

Abstract
Antibiotic resistance has emerged as a serious threat to global public health in recent years. Lack of novel antimicrobials, especially new classes of compounds, further aggravates the situation. For Gram-negative bacteria, their double layered cell envelope and an array of efflux pumps act as formidable barriers for antimicrobials to penetrate. While cytoplasmic targets are hard to reach, proteins in the periplasm are clearly more accessible, as the drug only need to breach the outer membrane. In this review, we discuss recent efforts on the validation and testing of periplasmic proteins as potential antimicrobial targets and the development of related inhibitors that either inhibit the growth of a bacterial pathogen or reduce its virulence during interaction with host cells. We conclude that the periplasm contains a promising pool of novel antimicrobial targets that should be scrutinized more closely for the development of effective treatment against multidrug-resistant Gram-negative bacteria.
Keywords: antimicrobial resistance, periplasm, antimicrobial target, Gram-negative bacterium, inhibitor, efflux pump, chaperone, virulence
Graphical Abstract
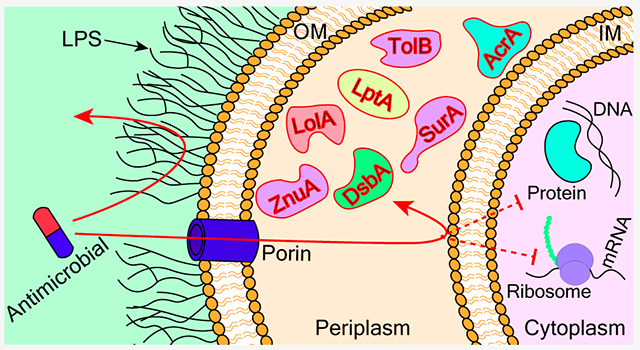
1. ANTIMICROBIAL RESISTANCE AND THE PERMEATION BARRIER IN GRAM-NEGATIVE BACTERIA
Multidrug resistance (MDR) in bacteria has become a major global health challenge. The United Nation’s Interagency Coordination Group Report indicates that approximately 700 000 people lose their lives worldwide each year because of infections caused by multidrug resistant bacteria, and it further predicts that this number could reach a staggering 10 million deaths per year by the year 2050.1 The development of new classes of effective antimicrobials is an urgent need. The availability of antimicrobials has made modern medicine possible, since the discovery of penicillin and its antibacterial properties by Alexander Fleming in 1928.2,3 Immediately after the discovery, Fleming warned the world about the possibility of the development of bacterial resistance to the drug if it was not used properly.3 Antimicrobial development underwent rapid growth in the 1940s through 1960s, when most of the currently used antimicrobials were marketed. However, following this period, very few new classes of antimicrobials have been developed, despite efforts from many researchers.4 Because the pace of antimicrobial development is not keeping up with the fast development of resistance among bacterial pathogens, resistant bacteria have been identified for all currently available antimicrobials. Drug resistance is normally observed only a few years after the introduction of an antimicrobial into market.3,5 We are facing the challenges of a rapidly growing list of drug resistant pathogens coupled with a dwindling supply of effective antimicrobials. Initially coined by Louis B. Rice and later recognized by WHO, the “ESKAPE pathogens”, which include Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp, are a group of deadly bacterial pathogens with rapidly growing multidrug resistant properties.6,7 These bugs were chosen based on the rate of infection, mortality, occurrence of resistance, and availability of treatment options.8 Four out of the six pathogens are Gram-negative bacteria. In addition, in 2017 WHO published a list of 12 pathogens categorized in critical, high, and medium crisis. The critical category contains three bacteria, all of which are Gram-negative. Altogether nine out of the 12 pathogens are Gram-negative.9
The differentiation of the Gram-negative and Gram-positive bacteria started with the development of a staining technique in 1884, later modified by Hucker in 1921 to classify bacteria into two groups.10 This classification basically differentiates bacteria according to their cell envelope structure. Gram-positive bacteria have a very thick peptidoglycan cell wall on the outside of a plasma membrane, whereas Gram-negative bacteria have a thin peptidoglycan layer between an outer membrane (OM) and an inner membrane (IM). Development of effective antimicrobials against Gram-negative bacteria is especially difficult due to its cell envelope structure.
The structure of the Gram-negative bacterial cell envelope is illustrated in Figure 1. The first clear picture of this double membrane structure emerged with the development of electron microscopy during the 1950s and 1960s, which revealed three different layers in the Gram-negative bacterial cell envelope, an OM, an IM, and a peptidoglycan layer between them.11 The OM is an asymmetric lipid bilayer comprising phospholipids in the inner leaflet and glycolipids called lipopolysaccharides (LPSs) in the outer leaflet.11,12 LPSs are composed of lipid A, a nonrepeating chain of core oligosaccharides extending to the cell surface, and a polysaccharide called O-antigen on the top.12,13 Lipid A is found to be relatively conserved among bacteria, whereas the core oligosaccharides are variable between different bacterial species. Similarly, the O-antigen is much more variable and can be largely different even among different strains of the same bacteria.12 The LPS is a major contributor to the toxic and lethal effects in different host organisms.13 Apart from LPS and phospholipid, the OM is also composed of a large number of proteins. Most of the proteins present in the OM can be broadly classified into lipoproteins and β-barrel proteins.11 β-Barrel proteins are mainly responsible for the passage of various molecules across the OM. These could be essential nutrients, harmful antimicrobials, secreted virulence factors, or surface proteins. Lipoproteins, however, are thought to be responsible for a large variety of functions including biogenesis of other surface structures, maintenance of OM integrity, signal transduction, cell wall metabolism, conjugation, substrate transport, and efflux.11,14,15 Apart from the IM and OM, peptidoglycan cell wall serves as an important component of the cell envelope. It consists of a complex scaffold structure made up of carbohydrate strands cross-linked by short pentapeptides to provide mechanical strength to the cell envelope.16
Figure 1.
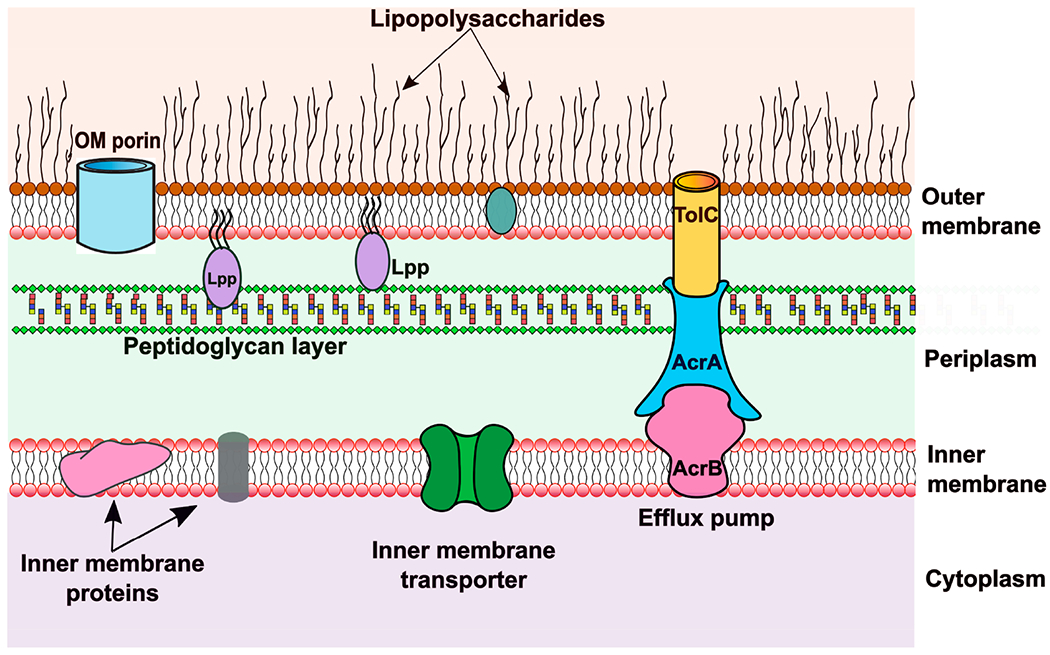
Gram-negative cell envelope. Representative figure of the Gram-negative bacterial cell envelope showing the outer membrane, inner membrane, and peptidoglycan layer. The outer membrane contains lipopolysaccharides, proteins such as porins, and lipoproteins (Lpp). The inner membrane also contains integral membrane proteins and transporters. Several representative transporters are presented including ones spanning both membranes (such as AcrAB-TolC) and ones that only transport across the inner membrane.
The double-layered cell envelope of Gram-negative bacteria presents a formidable barrier for the entry of antimicrobials.17,18 The LPS outer leaflet and phospholipid inner leaflet of the OM effectively block most hydrophobic and large hydrophilic molecules. Porins enable the entrance of only selected small hydrophilic molecules across the OM. The inner cytoplasmic membrane is hydrophobic in nature and lacks porin-like channels, hindering the entry of hydrophilic compounds but not hydrophobic or amphiphilic compounds.18–20 This “orthogonal selectivity” has been proposed to be responsible for the difficulty in penetration into Gram-negative bacteria. However, Richter and Hergenrother recently reported that intact cells and protoplasts accumulated their test compounds similarly, arguing against the “orthogonal selectivity” mechanism of impermeability.17,21 The presence of multidrug efflux pumps in the inner membrane help the bacteria to efflux out the compounds that were able to penetrate the outer membrane.20,22 Krishnamoorthy et al. reported that there is a synergistic relationship between the OM barrier and active efflux in Gram-negative bacteria.22 Cooper et al. suggested that the OM impermeability and active efflux across the IM are the two major contributors to the Gram-negative cell envelope as a penetration barrier.23 Even though several novel compounds have been found to inhibit various cellular targets in vitro, they could not be developed into lead compounds for antimicrobial development because of their poor bacterial penetration.19,24 While cytosolic targets are difficult to access in Gram-negative bacteria, an alternative strategy is to avoid the problem by disrupting targets in the periplasm.
2. TARGETING PERIPLASMIC PROTEINS
The volume of the periplasmic space ranges from 7% to up to 40% in Gram-negative bacteria, according to different studies.25–28 The periplasm is now recognized as a viscous and gel-like space due to the presence of a large number of proteins and unpolymerized peptidoglycan.29,30 The periplasm is highly rich in various proteins including oxidases, nucleases, proteases, and transport machineries. All enzymes in the periplasm are ATP-independent, due to the lack of ATP in this cellular compartment. Proteomic analysis of bacteria revealed that the periplasm contains up to 30% of total cellular proteins.31
Miller et al. reviewed various functions of the periplasm and listed protein transport, protein folding, oxidation and quality control of membrane and secreted proteins, integrity of cell envelope, maintenance of Donnan potential, nutrient uptake, essential for cell division, electron transport, and assembly of locomotion machinery.28 Some components are essential for the survival of bacteria, while others responsible for maintaining bacterial virulence and communication.32–34 These periplasmic proteins could be potential antimicrobial targets.
A good example is the peptidoglycan synthesis and cross-linking pathway. The well-known penicillin and the entire family of β-lactams are among the most successful class of antimicrobials targeting this pathway in the periplasm. The biogenesis of OM proteins is another example. Inhibiting the proper folding of OM proteins and virulence factors in the periplasm severely compromises the OM integrity and reduces bacterial virulence.32,34,35 Dickey et al.36 summarized the benefits of using antivirulence agents in a recent review. First, antivirulence agents are less likely to develop evolutionary resistance as they do not affect the normal cellular growth. This could be the greatest advantage. Since they only affect the pathogenicity, normal commensals should not be affected by the treatment, which helps in preserving the beneficial body microbiome. Antivirulence compounds could be used as synergistic compounds in combination with otherwise ineffective antibiotics.36 However, the antivirulence agents have their own problems as well. Although considered less likely to develop resistance, for some targets such as the quorum sensing mechanism, resistance has been shown to develop readily by mechanisms such as mutations with increased efflux.37–40 They might be less effective and need to be used in combination with other therapeutics in treatment. Another concern is that since they do not act to eliminate the bacteria, reoccurrence of the infection could be very likely.36 Safety concerns and adverse side effects are other factors hindering these compounds from going into clinical trials.41 However, Allen et al. suggested that while bacteria could develop resistance against antivirulence reagents, selection for resistance can be reduced or even reversed with appropriate combinations of target and treatment environment.42 Despite of these concerns, several antivirulence therapies have been approved by the FDA for bacteria that cause acute toxin-mediated diseases including Clostridium botulinum, Bacillus anthracis, and Clostridium difficile, and there are candidates in clinical trials for a few other Gram-negative pathogens.36
Other than inhibiting bacterial growth and reducing virulence, another strategy is to reduce the intrinsic resistance of pathogens to antimicrobials. Efflux is a major mechanism of multidrug resistance in Gram-negative bacteria. Targeting the periplasmic component of efflux pumps has been shown to sensitize the pathogen to various antimicrobials.43 Below we discuss recent progress in the validation of and inhibitor development for specific periplasmic proteins as targets for antimicrobials.
3. RECENTLY EXPLOITED PERIPLASMIC ANTIMICROBIAL TARGETS
With the goal of finding an effective and novel target in the periplasm of Gram-negative bacteria, many studies have been reported in recent years.44 In this review, we focus on studies in the development of new antivirulence agents or antibiotic adjuvants by exploiting the periplasmic targets in Gram-negative bacteria as summarized in Table 1.
Table 1.
Summary of Periplasmic Targets Discussed in This Review and Effects of Their Inhibitiona
| Inhibition of Periplasmic Protein’s Function | ||||||
|---|---|---|---|---|---|---|
| AcrA | DsbA | SurA | LolA | LptA/LptH | ZnuA | TolB |
| 201752 | 201598,99 | 2018118 | 2009120 | 2018158 | 2016166 | |
| disruption of drug efflux | disruption of the folding of proteins containing disulfide bonds | disruption of the folding of OM proteins | interruption of lipoprotein transport to the OM | inhibition of LPS transport to OM | inhibition of zinc uptake in zinc deficient environment | disruption of the OM integrity |
| increased sensitivity to selected antimicrobials | reduction of virulence | leakage of OM and reduction of virulence | deposition of OM lipoproteins in the IM and leakage of the OM | compromised OM integrity, decreased virulence, and increased sensitivity to antimicrobials | reduction of virulence | leakage of cell envelope and increase in antimicrobial sensitivity |
Year presented indicates the first publication of inhibitors for the respective protein.
3.1. AcrA.
Acriflavine resistance protein A (AcrA) is the periplasmic component45 of the major multidrug efflux system AcrAB-TolC, which also consists of the IM protein acriflavine resistance B (AcrB)46 and OM protein channel TolC.47,48 AcrAB-TolC work together to transport a broad range of compounds, including dyes, detergents, and antimicrobials, out of the Gram-negative bacterial cells into the external environment, thereby contributing to intrinsic antibiotic resistance.49,50 Serving as a bridge between AcrB and TolC, AcrA is structurally flexible and forms a hexameric ring once the AcrAB-TolC pump assembles51,52 (Figure 2a). Knowledge of the structure and function of AcrA enabled the discovery of inhibitors that can potentiate antimicrobial activities.53–55
Figure 2.
