

Abstract
More than two decades after the natural gene-silencing mechanism of RNA interference was elucidated, small interfering RNA (siRNA)-based therapeutics have finally broken into the pharmaceutical market. With three agents already approved and many others in advanced stages of the drug development pipeline, siRNA drugs are on their way to becoming a standard modality of pharmacotherapy. The majority of late-stage candidates are indicated for rare or orphan diseases, whose patients have an urgent need for novel and effective therapies. Additionally, there are agents that have the potential to meet the need of a broader population. Inclisiran, for instance, is being developed for hypercholesterolemia and has shown benefit in patients who are uncontrolled even after maximal statin therapy. This review provides a brief overview of mechanisms of siRNA action, physiological barriers to its delivery and activity, and the most common chemical modifications and delivery platforms used to overcome these barriers. Furthermore, this review presents comprehensive profiles of the three approved siRNA drugs (patisiran, givosiran, and lumasiran) and the seven other siRNA candidates in Phase 3 clinical trials (vutrisiran, nedosiran, inclisiran, fitusiran, teprasiran, cosdosiran, and tivanisiran), summarizing their modifications and delivery strategies, disease-specific mechanisms of action, updated clinical trial status, and future outlooks.
Keywords: siRNAs, drug development, FDA approval, clinical trial
1. Introduction
RNA interference (RNAi), a regulatory mechanism of most eukaryotic cells to directly control gene activity, has become a mechanism of drug action in the development of RNAi-based therapies [1, 2]. For over two decades, researchers have known of the phenomenon of RNAi, a natural mechanism by which short strands of RNA, such as small interfering RNAs (siRNAs), cause targeted gene suppression [3]. siRNAs are short double-stranded RNAs that dissociate to single strands and bind specifically to target messenger RNA (mRNA) sequences. Their binding triggers a series of actions that result in the cleavage and degradation of the target mRNA, preventing translation and any other subsequent steps of gene expression and function. siRNAs have major therapeutic potential, affording an opportunity to selectively target and silence the mRNA products of genes, previously considered “undruggable” targets [4]. Thanks to an extensive knowledge of the human genome, the majority of human protein-coding genes have been decoded and annotated [5]. After identifying a target mRNA sequence, it becomes relatively straightforward to create complementary siRNA molecules, leading to downstream silencing of the protein encoded by the mRNA [6]. Conversely, most conventional small molecule drugs act at the protein level, which requires a higher level of structural precision and therefore a more complex and challenging development process.
From conceptual to executable drug-related knowledge, development of siRNA-based drugs has taken nearly 20 years. In August 2018, 20 years after RNAi was first discovered, the FDA approved patisiran, the first siRNA drug [7]. The second, givosiran, was approved in November 2019 [8], and the third, lumasiran, in November 2020 (https://www.fda.gov/news-events/press-announcements/fda-approves-first-drug-treat-rare-metabolic-disorder). Currently, there are seven siRNA drugs in late stages of Phase 3 clinical trials, including vutrisiran, nedosiran, inclisiran, fitusiran, teprasiran, cosdosiran, and tivanisiran (Fig. 1), some of which are very close to FDA approval. Why did it take so long to fully realize and implement the therapeutic potential of RNA interference? The main challenge to siRNA drug development is site-specific delivery [9]. Large anionic siRNA molecules must overcome a variety of physiological barriers to reach the cytoplasm in target cells. siRNA drugs have been made possible by numerous chemical modifications and delivery systems utilized to increase their stability and specificity. This review provides a brief overview of the mechanisms of siRNA action, physiological barriers for delivery and efficacy, and the most common chemical modifications and delivery platforms used to overcome these barriers. Comprehensive profiles summarize the information on the three approved siRNA drugs and the seven siRNA candidates in Phase 3 clinical trials.
Figure 1.
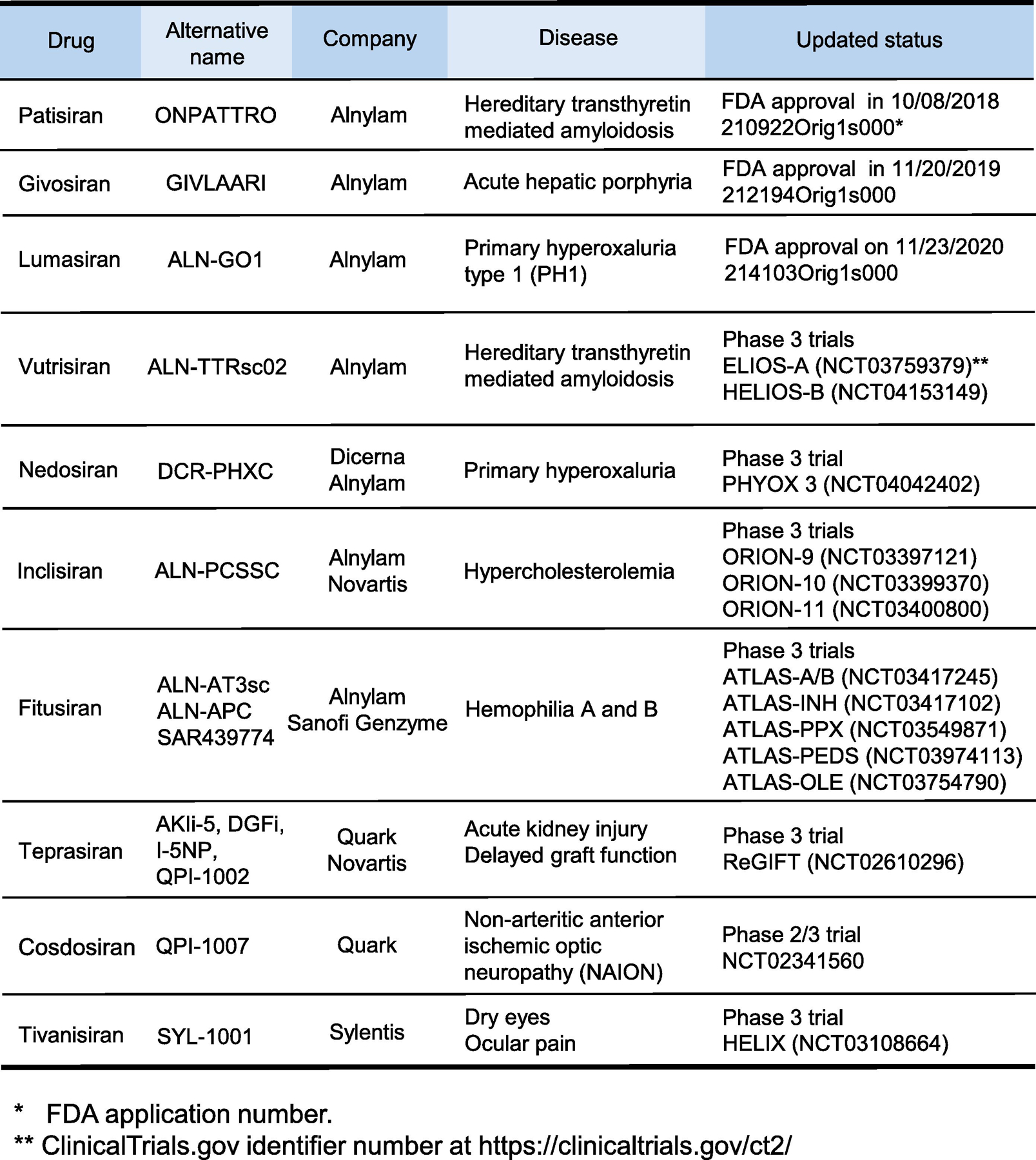
A summary of development of siRNA-based drugs with either FDA approval or in late phase 3 clinical trials.
2. General characterization of siRNA drugs
2.1. Mechanisms of action
The mechanisms of action of siRNA drugs are mainly through inhibition of expression of target genes by RNAi. The endogenous process of RNAi starts in the cytoplasm with the endoribonuclease Dicer, which produces mature siRNA by cleaving longer double-stranded RNA (dsRNA) or short hairpin RNA (shRNA) [10]. The resulting siRNA is 21–23 bases long and generally has 2 overhanging phosphorylated bases at the 3’ end of each strand. Following processing, mature siRNA is incorporated into the RNA-induced silencing complex (RISC), which is made up of a collection of integral proteins, including Dicer and Ago-2 [11]. The siRNA is then separated into the sense and antisense strands. The sense strand is merely a passenger that is released from the complex, forming mature RISC. The antisense strand remains, serving as a guide that leads and aligns the complex to the target mRNA sequence. Complementary binding of the guide to the target triggers cleavage of the target sequence, mediated by Ago-2 endonucleases in RISC [12].
When utilizing RNAi pathways for therapeutic gain, one can bypass the initial Dicer-mediated step of processing mature siRNA by directly administering artificially prepared siRNAs. Since the activity of RISC is ultimately determined by the guide strand, it is critical to synthesize an antisense strand that optimizes selectivity and potency. In addition to ensuring that the strand is complementary to the target mRNA, it is equally important to synthesize a strand that will not bind off-target, partially homologous mRNA sequences. Even a 7-base sequence complementary to the seed domain of the antisense strand can potentially trigger RISC [4]. Utilizing tools such as NCBI BLAST may allow for the determination of optimal target sequences that are unique within the human transcriptome.
2.2. Barriers to delivery
siRNAs have high therapeutic potential, but they pose notable delivery challenges [13]. For siRNAs to have therapeutic efficacy, they must be effectively delivered to and taken up into the intended target site. Unfortunately, siRNA molecules have low bioavailability due to their large size and anionic charge. At the systemic level, delivery is compromised by rapid clearance, especially via the kidneys. Naked unmodified agents may have a half-life as short as 5 minutes [14]. Nanocarrier-encapsulated drugs are often subject to serum protein adsorption. This opsonization results in uptake by the reticuloendothelial system (RES) and clearance by phagocytes [15]. siRNAs are also rapidly degraded by nucleases present in plasma, tissues, and the cytoplasm.
After surviving systemic clearance, the drugs must pass through the capillary endothelium into the tissues, which is particularly challenging due to the abundance of adherence and tight junctions. siRNAs may passively accumulate to fenestrated sites, such as the liver or tumor tissue, but this presents a challenge to delivering these therapeutics to other sites besides these organs that preferentially take up these molecules [16]. Even after successful transport into the target tissue compartment, siRNAs must be taken up into the target cells. However, RNAs do not spontaneously cross cellular membranes, which represents a barrier to cellular internalization. Furthermore, endosomal trapping after internalization can be another limiting factor in targeting siRNA molecules to their molecular sites. siRNA therapeutics may enter a cell through transfection or conjugation to a ligand with a high affinity receptor on the target cell [17]. After internalization, less than 1% of siRNA molecules escape the endosomal compartment. siRNA molecules trapped in endosomes are either degraded or recycled back to the surface for extrusion from the cell. Strategies are in development to increase the proportion of drugs released and/or escaping from endosomal trapping [18]. For example, endosome-disrupting small molecule compounds can efficiently increase release of siRNA drugs from vesicles and enhance target knockdown up to ~47 fold in tumor cells [19]. The strategy to endosomal escape can be applied to improve delivery of siRNA and other nucleic acid-based therapeutics.
Finally, siRNA may activate an undesired immunogenic response, as extracellular and intracellular immune mediators may falsely recognize these as viral RNA molecules [20]. For instance, Toll-like receptors may recognize certain sequences as immunostimulant motifs. These reactions result in unfavorable adverse reactions [21].
