type XXIV, formerly called Nagarse; Sigma-Aldrich) dissolved in 10 ml medium A for 8 min at room temperature while gently stirring. The protease reaction was stopped by adding 1 ml of 0.2 mg/ml of bovine serum albumin dissolved in medium A containing (in mmol/L): 225 mannitol, 70 sucrose, 1 EGTA and 10 HEPES, pH 7.2. The tissue was then homogenized with a Potter-Elvehjem homogenizer, and mitochondria were isolated by differential centrifugation as described by us before [8]. The final mitochondrial pellet was suspended in isolation medium B containing (in mmol/L): 225 mannitol, 70 sucrose, and 10 HEPES, pH 7.2. Mitochondrial protein concentration was determined by protein assay (Bradford, 1976) (Pierce BCA; Thermo Fisher Scientific). All isolation procedures were performed on wet ice.
2.5. Mitochondrial oxygen consumption
Mitochondrial oxygen consumption (respiration) was measured polarographically at 37°C with a Clark-type oxygen electrode (Model 782; Strathkelvin Instruments; Glasgow, UK) as previously described [28]. Freshly isolated mitochondria placed in 300 μl of MiRO5 respiration buffer containing (in mM): 110 Sucrose, 60 K-lactobionate, 20 HEPES, 3 MgCl2, 20 taurine, 10 KH2PO4, 0.5 EGTA, and 1 g/l BSA, pH = 7.1, and basal respiration rate was recorded. State 2 of respiration was initiated by the addition of 5 mmol/L glutamate and 5 mmol/L malate as substrates in the presence of 2 µM free Ca2+. Maximal respiration rate (State 3) was activated by adding 1 mmol/L ADP in the presence of 2 µM free Ca2+. Respiration rates were expressed as nmol O2 min−1 mg mitochondrial protein−1, and normalized to the no polyP treatment conditions. The respiratory control ratio (RCR) was calculated as state 3 divided by state 2 respiration rate (RCR=State3/State2) to estimate mitochondrial integrity and coupling efficiency.
2.6. polyP extraction, quantification, and size determination
polyP was extracted using a modified phenol/chloroform extraction protocol [6, 8]. The mitochondrial pellet (see above) was resuspended in 250 μl TELS buffer (100 mM LiCl, 10 mM EDTA, and 10 mM Tris, pH 8.0, 0.5% SDS) and mixed with 250 μl of acid phenol/chloroform, pH 4.5 (with isoamyl alcohol [IAA]; acid phenol/chloroform/IAA [125:24:1]; Invitrogen). 425–600-μm glass beads (Sigma-Aldrich) were added to the level right below the phenol fraction for extraction of polyP associated with the membrane fraction. Samples were vortexed for 5 min at 4°C, followed by centrifugation at 1,500 g for 5 min at 4°C. The water phase was transferred to a new tube and subjected to chloroform extraction with the equal volume of chloroform to remove traces of organic solvents from the water phase. polyP was precipitated from the water phase by adding 2.5 volumes of ethanol, followed by overnight incubation at 20°C. The water-ethanol mixture was centrifuged for 10 min at 10,000 g. The resulting pellet containing polyP was resuspended in 50 μl of a buffer (0.1% SDS, 1 mM EDTA, and 10 mM Tris-HCl, pH 7.4) treated with RNAse and DNAse to remove nucleic acid contamination and loaded on a 30% polyacrylamide gel prepared in the following solution (in mM): 90 Tris-KOH, pH 8.3, 90 boric acid, and 2.7 EDTA in the presence of 7 M urea. A gel size of 70 cm was run for 30 min at 80 V, followed by 6 h at 40 V. polyP was visualized by DAPI staining [31].
2.7. Synthetic polyP synthesis and fractionation
Synthetic polyP with 14 phosphate residues linked together (short chain), 60 (medium chain), and 130 (long chain) were synthesized and purified as described before in [32]. To synthesize polyP, sodium phosphate was rapidly heated to the temperature higher than 600°C and the melted phosphate solution was rapidly cooled to the room temperature (RT). Depending on the heating temperature and time, polyP of various chain lengths could be synthesized. To extract polyP, sodium polyP was dissolved in purified water (10 w/v%), and then 96% ethanol was gradually added to the solution at final concentration of 14 w/v%. The solution was vigorously stirred and allowed to stand at RT for approximately 30 minutes. Then centrifugation (10,000 g, 20 minutes at RT) was performed to separate the precipitate from the aqueous solution. The aqueous solution fraction was discarded, and 70% ethanol was added to the collected precipitate for washing, and then the precipitate was vacuum-dried. The resultant polyP solution had high viscosity and normally contained more than 60% polyP. Gel permeation chromatography (HPLC) was performed to separate polyP by size using Shimadzu LC-2010C with refractive index detector (RID-10A) and Ohpak SB-803 HQ column (Shodex, column size 30 cm with 8 mm internal diameter) that had been equilibrated with 100 mM NaCl (pH 7.5). Samples were run at a flow rate of 1 ml per minute at a temperature of 25°C. Protein absorbance was used to determine protein elution. The fractions were tested for polyP by DAPI staining [31]. Combining of this heating method, subsequent ethanol extraction and size fractionation protocol, we obtained and purified short, medium and long-chain polyP with ~14, 60 and 130 phosphate residues, respectively. Stock solutions of polyP standards (sodium salt with polyP content of 60%) were prepared in distilled water.
2.8. Confocal microscopy
Laser scanning confocal microscopy (Nikon A1 and a Zeiss LSM 780) was used to follow the changes in mitochondrial polyP accumulation, mitochondrial permeability transition pore (mPTP), mitochondrial membrane potential (ΔΨm), and mitochondrial ATP generation using specific fluorescent indicators. For measurement the myocytes were plated on laminin-covered coverslips and incubated in Tyrode solution containing 1 mM Ca2+ at room temperature. The fluorescence image was recorded every 5 sec. Fluorescence levels were corrected for background fluorescence and normalized as described below.
2.8.1. Mitochondrial polyP accumulation
These were estimated in intact cells loaded with 5 μg/ml 4’,6-diamidino-2-phenylindole, dihydrochloride (DAPI) for 30 min at 37°C [8, 25]. DAPI was excited with 408-nm laser light, and emitted fluorescence was measured at 552–617 nm. For DAPI emission spectrum recording, cells were excited at 408 nm, and the emission spectrum was collected between 500 and 675 nm. Data are presented as background subtracted fluorescence in arbitrary fluorescence units collected from the whole cell.
2.8.2. ATP Measurements
ATP measurements were performed indirectly via the free magnesium (Mg2+) concentration using the fluorescent dye mag-fluo-4 [33, 34]. Since free [Mg2+]i is kept constant within a rather narrow range, ATP hydrolysis leads to concomitant increase in free [Mg2+]i as measured with fluorescent Mg2+ indicators such as mag-fluo-4 [33]. Therefore, an increase in mag-fluo-4 fluorescence indicates a decrease in ATP concentration. For ATP measurements myocytes were loaded with 10 µM mag-fluo-4/AM (λex=488 nm, λem= 565–605 nm) for 30 min at 37°C. All data from these measurements are expressed as R=1-F/F0.
2.8.3. mPTP activity
mPTP activity was monitored in permeabilized myocytes loaded with 5 μM calcein/AM (λex=488 nm, λem=510 nm) for 40 min at 37°C [34]. Opening of mPTP resulted in the loss of mitochondria-trapped calcein (620 Da) and a decrease of fluorescence. At the end of each recording 10 μg/ml of the pore-forming antibiotic alamethicin was applied to provide a control measure for maximum calcein release from mitochondria. Loss of mitochondrial calcein induced by elevating [Ca2+]em was quantified as the rate of decline of fluorescence (d(ΔF)/dt) calculated from the linear fit to the initial decrease of calcein fluorescence. The rate of decline was normalized (%) to the basal decline of calcein fluorescence addition (taken as 100%) before [Ca2+]em elevation. In experiments performed in intact cells, cardiomyocytes were additionally incubated with 1 mM CoCl2 to quench cytosolic calcein fluorescence as described in [25, 35].
2.8.4. Mitochondrial Membrane Potential
Changes in mitochondrial membrane potential (ΔΨm) were followed using the potential-sensitive dye tetramethylrodamine methyl ester (TMRM; λex=514 nm, λem=590 nm) [25, 34]. Cells were exposed to 5 nM TMRM for 30 min at 37°C prior to experiments. All solutions contained 5 nM TMRM during recordings. In the end of each experiment 10 µM carbonyl cyanide p-(trifluromethoxy)phenylhydrazone (FCCP) was added to calibrate the signal. All data were background corrected.