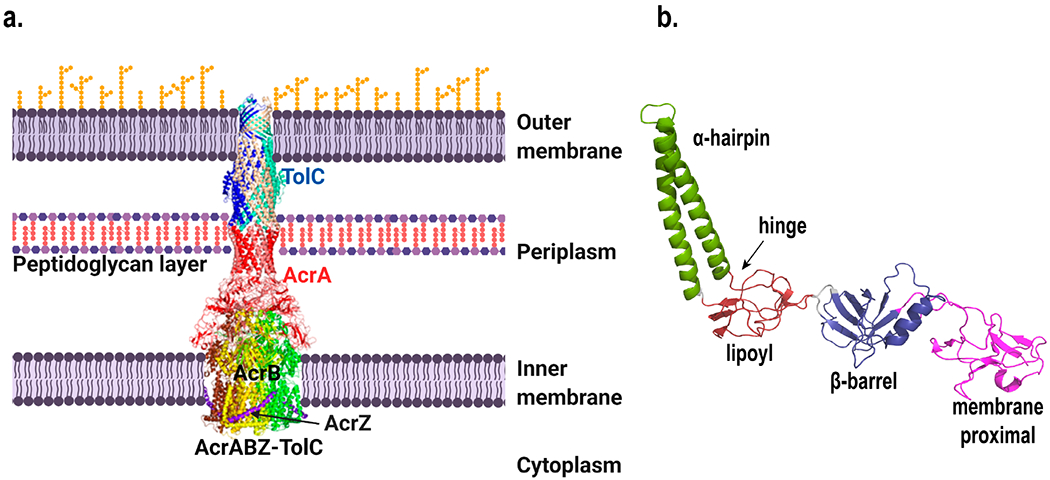
(a) Structure of the AcrABZ–TolC complex. Figure was created using Biorender. (b) Structure of E. coli AcrA with domains color coded and labeled. Both images were created with Pymol73 using the cryo-EM structure 5O66.pdb.74
3.1.1. Structure of AcrA.
AcrA is arranged linearly into four domains, the α-helical hairpin, lipoyl, β-barrel, and membrane proximal (MP) domains (Figure 2b)46,56–58 According to cross-linking and mutagenesis studies and more recently cryo-EM imaging of the assembled complex, while the α-helical hairpin domain interacts with TolC, the other domains interact with AcrB.59–63 The flexibility of the α-helical hairpin can be attributed to the hinge region that connects it to the lipoyl domain.58 The β-barrel domain is known to be involved in binding to ligand with a change in conformation that affects the MP domain.64
AcrA assembles as a trimer of dimers and brings in close-proximity the OM and IM proteins when the AcrAB-TolC complex forms.65 The first 24 amino acid residues of AcrA comprise the signal peptide,47 which is removed by signal peptidase after transportation of AcrA to the periplasm.66 The subsequent lipid acylation at Cys25 attaches AcrA to the outer leaflet of the IM.67 Studies have revealed that the function of AcrA does not depend on membrane anchoring though.58
AcrA couples the process of transport between the IM and the OM.52 When any of the components of AcrAB-TolC is inactivated, the function of the pump is annulled, thus making the bacteria sensitive to many antimicrobials.68
3.1.2. AcrA Is Important for Multidrug Resistance.
Disruption of the acrA gene in E. coli leads to hypersensitivity to antimicrobials, dyes, and detergents.69,70 Blair et al. showed that inactivation of AcrA alone in Salmonella typhimurium inactivates the efflux pump, making the bacteria highly susceptible to several compounds including ampicillin, nalixidic acid, chloramphenicol, tetracycline, triclosan, acriflavine, fusidic acid, ciprofloxacin, ethidium bromide, bile, SDS, and Triton.71 Accumulation of the fluorescent dye Hoechst H33342 in the AcrA mutant strain was 2-fold higher than in the corresponding AcrB and TolC mutant strains. This implies that AcrA is pivotal for the efficient functioning of the AcrAB-TolC pump.
Furthermore, Ge et al. found that the multidrug efflux activity of AcrAB-TolC in E. coli was impaired by a single AcrA G363C substitution.72 In a more recent study, Hazel et al.65 explored the conformational dynamics of AcrA. Through simulation studies, two conformational basins were identified: a trans-like conformation where the MP and α-helical domains of AcrA pointed in opposite directions and a cis-like conformation where they pointed in the same direction. Double cysteine mutations were introduced into AcrA to promote the cis-like conformation, which was not compatible with pump assembly. As expected, drug efflux was compromised in the mutant.65
3.1.3. Inhibitors of AcrA and Their Effects on Drug Resistance.
Efflux pump inhibitors (EPIs) that target AcrB have been the focus of many studies.53,75–77 These inhibitors include DA-13-1809, MC207110 (PAβN), and MBX2319, but clinical trials of these inhibitors have been hampered by a variety of factors,52,53,77–79 including the structural complexity of the RND pump, the difficulty in tailoring the pharmacokinetics of the EPI to those of antibiotics to ensure efficacy, and the potential cytotoxicity of the EPI.53,75 By diversifying the chemistries and mechanisms of action, the chance of obtaining clinically useful EPIs could be greatly increased.
A combination of computational and experimental methods was used by Abdali et al. to find compounds that can potentiate the activities of antimicrobials through inhibiting the activity of AcrA.52 A computational method called “ensemble docking” was used to rank compounds by their ability to bind to AcrA. Top scorers were then analyzed for drug potentiation experimentally and their mechanisms of action were determined.52 EPIs were identified as compounds that potentiated the activity of novobiocin in a strain with an active AcrAB-TolC efflux but not in the corresponding ΔtolC strain.
After the experimental screens, computational docking, and identification of AcrA binders, seven compounds were found to bind at the hinge region or the membrane proximal sites or both. Among them NSC 60339, NSC 227186, NSC 33353, and NSC 305798 were in the top 5% among all hits from computational prediction, and they were able to potentiate the activity of novobiocin (Figure S1). The MPC4 (minimal potentiation concentration at which the inhibitory activity of an antibiotic is potentiated 4-fold) values of these four compounds for novobiocin were 25, 6.25, 1.56, and 12.5 μM, respectively. NSC 227186, also known as clorobiocin, is structurally similar as novobiocin and binds to AcrA. NSC 60339 (SLU-258) led to conformational changes in AcrA upon binding as revealed from limited proteolysis studies.80 Several SLU-258 analogs were then synthesized and characterized, including SLU-417 and SLU-225 (Figure S2).54 While the MPC4 values of the analogs were not drastically improved compared to SLU-258, they showed favorable properties as potential EPIs including increased outer membrane permeability and being less prone to efflux.54 Computational modeling was coupled with experimental measurement of tryptophan fluorescence to locate the binding sites of clorobiocin and SLU-258 to the interface between the lipoyl and β-barrel domains.80
More recently, Green et al. identified a new set of EPIs through exploring existing physicochemical guidelines for permeability of OM and efflux in E. coli, together with potentiation and binding assays using bacteria strains whose permeability barriers could be modulated or controlled.81 They used the ZINC database and filtered the compounds based on the properties matching existing EPIs and performed a docking experiment to find AcrA binders. Based on the docking scores, they purchased 34 commercially available predicted binders and searched for potential EPIs. Six molecules, 24123034, 36287038, 56871955, 58997260, 65071797, and 98577577 (Figure S3) with a shared scaffold were determined to potentiate the antibiotic activity of novobiocin and erythromycin up to 16-fold at 50 μM in hyperporinated E. coli cells. But in wild-type E. coli cells, the MPC4 values increased to 100–200 μM suggesting that these EPIs did not penetrate the E. coli OM readily. Notably, these molecules could potentiate the activity of antimicrobials in wild-type strains of Acinetobacter baumannii with MPC4 of 25–100 μM (novobiocin) and 50–200 μM (erythromycin). They also potentiate these two drugs in Klebsiella pneumoniae at concentrations of 100–200 μM. The binding of these EPIs to E. coli AcrA was confirmed using surface plasmon resonance (SPR) measurements.
3.2. Disulfide Bond Forming Protein (DsbA).
DsbA belongs to the family of disulfide bond forming (Dsb) proteins in bacteria. It is a periplasmic protein involved in catalyzing disulfide bond formation during the folding of secreted proteins.32,82,83 Apart from acting as a thiol oxidoreductase, it also plays a role in maintaining the redox potential in the periplasm.32 DsbA catalyzes the oxidation of two thiol groups in its substrate protein to form a disulfide bond, while the disulfide bond in itself is reduced into two free Cys. A partner protein DsbB subsequently oxidizes DsbA back into its original form (Figure 3a). The active site of DsbA consists the signature CXXC sequence, similar to other thiol–disulfide oxidoreductase enzymes.83,84 Apart from the DsbA/DsbB system, there are other Dsb proteins including DsbC and DsbG that correct disulfide bonds formed by mistake. The isomerization of incorrectly folded substrate to form correct disulfide bonds is catalyzed by DsbC/DsbG, which are maintained in the reduced form by cytoplasmic membrane protein DsbD.83 Among all these disulfide bond forming proteins, DsbA is the most common one whereas other Dsb proteins are not as widespread.83,85–87
Figure 3.
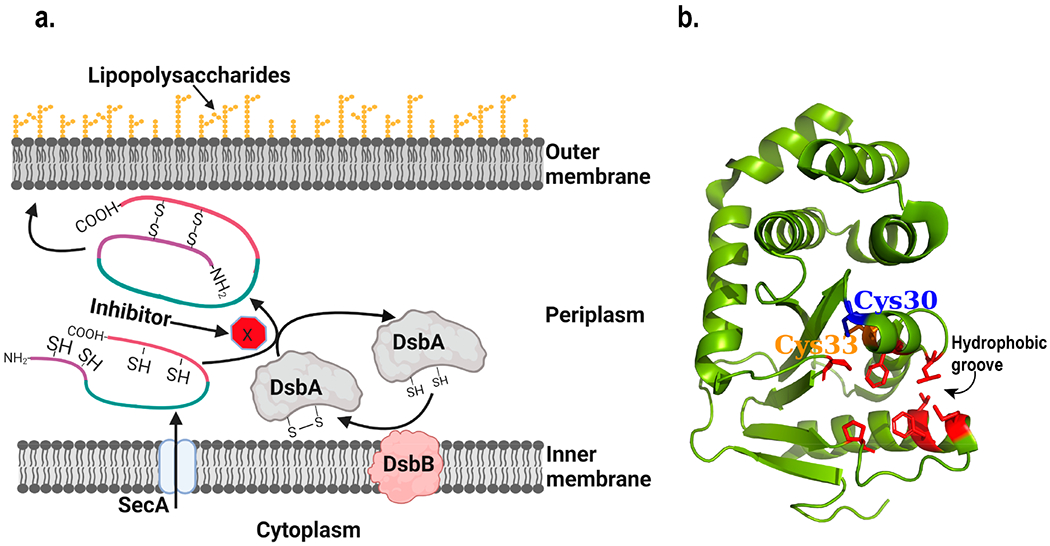
Structure and function of DsbA. (a) Schematic diagram of the working mechanism of DsbA. The process blocked by inhibitors is highlighted. Figure was created using Biorender. (b) Structure of E. coli DsbA (Created with Pymol73 using 1FVK.pdb). Cysteines present in the active site in the CXXC motif are shown. Residues lining the hydrophobic groove are highlighted in red and with their side chains shown.88
3.2.1. DsbA Is Essential for Bacterial Virulence.
By definition, virulence factors are molecules that play important roles during infection by assisting bacterial invasion into the host, attach to host cells, escape through host defense mechanisms, and also sometimes act as toxins.89 Since many virulence factors are secreted out of the cell, DsbA is responsible for the proper folding of ones containing disulfide bonds, such as in pertussis toxin, cholera toxin, and heat labile E. coli toxin.83,90,91 In addition, DsbA is responsible for the proper folding of the secretory machineries such as the type III secretion system, which is responsible for secreting effector proteins for virulence.83 Therefore, DsbA has been explored as a potential target for the reduction of bacterial virulence. DsbA has been found to be essential for virulence of a wide range of bacteria including E. coli, Pseudomonas, Shigella, Burkholderia, Salmonella, and Vibrio cholerae.32,85,92–95
Initial studies showed that mutations in DsbA caused improper folding of proteins such as alkaline phosphatases, β-lactamases, and OmpA. It was shown that in the absence of DsbA, these proteins were secreted without disulfide bonds.82 In the absence of DsbA, alkaline phosphatase activity was reduced by approximately 60% and a nonmotile phenotype was observed in E. coli.93 Since DsbA and DsbB are involved in the formation of different toxins such as cholera toxin and heat stable toxin of E. coli, secretion systems, pili, adhesins, and motility machineries, defects in the DsbA/DsbB system cause the bacteria to lose those virulence factors.32,83,92 Apart from virulence, DsbA has been observed to play a role in biofilm formation in E. coli, as deletion of the dsbA gene leads to defective biofilm formation, presumably due to the loss in surface attachment machinery.93 Ha et al.32 in 2003 conducted a study in Pseudomonas aeruginosa where they created a transposon-based insertion library and identified cells that were noninfective toward HeLa cells. They found that the loss in pathogenesis was mainly caused by the loss of DsbA protein, which subsequently led to the loss of the type III secretion system, cell adhesion, and twitching motility due to low levels of pili expression. Upon expression of plasmid encoded DsbA, all lost functions in the mutant strains were fully restored. Hence, they concluded that DsbA is a good target for the development of anti-infective therapeutics.32 Similar studies in enteropathogenic E. coli also demonstrated that the type III secretion system became defective without DsbA.96
Ireland et al. in 2014 studied the role of DsbA in Burkholderia pseudomallei and observed that its deletion led to reduced virulence of this bacteria in macrophages and a mouse infection model.85 In the mouse model, infection with a dsbA deletion strain did not result in death, but the presence of bacteria could still be observed in organs, suggesting the loss of virulence. Similar studies in Burkholderia cepacia have also revealed that DsbA was involved in the virulence and resistance against antimicrobials and heavy metals.97
3.2.2. Inhibitors of DsbA.
DsbA has been pursued as an antivirulence target by several research groups including those of Jennifer L. Martin, Begona Heras, and Martin J Scanlon.84,85,98–101 In 2015, four important studies were published on the search for DsbA inhibitors. Duprez et al. showed that short peptide can be used to inhibit the function of DsbA.99 They analyzed the binding of DsbB to DsbA and designed the peptides to be similar to the DsbB P2 loop, which binds into a hydrophobic groove of DsbA (Figure 3b). A nonapeptide, 1PSPFATCDF9, was found to bind DsbA with a stoichiometry of 1:1 and dissociation constant, Kd of 4 μM. The peptide sequence was further optimized, and 1PSPWATCDF9 was found to have a Kd of ~2.9 μM and IC50 of ~5.1 μM. A reversible binding mechanism was proposed in which the Cys-containing peptide catalyzed the oxidation of DsbA. In conclusion, the study indicates that Cys-containing short peptides derived from the DsbB P2 loop could bind to DsbA and inhibit its activity.99
Duprez et al. published another study in the same year on the development of peptidomimetic DsbA inhibitors that inhibited noncovalently.102 Computer modeling was used to design peptidomimetic molecules that bound DsbA in the hydrophobic groove. Peptidomimetic compound 1 was chosen as the best hit, and nine derivatives were then synthesized and tested experimentally for their affinity and inhibition of DsbA. The peptidomimetic compound 1 contained a tryptophan residue with a morpholine group at the C-terminus and a benzyl group at the N-terminus (Figure S4). This compound was selected as a good therapeutic candidate because it possessed drug-like properties such as a clogP value of 2.6, with six rotatable bonds, three each of hydrogen bond donors and acceptors, a molecular weight of 406 Da, and being neutral at physiological pH. Compounds 1–10 did not bind DsbA significantly as determined using differential scanning fluorimetry and isothermal calorimetry. However, compound 10 (Figure S6) showed inhibition of DsbA in an enzyme assay with an IC50 of 1.1 mM, which was approximately 200-fold higher than that of the peptide inhibitors. The low effectiveness of these peptidomimetic compounds has been attributed to the weak and reversible binding in the hydrophobic groove. These are valuable proof-of-principle studies, advocating for further development of peptidomimetic DsbA inhibitors.102
Adams et al. did a fragment-based screening to search for inhibitors for E. coli DsbA. Their inhibitors were derivatives of the phenylthiazole class of compounds.98 Through measuring the chemical shift perturbations (CSP) in NMR, they identified 37 compounds that bound to DsbA in the hydrophobic groove. Derivatives of the most promising hits, the phenylthiazoles, were synthesized and characterized (compound 4, Figure S5). Various amino acid derivatives of compound 4 were then synthesized and characterized. Aromatic amino acid substitution led to an increase of affinity to DsbA as showed in compounds 39 and 40 (Figure S5). Short polar amino acid substituents also improved the binding (compound 41 and 42, Figure S5), whereas affinity was lost with lysine substitution. Compound 40 was cocrystallized with DsbA, and it formed hydrogen bonds with His32 and Gln164 of DsbA. Compound 40 was then used to study whether the presence of the inhibitor affected bacterial motility. At a concentration of 600 μM, compound 40 reduced the motility by 59%, as determined by observation of swarming in agar plates.98 This observation was consistent with DsbA’s role in the secretion of proteins related to motility.98 A few years later, Totsika et al.101 studied the inhibitory activity of structurally similar phenylthiophene and phenoxy phenyl derivatives (Figure S6) toward different homologues of E. coli DsbA, DsbL, and SrgA in different uropathogenic E. coli strains as well as in Salmonella typhimurium. They observed that the motility of these bacteria was inhibited, while the growth was normal. Binding studies revealed that these inhibitors bind in the hydrophobic grooves of the enzymes, similar as their binding site in E. coli DsbA. Hence, they demonstrated that these inhibitors can be used in structurally diverse homologues of DsbA in pathogenic E. coli and Salmonella and paved a path forward to develop DsbA inhibitors for different pathogenic bacteria.