There are two broad strategies that have been utilized to address these challenges in order to enhance the clinical utility of siRNA drugs: chemically modifying the siRNA itself and employing various potential delivery strategies. Ultimately, the goal is to maintain the drug’s functionality while improving its pharmacokinetics, pharmacodynamics, and safety profile. Such modifications are intended to increase efficiency and potency, while minimizing toxicity and cost. Many outstanding reviews have thoroughly described the chemical modifications and delivery systems used in siRNA development [4, 13, 16, 22]. Therefore, here, we will focus largely on the strategies most commonly used in approved siRNA drugs and late-stage candidates in clinical trials.
2.3. Chemical modifications
siRNA molecules may be chemically modified upon their sugar-phosphate backbones and also upon their purine and pyrimidine bases. One of the earliest-discovered and most commonly utilized modifications is the replacement of the highly charged, unstable phosphodiester backbone with a phosphorothioate (PS) backbone [23]. Replacing one of the non-bridge oxygen atoms on the phosphate group with a sulfur atom significantly increases resistance to nuclease and phosphodiesterase activities [24]. Additionally, this increases the molecule’s hydrophobicity, which promotes binding to carrier plasma proteins, such as albumin, resulting in increased circulation time, slower degradation, and more favorable pharmacokinetics. PS-modified RNAi drugs have demonstrated notably improved and reproducible stability and efficacy. This has been one of the most important and successful modifications in the field [13, 16, 25]. However, some studies have suggested that fully PS-modified siRNAs have decreased gene-silencing effect, resulting in decreased efficacy, likely because PS in the central part of the strand interferes with RISC recognition and activation. Partially PS-modified siRNA molecules retain their efficacy while displaying improved pharmacokinetics. PS modifications at the end of the strands appear to be the best-tolerated [16, 26]. PS-containing enriched CpG dinucleotide repeats also activate toll-like receptors and generate an immune response, so caution must be exercised while designing CpG containing PS.
Sugar modifications are most common at the 2’ position, because the existing 2’-OH group is not essential for siRNA’s silencing effect [16, 26]. Additionally, the 2’-OH participates in nuclease-mediated cleavage, so replacing the OH with other functional groups can protect against endoribonuclease degradation [27]. Most commonly, the OH group is replaced with a 2’-fluoro (2’-F), 2’-O-methyl (2’-OMe), or a 2’-O-methoxyethyl (2ʹ-MOE), all of which enhance stability and increase resistance to nucleases. Substitutions with 2’-F and 2’-OMe also decrease immunogenicity and subsequent immune-mediated off-target effects [4, 16, 25, 27]. However, as in the case of PS overuse, extensive use of any single 2’-modification may decrease siRNA’s gene-silencing effect. For instance, a fully 2’-O-methylated siRNA renders the molecule biologically inactive [16]. However, alternating different 2’-substitutions, such as 2’-OMe and 2’-F, increases nuclease resistance while maintaining gene-silencing activity [16, 28]. Over-using bulkier substituents, such as 2’-MOE, may also decrease gene-silencing activity due to steric hindrance [29]. Most approved and late-stage siRNA drugs contain PS and multiple 2’-OMe and 2’-F modifications (Fig. 2).
Figure 2:
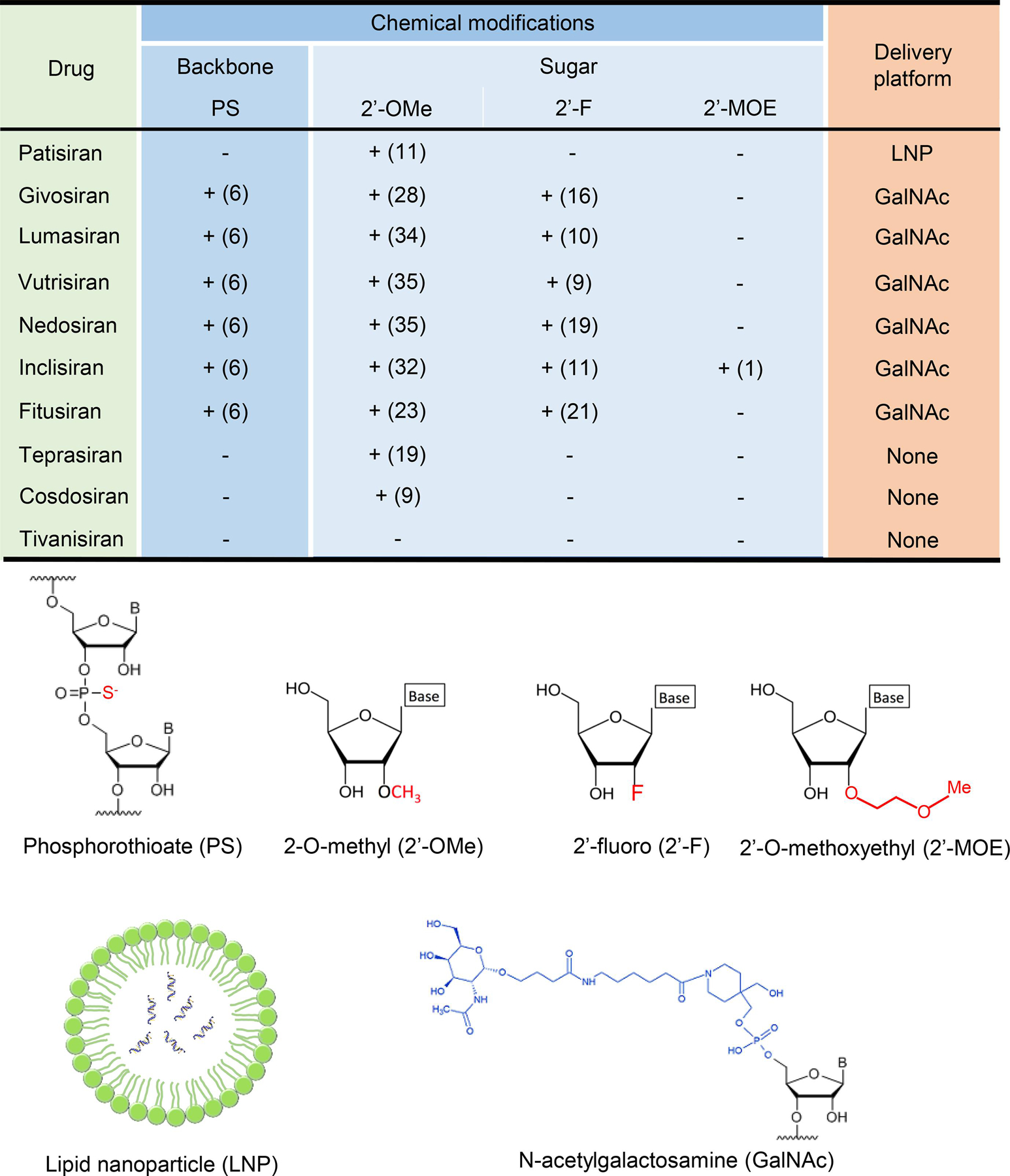
A summary of chemical modifications in backbone and sugar and delivery platforms of the siRNA drugs.
siRNA drug design and modifications can now be rationally designed by use of algorithms. Years of research and testing have yielded substitution and modification predictions that increase stability, protection, and bioavailability while maintaining efficacy and potency. For example, Alnylam Pharmaceuticals, a company at the forefront of the RNAi therapeutic race, has developed five generations of siRNA designs from 1) partially modified, 2) standard template chemistry, 3) enhanced stabilization chemistry (ESC), 4) advanced ESC, and 5) ESC+. Multiple ESC+ conjugates are currently in the clinical pipeline [30].
Several other base modifications are currently under development, but are not reviewed here since they have not yet been widely utilized in preclinical and clinical studies with limited published information.
2.4. Delivery systems
Delivery systems are critical for the drug discovery and development pipelines of siRNA drugs. Since siRNA molecules are fairly large (13–14 kDa) and hydrophilic, they are unable to passively cross the cell membrane. While chemical modifications and the addition of functional groups may increase stability and resistance to nucleases, they do not address permeability through lipid bilayers. To address this issue, two major delivery strategies are used: 1) formulation of the siRNA into nanocarriers that allow for transfection into target cells, and 2) conjugation of the siRNA to a targeting ligand that binds to a specific, high-capacity receptor on target cells. An ideal delivery system is biocompatible and non-immunogenic, allowing for specific cellular transport and entry [13, 18].
Lipid nanoparticles (LNPs) are the most successful formulation-based siRNA delivery strategy. They were used to deliver patisiran, the first approved siRNA drug [31]. LNPs are made up of cationic, ionizable, and helper lipids, such as cholesterol, 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), 1,2-dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-200, and D-Lin-MC3-DMA, which promote RNA packing, increase stability, and allow for passage through the lipid bilayer [6, 16]. LNP surfaces are PEGylated to reduce aggregation, opsonization, and RES clearance [16]. siRNA molecules are encapsulated within the LNP, protecting them from degradation. This greatly improves their pharmacokinetics and bioavailability, allowing for lower dose requirements [31]. LNPs are large, roughly 100-nm nanoparticles and can therefore only pass through fenestrated endothelium, making them optimal for targeting the liver [16, 25]. Due to the leaky vasculature at cancer sites, LNPs may also potentially be used to target certain tumors [6, 16].
Other formulation-based strategies are currently in development, including complexes with cationic transfection agents, polymeric nanoparticles, liposomes, micelles, dendrimers, niosomes, metallic nanoparticles, human serum albumin-based nanoparticles, and oligonucleotide nanoparticles [32]. However, these all have various drawbacks and difficulties, rendering them at the present time as non-feasible pharmaceutical strategies.
Although LNPs have demonstrated clinical utility, there are numerous drawbacks associated with their use. The main issue is the toxicity of its excipients [13, 18], which manifests as administration-associated inflammation. This can be so severe, that patients must be pre-treated with a cocktail of anti-inflammatory drugs. Additionally, LNP formulations must be administered by a medical professional through an intravenous infusion, which is an invasive and time-consuming method less favorable to patients [18]. Finally, due to the large size of LNPs, they predominantly target only fenestrated tissues, such as the liver. Their clinical utility to target other tissues is extremely limited [13]. Researchers are developing improved formulations that circumvent these issues, so LNPs may still have applications in the future [4]. However, the future of delivery seems to lie in bioconjugation to promising new ligands [27].
Bioconjugates are created by covalently conjugating siRNA molecules or their nanocarriers to specific molecules that enhance both delivery and uptake. The most promising conjugates are targeting ligands. In contrast to LNPs, these active targeting modifications allow for specific delivery by conjugating the drug to a cell-specific ligand, such as targeting peptides or antibodies [25]. This increases the concentration of the drug at the target site and often mediates internalization, resulting in increased bioavailability and efficacy and decreased off-target effects. Additionally, bioconjugates tend to be less toxic and less immunogenic, due to their relatively smaller size [27].
A typical example of bioconjugates is glycoproteins terminating with N-acetylgalactosamine (GalNAc) sugars with high binding affinity and specificity to asialoglycoprotein (ASGPR), a receptor abundantly expressed in hepatocytes. Triantennary GalNAc (tri-GalNAc) has the highest affinity toward ASGPR [33]. Tri-GalNAc-conjugated antisense oligos display highly specific delivery to and internalization into hepatocytes [16, 25, 34]. Since ASGPR is an abundant, high-capacity receptor with rapid recycling and turnover, a single administration of GalNAc-siRNA conjugate yields very high siRNA uptake [35]. Tri-GalNAc was successfully used to deliver givosiran and lumasiran [36] and it has rapidly become the most popular platform for siRNA bioconjugation. Compared to LNPs, GalNAc-siRNA conjugates are much more straightforward to synthesize and refine [13]. Additionally, GalNAc conjugates are clinically convenient as they can be self-administered subcutaneously, resulting in rapid absorption, high uptake, and long half-life [35]. Unlike LNPs, GalNAc conjugates have a very favorable toxicity profile and therefore do not require the pre-infusion anti-inflammatory treatment that LNPs formulations do. GalNAc conjugates have been so successful that up to 2/3 of all RNAi drugs in clinical trials are GalNAc conjugates, including givosiran, vutrisiran, nedosiran, inclisiran, and fitusiran [4]. It is likely that these will far outpace and overshadow the use of LNPs, which has only been used in patisiran (Fig. 2).