2.8.5. Chemical in-vitro Ischemia/Repefusion Protocol
Ischemia was simulated by acidosis (pH 6.4), inhibition of glycolysis (glucose replaced with 20 mM deoxyglucose), and inhibition of mitochondrial respiration (with complex IV inhibitor sodium cyanide, NaCN) by cell exposure to glucose-free modified Tyrode solution containing (in mM) 20 2-deoxyglucose, 2 NaCN, 135 NaCl, 4 KCl, 1 MgCl2, 2 CaCl2, and 10 Hepes, pH 6.4 for 20 min [25]. Reperfusion was simulated by 15 min superfusion with standard Tyrode solution consisting of (in mM) 135 NaCl, 4 KCl, 10 glucose, 10 Hepes, 1 MgCl2, and 2 CaCl2; pH 7.4.
2.8.6. Superoxide Generation during in-vitro Chemical Ischemia and Repefusion
Reactive oxygen species (ROS) measurements were performed in intact cells loaded with the fluorescent probe MitoSOX Red that preferentially detects superoxide, O2.−. Cells were loaded with 0.5 µM MitoSox Red for 30 min at 37°C [25]. Changes in [ROS] are expressed as rates of ROS production (d(ΔF/F0)/dt) estimated from the initial linear phase of the MitoSOX Red (λex=543 nm, λem=555–617 nm) fluorescence increase normalized to basal rate.
2.9. Statistical Analysis
All data are expressed as mean ± standard error. Statistical significance of differences between experimental groups was determined using Student paired t test or two-way ANOVA, followed by Tukey’s post-test when appropriate. A value of p<0.05 was considered statistically significant.
3. Results
3.1. Ischemia-reperfusion leads to the decrease in polyP chain-lenght
We have previously shown that inorganic polyP is a potent activator of Ca2+-induced mitochondrial permeability transition pore (mPTP) in rabbit cardiac myocytes and that polyP depletion by expression of yeast exopolyphosphatase (PPX) prevents mPTP activation [8]. However, when experiments were performed to simulate ischemia-reperfusion (I/R) injury, polyP depletion failed to protect from oxidative stress-induced mPTP opening and even exacerbated cell death following I/R [25]. We, therefore, aimed to determine the reason for this discrepancy and evaluate whether polyP has an additional, mPTP independent mechanism of action in the heart. To achieve this goal, we subjected Langendorff-perfused mouse hearts to I/R injury using the protocol shown in Fig. 1B.
Specifically, hearts were perfused with Krebs-Henseleit buffer for 5 min and then exposed to global no-flow ischemia for 20 min, followed by restoration of flow for 15 min (reperfusion). Mitochondria were isolated from 3 groups of hearts (control perfusion with no exposure to I/R, ischemia exposure only, and hearts exposed to sequential I/R) and polyP was detected as described before [8]. Fig. 1C illustrates a decrease in polyP chain length in mitochondria which where subjected to ischemia and I/R (left image), and the presence of polyP is evident by its characteristic ladder-like polyP distribution on high-resolution polyacrilamide gel (right image) [36]. The appearance of this ladder is caused by the fact that both polyP standard and polyP sample from mitochondria are composed of a heterogeneous mixture of polymers of various sizes. Each band seen on the gel represents a polyP polymer of a certain size, with every next band representing the polymer that differs by a single Pi group (Fig. 1 C, right). Furthermore, we detected that total polyP concentration was actually increased from 230 ± 30 pmol/mg of protein in control (untreated conditions) to 350 ± 50 pmol/mg polyP (Pi units released by PPX treatment), n=4, P<0.05 following I/R (Fig. 1D). The amount of polyP was quantified using a highly specific enzymatic assay that relies on measurements of Pi released from the sample upon treatment with recombinant PPX [37]. Overall, these ex-vivo experiments suggest that both amount and size of polyP change dramatically during I/R, and are in agreement with our previous in-vitro study where polyP levels were increased in conditions of simulated chemically-induced ischemia and reperfusion [25]. PolyP depletion achieved by expression of the mitochondrially-targeted PPX resulted in nearly ~80% decrease in total mitochondrial polyP levels. While polyP depletion with PPX led to the decrease in mPTP activity, it also increased cell death following I/R [25].
Since polyP depletion with PPX eventually leads to the decrease of both long and short polyPs [8], we hypothesized that the observed shortening in polyP chain length during I/R could be responsible for its different effects on mitochondrial function. To test this hypothesis, we proceeded with in-vitro experiments to examine the effect of synthetic polyPs with different chain length on mitochondria. In-vivo experiments with synthetic polyPs are not feasible because polyP is known to enhance blood coagulation and clotting [38, 39] which will make data interpretation difficult. Furthermore, at the moment there are no known plasma membrane transporters of polyP that also precludes us from performing experiments in intact hearts or cardiomyocytes with synthetic polyPs. Polyphosphates with different chain length: short (14 phosphate residues linked together), medium (60 phosphate residues), and long-chain (130 phosphate residues) were synthesized (Fig. 1E) and purified as described before [32] and the purity of preparation is shown in Figs. 1F and G.
3.2. Calcium dependent uptake of polyP by the energized mitochondria
First, we tested whether mitochondria are capable of polyP uptake under energized conditions in permeabilized cardiac myocytes. To do this, we used DAPI fluorescent probe and confocal settings optimized for polyP detection as we described before [8, 12, 25]. As shown in Figs. 2A and B, addition of 100 µM short (polyP-14) or long (polyP-130) did not lead to a significant polyP accumulation inside of mitochondria in the conditions of low extramitochondrial Ca2+ ([Ca2+]em). However, when [Ca2+]em was elevated from 0.1 to 2 µM, both short and long polyP accumulated inside mitochondria but at different rates and extents. As demonstrated in Fig. 2C, more short-length polyP was accumulated inside of mitochondria compared to long polyP. Interestingly, in both cases the polyP uptake was inhibited by F0/F1-ATP synthase inhibitor oligomycin, indicating a link between ATPase activity and polyP uptake. Furthermore, mPTP inhibition with 1 µM cyclosporin A (CsA) significantly elevated accumulation of long-chain polyP but did not affect accumulation of short chain polyP. These observations can be explained either by chain-length dependent differences in the rate of polyP membrane transport or in rates of polyP consumption inside mitochondria.
Figure 2. Inorganic polyphosphate accumulation in mitochondria depends on the activity of the mitochondrial ATP synthase.