Halili et al. reported that synthetic analogs of ubiquinone, a cofactor of DsbB, inhibit disulfide bond formation in E. coli cells through targeting the DsbA/DsbB enzyme complex.100 A series of ubiquinone derivatives were synthesized and characterized, with IC50 ranging from 1.1 to 43 μM. The most potent compounds were identified as compound 8 and compound 19 (Figure S7). Interestingly, the inhibitors were found to covalently modify Cys33 of DsbA, inhibiting its function with an IC50 of 0.8 μM for compound 19. NMR studies revealed that compound 19 bound covalently to reduced DsbA but noncovalently to the oxidized DsbA in the hydrophobic groove near the active site. Compound 19 was found to inhibit the function of DsbA but not the human thioredoxin (hTRX1). In addition, compound 19 did not react with free cysteines in human serum albumin, and only reacted with reduced glutathione at high pH and high concentration. Despite these promising results, compound 19 could not be used for in vivo assays due to its poor water solubility.100 An increasing number of drugs that function through covalent modification have been developed recently.103 The inhibitors from this study could be promising lead compounds if their solubility could be improved without compromising their activities100.
More recently in 2019, Duncan et al. used a fragment-based method to search for compounds that bind and inhibit E. coli DsbA.84 They synthesized a series of compounds based on a scaffold identified from in silico screening, 2-(6-bromobenzo-furan-3-yl) acetic acid. NMR was used to identify compounds that interacted with DsbA and later used to determine the binding affinity. Compounds 25 and 28 had the best binding affinities in the group, with dissociation constants of 326 ± 25 and 341 ± 57 αM respectively (Figure S8).84 These inhibitors were also found to bind in the same hydrophobic groove in the DsbA structure.
3.3. Periplasmic Chaperone SurA.
3.3.1. SurA Plays an Important Role in OM Protein (OMP) Folding.
SurA assists with the transport and delivery of nascent OMPs to the OM, where the BAM complex inserts them into the membrane.33,104–108 There are three chaperones present in the periplasm, SurA, Skp, and DegP. SurA is the major one responsible for the folding of most OMPs, while the other chaperones rescue OMPs that fall off the SurA pathway, especially when the bacteria are under stress.33,104,105 SurA recognizes the signature sequence Ar-X-Ar, where Ar is an aromatic amino acid and X is any other amino acid, which is abundant in OMPs.107,109
3.3.2. Structure of SurA.
SurA consists of four domains: an N terminal domain, two peptidyl-prolyl isomerase (PPIase) domains, P1 and P2, and a C terminal domain (Figure 4).33,107 The N- and C-terminal domains form a core domain. The P1 domain lies in close interaction with the core domain, whereas P2 lies at a satellite position far from the core domain.110 Studies have shown that the two PPIase domains were not involved in the chaperone activity in vivo, as a truncated SurA mutant missing the P1 and P2 domains was functional.111 The observation that SurA homologues in some species only contained one PPIase domain also suggested that the PPIase domain is not essential for SurA function.112 Webb et al. suggested that binding of substrate to SurA was dependent on only the N terminal 150 residues.113 However, later studies revealed that the P1 domain was involved in substrate binding. Substrate binding led to dimerization of SurA, where the substrate was located between the two P1 domains of the SurA dimer.114 The exact role and importance of the PPIase domains is still under debate. Ricci et al. found that in a BAM null-like mutant strain where there was no interaction between SurA and the BAM complex, a gain-of-function mutation in SurA was present in the P1 domain. This observation suggested that when there was no interaction between SurA and the BAM complex, the P1 domain played an auto-inhibitory role in SurA.115 Furthermore, a recent study by Soltes et al. published in 2016, demonstrated that the P1 domain was involved in the inhibition of SurA activity in the absence of P2 domain when the function of its partner protein BAM was compromised.112 The presence of the P2 domain however relieved this autoinhibition. In the absence of the P2 domain, the P1 domain was locked together with the core NC-terminal domain, inhibiting SurA function. The presence of the P2 domain unlocked the tight interaction between the P1 and core NC-terminal domains.112 Thus, they proposed that SurA existed in equilibrium between a more disorganized, active form and a more structured, less active form. The BAM complex seemed to play a regulatory role in this equilibrium. In addition, we found that structural flexibility of the core domain was not critical for SurA activity, as the introduction of four pairs of disulfide bond as staples to restrict SurA conformational change did not impair the function of SurA.33
Figure 4.
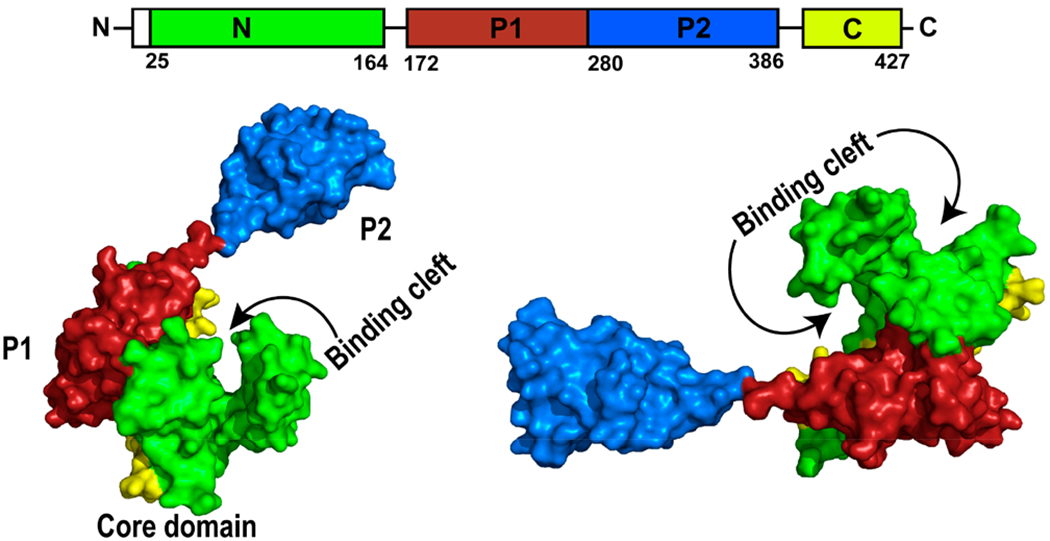
Structure of E. coli SurA (Created with Pymol73 using M5Y.pdb) showing each domain and the substrate binding cleft. SurA sequence is illustrated to show the sequential organization of the domains. Green and yellow represent N and C terminal domains, which form the core domain. Red represents the P1 domain, and blue represents the satellite P2 domain.110
3.3.3. Inhibition of SurA Increased Antibiotic Susceptibility and Decreased Virulence.
Although first known as essential for survival in the stationary phase, SurA has been extensively studied to determine its essentiality in membrane integrity and selective entry of various toxic substances lethal to the bacteria.107,116 As it has been shown that lack of SurA or inhibition of its function made the bacteria more sensitive toward antimicrobials.33,35 Studies from Watts and Hunstad108 and Justice et al.117 showed that loss of SurA led to increased susceptibility of E. coli toward novobiocin and loss in pilus generation, both detrimental for the viability and virulence of E. coli. For novobiocin, the SurA deficient mutant showed an increase of more than 10 mm in zone diameter of inhibition compared to the wild-type strain, and the growth of the mutant was completely lost at a concentration of 10 μ/mL whereas the wild-type strain showed normal growth.108,117
In 2019, Klein et al. studied the effect of deletion of periplasmic chaperones as well as components of the OMP assembly machinery in Pseudomonas aeruginosa.35 They found that the depletion of SurA led to a significant difference in the protein composition of the OM and compromised integrity of the membrane.35 Depletion and deletion of SurA and BamC led to an increase in susceptibility of the otherwise resistant strain of Pseudomonas aeruginosa even for some Gram-positive specific antimicrobials. Drug resistance was restored when the disrupted protein was expressed from a plasmid. These observations led to the conclusion that SurA could be targeted to increase bacterial susceptibility for antimicrobials, through compromising OM integrity.35 In addition, SurA-deficient strains were less virulent when tested in a Galleria mellonella infection model, as SurA is critical for the biogenesis of several secretion systems that are important in virulence and infection.35 Proteome analysis showed that the OM lacked the receptors for siderophores including FpvA, FecA, and FiuA, which prevented the pathogen from obtaining iron, which is essential for its growth.35
3.3.4. Inhibitors of SurA.
Bell et al. screened over 10 million compounds from two subsets of the ZINC12 database, “Fragment Like” and “Drug Now”, through computationally docking onto the structure of SurA lacking the P2 domain (SurAΔP2).118 The top hits from the in silico screening were later characterized experimentally. ZINC22055514, ZINC26894893 and ZINC19200219 were identified from the “Fragment Like” subset as the top three compounds with the highest binding affinities (Figure S9). In agreement with previous findings, these compounds possessed high aromaticity and hydrophobicity, resembling the Ar-X-Ar motif in natural SurA substrates. Nitrogen was another feature seen in the compounds that showed high binding affinity. Additional compounds from the “Drug Now” subset were screened. Based on the structures of the hit compounds, features that favor or disfavor SurA binding were summarized. The authors also experimentally measured SurA binding affinity of 12 hits that are commercially available using a competitive fluorescence anisotropy assay. Among them, Fmoc-β-(2-quinolyl)-d-alanine had the best IC50 of ~56.98 μM. This discovery prompted them to test additional Fmoc protected aromatic amino acids. The IC50 values of Fmoc-L-tryptophan and Fmoc-L-phenylalanine were 23.33 and 156.6 μM, respectively. Other Fmoc modified amino acids tested did not bind to SurA effectively. While these compounds are not potent enough to serve as SurA inhibitors, they could be a promising scaffold for the development of useful SurA inhibitors.118
3.4. Lipoprotein Carrier Protein (LolA).
LolA is a small 20 kDa periplasmic protein responsible for the transport of lipoproteins from the IM to the OM.119 It is part of the lipoprotein transporting Lol complex, which consists of LolA, LolB, LolC, LolD, and LolE. The function of LolA is to receive nascent lipoprotein from the IM protein complex LolCDE and pass it on to the OM protein LolB, which then inserts it into the OM (Figure 5a).120,121
Figure 5.
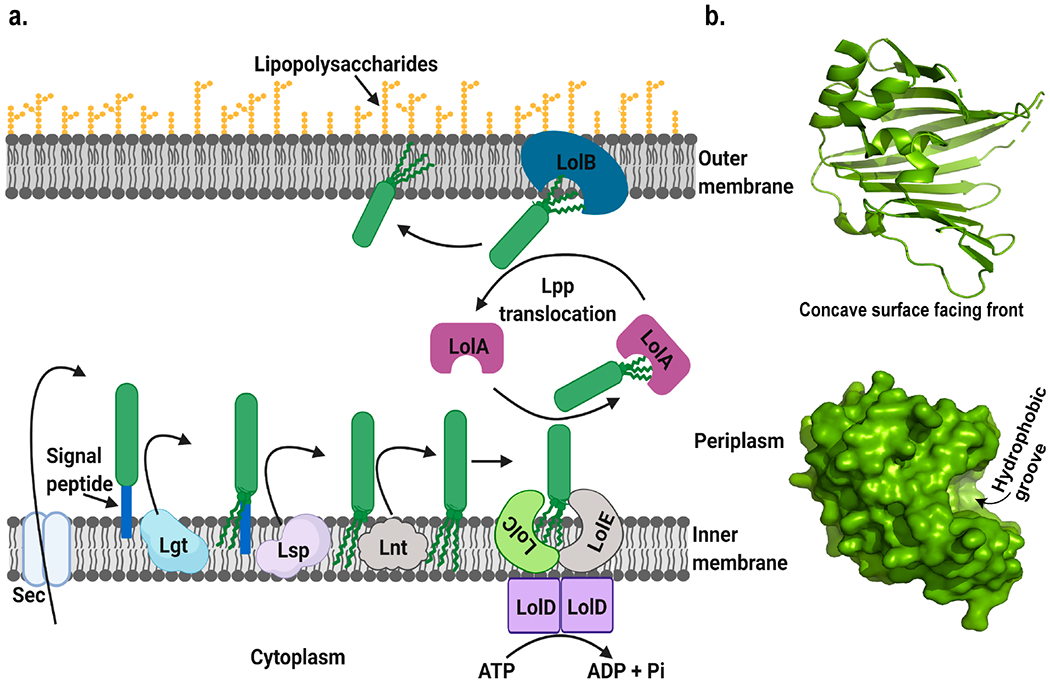
(a) Schematic representation of the transport mechanism of OM lipoprotein. Lpp represents OM lipoprotein. After translocation into the periplasmic side by the sec machinery, it is diacylated by dicaylglyceryl transferase (Lgt), followed by the removal of the signal peptide by lipoprotein signal peptidase (Lsp) and acylation by N-acyl transferase (Lnt), and finally translocated across the periplasm by the Lol complex. Figure was created using Biorender. (b) Structure of E. coli LolA (Created with Pymol73 using 1UA8.pdb) showing the concave surface and hydrophobic groove.119
Lipoproteins are synthesized with a signal peptide in their N-terminal end. A short, conserved region is present in the C-terminal region of the signal peptide called the lipobox. After translocation of the nascent polypeptide chain by the sec machinery, diacylglycerol is attached to a specific cysteine present in the lipobox by lipoprotein diacylglyceryl transferase (Lgt) to form pro-lipoprotein. During maturation, the signal peptide is cleaved by lipoprotein signal peptidase (Lsp) to form mature lipoproteins. In Gram-negative bacteria an extra acylation step occurs at the N-terminus of mature lipoproteins catalyzed by lipoprotein N-acyl transferase (Lnt). The lipoprotein is then sorted and transported to the destined region.121,122 Selection and sorting of lipoproteins follow a “+2 rule”. Presence of aspartate at the +2 position after the acylated cysteine at +1 position signals the lipoprotein to be delivered to the IM, otherwise it is transported to the OM by the Lol pathway.123
The LolCDE complex catalyzes the release of the lipoproteins from the IM in an ATP dependent manner. LolA then receives the lipoprotein, crosses the periplasm as a LolA–Lpp complex, and presents the lipoprotein cargo to the protein LolB, which then inserts the lipoprotein into the OM.121 LolA is essential to the release of lipoproteins from the IM. Without LolA, lipoproteins that should be associated with the OM remain deposited in the IM.121
LolA consists of 11 antiparallel strands of β-sheet and 3 α-helices, forming an unclosed β-barrel with α-helices sitting on top of the concave side (Figure 5b). A hydrophobic cavity formed between the unclosed β-barrel the α-helical lid is the putative binding site for lipoproteins. A similar type of cavity also exists in LolB but with higher affinity of binding.119 Although lipoproteins have highly hydrophobic N-terminal lipids, the lipoprotein–LolA complexes are water-soluble and diffuse freely in the periplasm. Cargo delivery from LolA to LolB relies on the affinity difference.121 Recent simulation studies also suggest that the transfer of lipoproteins from LolA to LolB is thermodynamically favorable.124
3.4.1. Inhibition or Depletion of LolA Is Lethal.
Lack of lipoproteins could alter the OM assembly and make it leaky, subsequently sensitizing or even killing the bacteria.24 In addition to the maintenance of OM stability and integrity, lipoproteins are also involved in cell wall synthesis, antibiotic efflux, protein secretion and synthesis of motility machinery.123 Since the Lol system is involved in the transport of OM lipoproteins across the periplasm, defects in LolA function led to the accumulation of lipoproteins in the IM.120 In addition, depletion of essential lipoproteins in the OM also severely impaired bacterial fitness and survival.