Other ligand-receptor pairs in development include glucagon-like peptide-1 and its receptor in pancreatic beta cells, transferrin and its receptor protein 1 in skeletal and cardiac muscle, cyclic arginyl-glycyl-aspartic acid and integrins on cancer cells, folate and folate receptors on cancer cells, and antibodies and their cell-specific receptors [16, 25, 27]. siRNA drugs may also be conjugated to cationic peptide moieties, such as penetratin and Endo-Porter, which effectively penetrate tissue barriers and cell membranes [16]. Conjugation to lipophilic moieties, such as cholesterol, improves pharmacokinetic properties and increases serum stability [27, 37].
With recent advances in the development and/or adoption of novel chemical modifications and delivery systems to overcome these barriers, siRNA-based therapy is now a reality for two FDA-approved drugs and nearly a dozen others on late stages of clinical trials. For the remainder of this review, we will discuss three FDA-approved drugs (patisiran, givosiran, and lumasiran) and seven candidates in Phase 3 trials (vutrisiran, nedosiran, inclisiran, fitusiran, teprasiran, cosdosiran, and tivanisiran).
3. siRNA drugs in the clinic and late-stage development
In this section, we will discuss the mRNA target, chemical modifications, delivery platform, clinical trial results, and current status for each approved and late Phase-3 siRNA drug.
3.1. Patisiran
Patisiran, branded as ONPATTRO by Alnylam Pharmaceuticals, is the first commercialized siRNA therapeutic drug [38]. Approved by the FDA on August 10, 2018, it treats peripheral nerve disease secondary to hereditary transthyretin-mediated amyloidosis (hATTR). This is a progressive, fatal orphan disease caused by mutations in the hepatocyte-derived protein transthyretin (TTR), which results in the systemic buildup of amyloid deposits in key organs, including the peripheral and central nervous systems, heart, kidneys, and gastrointestinal tract [39]. This causes debilitating symptoms including severe neuropathy, cardiomyopathy, and death within 5–15 years of diagnosis [40].
Patisiran is an siRNA molecule that targets a conserved sequence on all variants of TTR mRNA, thereby reducing the production of TTR protein in amyloid deposits [41]. Preclinical evaluation of patisiran showed robust silencing of TTR in the liver [42]. The drug is made up of two 21-base strands, with eleven 2’-OMe modifications present on all pyrimidines in the sense strand and two of the uridines in the antisense strand. All other ribonucleotides are unmodified. There are no modifications to the backbone; all of the linkages are unmodified phosphodiesters [31]. Due to its limited chemical modifications, patisiran must be formulated in second-generation LNPs that contain cholesterol, a polar lipid DSPC, a PEGylated lipid PEG2000-C-DMG, and an ionizable amino lipid DLin-MC3-DMA [31, 42]. Cholesterol and DSPC increase the stability of the formulation, PEG2000-C-DMG increases stability and circulation time, and DLin-MC3-DMA facilitates the LNP’s formation, uptake, and release [31]. These large LNPs have high affinity to the fenestrated tissue of the liver, binding to ApoE receptors and endocytosing into hepatocytes, the primary producers of TTR [31].
Efficacy and safety of patisiran have been determined in numerous clinical trials [43–50]. The landmark Phase 3 APOLLO study (ClinicalTrials.gov identifier: NCT01960348) was an 18-month placebo-controlled study that randomized 225 hATTR subjects in a 2:1 ratio to patisiran or placebo [45]. The primary outcome measure was the modified Neuropathy Impairment Score (mNIS+7). Secondary measures included various scoring systems to measure quality of life, including the Norfolk Quality-of-Life Diabetic Neuropathy (QOL-DN) score, motor strength, level of disability, mobility, body mass index (BMI), and autonomic symptoms. Patisiran treatment resulted in a rapid robust reduction (81%) in serum TTR levels sustained over the 18-month study period. Compared to placebo, patisiran-treated participants demonstrated a statistically significant improvement in polyneuropathy (measured by the mNIS+7). Patisiran treatment also statistically improved all secondary endpoints compared to placebo within 9 months. Patisiran was generally well-tolerated with a consistent safety profile and adverse effects (AEs) and mortality rates comparable to the placebo group [51]. There were no deaths considered attributable to patisiran treatment and AEs were mild in both severity and frequency. The most common AEs were peripheral edema, upper respiratory tract infections, and infusion-related reactions, which may be mitigated with pre-infusion treatment and a slower infusion rate [45, 51, 52].
Patisiran is commercially available as a 10 mg/5 mL lipid complex injection with a recommended dosage of 0.3 mg/kg every 3 weeks for patients weighing less than 100 kg by the FDA. It must be administered through intravenous infusion over 80 minutes due to the inflammatory infusion-related reactions and requires premedication with a corticosteroid, acetaminophen, and antihistamines [51]. There are no contraindications for the use of patisiran. Although the approval of patisiran was a landmark event and a major success for RNAi therapeutics, further studies should be done to determine its long-term safety and efficacy profile [51]. Additionally, with the development of other hATTR treatments, such as inotersen and vutrisiran, patisiran’s future place in therapy may be limited.
3.2. Givosiran
Givosiran, branded by Alnylam Pharmaceuticals as GIVLAARI, is the second commercialized siRNA therapeutic drug [8]. After receiving a priority review as an orphan product and breakthrough therapy designation, Givosiran was approved by the FDA in November 20, 2019 for adults with acute hepatic porphyria (AHP). AHP is a rare genetic disorder [53] where mutations in the heme synthetic pathway result in feedback up-regulation of aminolevulinate synthase 1 (ALAS1), the first enzyme in this biosynthetic pathway [54]. Induction of ALAS1 results in increased production and accumulation of the neurotoxic metabolites aminolevulinic acid (ALA) and porphobilinogen (PBG), which cause debilitating and sometimes life-threatening symptoms, including severe abdominal pain, vomiting, hypertension, tachycardia, mental status changes, seizures, muscle weakness, and paralysis. AHP is also associated with chronic comorbidities, including chronic kidney disease, hypertension, neuropathy, and liver disease [55, 56].
Givosiran binds to a target sequence on ALAS1 mRNA, resulting in decreased ALAS1 synthesis and therefore decreased production of ALA and PBG, the mediators of the disease’s symptoms [57]. Givosiran consists of a 21-base sense strand and a 23-base antisense strand and utilizes Alnylam’s ESC GalNAc technology. It is fully modified with 16 nucleotides containing a 2’-F substitution and the remaining 2’-OMe substituted nucleotides. Six of its backbone linkages distributed at the ends of the strands are PS-modified. Conjugation to tri-GalNAc allows for high-uptake into the liver, the major site of ALAS1 synthesis, through subcutaneous administration [57].
An initial Phase 1 trial (NCT02452372) in patients with acute intermittent porphyria (AIP), the most common subtype of AHP, compared givosiran to placebo (normal saline) in 40 randomized participants [58]. Once-monthly injections resulted in rapid and sustained reductions in ALAS1 mRNA, ALA, and PBG levels, notably decreasing the rate of disease attacks. Encouraged by these results, the company launched the Phase 3 ENVISION trial (NCT03338816), in which 94 AHP patients (89 of whom had AIP) were randomized 1:1 to receive placebo or 2.5 mg/kg once per month subcutaneously for a 6-month period [59]. The primary endpoint was the annualized rate of porphyria attacks in patients with AIP. Givosiran-treated patients demonstrated a 74% lower attack rate. Patients in the treatment arm also met key secondary end points, including sustainably lowered levels of urinary ALA and PBG, a lower rate of hemin use (an existing treatment to manage porphyria attacks), a lower daily pain score, and a higher quality of life (measured by the SF-12) [59]. The most common AEs associated with givosiran were mild to moderate injection site reactions and nausea [58, 59]. However, more concerningly, givosiran administration was associated with severe renal and hepatic AEs, characterized by increased serum creatinine, reduced eGFR, worsening of chronic kidney disease, and alanine aminotransferase (ALT) elevations > 3 × upper limit of the normal range [58, 60]. No deaths were reported [58, 59].
Based on these results, givosiran was marketed under the FDA approval as a 189 mg/mL injection subcutaneously with a recommended dose of 2.5 mg/kg once per month. Although the findings from the initial double-blind phase of ENVISION results were extremely promising and encouraging to patients with ALP, 6 months is a fairly short period to assess its clinical utility for a disease with chronic nature [60]. Seventeen patients who completed the initial Phase 1 study are now enrolled in a Phase 2 open-label extension (OLE) and the ENVISION trial (NCT03338816) is in its 29-month OLE period. Givosiran’s long term safety and efficacy should be carefully monitored, especially given its association with severe adverse effects.
3.3. Lumasiran
Lumasiran, branded by Alnylam Pharmaceuticals as OXLUMO, was the third (and most recent) FDA-approved siRNA therapeutic. On November 23, 2020, it became the first FDA-approved treatment for the orphan disease primary hyperoxaluria type 1 (PH1) [61]. Primary hyperoxalurias are characterized by deficiencies in hepatic enzymes and subsequent accumulation of oxalate, a toxic metabolite [62]. Glyoxylate, the major precursor of oxalate, normally undergoes several metabolic pathways. In PH1, mutations in the AGT gene that encodes an enzyme that metabolizes glyoxylate to pyruvate and glycine, result in accumulation of glyoxylate. The glyoxylate is diverted into another metabolic pathway and converted to oxalate. Oxalate is efficiently eliminated through the kidneys, but its overproduction and increasing concentrations ultimately trigger crystallization of calcium oxalate, which deposits as kidney and urinary tract stones. Eventually, sustained kidney damage progresses to chronic kidney disease and end-stage renal disease; as renal function and excretion decline, oxalate accumulates in extrarenal tissue, resulting in often-fatal systemic oxalosis [63, 64].
Lumasiran targets the mRNA that translates the hepatic enzyme glyoxylate oxidase (GO), which synthesizes glyoxylate. By decreasing the expression of GO, lumasiran decreases the production of glyoxylate and subsequently decreases oxalate synthesis [63, 64]. Lumasiran is made up of 2 subunits: one 21-base strand and another 23-base strand. It utilizes Alnylam’s ESC-GalNAc platform and is fully modified with 10 2’-F substituted nucleotides and the other 34 2’-OMe substituted nucleotides. Like other ESC compounds, it has six PS linkages at the strand extremities. Conjugation to GalNAc allows for specifically efficient hepatic uptake through subcutaneous administration.
In preclinical studies with rat and monkey at supratherapeutic doses, lumasiran shared similar safety signals with other siRNA drugs in the organ of pharmacodynamic effect (liver), the organ of elimination (kidney), and the reticuloendothelial system (lymph nodes) [65]. The majority of AEs were non-adverse, partially to completely reversible, correlate well with pharmacokinetic parameters and tissue distribution, and often reflect drug accumulation.