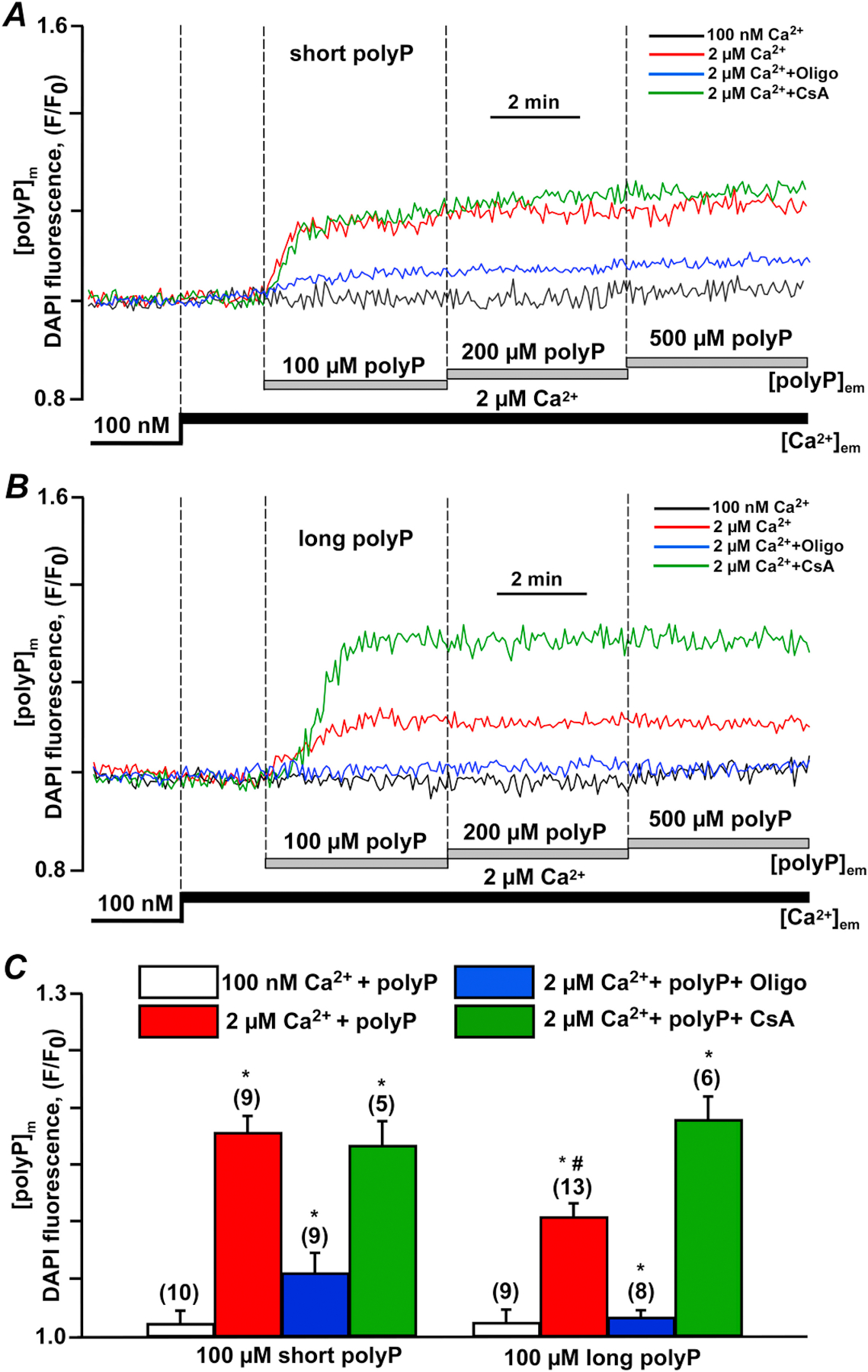
(A) Inorganic polyphosphate (polyP) accumulation in mitochondria of permeabilized mouse cardiac myocytes was induced by addition of 100–500 µM short (14 phosphate residues) polyPs upon elevation of extramitochondrial Ca2+ ([Ca2+]em) from 0.1 to 2 µM in the absence (red) or presence of 5 µg/ml Oligomycin (Oligo, blue) or 1 µM cyclosporin A (CsA, green). (B) polyP accumulation in mitochondria of permeabilized mouse cardiac myocytes was induced by addition of 100–500 µM long (130 phosphate residues) polyPs upon [Ca2+]em elevation from 0.1 to 2 µM in the absence (red) or presence of 5 µg/ml Oligo (blue) or 1 µM CsA (green). (C) Summary bars of total accumulation of 100 µM short (14 phosphate residues) and long (130 phosphate residues) polyPs in mitochondria of permeabilized mouse cardiac myocytes in the absence or presence of 5 µg/ml Oligo or 1 µM CsA. Data expressed as mean±SEM. n=number of cells tested from 3 to 5 wild type (WT) mice. *p<0.05 vs corresponding addition of polyP in low [Ca2+]em conditions for short and long polyPs.
3.3. PolyP stimulates ATP production in a chain-length and CypD dependent manner
Next, we investigated the effect of added polyPs on mitochondrial energy metabolism by monitoring ATP generation with the Mg2+-sensitive fluorescent dye mag-fluo-4 (Fig. 3). This method takes advantage of the fact that ATP is the main intracellular Mg2+ buffer, such that reduced [ATP]i raises free [Mg2+]i,, and a decline in ATP levels leads to an increase in [Mg2+]i, resulting in corresponding changes in mag-fluo-4 fluorescence [33]. To reflect [ATP]i changes, we inverted mag-fluo-4 signals in Fig. 3. As shown in Fig. 3A, long-chain and to a lesser degree, medium polyPs increased ATP levels in permeabilized WT myocytes, while short chain polyP did not. Addition of the mitochondrial uncoupler 10 µM FCCP led to a significant decrease in ATP levels in myocytes treated with long and medium-chain polyPs, however did not have any effect on cells treated with short-chain polyP consistent with mitochondrial already being uncoupled. Importantly, short chain polyP was able to increase ATP levels in myocytes lacking cyclophilin D (CypD KO) that are resistant to mPTP (Fig. 3B, C). This potentially suggests that the lack of stimulation by short chain polyP in Fig. 3A might be due to mPTP activation (and uncoupling), rather than its inability to stimulate ATP production (which was preserved when mPTP was inhibited).
Figure 3. Inorganic polyphosphate effect on ATP generation depends on polymer chain length and cyclophilin D presence.

(A) Representative traces of normalized inverted fluorescent mag-fluo-4 intensities reflecting ATP generation inside permeabilized WT cardiac myocytes upon [Ca2+]em elevation from 0.1 to 2 µM in presence of short (14), medium (60), and long (130) polyP. 10 μM FCCP was added to uncouple mitochondria and normalize the signal. (B) Representative traces of normalized inverted fluorescent mag-fluo-4 intensities reflecting ATP generation inside permeabilized cardiac myocytes from cyclophilin D (CypD) KO mice upon [Ca2+]em elevation from 0.1 to 2 µM in presence of short (14), medium (60), and long (130) polyP. (C) Normalized ATP levels in cardiac myocytes from WT and CypD KO mice treated with short, medium and long chain polyP. n=number of cells obtained from 3 to 5 WT and CypD KO animals. Data expressed as mean±SEM. *p<0.05 WT vs CypD KO groups, **p<0.05 short polyP vs medium and long polyPs.
3.4. Long-chain polyPs enhance ADP-linked respiration and respiratory reserve capacity while short-chain polyPs induce proton leak and mitochondrial uncoupling
Next, using a Clark type electrode, we monitored oxygen consumption in isolated cardiac mitochondria from WT and CypKO animals. Mitochondria were placed in MiRO5 respiration buffer and basal respiration was recorded for 1 min. Then state 2 respiration was activated by addition of the mitochondrial complex I substrates 5 mmol/L malate and 5 mmol/L glutamate in the presence of 2 µM free Ca2+ and rate of oxygen consumption was measured. Addition of extramitochondrial Ca2+ was required since synthetic polyPs were entering mitochondria only in the presence of Ca2+ as shown in Fig. 2. Then ADP-induced respiration (State 3) linked to oxidative phosphorylation was measured by subsequent addition of 1 mmol/L ADP to the respiration chamber. Respiration rates (in nmol O2 min−1 mg mitochondrial protein−1) were normalized to no-polyP treatment conditions for Fig. 4. As shown in Fig. 4A, short polyP significantly increased basal respiration, while state 2 (Mal/Glut) and state 3 (ADP) respiration was decreased indicating a possible proton leak and uncoupling of the mitochondrial respiratory chain resulting in the decline in the respiratory control ratio (RCR; Fig. 4C). RCR is calculated as state 3 divided by state 2 respiration rate and represents a measure of the mitochondrial integrity and coupling efficiency. Interestingly, in agreement with data presented in Fig. 3, long chain polyPs significantly increased ADP-activated state 3 respiration in WT mitochondria leading to the enhancement in RCR and therefore mitochondrial coupling. The effect of long-chain polyP on the increase in state 3 and RCR was not different in WT and CypD KO mitochondria, however the uncoupling effect of short-chain polyP was prevented in mitochondria from CypD KO hearts, suggesting that uncoupling effect of short-chain polyP most probably was due to mPTP activation, and the effect of long polyP on energetics did not involve CypD.
Figure 4. Long-chain length polyphosphate but not short-chain length polyphosphate enhances ADP-dependent respiration and mitochondrial respiratory control ratio.

(A) Oxygen consumption in isolated mitochondria from WT mouse hearts at baseline (basal) and upon subsequent addition of the mitochondrial complex I substrates (5 mmol/L malate and 5 mmol/L glutamate) in the presence of 2 µM free Ca2+ to induce State 2 of respiration followed by addition of 1 mmol/L ADP to induce State 3 of respiration in the absence or presence of 100 µM short or long chain polyPs. (B) Oxygen consumption in isolated mitochondria from CypD KO mouse hearts at baseline (basal) and upon subsequent addition of the mitochondrial complex I substrates (5 mmol/L malate and 5 mmol/L glutamate) in the presence of 2 µM free Ca2+ to induce State 2 of respiration followed by addition of 1 mmol/L ADP to induce State 3 of respiration in the absence or presence of 100 µM short or long chain polyPs. (C) Respiratory control ratio (RCR) was decreased in mitochondria treated with short polyPs in CypD-dependent manner while long chain polyPs increased RCR independently of CypD presence. n=mitochondrial preparations from 3–5 WT and CypD KO animals. Data expressed as mean±SEM. *p<0.05 vs corresponding no polyP conditions.