Mutagenesis study showed that formation of a disulfide bond between I93C and F140C inactivated the function of LolA and thereby severely inhibited the growth of E. coli.125 Oxidized LolA triggered the stress response mechanism, which substantiated the importance of LolA for the normal growth of bacteria.126 Liao et al. demonstrated that a lolA mutation in Xanthomonas campestris led to reduced virulence, adhesion, biofilm formation, and tolerance.127 However, a recent study showed that in a stress-maintained system E. coli could still grow when both lolA and lolB genes were deleted.128 This suggested the existence of an alternative route for lipoprotein transport that involves other periplasmic proteins.
3.4.2. Inhibitors of LolA.
Several inhibitors target the maturation steps of lipoproteins before and during transport. In this review, we focus on the periplasmic component of the lipoprotein transport system, that is, LolA and its inhibitors. Readers can refer to other excellent reviews for the IM targets involved in maturation as well as transport of lipoproteins.24 Pathania et al. found that the inhibition of cell growth by compound MAC13243 (Figure S10) in E. coli could be suppressed by the overexpressing the lolA gene.120 MAC13243 was identified in a screening for molecules that inhibit the growth of E. coli at 50 μM. Using chemical genomics and overexpression suppression studies, the potential target of MAC13243 was identified to be LolA. Overexpressing lolA gene led to an increase of minimum inhibitory concentration (MIC) of MAC13243 by 16-fold.120 It was observed that new Lpps accumulated in the IM when the cells were treated with the compound, indicating that the presence of the compound inhibited the transport of Lpp from the IM to the OM. The binding dissociation constant of MAC13243 to LolA was determined to be 7.5 μM. Structure–activity relationship studies revealed the importance of the benzyl moiety substitution and the preference of an intermediate electron-withdrawing substituent group such as chlorine or bromine. The location of the halogen substituent is another important factor with a para position being ideal. In contrast, the dimethoxyphenylethyl group was less important to LolA interaction. MAC13243 and its analogs were tested in several bacteria strains, and they were found to be active in Gram-negative bacteria only. Finally, this family of compounds were shown not to be affected by the AcrAB-TolC efflux pump.120
Barker et al. found that MAC13243 was prone to degradation in an aqueous solution under acidic conditions.129 One of the degradation products, S-(4-chlorobenzyl) isothiourea, was responsible for the antibiotic activity and appeared to be closely related to another compound, A22 (Figure S10). A22 has previously been shown to inhibit MreB. Hence, inhibition of MreB was suspected to be the mechanism of cell growth inhibition by MAC13243. Further investigation revealed that the overexpression of lolA restored cell growth in the presence of the degradation products and analogs such as A22. NMR studies confirmed the interaction between the analogs with LolA. The phenotype of lolA deleted cells was very similar to that of the mreB deleted cells. Although the relationship between these two proteins is unclear, inhibition of LolA activity likely contributed to the observed antimicrobial activity.129
Later in 2017, Muheim et al. studied the performance of MAC13243 as a potentiator.34 They investigated the effect of the sublethal concentration of this compound on the OM permeability of E. coli. Results showed that the depletion of LolA as well as the use of sublethal concentration of MAC13243 increased the permeability of the OM. Compared to a known permeabilizing agent colistin, MAC13243 caused more severe OM leakage. Antimicrobials with high molecular weight, such as vancomycin, erythromycin, rifampicin, and novobiocin, were potentiated when LolA was depleted or MAC13243 was used. Calculations of the fractional inhibitory concentration index (FICI) showed a synergistic relationship between MAC13243 and erythromycin or novobiocin but not with vancomycin or rifampicin.
However, one recent article argued against the inhibition of LolA by A22-like molecules. Buss et al. suggested that the overexpression of LolA suppressed the lethal effects of A22-like molecules through induction of the Rcs stress response system. They demonstrated that in ΔrcsF mutants, overexpression of LolA did not inhibit the lethal effect of A22. This suggested there could be an alternative target of A22 aside from LolA.130
3.5. Lipopolysaccharide Transport Protein (LptA).
3.5.1. LptA and LPS Transport.
LPS transport protein A (LptA) is involved in the transport of LPS from the IM to the OM.131 It is part of a pathway that consists of seven proteins (LptABCDEFG) responsible for LPS assembly and transport to the cell surface (Figure 6a).132 The Lpt machinery is segmented into two functional assemblies.133,134 At the IM, LptB2FG and LptC provide the energy for transport. LptD and LptE form the OM translocon responsible for LPS assembly at the last steps of transport.135,136 LptA links the functions of the ATP driven LptB2FGC and the LptDE complexes, delivering LPS across the periplasm.137
Figure 6.
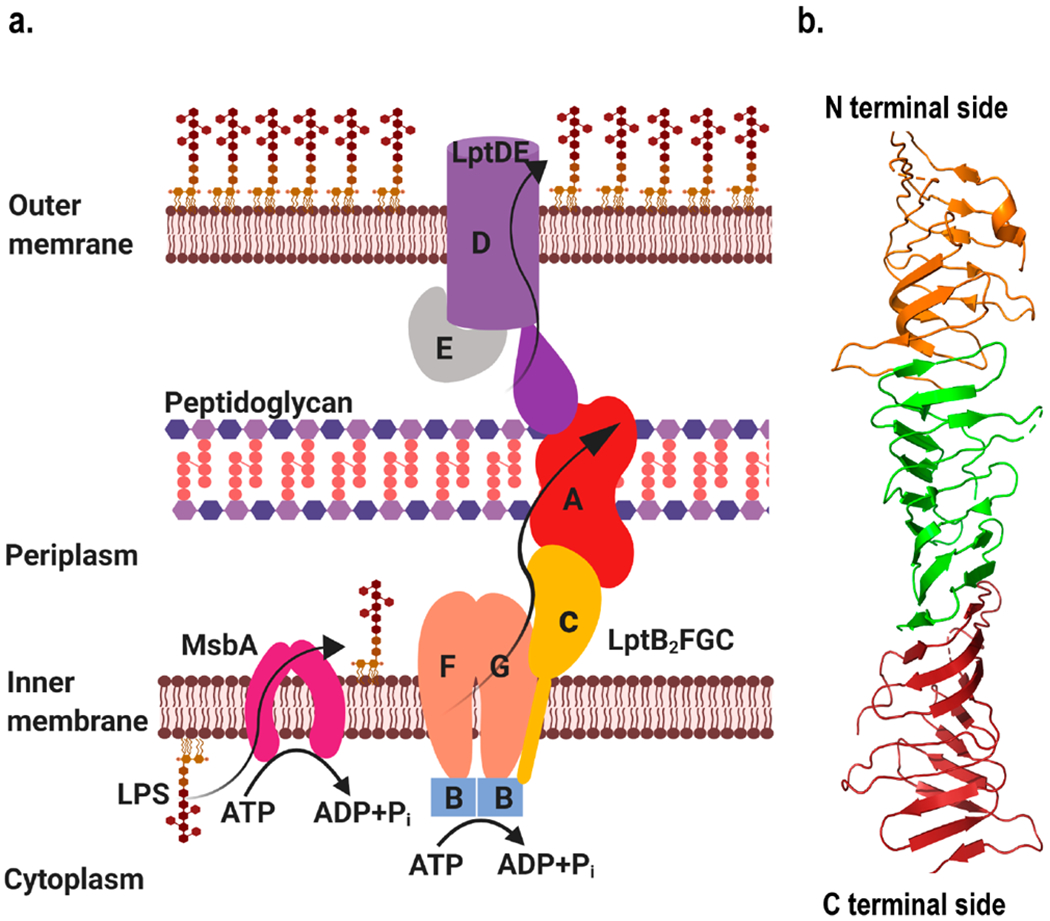
(a) LPS transport pathway in E. coli where an ATP-binding cassette (ABC) transporter and a flippase, MbsA, are involved in flipping LPS synthesized in the cytoplasm to the periplasmic side, which is then moved to the cell surface through the Lpt pathway. The pathway comprises seven proteins LptA, LptB, LptC, LptD, LptE, LptF, and LptG. The LptB2FG transfers LPS to LptC, driven by ATP hydrolysis. LPS was transported to the OM through a protein bridge formed by LptC and LptA and integrated into the OM by LptDE. Figure was created using Biorender. (b) Structure of E. coli LptA (Created with Pymol73 using 2R1A.pdb) where each color represents a monomer of LptA interacting with neighboring monomer in head to tail fashion.131
LPS plays an importance role in forming the permeation barrier in Gram-negative bacteria.138 The mechanism of transport of LPS through the periplasmic space is yet to be fully understood.139 Several reports showed that LptA oligomerization was essential for the interaction with LPS and the delivery through periplasm.140,141 Defects in LptA function impaired transport of LPS and assembly at the OM, with the consequence of increased susceptibility to antimicrobials.142
3.5.2. Structure and Function of LptA.
Two models have been proposed to explain the transport of LPS across the membrane, the shuttling model and the trans-envelope model.12 In the shuttling model, transport of LPS across the periplasm occurs via a chaperone system.131 LptA is proposed to work similarly to LolA, acting as a chaperone protein and shielding the acyl chains of LPS during periplasmic transport.15,143 For the trans-envelope model, a multiprotein complex mediates the LPS transport.144,145 Later LptA was found to form part of a trans-envelope bridge spanning the periplasmic space during LPS transport, lending credence to the trans-envelope model.133,146
LptA contains 16 antiparallel β-strands, folding into a twisted β-jellyroll. In the crystal structure, LptA molecules form long fibers, with each LptA interacting with an adjacent LptA molecule in a head-to-tail fashion (Figure 6b).131 Later Freinkman et al. used an in vivo photo-cross-linking method to probe the protein–protein interaction sites among the Lpt components that form the Lpt bridge.147 Both LptC and the N-terminal domain of LptD are structural homologous of LptA. They were found to assemble with LptA in a head-to-tail fashion, similar to the LptA assembly observed in its crystal structure. In addition, the in vivo cross-linking data was compatible with LptA functioning either as a single monomer or as a head-to-tail dimer to interface between LptC and LptD.
3.5.3. LptA Mutations and Their Effects.
Suits et al. reported that the overexpression of certain LptA mutants was deleterious to the transport of LPS.131 Several conserved residues in the N-terminal region of LptA were mutated to probe their impact on LPS transport. Ile36 and Ile38 were mutated to Asp or Glu, Arg76 was mutated to Asp, and Phe95 was mutated to Ala. These mutations did not impair the function of the protein, although their overexpression appeared to be detrimental to LPS transport. Overexpression of LptA I36D, I36E, I38D, and I38E in cells was more lethal than overexpression of the wild-type LptA. Overexpression of LptA R76D, R76D and F95A showed no negative effects on function. These mutations likely disrupted the head-to-tail stacking of LptA molecules, thereby blocking LPS transport. In another study, Falchi et al. mutated four conserved amino acid residues (I36A, I38A, R76, and K83). I36 and I38 are found in a location implicated in LptA–LptC or LptA–LptA interaction.132 These mutations led to defective Lpt assembly and increased susceptibilities to many antimicrobials including novobiocin, rifampicin, and bacitracin.
3.5.4. LptA Inhibitors Alter or Block LPS Transport.
The inhibition of LPS biogenesis leads to cell death.148 Cell death is likely due to compromised OM assembly.146,149 Compounds including 4-phenylpyrrolcarbazole (PPC) derivatives have been shown to be potent against the Lpt pathway.150–152 These compounds were previously known to inhibit Wee1, a kinase that controls mitosis in eukaryotes. They were found to inhibit the ATPase activity of LptB. ATPases have been used routinely as important targets in eukaryotes with their inhibitors used to treat cancer.153 Many of the studies on inhibitors have been on LptB, LptD, and the LptB2FCG complex, but not LptA specifically.150,152,154–156 Vetterli et al. showed that LptA was inhibited by thanatin, a peptide of 21-residues containing a disulfide bond (GSKKPVPIIYCNRRTGKCQRM) (Figure S11) that is naturally derived from an insect, Podisus maculiventris.157,158 NMR structure of the LptA–thanatin complex was obtained and overlaid on the crystal structure of LptA’s head-to-tail oligomer, revealing that the binding site of thanatin on LptA overlapped with the LptA–LptA interface. This result suggests that thanatin worked through blocking the interaction between the LptA and its function partner LptD or preventing the formation of the functional LptA dimer. However, more recently Ma et al. reported that thanatin disrupted the OM by competitively displacing divalent cations on the OM and inducing the release of lipopolysaccharides.159
3.6. ZnuA.
ZnuA is the periplasmic component of the high affinity zinc specific uptake system ABC transporter (ZnuABC), which is responsible for the uptake of zinc in many Gram-negative bacteria in zinc limited environments.160,161 ZnuABC transporter consists of three components, a periplasmic binding protein (ZnuA), an integral membrane transporter (ZnuB), and an ATPase (ZnuC). ZnuA captures zinc in the periplasm, and ZnuB and ZnuC work together to transport zinc across the IM powered by ATP hydrolysis.160,161 Although some Gram-negative bacteria, including Pseudomonas aeruginosa,162 Neisseria meningitidis,163 Yersinia pestis,164 and Acinetobacter baumannii,165 have multiple zinc uptake systems, ZnuABC has been found to be the major uptake system in zinc deficient environments during infection.166 The crystal structures of ZnuA from different Gram-negative bacteria revealed highly conserved structure with two (α/β)4 domains connected by a flexible α-helix (Figure 7a). The primary zinc binding site is located in the interdomain cleft, where zinc is coordinated by three conserved histidine residues and a glutamate or water molecule.167 Apart from this site, a second zinc binding site has been observed in E. coli ZnuA, 12 Å away from the primary site.160,168 Moreover, another distinct feature of this protein is the presence of a histidine rich loop, which has not been properly resolved in crystal structures. It is located near the primary metal binding site, comprises sequentially diverse acidic residues, and is believed to have a role in zinc sequestration.168
Figure 7.