In an initial Phase 1/2 trial (NCT02706886), healthy volunteers and PH1 patients were randomized to receive lumasiran or placebo followed by lumasiran. Lumasiran dosing caused a 75% mean maximal reduction in urinary oxalate (UOx) levels. Patients who received 3 mg/kg of lumasiran monthly or quarterly were able to achieve normal UOx levels. The majority of AEs were mild to moderate. Most patients subsequently enrolled in a Phase 2 OLE, which is ongoing. To further assess lumasiran’s safety and efficacy, Alnylam launched ILLUMINATE-A, a multi-pronged Phase 3 trial (NCT03681184). ILLUMINATE-A studied the effects of 6-month dosing in 39 PH1 patients ≥ 6 years old. Patients were randomized 2:1 to receive subcutaneous lumasiran or placebo, with an initial dose of lumasiran 3 mg/kg per month x 3 months, followed by a maintenance dose of 3 mg/kg per 3 months. The primary endpoint was reduction in 24-hour UOx levels over months 3 – 6. Lumasiran treatment caused a 65.4% mean reduction relative to baseline and 53.5% reduction relative to placebo. Lumasiran also met all secondary endpoints, including 1) the proportion of lumasiran patients who achieved near-normalization (84%) or normalization (52%) of UOx vs. 0% in the placebo group and 2) change in plasma oxalate (−39.8% with lumasiran vs. −0.3% with placebo). Lumasiran had a favorable safety profile. All reported AEs were mild to moderate in severity and the most common of which were transient injection site reactions.
Most ILLUMINATE-A participants have rolled over into the extension period of the trial in ILLUMINATE-B (NCT03905694) with single-arm open-label lumasiran treatment in PH1 patients < 6 years old and ILLUMINATE-C (NCT04152200) with single-arm open-label lumasiran treatment in advanced PH1 patients, which are pending. Based on ILLUMINATE-A’s promising results, Alnylam’s new drug application (NDA) was granted Priority Review designation; lumasiran had also previously received both Orphan Drug and Breakthrough Therapy designations. Given this status, lumasiran’s November 2020 approval was celebrated, but expected.
3.4. Vutrisiran
Like patisiran, vutrisiran is indicated to treat hATTR. It binds to a conserved sequence on all TTR variants. However, vutrisiran’s modifications and delivery system represent a more advanced stage of drug development. Like the majority of Alnylam Pharmaceutical’s siRNA candidates, vutrisiran utilizes the company’s enhanced stabilization chemistry (ESC)-GalNAc delivery platform, a third-generation GalNAc-siRNA conjugation design [30, 66, 67]. ESC conjugates optimize the incorporation, distribution, and placement of 2’-OMe, 2’F, and PS modifications [66, 67]. Vutrisiran is made up of one 21-base strand and another 23-base strand, both of which are fully modified. Of the 44 nucleotides in total, 35 contain a 2’-OMe substitution, and 9 contain a 2’-F substitution. The compound contains 6 PS linkages, distributed at the ends of the strands. Its conjugation to GalNAc allows for specific, high-uptake delivery to hepatocytes, the primary site of TTR production.
Vutrisiran is currently in late-stage clinical trials. An initial Phase 1 trial, completed in January 2018, assessed its safety, tolerability, pharmacokinetics, and pharmacodynamics vs. placebo (normal saline) in 80 healthy participants [68]. In this single ascending dose study, participants were randomized 6:2 to receive a single subcutaneous dose of vutrisiran (5 – 300 mg) or placebo. A single dose of the drug caused rapid, robust (57–97%), and durable (≥ 90 days) reductions in serum TTR levels. A quarterly dose of 25 mg every 3 months caused similar or greater efficacy than patisiran’s dosing regimen with a predicted TTR knockdown of 88–90%. Vutrisiran was very well tolerated and all AEs were mild to moderate. The most common treatment-related side effect was mild injection site pain, which was seen in less than 7% of treated participants, all of whom had received relatively high doses (≥ 50 mg) [68].
Based on these results, the company launched two Phase 3 trials: HELIOS-A (NCT03759379) and HELIOS-B (NCT04153149). HELIOS-A is an open-label study that will compare vutrisiran to patisiran in patients with hATTR amyloidosis with polyneuropathy. One hundred and sixty-four participants have been enrolled for an 18-month treatment period to be followed by a lengthier extension period. During the initial treatment period, participants were randomized to receive either 25 mg of vutrisiran subcutaneously every 12 weeks or 0.3 mg/kg of patisiran intravenously every 3 weeks. In the following extension period, all participants were eligible to receive vutrisiran. Much like the APOLLOS trial, the primary endpoints of the study are the change in baseline from the mNIS+7 and QOL-DN scores to measure neurologic impairment and quality of life, respectively. Secondary endpoints include measures of mobility, BMI, disability, and serum TTR. In addition to inter-arm comparisons, results from the vutrisiran arm will also be compared to the placebo arm from the APOLLOS trial. Preliminary results are expected to be released in late 2020 or early 2021.
HELIOS-B will evaluate vutrisiran vs. placebo for 600 participants suffering from hATTR with cardiomyopathy. Patients will be randomized to receive placebo or 25 mg of vutrisiran subcutaneously once every 3 months during a 36-month treatment period. The primary endpoint will be all-cause mortality and recurrent cardiovascular hospitalizations at 30 months. Secondary endpoints include measures of functional exercise capacity, self-perception of health status, and cardiac structure and function. The study is not expected to conclude until June 2024, but if successful, it could broaden Alnylam’s hATTR treatment scope beyond just neuropathy.
In April 2020, the FDA awarded vutrisiran a fast-track designation, anticipating positive results from the HELIOS trials. If vutrisiran lives up to its expectations, it may prove to be a more clinically utile, effective treatment option for hATTR than patisiran.
3.5. Nedosiran
Another company, Dicerna Pharmaceuticals, is developing another potential siRNA solution for primary hyperoxaluria. Their candidate, nedosiran, is in Phase 3 trials. Unlike lumasiran, it targets the hepatic enzyme lactate dehydrogenase (LDH), which controls the final step in glyoxylate metabolism and may be a more potent target than GO. Since it controls the final common step in oxalate synthesis, LDH inhibition is effective not only for PH1, but also for subtypes 2 and 3. Although they are characterized by different mutations, the end result of oxalate overproduction is the same [64, 69]. Preclinical in vivo evidence in mammals supported LDH as the key enzyme responsible for converting glyoxylate to oxalate [69]. Reduction of hepatic LDH expression by nedosiran achieved efficient reduction in oxalate production and prevented calcium oxalate crystal deposition in genetically engineered mouse models of PH types 1 (PH1) and 2 (PH2), as well as in chemically-induced PH mouse models.
Nedosiran utilizes Dicerna’s proprietary GalXC platform, a GalNAc-siRNA conjugation technology analogous to Alnylam’s ESC. The molecule consists of a 22-base antisense strand and a 36-base sense strand, which doubles over to form a tetraloop configuration. Almost all the bases are modified; 19 have a 2’-F substitution, and 35 have a 2’-OMe substitution. Six backbone linkages at the extremities of the molecule have been replaced with PS groups. Conjugation to GalNAc results in high hepatic uptake after subcutaneous administration [22].
An initial Phase 1 trial, PHYOX 1 (NCT03392896), measured the safety and efficacy of a single nedosiran dose in healthy volunteers and PH patients. In PH1 patients, a single 1.5 mg/kg dose was associated with a 48% mean maximal reduction in UOx, a single 3 mg/kg dose with a 71% reduction, and a 6 mg/kg dose with a 66% reduction. Most recently, Dicerna has presented preliminary data from PHYOX 3 (NCT04042402), an OLE study in which PH patients receive monthly fixed subcutaneous dosing. While no information about the drug’s efficacy or specific doses have been disclosed, the company reported a favorable AE profile with no drug-related severe AEs reported as of 3/21/20.
In April 2020, Dicerna and Alnylam announced that they had formed a collaboration for their PH programs, granting each other non-exclusive cross-licenses to lumasiran and nedosiran. This ensures that each company can continue to develop and commercialize its respective products. Lumasiran was the first to be approved, but nedosiran will have a potentially wider scope, since it is not limited to just PH1 patients. Additionally, Dicerna has received both Breakthrough Therapy and Rare Pediatric Disease designations from the FDA, the latter of which will allow the FDA to grant nedosiran priority review once Dicerna submits an NDA.
3.6. Inclisiran
Inclisiran is arguably the most promising siRNA therapeutic drug in development in terms of its potential impact. Unlike the majority of approved and late-stage siRNA agents, it is designed to treat a very prevalent indication, hypercholesterolemia. This could dramatically expand siRNA’s clinical portfolio beyond just orphan diseases, cementing RNAi’s place as a pillar of modern pharmacotherapy. Inclisiran is a collaborative project between Alnylam and Novartis Pharmaceuticals (formerly Alnylam and The Medicines Company, the latter of which was acquired by Novartis in January 2020).
Managing cholesterol levels is critical to prevent further complications, especially cardiovascular events. Statins are the current standard of care, but many patients do not reach their therapeutic goals even at maximal statin doses and adherence can be challenging [70]. There is an increasing need for non-statin therapies with novel mechanisms of action. Inclisiran provides a new hope in the management of cholesterol homeostasis disorders [71].
As its mechanism of action, inclisiran targets a key protein in the low-density lipoprotein cholesterol (LDL-C) metabolic pathway called proprotein convertase subtilisin/kexin type 9 (PCSK9). Typically, upon binding by LDL-C, the bound receptor is internalized. The LDL particle is transferred to a lysosome for digestion and the receptor is recycled and re-inserted back into the hepatocyte plasma membrane. When PCSK9 is present, it binds the LDL receptors, which triggers the internalization and degradation of this protein complex. Inhibition of PCSK9 results in increased receptor recycling and higher plasma membrane receptor density, subsequently increasing LDL-C binding and decreasing circulating LDL-C levels [71]. Inclisiran, an siRNA therapeutic, is a first-in-class PCSK9 inhibitor. It consists of a 23-base guide strand and a 21-base passenger strand and utilizes Alnylam’s ESC-GalNAc delivery platform. Inclisiran is fully modified with one 2’-MOE, eleven 2’-F, and thirty-two 2’-OMe substitutions. Six PS modifications are distributed across the termini of the strands and GalNAc conjugation allows for high hepatocyte uptake after subcutaneous administration [72].
In preclinical studies involving in non-human primates, treatment with inclisiran resulted in reductions of more than 80% in plasma PCSK9 levels and approximately 60% lowering of the serum level of LDL-C with peak effects lasting >30 days and with a very slow return to baseline levels over 90 ~ 120 days after administration [73].
Because of its much more common indication, inclisiran’s clinical trials have been significantly larger-scale and more highly powered than siRNAs that treat orphan diseases. An initial Phase 1 trial in healthy volunteers demonstrated impressive and durable reductions in PCSK9 and LDL-C concentrations [73]. After these promising results, the company launched the ORION program to assess the efficacy and safety of inclisiran in patients with atherosclerotic cardiovascular disease (ASCVD) and familial hypercholesterolemia (FH). ORION-2 (NCT02963311), inclisiran’s first Phase 2 trial, enrolled 501 patients with elevated serum LDL-C and high ASCVD risk, who were on a maximally tolerated dose of statin and/or ezetimibe [74]. Patients received single or double doses of placebo or inclisiran. A double 300 mg dose of inclisiran at the first and ninetieth days resulted in the highest efficacy, a mean PCSK9 reduction of 69.1% and an LDL-C reduction of 52.6% at Day 180 with sustained reduction even at Day 240. Additionally, the drug was well-tolerated with an AE rate comparable to that of the placebo group.