3.5. Short-chain polyP activates mitochondrial permeability transition pore
Our data suggest that polyP can be taken up by mitochondria in a calcium dependent manner and stimulate mitochondrial ATP production. At the same time short chain polyP causes uncoupling effect on mitochondria that is prevented by KO of mPTP activator CypD. This is consistent with our previous reports that demonstrate that polyP can act as a potent activation of mPTP. To investigate whether the length of polyP polymer is important for previously observed effect of polyP on mPTP activity, we directly monitored mPTP activity in permeabilized cardiac myocytes with mitochondrially-entrapped fluorescent probe calcein. Mitochondrially-entrapped calcein green with a molecular weight of 620 Da can be released from mitochondria only when mPTP opens which allows ions and compounds with the molecular weight less than 1500 Da to pass through [28, 34]. In these experiments, mPTP activity was quantified as % change in the rate of calcein fluorescence decline compared to basal conditions (defined as 100% in [Ca2+]em = 0.1 μM). As shown in Figs. 5A and D, an elevation of [Ca2+]em from 0.1 to 2 μM caused calcein release from mitochondria (rate of fluorescence decline was 310 ± 55%; n = 20) that was significantly inhibited in both CsA-treated WT (102 ± 3%; n = 10, P <0.01 compared to untreated WT; Figs. 5B and D) and CypD-KO (123 ± 8%; n = 19, P < 0.01 compared to WT; Figs. 5C and D) myocytes confirming the fidelity of this assay for mPTP detection. As shown in Fig. 5A and summarized in Fig. 5D, short chain polyPs significantly enhanced the rate of calcein release (524 ± 52%; n =27, P<0.05 compared to control), and therefore mPTP activity in WT myocytes. Medium chain polyP did not have a significant effect (385 ± 58%; n =20, P=0.18), while long-chain polyP decreased mPTP opening (158 ± 13%; n =21, P<0.05 compared to control; Fig. 5A and D). The effect of short chain polyP was abrogated in WT myocytes in the presence of CsA (185 ± 26%; n =13, P<0.05 compared to control; Fig. 5B, D) or in CypD KO myocytes (157 ± 16%; n =14, P<0.01 compared to control; Fig. 5C, D) further confirming mPTP involvement.
Figure 5. Short-chain length polyphosphate (polyP) but not long-chain length polyP increased opening of the mitochondrial permeability transition pore.
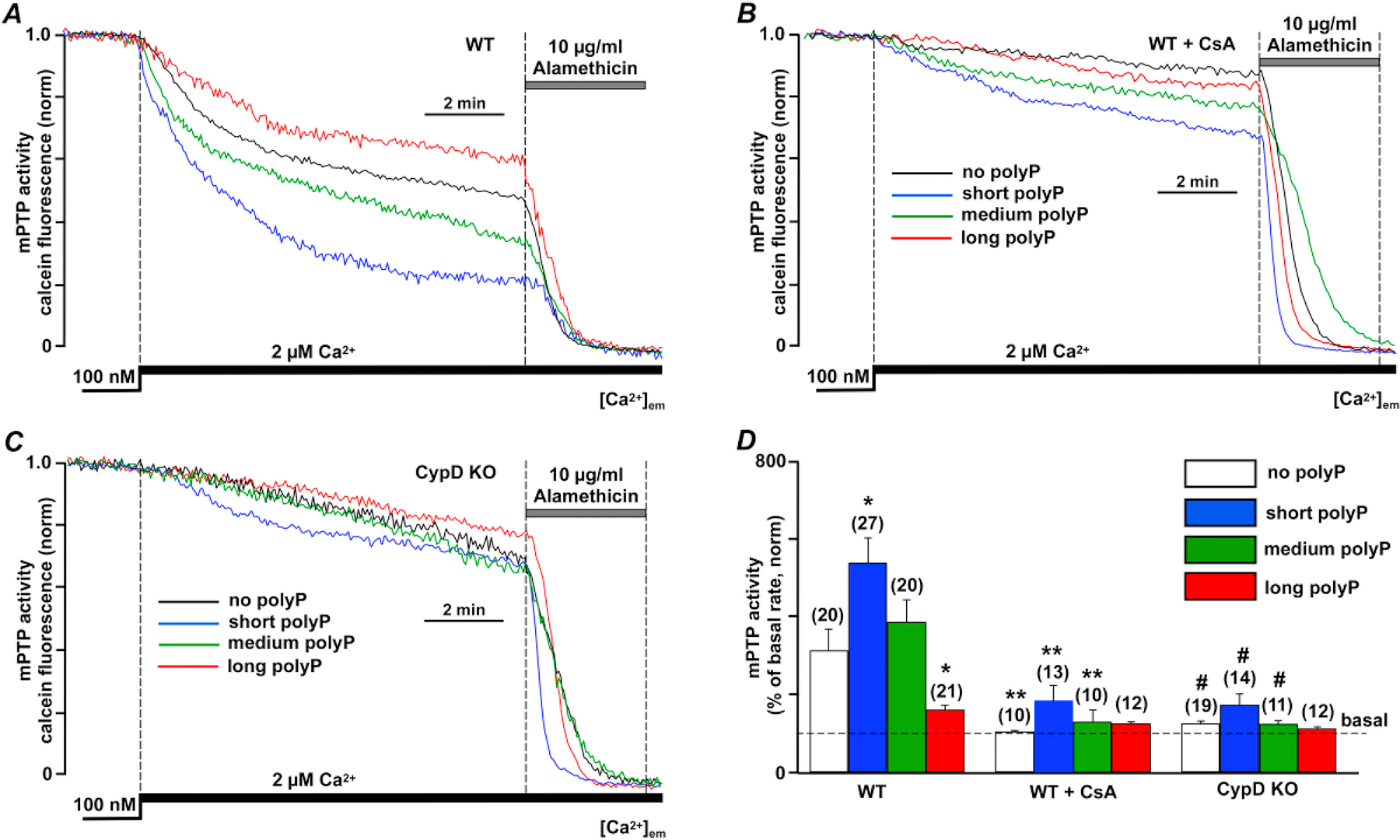
(A) Representative traces of normalized calcein release from mitochondria in permeabilized WT cardiac myocytes reflecting opening of the mitochondrial permeability transition pore (mPTP) upon elevation of extramitochondrial Ca2+ ([Ca2+]em) from 0.1 to 2 µM in the absence and presence of short (blue), medium (green) and long chain (red) polyPs. 10 μg/ml alamethicin was added in the end of each recording to release all calcein from mitochondria and obtain the minimal fluorescence level required to calculate total calcein signal. (B) Representative traces of normalized calcein release from mitochondria in permeabilized WT cardiac myocytes treated with 1 µM CsA upon [Ca2+]em elevation from 0.1 to 2 µM in the absence and presence of short (blue), medium (green) and long chain (red) polyPs. (C) Representative traces of normalized calcein release from mitochondria in permeabilized CypD KO cardiac myocytes upon [Ca2+]em elevation from 0.1 to 2 µM in presence of short, medium and long chain polyPs. (D) Summary data reflecting mPTP activity in cells from 3 animal groups. n=number of cells from 3 to 5 animals. Data expressed as mean±SEM. *P<0.05 in untreated vs poly P treated cells in WT myocytes, **P<0.05 in WT+CsA group vs WT myocytes, and #P<0.05 in CypD KO group vs WT myocytes
3.6. PolyPs with short chain-length but not with long-chain length induce a dissipation of the mitochondrial membrane potential
Since the opening of non-specific mPTP channel dissipates the mitochondrial membrane potential (ΔΨm), we tested how polyPs with different chain length affect ΔΨm. To monitor ΔΨm, we measured Ca2+-induced changes in ΔΨm using the voltage-sensitive dye TMRM in permeabilized WT cardiac myocytes. As shown in Fig. 6A, elevation of [Ca2+]em from 0.1 to 2 µM is associated with small ΔΨm depolarization (degree of ΔΨm depolarization normalized to total depolarization achieved after addition of the mitochondrial uncoupler 10 µM FCCP was 22 ± 2%; n = 6). Addition of short polyP, however, dramatically increased ΔΨm depolarization (62 ± 5%; n =6, P<0.01 compared to control; Fig. 6A, B) which corresponds to the increase in mPTP opening during mitochondrial exposure to high Ca2+ presented in Fig. 5A. Medium-chain polyP did not affect ΔΨm significantly (18 ± 2%; n =21, P=0.08 compared to control) while long-chain polyP actually diminished the degree of ΔΨm depolarization induced by Ca2+ (15 ± 2%; n=6, P<0.01 compared to control; Figs. 6A, B). To conclude, these data indicate that only short chain polyPs enhance mPTP activation which leads to dissipation of ΔΨm across mitochondria membrane.