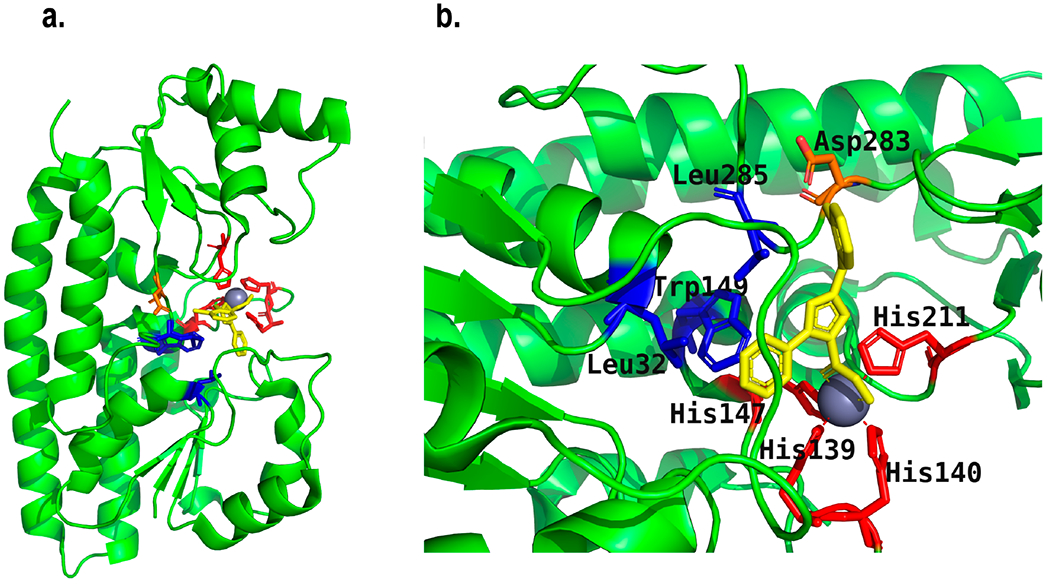
(a) Structure of Salmonella ZnuA (SeZnuA) (Created with Pymol73 using 4BBP.pdb) showing the binding of its inhibitor RDS51 (yellow) along with zinc.166 (b) Enlarged view of structure a showing the coordination of zinc with four histidine residues (red) and two oxygens of RDS51. Blue residues have hydrophobic interactions with RDS51, and the orange residue forms a H-bond with RDS51.
3.6.1. ZnuA Is Essential for Virulence in Zinc Limited Environments.
Zinc is an essential metal for bacteria as it is a cofactor for many proteins including DNA polymerases, proteases, and ribosomal proteins.169 Studies have shown that deletion or mutation in the ZnuABC system or the periplasmic component ZnuA led to growth defects and reduction in virulence in many Gram-negative bacteria including E. coli,161 Salmonella,170 Brucella abortus,171 Campylobacter jejuni,172 Haemophilus ducreyi,173 and Moraxella catharralis.174 For example, Sabri et al. reported that deletion of the Znu transporter inhibited the growth of a uropathogenic strain of E. coli in zinc limited conditions.161 Furthermore, they found that on complementation by plasmid containing Znu or addition of zinc, growth was restored. Also, in case of ZnuA deficient strains, the ability to infect and grow in the urinary tract and kidneys in the murine infection model was compromised. Deletion of ZnuA also compromised the motility of bacteria and made bacteria more susceptible to oxidative stress. Ammendola et al. studied the role of ZnuABC transporter in the virulence of Salmonella enterica ser. Typhimurium and Enteritidis.170 They discovered a loss of virulence when this transporter was compromised, and the defect was similar when the entire gene cluster or just the znuA gene was compromised. Pathogenicity was also reduced in mouse model. Because of this loss in virulence, znuA deleted mutants of Brucella abortus had been studied as a potential candidate for the development of live vaccine.171
3.6.2. Inhibitors of ZnuA.
Studies showed that LPS treatment led to a decrease of zinc level in serum of an animal model, while the overall zinc availability at the infection site was further reduced by the host system through the secretion of calprotectin, a protein that captures zinc.161,175 Sensing the scarcity of zinc at the infection site, bacteria rely on the highly efficient zinc uptake system ZnuABC to acquire zinc. Disruption of the zinc uptake pathway severely affected zinc homeostasis in bacteria during infection. Ilari et al. searched for inhibitors for ZnuA to reduce virulence of Salmonella.166 They screened an in-house chemical library at University of Rome for compounds that bound zinc and tested their antibacterial activity. Among those compounds, RDS50 and RDS51 were found to inhibit Salmonella growth at concentrations less than 500 μM. When presaturated with zinc, they had even higher growth inhibition effect. RDS51 and its zinc saturated form had remarkable inhibition toward Salmonella invasion of Caco-2 cells. The crystal structure of ZnuA showed that zinc was coordinated by Glu59, His147, His211, and His140 of the metal binding site. However, in the presence of RDS51, zinc was found to coordinate with four histidine residues, His139, His140, His147, and His211, as well as two oxygens from RDS51. Apart from binding with zinc via oxygen atoms, RDS51 was shown to form hydrophobic interactions with Leu285, Leu32, and Trp149 and hydrogen bond with Asp283 through its chloride (Figure 7b).166
3.7. TolB.
TolB is one of the most abundant proteins in the periplasm. It is a component of the Tol-PAL system, initially identified as a component of the colicin transport system. The Tol-PAL system is composed of four Tol proteins and an OM PAL (peptidoglycan associated lipoprotein) protein (Figure 8).176,177 Among the Tol proteins, TolB is the only protein entirely located in the periplasm. TolQ and TolR are integral IM proteins. TolA has an extended second domain and a C-terminal domain in the periplasm, with the N-terminal domain anchored to the IM.176 TolB has two domains, an N-terminal domain with two α-helices, a five stranded β-sheet, and a long loop on its back and a C-terminal domain with a six bladed β-propeller.178,179
Figure 8.
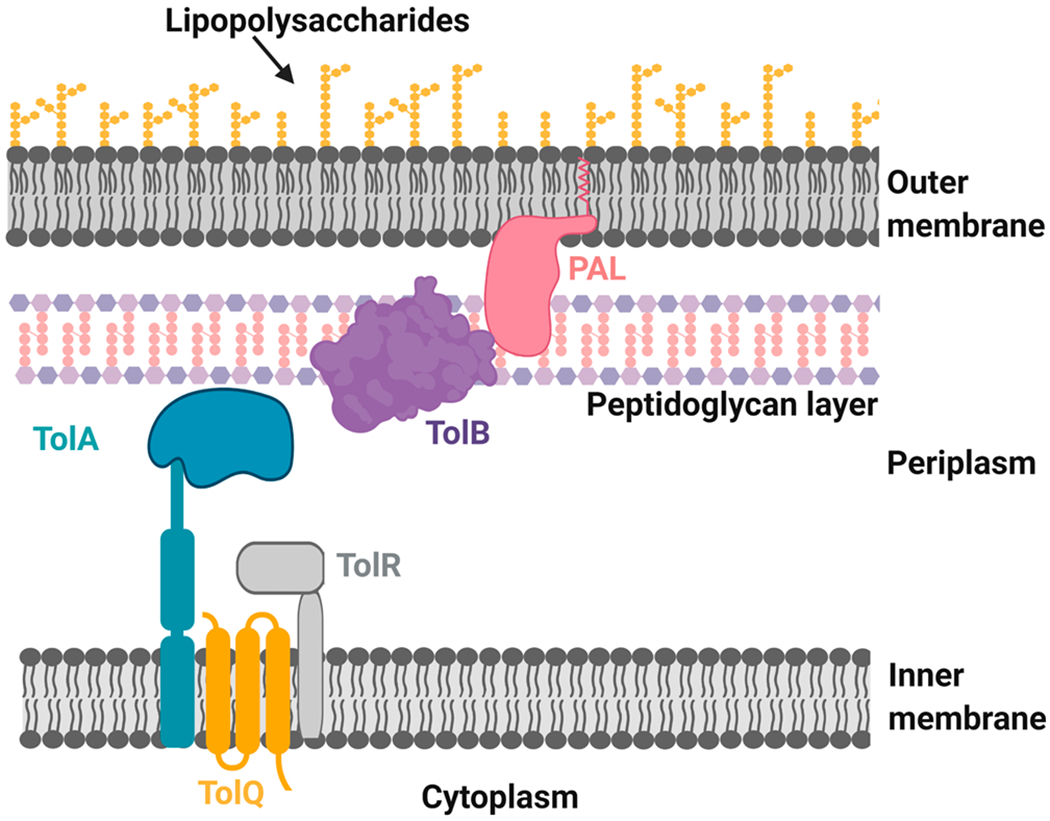
Assembly of Tol-PAL system in Gram-negative cell envelope. Figure was created using Biorender.
Originally, the Tol-PAL system was discovered for its role in the import of colicins, but subsequent studies revealed that it plays an essential role in the maintenance of cell envelope integrity. The Tol-PAL complex interacts with all three layers of the cell envelope: the OM, peptidoglycan, and the IM. The abundance of various porins, lipoproteins, and sugar content in LPS decreased in Tol inactive mutants.29,176,180,181 The role of this complex in the maintenance of cell envelope integrity makes it a potential antimicrobial target.
Lo Sciuto et al. reported in 2014 that TolB was essential for the survival and virulence of multidrug-resistant Pseudomonas aeruginosa.177 A tolB knockout strain of P. aeruginosa could not be created, suggesting its essential role in survival. A conditional tolB knockout strain, PAO1 ΔtolB araC-PBAD tolB, which only grew in the presence of arabinose, was created. On further analysis of those cells using a dual refresh strategy, it was observed that the tolB deficient strain was ~1000-fold more sensitive toward SDS than the wild-type or tolB expressing mutant strain. Electron microscopy studies revealed the presence of OM blebs and the release of cellular contents during cell elongation and membrane invagination. Compromised membrane integrity during cell division was inferred. Disk diffusion assay in the presence of a very low concentration of arabinose showed higher sensitivity of the mutant against clinically used antimicrobials ciprofloxacin, ofloxacin, imipenem, and ceftazidime, but not ampicillin and polymyxin. In vivo studies performed using a G. mellonella larvae model showed that the lethal dose represented by LD90 was around 600000-fold higher for the mutant than the wild-type strain. In conclusion, they suggested that TolB could be a good target to design antimicrobials against P. aeruginosa because of its abundance, accessibility, and importance in bacterial growth.177
4. CONCLUDING REMARKS
In this work, we reviewed recent studies on the exploration of several periplasmic proteins as antimicrobial targets. Other than TolB, small molecule inhibitors have been identified for all six of the other proteins discussed (Table 1). Most of these studies were published in the past five years, and the potential for the development of these inhibitors into clinically useful therapeutics remains to be explored. We would like to point out that it is always a possibility for inhibitors identified through design or screening to hit multiple target proteins, not just the intended one. A genetic approach can be helpful in validating the target in cells, for example, showing that resistance mutations reside in that target gene or compensation of growth defect through overexpression of the target protein. But that can be difficult with antivirulence targets, as it is often hard to create a selection in vitro for such mutations. Thus, selectivity of inhibitors against antivirulence targets has generally not been proven.
In this review, we focused on proteins located completely in the periplasm. Apart from these periplasmic proteins, there are several other IM proteins that contain domains (either active site or allosteric site) extended into the periplasm, such as the type I and II signal peptidase, LolC, LolE, DsbB, LptC, and TolA. Inhibitors could potentially be developed to target these domains. In addition to the ones discussed above, several less-studied periplasmic proteins could also be potential antimicrobial targets. One example is LptH, a LptA orthologue in Pseudomonas aeruginosa.182 Another example is the periplasmic component of other zinc uptake pathways such as AztC and metallochaperone AztD of Paracoccus denitrificans.183 The periplasmic Cu—Zn superoxide dismutase of Shigella dysenteriae also plays an important role in cell survival.184 Additional studies are necessary to further understand the structure and mechanism of these proteins.
Multidrug resistant Gram-negative bacteria are rapidly evolving as a global health threat. We need to act fast to win this war through the development of new classes of antimicrobials and novel targets. However, designing and developing new antimicrobials turns out to be a challenging task, especially for those that are effective against Gram-negative bacteria. The diverse functions of the periplasmic proteins make the periplasm a site of interest for identifying new drug targets. Periplasmic proteins play important roles in maintaining cell survival, membrane integrity, and cell division, as well as the synthesis of virulence factors, adhesion molecules, and signaling molecules. Periplasmic targets are more readily accessible with a lower permeation barrier. However, even with just the OM to breach, the task is by no means straightforward. For example, strains expressing engineered porins with a large pore have been successfully employed to further permeabilize the OM to enable the identification of early lead compounds.39 With the development of new techniques and alternative approaches such as targeting virulence and resistance rather than survival, the periplasm might turn out to be the spot where the next new antimicrobial comes from, just like the β-lactams that started the golden era of antimicrobial development.
Supplementary Material
ACKNOWLEDGMENTS
This work is supported by NIH grant numbers 1R56AI137020 and 1R21AI142063-01, and NSF grant number CHE-1709381.
Footnotes
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsinfecdis.0c00384.
Experimental and computation hits and synthesized analogs that bind to AcrA, EPIs with similar novel scaffold, structures of compounds 1 and 10, 4, 39, 40, 41, and 42, phenylthiophene and phenoxy phenyl inhibitors of DsbA, analogues of ubiquinone (1, 8, 18, and 19), benzofuran derivatives (25 and 28), binders and potential inhibitors of SurA, MAC14243 and A22, and thanatin (inhibitor of LptA) (PDF)
The authors declare no competing financial interest.
Complete contact information is available at: https://pubs.acs.org/10.1021/acsinfecdis.0c00384.
Contributor Information
Ankit Pandeya, Department of Chemistry, University of Kentucky, Lexington, Kentucky 40506, United States.
Isoiza Ojo, Department of Chemistry, University of Kentucky, Lexington, Kentucky 40506, United States.
Olaniyi Alegun, Department of Chemistry, University of Kentucky, Lexington, Kentucky 40506, United States.
Yinan Wei, Department of Chemistry, University of Kentucky, Lexington, Kentucky 40506, United States.