Novartis recently released the final results from its pivotal Phase 3 studies: ORION-9 (NCT03397121) [75], ORION-10 (NCT03399370) [76], and ORION-11 (NCT03400800) [76], which studied inclisiran in 482 patients with heterozygous FH, 1,561 patients with ASCVD from the US, and 1,617 patients with ASCVD or ASCVD risk equivalents outside of the US, respectively. The large majority of participants were already on statin therapy. In all 3 trials, participants received 300 mg inclisiran or placebo on Days 1, 90, 270, and 450. Results were followed up to Day 540. Each trial achieved the shared co-primary end points: the placebo-corrected percentage change in LDL-C from baseline to Day 510 and the time-adjusted percentage changes in LDL-C from baseline after Day 90 and up to Day 540. A pooled analysis of the data from all 3 trials revealed that inclisiran durably reduced LDL-C by 51% at Day 510 (placebo-adjusted) as well as a 51% reduction at Day 540 (time-adjusted and placebo-adjusted). Additionally, inclisiran decreased PCSK9 levels by 60.7% in ORION-9, 69.8% in ORION-10, and 63.6% in ORION-11 compared to placebo-group increases of 17.7%, 13.5%, and 15.6%, respectively. Inclisiran was also associated with lower levels of total cholesterol, non-HDL cholesterol, apolipoprotein B, and triglycerides compared to placebo [75, 76]. The drug showed a favorable safety and tolerability profile. Most AEs were mild to moderate and similarly frequent between treatment and placebo groups. Mild transient injection-site reactions were more frequent in inclisiran. Severe AEs were less common with inclisiran than with placebo [75, 76]. Patients who have completed ORION-9, ORION-10, and ORION-11 are eligible for enrollment into ORION-8 (NCT03814187), an ongoing OLE that will monitor inclisiran’s effects for up to 3 years.
Inclisiran has numerous advantages compared to other types of PCSK9 inhibitor drugs in development. The Medicines Company filed an NDA for inclisiran in December 2019 for secondary prevention in patients with ASCVD and FH. Given its positive Phase 3 results and strong potential, inclisiran is almost certain to receive FDA approval in the near future. In Europe, the drug has already been approved by the European Commission and commercialized as Leqvio (https://www.novartis.com/news/media-releases/novartis-receives-eu-approval-leqvio-inclisiran-first-class-sirna-lower-cholesterol-two-doses-year). Inclisiran is likely to do well against its main competitors, alirocumab and evolocumab, which are anti-PCSK9 monoclonal antibodies. Evolocumab (Repatha) is currently the most prevalent PCSK9-targeting drug and has been a commercial success with annual sales averaging $2.5 billion. However, although successful, it has several disadvantages compared to inclisiran. It is much more expensive to synthesize (and therefore prohibitively costly) and it has a more frequent dosing period, making it less convenient for patients [77].
Despite inclisiran’s projected success, however, some experts caution that it may not live up to expectations. Decreased LDL-C and PCSK9 levels may not necessarily translate into improved cardiovascular outcomes. Trials of evolocumab have shown no effects on cardiovascular mortality despite reduced LDL-C, suggesting that PCSK9 inhibition alone is insufficient for the prevention and treatment of dyslipidemias and cardiovascular disease [78]. In fact, a pooled analysis of ORION-9, ORION-10, and ORION-11 showed only a 2.5% decrease in major cardiovascular events with inclisiran than placebo [78]. Additionally, a recent meta-analysis did not associate inclisiran treatment in hypercholesterolemic patients with any significant decrease in cardiovascular ischemic end points [79]. Perhaps these findings will be refuted in ORION-4 (NCT03705234), an ongoing Phase 3 trial that aims to recruit 15,000 participants with pre-existing ASCVD who are unable to achieve LDL-C goals. The trial is expected to run until 2024 and evaluate cardiovascular outcomes for inclisiran vs. placebo. Since inclisiran is likely to be approved before the ORION-4 results are released, providers will have to utilize their clinical judgement and monitor patient outcomes carefully to determine inclisiran’s place in therapy.
3.7. Fitusiran
Fitusiran is a collaboration between Alnylam and Sanofi Genzyme, currently in Phase 2 and 3 trials for hemophilia A and B. Hemophilia A and B are X-linked bleeding disorders caused by deficiencies in clotting factors VIII and IX, respectively [80]. This leads to a clinical presentation of life-threatening, recurrent, and spontaneous bleeding, especially in joints and muscles. Long-term complications include chronic arthritis, disability, and pain. The current standard of care consists of frequent (2–3 times weekly) prophylactic infusions of concentrated clotting factor products to replace the depleted factor [81]. This prophylactic regimen is costive, invasive, and burdensome, leading to a high rate of nonadherence as patients opt to treat with on-demand therapy instead, which is less effective. Of even more concern is the fact that up to 30% of factor-treated hemophilia A patients and 3–5% of hemophilia B patients develop neutralizing anti-factor alloantibodies, also known as inhibiting antibodies, that reduce the treatment’s efficacy. These patients may need to resort to second-line therapies with bypassing agents, which are less preferable.
siRNA-based therapy targeting antithrombin for the treatment of hemophilia A and B is in development. Rather than replacing clotting factors, fitusiran inhibits the production of anti-clotting factors, ultimately causing the same end result, decreased bleeding events [82, 83]. Fitusiran binds to the mRNA of antithrombin, an endogenous anticoagulant, resulting in increased generation of thrombin (a pro-clotting enzyme) and enhanced hemostasis. In theory, this mechanism should be effective regardless of the presence or absence of inhibiting antibodies. Fitusiran is yet another representative of Alnylam’s ESC-GalNAc delivery platform. Like the other ESC candidates, it is fully modified with twenty-one 2’-F substitutions and twenty-three 2’-OMe substitutions. It consists of a 21-base strand and a 23-base strand and has 6 PS linkages at the ends of the strands [84]. Conjugation to a tri-GalNAc moiety directs it to the liver, the major site of antithrombin synthesis.
Efficacy and safety of fitusiran have been tested in a series of clinical trials. An initial four-part Phase 1 (NCT02554773) study assessed the drug’s effects in 37 healthy volunteers, patients with hemophilia A or B and no inhibitors, and patients with hemophilia A or B and inhibitors [85]. The results were promising. Fitusiran-treated patients demonstrated dose-dependent decreases of antithrombin levels and a lower rate of bleeding episodes. Most AEs were mild to moderate in severity. No participants developed antibodies to fitusiran during the study. In a follow-up Phase 2 OLE, open to all patients with hemophilia who had been previously dosed during the Phase 1 study, participants received fixed monthly doses of 50 or 80 mg fitusiran subcutaneously. The initial safety profile was promising with most AEs mild to moderate in severity. However, the study was suspended in September 2017 after a severe thromboembolic event occurred, resulting in a patient’s death. The patient had developed exercise-induced hip pain, which was treated with 3 doses of factor products. The patient subsequently experienced a cerebral venous sinus thrombosis. Unfortunately, this was initially misdiagnosed as a subarachnoid hemorrhage not attributable to fitusiran. To treat this apparent hemorrhage, the patient was given still more factor products over a 14-day hospitalization. He passed away from cerebral edema. Upon discovering that the initiating event was, in fact, a thrombosis and therefore potentially related to fitusiran administration, the Phase 2 study was suspended. Afterwards, Alnylam developed new risk mitigation measures to manage bleeding events and the FDA lifted the suspension in December 2017. As of the data cutoff in March 2020, monthly subcutaneous dosing resulted in sustained lowering of antithrombin (~75% decrease) in both patients with and without inhibitors, a decreased annualized bleeding rate, most notable in the inhibitor subgroup (pre-study rate of 42.0 vs. treatment rate of 0.44), and no detection of anti-fitusiran antibody formation. After implementing the new bleed event management guidelines, all treated bleeds were successfully managed and no further thromboembolic events have been reported in patients compliant to the guidelines. The most common AEs were increased ALTs and injection site erythema.
Alnylam and Sanofi are currently awaiting results from the Phase 3 trial ATLAS, which has multiple branches, including 1) ATLAS-A/B (NCT03417245), which will assess patients ≥ 12 years of age with hemophilia A or B without inhibitors, who currently manage their bleeds with on-demand factor replacement therapy (these patients will receive fitusiran or on-demand factor replacement therapy); 2) ATLAS-INH (NCT03417102), which will assess patients ≥ 12 years of age with hemophilia A or B with inhibitors, who currently manage their bleeds with on-demand bypassing agent therapy (these patients will receive fitusiran or on-demand bypassing agent therapy); 3) ATLAS-PPX (NCT03549871), which will assess patients ≥ 12 years of age with hemophilia A or B with or without inhibitors, who currently manage their bleeds prophylactically (these patients will receive fitusiran or prophylactic factor therapy/bypassing agent therapy); 4) ATLAS-PEDS (NCT03974113), which will assess patients < 12 years of age with hemophilia A or B with inhibitors (these patients will receive a selected dose of fitusiran per study protocol); and 5) ATLAS-OLE (NCT03754790), which will measure the long-term safety and efficacy of once-monthly subcutaneous fitusiran in patients with hemophilia A or B, with or without inhibitors. Patients who complete prior ATLAS trials may be eligible for ATLAS-OLE. Alnylam and Sanofi continue to investigate the clinical profiles of fitusiran in their Phase 3 ATLAS program with results expected during the first half of 2021.
3.8. Teprasiran
Teprasiran, more commonly known as QPI-1002, was the first systemically administered siRNA drug to enter human clinical trials. Designed by Quark Pharmaceuticals and licensed to Novartis, it is being developed as a prophylactic treatment for acute kidney injury (AKI) following kidney transplant or cardiovascular surgery [86]. AKI, characterized by an abrupt decrease in kidney function, is associated with high mortality due to a severe clinical presentation coupled with a lack of effective treatment [87]. In some situations, AKI is predictable, warranting the use of preventive therapies. For instance, patients who receive kidney transplants often develop a form of AKI called delayed graft function (DGF). Similarly, high-risk patients who undergo cardiovascular surgery are prone to developing AKI at post-operation. Since p53, a pro-apoptotic transcription factor that is activated in response to physiological stress, is a key mediator of AKI, inhibiting its production produces significant renal benefits, including reduced necrosis, apoptosis, and inflammation [88, 89].
Teprasiran works by targeting the mRNA of p53. Unlike the majority of previously discussed siRNA therapeutics, teprasiran is only minimally modified. It consists of two 19-base strands with 19 2’-OMe substituted bases. There are no modifications to its phosphate backbone. Strikingly, teprasiran is administered as a naked siRNA molecule with no delivery system or conjugated targeting ligand. As previously discussed, systemically administered naked siRNA is largely internalized and cleared by the kidney. For teprasiran as therapy for AKI, this preferential renal localization is highly advantageous. Intravenously injected naked teprasiran is rapidly renally excreted, then reabsorbed by the proximal tubular cells (PTC), where it accumulates. Despite its lack of modifications and targeting ligands, teprasiran attains significant cellular internalization thanks to specialized PTC anatomy. The siRNA binds extensively to the proximal tubule brush border and is subsequently endocytosed [90, 91]
Teprasiran has undergone several clinical trials to assess its use in AKI prophylaxis after cardiac surgery or kidney transplant. A dose-escalation Phase 1 trial in 16 patients undergoing on-pump cardiac surgery showed encouraging safety and tolerability. A subsequent Phase 2 trial (NCT02610283) randomized 341 subjects undergoing cardiac surgery to receive teprasiran or placebo. The primary endpoint was the proportion of subjects who developed AKI through Day 5. The trial met this endpoint with teprasiran treatment significantly reducing AKI incidence, severity, and duration. Additionally, teprasiran treatment was associated with a 29% relative risk reduction in the major adverse kidney events at Day 90 (MAKE90) endpoint. Once again, teprasiran was well-tolerated with a similar AE profile between treatment and placebo. The most common AEs in both groups were those typical of post-cardiac surgery complications. Quark is currently recruiting participants for a Phase 3 trial (NCT03510897) that will test teprasiran vs. placebo in 1,088 patients at high risk for AKI following cardiovascular surgery. Its primary endpoint will be the proportion of subjects who develop any MAKE90 components.