Figure 6. Inorganic polyphosphate with short-chain length induced a profound drop in mitochondrial membrane potential.
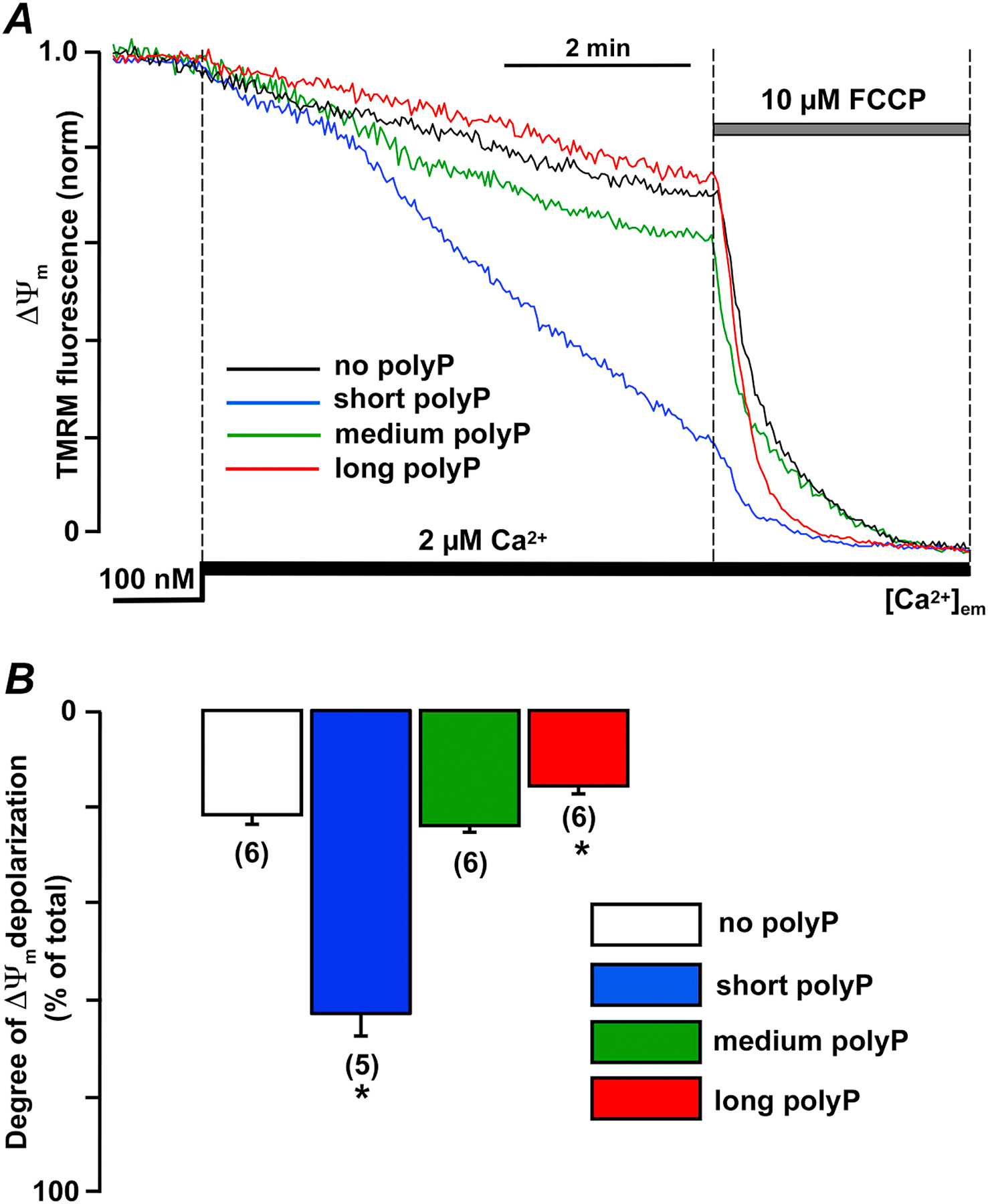
(A) Representative traces of normalized TMRM fluorescence reflecting changes in mitochondrial membrane potential in permeabilized WT cardiac myocytes upon elevation of extramitochondrial Ca2+ ([Ca2+]em) from 0.1 to 2 µM in presence of short (14), medium (60), and long (130) polyP. 10 μM FCCP was added to uncouple mitochondria and obtain the minimal fluorescence level to calculate total TMRM signal. (B) Shown are normalized drop in TMRM levels upon [Ca2+]em elevation from 0.1 to 2 µM in the absence or presence of short (blue), medium (green), and long (red) polyP. n=number of cells from 3 to 5 WT animals. Data expressed as mean±SEM. * P<0.05 between no polyP vs polyP
3.7. Elevated superoxide generation led to mPTP opening during ischemia in mouse cardiac myocytes which was further exacerbated in reperfusion
Since polyP is known to enhance blood coagulation and clotting [38, 39] we were not able to perfuse the whole mouse hearts with synthetic polyPs. To verify that conditions of ischemia and reperfusion (as shown in Fig. 1) are associated with excessive oxidative stress and mPTP opening, we exposed intact mouse cardiac myocytes to 20 min of simulated ischemia followed by 15 min of reperfusion with a similar protocol as we used before [25]. As shown in Figs. 7A, B, mPTP opening as monitored by calcein release from mitochondria was already observed under ischemic conditions, but was further exacerbated in reperfusion. Superoxide generation monitored by changes in MitoSOX Red fluorescence (Fig. 7C) was already elevated to a maximal degree during ischemia, and remained elevated during I/R. Despite the observed protection against mPTP opening in WT myocytes treated with CsA or CypD KO mice (Figs. 7A, B), the levels of superoxide were not changed under these conditions suggesting that oxidative stress was causing mPTP opening under ischemic conditions. Based on the fact that ischemia was associated with the increased polyP generation in the heart (see Figs. 1C,D), we suggest that similar to bacteria [40] mammalian cells produce polyP in response to the oxidative stress. Under conditions of the excessive mPTP opening in reperfusion (red line in Fig. 7A), long polyP could be used to support ATP generation leading to generation of short-chained polyPs capable of mPTP activation (see Fig. 5A). This, in fact, is indirectly supported by our data presented in Fig. 3A where addition of the mitochondrial uncoupler FCCP which dissipates ΔΨm (see Fig. 6A) similar to that observed in I/R, was associated with dramatic decrease in ATP levels when long polyP was used to support respiration. Addition of FCCP had no effect on ATP levels when short polyP was used, suggesting that mitochondrial respiratory chain was already uncoupled. These data, together with other results presented in this study suggest that polyP effects could be different depending on the metabolic status of cell.
Figure 7. mPTP opening and ROS generation was observed in mouse cardiac myocytes exposed to simulated chemical ischemia and reperfusion.