REFERENCES
- (1).IACG (2019) No Time to Wait-Securing the Future from Drug-resistant Infections, Report to the Secretary General of the United Nations, pp 1–28. [Google Scholar]
- (2).Ligon BL (2004) Penicillin: its discovery and early development. Seminars in pediatric infectious diseases 15, 52–57. [DOI] [PubMed] [Google Scholar]
- (3).Zaman SB, Hussain MA, Nye R, Mehta V, Mamun KT, and Hossain N (2017) A review on antibiotic resistance: alarm bells are ringing. Cureus 9 (6), e1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Lewis K (2017) New approaches to antimicrobial discovery. Biochem. Pharmacol . 134, 87–98. [DOI] [PubMed] [Google Scholar]
- (5).Li B, and Webster TJ (2017) Bacteria antibiotic resistance: New challenges and opportunities for implant-associated orthopedic infections. J. Orthop. Res. 36 (1), 22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Mulani MS, Kamble EE, Kumkar SN, Tawre MS, and Pardesi KR (2019) Emerging strategies to combat ESKAPE pathogens in the era of antimicrobial resistance: a review. Front. Microbiol. 10, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Rice LB (2008) Federal Funding for the Study of Antimicrobial Resistance in Nosocomial Pathogens: No ESKAPE. J. Infect. Dis. 197 (8), 1079–1081. [DOI] [PubMed] [Google Scholar]
- (8).González-Bello C (2017) Antibiotic adjuvants-A strategy to unlock bacterial resistance to antibiotics. Bioorg. Med. Chem. Lett 27 (18), 4221–4228. [DOI] [PubMed] [Google Scholar]
- (9).Breijyeh Z, Jubeh B, and Karaman R (2020) Resistance of Gram-Negative Bacteria to Current Antibacterial Agents and Approaches to Resolve It. Molecules 25 (6), 1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Pukhrambam N (2019) Comparison of original gram stain and its modification in the gingival plaque samples. J. Bacteriol Mycol Open Access 7 (1), 1–3. [Google Scholar]
- (11).Silhavy TJ, Kahne D, and Walker S (2010) The bacterial cell envelope. Cold Spring Harbor Perspect. Biol 2 (5), No. a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Bos MP, Robert V, and Tommassen J (2007) Biogenesis of the gram-negative bacterial outer membrane. Annu. Rev. Microbiol 61, 191–214. [DOI] [PubMed] [Google Scholar]
- (13).Matsuura M (2013) Structural Modifications of Bacterial Lipopolysaccharide that Facilitate Gram-Negative Bacteria Evasion of Host Innate Immunity. Front. Immunol 4, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Braun V, and Hantke K (2019) Lipoproteins: Structure, Function, Biosynthesis, Bacterial Cell Walls and Membranes, pp 39–77, Springer. [DOI] [PubMed] [Google Scholar]
- (15).Okuda S, and Tokuda H (2011) Lipoprotein Sorting in Bacteria. Annu. Rev. Microbiol 65 (1), 239–259. [DOI] [PubMed] [Google Scholar]
- (16).Morè N, Martorana AM, Biboy J, Otten C, Winkle M, Serrano CKG, Silva AM, Atkinson L, Yau H, Breukink E, et al. (2019) Peptidoglycan remodeling enables Escherichia coli to survive severe outer membrane assembly defect. mBio 10 (1), No. e02729–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Richter MF, Drown BS, Riley AP, Garcia A, Shirai T, Svec RL, and Hergenrother PJ (2017) Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature 545 (7654), 299–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Silver LL (2016) A Gestalt approach to Gram-negative entry. Bioorg. Med. Chem 24 (24), 6379—6389. [DOI] [PubMed] [Google Scholar]
- (19).Silver LL (2011) Challenges of antibacterial discovery. Clin Microbiol Rev . 24 (1), 71–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Zgurskaya HI, and Rybenkov VV (2020) Permeability barriers of Gram-negative pathogens. Ann. N. Y. Acad. Sci . 1459 (1), 5—18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Richter MF, and Hergenrother PJ (2019) The challenge of converting Gram-positive-only compounds into broad-spectrum antibiotics. Ann. N. Y. Acad. Sci 1435 (1), 18–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Krishnamoorthy G, Leus IV, Weeks JW, Wolloscheck D, Rybenkov VV, and Zgurskaya HI (2017) Synergy between Active Efflux and Outer Membrane Diffusion Defines Rules of Antibiotic Permeation into Gram-Negative Bacteria. mBio 8 (5), No. e01172–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Cooper SJ, Krishnamoorthy G, Wolloscheck D, Walker JK, Rybenkov VV, Parks JM, and Zgurskaya HI (2018) Molecular Properties That Define the Activities of Antibiotics in Escherichia coli and Pseudomonas aeruginosa. ACS Infect. Dis 4 (8), 1223–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Lehman KM, and Grabowicz M (2019) Countering Gram-Negative Antibiotic Resistance: Recent Progress in Disrupting the Outer Membrane with Novel Therapeutics. Antibiotics 8 (4), 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Prochnow H, Fetz V, Hotop S-K, García-Rivera MA, Heumann A, and Brönstrup M (2019) Subcellular quantification of uptake in Gram-negative bacteria. Anal. Chem 91 (3), 1863–1872. [DOI] [PubMed] [Google Scholar]
- (26).Stock JB, Rauch B, and Roseman S (1977) Periplasmic space in Salmonella typhimurium and Escherichia coli. J. Biol. Chem 252 (21), 7850–7861. [PubMed] [Google Scholar]
- (27).Pilizota T, and Shaevitz JW (2012) Fast, multiphase volume adaptation to hyperosmotic shock by Escherichia coli. PLoS One 7 (4), e35205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Miller SI, and Salama NR (2018) The gram-negative bacterial periplasm: Size matters. PLoS Biol. 16 (1), e2004935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Lazzaroni JC, Germon P, Ray M-C, and Vianney A (1999) The Tol proteins of Escherichia coli and their involvement in the uptake of biomolecules and outer membrane stability. FEMS Microbiol. Lett 177 (2), 191–197. [DOI] [PubMed] [Google Scholar]
- (30).Hobot J, Carlemalm E, Villiger W, and Kellenberger E (1984) Periplasmic gel: new concept resulting from the reinvestigation of bacterial cell envelope ultrastructure by new methods. J. Bacteriol 160 (1), 143–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Weiner JH, and Li L (2008) Proteome of the Escherichia coli envelope and technological challenges in membrane proteome analysis. Biochim. Biophys. Acta, Biomembr. 1778 (9), 1698–1713. [DOI] [PubMed] [Google Scholar]
- (32).Ha UH, Wang Y, and Jin S (2003) DsbA of Pseudomonas aeruginosa is essential for multiple virulence factors. Infect. Immun 71 (3), 1590–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Zhong M, Ferrell B, Lu W, Chai Q, and Wei Y (2013) Insights into the function and structural flexibility of the periplasmic molecular chaperone SurA. J. Bacteriol 195 (5), 1061–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Muheim C, Gotzke H, Eriksson AU, Lindberg S, Lauritsen I, Norholm MHH, and Daley DO (2017) Increasing the permeability of Escherichia coli using MAC13243. Sci. Rep 7 (1), 17629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Klein K, Sonnabend MS, Frank L, Leibiger K, Franz-Wachtel M, Macek B, Trunk T, Leo JC, Autenrieth IB, Schutz M, and Bohn E (2019) Deprivation of the Periplasmic Chaperone SurA Reduces Virulence and Restores Antibiotic Susceptibility of Multidrug-Resistant Pseudomonas aeruginosa. Front. Microbiol 10, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Dickey SW, Cheung GYC, and Otto M (2017) Different drugs for bad bugs: antivirulence strategies in the age of antibiotic resistance. Nat. Rev. Drug Discovery 16 (7), 457–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Imperi F, Fiscarelli EV, Visaggio D, Leoni L, and Visca P (2019) Activity and Impact on Resistance Development of Two Antivirulence Fluoropyrimidine Drugs in Pseudomonas aeruginosa. Front. Cell. Infect. Microbiol 9, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).García-Contreras R, Maeda T, and Wood TK (2013) Resistance to Quorum-Quenching Compounds. Appl. Environ. Microbiol . 79 (22), 6840–6846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Maeda T, García-Contreras R, Pu M, Sheng L, Garcia LR, Tomás M, and Wood TK (2012) Quorum quenching quandary: resistance to antivirulence compounds. ISME J. 6 (3), 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Maura D, Ballok AE, and Rahme LG (2016) Considerations and caveats in anti-virulence drug development. Curr. Opin. Microbiol 33, 41–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Rasko DA, and Sperandio V (2010) Anti-virulence strategies to combat bacteria-mediated disease. Nat. Rev. Drug Discovery 9 (2), 117–128. [DOI] [PubMed] [Google Scholar]
- (42).Allen RC, Popat R, Diggle SP, and Brown SP (2014) Targeting virulence: can we make evolution-proof drugs? Nat. Rev. Microbiol 12 (4), 300–308. [DOI] [PubMed] [Google Scholar]
- (43).Colclough AL, Alav I, Whittle EE, Pugh HL, Darby EM, Legood SW, McNeil HE, and Blair JM (2020) RND efflux pumps in Gram-negative bacteria; regulation, structure and role in antibiotic resistance. Future Microbiol. 15 (2), 143–157. [DOI] [PubMed] [Google Scholar]
- (44).Frieri M, Kumar K, and Boutin A. J. J. o. i. (2017) Antibiotic resistance. Journal of infection and public health 10 (4), 369–378. [DOI] [PubMed] [Google Scholar]
- (45).Fralick JA (1996) Evidence that TolC is required for functioning of the Mar/AcrAB efflux pump of Escherichia coli. Journal of bacteriology 178 (19), 5803–5805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Zgurskaya HI, and Nikaido H (1999) Bypassing the periplasm: reconstitution of the AcrAB multidrug efflux pump of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A 96 (13), 7190–7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Dinh T, Paulsen IT, and Saier MH Jr. (1994) A family of extracytoplasmic proteins that allow transport of large molecules across the outer membranes of gram-negative bacteria. J. Bacteriol 176 (13), 3825–3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Okusu H, Ma D, and Nikaido H (1996) AcrAB efflux pump plays a major role in the antibiotic resistance phenotype of Escherichia coli multiple-antibiotic-resistance (Mar) mutants. Journal of bacteriology 178 (1), 306–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Ma D, Cook DN, Alberti M, Pon NG, Nikaido H, and Hearst JE (1993) Molecular cloning and characterization of acrA and acrE genes of Escherichia coli. J. Bacteriol . 175 (19), 6299–6313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Nikaido H, and Takatsuka Y (2009) Mechanisms of RND multidrug efflux pumps. Biochim. Biophys. Acta, Proteins Proteomics 1794 (5), 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Zgurskaya HI, Yamada Y, Tikhonova EB, Ge Q, and Krishnamoorthy G (2009) Structural and functional diversity of bacterial membrane fusion proteins. Biochim. Biophys. Acta, Proteins Proteomics 1794 (5), 794–807. [DOI] [PubMed] [Google Scholar]
- (52).Abdali N, Parks JM, Haynes KM, Chaney JL, Green AT, Wolloscheck D, Walker JK, Rybenkov VV, Baudry J, Smith JC, and Zgurskaya HI (2017) Reviving Antibiotics: Efflux Pump Inhibitors That Interact with AcrA, a Membrane Fusion Protein of the AcrAB-TolC Multidrug Efflux Pump. ACS Infect. Dis. 3 (1), 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Opperman TJ, and Nguyen ST (2015) Recent advances toward a molecular mechanism of efflux pump inhibition. Front. Microbiol. 6, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Haynes KM, Abdali N, Jhawar V, Zgurskaya HI, Parks JM, Green AT, Baudry J, Rybenkov VV, Smith JC, and Walker JK (2017) Identification and Structure-Activity Relationships of Novel Compounds that Potentiate the Activities of Antibiotics in Escherichia coli. J. Med. Chem. 60 (14), 6205–6219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Zgurskaya HI, Weeks JW, Ntreh AT, Nickels LM, and Wolloscheck D (2015) Mechanism of coupling drug transport reactions located in two different membranes. Front. Microbiol. 6, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Akama H, Kanemaki M, Yoshimura M, Tsukihara T, Kashiwagi T, Yoneyama H, Narita S, Nakagawa A, and Nakae T (2004) Crystal structure of the drug discharge outer membrane protein, OprM, of Pseudomonas aeruginosa: dual modes of membrane anchoring and occluded cavity end. J. Biol. Chem. 279 (51), 52816–52819. [DOI] [PubMed] [Google Scholar]
- (57).Higgins MK, Bokma E, Koronakis E, Hughes C, and Koronakis V (2004) Structure of the periplasmic component of a bacterial drug efflux pump. Proc. Natl. Acad. Sci. U. S. A. 101 (27), 9994–9999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Mikolosko J, Bobyk K, Zgurskaya HI, and Ghosh P (2006) Conformational flexibility in the multidrug efflux system protein AcrA. Structure 14 (3), 577–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Lobedanz S, Bokma E, Symmons MF, Koronakis E, Hughes C, and Koronakis V (2007) A periplasmic coiled-coil interface underlying TolC recruitment and the assembly of bacterial drug efflux pumps. Proc. Natl. Acad. Sci. U. S. A. 104 (11), 4612–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Symmons MF, Bokma E, Koronakis E, Hughes C, and Koronakis V (2009) The assembled structure of a complete tripartite bacterial multidrug efflux pump. Proc. Natl. Acad. Sci. U. S. A. 106 (17), 7173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Kim J-S, Jeong H, Song S, Kim H-Y, Lee K, Hyun J, and Ha N-C (2015) Structure of the tripartite multidrug efflux pump AcrAB-TolC suggests an alternative assembly mode. Mol. Cells 38 (2), 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Jeong H, Kim J-S, Song S, Shigematsu H, Yokoyama T, Hyun J, and Ha N-C (2016) Pseudoatomic structure of the tripartite multidrug efflux pump AcrAB-TolC reveals the intermeshing. cogwheel-like interaction between AcrA and TolC. Structure 24 (2), 272–276. [DOI] [PubMed] [Google Scholar]
- (63).Du D, Wang Z, James NR, Voss JE, Klimont E, Ohene-Agyei T, Venter H, Chiu W, and Luisi BF (2014) Structure of the AcrAB-TolC multidrug efflux pump. Nature 509 (7501), 512–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).De Angelis F, Lee JK, O’Connell JD 3rd, Miercke LJ, Verschueren KH, Srinivasan V, Bauvois C, Govaerts C, Robbins RA, Ruysschaert JM, Stroud RM, and Vandenbussche G (2010) Metal-induced conformational changes in ZneB suggest an active role of membrane fusion proteins in efflux resistance systems. Proc. Natl. Acad. Sci. U. S. A. 107 (24), 11038–11043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Hazel AJ, Abdali N, Leus IV, Parks JM, Smith JC, Zgurskaya HI, and Gumbart JC (2019) Conformational Dynamics of AcrA Govern Multidrug Efflux Pump Assembly. ACS : Infect. Dis. 5 (11), 1926–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Paetzel M, and Strynadka N (2001) Signal peptide cleavage in the E. coli membrane. CSBMCB Bulletin 2, 60. [Google Scholar]
- (67).Buddelmeijer N (2015) The molecular mechanism of bacterial, lipoprotein modification-how, when and why? FEMS microbiology reviews 39 (2), 246–261. [DOI] [PubMed] [Google Scholar]
- (68).Ma D, Cook DN, Alberti M, Pon NG, Nikaido H, and Hearst JE (1995) Genes acrA and acrB encode a stress-induced efflux system of Escherichia coli. Mol. Microbiol. 16 (1), 45–55. [DOI] [PubMed] [Google Scholar]