A Phase 1/2 trial was also conducted for DGF prophylaxis in patients undergoing deceased donor kidney transplantation, who received a single 10 mg/kg IV injection 30 minutes after circulatory reperfusion was achieved to the transplant. The primary endpoint was to achieve at least a 30% relative risk reduction of DGF. Although the total study population only achieved a 15.1% reduction, two sub-populations achieved the primary endpoint: patients in the “Expanded Criteria Donor kidneys entirely Cold Stored” group and patients whose kidney donors were older than 35 years. Teprasiran-treated patients also had a statistically significantly better eGFR by Day 30. Treated patients had a 1.5-fold lower dialysis rate than placebo, but this was not statistically significant (p = 0.059). The drug was well-tolerated with similar AE rates in both treatment and placebo groups [89]. A Phase 3 trial, ReGIFT (NCT02610296), was conducted for DGF prophylaxis in transplantation after donor brain stem death with a focus on high-risk patients. The study was completed in January 2019, but results have not yet been released. In the meantime, teprasiran has received an orphan product designation from the FDA for its DGF prophylaxis indication.
3.9. Cosdosiran
Cosdosiran, more commonly known as QPI-1007, is another Quark Pharmaceuticals candidate. It is an siRNA therapeutic in development to treat nonarteritic anterior ischemic optic neuropathy (NAION) and primary angle glaucoma. NAION and glaucoma are ocular diseases characterized by loss of retinal ganglion cells (RGCs) [92]. Since RGCs are unable to divide and replenish themselves, their progressive loss ultimately results in visual impairment and blindness. As apoptosis is thought to be the main cause of RGC death, proteins involved in the apoptotic pathway have high potential as therapeutic targets. Cosdosiran inhibits the mRNA of caspase-2, a pro-apoptotic RGC protein that plays a key role in the pathway [93].
Like teprasiran, cosdosiran is a naked and barely modified agent. It is a duplex made up of two 19-base strands, with nine 2’-OMe modified bases in the guide strand and an L-DNA cytidine and inverted deoxyabasic moiety on opposite ends of the passenger strand [94]. It has no backbone modifications and all the phosphate linkages are intact. Lack of a delivery system for cosdosiran is logical given its local administration to the target site; it is injected intravitreally directly into the eye. Coupled with the eye’s immune privilege, this route of administration allows cosdosiran to bypass many of the pharmacokinetic barriers associated with systemic administration. Although its modifications are fewer than for other siRNA therapeutics, they seem to be sufficient for efficacy. The molecule does not have any detectable immune-stimulatory activity and remains chemically stable in intravitreal fluid [95]. Additionally, models have shown that retinal ganglion cells readily take up the siRNA molecule, even without the use of complex delivery techniques. This may be partially attributable to the drug’s prolonged exposure in the vitreous humor, which is in direct contact with the retinal ganglion cell layer [94].
Quark conducted a Phase 1/2a clinical trial (NCT01064505) with single-injection and dose escalation in 48 subjects who had low visual acuity or acute NAION. Participants received a single intravitreal injection of cosdosiran at doses ranging from 0.2 to 6.0 mg. The drug was well-tolerated with no serious AEs and the most common AEs were typical of intravitreal injections (conjunctival hemorrhage, conjunctival chemosis, and eye pain). In terms of efficacy, 51.7% of participants had improved visual acuity. Additionally, compared to natural history controls from a previous gold standard trial, cosdosiran treatment decreased loss of visual acuity. Based on these data, the researchers concluded that the drug did have a neuroprotective effect. However, no significant changes were seen on secondary outcome measures, including visual field and retinal nerve fiber thickness [96]. Following these results, Quark launched a Phase 2b/3 trial (NCT02341560) to assess safety and efficacy of cosdosiran in 732 subjects with recent-onset NAION. In this placebo-controlled trial, participants were randomized to five groups: Cohort 1, who received one 1.5 mg dose of cosdosiran and 2 subsequent doses of placebo; Cohort 2, who received one 3 mg dose of cosdosiran and 2 subsequent doses of placebo; Cohort 3, who received three 1.5 mg doses of cosdosiran; Cohort 4, who received three 3 mg doses of cosdosiran; and Cohort 5, who received three doses of placebo [96]. Unfortunately, as of August 2020, ClinicalTrials.gov reports that the study has been terminated after an interim analysis did not warrant its continuation. The company has not released any statements on the matter.
Quark also conducted a placebo-controlled Phase 2 trial in ~60 subjects with acute primary angle glaucoma. Participants received a single 1.5 mg dose of cosdosiran or placebo via intravitreal injection. The study’s primary objective was to study the drug’s safety, tolerability, and pharmacokinetics in this patient population; however, although the study concluded in July 2015, results have not yet been released.
Cosdosiran has been granted orphan designation by the FDA for its NAION indication. Despite this, however, its future success is not assured. It is concerning and somewhat questionable that no results have been published since the initial report of the first Phase 1/2a trial and even those findings have not resulted in peer-reviewed publications. The data from this trial, while apparently promising, also have significant limitations. The sample size was very small. Since the study was not placebo-controlled, results could only be compared to historical controls [9, 97]. More abundant and high-quality data are certainly needed to assess whether or not cosdosiran should continue to be developed.
3.10. Tivanisiran
Tivanisiran, developed by Sylentis S.A., is an siRNA candidate in Phase 3 trials for the treatment of ocular pain and dry eye disease (DED). DED is a multifactorial ophthalmic surface disease characterized by a disruption in tear film homeostasis, ocular inflammation, and neurosensory abnormalities [98]. These result in a clinical presentation of ocular pain/discomfort, dryness, itching, burning, and photophobia. Current management of DED include treatment forms ranging from artificial tear drops, the primary conventional treatment method, to the latest surgical applications [99]. The aims of treatment are to restore homeostasis in the ocular surface, break the vicious cycle of inflammation, and ensure long-term ocular surface comfort.
Unlike the current available therapies, which largely target only the inflammatory aspect of DED, tivanisiran inhibits additional key pathways. It targets the mRNA of transient receptor potential cation channel subfamily V member 1 (TRPV1), a channel involved in pain signal transduction, fibrogenesis modulation, the stress response, and the innate inflammatory response [100]. TRPV1 has been proven to play a significant role in mediating enhanced nocifensive behavior in DED [101]. Strategies to target specific transducer molecules on corneal nerves may prove beneficial as adjunct therapies in managing ocular pain in moderate to severe cases of DED. Tivanisiran has been designed to reduce ocular discomfort and pain and to improve ocular hyperemia and tear quality.
Tivanisiran is unique in that it is formulated as a completely non-modified, naked siRNA molecule. Composed of two 19-base strands, it has neither base substitutions, backbone replacements, nor a delivery system. It is administered as a topical eye drop. Like cosdosiran, tivanisiran has less need for modifications and delivery strategies due the ease with which it can be administered directly to its target site, allowing for direct local delivery and bypassing of systemic pharmacokinetic barriers. Indeed, despite its lack of modifications, biodistribution studies have shown that tivanisiran is rapidly absorbed into the eye and detectable within the cytoplasm of cells [100]. Based on the drug’s distribution pattern, experts believe it may be absorbed via the conjunctiva-scleral pathway, a route that provides less resistance for hydrophilic molecules such as siRNAs [102].
Sylentis evaluated tivanisiran’s efficacy and safety in one Phase 1 and two Phase 2 clinical trials, testing various doses of the drug in a total of 30 healthy volunteers (Phase 1) and 126 subjects with DED (Phase 2) [103]. In the Phase 1 trial, subjects received single or multiple doses of tivanisiran daily for seven consecutive days. No clinically significant changes or drug-related AEs were detected. In the Phase 2 trials, subjects received placebo or tivanisiran once a day for ten consecutive days. The primary endpoints were DED symptomatic scores, measured by the Visual Analog Scale (VAS) and the Ocular Surface Disease Index (OSDI), and ocular tolerance, measured by the frequency of conjunctival hyperemia and change in total corneal staining. Tivanisiran at a dose of 11.25 mg/mL (1.125%) significantly decreased VAS scores compared to placebo, indicating a decreased level of pain in treated patients. However, the OSDI scores decreased equivalently in both treatment and placebo groups, indicating no significant impact on disease severity. Conjunctival hyperemia and corneal staining scores were also improved in the 11.25 mg/mL treatment group. In all 3 trials, the drug displayed an excellent safety profile, with a very low AE rate and similar AE rate between placebo and treatment groups [103].
In the pivotal Phase 3 trial HELIX (NCT03108664), 330 subjects with moderate to severe DED were treated with 11.25 mg/mL tivanisiran or placebo (artificial tears) once a day for 28 days. The primary endpoints of the study were the change in ocular pain as measured by VAS scores, total corneal staining, and conjunctival hyperemia. The trial failed to meet these endpoints. Although tivanisiran-treated participants showed clinically relevant improvements in pain and total corneal staining relative to baseline, there was no statistically significant difference compared to placebo. However, the trial did meet one of its secondary endpoints. Treated participants demonstrated reduced central corneal staining, indicating a reduction in central corneal damage. Additionally, in a subpopulation of 30 participants who had DED secondary to Sjögren’s Syndrome, tivanisiran treatment was associated with statistically significant improvements in VAS scores, total and central corneal staining, and quality of life compared to placebo. Once again, the drug’s safety and tolerability were very favorable. There were no serious AEs reported and no difference in the AE rate between groups.
Although tivanisiran missed its primary endpoints in the total HELIX population, its efficacy in patients with Sjögren’s Syndrome makes it a promising candidate for this population, who suffer from a more extreme DED pathology. For patients with mild or moderate DED, it may still provide a clinically relevant benefit, but other options may prove to be more effective.
4. Conclusion
Although only three siRNA therapeutics have been approved by the FDA to date, more are sure to follow in the coming years. With the refinement of GalNAc and the development of other novel strategies for targeting and delivery of siRNAs to other organs than liver, researchers have largely overcome the barriers of clinical utility of siRNA molecules, which are the main rate-limiting step in commercializing siRNA therapeutics. Besides siRNA products, other oligonucleotide-based therapies like miRNAs and antisense oligonucleotides are gaining prominence as well. In today’s post-genome era, such products are becoming increasingly feasible and utile. Additionally, compared to other novel therapeutic classes, such as monoclonal antibodies, siRNA products have several advantages. First of all, they are relatively less expensive to synthesize and manufacture than their antibody rivals, so they can theoretically be priced at a more competitive rate [77]. Secondly, most late-stage products offer convenient dosing with regimens as infrequent as bi-annual treatments (e.g. inclisiran) and are potentially self-administrable (e.g. subcutaneous and topical products). Factors like these will become increasingly important as novel therapies are developed for rare indications and patients gain consumer power and choice as to which treatment they utilize.