(A) mPTP activity monitored by changes in calcein fluorescence during 20 min ischemia and 15 min of reperfusion in control wild type cardiac myocytes in the absence (WT) or presence of 1 µM of CsA (WT + CsA), and myocytes from cyclophilin D knockout (CypD KO) mice. (B) Summary of the mPTP activity during ischemia and reperfusion calculated as the rate of calcein release from mitochondria normalized to the basal rate. (C) Superoxide generation during ischemia and reperfusion in 3 experimental groups (WT, WT + CsA, and CypD KO) calculated as the rate of MitoSOX Red fluorescence increase relative the basal rate. Data expressed as mean±SEM. n=number of cells from 3 to 5 WT and CypD KO animals. * P<0.05 between no treatment (basal level) vs ischemia and ischemia-reperfusion, ** P<0.05 between ischemia vs ischemia-reperfusion, # P<0.05 between WT vs WT +CsA and CypD KO
4. Discussion
In this study we investigated the differential effects of polyP on cardiac mitochondrial function. The main motivation was to find the solution to the prior dichotomy that mitochondrial polyP depletion protected hearts from Ca2+-overload induced mPTP opening [8, 12], but failed to protect against mPTP opening during I/R and even exacerbated cell death [25]. In this study, we found that differences in the functional effects of short vs. long polyP chains (14 vs. 130 phosphates) may resolve this dichotomy. Specifically, short polyP promotes mPTP and proton leak (bad), while long polyP suppresses mPTP and increases both ATP production and RCR (good). Moreover, ischemia-reperfusion caused a shift in endogenous polyP from longer to shorter forms (Fig. 1). This would worsen mPTP and mitochondrial dysfunction because of both the higher maladaptive effects of short and the loss of beneficial effects of long polyP. The depletion of long polyP by mitochondrially-targeted exopolyphosphatase (PPX) would remove the benefit of long polyP on mPTP suppression and may explain why polyP depleted hearts fared somewhat worse than controls during I/R.
To elucidate this duality of polyP effects, we employed synthetic polyPs with different chain length (Fig. 1), tested their ability to enter mitochondria (Fig. 2), influence energy metabolism (Figs. 3 and 4) and modulate Ca2+-induced mPTP and mitochondrial membrane potential (Figs. 5 and 6). We found that these synthetic polyPs could be taken up by mitochondria when added to permeabilized ventricular myocytes (Fig. 2), but that mPTP activation could limit polyP uptake and that the mitochondrial ATP synthase might be involved in polyP transfer across the inner mitochondrial membrane. The potential molecular mechanism for this ATP synthase involvement may merit further study. The use of polyP of different lengths allowed us to distinguish the aforementioned differential functional effects of short vs. long polyP. That is, short chain polyPs led to the activation of the mPTP (Fig. 5), induced proton leak in the oxidative phosphorylation pathway (Fig. 4), mitochondrial uncoupling (Figs. 3 and 6) and failed to increase ATP levels in cardiac myocytes (Fig. 3). In contrast, longer chain polyPs demonstrated a mainly protective effect against mPTP opening and also enhanced energy generation. These data further support the idea that involvement of polyP in mitochondrial function should be considered in connection with specific metabolic and functional state of the organelle. Indeed, a prior study performed in human leukemic cell line HL-60 demonstrated that apoptosis induction by cell treatment with actinomycin D resulted in degradation of long chain polyP and accumulation of short chain polyPs [10]. Furthermore, their study has also reported a significant decrease in long chain polyPs in rat brain and liver with aging, again emphasizing the importance of long polyPs for cell survival [10]. This might be particularly important due to the link between polyP and levels of ATP and ADP which are known modulators of mPTP [30]. Indeed, our work supports earlier studies regarding the close link between mitochondrial energetics and levels of polyP.
The possible contribution of polyP towards energy production in mammalian cells has been suggested by us before [11, 12]. In these experiments stimulation of energy metabolism led to increased mitochondrial polyP, while in cultured cells polyP depletion led to energetic failure. Interestingly, it was also demonstrated that mitochondrial depolarization leads to the decrease in polyP chain length – an effect similar to what we observed in this work during I/R. Studies performed in bone-forming osteoblast SaOS-2 cells demonstrated that addition of inorganic polyP stimulated bone maturation which was associated with an increase in ATP levels [41, 42]. Ca2+ ions were required for polyP to be effective for stimulation of bone mineralization and energy production in SaOS-2 cells. Notably, another study found that only medium and long-chain polyPs stimulated tissue regeneration and bone formation while short-chain polyPs did not have this positive effect [32].
While, as mentioned above polyP involvement in metabolism can be important contributor to the regulation of mPTP, our data do not indicate such a direct link. We hypothesize that in case of mPTP activation, polyP might be involved as a chaperone or a structural component of the mPTP channel. Previous studies have demonstrated that polyP forms complexes with poly-β-hydroxybutyrate (PHB) and Ca2+ on bacterial plasma membranes, and participate in Ca2+ transport inside bacteria which is responsible for initiation of bacterial competence [22, 43]. Pavlov et al. [44] were able to isolate similar polyP-Ca2+-PHB complexes from rat liver mitochondria which upon incorporation into lipid bilayers induced a current with characteristics similar to that observed in mPTP channels. Interestingly, the study of Baev et al. [45] did not find any effects of polyP on permeability of lipid bilayers when polyP was added alone to the artificial membranes but demonstrated modulatory effects of polyP on the membranes from the native de-energized mitochondria. This is a very important observation that suggests that polyP needs a co-partner to activate mPTP in mitochondria. We further confirmed the presence of polyP in cardiac mitochondria and their contribution toward mPTP activation upon mitochondrial Ca2+ overload [8]. PolyP which are present in mammalian cells are typically longer [3, 6] compared to polyPs found in bacteria and which are used for commercial purposes [46]. This is consistent with our data presented in Fig. 1 where under physiological conditions polyPs with medium to long chain length were present in cardiac mitochondria. However, a significant shift towards short polyPs was detected under pathological conditions such as I/R.
The mPTP activation process is complicated and the molecular details are not completely understood, with several possibilities currently been investigated [19, 47–49]. What appears to be an essential condition for mPTP activation is Ca2+ uptake by energized mitochondria in the presence of orthophosphate. Further, while molecular composition of the pore part of mPTP is not well established [49–51], mPTP activity strongly depends on the interaction with protein cyclophilin D (CypD) which belongs to the family of peptidyl-prolyl cis-trans isomerases whose enzymatic activity can be inhibited by cyclosporin A [52].
The mPTP effects of polyP here are not non-specific polyP effects on lipid bilayer membranes because it is strongly dependent on CypD and presence of mitochondrial Ca2+ uptake. Together with previous reports, the polyP involvement in mPTP seen here could occur by several, non-mutually excluding, mechanisms: 1) polyP might be directly involved in mPTP channel formation through Ca2+-mediated interaction with PHB and ATP synthase; 2) polyP effects could be linked to its ability to regulate mitochondrial Ca2+ buffering capacity thereby regulating the mPTP activation threshold; 3) polyP might be involved in the mPTP through its chaperone activity [40, 53] and it’s ability to modulate other proteins involved in mPTP activation and/or complex assembly. Specifically, the study of Gray et al. demonstrated that bacteria exposed to the oxidative stress respond by a dramatic accumulation of long-chain polyPs which are required for bacterial surviving in proteotoxic stress conditions [40]. This protective effect of polyP was related to its chaperone’s action and the ability of polyP to protect certain proteins against stress-induced unfolding and aggregation [40]. The concept of misfolded proteins as a key trigger of ROS-induced mPTP opening is well known [54]. Lemasters’s group [54] introduced the concept that mPTP forms by aggregation of misfolded integral membrane proteins damaged by oxidative stress. According to their model [54], mitochondrial damage after the oxidative stress causes misfolding and clustering of native mitochondrial membrane proteins. This misfolding can potentially expose hydrophilic residues to the bilayer phase to enclose aqueous channels conducting low molecular weight solutes. Initially, chaperone-like proteins are able to block conductance through these misfolded protein clusters [54]. However, when protein clusters exceed the ability of chaperons to block conductance [55] during increased oxidative stress observed in reperfusion, mPTP opens in high-conductance mode [54] leading to Ca2+, other ions and metabolites release from mitochondria. In our 2015 study [25], we demonstrated that total polyP levels in control rabbit ventricular myocytes were increased under conditions of simulated I/R suggesting that polyP is formed under the stress conditions. PolyP depletion by expression of mitochondria-targeted exopolyphoshatase (PPX) that specifically hydrolyzes polyP into inorganic phosphate resulted in nearly 80% decrease in mitochondrial polyP levels. While polyP depletion by PPX overexpression led to the decrease in mPTP activity, it also increased cell death following I/R [25]. Since polyP depletion with PPX eventually leads to the decrease of both long and short polyPs [8], we hypothesized that the observed shortening in polyP chain length (see Fig. 1 in the current manuscript) during I/R could be responsible for its different effects on mitochondrial function. Similar to the results obtained in rabbits, mPTP opening was observed in both ischemia (in a smaller degree) and reperfusion (more pronounced) conditions in isolated mouse cardiomyocytes (Figs. 7A, B). The decrease in calcein fluorescence (used as a measure for mPTP) was attenuated by cell treatment with CsA and was not observed in CypD KO mice confirming that the observed decrease in calcein fluorescence was indeed due to mPTP activity. Even though conditions of ischemia (in particular low pH) do not favor mPTP opening [56], other studies beside us showed mPTP opening during ischemia (see [57] for the review). However, in agreement with our previous study [25] and data from the Schumacker’s group [58, 59], mitochondrial oxidative stress was already observed during ischemia (see also Fig. 7C) which could contribute to polyP formation, mPTP opening and prime cardiomyocytes for cell death during reperfusion.