- (69).Nakamura H (1965) Gene-Controlled Resistance to Acriflavine and Other Basic Dyes in Escherichia coli. J. Bacteriol 90 (1), 8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Nakamura H (1968) Genetic determination of resistance to acriflavine, phenethyl alcohol, and sodium dodecyl sulfate in Escherichia coli. J. Bacteriol. 96 (4), 987–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Blair JM, La Ragione RM, Woodward MJ, and Piddock LJ (2009) Periplasmic adaptor protein AcrA has a distinct role in the antibiotic resistance and virulence of Salmonella enterica serovar Typhimurium. J. Antimicrob. Chemother. 64 (5), 965–972. [DOI] [PubMed] [Google Scholar]
- (72).Ge Q, Yamada Y, and Zgurskaya H (2009) The C-terminal domain of AcrA is essential for the assembly and function of the multidrug efflux pump AcrAB-TolC. J. Bacteriol. 191 (13), 4365–4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Schrodinger LLC (2020) PyMOL Molecular Graphics System, version 2.4.0. [Google Scholar]
- (74).Wang Z, Fan G, Hryc CF, Blaza JN, Serysheva II, Schmid MF, Chiu W, Luisi BF, and Du D (2017) An allosteric transport mechanism for the AcrAB-TolC multidrug efflux pump. eLife 6, e24905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Lomovskaya O, and Bostian KA (2006) Practical applications and feasibility of efflux pump inhibitors in the clinic–a vision for applied use. Biochem. Pharmacol. 71 (7), 910–918. [DOI] [PubMed] [Google Scholar]
- (76).Lomovskaya O, Warren MS, Lee A, Galazzo J, Fronko R, Lee M, Blais J, Cho D, Chamberland S, Renau T, Leger R, Hecker S, Watkins W, Hoshino K, Ishida H, and Lee VJ (2001) Identification and characterization of inhibitors of multidrug resistance efflux pumps in Pseudomonas aeruginosa: novel agents for combination therapy. Antimicrob. Agents Chemother. 45 (1), 105–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Nakashima R, Sakurai K, Yamasaki S, Hayashi K, Nagata C, Hoshino K, Onodera Y, Nishino K, and Yamaguchi A (2013) Structural basis for the inhibition of bacterial multidrug exporters. Nature 500 (7460), 102–106. [DOI] [PubMed] [Google Scholar]
- (78).Tikhonova EB, Yamada Y, and Zgurskaya HI (2011) Sequential mechanism of assembly of multidrug efflux pump AcrAB-TolC. Chem. Biol. 18 (4), 454–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Lomovskaya O, and Watkins WJ (2001) Efflux pumps: their role in antibacterial drug discovery. Curr. Med. Chem. 8 (14), 1699–711. [DOI] [PubMed] [Google Scholar]
- (80).Darzynkiewicz ZM, Green AT, Abdali N, Hazel A, Fulton RL, Kimball J, Gryczynski Z, Gumbart JC, Parks JM, Smith JC, and Zgurskaya HI (2019) Identification of Binding Sites for Efflux Pump Inhibitors of the AcrAB-TolC Component AcrA. Biophys. J. 116 (4), 648–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Green AT, Moniruzzaman M, Cooper CJ, Walker JK, Smith JC, Parks JM, and Zgurskaya HI (2020) Discovery of multidrug efflux pump inhibitors with a novel chemical scaffold. Biochim. Biophys. Acta, Gen. Subj. 1864 (6), 129546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Bardwell JC, McGovern K, and Beckwith J (1991) Identification of a protein required for disulfide bond formation in vivo. Cell 67 (3), 581–589. [DOI] [PubMed] [Google Scholar]
- (83).Smith RP, Paxman JJ, Scanlon MJ, and Heras B (2016) Targeting Bacterial Dsb Proteins for the Development of Anti-Virulence Agents. Molecules 21 (7), 811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Duncan LF, Wang G, Ilyichova OV, Scanlon MJ, Heras B, and Abbott BM (2019) The Fragment-Based Development of a Benzofuran Hit as a New Class of Escherichia coli DsbA Inhibitors. Molecules 24 (20), 3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Ireland PM, McMahon RM, Marshall LE, Halili M, Furlong E, Tay S, Martin JL, and Sarkar-Tyson M (2014) Disarming Burkholderia pseudomallei: structural and functional characterization of a disulfide oxidoreductase (DsbA) required for virulence in vivo. Antioxid. Redox Signaling 20 (4), 606–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Heras B, Shouldice SR, Totsika M, Scanlon MJ, Schembri MA, and Martin JL (2009) DSB proteins and bacterial pathogenicity. Nat. Rev. Microbiol. 7 (3), 215–225. [DOI] [PubMed] [Google Scholar]
- (87).Dutton RJ, Boyd D, Berkmen M, and Beckwith J (2008) Bacterial species exhibit diversity in their mechanisms and capacity for protein disulfide bond formation. Proc. Natl. Acad. Sci. U. S. A. 105 (33), 11933–11938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Guddat LW, Bardwell JC, Glockshuber R, Huber-Wunderlich M, Zander T, and Martin JL (1997) Structural analysis of three His32 mutants of DsbA: support for an electrostatic role of His32 in DsbA stability. Protein Sci. 6 (9), 1893–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Peterson JW (1996) Bacterial pathogenesis. in Medical Microbiology, 4th ed. (Baron S, Ed.), University of Texas Medical Branch at Galveston. [PubMed] [Google Scholar]
- (90).Yu J, Webb H, and Hirst TR (1992) A homologue of the Escherichia coli DsbA protein involved in disulphide bond formation is required for enterotoxin biogenesis in Vibrio cholerae. Mol. Microbiol. 6 (14), 1949–1958. [DOI] [PubMed] [Google Scholar]
- (91).Stenson TH, and Weiss AA (2002) DsbA and DsbC are required for secretion of pertussis toxin by Bordetella pertussis. Infect. Immun. 70 (5), 2297–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Yu J, and Kroll JSJM (1999) infection, DsbA: a protein-folding catalyst contributing to bacterial virulence. Microbes Infect. 1 (14), 1221–1228. [DOI] [PubMed] [Google Scholar]
- (93).Lee Y, Kim Y, Yeom S, Kim S, Park S, Jeon CO, and Park W (2008) The role of disulfide bond isomerase A (DsbA) of Escherichia coli O157:H7 in biofilm formation and virulence. FEMS Microbiol. Lett. 278 (2), 213–222. [DOI] [PubMed] [Google Scholar]
- (94).Yu J (1998) Inactivation of DsbA, but Not DsbC and DsbD, Affects the Intracellular Survival and Virulence of Shigella flexneri. Infect. Immun. 66 (8), 3909–3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Miki T, Okada N, and Danbara H (2004) Two periplasmic disulfide oxidoreductases, DsbA and SrgA, target outer membrane protein SpiA, a component of the Salmonella pathogenicity island 2 type III secretion system. J. Biol. Chem. 279 (33), 34631–34642. [DOI] [PubMed] [Google Scholar]
- (96).Miki T, Okada N, Kim Y, Abe A, and Danbara H (2008) DsbA directs efficient expression of outer membrane secretin EscC of the enteropathogenic Escherichia coli type III secretion apparatus. Microb. Pathog. 44 (2), 151–158. [DOI] [PubMed] [Google Scholar]
- (97).Hayashi S, Abe M, Kimoto M, Furukawa S, and Nakazawa T (2000) The dsbA-dsbB disulfide bond formation system of Burkholderia cepacia is involved in the production of protease and alkaline phosphatase, motility, metal resistance, and multi-drug resistance. Microbiol. Immunol. 44 (1), 41–50. [DOI] [PubMed] [Google Scholar]
- (98).Adams LA, Sharma P, Mohanty B, Ilyichova OV, Mulcair MD, Williams ML, Gleeson EC, Totsika M, Doak BC, Caria S, Rimmer K, Horne J, Shouldice SR, Vazirani M, Headey SJ, Plumb BR, Martin JL, Heras B, Simpson JS, and Scanlon MJ (2015) Application of fragment-based screening to the design of inhibitors of Escherichia coli DsbA. Angew. Chem., Int. Ed. 54 (7), 2179–2184. [DOI] [PubMed] [Google Scholar]
- (99).Duprez W, Premkumar L, Halili MA, Lindahl F, Reid RC, Fairlie DP, and Martin JL (2015) Peptide inhibitors of the Escherichia coli DsbA oxidative machinery essential for bacterial virulence. J. Med. Chem. 58 (2), 577–587. [DOI] [PubMed] [Google Scholar]
- (100).Halili MA, Bachu P, Lindahl F, Bechara C, Mohanty B, Reid RC, Scanlon MJ, Robinson CV, Fairlie DP, and Martin JL (2015) Small molecule inhibitors of disulfide bond formation by the bacterial DsbA-DsbB dual enzyme system. ACS Chem. Biol. 10 (4), 957–964. [DOI] [PubMed] [Google Scholar]
- (101).Totsika M, Vagenas D, Paxman JJ, Wang G, Dhouib R, Sharma P, Martin JL, Scanlon MJ, and Heras B (2018) Inhibition of Diverse DsbA Enzymes in Multi-DsbA Encoding Pathogens. Antioxid. Redox Signaling 29 (7), 653–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Duprez W, Bachu P, Stoermer MJ, Tay S, McMahon RM, Fairlie DP, and Martin JL (2015) Virtual Screening of Peptide and Peptidomimetic Fragments Targeted to Inhibit Bacterial Dithiol Oxidase DsbA. PLoS One 10 (7), No. e0133805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Singh J, Petter RC, Baillie TA, and Whitty A (2011) The resurgence of covalent drugs. Nat. Rev. Drug Discovery 10 (4), 307–317. [DOI] [PubMed] [Google Scholar]
- (104).Sklar JG, Wu T, Kahne D, and Silhavy TJ (2007) Defining the roles of the periplasmic chaperones SurA, Skp, and DegP in Escherichia coli. Genes Dev. 21 (19), 2473–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Fardini Y, Trotereau J, Bottreau E, Souchard C, Velge P, and Virlogeux-Payant I (2009) Investigation of the role of the BAM complex and SurA chaperone in outer-membrane protein biogenesis and type III secretion system expression in Salmonella. Microbiology 155 (5), 1613–1622. [DOI] [PubMed] [Google Scholar]
- (106).Rouvière PE, and Gross CAJG (1996) development, SurA, a periplasmic protein with peptidyl-prolyl isomerase activity, participates in the assembly of outer membrane porins. Genes Dev. 10 (24), 3170–3182. [DOI] [PubMed] [Google Scholar]
- (107).Bitto E, and McKay DB (2003) The periplasmic molecular chaperone protein SurA binds a peptide motif that is characteristic of integral outer membrane proteins. J. Biol. Chem. 278 (49), 49316–49322. [DOI] [PubMed] [Google Scholar]
- (108).Watts KM, and Hunstad DA (2008) Components of SurA required for outer membrane biogenesis in uropathogenic Escherichia coli. PLoS One 3 (10), No. e3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Hennecke G, Nolte J, Volkmer-Engert R, Schneider-Mergener J, and Behrens S (2005) The periplasmic chaperone SurA exploits two features characteristic of integral outer membrane proteins for selective substrate recognition. J. Biol. Chem. 280 (25), 23540–23548. [DOI] [PubMed] [Google Scholar]
- (110).Bitto E, and McKay DB (2002) Crystallographic structure of SurA, a molecular chaperone that facilitates folding of outer membrane porins. Structure 10 (11), 1489–1498. [DOI] [PubMed] [Google Scholar]
- (111).Behrens S, Maier R, de Cock H, Schmid FX, and Gross CA (2001) The SurA periplasmic PPIase lacking its parvulin domains functions in vivo and has chaperone activity. EMBO journal 20 (1-2), 285–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Soltes GR, Schwalm J, Ricci DP, and Silhavy TJ (2016) The Activity of Escherichia coli Chaperone SurA Is Regulated by Conformational Changes Involving a Parvulin Domain. J. Bacteriol. 198 (6), 921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Webb HM, Ruddock LW, Marchant RJ, Jonas K, and Klappa P (2001) Interaction of the periplasmic peptidylprolyl cis-trans isomerase SurA with model peptides. The N-terminal region of SurA id essential and sufficient for peptide binding. J. Biol. Chem. 276 (49), 45622–45627. [DOI] [PubMed] [Google Scholar]
- (114).Xu X, Wang S, Hu Y-X, and McKay DB (2007) The periplasmic bacterial molecular chaperone SurA adapts its structure to bind peptides in different conformations to assert a sequence preference for aromatic residues. J. Mol. Biol. 373 (2), 367–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Ricci DP, Schwalm J, Gonzales-Cope M, and Silhavy TJ (2013) The activity and specificity of the outer membrane protein chaperone SurA are modulated by a proline isomerase domain. mBio 4 (4), e00540–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Tormo A, Almiron M, and Kolter R (1990) surA, an Escherichia coli gene essential for survival in stationary phase. J. Bacteriol. 172 (8), 4339–4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Justice SS, Hunstad DA, Harper JR, Duguay AR, Pinkner JS, Bann J, Frieden C, Silhavy TJ, and Hultgren SJ (2005) Periplasmic peptidyl prolyl cis-trans isomerases are not essential for viability, but SurA is required for pilus biogenesis in Escherichia coli. J. Bacteriol. 187 (22), 7680–7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Bell EW, Zheng EJ, and Ryno LM (2018) Identification of inhibitors of the E. coli chaperone SurA using in silico and in vitro techniques. Bioorg. Med. Chem. Lett. 28 (22), 3540–3548. [DOI] [PubMed] [Google Scholar]
- (119).Takeda K, Miyatake H, Yokota N, Matsuyama S.-i., Tokuda H, and Miki K (2003) Crystal structures of bacterial lipoprotein localization factors, LolA and LolB. THE EMBO J. 22 (13), 3199–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Pathania R, Zlitni S, Barker C, Das R, Gerritsma DA, Lebert J, Awuah E, Melacini G, Capretta FA, and Brown ED (2009) Chemical genomics in Escherichia coli identifies an inhibitor of bacterial lipoprotein targeting. Nat. Chem. Biol. 5 (11), 849–856. [DOI] [PubMed] [Google Scholar]
- (121).Tokuda H, and Matsuyama S. -i. (2004) Sorting of lipoproteins to the outer membrane in E. coli. Biochim. Biophys. Acta, Mol. Cell Res. 1693 (1), 5–13. [DOI] [PubMed] [Google Scholar]
- (122).Kovacs-Simon A, Titball R, and Michell SLJI (2011) immunity, Lipoproteins of bacterial pathogens. Infect. Immun. 79 (2), 548–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Grabowicz MJA (2018) Lipoprotein Transport: Greasing the Machines of Outer Membrane Biogenesis: Re-Examining Lipoprotein Transport Mechanisms Among Diverse Gram-Negative Bacteria While Exploring New Discoveries and Questions. BioEssays 40 (4), 1700187. [DOI] [PubMed] [Google Scholar]
- (124).Rao S, Bates GT, Matthews CR, Newport TD, Vickery ON, and Stansfeld PJ (2020) Characterizing Membrane Association and Periplasmic Transfer of Bacterial Lipoproteins through Molecular Dynamics Simulations. Structure 28 (4), 475–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Tajima T, Yokota N, Matsuyama S. -i., and Tokuda H (1998) Genetic analyses of the in vivo function of LolA, a periplasmic chaperone involved in the outer membrane localization of Escherichia coli lipoproteins. FEBS Lett. 439 (1–2), 51–54. [DOI] [PubMed] [Google Scholar]
- (126).Tao K, Watanabe S, Narita S. -i., and Tokuda H (2010) A periplasmic LolA derivative with a lethal disulfide bond activates the Cpx stress response system. J. Bacteriol. 192 (21), 5657–5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Liao C-T, Chiang Y-C, and Hsiao Y-M (2019) Functional characterization and proteomic analysis of lolA in Xanthomonas campestri s pv. campestris. BMC Microbiol. 19 (1), 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Grabowicz M, and Silhavy TJ (2017) Redefining the essential trafficking pathway for outer membrane lipoproteins. Proc. Natl. Acad. Sci U.S.A.114 (18), 4769–4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Barker CA, Allison SE, Zlitni S, Nguyen ND, Das R, Melacini G, Capretta AA, and Brown ED (2013) Degradation of MAC13243 and studies of the interaction of resulting thiourea compounds with the lipoprotein targeting chaperone LolA. Bioorg. Med. Chem. Lett. 23 (8), 2426–2431. [DOI] [PubMed] [Google Scholar]