Fortunately for the pharmaceutical companies that have invested so much time into siRNA development, the payoff promises to be high. Inclisiran was included on Cortellis’ shortlist of Drugs to Watch in 2020. It is projected to generate $1.16 billion in sales by 2024. Givosiran is forecasted to reach $560 million by 2025 [104]. Patisiran’s 2019 revenue was $166.4 million, its estimated 2020 revenue is $280 – $300 million, and sales are estimated to peak at $1 billion. Shares in Alnylam gained 41.9% in 2019, contrary to a 1.2% industry-wide decline. Only time will tell if these RNAi therapies are deserving of their new blockbuster status.
Acknowledgement
This work was supported in part by the National Institute of General Medical Sciences (R01GM118367) for X.B.Z and the National Cancer Institute (R01CA241194) for R.B.
Abbreviations:
- AEs
adverse effects
- AHP
acute hepatic porphyria
- AIP
acute intermittent porphyria
- AKI
acute kidney injury
- ALA
aminolevulinic acid
- ALAS1
aminolevulinate synthase 1
- ALT
alanine aminotransferase
- ASCVD
atherosclerotic cardiovascular disease
- ASGPR
asialoglycoprotein
- BMI
body mass index
- DED
dry eye disease
- DGF
delayed graft function
- dsRNA
double-stranded RNA
- DSPC
1,2-distearoyl-sn-glycero-3-phosphocholine
- eGFR
estimated glomerular filtration rate
- ESC
enhanced stabilization chemistry
- FH
familial hypercholesterolemia
- GalNAc
N-acetylgalactosamine
- GO
glyoxylate oxidase
- hATTR
hereditary transthyretin-mediated amyloidosis
- HDL-C
high-density lipoprotein cholesterol
- LDH
lactate dehydrogenase
- LDL-C
low-density lipoprotein cholesterol
- LNP
lipid nanoparticle
- MAKE90
major adverse kidney events at day 90
- mRNA
messenger RNA
- NAION
nonarteritic anterior ischemic optic neuropathy
- NDA
new drug application
- OLE
open-label extension
- OSDI
ocular surface disease index
- PCSK9
proprotein convertase subtilisin/kexin type 9
- PBG
porphobilinogen
- PH1
primary hyperoxaluria type 1
- PS
phosphorothioate
- PTC
proximal tubular cell
- QOL-DN
quality-of-life diabetic neuropathy
- RES
reticuloendothelial system
- RGCs
retinal ganglion cells
- RISC
RNA-induced silencing complex
- shRNA
short hairpin RNA
- siRNA
small interfering RNA
- TTR
transthyretin
- UOx
urinary oxalate
- VAS
visual analog scale
- 2’-F
2’-fluoro
- 2’-MOE
2’-O-methoxyethyl
- 2’-OMe
2-O-methyl
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors declare that there are no conflicts of interest.
References
- 1.Kim D and Rossi J, RNAi mechanisms and applications. Biotechniques, 2008. 44(5): p. 613–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ozcan G, et al. , Preclinical and clinical development of siRNA-based therapeutics. Adv Drug Deliv Rev, 2015. 87: p. 108–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fire A, et al. , Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature, 1998. 391(6669): p. 806–11. [DOI] [PubMed] [Google Scholar]
- 4.Setten RL, Rossi JJ, and Han SP, The current state and future directions of RNAi-based therapeutics. Nat Rev Drug Discov, 2019. 18(6): p. 421–446. [DOI] [PubMed] [Google Scholar]
- 5.Frazer KA, Decoding the human genome. Genome Res, 2012. 22(9): p. 1599–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu B, et al. , Clinical advances of siRNA therapeutics. J Gene Med, 2019. 21(7): p. e3097. [DOI] [PubMed] [Google Scholar]
- 7.Wood H, FDA approves patisiran to treat hereditary transthyretin amyloidosis. Nat Rev Neurol, 2018. 14(10): p. 570. [DOI] [PubMed] [Google Scholar]
- 8.Scott LJ, Givosiran: First Approval. Drugs, 2020. 80(3): p. 335–339. [DOI] [PubMed] [Google Scholar]
- 9.Titze-de-Almeida R, David C, and Titze-de-Almeida SS, The Race of 10 Synthetic RNAi-Based Drugs to the Pharmaceutical Market. Pharm Res, 2017. 34(7): p. 1339–1363. [DOI] [PubMed] [Google Scholar]
- 10.Ketting RF, et al. , Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C. elegans. Genes Dev, 2001. 15(20): p. 2654–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zamore PD, et al. , RNAi: double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell, 2000. 101(1): p. 25–33. [DOI] [PubMed] [Google Scholar]
- 12.Dana H, et al. , Molecular Mechanisms and Biological Functions of siRNA. Int J Biomed Sci, 2017. 13(2): p. 48–57. [PMC free article] [PubMed] [Google Scholar]
- 13.Dowdy SF, Overcoming cellular barriers for RNA therapeutics. Nat Biotechnol, 2017. 35(3): p. 222–229. [DOI] [PubMed] [Google Scholar]
- 14.Gao S, et al. , The effect of chemical modification and nanoparticle formulation on stability and biodistribution of siRNA in mice. Mol Ther, 2009. 17(7): p. 1225–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blanco E, Shen H, and Ferrari M, Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol, 2015. 33(9): p. 941–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun Y, et al. , Enhancing the Therapeutic Delivery of Oligonucleotides by Chemical Modification and Nanoparticle Encapsulation. Molecules, 2017. 22(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang J, et al. , Delivery of siRNA therapeutics: barriers and carriers. AAPS J, 2010. 12(4): p. 492–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crooke ST, et al. , RNA-Targeted Therapeutics. Cell Metab, 2018. 27(4): p. 714–739. [DOI] [PubMed] [Google Scholar]
- 19.Du Rietz H, et al. , Imaging small molecule-induced endosomal escape of siRNA. Nat Commun, 2020. 11(1): p. 1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meng Z and Lu M, RNA Interference-Induced Innate Immunity, Off-Target Effect, or Immune Adjuvant? Front Immunol, 2017. 8: p. 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dalpke A and Helm M, RNA mediated Toll-like receptor stimulation in health and disease. RNA Biol, 2012. 9(6): p. 828–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu B, et al. , Therapeutic siRNA: state of the art. Signal Transduct Target Ther, 2020. 5(1): p. 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Micklefield J, Backbone modification of nucleic acids: synthesis, structure and therapeutic applications. Curr Med Chem, 2001. 8(10): p. 1157–79. [DOI] [PubMed] [Google Scholar]
- 24.Nguyen D, et al. , Stereospecific Effects of Oxygen-to-Sulfur Substitution in DNA Phosphate on Ion Pair Dynamics and Protein-DNA Affinity. Chembiochem, 2016. 17(17): p. 1636–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seth PP, Tanowitz M, and Bennett CF, Selective tissue targeting of synthetic nucleic acid drugs. J Clin Invest, 2019. 129(3): p. 915–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chiu YL and Rana TM, siRNA function in RNAi: a chemical modification analysis. RNA, 2003. 9(9): p. 1034–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chernikov IV, Vlassov VV, and Chernolovskaya EL, Current Development of siRNA Bioconjugates: From Research to the Clinic. Front Pharmacol, 2019. 10: p. 444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lam JK, et al. , siRNA Versus miRNA as Therapeutics for Gene Silencing. Mol Ther Nucleic Acids, 2015. 4: p. e252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prakash TP, et al. , Positional effect of chemical modifications on short interference RNA activity in mammalian cells. J Med Chem, 2005. 48(13): p. 4247–53. [DOI] [PubMed] [Google Scholar]
- 30.Weng Y, et al. , RNAi therapeutic and its innovative biotechnological evolution. Biotechnol Adv, 2019. 37(5): p. 801–825. [DOI] [PubMed] [Google Scholar]
- 31.Zhang X, Goel V, and Robbie GJ, Pharmacokinetics of Patisiran, the First Approved RNA Interference Therapy in Patients With Hereditary Transthyretin-Mediated Amyloidosis. J Clin Pharmacol, 2019. [DOI] [PMC free article] [PubMed]
- 32.Chenthamara D, et al. , Therapeutic efficacy of nanoparticles and routes of administration. Biomater Res, 2019. 23: p. 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cedillo I, et al. , Synthesis of 5’-GalNAc-Conjugated Oligonucleotides: A Comparison of Solid and Solution-Phase Conjugation Strategies. Molecules, 2017. 22(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prakash TP, et al. , Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N-acetyl galactosamine improves potency 10-fold in mice. Nucleic Acids Res, 2014. 42(13): p. 8796–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Springer AD and Dowdy SF, GalNAc-siRNA Conjugates: Leading the Way for Delivery of RNAi Therapeutics. Nucleic Acid Ther, 2018. 28(3): p. 109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de Paula Brandao PR, Titze-de-Almeida SS, and Titze-de-Almeida R, Leading RNA Interference Therapeutics Part 2: Silencing Delta-Aminolevulinic Acid Synthase 1, with a Focus on Givosiran. Mol Diagn Ther, 2020. 24(1): p. 61–68. [DOI] [PubMed] [Google Scholar]
- 37.Chen C, Yang Z, and Tang X, Chemical modifications of nucleic acid drugs and their delivery systems for gene-based therapy. Med Res Rev, 2018. 38(3): p. 829–869. [DOI] [PubMed] [Google Scholar]
- 38.Hoy SM, Patisiran: First Global Approval. Drugs, 2018. 78(15): p. 1625–1631. [DOI] [PubMed] [Google Scholar]
- 39.Hund E, Familial amyloidotic polyneuropathy: current and emerging treatment options for transthyretin-mediated amyloidosis. Appl Clin Genet, 2012. 5: p. 37–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adams D, et al. , Rapid progression of familial amyloidotic polyneuropathy: a multinational natural history study. Neurology, 2015. 85(8): p. 675–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hawkins PN, et al. , Evolving landscape in the management of transthyretin amyloidosis. Ann Med, 2015. 47(8): p. 625–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Butler JS, et al. , Preclinical evaluation of RNAi as a treatment for transthyretin-mediated amyloidosis. Amyloid, 2016. 23(2): p. 109–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Suhr OB, et al. , Efficacy and safety of patisiran for familial amyloidotic polyneuropathy: a phase II multi-dose study. Orphanet J Rare Dis, 2015. 10: p. 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Adams D, et al. , Trial design and rationale for APOLLO, a Phase 3, placebo-controlled study of patisiran in patients with hereditary ATTR amyloidosis with polyneuropathy. BMC Neurol, 2017. 17(1): p. 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Adams D, et al. , Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N Engl J Med, 2018. 379(1): p. 11–21. [DOI] [PubMed] [Google Scholar]