We think the third scenario is most likely. While we could not exclude the direct involvement of polyP in channel formation, earlier reports suggested that medium length polyPs were forming complexes with PHB in bacteria [22, 43, 60], but our strongest effects were with short-chain polyP chains. It is possible that the effect of polyPs with different chain length could be tissue, species, and concentration-dependent. Indeed, the study of Baev et al. [45] revealed that polyPs with longest chain length had the most prominent effect on mitochondrial swelling in de-energized rat liver mitochondria. These experiments, however, were performed in highly isosmotic conditions in the presence of 40 mM Ca(NO3)2 and 1 µM rotenone, a known inhibitor of the mitochondrial complex I. These experimental conditions would prevent possible beneficial effects of long polyPs to enhance mitochondrial bioenergetics since rotenone inhibits mitochondrial respiratory chain, and therefore ATP generation. While it is possible that such a difference is due to the experimental conditions it is tantalizing to propose that effects of polyP could be also tissue specific. Earlier studies from Kornberg’s lab [6] reported that heart and brain tissues contained the highest amount of polyP compared to other tissues. For examples, approximately 95 and 114 µM were extracted from brain and heart tissue, respectively, while only 38 µM polyP was extracted from liver tissues with similar protein content [6]. Furthermore, electrophoretic analysis revealed that all tissues samples examined in this study except the liver contained a mix of short, medium and long polyPs. Strikingly, only long chain polyPs were detected in liver samples [6, 9]. The physiological importance of this observation needs further evaluation but possibly can explain the difference between our study and the study of Baev et al. [45] which was performed using isolated liver mitochondria.
Ca2+ buffering effects are non-specific and thus are not expected to be chain dependent. Furthermore, our prior study did not find any significant changes in mitochondrial Ca2+ levels in cardiac myocytes treated with exopolyphosphatase PPX [8]. In contrast, polyP binding to molecular protein targets can be chain-length dependent [40, 61]. This interpretation is consistent with our observation that short chain polyP stimulates ATP production only in CypD KO cells (but not in WT cells) suggesting that in the absence of a mPTP inhibitor short chain polyP is bound to its putative molecular target. Whether the chain-length dependent target is CypD or some other protein remains to be established. Intriguingly, a recent study discovered that several human proteins are being post-translationally modified by polyP in the process called polyphosphorylation [62]. However, further studies are required to determine how this polyphosphorylation affects protein function and/or conformation.
We should also emphasize that long chain polyPs had no stimulating effect on mPTP activity, but rather inhibited mPTP opening (Fig. 5), and the effect of long polyP on ATP generation was not dependent on CypD. This may indicate that long chain polyPs lack the ability to bind to CypD and are thus potentially cardioprotective during I/R. While the enzyme responsible for polyP formation in mammalian cells is still unknown, it was demonstrated that human gastrointestinal tract bacteria (probiotics) produce polyP, and that polyP is responsible for probiotic actions that protect the intestinal epithelia from oxidant stress and epithelial injury [63]. Intriguingly, several studies [64, 65] demonstrated that probiotic administration attenuates myocardial infarction following I/R injury and prevents myocardial hypertrophy and heart failure development following myocardial infarction in the rat. Our data revealed that similar to bacteria, cardiac myocytes exposed to the oxidative stress during ischemia respond with compensatory increase in polyP formation (as shown in Fig. 1C, D). During reperfusion, however, polyP is used by cardiac myocytes to support cellular bioenergetics leading to the depletion of long polyP and accumulation of short chain polyPs which promote mPTP opening. From this point of view, promoting long polyP generation in reperfusion could be cardioprotective.
5. Conclusions
Taken together, these experiments have shown that inorganic polyP is actively generated and metabolized under conditions of stress such as ischemia and reperfusion. During I/R a significant reduction in polyP chain length is observed (from long to short). Using synthetic polyPs, we found that polyP has a dual effect on mitochondrial function in cardiac myocytes depending on polyP chain length. Short chain polyP (14 phosphate residues) strongly activated Ca2+-induced mPTP opening in cyclosporin A and cyclophilin D-dependent manner, indicating possible interactions of polyP with cyclophilin D. Long-chain polyP (130 phosphate residues) actually decreased mPTP opening and enhanced ADP-linked respiration and ATP levels in mitochondria. This could explain the fact that polyP depletion did not prevent mPTP opening and even exacerbated cell death following I/R in our previous study [25]. Therefore, promoting long chain polyP formation during I/R could be beneficial for cardiac function. Overall, these findings support the idea that the involvement of polyP in mitochondrial function should be considered in connection with the specific metabolic and functional state of the organelle.
Highlights.
Inorganic polyphosphate has a dual effect on mitochondrial function in the heart.
Ischemia and reperfusion decreases polyP chain length from long to short.
Short polyP activates mPTP opening in CsA and cyclophilin D-dependent manner.
Long polyP decreases mPTP opening and enhances ATP generation in mitochondria.
This could explain the fact that polyP depletion exacerbated cell death during I/R.
Acknowledgments
Financial supports
This work was supported by an American Heart Association Grant-In-Aid 15GRNT25090220 (to END) and NIH R01HL132831 (to DMB).
Nonstandard Abbreviations and Acronyms
- Ca2+
free calcium
- [Ca2+]em
extramitochondrial free calcium
- CsA
cyclosprorin A
- CypD
Cyclophillin D
- ETC
electron transport chain
- FCCP
carbonyl cyanide 4-(trifluoromethyhoxy)-phenylhydrazone
- I/R
ischemia-reperfusion
- KO
knockout
- mPTP
mitochondrial permeability transition pore
- PHB
poly-β-hydroxybutyrate
- polyP
inorganic polyphosphate
- PPX
exopolyphosphatase
- ROS
reactive oxygen species
- WT
wildtype
- ΔΨm
mitochondrial membrane potential
Footnotes
Conflicts of Interests
None
References
- [1].Dedkova EN, Biochem Soc Trans 44 (2016) 25–34. [DOI] [PubMed] [Google Scholar]
- [2].Kornberg A, Rao NN, Ault-Riche D, Annu Rev Biochem 68 (1999) 89–125. [DOI] [PubMed] [Google Scholar]
- [3].Seidlmayer LK, Dedkova EN, in: Kulakovskaya T, Pavlov E, Dedkova EN (Eds.), Inorganic polyphosphates in eukaryotic cells, Springer International Publishing, Switzerland, 2016, pp. 91–114. [Google Scholar]
- [4].Kulaev IS, Kulakovskaya TV, Andreeva NA, Lichko LP, Prog Mol Subcell Biol 23 (1999) 27–43. [DOI] [PubMed] [Google Scholar]
- [5].Kulaev I, Vagabov V, Kulakovskaya T, New York, NY: John Wiley & Sons Inc. Ltd ISBN: 0–470-85810–9 (2004). [Google Scholar]
- [6].Kumble KD, Kornberg A, J Biol Chem 270 (1995) 5818–5822. [DOI] [PubMed] [Google Scholar]
- [7].Pisoni RL, Lindley ER, J Biol Chem 267 (1992) 3626–3631. [PubMed] [Google Scholar]
- [8].Seidlmayer LK, Gomez-Garcia MR, Blatter LA, Pavlov E, Dedkova EN, J Gen Physiol 139 (2012) 321–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lynn WS, Brown RH, Biochem Biophys Res Commun 11 (1963) 367–371. [DOI] [PubMed] [Google Scholar]