- (130).Buss JA, Baidin V, Welsh MA, Flores-Kim J, Cho H, Wood BM, Uehara T, Walker S, Kahne D, and Bernhardt TG (2019) Pathway-directed screen for inhibitors of the bacterial cell elongation machinery. Antimicrob. Agents Chemother. 63 (1), No. e01530–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (131).Suits MD, Sperandeo P, Deho G, Polissi A, and Jia Z (2008) Novel structure of the conserved gram-negative lipopolysaccharide transport protein A and mutagenesis analysis. J. Mol. Biol. 380 (3), 476–488. [DOI] [PubMed] [Google Scholar]
- (132).Villa R, Martorana AM, Okuda S, Gourlay LJ, Nardini M, Sperandeo P, Deho G, Bolognesi M, Kahne D, and Polissi A (2013) The Escherichia coli Lpt transenvelope protein complex for lipopolysaccharide export is assembled via conserved structurally homologous domains. J. Bacteriol. 195 (5), 1100–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Okuda S, Sherman DJ, Silhavy TJ, Ruiz N, and Kahne D (2016) Lipopolysaccharide transport and assembly at the outer membrane: the PEZ model. Nat. Rev. Microbiol. 14 (6), 337–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).Sperandeo P, Martorana AM, and Polissi A (2017) Lipopolysaccharide biogenesis and transport at the outer membrane of Gram-negative bacteria. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 1862 (11), 1451–1460. [DOI] [PubMed] [Google Scholar]
- (135).Chimalakonda G, Ruiz N, Chng SS, Garner RA, Kahne D, and Silhavy TJ (2011) Lipoprotein LptE is required for the assembly of LptD by the beta-barrel assembly machine in the outer membrane of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 108 (6), 2492–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Qiao S, Luo Q, Zhao Y, Zhang XC, and Huang Y (2014) Structural basis for lipopolysaccharide insertion in the bacterial outer membrane. Nature 511 (7507), 108–111. [DOI] [PubMed] [Google Scholar]
- (137).Okuda S, Freinkman E, and Kahne D (2012) Cytoplasmic ATP hydrolysis powers transport of lipopolysaccharide across the periplasm in E. coli. Science (Washington, DC, U. S.) 338 (6111), 1214–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Nikaido H (2003) Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 67 (4), 593–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Raetz CR, and Whitfield C (2002) Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 71, 635–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Laguri C, Sperandeo P, Pounot K, Ayala I, Silipo A, Bougault CM, Molinaro A, Polissi A, and Simorre JP (2017) Interaction of lipopolysaccharides at intermolecular sites of the periplasmic Lpt transport assembly. Sci. Rep. 7 (1), 9715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (141).Schultz KM, Lundquist TJ, and Klug CS (2017) Lipopolysaccharide binding to the periplasmic protein LptA. Protein Sci. 26 (8), 1517–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (142).Falchi FA, Maccagni EA, Puccio S, Peano C, De Castro C, Palmigiano A, Garozzo D, Martorana AM, Polissi A, Deho G, and Sperandeo P (2018) Mutation and Suppressor Analysis of the Essential Lipopolysaccharide Transport Protein LptA Reveals Strategies To Overcome Severe Outer Membrane Permeability Defects in Escherichia coli. J. Bacteriol. 200 (2), e00487–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (143).Matsuyama S, Tajima T, and Tokuda H (1995) A novel periplasmic carrier protein involved in the sorting and transport of Escherichia coli lipoproteins destined for the outer membrane. EMBO J. 14 (14), 3365–3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (144).Bayer ME (1968) Areas of adhesion between wall and membrane of Escherichia coli. J. Gen. Microbiol. 53 (3), 395–404. [DOI] [PubMed] [Google Scholar]
- (145).Bayer ME (1991) Zones of membrane adhesion in the cryofixed envelope of Escherichia coli. J. Struct. Biol. 107 (3), 268–280. [DOI] [PubMed] [Google Scholar]
- (146).Tefsen B, Geurtsen J, Beckers F, Tommassen J, and de Cock H (2005) Lipopolysaccharide transport to the bacterial outer membrane in spheroplasts. J. Biol. Chem. 280 (6), 4504–4509. [DOI] [PubMed] [Google Scholar]
- (147).Freinkman E, Okuda S, Ruiz N, and Kahne D (2012) Regulated assembly of the transenvelope protein complex required for lipopolysaccharide export. Biochemistry 51 (24), 4800–4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Sampson BA, Misra R, and Benson SA (1989) Identification and characterization of a new gene of Escherichia coli K-12 involved in outer membrane permeability. Genetics 122 (3), 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (149).Chng SS, Gronenberg LS, and Kahne D (2010) Proteins required for lipopolysaccharide assembly in Escherichia coli form a transenvelope complex. Biochemistry 49 (22), 4565–4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (150).Sherman DJ, Okuda S, Denny WA, and Kahne D (2013) Validation of inhibitors of an ABC transporter required to transport lipopolysaccharide to the cell surface in Escherichia coli. Bioorg. Med. Chem. 21 (16), 4846–4851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (151).Choi U, and Lee CR (2019) Antimicrobial Agents That Inhibit the Outer Membrane Assembly Machines of Gram-Negative Bacteria. J. Microbiol. Biotechnol 29 (1), 1–10. [DOI] [PubMed] [Google Scholar]
- (152).Gronenberg LS, and Kahne D (2010) Development of an Activity Assay for Discovery of Inhibitors of Lipopolysaccharide Transport. J. Am. Chem. Soc. 132 (8), 2518–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (153).Chene P (2002) ATPases as drug targets: learning from their structure. Nat. Rev. Drug Discovery 1 (9), 665–673. [DOI] [PubMed] [Google Scholar]
- (154).Srinivas N, Jetter P, Ueberbacher BJ, Werneburg M, Zerbe K, Steinmann J, Van der Meijden B, Bernardini F, Lederer A, Dias RL, Misson PE, Henze H, Zumbrunn J, Gombert FO, Obrecht D, Hunziker P, Schauer S, Ziegler U, Kach A, Eberl L, Riedel K, DeMarco SJ, and Robinson JA (2010) Peptidomimetic antibiotics target outer-membrane biogenesis in Pseudomonas aeruginosa. Science (Washington, DC, U. S.) 327 (5968), 1010–3. [DOI] [PubMed] [Google Scholar]
- (155).Vetterli SU, Moehle K, and Robinson JA (2016) Synthesis and antimicrobial activity against Pseudomonas aeruginosa of macrocyclic β-hairpin peptidomimetic antibiotics containing N-methylated amino acids. Bioorg. Med. Chem. 24 (24), 6332–6339. [DOI] [PubMed] [Google Scholar]
- (156).Urfer M, Bogdanovic J, Lo Monte F, Moehle K, Zerbe K, Omasits U, Ahrens CH, Pessi G, Eberl L, and Robinson JA (2016) A Peptidomimetic Antibiotic Targets Outer Membrane Proteins and Disrupts Selectively the Outer Membrane in Escherichia coli. J. Biol. Chem. 291 (4), 1921–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (157).Fehlbaum P, Bulet P, Chernysh S, Briand JP, Roussel JP, Letellier L, Hetru C, and Hoffmann JA (1996) Structure-activity analysis of thanatin, a 21-residue inducible insect defense peptide with sequence homology to frog skin antimicrobial peptides. Proc. Natl. Acad. Sci. U. S. A. 93 (3), 1221–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (158).Vetterli SU, Zerbe K, Müller M, Urfer M, Mondal M, Wang S-Y, Moehle K, Zerbe O, Vitale A, Pessi G, Eberl L, Wollscheid B, and Robinson JA (2018) Thanatin targets the intermembrane protein complex required for lipopolysaccharide transport in Escherichia coli. Science Advances 4 (11), No. eaau2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Ma B, Fang C, Lu L, Wang M, Xue X, Zhou Y, Li M, Hu Y, Luo X, and Hou Z (2019) The antimicrobial peptide thanatin disrupts the bacterial outer membrane and inactivates the NDM-1 metallo-beta-lactamase. Nat. Commun. 10 (1), 3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (160).Yatsunyk LA, Easton JA, Kim LR, Sugarbaker SA, Bennett B, Breece RM, Vorontsov II, Tierney DL, Crowder MW, and Rosenzweig AC (2008) Structure and metal binding properties of ZnuA, a periplasmic zinc transporter from Escherichia coli. JBIC, J. Biol. Inorg. Chem. 13 (2), 271–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (161).Sabri M, Houle S, and Dozois CM (2009) Roles of the Extraintestinal Pathogenic Escherichia coli ZnuACB and ZupT Zinc Transporters during Urinary Tract Infection. Infect. Immun. 77 (3), 1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (162).D’Orazio M, Mastropasqua MC, Cerasi M, Pacello F, Consalvo A, Chirullo B, Mortensen B, Skaar EP, Ciavardelli D, Pasquali P, and Battistoni A (2015) The capability of Pseudomonas aeruginosa to recruit zinc under conditions of limited metal availability is affected by inactivation of the ZnuABC transporter. Metallomics 7 (6), 1023–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (163).Stork M, Bos MP, Jongerius I, de Kok N, Schilders I, Weynants VE, Poolman JT, and Tommassen J (2010) An outer membrane receptor of Neisseria meningitidis involved in zinc acquisition with vaccine potential. PLoS Pathog. 6 (7), e1000969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (164).Bobrov AG, Kirillina O, Fetherston JD, Miller MC, Burlison JA, and Perry RD (2014) The Yersinia pestis siderophore, yersiniabactin, and the ZnuABC system both contribute to zinc acquisition and the development of lethal septicaemic plague in mice. Mol. Microbiol. 93 (4), 759–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (165).Hood MI, Mortensen BL, Moore JL, Zhang Y, Kehl-Fie TE, Sugitani N, Chazin WJ, Caprioli RM, and Skaar EP (2012) Identification of an Acinetobacter baumannii Zinc Acquisition System that Facilitates Resistance to Calprotectin-mediated Zinc Sequestration. PLoS Pathog. 8 (12), No. e1003068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Ilari A, Pescatori L, Di Santo R, Battistoni A, Ammendola S, Falconi M, Berlutti F, Valenti P, and Chiancone E (2016) Salmonella enterica serovar Typhimurium growth is inhibited by the concomitant binding of Zn(II) and a pyrrolyl-hydroxamate to ZnuA, the soluble component of the ZnuABC transporter. Biochim. Biophys. Acta, Gen. Subj. 1860 (3), 534–541. [DOI] [PubMed] [Google Scholar]
- (167).Castelli S, Stella L, Petrarca P, Battistoni A, Desideri A, and Falconi M (2013) Zinc ion coordination as a modulating factor of the ZnuA histidine-rich loop flexibility: a molecular modeling and fluorescence spectroscopy study. Biochem. Biophys. Res. Commun. 430 (2), 769–773. [DOI] [PubMed] [Google Scholar]
- (168).Hecel A, Kola A, Valensin D, Kozlowski H, and Rowinska-Zyrek M (2020) Metal Complexes of Two Specific Regions of ZnuA, a Periplasmic Zinc(II) Transporter from Escherichia coli. Inorg. Chem. 59 (3), 1947–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Ciavardelli D, Ammendola S, Ronci M, Consalvo A, Marzano V, Lipoma M, Sacchetta P, Federici G, Di Ilio C, Battistoni A, and Urbani A (2011) Phenotypic profile linked to inhibition of the major Zn influx system in Salmonella enterica: proteomics and ionomics investigations. Mol. BioSyst. 7 (3), 608–619. [DOI] [PubMed] [Google Scholar]
- (170).Ammendola S, Pasquali P, Pistoia C, Petrucci P, Petrarca P, Rotilio G, and Battistoni A (2007) High-affinity Zn2+ uptake system ZnuABC is required for bacterial zinc homeostasis in intracellular environments and contributes to the virulence of Salmonella enterica. Infect. Immun. 75 (12), 5867–5876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (171).Yang X, Becker T, Walters N, and Pascual DW (2006) Deletion of znuA virulence factor attenuates Brucella abortus and confers protection against wild-type challenge. Infect. Immun. 74 (7), 3874–3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (172).Davis LM, Kakuda T, and DiRita VJ (2009) A Campylobacter jejuni znuA orthologue is essential for growth in low-zinc environments and chick colonization. J. Bacteriol 191 (5), 1631–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (173).Lewis DA, Klesney-Tait J, Lumbley SR, Ward CK, Latimer JL, Ison CA, and Hansen EJ (1999) Identification of the znuA-encoded periplasmic zinc transport protein of Haemophilus ducreyi. Infect. Immun. 67 (10), 5060–5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (174).Murphy TF, Brauer AL, Kirkham C, Johnson A, Koszelak-Rosenblum M, and Malkowski MG (2013) Role of the zinc uptake ABC transporter of Moraxella catarrhalis in persistence in the respiratory tract. Infect. Immun. 81 (9), 3406–3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (175).Liuzzi JP, Lichten LA, Rivera S, Blanchard RK, Aydemir TB, Knutson MD, Ganz T, and Cousins R (2005) Interleukin-6 regulates the zinc transporter Zip14 in liver and contributes to the hypozincemia of the acute-phase response. Proc. Natl. Acad. Sci. U. S. A. 102 (19), 6843–6848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (176).Lazzaroni J-C, Dubuisson J-F, and Vianney A (2002) The Tol proteins of Escherichia coli and their involvement in the translocation of group A colicins. Biochimie 84 (5), 391–397. [DOI] [PubMed] [Google Scholar]
- (177).Lo Sciuto A, Fernandez-Pinar R, Bertuccini L, Iosi F, Superti F, and Imperi F (2014) The periplasmic protein TolB as a potential drug target in Pseudomonas aeruginosa. PLoS One 9 (8), No. e103784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (178).Abergel C, Bouveret E, Claverie J-M, Brown K, Rigal A, Lazdunski C, and Bénédetti H (1999) Structure of the Escherichia coli TolB protein determined by MAD methods at 1.95 Å resolution. Structure 7 (10), 1291–1300. [DOI] [PubMed] [Google Scholar]
- (179).Carr S, Penfold CN, Bamford V, James R, and Hemmings AM (2000) The structure of TolB, an essential component of the tol-dependent translocation system, and its protein-protein interaction with the translocation domain of colicin E9. Structure 8 (1), 57–66. [DOI] [PubMed] [Google Scholar]
- (180).Cascales E, Bernadac A, Gavioli M, Lazzaroni J-C, and Lloubes R (2002) Pal lipoprotein of Escherichia coli plays a major role in outer membrane integrity. J. Bacteriol. 184 (3), 754–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (181).Lloubès R, Cascales E, Walburger A, Bouveret E, Lazdunski C, Bernadac A, and Journet L (2001) The Tol-Pal proteins of the Escherichia coli cell envelope: an energized system required for outer membrane integrity? Res. Microbiol. 152 (6), 523–529. [DOI] [PubMed] [Google Scholar]
- (182).Bollati M, Villa R, Gourlay LJ, Benedet M, Dehò G, Polissi A, Barbiroli A, Martorana AM, Sperandeo P, Bolognesi M, and Nardini M (2015) Crystal structure of LptH, the periplasmic component of the lipopolysaccharide transport machinery from Pseudomonas aeruginosa. FEBS J. 282 (10), 1980–1997. [DOI] [PubMed] [Google Scholar]
- (183).Neupane DP, Avalos D, Fullam S, Roychowdhury H, and Yukl ET (2017) Mechanisms of zinc binding to the solute-binding protein AztC and transfer from the metallochaperone AztD. J. Biol. Chem. 292 (42), 17496–17505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (184).Raja SB, Murali MR, Roopa K, and Devaraj SNJB (2011) Pharmacotherapy, Imperatorin a furocoumarin inhibits periplasmic Cu-Zn SOD of Shigella dysenteriae their by modulates its resistance towards phagocytosis during host pathogen interaction. Biomed. Pharmacother. 65 (8), 560–568. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.