- 46.Minamisawa M, et al. , Association of Patisiran, an RNA Interference Therapeutic, With Regional Left Ventricular Myocardial Strain in Hereditary Transthyretin Amyloidosis: The APOLLO Study. JAMA Cardiol, 2019. 4(5): p. 466–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Solomon SD, et al. , Effects of Patisiran, an RNA Interference Therapeutic, on Cardiac Parameters in Patients With Hereditary Transthyretin-Mediated Amyloidosis. Circulation, 2019. 139(4): p. 431–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Obici L, et al. , Quality of life outcomes in APOLLO, the phase 3 trial of the RNAi therapeutic patisiran in patients with hereditary transthyretin-mediated amyloidosis. Amyloid, 2020. 27(3): p. 153–162. [DOI] [PubMed] [Google Scholar]
- 49.Gonzalez-Duarte A, et al. , Analysis of autonomic outcomes in APOLLO, a phase III trial of the RNAi therapeutic patisiran in patients with hereditary transthyretin-mediated amyloidosis. J Neurol, 2020. 267(3): p. 703–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Coelho T, et al. , A phase II, open-label, extension study of long-term patisiran treatment in patients with hereditary transthyretin-mediated (hATTR) amyloidosis. Orphanet J Rare Dis, 2020. 15(1): p. 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Coelho T, et al. , Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med, 2013. 369(9): p. 819–29. [DOI] [PubMed] [Google Scholar]
- 52.Kristen AV, et al. , Patisiran, an RNAi therapeutic for the treatment of hereditary transthyretin-mediated amyloidosis. Neurodegener Dis Manag, 2019. 9(1): p. 5–23. [DOI] [PubMed] [Google Scholar]
- 53.Whatley SD and Badminton MN, Acute Intermittent Porphyria, in GeneReviews((R)), Adam MP, et al. , Editors. 1993: Seattle (WA). [Google Scholar]
- 54.Anderson KE, Acute hepatic porphyrias: Current diagnosis & management. Mol Genet Metab, 2019. 128(3): p. 219–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Besur S, et al. , Clinically important features of porphyrin and heme metabolism and the porphyrias. Metabolites, 2014. 4(4): p. 977–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pallet N, et al. , Porphyria and kidney diseases. Clin Kidney J, 2018. 11(2): p. 191–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Agarwal S, et al. , Pharmacokinetics and Pharmacodynamics of the Small Interfering Ribonucleic Acid, Givosiran, in Patients With Acute Hepatic Porphyria. Clin Pharmacol Ther, 2020. 108(1): p. 63–72. [DOI] [PubMed] [Google Scholar]
- 58.Sardh E, et al. , Phase 1 Trial of an RNA Interference Therapy for Acute Intermittent Porphyria. N Engl J Med, 2019. 380(6): p. 549–558. [DOI] [PubMed] [Google Scholar]
- 59.Balwani M, et al. , Phase 3 Trial of RNAi Therapeutic Givosiran for Acute Intermittent Porphyria. N Engl J Med, 2020. 382(24): p. 2289–2301. [DOI] [PubMed] [Google Scholar]
- 60.Gonzalez-Aseguinolaza G, Givosiran - Running RNA Interference to Fight Porphyria Attacks. N Engl J Med, 2020. 382(24): p. 2366–2367. [DOI] [PubMed] [Google Scholar]
- 61.Scott LJ and Keam SJ, Lumasiran: First Approval. Drugs, 2021. [DOI] [PubMed]
- 62.Harambat J, et al. , Primary hyperoxaluria. Int J Nephrol, 2011. 2011: p. 864580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dindo M, et al. , Molecular basis of primary hyperoxaluria: clues to innovative treatments. Urolithiasis, 2019. 47(1): p. 67–78. [DOI] [PubMed] [Google Scholar]
- 64.Kletzmayr A, Ivarsson ME, and Leroux JC, Investigational Therapies for Primary Hyperoxaluria. Bioconjug Chem, 2020. 31(7): p. 1696–1707. [DOI] [PubMed] [Google Scholar]
- 65.Janas MM, et al. , The Nonclinical Safety Profile of GalNAc-conjugated RNAi Therapeutics in Subacute Studies. Toxicol Pathol, 2018. 46(7): p. 735–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Janas MM, et al. , Safety evaluation of 2’-deoxy-2’-fluoro nucleotides in GalNAc-siRNA conjugates. Nucleic Acids Res, 2019. 47(7): p. 3306–3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Foster DJ, et al. , Advanced siRNA Designs Further Improve In Vivo Performance of GalNAc-siRNA Conjugates. Mol Ther, 2018. 26(3): p. 708–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Habtemariam BA, et al. , Single Dose Pharmacokinetics and Pharmacodynamics of Transthyretin Targeting GalNAc-siRNA Conjugate, Vutrisiran, in Healthy Subjects. Clin Pharmacol Ther, 2020. [DOI] [PubMed]
- 69.Lai C, et al. , Specific Inhibition of Hepatic Lactate Dehydrogenase Reduces Oxalate Production in Mouse Models of Primary Hyperoxaluria. Mol Ther, 2018. 26(8): p. 1983–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Maningat P, Gordon BR, and Breslow JL, How do we improve patient compliance and adherence to long-term statin therapy? Curr Atheroscler Rep, 2013. 15(1): p. 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dyrbus K, et al. , Inclisiran-New hope in the management of lipid disorders? J Clin Lipidol, 2020. 14(1): p. 16–27. [DOI] [PubMed] [Google Scholar]
- 72.Khvorova A, Oligonucleotide Therapeutics - A New Class of Cholesterol-Lowering Drugs. N Engl J Med, 2017. 376(1): p. 4–7. [DOI] [PubMed] [Google Scholar]
- 73.Fitzgerald K, et al. , A Highly Durable RNAi Therapeutic Inhibitor of PCSK9. N Engl J Med, 2017. 376(1): p. 41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hovingh GK, et al. , Inclisiran Durably Lowers Low-Density Lipoprotein Cholesterol and Proprotein Convertase Subtilisin/Kexin Type 9 Expression in Homozygous Familial Hypercholesterolemia: The ORION-2 Pilot Study. Circulation, 2020. 141(22): p. 1829–1831. [DOI] [PubMed] [Google Scholar]
- 75.Raal FJ, et al. , Inclisiran for the Treatment of Heterozygous Familial Hypercholesterolemia. N Engl J Med, 2020. 382(16): p. 1520–1530. [DOI] [PubMed] [Google Scholar]
- 76.Ray KK, et al. , Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N Engl J Med, 2020. 382(16): p. 1507–1519. [DOI] [PubMed] [Google Scholar]
- 77.Sheridan C, PCSK9-gene-silencing, cholesterol-lowering drug impresses. Nat Biotechnol, 2019. 37(12): p. 1385–1387. [DOI] [PubMed] [Google Scholar]
- 78.Bamji AN, Do PCSK9 inhibitors do anything more than reduce LDL cholesterol? BMJ, 2020. 368: p. m1159. [DOI] [PubMed] [Google Scholar]
- 79.Asbeutah AAA, Asbeutah SA, and Abu-Assi MA, A Meta-Analysis of Cardiovascular Outcomes in Patients With Hypercholesterolemia Treated With Inclisiran. Am J Cardiol, 2020. 128: p. 218–219. [DOI] [PubMed] [Google Scholar]
- 80.Castaman G and Matino D, Hemophilia A and B: molecular and clinical similarities and differences. Haematologica, 2019. 104(9): p. 1702–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sorensen B, et al. , Rationale for individualizing haemophilia care. Blood Coagul Fibrinolysis, 2015. 26(8): p. 849–57. [DOI] [PubMed] [Google Scholar]
- 82.Machin N and Ragni MV, An investigational RNAi therapeutic targeting antithrombin for the treatment of hemophilia A and B. J Blood Med, 2018. 9: p. 135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sehgal A, et al. , An RNAi therapeutic targeting antithrombin to rebalance the coagulation system and promote hemostasis in hemophilia. Nat Med, 2015. 21(5): p. 492–7. [DOI] [PubMed] [Google Scholar]
- 84.Berk C, et al. , Pharmacodynamic and Pharmacokinetic Properties of Full Phosphorothioate Small Interfering RNAs for Gene Silencing In Vivo. Nucleic Acid Ther, 2020. [DOI] [PMC free article] [PubMed]
- 85.Pasi KJ, et al. , Targeting of Antithrombin in Hemophilia A or B with RNAi Therapy. N Engl J Med, 2017. 377(9): p. 819–828. [DOI] [PubMed] [Google Scholar]
- 86.Davidson BL and McCray PB Jr., Current prospects for RNA interference-based therapies. Nat Rev Genet, 2011. 12(5): p. 329–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Makris K and Spanou L, Acute Kidney Injury: Definition, Pathophysiology and Clinical Phenotypes. Clin Biochem Rev, 2016. 37(2): p. 85–98. [PMC free article] [PubMed] [Google Scholar]
- 88.Hulse M and Rosner MH, Drugs in Development for Acute Kidney Injury. Drugs, 2019. 79(8): p. 811–821. [DOI] [PubMed] [Google Scholar]
- 89.Gallagher KM, et al. , Recent early clinical drug development for acute kidney injury. Expert Opin Investig Drugs, 2017. 26(2): p. 141–154. [DOI] [PubMed] [Google Scholar]
- 90.Thompson JD, et al. , Toxicological and pharmacokinetic properties of chemically modified siRNAs targeting p53 RNA following intravenous administration. Nucleic Acid Ther, 2012. 22(4): p. 255–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Molitoris BA, et al. , siRNA targeted to p53 attenuates ischemic and cisplatin-induced acute kidney injury. J Am Soc Nephrol, 2009. 20(8): p. 1754–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gaier ED and Torun N, The enigma of nonarteritic anterior ischemic optic neuropathy: an update for the comprehensive ophthalmologist. Curr Opin Ophthalmol, 2016. 27(6): p. 498–504. [DOI] [PubMed] [Google Scholar]
- 93.Ahmed Z, et al. , Ocular neuroprotection by siRNA targeting caspase-2. Cell Death Dis, 2011. 2: p. e173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Solano EC, et al. , Toxicological and pharmacokinetic properties of QPI-1007, a chemically modified synthetic siRNA targeting caspase 2 mRNA, following intravitreal injection. Nucleic Acid Ther, 2014. 24(4): p. 258–66. [DOI] [PubMed] [Google Scholar]
- 95.Jiang J, et al. , Progress on ocular siRNA gene-silencing therapy and drug delivery systems. Fundam Clin Pharmacol, 2020. [DOI] [PubMed]
- 96.Kupersmith MJ and Miller NR, A Nonarteritic Anterior Ischemic Optic Neuropathy Clinical Trial: An Industry and NORDIC Collaboration. J Neuroophthalmol, 2016. 36(3): p. 235–7. [DOI] [PubMed] [Google Scholar]
- 97.Hayreh SS, Controversies on neuroprotection therapy in non-arteritic anterior ischaemic optic neuropathy. Br J Ophthalmol, 2020. 104(2): p. 153–156. [DOI] [PubMed] [Google Scholar]
- 98.Shimazaki J, Definition and Diagnostic Criteria of Dry Eye Disease: Historical Overview and Future Directions. Invest Ophthalmol Vis Sci, 2018. 59(14): p. DES7–DES12. [DOI] [PubMed] [Google Scholar]
- 99.Simsek C, et al. , Current Management and Treatment of Dry Eye Disease. Turk J Ophthalmol, 2018. 48(6): p. 309–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Moreno-Montanes J, Bleau AM, and Jimenez AI, Tivanisiran, a novel siRNA for the treatment of dry eye disease. Expert Opin Investig Drugs, 2018. 27(4): p. 421–426. [DOI] [PubMed] [Google Scholar]
- 101.Bereiter DA, et al. , TRPV1 and TRPM8 Channels and Nocifensive Behavior in a Rat Model for Dry Eye. Invest Ophthalmol Vis Sci, 2018. 59(8): p. 3739–3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Schoenwald RD, et al. , Penetration into the anterior chamber via the conjunctival/scleral pathway. J Ocul Pharmacol Ther, 1997. 13(1): p. 41–59. [DOI] [PubMed] [Google Scholar]
- 103.Benitez-Del-Castillo JM, et al. , Safety and Efficacy Clinical Trials for SYL1001, a Novel Short Interfering RNA for the Treatment of Dry Eye Disease. Invest Ophthalmol Vis Sci, 2016. 57(14): p. 6447–6454. [DOI] [PubMed] [Google Scholar]
- 104.Second RNAi drug approved. Nat Biotechnol, 2020. 38(4): p. 385. [DOI] [PubMed] [Google Scholar]