- [10].Lorenz B, Munkner J, Oliveira MP, Kuusksalu A, Leitao JM, Muller WE, Schroder HC, Biochim Biophys Acta 1335 (1997) 51–60. [DOI] [PubMed] [Google Scholar]
- [11].Pavlov E, Aschar-Sobbi R, Campanella M, Turner RJ, Gomez-Garcia MR, Abramov AY, J Biol Chem 285 (2010) 9420–9428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Seidlmayer LK, Blatter LA, Pavlov E, Dedkova EN, Channels (Austin) 6 (2012) 463–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Holmstrom KM, Marina N, Baev AY, Wood NW, Gourine AV, Abramov AY, Nature communications 4 (2013) 1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Dedkova EN, Blatter LA, Front Physiol 5 (2014) 260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Abramov AY, Fraley C, Diao CT, Winkfein R, Colicos MA, Duchen MR, French RJ, Pavlov E, Proc Natl Acad Sci U S A 104 (2007) 18091–18096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hunter DR, Haworth RA, Southard JH, J Biol Chem 251 (1976) 5069–5077. [PubMed] [Google Scholar]
- [17].Di Lisa F, Bernardi P, J Mol Cell Cardiol 46 (2009) 775–780. [DOI] [PubMed] [Google Scholar]
- [18].Pavlov E, Zakharian E, Bladen C, Diao CT, Grimbly C, Reusch RN, French RJ, Biophys J 88 (2005) 2614–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Elustondo PA, Nichols M, Negoda A, Thirumaran A, Zakharian E, Robertson GS, Pavlov EV, Cell Death Discov 2 (2016) 16070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Reusch RN, Sadoff HL, J Bacteriol 156 (1983) 778–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Reusch RN, Huang R, Bramble LL, Biophys J 69 (1995) 754–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Reusch RN, Sadoff HL, Proc Natl Acad Sci U S A 85 (1988) 4176–4180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Reusch RN, Hiske TW, Sadoff HL, J Bacteriol 168 (1986) 553–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Huang R, Reusch RN, J Bacteriol 177 (1995) 486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Seidlmayer LK, Juettner VV, Kettlewell S, Pavlov EV, Blatter LA, Dedkova EN, Cardiovasc Res 106 (2015) 237–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Angelova PR, Baev AY, Berezhnov AV, Abramov AY, Biochem Soc Trans 44 (2016) 40–45. [DOI] [PubMed] [Google Scholar]
- [27].Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD, Nature 434 (2005) 658–662. [DOI] [PubMed] [Google Scholar]
- [28].Dedkova EN, Seidlmayer LK, Blatter LA, J Mol Cell Cardiol 59 (2013) 41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Rehncrona S, Mela L, Siesjo BK, Stroke 10 (1979) 437–446. [DOI] [PubMed] [Google Scholar]
- [30].Sokolova N, Pan S, Provazza S, Beutner G, Vendelin M, Birkedal R, Sheu SS, PLoS One 8 (2013) e83214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Smith SA, Morrissey JH, Electrophoresis 28 (2007) 3461–3465. [DOI] [PubMed] [Google Scholar]
- [32].Shiba T, in: Kulakovskaya T, Pavlov E, Dedkova EN (Eds.), Inorganic polyphosphates in eukariotic cells, Spinger International Publishing, Switzerland, 2016, pp. 139–158. [Google Scholar]
- [33].Leyssens A, Nowicky AV, Patterson L, Crompton M, Duchen MR, J Physiol 496 ( Pt 1) (1996) 111–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Dedkova EN, Blatter LA, J Mol Cell Cardiol 52 (2012) 48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Petronilli V, Miotto G, Canton M, Brini M, Colonna R, Bernardi P, Di Lisa F, Biophys J 76 (1999) 725–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Cowling RT, Birnboim HC, J Biol Chem 269 (1994) 9480–9485. [PubMed] [Google Scholar]
- [37].Ahn K, Kornberg A, J Biol Chem 265 (1990) 11734–11739. [PubMed] [Google Scholar]
- [38].Muller F, Mutch NJ, Schenk WA, Smith SA, Esterl L, Spronk HM, Schmidbauer S, Gahl WA, Morrissey JH, Renne T, Cell 139 (2009) 1143–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Smith SA, Gajsiewicz JM, Morrissey JH, Front Med (Lausanne) 5 (2018) 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Gray MJ, Wholey WY, Wagner NO, Cremers CM, Mueller-Schickert A, Hock NT, Krieger AG, Smith EM, Bender RA, Bardwell JC, Jakob U, Mol Cell 53 (2014) 689–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Muller WE, Tolba E, Feng Q, Schroder HC, Markl JS, Kokkinopoulou M, Wang X, J Cell Sci 128 (2015) 2202–2207. [DOI] [PubMed] [Google Scholar]
- [42].Muller WEG, Wang S, Neufurth M, Kokkinopoulou M, Feng Q, Schroder HC, Wang X, J Cell Sci 130 (2017) 2747–2756. [DOI] [PubMed] [Google Scholar]
- [43].Castuma CE, Huang R, Kornberg A, Reusch RN, J Biol Chem 270 (1995) 12980–12983. [DOI] [PubMed] [Google Scholar]
- [44].Pavlov E, Grimbly C, Diao CT, French RJ, FEBS Lett 579 (2005) 5187–5192. [DOI] [PubMed] [Google Scholar]
- [45].Baev AY, Negoda A, Abramov AY, J Bioenerg Biomembr 49 (2017) 49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kulakovskaya TV, Vagabov VM, Kulaev IS, Process Biochemistry 47 (2012) 1–10. [Google Scholar]
- [47].Bonora M, Bononi A, De Marchi E, Giorgi C, Lebiedzinska M, Marchi S, Patergnani S, Rimessi A, Suski JM, Wojtala A, Wieckowski MR, Kroemer G, Galluzzi L, Pinton P, Cell cycle 12 (2013) 674–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, Glick GD, Petronilli V, Zoratti M, Szabo I, Lippe G, Bernardi P, Proc Natl Acad Sci U S A 110 (2013) 5887–5892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park HA, Licznerski P, Li H, Nabili P, Hockensmith K, Graham M, Porter GA Jr., Jonas EA, Proc Natl Acad Sci U S A 111 (2014) 10580–10585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].He J, Carroll J, Ding S, Fearnley IM, Walker JE, Proc Natl Acad Sci USA 114 (2017) 3409–3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].He J, Carroll J, Ding S, Fearnley IM, Walker JE, Proc Natl Acad Sci U S A 114 (2017) 9086–9091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Giorgio V, Soriano ME, Basso E, Bisetto E, Lippe G, Forte MA, Bernardi P, Biochim Biophys Acta 1797 (2010) 1113–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Kampinga HH, Mol Cell 53 (2014) 685–687. [DOI] [PubMed] [Google Scholar]
- [54].He L, Lemasters JJ, FEBS Lett 512 (2002) 1–7. [DOI] [PubMed] [Google Scholar]
- [55].He L, Lemasters JJ, J Biol Chem 278 (2003) 16755–16760. [DOI] [PubMed] [Google Scholar]
- [56].Griffiths EJ, Halestrap AP, Biochem J 307 ( Pt 1) (1995) 93–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Perrelli MG, Pagliaro P, Penna C, World J Cardiol 3 (2011) 186–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Robin E, Guzy RD, Loor G, Iwase H, Waypa GB, Marks JD, Hoek TL, Schumacker PT, J Biol Chem 282 (2007) 19133–19143. [DOI] [PubMed] [Google Scholar]
- [59].Loor G, Kondapalli J, Iwase H, Chandel NS, Waypa GB, Guzy RD, Vanden Hoek TL, Schumacker PT, Biochim Biophys Acta 1813 (2011) 1382–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Das S, Lengweiler UD, Seebach D, Reusch RN, Proc Natl Acad Sci U S A 94 (1997) 9075–9079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Yoo NG, Dogra S, Meinen BA, Tse E, Haefliger J, Southworth DR, Gray MJ, Dahl JU, Jakob U, J Mol Biol (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Azevedo C, Singh J, Steck N, Hofer A, Ruiz FA, Singh T, Jessen HJ, Saiardi A, ACS Chem Biol 13 (2018) 1958–1963. [DOI] [PubMed] [Google Scholar]
- [63].Segawa S, Fujiya M, Konishi H, Ueno N, Kobayashi N, Shigyo T, Kohgo Y, PLoS One 6 (2011) e23278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Lam V, Moulder JE, Salzman NH, Dubinsky EA, Andersen GL, Baker JE, Radiat Res 177 (2012) 573–583. [DOI] [PubMed] [Google Scholar]
- [65].Gan XT, Ettinger G, Huang CX, Burton JP, Haist JV, Rajapurohitam V, Sidaway JE, Martin G, Gloor GB, Swann JR, Reid G, Karmazyn M, Circ Heart Fail (2014). [DOI] [PubMed] [Google Scholar]